Abstract
The location of the immediate eastern Polynesian origin for the settlement of Easter Island (Rapa Nui), remains unclear with conflicting archeological and linguistic evidence. Previous genetic commensal research using the Pacific rat, Rattus exulans; a species transported by humans across Remote Oceania and throughout the Polynesian Triangle, has identified broad interaction spheres across the region. However, there has been limited success in distinguishing finer-scale movements between Remote Oceanic islands as the same mitochondrial control region haplotype has been identified in the majority of ancient rat specimens. To improve molecular resolution and identify a pattern of prehistoric dispersal to Easter Island, we sequenced complete mitochondrial genomes from ancient Pacific rat specimens obtained from early archeological contexts across West and East Polynesia. Ancient Polynesian rat haplotypes are closely related and reflect the widely supported scenario of a central East Polynesian homeland region from which eastern expansion occurred. An Easter Island and Tubuai (Austral Islands) grouping of related haplotypes suggests that both islands were established by the same colonization wave, proposed to have originated in the central homeland region before dispersing through the south-eastern corridor of East Polynesia.
Introduction
The expansion of modern humans into the wider Pacific, culminating with the colonization of eastern Polynesia within the last 1,000 years, represents one of the last major migrations in human history (Duggan et al., 2014; Matisoo-Smith, 2015). Aided by the development of new watercraft technologies and navigational abilities, the appearance of the Lapita peoples in the Bismarck Archipelago between 3,400 and 3,200 B.P. (Green et al., 2008; Summerhayes et al., 2010) and successful movement of humans beyond the Solomon Islands instigated a phase of long-distance voyaging and settlement across Remote Oceania (see Figure 1; Irwin, 2008). Eastward expansion led to the colonization of Vanuatu and Fiji by 3,100 and 3,000 B.P. respectively (Denham et al., 2012), Tonga by 2,850 B.P. (Burley et al., 2015) and Samoa by 2,750 B.P. (Petchey, 2001; Clark et al., 2016).
Figure 1
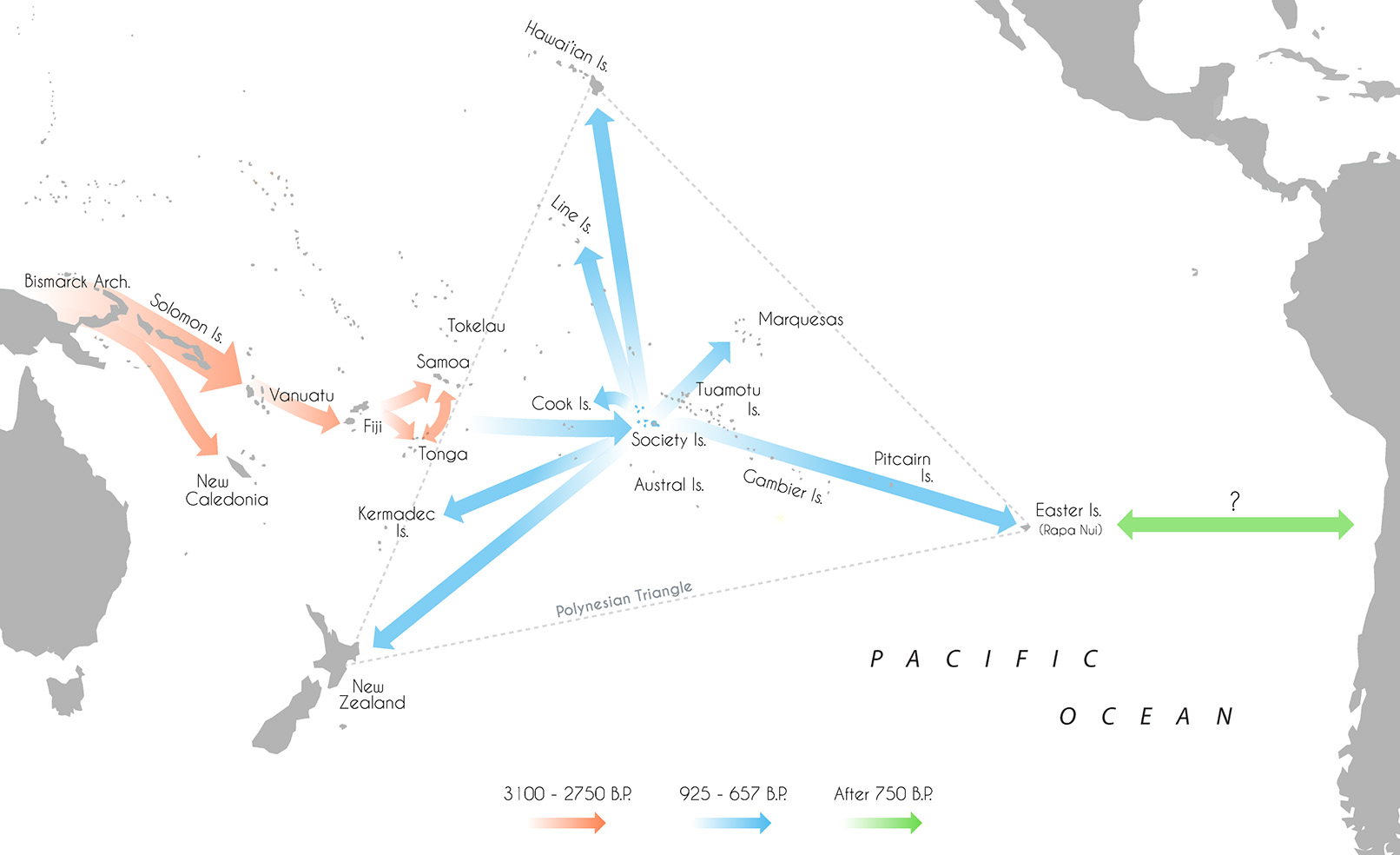
Map of the Pacific islands with Lapita/Polynesian dispersal trajectories and associated dates.
After some period of time, the descendants of these migrants proceeded to move further east into what is now denoted “The Polynesian Triangle;” a 20 million sq. km stretch of ocean comprised of over 500 islands scattered between Hawai'i, Easter Island (Rapa Nui) and New Zealand (Matisoo-Smith, 2015). Nearly every habitable island in this region was colonized, some subsequently used for permanent settlements and others abandoned (Kirch, 2000). The timing and sequence of settlement across many of the major archipelagos of central East Polynesia however remain largely unresolved, with conflicting estimates of initial colonization varying by more than 1,000 years (Wilmshurst et al., 2008, 2011). Recent research has generally supported a short and rapid colonization of eastern Polynesia in the early second millennium A.D (after 950 B.P.; Hunt and Lipo, 2006; Petchey et al., 2010; Molle and Conte, 2011; Rieth et al., 2011; Wilmshurst et al., 2011; Kahn, 2012; Mulrooney, 2013; Commendador et al., 2014; Kahn et al., 2015; Kahn and Sinoto, 2017). A two-phase sequence of settlement has been proposed—establishment in the Society Islands ~925–830 B.P. before a widespread radiation toward the Marquesas, ~750–673 B.P.; Easter Island, ~750–697 B.P.; Hawai'i, ~731–684 B.P.; New Zealand, ~720–668 B.P.; Southern Cooks, ~700–669 B.P.; and the Line Islands, ~675–657 B.P. (Wilmshurst et al., 2011). However, there are still irreconcilable differences in the chronological boundaries of some island groups, with some researchers advocating for an earlier settlement of the Cook Islands, the Marquesas, Hawai'i and Mangareva (Gambier Islands; Kirch et al., 2010; Allen and McAlister, 2013; Athens et al., 2014; Conte and Molle, 2014; Weisler et al., 2015).
The settlement of Easter Island, in the eastern-most corner of the Polynesian Triangle, has long held a fascination in prehistoric Pacific studies and popular literature. Paleoenvironmental evidence of severe deforestation and a proposed societal collapse (Mieth et al., 2002; Diamond, 2005; Butler and Flenley, 2010; Delhon and Orliac, 2010), have prompted many questions in relation to the immediate origin and timing of initial settlement, the potential of multiple colonization and/or interactions, and the long-term isolation of the Rapanui people. Faunal remains of the Pacific rat (Rattus exulans), a commensal species transported by ancient Polynesians, uncovered in the earliest archeological layers of human occupation, provide support for an initial Polynesian colonization of Easter Island (Martinsson-Wallin and Wallin, 2000; Barnes et al., 2006). Material remains, such as one-piece fish hooks and harpoon heads, are also distinctly Polynesian in origin (Weisler, 1996; Green, 1998). Based on material assemblages, potential migration routes through the Marquesas-Tuamotu-Mangareva area, the Mangareva-Pitcairn-Henderson area or directly via the Marquesas have been suggested (Green, 1998; Martinsson-Wallin and Crockford, 2001). However, Irwin (1992) and Green (1998) have noted potential difficulties in directly sailing from the Marquesas to Easter Island.
There is also increasing evidence for an Amerindian connection to Easter Island and wider Polynesia, particularly with regards to the presence of the kumara, or sweet potato, a South American tuber which was distributed throughout East Polynesia prior to European arrival in the region. Whether this reflects a post-settlement arrival of Amerindians to Easter Island and/or an eastern Polynesian return voyage to South America, remains unclear. Human ancient DNA studies have attested to the Rapanui people being of Polynesian descent (Hagelberg et al., 1994), yet recent research has identified Native American admixture—attributed to pre-European Native American contact (Moreno-Mayar et al., 2014; Thorsby, 2016). Technical and cultural concerns toward the sequencing, storage and interpretation of indigenous human DNA in the Pacific however, has limited access to human genetic material in this region, leaving such questions unresolved for the time being.
Subsequently, the commensal approach, using the genomes of transported (usually domesticated) plant and animal species as a proxy for human migration, has been increasingly applied to investigate human dispersal and interaction patterns across the Pacific (see Matisoo-Smith, 2015). The Pacific rat was the first commensal species studied in relation to human migration in Oceania (Matisoo-Smith, 1994). Proposed to have originated in Flores, Indonesia (Thomson V. et al., 2014), Pacific rats were transported by ancestral Polynesians and are now widely distributed across the region, including Hawai'i, New Zealand and Easter Island. Initial research investigating genetic variation in 400 bp (base pairs) of the hypervariable mitochondrial control region in modern Pacific rats, identified a central East Polynesian interaction sphere comprised of the southern Cook and Society Islands (Matisoo-Smith et al., 1998). This broad interaction sphere was central to a northern (i.e., the Marquesas and Hawai'i), and a southern Polynesian sphere (i.e., Kermadec Islands and New Zealand). The authors suggested that this likely reflects a central homeland region from which major East Polynesian islands were colonized.
A later phylogeographic analysis targeting ~200 bp of the mitochondrial control region in both ancient and modern Pacific rats across the Pacific (see Figure 2), found three major control region haplogroups; the first, an isolated group within Southeast Asia; the second, a dispersed group from Southeast Asia to Near Oceania; and the third, a primarily Remote Oceanic group (Matisoo-Smith et al., 2004). Haplogroup III is associated with the Lapita/Polynesian movement through Remote Oceania; its distribution extending from Vanuatu across to Central and East Polynesia, to Hawai'i, New Zealand and Easter Island, and from likely back migrations, into the Caroline and Marshall Islands. Haplogroup III is further divided into subgroups IIIA and IIIB, the latter derived from IIIA. These subgroups overlap in West Polynesia, however only subgroup IIIB has been found in East Polynesia (Matisoo-Smith et al., 2004). Samples from the haplogroup IIIB either belong to or are derived from the haplotype R9, which has been found across central East Polynesia. Unfortunately, given that most East Polynesian ancient rat specimens adhere to the R9 haplotype, there has been limited success in distinguishing finer-scale movements between the islands within the Polynesian Triangle. This was evident in a control region study on ancient Pacific rat skeletal remains from Anakena, Easter Island (Barnes et al., 2006). All samples presented the R9 control region haplotype and therefore it was not possible to distinguish the immediate origin of the Easter Island population from within eastern Polynesia.
Figure 2
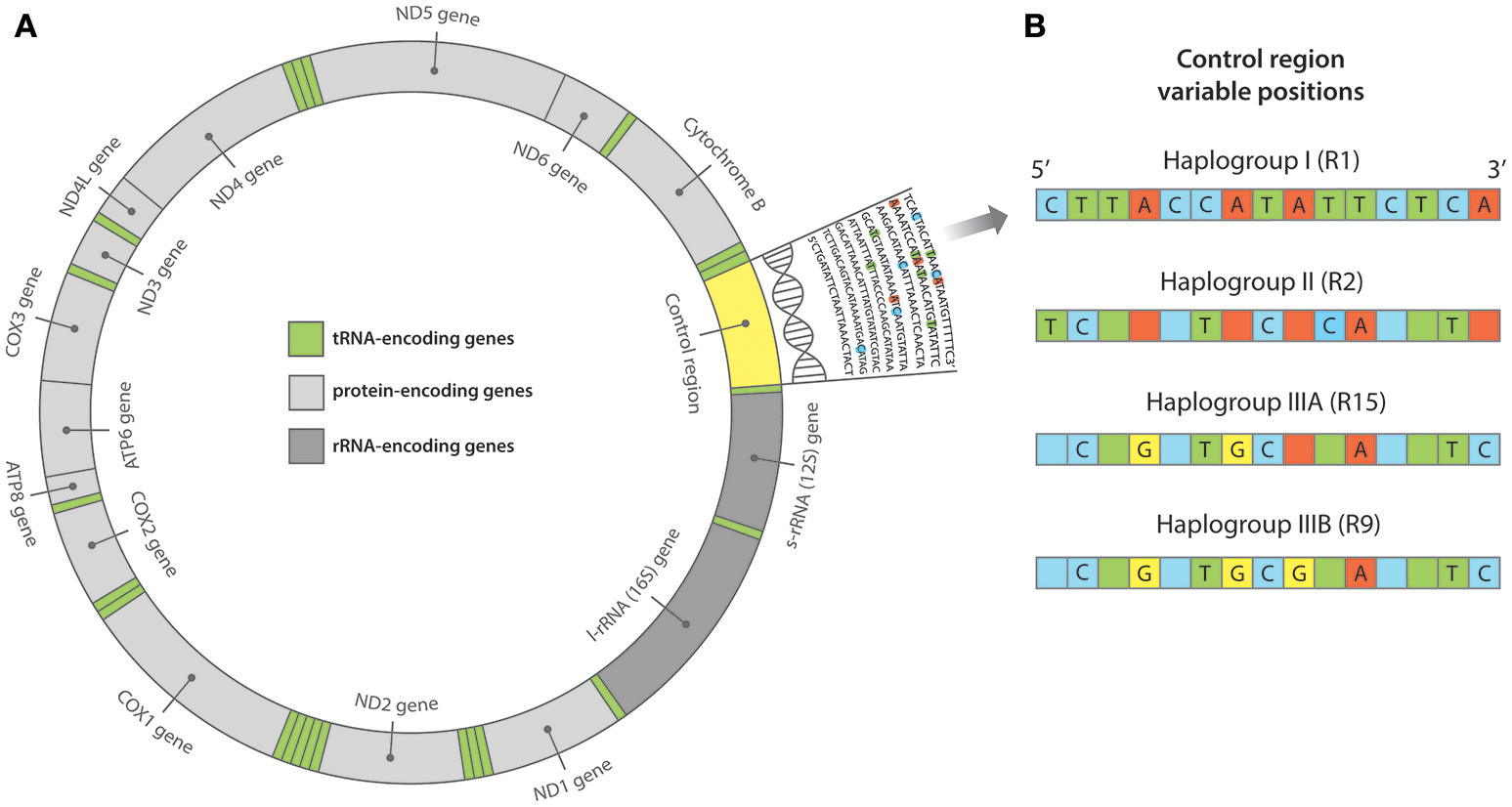
(A) Structure of the mitochondrial genome. At ~16,300 base pairs (bp) in length, the Pacific rat mitochondrial genome is comprised of protein, transfer RNA (tRNA) and ribosomal RNA (rRNA) encoding regions, in addition to the non-coding control region. The control region is often referred to as “hypervariable” as it accumulates mutations at a much higher rate than both the nuclear genome and the remaining regions of the mitochondrial genome. As such, the control region is often targeted for its variation in population genetic studies. (B) Pacific rat control region haplogroups as outlined in Matisoo-Smith et al. (2004). Variable positions across ~200 bp of the control region (from position 15,382 to 15,584 bp) have distinguished multiple haplotypes belonging to three major haplogroups—I, II, and III (A & B).
Recent advances in second-generation sequencing technologies and bioinformatics processing however, have made it viable to recover complete ancient mitochondrial genomes from preserved archaological and paleontological samples (Knapp and Hofreiter, 2010). Recent applications of complete mitochondrial sequencing in the Pacific have documented an increase in molecular resolution that can be utilized for fine-scale phylogenetic analyses (Miao et al., 2013; Duggan et al., 2014; Greig et al., 2015). This is of particular importance in Pacific research, as multiple population bottlenecks, the result of successive island migrations, may have severely reduced genetic variation in study species. Here we document complete and partial mitochondrial genome sequences from 13 ancient Pacific rats, sourced from early archeological deposits across West and East Polynesia. It was anticipated that with greater molecular resolution provided by complete mitochondrial genome sequencing, a greater number of Pacific rat lineages would be distinguished and could be used as a proxy to investigate the origins and dispersals of East Polynesian peoples. Our chief objective was to identify a potential immediate origin for the Polynesian settlers who introduced these rats to Easter Island.
Materials and methods
Sample collection and DNA extraction
A total of 77 ancient Pacific rat skeletal remains from Easter Island, Tonga, Tokelau, American Samoa, Austral Islands, Cook Islands and the Society Islands were collected for mitochondrial genome extraction and sequencing. These samples were sourced from multiple archeological excavations (see SI Table 1) and were placed within a dark and dry environment for long-term storage. All ancient DNA (aDNA) extractions and library building prior to PCR amplifications were conducted in a purpose-built aDNA laboratory in the Richardson Building at the University of Otago, following recommendations in Knapp et al. (2012a). Access and use of the aDNA laboratory requires adherence to strict protocols in place to minimize contamination risks, particularly from areas where amplified DNA is present, i.e., modern DNA laboratories. Femora were common in the acquired Pacific rat bone assemblage and were targeted for further processing. Prior to DNA extractions, bone samples were soaked in 5% bleach for 10 min, rinsed several times with ultrapure water to remove residual bleach and left to dry overnight. Each bone sample was ground into a fine powder using a sterile mortar and pestle and extracted using a silica based extraction protocol (Rohland and Hofreiter, 2007). Samples were processed in sets of nine in conjunction with one extraction blank.
Library preparation
Double-stranded barcoded libraries were generated from aDNA extracts and extraction blanks as described in Knapp et al. (2012b), in preparation for hybridization capture and paired end sequencing on an Illumina MiSeq platform. A quantitative PCR (qPCR) was then performed to determine the number of cycles required for the sufficient amplification of each library for immortalization. If libraries presented a significant amount of adapter dimer, a second attempt at library preparation was made or the sample was removed from further processing. Libraries were immortalized by PCR with the following reagent concentrations: 19 μl of adapter ligated library; 1xTaq Buffer; 2.5 mM MgCl2; 1 mM of dNTPs; 3.75 U of AmpliTaq Gold polymerase (ThermoFisher Scientific); 0.2 μM of extension primer, Sol_ext_P5; and 10 μM of a barcoded index primer, Sol_ext_P7. The following immortalization conditions were used: 95°C, 12 min, 22–30 cycles (determined by when the plateau was reached during the previous qPCR) of: 94°C, 30 s; 58°C, 30 s; 72°C, 1 min, followed by a final extension of 72°C for 10 min. Following immortalization, samples were purified with a MinElute PCR Purification Kit (QIAGEN) with the following modification: 2x PE washes. Immortalized libraries were then amplified using a KAPA HiFi PCR Kit (KAPA Biosystems) in preparation for hybridization capture, each 50 μl reaction containing: 1 μl of immortalized library; 1x KAPA HiFi Buffer; 0.3 mM dNTPs; 0.3 mM each of amplification primers, Sol_amp_p5 and Sol_amp_p7; and 1 U of KAPA HiFi DNA Polymerase. The following amplification conditions were used: 94°C, 5 min, 10 cycles of: 94°C, 20 s; 55°C, 55 s; 72°C, 15 s, followed by a final extension of 72°C for 5 min. Following amplification, samples were once again purified.
Bait production
Bait was produced for hybridization capture using DNA extracted from tissue of a laboratory rat (Rattus norvegicus), sourced from the Department of Anatomy, University of Otago, following the protocol described by Maricic et al. (2010). The complete mitochondrial genome of the R. norvegicus specimen was targeted using primers designed to amplify two 8–9.5 kb overlapping mitochondrial fragments (see Table 1).
Table 1
| Region amplified | Primer pairs | Primer sequences (5′–3′) | Amplicon length | Annealing temperature |
|---|---|---|---|---|
| COI → Cyt b | BatL5310 & RCb9H | CCTACTCRGCCATTTTACCTATG | 9.5 kb | 47°C |
| TACACCTAGGAGGTCTTTAATTG | ||||
| Cyt b → COI | RGlu2L & R6036R | CAGCATTTAACTGTGACTAATGAC | 8.3 kb | 49°C |
| ACTTCTGGGTGTCCAAAGAATCA |
Long-range PCR amplification for bait production.
Hybridization capture and sequencing
Ancient libraries were captured using a modified hybridization protocol, adapted from Maricic et al. (2010). A hybridization mixture containing: 2 μg of barcoded library; 1 μM each of blocking oligonucleotides; 0.6x Agilent blocking agent; and 0.6x Agilent hybridization buffer was prepared for each sample and treated to the following conditions: 95°C, 3 min; 37°C, 30 min. The hybridization mixture was then added to the pre-prepared baited beads and rotated at 12 rpm for 48 h at 65°C. The streptavidin beads were then isolated and washed as described in Maricic et al. (2010), re-suspended in 15 μl of 1x TE solution and heated at 95°C, 3 min, denaturing the captured libraries from the beads. Captured libraries were re-amplified using a KAPA HiFi PCR Kit (KAPA Biosystems) with each 50 μl reaction containing: 15 μl of post-captured library; 1x KAPA Buffer; 0.3 mM dNTPs; 0.3 mM each of the amplification primers, Sol_amp_p5 and Sol_amp_p7; and 1 U of KAPA HiFi DNA Polymerase. The following amplification conditions were used: 94°C, 5 min, 20 cycles of: 94°C, 20 s; 55°C, 55 s; 72°C, 15 s, followed by a final extension of 72°C for 5 min. Libraries were then purified with a MinElute PCR Purification Kit (QIAGEN), quantified with a Qubit 2.0 Fluorometer (Invitrogen), pooled in equimolar concentrations and sequenced using 2 × 75 base pair-end runs on an Illumina MiSeq sequencing platform, conducted by New Zealand Genomics Limited (NZGL) at Massey University, New Zealand. Libraries prepared from extraction blanks were sequenced separately.
Raw data processing
Sequencing reads were initially processed in AdapterRemoval (v.2; Lindgreen, 2012) to remove adapters, merge paired-end fragments (overlapping by at least 11 base pairs), and remove stretches of Ns, bases with low quality scores (<30) and short reads (<25). Reads were then mapped to a reference genome (R. exulans, GenBank accession NC_012389) in BWA v.0.7.12 (Li and Durbin, 2009), following recommended settings for ancient DNA, i.e., −n 0.03 (allow for more substitutions), -o 2 (allow more gaps at the beginning) and −l 1,024 (deactivate seed mapping; Schubert et al., 2012). Reads were also mapped to mitochondrial reference genomes from cow (Bos Taurus, GenBank accession NC_006853.1), pig (Sus scrofa, NC_0012095.1), human (Homo sapiens, GenBank NC_012920.1), chicken (Gallus gallus, GenBank NC_001323.1) and dog (Canis lupus familiaris, GenBank NC_002008.4) to detect any contamination. Unmapped reads were removed using SAMtools (Li et al., 2009; See Supplementary Materials).
PCR duplicates were then removed from unmerged reads using Picard's MarkDuplicates tool and from merged reads using script from PaleoMix (Schubert et al., 2014). Merged and unmerged reads were combined into one BAM file and coverage plots were produced for each sample using SAMtools (Li et al., 2009). The program mapDamage (v.2.0; Jónsson et al., 2013) was then implemented to assess damage patterns and lower the quality score of these damaged sites using the “–rescale” option. Plots to assess characteristic aDNA damage patterns were produced for each sample (see Supplementary Materials). A variant call file was generated and a consensus sequence produced, that was then converted to a FASTA file using GATK's FastaAlternateReferenceMaker (McKenna et al., 2010), masking positions with a read depth of <2. SNPs were visually inspected using the Integrative Genomics Viewer (IGV; Robinson et al., 2011; Thorvaldsdóttir et al., 2013) to ensure consistency with the variant calling criteria. Consensus sequence coverage was evaluated in Geneious (v.8.1.8; http://www.geneious.com, Kearse et al., 2012); sequences with large fragments missing (more than 50% of the reference genome) and/or that exhibit read depth <2 were removed from subsequent phylogenetic analyses.
Phylogenetic analyses
Ancient mitochondrial sequences were aligned using MUSCLE in Geneious (v.8.1.8). Two phylogenetic analyses were conducted: 1. with near complete sequences (>90% of the reference sequence), and 2. with all near complete and partial sequences (>50% of the reference genome). Sites were masked where any of the sequences provided missing data, using the Mask Alignment tool in Geneious.
A Bayesian inference analysis of phylogeny was conducted using the MrBayes plugin (v.2.2.2: Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003) implemented in Geneious (v.8.1.8). This consisted of running four chains with the heated chain temperature set at 0.2 to allow sufficient chain-swapping over 20 million generations, with a burn-in length of 100,000 and sampling every 2,000 generations. The best-fit model for nucleotide substitution was determined using jModeltest2 (https://github.com/ddarriba/jmodeltest2, Guindon and Gascuel, 2003; Darriba et al., 2012) with 11 substitution schemes. Model selection was computed using the Bayesian and Akaike information criteria (BIC and AIC). The HKY85 substitution model (Nst = 2; Hasegawa et al., 1985) with invariable rate variation (rate variation command “propinv”) was applied for the ancient sequence analysis. Complete mitochondrial sequences from Thailand, Papua New Guinea and New Zealand (Genbank EU273710.1, EU273709.1, EU273711.1; Robins et al., 2008) were added for the purposes of outgrouping (Thailand) and to assess where they position in relation to the ancient sequence assemblage. The resulting Bayesian consensus trees were visualized and exported from Geneious (v.8.1.8). Convergence was assessed using the Trace tool in Geneious; there were no obvious trend lines to suggest that the MCMC was still converging and there were no large-scale fluctuations that would suggest poor mixing. Effective sample sizes (ESS) for all traces exceeded 100.
Maximum likelihood (ML) analyses (Felsenstein, 1981) were conducted using the PhyML plugin (v.3.0: Guindon et al., 2010) implemented in Geneious (v.8.1.8). Substitution rate categories were set to 4 and bootstrap support was estimated using 1,000 replicates. The HKY85 substitution model (Hasegawa et al., 1985), with software-optimized estimates of the proportion of invariable sites, the gamma shape parameter and the transition/transversion ratio was applied. Convergence was assessed by comparing multiple runs and ensuring that bootstrap support was consistent at a determined level of replicates.
The population structure of the ancient Polynesian rat assemblage was evaluated using a median-joining network analysis, generated in PopArt (v 1.7; Bandelt et al., 1999; Leigh and Bryant, 2015) with default settings. In addition, the control region fragment (15,387–15,590 bp) was extracted from the complete mitochondrial sequences, in order to assign each sample into the three major control region haplogroups—I, II, III (A & B) as described in Matisoo-Smith et al. (2004). This was used to compare the molecular resolution between control region and complete mitochondrial genome analyses.
Results
DNA preservation and sequence recovery
Forty of the 77 samples produced adequately preserved DNA that amplified and was subsequently sequenced. However, 27 samples were removed post-sequencing as a result of missing large fragments and low read depth (<2) when mapped to a Pacific rat mitochondrial reference genome. This resulted in a total of 13 complete and partial sequences to be used in subsequent phylogenetic analyses. Complete mitochondrial genomes were obtained from six samples, with the remaining seven samples providing partial coverage (ranging from 53 to 94%; see Table 2). The average read depth for the thirteen samples is 135.6x (ranging between 1.8x and 657.8x; see SI Figure 1). Ignoring missing data across the sequences (designated “N”), the average read depth for the 13 samples is 136.2x (ranging between 2.8x and 657.8x).
Table 2
| Location | Site | Sample code | GenBank accession number | Associated layer date |
|---|---|---|---|---|
| Tonga | Faleloa, Foa | MS20596 | KY814713 | 2,600 ± 50 B.P. |
| Tokelau | Fakaofo | MS10591 | KY814711 | 1,090 ± 60 B.P. |
| MS10592 | KY814712 | 1,090 ± 60 B.P. | ||
| Austral Islands | Atiahara, Tubuai | MS10600 | KY814714 | 769–675 B.P. |
| MS10601 | KY814715 | 769–675 B.P. | ||
| Society Islands | Terre Namotu, Maupiti | MS10502 | KY814709 | 495–320 B.P. |
| Haumi archeological site, Mo'orea | MS10523 | KY814710 | 911–652 B.P. | |
| Easter Island | Anakena | MS10602 | KY814716 | 610–560 B.P. |
| MS10604 | KY814717 | 610–560 B.P. | ||
| MS10605 | KY814718 | 610–560 B.P. | ||
| MS10606 | KY814719 | 610–560 B.P. | ||
| MS10609 | KY814720 | 610–560 B.P. | ||
| MS10610 | KY814721 | 610–560 B.P. |
Ancient samples used in the phylogenetic analyses.
Associated layer dates are as reported in archeological excavation publications (see SI Table 1).
DNA preservation and recovery is largely dependent on environmental factors (i.e., temperature, moisture, and acidity of the surrounding environment) and the age of the sample. Pacific preservation varies across tropical to temperate climates, and whether the sample was recovered from a protected, open and/or coastal context (Robins et al., 2001). Specimens recovered from East Polynesia generally provided greater DNA preservation than those from West Polynesia. It is clear from the read fragment distributions (see SI Figure 2), that the Easter Island samples in particular were less fragmented, providing peak read length between 100 and 120 bp. Rat specimens retrieved from Easter Island provided greater DNA preservation than all other surveyed locations in central East Polynesia. This is consistent with previous commensal research that has recovered complete ancient mitochondrial genomes from another temperate location in the Pacific, i.e., New Zealand (Greig et al., 2015).
Sequence authenticity
To estimate the degree of contamination, reads from each sample were mapped to mitochondrial reference genomes that represent common contaminants i.e., pig, human, cow, chicken, and dog. Only one ancient sample (MS10605) exhibited a small amount of human contamination equating to ~0.03%. A lack of exogenous DNA, particularly from cow, pig, and chicken, indicates that reagents used in this study were free from contamination. It has previously been asserted that laboratory reagents, such as dNTPs, can contain DNA from domestic and laboratory animals that may obscure endogenous DNA amplification (Leonard et al., 2007; Thomson V. A. et al., 2014). No blank processed with any of the modern and ancient samples exhibited contamination, providing no indication of cross-contamination during the processing of samples. The deamination patterns exhibited in the reads from each ancient sample were consistent with expected damage in ancient DNA sequences (see SI Figures 3–6). The observed C to T misincorporation rate of 0.20 for the first nucleotide position of the 5′ end exceeds the maximum rate of 0.05 for samples <117 years old, and is consistent within samples older than 500 years (Sawyer et al., 2012). This indicates that the sequences are endogenous mtDNA of rats obtained from early archeological deposits across Polynesia.
Phylogenetic analyses
Across the 13 ancient mitochondrial genomes, a total of 88 variable sites were identified. Interestingly, only three sites of variation were identified in the hypervariable (HVR) I region (positions 16,024–16,383 bp) of the control region. This hypervariable region is commonly targeted for population studies as it is considered fast mutating. However, in the ancient R. exulans sequences assessed in the current study, a higher degree of variation is observed across the protein-coding regions.
The consensus maximum likelihood (ML) and the Bayesian Inference trees for the complete ancient sequences were congruent and illustrated two major groupings—the first consisting of the Papua New Guinea and Thailand sequences, and the second consisting of the ancient rats sampled from across Remote Oceania (see Figure 3A). The Fakaofo (Tokelau; MS10592), Maupiti (Society Islands; MS10502), New Zealand (GenBank EU273711.1) and Easter Island sequences are similar, however there is high bootstrap and Bayesian posterior probability support for a distinct grouping of the Easter Island samples. In the median-joining (MJ) network, the Remote Oceanic samples radiate off two central nodes within 10 mutational steps (see Figure 3B). The Easter Island samples form a distinct cluster, whereby three of the samples (MS10604, 605, and 609) share the same mitochondrial haplotype.
Figure 3
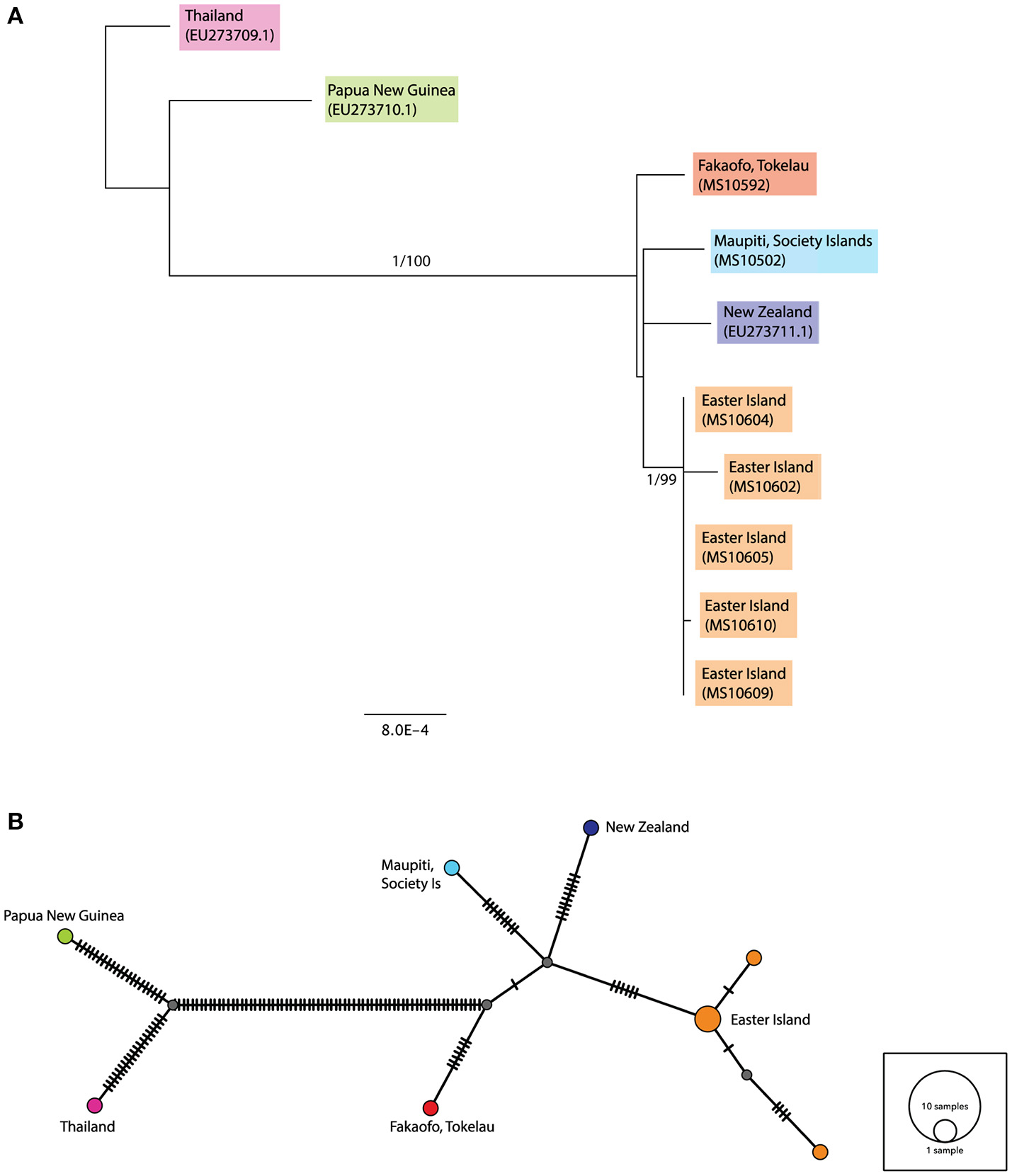
(A) Rooted maximum likelihood (ML) tree inferred from the complete (>90%) mitochondrial sequence dataset. Values above each node represent Bayesian posterior probabilities (obtained using MrBayes) and ML bootstrap values (obtained from PhyML). Only posterior probabilities/bootstrap values, where one or both are above 0.7 and 70, respectively, are included. Individual sample names are included and geographic populations defined by color. Scale bar represents amino acid substitutions per site. Additional R. exulans sequences (Thailand, Papua New Guinea (outgroup) and New Zealand; Genbank EU273709.1, EU273710.1, EU273711.1; Robins et al., 2008) were included. (B) Median-joining (MJ) network inferred from the complete (>90%) mitochondrial sequence dataset. The diameter of each circle is proportional to the frequency of a specific haplotype. Small gray circles indicate missing haplotypes. Nucleotide substitutions between haplotypes are represented by dashes.
In the ML tree containing both complete and partial ancient mitochondrial sequences (see Figure 4A), a sample from Fakaofo (Tokelau; MS10591) occupies a branch outside of the remaining Remote Oceanic sequences. In the corresponding MJ network (Figure 4B), MS10591 is clearly distinct, radiating from a central Remote Oceanic node by six mutational steps. Within the remaining Remote Oceanic samples, there is high Bayesian posterior probability support for four distinct groups, the first solely containing the New Zealand sequence; the second containing the Foa, Tonga (MS10596) and the Fakaofo, Tokelau (MS10592) sequences; the third containing the Society Island sequences (MS10523 and MS10502); and the fourth, an Easter Island/Tubuai (Austral Is) group of eight sequences. In the MJ network, these groups are not highly distinct, radiating from the central node by one mutation. However, its needs to be clarified that by including partial sequences in a PopArt network analysis, missing sites may potentially mask phylogenetically informative sites that were present in the MJ network of complete mitochondrial genomes.
Figure 4
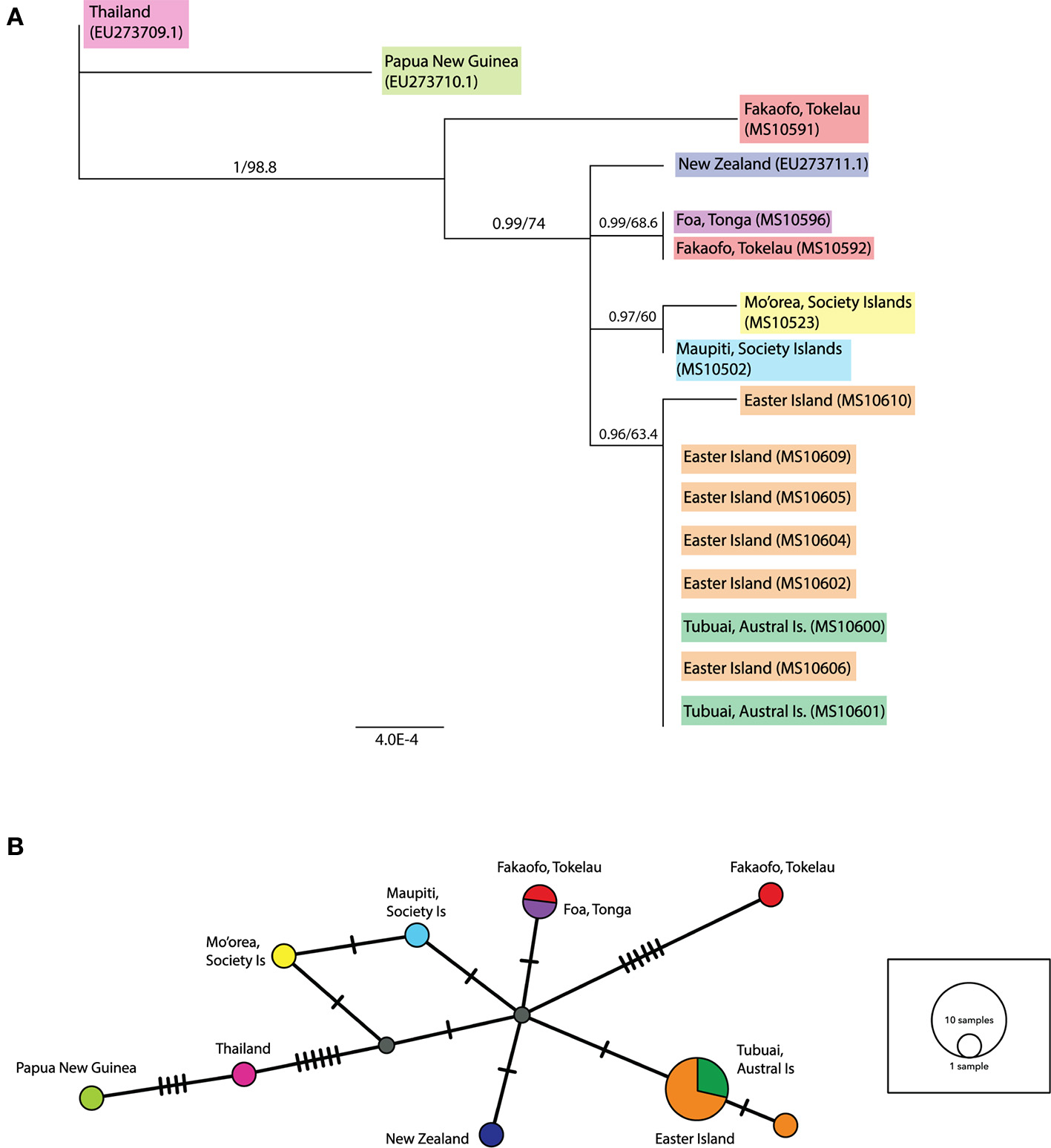
(A) Rooted maximum likelihood (ML) tree inferred from the complete and partial mitochondrial sequence dataset. Values above each node represent Bayesian posterior probabilities (obtained using MrBayes) and ML bootstrap values (obtained from PhyML). Only posterior probabilities/bootstrap values, where one or both are above 0.7 and 70, respectively, are included. Individual sample names are included and geographic populations defined by color. Scale bar represents amino acid substitutions per site. Additional R. exulans sequences (Thailand, Papua New Guinea (outgroup) and New Zealand; Genbank EU273709.1, EU273710.1, EU273711.1; Robins et al., 2008) were included. (B) Median-joining (MJ) network inferred from the complete and partial mitochondrial sequence dataset. The diameter of each circle is proportional to the frequency of a specific haplotype. Small gray circles indicate missing haplotypes. Nucleotide substitutions between haplotypes are represented by dashes.
A network analysis of 203 bp of the control region was undertaken to categorize samples into the three major haplogroups—I, II, III as described in Matisoo-Smith et al. (2004). All of the ancient Polynesian samples that were not missing fragments in the control region, clustered within Haplogroup IIIB, in particular adhering to the R9 haplotype (see Figure 5). It is clear that there is little nucleotide variation in the control region that can be used to distinguish ancient Pacific rat populations in Remote Oceania. Interestingly, the Thailand sequence (GenBank EU273710.1), clustered outside all three major haplogroups, indicating the existence of potentially another major Pacific rat haplogroup.
Figure 5
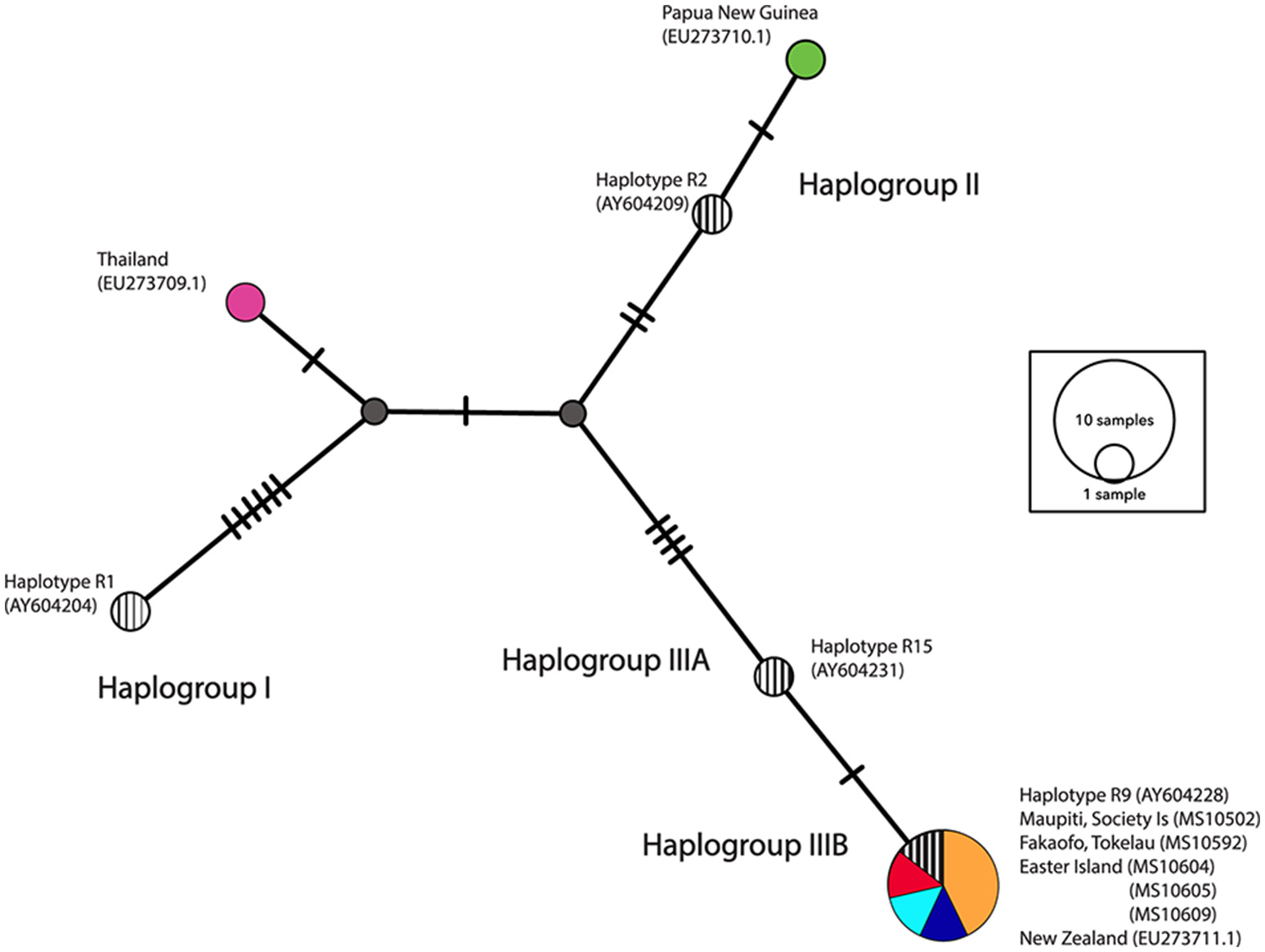
Median-joining (MJ) network of control region haplotypes. Striped circles represent four central haplotypes chosen to represent each haplogroup (I, II, IIIA, and IIIB). The diameter of each circle is proportional to the frequency of a specific haplotype.
Discussion
The application of complete mitochondrial genome sequencing
Mitochondrial hypervariable regions accumulate mutations at a faster rate than other mitochondrial regions (Stoneking, 2000), producing a greater amount of population variation that can be utilized for phylogenetic research. As such, the majority of commensal studies in the Pacific, particularly those using archeological specimens, have targeted fragments of 100–600 bp in the mitochondrial control region (e.g., Barnes et al., 2006; Larson et al., 2007; Storey et al., 2007, 2010; Oskarsson et al., 2012). As ancient DNA is highly fragmented, it is practical to target fragments of the mitochondrial control region that produce the most usable variation. The emergence of second-generation sequencing however has made it viable to generate longer DNA sequences, e.g., complete mitochondrial genomes, from fragmented aDNA (Knapp and Hofreiter, 2010). The use of hybridization capture for target DNA enrichment prior to second-generation sequencing (Ng et al., 2009), is also beginning to supersede the traditional primer-based PCR amplification that can often amplify large amounts of exogenous contaminant DNA (Burbano et al., 2010; Knapp et al., 2012b; Li, 2013; Greig et al., 2015). Recent studies using complete mitochondrial genomes in the Pacific have reported a higher level of molecular resolution that can distinguish phylogenetic groups and individuals with greater accuracy than use of the control region alone (Miao et al., 2013; Duggan et al., 2014; Greig et al., 2015).
The control region analysis conducted in this study showed that all ancient Polynesian samples (that did not contain missing sites in the control region) clustered within Haplogroup IIIB, in particular, possessing the R9 haplotype. The use of this control region fragment alone did not produce enough variation to distinguish island groups or specific lineages. The low variation in the hypervariable regions of the ancient Pacific rat genomes may reflect a number of bottlenecks in the establishment of populations. Pacific rats, carried by migrating humans, would have been established on Pacific islands in a stepping-stone manner. A subset of the population (and therefore haplotypes) would have been carried on each migratory journey, as humans moved eastwards into the far reaches of Polynesia. Furthermore, as populations were established on islands, gene flow would have been heavily restricted and subsequently prolonged low variation. Our study indicates that targeting one region (e.g., HVR I) may not provide enough variation across all samples to sufficiently distinguish populations. In conjunction with previous Pacific commensal research using complete mitochondrial genomes, our results suggest that complete mitochondrial sequencing is necessary to assess all mitochondrial variation and gain the highest molecular resolution for subsequent phylogenetic analyses.
Implications for pacific rat and associated human dispersal
Archaeological and linguistic evidence suggests that Tonga and Samoa form the West Polynesian homeland from which East Polynesian populations are derived (Emory, 1946; Green, 1966, 1981; Pawley, 1966; Groube, 1971). The median-joining network analyses conducted in this study illustrate a star-shaped pattern whereby the Remote Oceanic sequences radiate off two shared central nodes. This is consistent with the scenario of an eastern dispersal from West Polynesia into the central East Polynesian archipelagos, before a widespread expansion to the extremes of Easter Island, Hawai'i and New Zealand. In the MJ network containing both complete and partial ancient rat sequences, the Tongan sequence (MS10596) shares the same haplotype as a sequence from Fakaofo, Tokelau (MS10592). Tokelau has previously been described as an intermediary between West and East Polynesia (Burrows, 1939). Whilst it has not yet been established whether key Tokelauan islands were directly settled from the West Polynesian homeland or from a back migration originating from central East Polynesia, re-analyzed radiocarbon determinations suggest that Fakaofo and Atafu were colonized by 750 B.P (Petchey et al., 2010). This coincides with the widespread expansion from central East Polynesia to the outliers of the Polynesian Triangle. Regardless of its immediate origin, the Tokelauan (MS10592) haplotype is present in an early Tongan deposit that predates East Polynesian expansion. Therefore, this shared haplotype is established to have been present in a Tongan population and subsequently carried with either West Polynesian or descendent East Polynesian settlers into Fakaofo, Tokelau. The other ancient Tokelauan haplotype, MS10591, is highly diverged in both the ML and MJ analyses. This haplotype may either represent another form carried by Tongan/Samoan populations—presently unsampled in West Polynesia, or a later evolved haplotype that diverged post-settlement of Tokelau.
The earliest radiocarbon dates associated with human settlement in central East Polynesia are found in the Society Islands (~925–830 B.P.; Wilmshurst et al., 2011). The Society Islands are often represented as the central East Polynesian homeland from which widespread eastern expansion occurred after 750 B.P. The ancient Society Island sequences obtained from Maupiti and Mo'orea (MS10502 and MS10592) represent two closely related haplotypes that radiate from the same central nodes in the MJ network as the rest of the Remote Oceanic assemblage. These two haplotypes however, are not ancestral to the remaining East Polynesian samples. As no other recovered ancient central East Polynesian haplotypes are derived from the haplotype found on Mo'orea, which given its associated layer date of 911–652 B.P. (Kahn, 2016) would have existed at the time of widespread expansion, or the haplotype from Maupiti, it could be argued that the continued East Polynesian expansion did not originate from these islands. However, given our limited success in recovering complete mitochondrial genomes from the Society Islands, we may have not sampled potential ancestral haplotypes present in archeological deposits in this region. Further sampling of ancient Pacific rats across the Society Islands and nearby Cook Islands is required to assess whether an ancestral haplotype, corresponding to the central nodes in the MJ networks, were established in these central island groups and whether it existed concurrently with derived haplotypes, such as those found in Mo'orea and Maupiti.
The Wilmshurst et al. (2011) re-analysis of East Polynesian radiocarbon determinations from early settlement deposits, suggests that Easter Island was colonized between ~750 and 650 B.P. The initial colonization is supported to be of Polynesian origin (Weisler, 1996; Green, 1998; Martinsson-Wallin and Wallin, 2000; Barnes et al., 2006), however the immediate origin remains unclear. The grouping of the Tubuai (Austral Is) and Easter Island haplotypes in this study suggests that both island groups were established by the same colonization wave, that would have carried the haplotype that the Easter Island and Tubuai rats share or are derived from. Whilst haplotype networks, such as the median-joining network, do not indicate ancestor-descendent relationships and therefore the direction of migration, it is unlikely that the Tubuai haplotypes are derived from the Easter Island population. Geographically, Easter Island is situated in the eastern-most corner of Polynesia. Migratory voyages are likely to have passed through other island archipelagos in a stepping-stone manner before reaching Easter Island. Therefore, it is highly likely that the Austral Islands were colonized prior to Easter Island. Preliminary radiocarbon dating on Tubuai suggests that it had been occupied earlier than Easter Island (Worthy and Bollt, 2011) and was contemporaneous with sites in the southern Cooks (Walter, 1998). This is the first commensal study to specify a potential immediate origin for the Easter Island population, providing support for the colonization of Easter Island through a south-eastern voyaging corridor, originating from the Austral Islands and potentially moving through the Gambier, Pitcairn and Henderson Island regions. Similar cultural assemblages documented by Weisler (1996), indicate the existence of an interaction sphere between Mangareva (Gambier Islands), Pitcairn and Henderson Island. Further sampling would be required in the Gambier, Pitcairn and Henderson Islands however to establish haplotype continuity in this south-eastern corridor.
The absence of variation in the Barnes et al. (2006) study of Easter Island rats, was attributed to a single or limited introduction of rats, followed by long-term isolation. The early divergence of the Rapanui language from the East Polynesian language group (Trudgill, 2004) is consistent with an early Easter Island colonization followed by extreme isolation from all other East Polynesian groups. The adherence of the ancient Easter Island rats sampled in this project to three haplotypes suggests that there may have been limited variation in the colonizing population of rats, however the sample size is too small to assess population variation. Notably, all Easter Island rat samples in this study were sourced from the earliest occupation layers at Anakena; described in traditional history as the landing place of the initial colonists (Métraux, 1940). Further sampling of ancient Easter Island rats from various time periods and archeological contexts would determine, from the absence or introduction of new haplotypes, whether inter-island contact was prevalent post-colonization.
Conclusion
Recent developments in target DNA enrichment and sequencing technologies have improved the cost-effectiveness and viability of recovering complete mitochondrial genomes from ancient fragmented DNA. Complete mitochondrial genome sequencing provides a higher degree of molecular resolution that can be used to distinguish Pacific rat populations across Remote Oceania. Our results indicate that Remote Oceanic rat haplotypes are closely related and are consistent with the scenario of a central East Polynesian homeland from which widespread expansion occurred. We also document the clustering of Tubuai and Easter Island haplotypes, which suggest that Easter Island may have been settled via a south-east voyaging corridor through the Austral Islands, Gambier, Pitcairn and Henderson regions. While the 13 mitogenome sequences presented here provide new details regarding relationships between various Pacific rat populations, further sequencing of ancient R. exulans samples and similar analyses of genetic variation of other commensal species in East Polynesia will refine our understanding of human dispersal and interaction across this vast region.
Statements
Author contributions
KW and EM conceived the research and wrote the manuscript with contributions from all co-authors; KW, CC, and OK performed the experiments and analyzed data; JK, TH, and DB obtained and provided samples and important site information; all authors reviewed the manuscript.
Funding
Funding for the research presented here was provided to EM by the Department of Anatomy, University of Otago. National Science Foundation Grant CNH Award Number 1313830 provided funds to JK for the Mo'orea and Maupiti archaeological fieldwork.
Acknowledgments
We would like to thank Dr. Simon Best for access to the Rattus exulans sample from Fakaofo, Tokelau.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2017.00052/full#supplementary-material
References
1
Allen M. S. McAlister A. (2013). Early Marquesan settlement and patterns of interaction: new insights from Hatiheu Valley, Nuku Hiva Island. J. Pac. Archaeol.4, 90–109.
2
Athens J. S. Rieth T. Dye T. (2014). A paleoenvironmental and archaeological model-based age estimate for the colonization of Hawai'i. Am. Antiq.79, 144–155. 10.7183/0002-7316.79.1.144
3
Bandelt H. J. Forster P. Röhl A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol.16, 37–48. 10.1093/oxfordjournals.molbev.a026036
4
Barnes S. S. Matisoo-Smith E. Hunt T. L. (2006). Ancient DNA of the Pacific rat (Rattus exulans) from Rapa Nui (Easter Island). J. Archaeol. Sci.33, 1536–1540. 10.1016/j.jas.2006.02.006
5
Burbano H. A. Hodges E. Green R. E. Briggs A. W. Krause J. Meyer M. et al . (2010). Targeted investigation of the Neandertal genome by array-based sequence capture. Science328, 723–725. 10.1126/science.1188046
6
Burley D. Edinborough K. Weisler M. Zhao J.-X. (2015). Bayesian modeling and chronological precision for Polynesian settlement of Tonga. PLoS ONE10:e0120795. 10.1371/journal.pone.0120795
7
Burrows E. G. (1939). Western Polynesia: a study in cultural differentiation. Gothenburg Ethnol. Stud.7, 85–88.
8
Butler K. R. Flenley J. R. (2010). The Rano Kau 2 pollen diagram: palaeoecology revealed. Rapa Nui J.24, 5–10.
9
Clark J. T. Quintus S. Weisler M. St Pierre E. Nothdurft L. Feng Y. (2016). Refining the chronology for West Polynesian colonization: new data from the Samoan archipelago. J. Archaeol. Sci. Rep.6, 266–274. 10.1016/j.jasrep.2016.02.011
10
Commendador A. S. Dudgeon J. V. Fuller B. T. Finney B. P. (2014). Radiocarbon dating human skeletal material on Rapa Nui: evaluating the effect of uncertainty in marine-derived carbon. Radiocarbon56, 277–294. 10.2458/56.16923
11
Conte E. Molle G. (2014). Reinvestigating a key site for Polynesian prehistory: new results from the Hane dune site, Ua Huka (Marquesas). Archaeol. Ocean.49, 121–136. 10.1002/arco.5037
12
Darriba D. Taboada G. L. Doallo R. Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods9, 772–772. 10.1038/nmeth.2109
13
Delhon C. Orliac C. (2010). The vanished palm trees of Easter Island: new radiocarbon and phytolith data, in The Gotland Papers-Selected Papers from the VII International Conference on Easter Island and the Pacific: Migration, Identity, and Cultural Heritage. (Gotland University Press), 97–110.
14
Denham T. Ramsey C. B. Specht J. (2012). Dating the appearance of Lapita pottery in the Bismarck Archipelago and its dispersal to Remote Oceania. Archaeol. Ocean.47, 39–46. 10.1002/j.1834-4453.2012.tb00113.x
15
Diamond J. (2005). Collapse: How Societies Choose to Fail or Succeed. New York, NY Viking.
16
Duggan A. T. Evans B. Friedlaender F. R. Friedlaender J. S. Koki G. Merriwether D. A. et al . (2014). Maternal history of Oceania from complete mtDNA genomes: contrasting ancient diversity with recent homogenization due to the austronesian expansion. Am. J. Hum. Gen.94, 721–733. 10.1016/j.ajhg.2014.03.014
17
Emory K. P. (1946). Eastern Polynesia: Its Cultural Relationships. Ph.D. thesis, Yale University.
18
Felsenstein J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol.17, 368–376. 10.1007/BF01734359
19
Green R. (1966). Linguistic subgrouping within Polynesia: the implications for prehistoric settlement. J. Polyn. Soc.75, 6–38.
20
Green R. C. (1981). Location of the “Polynesian homeland: a continuing problem, in Studies in Pacific languages and cultures in honour of Bruce Biggs, eds HollymanJ.PawleyA. (Auckland: Linguistic Society of New Zealand), 133–158.
21
Green R. C. (1998). Rapanui origins prior to European contact: the view from eastern Polynesia, in Easter Island and East Polynesian Prehistory, ed VargasP. (Santiago: Universidad de Chile), 87–110.
22
Green R. C. Jones M. Sheppard P. (2008). The reconstructed environment and absolute dating of SE-SZ-8 Lapita site on Nendö, Santa Cruz, Solomon Islands. Archaeol. Ocean.43, 49–61. 10.1002/j.1834-4453.2008.tb00030.x
23
Greig K. Boocock J. Prost S. Horsburgh K. A. Jacomb C. Walter R. et al . (2015). Complete mitochondrial genomes of New Zealand's first dogs. PLoS ONE10:e0138536. 10.1371/journal.pone.0138536
24
Groube L. M. (1971). Tonga, Lapita pottery, and Polynesian origins. J. Polyn. Soc.80, 278–316.
25
Guindon S. Dufayard J.-F. Lefort V. Anisimova M. Hordijk W. Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol.59, 307–321. 10.1093/sysbio/syq010
26
Guindon S. Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol.52, 696–704. 10.1080/10635150390235520
27
Hagelberg E. Quevedo S. Turbon D. Clegg J. B. (1994). DNA from ancient Easter Islanders. Nature369, 25–26. 10.1038/369025a0
28
Hasegawa M. Kishino H. Yano T. A. (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol.22, 160–174. 10.1007/BF02101694
29
Huelsenbeck J. P. Ronquist F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics17, 754–755. 10.1093/bioinformatics/17.8.754
30
Hunt T. L. Lipo C. P. (2006). Late colonization of Easter Island. Science311:1603. 10.1126/science.1121879
31
Irwin G. (1992). The Prehistoric Exploration and Colonization of the Pacific. Cambridge: Cambridge University Press.
32
Irwin G. (2008). Pacific seascapes, canoe performance, and a review of lapita voyaging with regard to theories of migration. Asian Perspect.47, 12–27. 10.1353/asi.2008.0002
33
Jónsson H. Ginolhac A. Schubert M. Johnson P. L. F. Orlando L. (2013). mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics29, 1682–1684. 10.1093/bioinformatics/btt193
34
Kahn J. G. (2012). Coastal Occupation at the GS-1 Site, Cook's Bay, Mo‘orea, Society Islands. J. Pac. Archaeol.3, 52–61.
35
Kahn J. G. Dotte-Sarout E. Molle G. Conte E. (2015). Mid-to late prehistoric landscape change, settlement histories, and agricultural practices on Maupiti, Society Islands (Central Eastern Polynesia). J. Island Coast. Archaeol.10, 363–391. 10.1080/15564894.2014.1001922
36
Kahn J. G. (2016). Annual Report of Archaeological Research Activities Between June 6-August 15, 2016.Ra'iātea and Mo'orea Island: Service de la Culture et du Patrimoine.
37
Kahn J. G. Sinoto Y. (2017). Refining the Society Islands cultural sequence: colonization phase and developmental phase coastal occupation on Mo'orea island. J. Polyn. Soc.126, 33–60. 10.15286/jps.126.1.33-60
38
Kearse M. Moir R. Wilson A. Stones-Havas S. Cheung M. Sturrock S. et al . (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics28, 1647–1649. 10.1093/bioinformatics/bts199
39
Kirch P. V. (2000). On the Road of the Winds: An Archaeological History of the Pacific Islands before European Contact. Berkeley, CA: University of California Press.
40
Kirch P. V. Conte E. Sharp W. Nickelsen C. (2010). The Onemea site (Taravai Island, Mangareva) and the human colonization of southeastern Polynesia. Archaeol. Ocean.66–79. 10.1002/j.1834-4453.2010.tb00081.x
41
Knapp M. Clarke A. C. Horsburgh K. A. Matisoo-Smith E. A. (2012a). Setting the stage –Building and working in an ancient DNA laboratory. Ann. Anat. Anat. Anz.194, 3–6. 10.1016/j.aanat.2011.03.008
42
Knapp M. Hofreiter M. (2010). Next generation sequencing of ancient DNA: requirements, strategies and perspectives. Genes1:227. 10.3390/genes1020227
43
Knapp M. Stiller M. Meyer M. (2012b). Generating barcoded libraries for multiplex high-throughput sequencing, in Ancient DNA: Methods and Protocols, eds ShapiroB.HofreiterM. (New York, NY: Humana Press), 155–170.
44
Larson G. Cucchi T. Fujita M. Matisoo-Smith E. Robins J. Anderson A. et al . (2007). Phylogeny and ancient DNA of Sus provides insights into neolithic expansion in Island Southeast Asia and Oceania. Proc. Natl. Acad. Sci. U.S.A.104, 4834–4839. 10.1073/pnas.0607753104
45
Leigh J. W. Bryant D. (2015). PopArt: full-feature software for haplotype network construction. Methods Ecol. Evol.6, 1110–1116. 10.1111/2041-210X.12410
46
Leonard J. A. Shanks O. Hofreiter M. Kreuz E. Hodges L. Ream W. et al . (2007). Animal DNA in PCR reagents plagues ancient DNA research. J. Archaeol. Sci.34, 1361–1366. 10.1016/j.jas.2006.10.023
47
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997.
48
Li H. Durbin R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics25, 1754–1760. 10.1093/bioinformatics/btp324
49
Li H. Handsaker B. Wysoker A. Fennell T. Ruan J. Homer N. et al . (2009). The sequence alignment/map format and SAMtools. Bioinformatics25, 2078–2079. 10.1093/bioinformatics/btp352
50
Lindgreen S. (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes5:337. 10.1186/1756-0500-5-337
51
Maricic T. Whitten M. Pääbo S. (2010). Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE5:e14004. 10.1371/journal.pone.0014004
52
Martinsson-Wallin H. Crockford S. J. (2001). Early settlement of Rapa Nui (Easter Island). Asian Perspect.244–278. 10.1353/asi.2001.0016
53
Martinsson-Wallin H. Wallin P. (2000). Ahu and settlement: archaeological excavations at Anakena and La Perouse, in Easter Island Archaeology: Research on Early Rapanui Culture eds StevensonC. M.AyresW. S. (Los Osos, CA: Easter Island Foundation), 27–44.
54
Matisoo-Smith E. (1994). The human colonisation of Polynesia. A novel approach: genetic analyses of the Polynesian rat (Rattus exulans). J. Polyn. Soc.103, 75–87.
55
Matisoo-Smith E. (2015). Ancient DNA and the human settlement of the Pacific: a review. J. Hum. Evol.79, 93–104. 10.1016/j.jhevol.2014.10.017
56
Matisoo-Smith E. Roberts R. M. Irwin G. J. Allen J. S. Penny D. Lambert D. M. (1998). Patterns of prehistoric human mobility in Polynesia indicated by mtDNA from the Pacific rat. Proc. Natl. Acad. Sci. U.S.A.95, 15145–15150. 10.1073/pnas.95.25.15145
57
Matisoo-Smith E. Robins J. H. Green R. C. (2004). Origins and dispersals of Pacific peoples: evidence from mtDNA phylogenies of the Pacific rat. Proc. Natl. Acad. Sci. U.S.A.101, 9167–9172. 10.1073/pnas.0403120101
58
McKenna A. Hanna M. Banks E. Sivachenko A. Cibulskis K. Kernytsky A. et al . (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res.20, 1297–1303. 10.1101/gr.107524.110
59
Métraux A. (1940). Ethnology of Easter Island. Bulletin No. 160. Honolulu: Bernice, P. Bishop Museum.
60
Miao Y. W. Peng M. S. Wu G. S. Ouyang Y. N. Yang Z. Y. Yu N. et al . (2013). Chicken domestication: an updated perspective based on mitochondrial genomes. Heredity110, 277–282. 10.1038/hdy.2012.83
61
Mieth A. Bork H. R. Feeser I. (2002). Prehistoric and recent land use effects on Poike peninsula, Easter Island (Rapa Nui). Rapa Nui J.16, 89–95.
62
Molle G. Conte E. (2011). New perspectives on the occupation of Hatuana Dune Site, Ua Huka, Marquesas Islands. J. Pac. Archaeol.2, 103–108.
63
Moreno-Mayar J. V. Rasmussen S. Seguin-Orlando A. Rasmussen M. Liang M. Flåm S. T. et al . (2014). Genome-wide ancestry patterns in rapanui suggest pre-European admixture with Native Americans. Curr. Biol.24, 2518–2525. 10.1016/j.cub.2014.09.057
64
Mulrooney M. A. (2013). An island-wide assessment of the chronology of settlement and land use on Rapa Nui (Easter Island) based on radiocarbon data. J. Archaeol. Sci.40, 4377–4399. 10.1016/j.jas.2013.06.020
65
Ng S. B. Turner E. H. Robertson P. D. Flygare S. D. Bigham A. W. Lee C. et al . (2009). Targeted capture and massively parallel sequencing of 12 human exomes. Nature461, 272–276. 10.1038/nature08250
66
Oskarsson M. C. R. Klütsch C. F. C. Boonyaprakob U. Wilton A. Tanabe Y. Savolainen P. (2012). Mitochondrial DNA data indicate an introduction through Mainland Southeast Asia for Australian dingoes and Polynesian domestic dogs. Proc. R. Soc. Lond. B Biol. Sci.279, 967–974. 10.1098/rspb.2011.1395
67
Pawley A. (1966). Polynesian languages: a subgrouping based on shared innovations in morphology. J. Polyn. Soc.75, 39–64.
68
Petchey F. (2001). Radiocarbon determinations from the Mulifanua Lapita site, Upolu, western Samoa. Radiocarbon43, 63–68. 10.1017/S0033822200031635
69
Petchey F. Addison D. J. McAlister A. (2010). Re-interpreting old dates: Radiocarbon determinations from the Tokelau Islands (South Pacific). J. Pac. Archaeol.1, 161–167.
70
Rieth T. M. Hunt T. L. Lipo C. Wilmshurst J. M. (2011). The 13th century Polynesian colonization of Hawai'i Island. J. Archaeol. Sci.38, 2740–2749. 10.1016/j.jas.2011.06.017
71
Robins J. H. McLenachan P. A. Phillips M. J. Craig L. Ross H. A. Matisoo-Smith E. (2008). Dating of divergences within the Rattus genus phylogeny using whole mitochondrial genomes. Mol. Phylogenet. Evol.49, 460–466. 10.1016/j.ympev.2008.08.001
72
Robins J. Matisoo-Smith E. Furey L. (2001). Hit or miss? Factors affecting DNA preservation in Pacific archaeological material, in Australasian Connections and New Directions: Proceedings of the 7th Australasian Archaeometry Conference, Vol. 5, eds JonesM.SheppardP. (Auckland: Department of Anthropology, University of Auckland), 295–305.
73
Robinson J. T. Thorvaldsdóttir H. Winckler W. Guttman M. Lander E. S. Getz G. et al . (2011). Integrative genomics viewer. Nat. Biotechnol.29, 24–26. 10.1038/nbt.1754
74
Rohland N. Hofreiter M. (2007). Ancient DNA extraction from bones and teeth. Nat. Protoc.2, 1756–1762. 10.1038/nprot.2007.247
75
Ronquist F. Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics19, 1572–1574. 10.1093/bioinformatics/btg180
76
Sawyer S. Krause J. Guschanski K. Savolainen V. Pääbo S. (2012). Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE7:e34131. 10.1371/journal.pone.0034131
77
Schubert M. Ermini L. Der Sarkissian C. Jónsson H. Ginolhac A. Schaefer R. et al . (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc.9, 1056–1082. 10.1038/nprot.2014.063
78
Schubert M. Ginolhac A. Lindgreen S. Thompson J. F. Al-Rasheid K. A. Willerslev E. et al . (2012). Improving ancient DNA read mapping against modern reference genomes. BMC Genomics13:178. 10.1186/1471-2164-13-178
79
Stoneking M. (2000). Hypervariable Sites in the mtDNA control region are mutational hotspots. Am. J. Hum. Genet.67, 1029–1032. 10.1086/303092
80
Storey A. A. Ramírez J. M. Quiroz D. Burley D. V. Addison D. J. Walter R. et al . (2007). Radiocarbon and DNA evidence for a pre-Columbian introduction of Polynesian chickens to Chile. Proc. Natl. Acad. Sci. U.S.A.104, 10335–10339. 10.1073/pnas.0703993104
81
Storey A. A. Spriggs M. Bedford S. Hawkins S. C. Robins J. H. Huynen L. et al . (2010). Mitochondrial DNA from 3000-year old chickens at the Teouma site, Vanuatu. J. Archaeol. Sci.37, 2459–2468. 10.1016/j.jas.2010.05.006
82
Summerhayes G. R. Leavesley M. Fairbairn A. Mandui H. Field J. Ford A. et al . (2010). Human adaptation and plant use in highland New Guinea 49,000 to 44,000 years ago. Science330:78. 10.1126/science.1193130
83
Thomson V. A. Lebrasseur O. Austin J. J. Hunt T. L. Burney D. A. Denham T. et al . (2014). Using ancient DNA to study the origins and dispersal of ancestral Polynesian chickens across the Pacific. Proc. Natl. Acad. Sci. U.S.A.111, 4826–4831. 10.1073/pnas.1320412111
84
Thomson V. Aplin K. P. Cooper A. Hisheh S. Suzuki H. Maryanto I. et al . (2014). Molecular genetic evidence for the place of origin of the Pacific rat, Rattus exulans. PLoS ONE9:e91356. 10.1371/journal.pone.0091356
85
Thorsby E. (2016). Genetic Evidence for a Contribution of Native Americans to the Early Settlement of Rapa Nui (Easter Island). Front. Ecol. Evol.4:118. 10.3389/fevo.2016.00118
86
Thorvaldsdóttir H. Robinson J. T. Mesirov J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinformatics14, 178–192. 10.1093/bib/bbs017
87
Trudgill P. (2004). Linguistic and social typology: the Austronesian migrations and phoneme inventories. Linguist. Typol.8, 305–320. 10.1515/lity.2004.8.3.305
88
Walter R. (1998). Anai'o: The Archaeology of a Fourteenth Century Polynesian Community in the Cook Islands. Auckland: New Zealand Archaeological Association.
89
Weisler M. I. (1996). Taking the mystery out of the Polynesian ‘mystery islands’: a case study from Mangareva and the Pitcairn group, in Oceanic Culture History: Essays in Honour of Roger Green, eds DavidsonJ.GreenR. C. (Auckland: New Zealand Archaeological Association Monograph 21), 615–629.
90
Weisler M. I. Mendes W. P. Hua Q. (2015). A prehistoric quarry/habitation site on Moloka‘i and a discussion of an anomalous early date on the Polynesian introduced Candlenut (kukui, Aleurites moluccana). J. Pac. Archaeol.6, 37–57.
91
Wilmshurst J. Hunt T. Lipo C. Anderson A. (2011). High-precision radiocarbon dating shows recent and rapid initial human colonization of East Polynesia. Proc. Natl. Acad. Sci. U.S.A.108, 1815–1820. 10.1073/pnas.1015876108
92
Wilmshurst J. M. Anderson A. J. Higham T. F. G. Worthy T. H. (2008). Dating the late prehistoric dispersal of Polynesians to New Zealand using the commensal Pacific rat. Proc. Natl. Acad. Sci. U.S.A.105, 7676–7680. 10.1073/pnas.0801507105
93
Worthy T. H. Bollt R. (2011). Prehistoric Birds and Bats from the Atiahara Site, Tubuai, Austral Islands, East Polynesia. Pac. Sci.65, 69–85. 10.2984/65.1.069
Summary
Keywords
ancient DNA, mitochondrial haplotypes, prehistoric dispersal, Polynesia, Easter Island, Rattus exulans
Citation
West K, Collins C, Kardailsky O, Kahn J, Hunt TL, Burley DV and Matisoo-Smith E (2017) The Pacific Rat Race to Easter Island: Tracking the Prehistoric Dispersal of Rattus exulans Using Ancient Mitochondrial Genomes. Front. Ecol. Evol. 5:52. doi: 10.3389/fevo.2017.00052
Received
24 March 2017
Accepted
09 May 2017
Published
31 May 2017
Volume
5 - 2017
Edited by
Valentí Rull, Institute of Earth Sciences Jaume Almera (CSIC), Spain
Reviewed by
Erik Thorsby, University of Oslo, Norway; Serge Morand, Centre National de la Recherche Scientifique (CNRS), France
Updates
Copyright
© 2017 West, Collins, Kardailsky, Kahn, Hunt, Burley and Matisoo-Smith.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Matisoo-Smith matisoo-smith@otago.ac.nz
This article was submitted to Paleoecology, a section of the journal Frontiers in Ecology and Evolution
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.