 Justin P. Shaffer1
Justin P. Shaffer1 Carolina Sarmiento2
Carolina Sarmiento2 Paul-Camilo Zalamea2
Paul-Camilo Zalamea2 Rachel E. Gallery3,4
Rachel E. Gallery3,4 Adam S. Davis5
Adam S. Davis5 David A. Baltrus1
David A. Baltrus1 A. Elizabeth Arnold1,4*
A. Elizabeth Arnold1,4*- 1School of Plant Sciences, University of Arizona, Tucson, AZ, USA
- 2Smithsonian Tropical Research Institute, Balboa, Panama
- 3School of Natural Resources and the Environment, University of Arizona, Tucson, AZ, USA
- 4Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ, USA
- 5Global Change and Photosynthesis Research Unit, United States Department of Agriculture - Agricultural Research Service, Urbana, IL, USA
Interactions between fungi and tropical trees help shape some of the most biodiverse communities on earth. These interactions occur in the presence of additional microbes that can modify fungal phenotypes, such as endohyphal bacteria (EHB). Here we examine the occurrence, diversity, and taxonomic composition of EHB in fungi that colonize seeds and leaves of plants in tropical forests. We use PCR and fluorescence microscopy to detect EHB in fungi, and a phylogenetic approach to explore evolutionary relationships among seed- and leaf-inhabiting fungi and their bacterial partners. Analyses focusing on two prevalent orders of fungi (Hypocreales and Xylariales) revealed that seed- and leaf-inhabiting fungi have a shared evolutionary history, yet differ in the prevalence, richness, and composition of their endohyphal symbionts. Phylogenetic analyses detected that the endohyphal habit is widespread, here encompassing members of seven phyla of bacteria (including three classes of Proteobacteria). Occurring in seed- vs. leaf-associated fungi has not resulted in detectable structure in the evolution of EHB, and no congruence was observed in the phylogenetic relationships of these apparently facultative, horizontally transmitted symbionts, and their fungal hosts. Our results are consistent with multiple origins of fungus-bacterium associations and argue for evaluating focal pairs to determine how particular EHB affect the establishment or maintenance of fungal symbioses in seeds and leaves.
Introduction
Interactions between fungi and tropical trees contribute directly to shaping some of the most biodiverse communities on earth (Arnold and Lutzoni, 2007; Zimmerman and Vitousek, 2012; Peay et al., 2013; Bagchi et al., 2014). Fungi that recruit to seeds in soil can alter seed fate by influencing secondary dispersal, dormancy, germination, and survival (Knoch et al., 1993; Gallery et al., 2007a,b; Dalling et al., 2011; Sarmiento et al., 2015). Fungi in sapwood and roots can change nutrient acquisition or transport, water transport, and tissue integrity (Blanchette, 1991; Agrios, 1997; Bonfante and Genre, 2010; Oliva et al., 2014). In leaves, fungi can alter photosynthetic rates, gas exchange, interactions with natural enemies, and growth (Pinto et al., 2000; Gilbert, 2002; Rodriguez et al., 2009; Grimmer et al., 2012). Together these interactions underlie a major component of forest dynamics and the maintenance of biological diversity in tropical plant communities (Gilbert, 2002).
Interactions of plants and fungi are influenced both by environmental factors and by the intricate processes of host-symbiont recognition (see Agrios, 1997; Schafer and Kotanen, 2003; Jones and Dangl, 2006; Kluger et al., 2008; Gallery et al., 2010). They also can be shaped by additional microbes that modify fungal phenotypes (Frey-Klett et al., 2007; Márquez et al., 2007). Plant-fungus interactions in complex, heterogeneous environments such as tropical forests may be especially subject to influence from other microbes (Frey-Klett et al., 2007; Bonfante and Anca, 2009). Such “symbio-modulatory” microbes may occur in the same microenvironments as the primary interactors, on their surfaces, or within their tissues, influencing the outcome of interactions through substrate modification, regulation of gene expression, or metabolite production (Partida-Martinez and Hertweck, 2005; Bonfante and Anca, 2009; Salvioli et al., 2010, 2016; Hoffman et al., 2013).
Recent work has shown that many plant-associated fungi harbor endosymbiotic bacteria that can alter plant-fungus interactions (Partida-Martinez and Hertweck, 2005; Partida-Martinez et al., 2007a; Salvioli et al., 2010, 2016; Hoffman et al., 2013). For example, the rice blast fungus Rhizopus microsporus (Mucoromycotina) can harbor Burkholderia rhizoxinica (Burkholderiaceae, Betaproteobacteria), which produces the virulence factor responsible for plant disease (Partida-Martinez and Hertweck, 2005; Partida-Martinez et al., 2007b). When cured of the bacterium, the fungus loses pathogenicity as well as the ability to produce asexual spores (Partida-Martinez and Hertweck, 2005; Partida-Martinez et al., 2007a). Similarly, the arbuscular mycorrhizal (AM) fungus Gigaspora margarita (Glomeromycota) can harbor the vertically transmitted bacterium Candidatus Glomeribacter gigasporarum (Burkholderiaceae) (Bianciotto et al., 1996, 2003, 2004). This bacterium enhances responsiveness to root-exuded strigolactones that influence hyphal elongation and branching in mycorrhizal establishment (Lumini et al., 2007; Anca et al., 2009). Genomic studies have shown that some bacterial endosymbionts can acquire carbon, nitrogen, and phosphorus from their fungal hosts, implying that they may compete for these often limiting resources (Salvioli et al., 2010; Ghignone et al., 2012). Such dynamics may be particularly important in environments such as nutrient-poor soils or the apoplast of living leaves (Vitousek and Sanford, 1986; Vorholt, 2012).
The ability to harbor endosymbiotic bacteria (hereafter endohyphal bacteria, EHB) appears to be phylogenetically and ecologically widespread in fungi. EHB have been observed in root-associated and soilborne fungi representing the Mucoromycotina (Rhizopus microsporus, Endogone spp.), Mortierellomycotina (Mortierella elongata), Glomeromycota (Gigaspora spp., Scutellospora spp.), Basidiomycota (Laccaria bicolor and Sebacina spp.), and Ascomycota (Tuber borchii) (Barbieri et al., 2000; Bianciotto et al., 2003; Bertaux et al., 2005; Partida-Martinez et al., 2007b; Sharma et al., 2008; Sato et al., 2010; Desirò et al., 2015). Growing evidence suggests that EHB also occur frequently in diverse foliar Ascomycota, including leaf-endophytic Eurotiomycetes, Dothideomycetes, Sordariomycetes, and Pezizomycetes (Hoffman and Arnold, 2010). One foliar endophyte that has been studied in some detail, Pestalotiopsis sp. 9143 (Xylariales, Sordariomycetes, Ascomycota), harbors an endohyphal Luteibacter sp. (Xanthomonadaceae, Gammaproteobacteria) that influences its ability to produce auxin and certain hydrolytic enzymes (Hoffman et al., 2013; Arendt, 2015).
Together these studies indicate that EHB can shape the phenotypes of plant-associated fungi with downstream effects on the fungus-plant interactions that contribute in turn to forest ecology. As a first step toward elucidating such interactions, we examined the occurrence, diversity, distribution, taxonomic placement, and evolutionary history of EHB in fungi associated with plants from lowland tropical forests. We focused specifically on seed-associated and foliar endophytic fungi, which are highly diverse functional groups that each encompass a gradient of beneficial to antagonistic interactions with seeds or leaves, respectively (Arnold et al., 2003; Arnold and Engelbrecht, 2007; Gallery et al., 2007a; Kluger et al., 2008). Here, we first determine the phylogenetic placement of these fungi and evaluate whether their use of distinctive host tissues (seeds vs. leaves) reflects divergent evolutionary histories. Upon establishing that they do not appear to be evolutionarily distinct, we next determine the frequency and diversity of EHB in representative fungal lineages. We infer the phylogenetic relationships of EHB to place these symbionts taxonomically, and then use measures of phylogenetic diversity and signal to inform their evolutionary history. Finally, we compare relationships among EHB with the phylogenetic relationships and ecological modes of their hosts.
Materials and Methods
Fungi Examined
We selected fungi isolated during previous studies from a living culture collection at the Robert L. Gilbertson Mycological Herbarium, University of Arizona, Tucson, USA (ARIZ) (Gallery et al., 2007a,b; Del Olmo-Ruiz and Arnold, 2014; Zalamea et al., 2015). We focused on two functional groups dominated by the species-rich Ascomycota: seed-associated fungi and foliar endophytic fungi. Both groups are highly diverse in lowland tropical forests, but are generally understudied with regard to their phylogenetic affinity, taxonomy, and ecological modes.
All fungi used in this study were isolated originally at Barro Colorado Island, Panama (BCI: 9° 10′N, 79° 51′W; 86 m.a.s.l.) or La Selva Biological Station, Heredia, Costa Rica (LS: 10° 26′N, 83° 59′W; 35 m.a.s.l.). Barro Colorado Island is located in a seasonally moist tropical forest (Holdridge, 1947) with an average rainfall of 2600 mm/y and a pronounced dry season from January to April (Leigh, 1999). The flora and vegetation of BCI have been described previously (Croat, 1978; Foster and Brokaw, 1982). La Selva is located in a wet tropical forest (Holdridge, 1947) with an average rainfall of 3962 mm/y (Sanford et al., 1994). The flora and vegetation of LS are described by La Flora Digital de La Selva1.
Seed-associated fungi were isolated originally from surface-sterilized seeds of trees following burial and incubation in soil in the forest understory at BCI (Supplementary Table 1). Seeds were retrieved from the soil at intervals (1–6 months following burial) and surface sterilized by sequential immersion in 95% ethanol (10 s), 0.7% sodium hypochlorite (NaClO; 2 min), and 70% ethanol (2 min). Each seed was allowed to surface-dry and cut in half under sterile conditions, and then plated on 2% malt extract agar (MEA), prepared without antibiotics, to isolate fungi from the seed interior (Gallery et al., 2007a; Sarmiento et al., 2015; Zalamea et al., 2015).
Foliar endophytic fungi were isolated originally from surface-sterilized, healthy leaves of diverse vascular plants at BCI and LS (Supplementary Table 1). Leaf pieces were washed with deionized water, patted dry, cut into small fragments (ca. 2-mm2), surface sterilized as above but using 0.525% NaClO, allowed to dry under sterile conditions, and plated on 2% MEA prepared without antibiotics (U'Ren et al., 2009; Del Olmo-Ruiz and Arnold, 2014; Del Olmo-Ruiz and Arnold, in revision; Arnold, unpublished data).
Emergent hyphae were isolated into pure culture and deposited as living vouchers at ARIZ. These vouchers were used in the present study. A diversity of fungi was obtained in culture (Gallery et al., 2007a,b; U'Ren et al., 2009; Del Olmo-Ruiz and Arnold, 2014; Del Olmo-Ruiz and Arnold, in revision; Sarmiento et al., 2015; Zalamea et al., 2015), but two genera were particularly common among isolates from seeds and leaves (putative Fusarium, Hypocreales; putative Xylaria, Xylariales). We therefore focused our work on these taxa, first using phylogenetic methods (below) to confirm the taxonomic placement and ecological diversity of isolates in each group.
Preliminary Taxonomic Placement of Fungal Strains
DNA Extraction and PCR
Total genomic DNA was extracted from axenic fungal cultures following Arnold and Lutzoni (2007) or with the Extract-N-Amp Plant Mini Kit (Sigma-Aldrich, St. Louis, MO, USA) following the manufacturer's instructions. The former is a phenol:chloroform based extraction and the latter a more rapid two-step kit that was adopted for convenience. Both methods provide comparable results (Sandberg et al., 2014). We used the forward primers ITS1F or ITS5 and reverse primers LR3 or ITS4 (10 μM) to amplify the nuclear ribosomal internal transcribed spacers and the 5.8S gene (ITS ribosomal DNA [rDNA]), and when possible, the first 600 base pairs (bp) of the large subunit (partial LSU rDNA). Fragment sizes ranged from ca. 600 (ITS rDNA) to 1200 bp (ITS rDNA–partial LSU rDNA). PCR methods followed Arnold and Lutzoni (2007) unless genomic DNA was obtained using the Extract-N-Amp kit, in which case methods followed the manufacturer's instructions.
Reactions were run on PTC-200 thermal cyclers (Bio-Rad, Hercules, CA, USA) with the following cycling parameters: 94°C for 3 min; 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min; and 72°C for 10 min. PCR products were evaluated by visualizing with SYBR Green I (Molecular Probes, Invitrogen, Carlsbad CA, USA) after electrophoresis on 1% agarose gels. Positive PCR products producing single bands were sequenced directly (see below). Negative controls included water instead of template and were always blank, indicating no contamination during the DNA extraction or amplification process.
Products producing multiple bands or displaying weak amplification were cloned with StrataClone cloning kits (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer's instructions, except that we used one-half the recommended reagent volumes for each reaction. Following blue/white screening, successful transformants were transferred to fresh plates and allowed to grow an additional 24 h to increase colony size. Eight positive clones per isolate were amplified using primers M13F(−20) and M13R(−27; numbers indicate specific primer variants) (10 μM) (Hoffman and Arnold, 2010). The cycling parameters were 94°C for 10 min; 35 cycles of 94°C for 1 min, 54°C for 1 min, and 72°C for 2 min; and 72°C for 10 min. Up to eight positive PCR products per isolate were selected for sequencing.
Positive PCR products showing one band of the appropriate length were cleaned with ExoSAP-IT (Affymetrix, Santa Clara, CA, USA) following the manufacturer's instructions. Cleaned products were diluted 1:2 with sterile water prior to sequencing.
Sequencing and Assembly
Positive products were sequenced bidirectionally with the original PCR primers (5 μM) at the University of Arizona Genetics Core (Applied Biosystems BigDye Terminator v3.1 cycle sequencing kits; Applied Biosystems AB3730XL DNA Analyzer; Foster City, CA, USA). An assembly pipeline consisting of phred and phrap (Ewing and Green, 1998; Ewing et al., 1998) driven by Chromaseq (Maddison and Maddison, 2005) in Mesquite v.2.75 (Maddison and Maddison, 2009) was used to call bases and assemble reads into contigs. Base calls were verified by manual inspection of chromatograms in Sequencher v.5.1 (Gene Codes Corp., Ann Arbor, MI, USA). ITS rDNA–partial LSU rDNA sequences were submitted to GenBank (accessions KU977534-KU978121, KU978403-KU978448, and KX530755-KX530763).
We used BLASTn comparisons with GenBank (nucleotide collection [nr/nt]) (Altschul et al., 1990) and the Ribosomal Database Project (RDP) Fungal ITS Classifier (Deshpande et al., 2015) to select 223 strains for further study. These were isolates that were tentatively identified as Fusarium, Xylaria, or close relatives of those genera. Together these fungi represented isolates from 29 species of vascular plants from BCI and LS (three species of ferns, foliage only; 26 woody angiosperms, foliage and/or seeds) (Supplementary Table 1).
Top matches in database searches are not always closest phylogenetic neighbors and consequently may be inappropriate for identification (see U'Ren et al., 2009; Koski and Golding, 2012; Porter and Golding, 2012; U'Ren et al., 2016). Therefore, we used phylogenetic analyses to clarify the taxonomic placement of these strains and the resulting phylogenies to inform the distribution of seed-associated vs. foliar endophytic habits in each focal group.
Phylogenetic Analyses of Fungal Strains
Phylogenetic Reconstructions Using ITS rDNA and ITS rDNA-Partial LSU rDNA
BLASTn- and RDP classification with parallel examination of preliminary sequence alignments led us to assemble 19 data sets that encompassed Fusarium and close relatives in the Hypocreales, and Xylaria and close relatives in the Xylariales (Supplementary Table 1). Care was taken to match the phylogenetic information provided by ITS rDNA or ITS rDNA–partial LSU rDNA with the appropriate breadth of taxon sampling in each focal data set. We delimited our taxon sampling in part by reviewing existing phylogenetic hypotheses for each group, and by evaluating the limitations imposed by ambiguous alignments of ITS rDNA at broad taxonomic levels (see Schroers, 2001; Zhang et al., 2006; O'Donnell et al., 2008; Hsieh et al., 2010; Lombard et al., 2010; Chaverri et al., 2011; Short et al., 2013; U'Ren et al., 2016).
For each dataset, we first used Muscle v3.7 (Edgar, 2004) to align sequences with well-curated references from GenBank (i.e., those from type specimens and/or published species descriptions, and with information on host, substrate, and geographic origin, if available; Supplementary Table 2). We then edited each alignment by eye in Mesquite v2.75 (Maddison and Maddison, 2009). Ambiguously aligned characters were excluded. Matrix details are listed in Supplementary Table 3 and alignments submitted were to TreeBASE2.
Maximum likelihood analyses were conducted in RAxML v.8.2.4 (Stamatakis, 2014) with bootstrap support determined on the basis of 1000 pseudoreplicates. Bayesian analyses were conducted in MrBayes v.3.2.6 (Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003) with six million generations, initiated with random trees, four chains, sampling every 1000th tree, and a burn-in of all trees with a standard deviation of split frequencies ≥ 0.01. Bayesian analyses did not converge after six million generations for the Fusarium solani species complex, so the run was extended to 30 million generations. For both ML and Bayesian analyses, we used jModelTest2 (Posada, 2008; Darriba et al., 2012) to select the appropriate model of sequence evolution. Models for each alignment are listed in Supplementary Table 3. Muscle, RAxML, Mr. Bayes, and jModelTest2 were implemented using the CIPRES Science Gateway (Miller et al., 2010).
Multilocus Analyses
Relationships remained unresolved in the analyses outlined above for two taxonomic groups: the Fusarium solani species complex (FSSC) and Gliocladiopsis. We therefore used multi-locus data to improve our inferences. For FSSC we used ITS rDNA–partial LSU rDNA, the RNA polymerase II second largest subunit gene (RPB2; 1500–1800 bp in length), and the translation elongation factor 1-alpha gene (TEF; 600–900 bp in length) following Short et al. (2014). For Gliocladiopsis we used ITS rDNA, the RNA polymerase II largest subunit gene (RPB1; 400 to 600 bp in length), and TEF following Castlebury et al. (2004) and Hirooka et al. (2013). PCR primers and cycling conditions are detailed in the Supplementary Materials and Methods. These sequences were submitted to GenBank (accessions KU978449-KU978453 [RPB1], KU978454-KU978547 [RPB2], KU978548-KU978605 [TEF]).
For each taxon, we aligned sequences with references, separately assembled and manually edited alignments, and conducted preliminary ML analyses for each gene as above. We explored topological congruence among the best ML gene trees for each taxon with the Wilcoxon signed-rank Templeton test (WT), implemented in PAUP*v.4.0a150 (Swofford, 2002). Following the exclusion of 13 isolates due to missing data (see Supplementary Table 1), we detected conflict among single-locus datasets for the FSSC (WT, p ≤ 0.05 for 2/3 comparisons). However, because the multilocus framework adopted here has been used to classify phylogenetically diverse and clinically relevant fusaria and members of the FSSC in recent studies, our dataset includes many of these previously described isolates, and ITS data did not yield well-resolved or well-supported topologies in this group, we proceeded with using all of the data in a combined analysis (see Chang et al., 2006; Zhang et al., 2006; O'Donnell et al., 2008; Short et al., 2013). For Gliocladiopsis, we included all isolates and detected no conflict among the three single-locus datasets (WT, p > 0.1 for all comparisons). Single-locus alignments and trees for the FSSC and Gliocladiopsis were submitted to TreeBASE2.
Following assessment of congruence of individual gene trees, we concatenated single-locus alignments for each taxon into a supermatrix, and carried out phylogenetic reconstructions as above while allowing for independent evolution of individual loci by specifying partitions in each analysis. Bayesian analyses did not converge after six million generations for the FSSC, so the run was extended to 30 million generations. Matrix details are listed in Supplementary Table 3, and final concatenated alignments were submitted to TreeBASE2.
Together these analyses clarified the phylogenetic placement of seed-associated fungi and foliar endophytic fungi, and showed that these functional groups have a shared evolutionary history (see below). Using this information we then structured our screening of EHB to evaluate whether bacterial endosymbionts differed in prevalence, diversity, or composition in seed-associated vs. foliar endophytic strains.
Detection of EHB
The presence or absence of bacteria in living hyphae was investigated initially using light microscopy for all fungi for which viable cultures were available. Once visual examination ruled out extrahyphal bacteria (i.e., contaminants in the medium or microbes on hyphal surfaces), we screened total genomic DNA from the fungal cultures for the bacterial 16S ribosomal RNA gene (16S rRNA).
PCR Screen for EHB
DNA extracted directly from apparently axenic fungal cultures (above) was screened for bacterial 16S rRNA through PCR using the primers 27F and 1492R following Hoffman and Arnold (2010). PCR products were visualized using gel electrophoresis as above. Positive PCR products producing single bands were prepared and sequenced directly as above. Positive products producing multiple bands or displaying weak or no amplification were cloned, and positive transformants were amplified and sequenced as above. All reads were processed as above. 16S rRNA gene sequences were submitted to GenBank (accessions KU978122-KU978402).
Live/Dead Visual Screen
Fungal cultures that were positive for bacterial 16S rRNA were examined by microscopy with the LIVE/DEAD BacLight™ Bacterial Viability Kit (Invitrogen, Carlsbad, CA, USA) following Hoffman and Arnold (2010). We examined exemplar fungi representing at least three unique bacterial 16S sequences for each of the major taxonomic groups of EHB recovered here (Supplementary Table 1).
We prepared fungal samples for visualization by removing a small piece of mycelium (≤ 2-mm2) from the growing edge of single colonies growing on 2% MEA. Fragments were aseptically transferred to glass slides containing 20 μL of 1:1:18 LIVE/DEAD stain (component A: component B: diH2O), teased apart using sterile insect mounting needles (size 00; BioQuip, Rancho Dominguez, CA, USA), covered with a coverslip, and incubated in darkness for 15 min. After incubation, we washed the mycelium by pulling sterile distilled water through the slide mounts with bibulous paper, and sealed the slides with two coats of nail polish. We used a Leica DM400B compound microscope with a 100-W mercury arc lamp for fluorescent imaging. Samples were viewed at room temperature with a Chroma Technology 35,002 filter set (480-nm excitation/520-nm emission) and 100 × APO oil objective.
Visible fluorescence of nucleic acids distinct from fungal mitochondrial or nuclear DNA, combined with the absence of extrahyphal bacteria and successful amplification of the bacterial 16S rRNA gene from fungal genomic DNA, provided evidence for the presence of viable EHB (Hoffman and Arnold, 2010; Arendt et al., 2016). We next evaluated the richness, phylogenetic placement, and composition of these EHB.
Analyses of EHB
OTU Clustering and Diversity
We used Sequencher v.5.1 (Gene Codes Corp., Ann Arbor, MI) to cluster EHB sequences at ≥97% sequence similarity in order to estimate EHB operational taxonomic units (OTUs) and calculate richness (Stackebrandt and Göbel, 1994; Kembel et al., 2014). We treated each fungal isolate as a distinct sample unit (i.e., containing an EHB community), and used the R package vegan to calculate diversity of EHB OTUs as Fisher's alpha (R Core Team, 2015; Oksanen et al., 2016). OTUs were classified tentatively by taking into account BLASTn comparisons with GenBank (16S rRNA collection; Altschul et al., 1990), the RDP Bacterial 16S Classifier (Wang et al., 2007), and the SILVA Incremental Aligner (“Search and Classify”; Pruesse et al., 2012; Quast et al., 2013). We looked for agreement among the three methods and carefully considered cases of disagreement. Results were used to frame taxon sampling for subsequent phylogenetic analyses.
Phylogenetic Reconstructions of 16S rRNA
We optimized (1) taxon sampling and (2) the taxonomic breadth appropriate for obtaining maximum resolution by assessing preliminary reconstructions at the level of bacterial phylum (or at the class level for Proteobacteria) (data not shown), and by considering published phylogenetic hypotheses for focal groups (Ludwig and Klenk, 2001; Vaneechoutte et al., 2004; Kämpfer et al., 2006; Tomitani et al., 2006; Nhung et al., 2007; Williams et al., 2007, 2010; Berrendero et al., 2011; Brady et al., 2013; Shih et al., 2013; Verma et al., 2013; Spring et al., 2015; Tank and Bryant, 2015). Overall, we generated 22 alignments that together contained 284 sequences representing EHB from our surveys. Our data sets also included 61 EHB sequences identified previously from endophytic Ascomycota (Hoffman and Arnold, 2010), 28 EHB sequences from root-associated Mucoromycotina, Mortierellomycotina, and Glomeromycota (Bianciotto et al., 2003; Partida-Martinez and Hertweck, 2005; Partida-Martinez et al., 2007a; Naumann et al., 2010; Sato et al., 2010; Lackner et al., 2011; Desirò et al., 2014, 2015), and 470 non-EHB reference sequences from GenBank representing the type or otherwise named strains with greatest homology to our unclassified EHB (Supplementary Table 4).
For each of the 22 datasets, we aligned 16S rRNA sequences with selected references from GenBank and inferred phylogenetic relationships as described above. Bayesian analyses did not converge after six million generations for Enterobacteriaceae, Pseudomonadales, and Pasteurellaceae (Gammaproteobacteria) such that these runs were extended to 20, 10, and 10 million generations, respectively. Matrix details are listed in Supplementary Table 3, and alignments were submitted to TreeBASE2.
Community Composition and Indicator Species
Treating each fungal isolate screened as a distinct sample, we evaluated whether communities of EHB differed as a function of the habit of the fungal strains from which they were isolated (i.e., seed-associated vs. foliar endophytic). We used Bray-Curtis dissimilarity, which was calculated using the relative abundances of all non-singleton EHB OTUs, and visualized results using non-metric multidimensional scaling (NMS, 999 runs). After finding a stable solution, we determined the proportion of variance explained by fungal functional group (seed-associated vs. foliar endophytic) with a post-hoc goodness-of-fit test and an independent analysis of similarity (ANOSIM, 999 permutations). Distance matrices, NMS, and ANOSIM were implemented using the R package vegan (R Core Team, 2015; Oksanen et al., 2016).
We used indicator species analysis (ISA) to explore whether certain taxa of EHB were strongly associated with the seed- or leaf-associated habit of host fungi. We first coded all non-singleton EHB OTUs by bacterial taxonomy (order or below except for the Bacteroidetes [phylum]) and the fungi in which they occur by habit (seed-associated or foliar endophytic), and then estimated the indicator value for each EHB taxon-host fungal habit combination. We assessed the significance of each indicator value by comparing with mean values obtained from a randomization test (999 permutations). Estimation of ISA parameters and indicator value significance were conducted using the R package indicspecies (De Cáceres and Legendre, 2009; R Core Team, 2015).
Analyses Based on Phylogenetic Inferences
Host Fungal Taxonomy, Host Plant Taxonomy, Geography, and the Identity of EHB
We annotated tips on bacterial phylogenies with data from original collections, GenBank, and/or published work to visualize patterns regarding fungal habit (i.e., seed-associated vs. foliar endophytic), fungal taxonomy (genus level or below), host plant species, and geography (Supplementary Tables 1, 2, 4, 5). We separately implemented multinomial logistic regression in JMP v.12.2 (SAS Institute, Cary, NC), combining those variables as predictors in an additive model to predict bacterial taxonomy at the level of phylum.
We measured congruence among the phylogenies of EHB, their fungal hosts, and the host plants from which the EHB-fungal associations were isolated. To obtain the host plant phylogeny, we used the Phylomatic tool implemented in Phylocom v.4.2 (Webb et al., 2008) to trim the Angiosperm Phylogeny Group megatree (APG III., 2009) to include only those plant lineages examined here. Cophylogeny between host plants and fungi or bacteria was assessed manually for all pairs of trees.
Ecological Modes of Fungal Hosts and EHB
We used metadata for EHB in each bacterial phylogeny to compare phylogenetic diversity (PD) based on the habits (occurring in seeds vs. leaves) and geographic origins (temperate vs. tropical) of host fungi. We further compared EHB detected in this study to those known from root-associated Mucoromycotina, Mortierellomycotina, and Glomeromycota (MMG), as the latter represent ecologically and evolutionarily distinct groups in which EHB have been well-documented (Bianciotto et al., 2004; Partida-Martinez et al., 2007a; Naumann et al., 2010; Sato et al., 2010; Desirò et al., 2015). We calculated PD for each analysis as the sum of branch lengths spanning the minimum path connecting all taxa belonging to defined groups (Vane-Wright et al., 1991; Faith, 1992; Humphries et al., 1995; Faith and Baker, 2006; Arnold et al., 2007). Because PD typically increases with sampling effort and comparisons can be misinterpreted if sample sizes of groups are not standardized, we reduced the number of sequences in each focal group to that of the group with the smallest sample size prior to PD calculations (i.e., rarefied PD; Nipperess and Matsen, 2013). We randomly selected sequences for removal during rarefaction, and performed this process three times for each comparison to obtain mean PD and standard deviation for each group. We used the R package picante for trimming trees and inferring PD (Kembel et al., 2010; R Core Team, 2015). Rarefied PD was compared between groups using Welch two-sample t-tests.
To quantify the degree to which EHB among fungi of a particular habit form phylogenetically distinct groups, we grouped EHB in each bacterial phylogeny as for PD above and calculated phylogenetic signal (Blomberg et al., 2003). Although phylogenetic signal in continuous traits can be quantified in many ways (see Gilbert and Webb, 2007; Hardy and Pavoine, 2012; Münkemüller et al., 2012), few methods are available for estimating phylogenetic signal in binary traits. We quantified phylogenetic signal using the sum of sister-clade differences (character dispersion, D, where D ≥ 1 indicates weak signal or convergent evolution and D ≤ 0 indicates strong signal; Fritz and Purvis, 2010). Strong signal implies relative phylogenetic clumping, or monophyly, and would indicate phylogenetically distinct groups. For each bacterial phylogeny, we estimated D for each group, and tested values for significant departure from a model in which traits have a phylogenetically random distribution (D = 1; 1000 permutations), and a model in which traits are clumped as if evolved by Brownian motion (D = 0; 1000 simulated walks). Analyses were conducted using the R package caper (Orm et al., 2013; R Core Team, 2015).
Results
Relationships among Fungi
We classified 223 fungal isolates representing two orders of Ascomycota (Hypocreales and Xylariales) from seeds and leaves of tropical plants into 19 currently recognized groups (Supplementary Table 1). Thirteen groups represented the majority of fungal strains (Figure 1 and Supplementary Figure 1): Allantonectria, Bionectria, Calonectria, Fusarium concolor, F. lateritium, F. “liseola,” the F. solani species complex (FSSC), and Gliocladiopsis (Hypocreales), and Hypoxylon, Entonaema, Nemania, Xylaria “PO,” and Xylaria “HY” (Xylariales). Phylogenetic analyses revealed that >10% of strains identified tentatively as Fusarium and Xylaria based on BLASTn analyses were identified incorrectly at the genus level or above (Supplementary Table 1).

Figure 1. Phylogenetic relationships among representative seed-associated and foliar endophytic fungi screened for EHB. (A) Fusarium solani species complex (FSSC, Nectriaceae) based on ITS rDNA–partial LS rDNA, RPB2, and TEF; (B) Xylaria “PO” (Xylarioideae) based on ITS rDNA. Topologies are best trees from maximum likelihood analyses. Branch support values are maximum likelihood bootstrap (MLBS; n = 1000 replicates) and Bayesian posterior probabilities (BPP; 6 million generations; burn-in of all trees with a standard deviation of split frequencies ≥0.01). Thickened branches are those with ≥70 MLBS and ≥95 BPP values. Branches lacking support values have values < 60 (MLBS) and < 80 (BPP). Taxon labels for tropical seed-associated fungi examined here are bolded and preceded by black circles, and those for tropical foliar endophytic fungi are preceded by white circles. Taxon labels for all fungi examined here include host plant species names, geographic locations, and GenBank accession numbers for top BLAST matches based on ITS rDNA. Signs indicate if EHB were detected (+) or not (−) during screening. Filled circles to the right of taxon labels indicate the presence of one of eight most common 16S OTUs, and numbers indicate the total number of unique 16S OTUs detected in each fungal isolate. Names for reference strains include hosts/substrates and geographic origins (when available) as well as GenBank accession numbers. C = Cecropia. Double- and triple-hash marks indicate branches shortened to one-half and one-quarter of their length, respectively. Named clades provide examples of the diversity and distribution of EHB OTUs among host fungi varying in ecological mode (e.g., seed-associated Fusarium keratoplasticum, foliar endophytic Xylaria cubensis), host plant species (e.g., within X. cubensis example clade A: fungi from Chrysophyllum cainito vs. other host plants), geographic location (e.g., within X. cubensis: fungi from Costa Rica vs. Panama), and close relatives (e.g., within FSSC haplotype 25: only two pairs of fungi share EHB OTUs). In general, we detected similar and diverse EHB OTUs among fungi consistent- and varying in each of the above factors.
These 13 topologies revealed that seed-associated and foliar endophytic fungi occurred together in many well-supported lineages (Figure 1 and Supplementary Figure 1). We initially treated these functional groups as distinct from one another based on their tissue of origin (seed vs. leaf; see below), but topologies in only 3 of 13 datasets suggested that they could be phylogenetically distinct (Allantonectria, Calonectria, and F. concolor, considering data for strains evaluated here and reference strains). In the remaining 10 topologies, there was no strong evidence for a distinctive evolutionary history of seed- vs. leaf-associated strains (Figure 1 and Supplementary Figure 1).
Occurrence and Diversity of EHB
EHB were detected in 167 of 223 fungal isolates (75%) (Figures 2, 3). Overall, EHB were detected in 17 of 19 fungal groups. Together these represented isolates from 27 host plant species from Panama and Costa Rica. We did not detect EHB in 46 strains (29.7%) of Hypocrealean fungi and 10 strains (14.7%) of Xylarialean fungi. Together these represented 49 strains (28.7%) of fungi isolated from seeds and seven strains (13.5%) of fungi isolated from leaves. In general, EHB were observed more frequently in fungi isolated from leaves vs. fungi isolated from seeds (Figure 3).
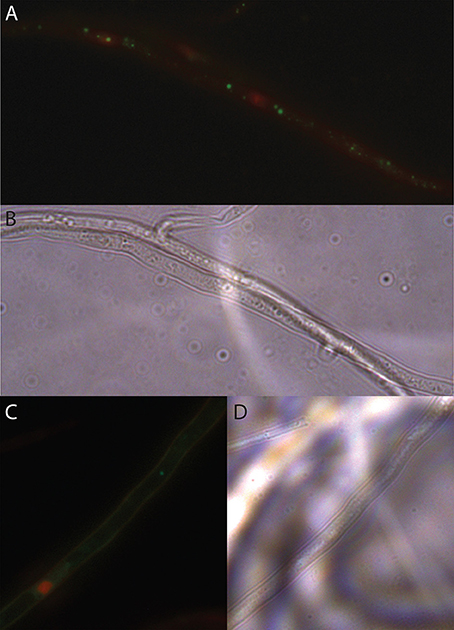
Figure 2. LIVE/DEAD staining of EHB in candidate fungi. (A) Seed-associated FSSC strain PS0362A. Photomicrograph of fluorescently tagged, viable, bacterial nucleic acids in green and compromised fungal organelles in red. Fungal mycelium was alive at the outset of the preparation process but was inactivated (B) same frame viewed with differential interference contrast (DIC). (C) Photomicrograph of foliar endophytic Fusarium concolor strain P0305, as above. (D) Same frame viewed with DIC. All images were taken at 1000 × magnification.
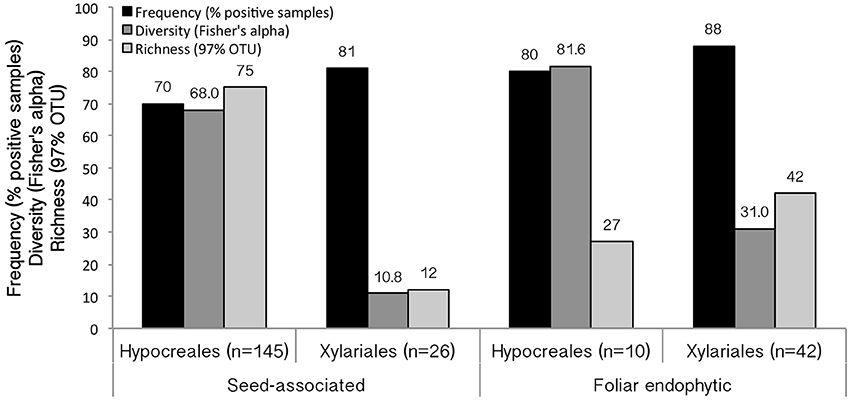
Figure 3. Results of screening for EHB among tropical seed-associated and foliar endophytic Hypocreales and Xylariales. Frequency (% of positive samples) indicates the number of fungal isolates observed to have EHB out of the total number screened. Diversity is calculated as Fisher's α. Richness indicates the number of EHB OTUs (97% 16S similarity). Samples sizes for each group are shown.
We detected 122 OTUs among a total of 284 EHB sequences (Figure 3, Supplementary Table 5). Of these, 80 OTUs were found only once (65.6%). Among the 42 OTUs found more than once, the majority were found in multiple fungal genera or species complexes (66.7%), or in fungi from multiple host plant species (66.7%). Overall, 42.9% occurred in more than one study site. In contrast, a minority (14 OTUs, 33%) were found in both seed-associated and foliar endophytic fungi.
EHB were observed more frequently among Xylariales than Hypocreales, but the overall diversity of EHB was greater among Hypocreales in both seed-associated and foliar endophytic fungi (as inferred using Fisher's alpha, which is robust to differences in sample size; Figure 3). For both orders the diversity of EHB among fungi isolated from leaves exceeded that for fungi isolated from seeds (Figure 3).
Relationships among EHB
EHB from seed-associated and foliar endophytic fungi examined here (N = 284 bacterial sequences) represented 22 alignable datasets corresponding to the Alphaproteobacteria (Rhizobiales, Rhodospirillaceae, Sphingomonadaceae), Betaproteobacteria (Burkholderiaceae, Comamonadaceae, Oxalobacteriaceae), Gammaproteobacteria (Chromatiales, Enterobacteriaceae, Pasteurellaceae, Pseudomonadales, Xanthomonadaceae), Acidobacteria (Acidobacteriaceae), Bacteroidetes, Cyanobacteria (Microchaetaceae + Rivulariaceae), Actinobacteria (Micrococcales + Streptomycetales, Micromonosporaceae, Nocardiaceae), Firmicutes (Bacillales excluding Paenibacillaceae, Lactobacillales, Negativicutes + Clostridiales, Paenibacillaceae), and Tenericutes (Mollicutes) (Figures 4, 5, and Supplementary Figure 2, Supplementary Table 5). Overall, Enterobacteriaceae (OTUs 4, 23), Burkholderiaceae (OTUs 2, 3), Rhizobiales (OTU 9), Clostridiales (OTU 19), and Comamonadaceae (OTU 10) were most common (Figure 1 and Supplementary Figure 1, Supplementary Table 5). The bacterial lineages observed here included novel taxa with regard to previously recognized EHB, as well as taxa that were closely related to but distinct from those groups (Figure 5 and Supplementary Figure 2). In total, we recovered EHB representing seven bacterial phyla, 23 orders, and 37 families (Figures 4, 5, Supplementary Figure 2, and Supplementary Table 5).
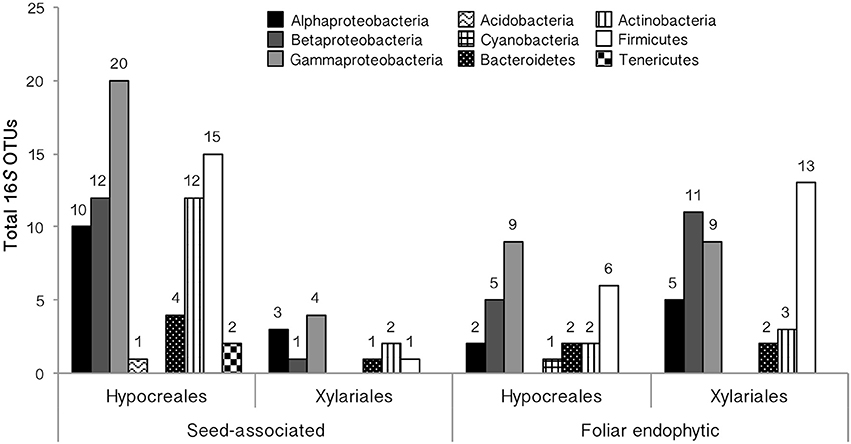
Figure 4. Taxonomic placement of EHB among tropical seed-associated and foliar endophytic Hypocreales and Xylariales. EHB OTUs are organized by phylum (or by class for Proteobacteria).
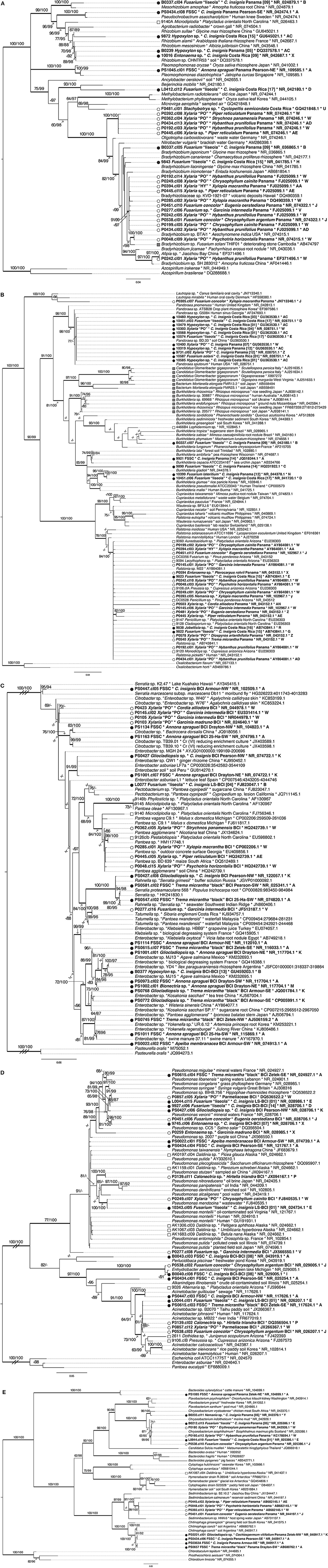
Figure 5. Phylogenetic relationships among EHB based on analysis of 16S rRNA. (A) Rhizobiales (Alphaproteobacteria); (B) Burkholderiales (Betaproteobacteria); (C) Enterobacteriaceae, (D) Pseudomonadales (Gammaproteobacteria); (E) Bacteroidetes. Topologies represent results from maximum likelihood analyses. Phylogenetic analyses and annotations of node support were carried out as for fungi. EHB from tropical seed-associated fungi are preceded by a black circle. EHB from tropical foliar endophytic fungi are preceded by a white circle. Taxon labels for all EHB observed in this study are bolded, show the host fungus, host plant, geographic origin, GenBank accession numbers for top BLAST matches, and letters indicating host fungal OTUs (95% ITS rDNA similarity). Reference EHB from temperate, foliar endophytic Ascomycota are preceded by white squares and taxon labels are as above but lack letters indicating host fungal OTUs. Reference EHB of root-associated Mucoromycotina, Mortierellomycotina, and Glomeromycota (MMG) are preceded by gray squares. Taxon labels for those latter sequences and for non-EHB references include hosts and geographic origins (when available) and GenBank accession numbers. C = Cecropia. FSSC = Fusarium solani species complex. Double- and triple-hash marks indicate branches shortened to one-half and one-quarter of their length, respectively.
We did not observe congruence among the phylogenies of EHB, their fungal hosts, and their host plants (Figure 5, Supplementary Figures 1–3). The habit of the fungal host, host fungal taxonomy, host plant species, and geographic origin (Panama vs. Costa Rica) were not significant predictors of EHB taxonomy at the phylum level ( = 1.2, p = 1.0; = 75.2, p = 1.0; = 217.2, p = 0.2; = 2.4, p = 1.0; respectively; likelihood ratio test).
EHB from seed-associated and foliar endophytic fungi often occurred together in well-supported lineages (Figure 5 and Supplementary Figure 2). However, at the OTU level, community composition of EHB differed significantly as a function of the habit of their host fungi (i.e., isolated from seed or leaf; Figure 6). Certain EHB OTU were significantly associated with either seed-associated (e.g., members of the Clostridiales) or foliar endophytic fungi (e.g., members of the Rhizobiales, Burkholderiaceae, and Bacillales) (Table 1).
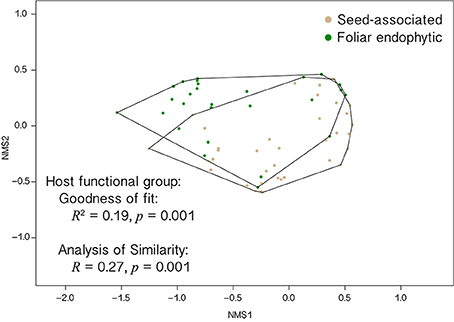
Figure 6. EHB communities differ as a function of the habit of their host fungi (seed-associated vs. foliar endophytic). Non-metric multidimensional scaling (NMS) plot shows fungi that screened positive for EHB separated by differences in their EHB community composition. Relative abundances of non-singleton EHB OTUs were used to calculate Bray-Curtis dissimilarity values among all fungi. This best solution among 999 runs has two dimensions and a stress = 0.11. Independent analyses (goodness of fit test and analysis of similarity) show that host fungal habit (seed-associated vs. foliar endophytic) is associated with significant differences in EHB community composition.
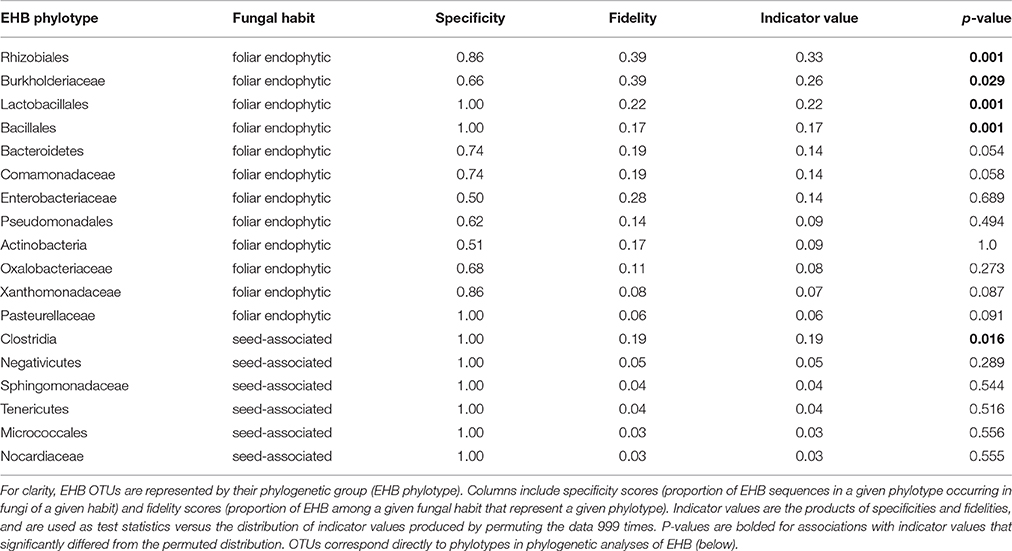
Table 1. Indicator species analysis identifies the EHB OTUs that are significantly associated with a given fungal habit (seed-associated or foliar endophytic).
Phylogenetic diversity of EHB was similar in fungi from tropical seeds vs. leaves for 5 of 11 phylogenies in which both host fungal habits were represented (rarefied PD; Table 2; see Supplementary Tables 6, 7 for additional comparisons). However, rarefied PD was significantly greater in foliar endophytic fungi for the Rhizobiales (Welch's t-test, t2 = 10.6, p = 0.01), Enterobacteriaceae (Welch's t-test, t2.38 = 4.8, p = 0.03), Pseudomonadales (Welch's t-test, t2 = 7.1, p = 0.02), Xanthomonadaceae (Welch's t-test, t2 = 6.7, p = 0.02), and Bacteroidetes (Welch's t-test, t2 = 1304.7, p < 0.00001). In contrast, phylogenetic diversity was significantly greater in seed-associated fungi for the Sphingomonadaceae (Welch's t-test, t2 = 6.1, p = 0.03).
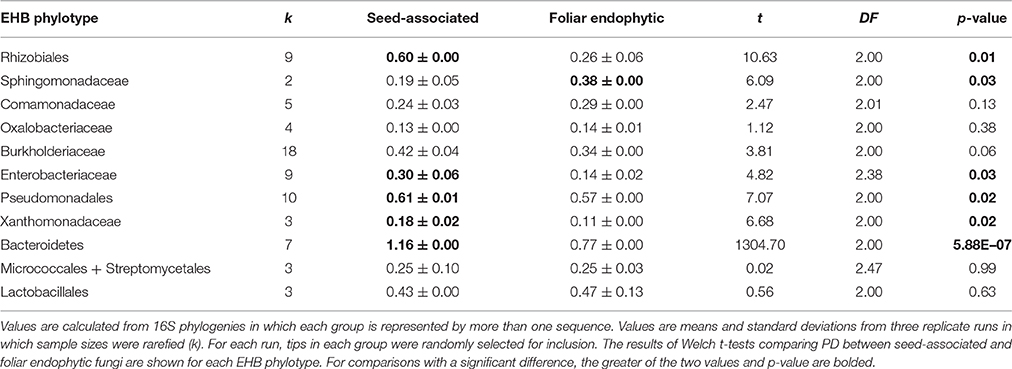
Table 2. Phylogenetic diversity (PD) among EHB of tropical seed-associated and foliar endophytic fungi.
Phylogenetic signal, defined as the degree to which EHB represent phylogenetically distinct groups relative to reference strains with different ecological modes, varied among taxonomic groups of bacteria and habits of host fungi (−2.22 ≤ D ≤ 2.99) but in general was weak (D ≥ 0 for 33 of 44 taxon-habit combinations [75%]). In general EHB were distributed across bacterial topologies in a manner that did not differ significantly from random (24 of 44 taxon-habit combinations [55%]) (Figure 5 and Supplementary Figure 2, Supplementary Table 8).
Discussion
We investigated the prevalence, diversity, composition, and phylogenetic relationships of endohyphal bacteria in two major clades of fungi, focusing on representative Hypocreales and Xylariales that colonized seeds and leaves of diverse hosts in lowland tropical forests. Our study provides a first perspective on the EHB associated with functionally distinct but closely related fungi in two major orders, and explores their diversity and phylogenetic history with respect to their host fungi and the plants in which these bacterial-fungal associations occur.
Seed-Associated and Foliar Endophytic Fungi Have a Shared Evolutionary History
Phylogenetic analyses revealed that seed-associated and foliar endophytic fungal habits were intermixed in many clades of Hypocreales and Xylariales. In general the topologies presented here are consistent with previous work, providing confidence in our assessments [e.g., Fusarium solani species complex (FSSC) agrees with O'Donnell et al. (2008) and Short et al. (2013), “Nectria” miltina, Xylaria venustula, and X. cubensis agrees with Del Olmo-Ruiz and Arnold (2014), Xylaria “HY” and Xylaria “PO” agree with Hsieh et al. (2010), and our placement of North American endophytes into Xylaria “HY,” Xylaria “PO,” Entonaema, and Nemania (Xylarioideae) agrees with U'Ren et al. (2016)] (Figure 1 and Supplementary Figure 1).
Our results agree with previous work (U'Ren et al., 2009) in suggesting an evolutionary and ecological continuity between fungi that recruit to seeds in soil and those that occur in healthy leaves. Although certain fungal taxa were represented by isolates from only one ecological mode (e.g., F. liseola and Hypoxylon, seed-associated fungi; Calonectria, F. concolor, foliar endophytic fungi), nearly all phylogenetic analyses included seed-associated or endophytic reference sequences from previous studies of forests in Panama (such that both seed-associated and foliar endophytic fungi were included; Figure 1 and Supplementary Figure 1). The majority of the remaining sequences are from fungi associated with wood or soil from other Central American, South American, or Asian tropical forests. Two exceptions are endolichenic references from boreal lichens common in all groups in the Xylarioideae, and human-associated strains in the FSSC. Future work using faster-evolving markers may detect evolutionary structure not visible in our analyses.
Endohyphal Bacteria Are Common among Seed-Associated and Foliar Endophytic Fungi
Overall, ca. 67% of EHB discovered here were observed in fungi that represented multiple phylogenetic lineages and plant hosts, and close to half were found in fungi that represented multiple geographic origins. However, only one-third were found in both seed-associated and foliar endophytic fungi, even though these fungi came from the same sites and in some cases, from the same plant species.
We speculate that differences in EHB could be linked with functional traits associated with growing in leaves vs. seeds or soil. Such differences could reflect differential selection by host fungi or distinctive host-symbiont recognition in focal pairings, potentially resulting in phylogenetic signal. However, such differences do not appear to be stable in an evolutionary sense: the broad habit of occurring in fungi that colonize seeds vs. leaves (or other plant tissues) has not resulted in detectable structure in the evolution of EHB. An exception is the EHB of root-associated Mucoromycotina, Mortierellomycotina, and Glomeromycota, which exhibited strong phylogenetic signal in all inclusive phylogenies (Figure 5B and Supplementary Figure 2Q, Supplementary Table 8).
All fungi surveyed here were isolated with a standard growth medium (2% MEA) that was not amended by antibiotics. Whether EHB influence the cultivability of fungi on certain media, and whether this could bias the isolation of fungi that differ in EHB as a function of occurring in different tissue types or hosts, remains an open question for future work.
Endohyphal Bacteria Are Horizontally Transmitted
Given that up to one third of EHB discovered here occurred in both seed-associated and foliar endophytic fungi, the question arises: where in the fungal life cycle do these infections arise? Comparisons between the phylogenies of bacteria and those of their fungal and plant hosts revealed a lack of concordance, consistent with facultative associations and horizontal transmission proposed previously for EHB of endophytic Ascomycota (Hoffman and Arnold, 2010; Arendt et al., 2016). Many species of Fusarium, Xylaria, and related fungi have a saprotrophic life phase, permitting colonization by EHB in soil or leaf litter. A route of infection from these substrates into fungi that then colonize seeds or leaves is plausible, and merits further exploration. Alternatively, it is possible that EHB infect foliar endophytes in the phyllosphere, accounting at least in part for the differences in EHB communities observed here.
The potential for horizontal transmission by EHB in Ascomycota has been verified experimentally in vitro in members of two classes (Sordariomycetes and Dothideomycetes) (Arendt et al., 2016). This is in contrast to endohyphal Burkholderiaceae observed in Rhizopus spp. and certain Glomeromycota, which can be vertically transmitted (Bianciotto et al., 2004; Partida-Martinez et al., 2007a). That these Burkholderiaceae are phylogenetically distinct from EHB observed among Ascomycota to date (Figure 5B) implies a diversity of ecological- or transmission modes in that bacterial family. More generally, whether fungi associate with bacteria prior to infecting plants or do so after co-infection may determine the degree to which EHB influence aspects of host specificity, pathogenicity, and/or ecological modes of the fungi they inhabit.
Endohyphal Bacteria Are Phylogenetically Diverse
Previous studies have documented EHB in the Rhizobiales and Sphingomonadaceae (Alphaproteobacteria), Burkholderiaceae, Comamonadaceae, and Oxalobacteriaceae (Betaproteobacteria), Enterobacteriaceae, Pseudomonadales, Xanthomonadaceae, and Pasteurellaceae (Gammaproteobacteria), Bacteroidetes, Micrococcales (Actinobacteria), Bacillales (Firmicutes), and Mollicutes (Tenericutes) (Barbieri et al., 2000; Bianciotto et al., 2003; Partida-Martinez et al., 2007b; Hoffman and Arnold, 2010; Desirò et al., 2015). Endohyphal bacteria of root-associated Mucoromycotina, Mortierellomycotina, and Glomeromycota are known only from Burkholderiaceae and Mollicutes. The bacterial lineages observed here included distinctive taxa with regard to previously recognized EHB, including Proteobacteria, Cyanobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, Firmicutes, and Tenericutes (Figures 4, 5, and Supplementary Figure 2). In addition to this taxonomic and phylogenetic diversity, EHB among these groups are related to a functionally diverse assemblage of reference bacteria. For example, four EHB of seed-associated fungi appear to be closely related to known Chitinophaga spp., some of which have been shown to have chitinolytic activity (Kämpfer et al., 2006) (Figure 5E).
In addition to contributing to the phylogenetic diversity of known groups, this work represents the first account of EHB belonging to the Rhodospirillaceae (Alphaproteobacteria), Chromatiales (Gammaproteobacteria), Microchaetaceae (Cyanobacteria), Streptomycetales, Micromonosporaceae, and Nocardiaceae (Actinobacteria), and Negativicutes, Clostridia, and Lactobacillales (Firmicutes). This phylogenetic richness and novelty was discovered among only two orders of Sordariomycetes. We anticipate that more sensitive detection methods and sampling of additional Ascomycota will likely reveal an even greater diversity of EHB. Similarly, examination of entire genomes of EHB will provide further insight with regard to relationships with known bacteria, the evolution of their symbioses with fungi (Baltrus et al., in revision), and the potential for these bacterial endosymbionts to influence the phenotypes of the fungi that, through complex and intriguing symbioses, contribute to the dynamics of tropical forests.
Author Contributions
JS characterized EHB and analyzed all data; CS, PZ, RG led isolation of fungi from seeds with collaboration from AD, AA; DB advised aspects of the bacterial analyses; JS, AA led the development of the manuscript, with contributions from all authors.
Funding
We thank the National Science Foundation (NSF DEB-1119758 to AA, NSF DEB-1120205 to James W. Dalling, NSF IOS-1354219 to DB, AA, and RG, NSF-IGERT Fellowship to JS), the Smithsonian Tropical Research Institute (STRI) (Short-term Fellowship to JS), the Mycological Society of America (Forest Fungal Ecology Award to JS), and the School of Plant Sciences (Pierson Fellowship to JS) for supporting this work. Additional support from the School of Plant Sciences and College of Agriculture and Life Sciences at the University of Arizona is gratefully acknowledged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Republic of Panama for access and the Smithsonian Tropical Research Institute for logistical support, J. Dalling for lab space, I. Quintero, D. Roche, C. Delevich, and J. Perez for field and lab assistance, J. U'Ren, K. Arendt, Y. Huang, J. DeVore, M. Shimabukuro, C. Smythe, and especially K. Garcia and A. Ndobegang for lab assistance, and J. Dalling, J. Wright, E. Leigh, G. Gilbert, and N. Zimmerman for helpful discussion. This paper represents a portion of the doctoral dissertation research of JS in Plant Pathology and Microbiology at the University of Arizona.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2016.00116
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anca, I.-A., Lumini, E., Ghignone, S., Salvioli, A., Bianciotto, V., and Bonfante, P. (2009). The ftsZ gene of the endocellular bacterium ‘Candidatus Glomeribacter gigasporarum’ is preferentially expressed during the symbiotic phases of its host mycorrhizal fungus. Mol. Plant Microbe Interact. 22, 302–310. doi: 10.194/MPMI-22-30302
APG III. (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowerng plants: APG III. Bot. J. Linn. Soc. 161, 105–121. doi: 10.1111/j.1095-8339.2009.00996.x
Arendt, K. A. (2015). Horizontally Acquired Endohyphal Bacteria Influence Enzyme Activity and Plant Decomposition by Foliar Fungi. master's thesis, University of Arizona, Tucson, AZ.
Arendt, K. A., Hockett, K. L., Araldi-Brondolo, S. J., Baltrus, D. A., and Arnold, A. E. (2016). Isolation of endohyphal bacteria from foliar Ascomycota and in vitro establishment of their symbiotic associations. Appl. Environ. Microbiol. 82, 2943–2949. doi: 10.1128/AEM.00452-16
Arnold, A. E., and Engelbrecht, B. M. J. (2007). Fungal endophytes nearly double minimum leaf conductance in seedlings of a neotropical tree species. J. Trop. Ecol. 23, 369–372. doi: 10.1017/S0266467407004038
Arnold, A. E., Henk, D. A., Eells, R. L., Lutzoni, F., and Vilgalys, R. (2007). Diversity and phylogenetic affinities of foliar fungal endophytes in loblolly pine inferred by culturing and environmental PCR. Mycologia 99, 185–206. doi: 10.3852/mycologia.99.2.185
Arnold, A. E., and Lutzoni, F. (2007). Diversity and host range of foliar fungal endophytes: are tropical leaves biodiversity hotspots? Ecology 88, 541–549. doi: 10.1890/05-1459
Arnold, A. E., Mejía, L. C., Kyllo, D., Royas, E. I., Maynard, Z., Robbins, N., et al. (2003). Fungal endophytes limit pathogen damage in a tropical tree. Proc. Natl. Acad. Sci. U.S.A. 26, 15649–15654. doi: 10.1073/pnas.2533483100
Bagchi, R., Gallery, R. E., Gripenberg, S., Gurr, S. J., Narayan, L., Addis, C. E., et al. (2014). Pathogens and insect herbivores drive rainforest plant diversity and composition. Nature 506, 85–88. doi: 10.1038/nature12911
Barbieri, E., Potenza, L., Rossi, I., Sisti, D., Giomaro, G., Rossetti, S., et al. (2000). Phylogenetic characterization and in situ detection of Cytophaga-Flexibacter-Bacteroides phylogroup bacterium in Tuber borchii Vittad. ectomycorrhizal mycelium Appl. Environ. Microbiol. 66, 5035–5042. doi: 10.1128/AEM.66.11.5035-5042.2000
Berrendero, E., Perona, E., and Mateo, P. (2011). Phenotypic variability and phylogenetic relationships of the genera Tolypothrix and Calothrix (Nostocales, Cyanobacteria) from running water. Int. J. Syst. Evol. Microbiol. 61, 3039–3051. doi: 10.1099/ijs.0.027581-0
Bertaux, J., Schmid, M., Hutzler, P., Hartmann, A., Garbaye, J., and Frey-Klett, P. (2005). Occurrence and distribution of endobacterial in the plant-associated mycelium of the ectomycorrhizal fungus Laccaria bicolor S238N. Environ. Microbiol. 7, 1786–1795. doi: 10.1111/j.1462-2920.2005.00867.x
Bianciotto, V., Bandi, C., Minerdi, D., Sironi, M., Tichy, H. V., and Bonfante, P. (1996). An obligately endosymbiotic mycorrhizal fungus itself harbors obligately intracellular bacteria. Appl. Environ. Microbiol. 62, 3005–3010.
Bianciotto, V., Genre, A., Jargeat, P., Lumini, E., Bécard, G., and Bonfante, P. (2004). Vertical transmission of endobacteria in the arbuscular mycorrhizal fungus Gigaspora margarita through generation of vegetative spores. Appl. Environ. Microbiol. 70, 3600–3608. doi: 10.1128/AEM.70.6.3600-3608.2004
Bianciotto, V., Lumini, E., Bonfante, P., and Vandamme, P. (2003). ‘Candidatus Glomeribacter gigasporarum’ gen. nov., sp. nov., an endosymbiont of arbuscular mycorrhizal fungi. Int. J. Syst. Evol. Microbiol. 53, 121–124. doi: 10.1099/ijs.0.02382-0
Blanchette, R. A. (1991). Delignification by wood-decay fungi. Annu. Rev. Phytopathol. 29, 381–398. doi: 10.1146/annurev.py.29.090191.002121
Blomberg, S. P., Garland, T. Jr., and Ives, A. R. (2003). Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745. doi: 10.1111/j.0014-3820.2003.tb00285.x
Bonfante, P., and Anca, I.-A. (2009). Plants, mycorrhizal fungi, and bacteria: a network of interactions. Ann. Rev. Microbiol. 63, 363–383. doi: 10.1146/annurev.micro.091208.073504
Bonfante, P., and Genre, A. (2010). Mechanisms underlying beneficial plant-fungus interactions in mycorrhizal symbiosis. Nat. Commun. 1, 48. doi: 10.1038/ncomms1046
Brady, C., Cleenwerck, I., Venter, S., Coutinho, T., and De Vos, P. (2013). Taxonomic evaluation of the genus Enterobacter based on multilocus sequence analysis (MLSA): proposal to reclassify E. nimipressuralis and E. amnigenus into Lelliottia gen. nov. as Lelliottia nimipressuralis comb. nov. and Lelliottia amnigena comb. nov., respectively, E. gergoviae and E. pyrinus into Pluralibacter gen. nov. as Pluralibacter gergoviae comb. nov. and Pluralibacter pyrinus comb. nov., respectively, E. cowanii, E. radicincitans, E. oryzae and E. arachidis into Kosakonia gen. nov. as Kosakonia cowanii comb. nov., Kosakonia radicincitans comb. nov., Kosakonia oryzae comb. nov. and Kosakonia arachidis comb. nov., respectively, and E. turicensis, E. helveticus and E. pulveris into Cronobacter as Cronobacter zurichensis nom. nov., Cronobacter helveticus comb. nov. and Cronobacter pulveris comb. nov., respectively, and emended description of the genera Enterobacter and Cronobacter. Syst. Appl. Microbiol. 36, 309–319. doi: 10.1016/j.syapm.2013.03.005
Castlebury, L. A., Rossman, A. Y., Sung, G.-H., Hyten, A. S., and Spatafora, J. W. (2004). Multigene phylogeny reveals new lineage for Stachybotrys chartarum, the indoor air fungus. Mycol. Res. 108, 864–872. doi: 10.1017/S0953756204000607
Chang, D. C., Grant, G. B., O'Donnell, K., Wannemuehler, K. A., Noble-Wang, J., Rao, C. Y., et al. (2006). Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. JAMA 296, 953–963. doi: 10.1001/jama.296.8.953
Chaverri, P., Salgado, C., Hirooka, Y., Rossman, A. Y., and Samuels, G. J. (2011). Delimitation of Neonectria and Cylindrocarpon (Nectriaceae, Hypocreales, Ascomycota) and related genera with Cylindrocarpon-like anamorphs. Stud. Mycol. 68, 57–78. doi: 10.3114/sim.2011.68.03
Dalling, J. W., Davis, A. S., Schutte, B. J., and Arnold, A. E. (2011). Seed survival in soil: interacting effects of predation, dormancy and the soil microbial community. J. Ecol. 99, 89–95. doi: 10.1111/j.1365-2745.2010.01739.x
Darriba, D., Toboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Del Olmo-Ruiz, M., and Arnold, A. E. (2014). Interannual variation and host affiliations of endophytic fungi associated with ferns at La Selva, Costa Rica. Mycologia 106, 8–21. doi: 10.3852/13-098
Deshpande, V., Wang, Q., Greenfield, P., Charleston, M., Porras-Alfaro, A., Kuske, C. R., et al. (2015). Fungal identification using a Bayesian classifier and the Warcup training set of internal transcribed spacer sequences. Mycologia 108, 1–5. doi: 10.3852/14-293
Desirò, A., Faccio, A., Kaech, A., Bidartondo, M. I., and Bonfante, P. (2015). Endogone, one of the oldest plant-associated fungi, host unique Mollicutes-related endobacteria. New Phytol. 205, 1464–1472. doi: 10.1111/nph.13136
Desirò, A., Salvioli, A., Ngonkeu, E. L., Mondo, S. J., Epis, S., Faccio, A., et al. (2014). Detection of a novel intracellular microbiome hosted in arbuscular mycorrhizal fungi. ISME J. 8, 257–270. doi: 10.1038/ismej.2013.151
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ewing, B., and Green, P. (1998). Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8, 186–194. doi: 10.1101/gr.8.3.186
Ewing, B., Hillier, L., Wendl, M., and Green, P. (1998). Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Faith, D. P. (1992). Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. doi: 10.1016/0006-3207(92)91201-3
Faith, D. P., and Baker, M. (2006). Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol. Bioinform. 2, 121–128.
Foster, R. B., and Brokaw, N. V. L. (1982). “Structure and history of vegetation of Barro Colorado Island,” in The Ecology of a Tropical Forest, eds A. Rand and E. G. Leigh Jr. (Washington DC: Smithsonian Institution Press), 67–81.
Frey-Klett, P., Garbaye, J., and Tarkka, M. (2007). The mycorrhiza helper bacteria revisited. New Phytol. 176, 22–36. doi: 10.1111/j.1469-8137.2007.02191.x
Fritz, S. A., and Purvis, A. (2010). Selectivity in mammalian extinction risk and threat types: a new measure of phylogenetic signal strength in binary traits. Conserv. Biol. 24, 1042–1051. doi: 10.1111/j.1523-1739.2010.01455.x
Gallery, R. E., Dalling, J. W., and Arnold, A. E. (2007a). Diversity, host affinity, and distribution of seed-infecting fungi: a case study with Cecropia. Ecology 88, 582–588. doi: 10.1890/05-1207
Gallery, R. E., Dalling, J. W., Wolf, B., and Arnold, A. E. (2007b). “Role of seed-infecting fungi in the recruitment limitation of neotropical pioneer species,” in Seed Dispersal: Theory and its Application in a Changing World, eds A. Dennis, R. Green, E. Schupp, and D. Westcott (Wallingford, CT: CABI Press), 479–498.
Gallery, R. E., Moore, D. J. P., and Dalling, J. W. (2010). Interspecific variation in susceptibility to fungal pathogens in seeds of 10 tree species in the neotropical genus Cecropia. J. Ecol. 98, 147–155. doi: 10.1111/j.1365-2745.2009.01589.x
Ghignone, S., Salvioli, A., Anca, I.-A., Lumini, E., Ortu, G., Petiti, L., et al. (2012). The genome of the obligate endobacterium of an AM fungus reveals an interphylum network of nutritional interactions. ISME J. 6, 136–145. doi: 10.1038/ismej.2011.110
Gilbert, G. S. (2002). Evolutionary ecology of plant diseases in natural ecosystems. Annu. Rev. Phytopathol. 40, 13–43. doi: 10.1146/annurev.phyto.40.021202.110417
Gilbert, G. S., and Webb, C. O. (2007). Phylogenetic signal in plant pathogen-host range. Proc. Natl. Acad. Sci. U.S.A. 104, 4979–4983. doi: 10.1073/pnas.0607968104
Grimmer, M. K., Foulkes, M. J., and Paveley, N. D. (2012). Foliar pathogenesis and plant water relations: a review. J. Exp. Bot. 63, 4321–4331. doi: 10.1093/jxb/ers143
Hardy, O. J., and Pavoine, S. (2012). Assessing phylogenetic signal with measurement error: a comparison of Mantel tests, Blomberg et al.'s K and phylogenetic distograms. Evolution 66, 2614–2621. doi: 10.1111/j.1558-5646.2012.01623.x
Hirooka, Y., Rossman, A. Y., Zhuang, W. Y., Salgado-Salazar, C., and Chaverri, P. (2013). Species delimitation for Neonectria coccinea group including the causal agents of beech bark disease in Asia, Europe, and North America. Mycosystems 32, 485–517.
Hoffman, M. T., and Arnold, A. E. (2010). Diverse bacteria inhabit living hyphae of phylogenetically diverse fungal endophytes. Appl. Environ. Microbiol. 76, 4063–4075. doi: 10.1128/AEM.02928-09
Hoffman, M. T., Gunatilaka, M. K., Wijeratne, K., Gunatilaka, L., and Arnold, A. E. (2013). Endohyphal bacterium enhances production of indole-3-acetic acid by a foliar fungal endophyte. PLoS ONE 8:e73132. doi: 10.1371/journal.pone.0073132
Holdridge, L. R. (1947). Determination of world plant formations from simple climatic data. Science 105, 367–368. doi: 10.1126/science.105.2727.367
Hsieh, H.-M., Lin, C.-R., Fang, M.-J., Rogers, J. D., Fournier, J., Lechat, C., et al. (2010). Phylogenetic status of Xylaria subgenus Pseudoxylaria among taxa of the subfamily Xylarioideae (Xylariaceae) and phylogeny of the taxa involved in the subfamily. Mol. Phylogenet. Evol. 54, 957–969. doi: 10.1016/j.ympev.2009.12.015
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Humphries, C. J., Williams, P. H., and Vane-Wright, R. I. (1995). Measuring biodiversity value for conservation. Annu. Rev. Ecol. Syst. 26, 93–111. doi: 10.1146/annurev.es.26.110195.000521
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kämpfer, P., Young, C.-C., Sridhar, K. R., Arun, A. B., Lai, W. A., Shen, F. T., et al. (2006). Transfer of [Flexibacter] sancti, [Flexibacter] filiformis, [Flexibacter] japonensis and [Cytophaga] arvensicola to the genus Chitinophaga and description of Chitinophaga skermanii sp. nov. Int. J. Syst. Evol. Microbiol. 56, 2223–2228. doi: 10.1099/ijs.0.64359-0
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kembel, S. W., O'Connor, T. K., Arnold, H. K., Hubbell, S. P., Wright, S. J., and Green, J. L. (2014). Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc. Natl. Acad. Sci. U.S.A. 111, 13715–13720. doi: 10.1073/pnas.1216057111
Kluger, C. G., Dalling, J. W., Gallery, R. E., Sanchez, E., Weeks-Galindo, C., and Arnold, A. E. (2008). Host generalists dominate fungal communities associated with four neotropical pioneer species. J. Trop. Ecol. 24, 351–354. doi: 10.1017/S0266467408005026
Knoch, T. R., Faeth, S. H., and Arnott, D. L. (1993). Endophytic fungi alter foraging and dispersal by desert seed-harvesting ants. Oecologia 95, 470–473. doi: 10.1007/BF00317429
Koski, L. B., and Golding, G. B. (2012). The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 52, 540–542. doi: 10.1007/s002390010184
Lackner, G., Moebius, N., Partida-Martinez, L., and Hertweck, C. (2011). Complete genome sequence of Burkholderia rhizoxinica, an endosymbiont of Rhizopus microsporus. J. Bacteriol. 193, 783–784. doi: 10.1128/JB.01318-10
Leigh, E. G. Jr. (1999). Tropical Forest Ecology: A View from Barro Colorado Island. New York, NY: Oxford University Press.
Lombard, L., Zhou, X. D., Crous, P. W., Wingfield, B. D., and Wingfield, M. J. (2010). Calonectria species associated with cutting rot of Eucalyptus. Persoonia 24, 1–11. doi: 10.3767/003158510X486568
Ludwig, W., and Klenk, H. P. (2001). “Overview: a phylogenetic backbone and taxonomic framework for procaryotic systematics,” in Bergey's Manual of Systematic Bacteriology 2nd Edn, Vol. 2, Part. A, eds D. J. Brenner, N. R. Krieg, and J. T. Staley (New York, NY: Springer), 49–66.
Lumini, E., Bianciotto, V., Jargeat, P., Novero, M., Salvioli, A., Faccio, A., et al. (2007). Presymbiotic growth and sporal morphology are affected in the arbuscular mycorrhizal fungus Gigaspora margarita cured of its endobacteria. Cell. Microbiol. 9, 1716–1729. doi: 10.1111/j.1462-5822.2007.00907.x
Maddison, D. R., and Maddison, W. P. (2005). ChromaSeq Module. Mesquite: a Modular System for Evolutionary Analysis. Version 1.06. Available online at: http://mesquiteproject.org/
Maddison, W. P., and Maddison, D. R. (2009). Mesquite: a Modular System for Evolutionary Analysis. Version 2.6. Available online at: http://mesquiteproject.org/
Márquez, L. M., Redman, R. S., Rodriguez, R. J., and Roossink, M. L. (2007). A virus in a fungus in a plant: three-way symbiosis required for thermal tolerance. Science 315, 513–515. doi: 10.1126/science.1136237
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE) (New Orleans, LA), 1–8.
Münkemüller, T., Lavergne, S., Bzeznik, B., Dray, S., Jombart, T., Schiffers, K., et al. (2012). How to measure and test phylogenetic signal. Methods Ecol. Evol. 3, 743–756. doi: 10.1111/j.2041-210X.2012.00196.x
Naumann, M., Schüßler, A., and Bonfante, P. (2010). The obligate endobacteria of arbuscular mycorrhizal fungi are ancient heritable components related to the Mollicutes. ISME J. 4, 862–871. doi: 10.1038/ismej.2010.21
Nhung, P. H., Ohkusu, K., Mishima, N., Noda, M., Shah, M. M., Sun, X., et al. (2007). Phylogeny and species identification of the family Enterobacteriaceae based on dnaJ sequences. Diagn. Microbiol. Infect. Dis. 58, 153–161. doi: 10.1016/j.diagmicrobio.2006.12.019
Nipperess, D. A., and Matsen, F. A. IV. (2013). The mean and variance of phylogenetic diversity under rarefaction. Methods Ecol. Evol. 4, 566–572. doi: 10.1111/2041-210X.12042
O'Donnell, K., Sutton, D. A., Fothergill, A., McCarthy, D., Rinaldi, M. G., Brandt, M. E., et al. (2008). Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J. Clin. Microbiol. 46, 2477–2490. doi: 10.1128/JCM.02371-07
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., et al. (2016). Vegan: Community Ecology Package. R Package Version 2.3-3. Available online at: http://CRAN.R-project.org/package=vegan
Oliva, J., Stenlid, J., and Martínez-Vilalta, J. (2014). The effect of fungal pathogens on the water and carbon economy of trees: implications for drought-induced mortality. New Phytol. 203, 1028–1035. doi: 10.1111/nph.12857
Orm, D., Freckleton, R., Thomas, G., Petzoldt, T., Frizt, S., Isaac, N., et al. (2013). Caper: comparative analyses of phylogenetics and evolution in R. R package version 0.5.2. Available online at: http://CRAN.R-project.org/package=caper
Partida-Martinez, L. P., Groth, I., Schmitt, I., Richter, W., Roth, M., and Hertweck, C. (2007b). Burkholderia rhizoxinica sp. nov. and Burkholderia endofungorum sp. nov., bacterial endosymbionts of the plant-pathogenic fungus Rhizopus microsporus. Int. J. Syst. Evol. Microbiol. 57, 2583–2590. doi: 10.1099/ijs.0.64660-0
Partida-Martinez, L. P., and Hertweck, C. (2005). Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 437, 884–888. doi: 10.1038/nature03997
Partida-Martinez, L. P., Monajembashi, S., Greulich, K.-O., and Hertweck, C. (2007a). Endosymbiont-dependent host reproduction maintains bacterial-fungal mutualism. Curr. Biol. 17, 773–777. doi: 10.1016/j.cub.2007.03.039
Peay, K. G., Baraloto, C., and Fine, P. V. A. (2013). Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J. 7, 1852–1861. doi: 10.1038/ismej.2013.66
Pinto, L. S. R. C., Azevedo, J. L., Pereira, J. O., Vieira, M. L. C., and Labate, C. A. (2000). Symptomless infection of banana and maize by endophytic fungi impairs photosynthetic efficiency. New Phytol. 147, 609–615. doi: 10.1046/j.1469-8137.2000.00722.x
Porter, T. M., and Golding, G. B. (2012). Factors that affect large subunit ribosomal DNA amplicon sequencing studies of fungal communities: classification method, primer choice, and error. PLoS ONE 7:e35749. doi: 10.1371/journal.pone.0035749
Posada, D. (2008). jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rodriguez, R. J., White, J. F. Jr., Arnold, A. E., and Redman, R. S. (2009). Fungal endophytes: diversity and functional roles. New Phytol. 182, 314–330. doi: 10.1111/j.1469-8137.2009.02773.x
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Salvioli, A., Chiapello, M., Fontaine, J., Hadj-Sahraoui, A. L., Grandmougin-Ferjani, A., Lanfranco, L., et al. (2010). Endobacteria affect the metabolic profile of their host Gigaspora margarita, an arbuscular mycorrhizal fungus. Environ. Microbiol. 12, 2083–2095. doi: 10.1111/j.1462-2920.2010.02246.x
Salvioli, A., Ghignone, S., Novero, M., Navazio, L., Venice, F., Bagnaresi, P., et al. (2016). Symbiosis with an endobacterium increases the fitness of a mycorrhizal fungus, raising its bioenergetic potential. ISME J. 10, 130–144. doi: 10.1038/ismej.2015.91
Sandberg, D. C., Battista, L. J., and Arnold, A. E. (2014). Fungal endophytes of aquatic macrophytes: diverse host-generalists characterized by tissue preferences and geographic structure. Microb. Ecol. 67, 735–747. doi: 10.1007/s00248-013-0324-y
Sanford, R. L., Paaby, P., Luvall, J. C., and Phillips, E. (1994). “Climate, geomorphology and aquatic systems,” in La selva: Ecology and Natural History of a Neotropical Rainforest, eds L. A. McDade, K. S. Bawa, H. A. Hespenheide, and G. S. Hartshorn (Chicago, IL: University of Chicago Press), 19–33.
Sarmiento, C., Zalamea, P.-C., Dalling, J. W., Davis, A. S., and Arnold, A. E. (2015). “Seed-associated fungi: effects on seed survival and germination of tropical pioneer species,” Association for Tropical Biology and Conservation Annual Meeting (Hawaii, HI).
Sato, Y., Narisawa, K., Tsuruta, K., Umezu, M., Nishizawa, T., Tanaka, K., et al. (2010). Detection of Betaproteobacteria inside the mycelium of the fungus Mortierella elongata. Microbes Environ. 25, 321–324. doi: 10.1264/jsme2.ME10134
Schafer, M., and Kotanen, P. M. (2003). The influence of soil moisture on losses of buried seeds to fungi. Acta Oecologica 24, 255–263. doi: 10.1016/j.actao.2003.09.001
Schroers, H.-J. (2001). A monograph of Bionectria (Ascomycota, Hypocreales, Bionectriaceae) and its Clonostachys anamorphs. Stud. Mycol. 46, 1–96. doi: 10.1017/S0269915X03272177
Sharma, M., Schmid, M., Rothballer, M., Hause, G., Zucarro, A., Imanl, J., et al. (2008). Detection and identification of bacteria intimately associated with fungi in the order Sebacinales. Cell. Microbiol. 10, 2235–2246. doi: 10.1111/j.1462-5822.2008.01202.x
Shih, P. M., Wu, D., Latifi, A., Axen, S. D., Fewer, D. P., Talla, E., et al. (2013). Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 1053–1058. doi: 10.1073/pnas.1217107110
Short, D. P. G., O'Donnell, K., and Geiser, D. M. (2014). Clonality, recombination, and hybridization in the plumbing-inhabiting human pathogen Fusarium keratoplasticum inferred from multilocus sequence typing. BMC Evol. Biol. 14:91. doi: 10.1186/1471-2148-14-91
Short, D. P. G., O'Donnell, K., Thrane, U., Nielson, K. F., Zhang, N., Juba, J. H., et al. (2013). Phylogenetic relationships among members of the Fusarium solani species complex in human infections and the descriptions of F. keratoplasticum sp. nov. and F. petroliphilum stat. nov. Fungal Genet. Biol. 53, 59–70. doi: 10.1016/j.fgb.2013.01.004
Spring, S., Scheuner, C., Göker, M., and Klenk, H.-P. (2015). A taxonomic framework for emerging groups of ecologically important marine gammaproteobacteria based on the reconstruction of evolutionary relationships using genome-scale data. Front. Microbiol. 6:281. doi: 10.3389/fmicb.2015.00281
Stackebrandt, E., and Göbel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Swofford, D. L. (2002). PAUP*. Phylogenetic Analysis Using Parsimony (*and other Methods). Version 4. Sunderland, MA: Sinauer Associates.
Tank, M., and Bryant, D. A. (2015). Chloracidobacterium thermophilum gen. nov., sp. nov.: an anoxygenic microaerophilic chlorophotoheterotrophic acidobacterium. Int. J. Syst. Evol. Microbiol. 65, 1426–1430. doi: 10.1099/ijs.0.000113
Tomitani, A., Knoll, A. H., Cavanaugh, C. M., and Ohno, T. (2006). The evolutionary diversification of cyanobacteria: Molecular-phylogenetic and paleontological perspectives. Proc. Natl. Acad. Sci. U.S.A. 103, 5442–5447. doi: 10.1073/pnas.0600999103
U'Ren, J. M., Dalling, J. W., Gallery, R. E., Maddison, D. R., Davis, E. C., Gibson, C. M., et al. (2009). Diversity and evolutionary origins of fungi associated with seeds of a neotropical pioneer tree: a case study for analyzing fungal environmental samples. Mycol. Res. 113, 432–449. doi: 10.1016/j.mycres.2008.11.015
U'Ren, J. M., Miadlikowska, J., Zimmerman, N. B., Lutzoni, F., Stajich, J. E., and Arnold, A. E. (2016). Contributions of North American endophytes to the phylogeny, ecology, and taxonomy of Xylariaceae (Sordariomycetes, Ascomycota). Mol. Phylogenet. Evol. 98, 210–232. doi: 10.1016/j.ympev.2016.02.010
Vaneechoutte, M., Kämpfer, P., De Baere, T., Falsen, E., and Verschraegen, G. (2004). Wautersia gen. nov., a novel genus accommodating the phylogenetic lineage including Ralstonia eutropha and related species, and proposal of Ralstonia [Pseudomonas] syzygii (Roberts et al. 1990) comb. nov. Int. J. Syst. Evol. Microbiol. 54(Pt 2), 317–327. doi: 10.1099/ijs.0.02754-0
Vane-Wright, R. I., Humphries, C. J., and Williams, P. H. (1991). What to protect? - Systematics and the agony of choice. Biol. Conserv. 55, 235–254. doi: 10.1016/0006-3207(91)90030-D
Verma, M., Lal, D., Kaur, J., Saxena, A., Kaur, J., Anand, S., et al. (2013). Phylogenetic analysis of phylum Actinobacteria based on whole genome sequences. Res. Microbiol. 164, 718–728. doi: 10.1016/j.resmic.2013.04.002
Vitousek, P. M., and Sanford, R. L. Jr. (1986). Nutrient cycling in moist tropical forest. Ann. Rev. Ecol. Syst. 17, 137–167. doi: 10.1146/annurev.es.17.110186.001033
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Webb, C. O., Ackerly, D. D., and Kembel, S. W. (2008). Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 24, 2098–2100. doi: 10.1093/bioinformatics/btn358
Williams, K. P., Gillespie, J. J., Sobral, B. W. S., Nordberg, E. K., Snyder, E. E., Shallom, J. M., et al. (2010). Phylogeny of Gammaproteobacteria. J. Bacteriol. 192, 2305–2314. doi: 10.1128/JB.01480-09
Williams, K. P., Sobral, B. W., and Dickerman, A. W. (2007). A robust species tree for the Alphaproteobacteria. J. Bacteriol. 189, 4578–4586. doi: 10.1128/JB.00269-07
Zalamea, P.-C., Sarmiento, C., Arnold, A. E., Davis, A. S., and Dalling, J. W. (2015). Do soil microbes and abrasion by soil particles influence persistence and loss of physical dormancy in seeds of tropical pioneers? Front. Plant Sci. 5:799. doi: 10.3389/fpls.2014.00799
Zhang, N., O'Donnell, K., Sutton, D. A., Nalim, F. A., Summerbell, R. C., Padhye, A. A., et al. (2006). Members of the Fusarium solani species complex that cause infections in both humans and plants are common in the environment. J. Clin. Microbiol. 44, 2186–2190. doi: 10.1128/JCM.00120-06
Keywords: Ascomycota, Barro Colorado Island, Cecropia, endobacteria, phylogenetic diversity, pioneers, symbiosis
Citation: Shaffer JP, Sarmiento C, Zalamea P-C, Gallery RE, Davis AS, Baltrus DA and Arnold AE (2016) Diversity, Specificity, and Phylogenetic Relationships of Endohyphal Bacteria in Fungi That Inhabit Tropical Seeds and Leaves. Front. Ecol. Evol. 4:116. doi: 10.3389/fevo.2016.00116
Received: 20 July 2016; Accepted: 20 September 2016;
Published: 13 October 2016.
Edited by:
Jeffrey Peter Townsend, Harvard University, USAReviewed by:
Donald Matthew Walker, Tennessee Technological University, USAJoão Alencar Pamphile, Universidade Estadual de Maringá, Brazil
Copyright © 2016 Shaffer, Sarmiento, Zalamea, Gallery, Davis, Baltrus and Arnold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Elizabeth Arnold, QXJub2xkQGFnLmFyaXpvbmEuZWR1