 Zhi-Zhong Li
Zhi-Zhong Li Shan-Shan Sun
Shan-Shan Sun Qing-Feng Wang
Qing-Feng Wang Jin-Ming Chen
Jin-Ming Chen- 1Key Laboratory of Aquatic Botany and Watershed Ecology, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, China
- 2Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan, China
Introduction
Distyly is a floral polymorphism whereby plants in a population display two floral morphs (referred to as the long- and short-styled morphs) that differ reciprocally in style length, anther height, and pollen size, etc., which together with a self-incompatibility (SI) system (Barrett et al., 2000; Barrett, 2002). The development of the two different morphs of flower is controlled by a single diallelic locus designated the “S locus” that is a multigene complex (Barrett, 1992). Although, progresses have been made to isolate and characterize the S locus in a number of distylous plant species (Barrett and Shore, 2008; Li et al., 2011; Nowak et al., 2015), the number of S locus genes and the molecular mechanism by which it operates still remain to be uncovered.
Heterostylous plants, including distylous or tristylous plants, have been used as a model for genetics and evolutionary biology (Barrett, 1992). Heterostyly is one of the most effective mechanisms to avoid selfing and promote outcrossing in flowering plant (Barrett et al., 2000; Nowak et al., 2015). Heterostyly represent remarkable example of convergent evolution via natural selection and have been found in a wide range of species belonged to at least 28 families (Barrett and Shore, 2008). However, little is known of the nature of evolutionary changes in genes determined by natural selection during the origin and evolution of the heterostyly in flowering plants.
Nymphoides peltata (Gmel.) O. Kuntze (Menyanthaceae) is a floating-leaf perennial aquatic plant. This species is a typical distylous plant with dimorphic herkogamy and intramorph- and self-incompatibility (Wang, 2005). N. peltata can produce a number of relatively large flowers, and these yellow and showy flowers are produced over a long period of time. This developmental pattern makes it easy to get rich flower organ materials and manipulate in subsequent experiments. Based on above-mentioned features, this species is very suitable for studying the molecular mechanism of heterostyly and has a potential to be a new study system. In this study, we sequenced two floral transcriptomes from long- and short-styled flowered plants of this distylous species using the RNA-Seq technique. We also idenfied the divergent orthologous genes between long- and short-styled flowers. Our aims were to provide genetic resources for screening genes involved in the development of distyly and for revealing the role of natural selection in driving the floral morph diversity in the evolution of distylous plant.
Experimental Design, Materials, and Methods
Plant Materials
Nymphoides peltata was planted in the greenhouse at the Wuhan Botanical Garden of the Chinese Academy of Sciences, China. The floral bud samples from four developmental stages (i.e., 1, 3, 5, and 8 mm) of the same individual for each morph were collected and stored subsequently at −80°C until RNA extraction.
Total RNA Extraction and Quality Control, Library Preparation, and RNA-Seq
For each developmental stage of each morph, total RNA was extracted using RNAiso Plus reagent (TaKaRa, Dalian, China) and was treated with RNase-free DNase I (TaKaRa, Dalian, China). Quality and integrity of the extracted total RNA was determined by using Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Concentrations of the total RNA were measured using Qubit® 2.0 Flurometer (Life Technologies, CA, USA). Equal amounts of total RNA (7 μ g) from each stage of long- or short-style flowers were pooled together, respectively. These two pooled RNA samples were sent to BGI (Beijing Genomic Institute) for RNA sequencing (BGI Inc., Shenzhen, China). Two cDNA libraries (referred as L1 and S1) from these two RNA samples were prepared using Illumina TruSeqRNA sample Preparation Kit (Illumina), and were sequenced using the Illumina Hiseq2000 (Illumina Inc., San Diego CA, USA) to generate 100 bp paired-end raw reads.
Transcriptome De novo Assembly
The raw reads were trimmed using the program Trimmomatic v0.32 (Bolger et al., 2014) by removing adapter sequences, sequences with unknown base call (N) more than 5%, and low quality sequences (<Q20). Transcriptome De novo assembly was carried out via a short read assembling program, Trinity (version trnityrnaseq_r2012-10-05) (Grabherr et al., 2011), with min_kmer_cov set to 2 by default and all other parameters set as their defaults. The statistics of the transcriptome assemblies are shown in Table 1.
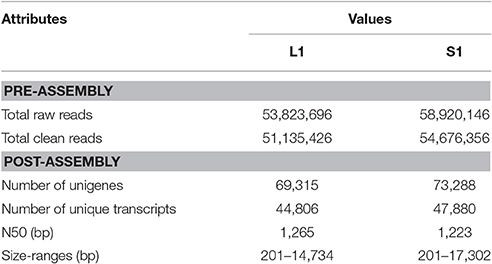
Table 1. Statistics of Nymphoides peltata transcriptome assemblies (L1, long-styled flowered plant; S1, short-styled flowered plant).
Identification of Orthologous Genes
To improve efficiency in searching orthologs between long- and short-styled flowered plants transcriptomes of N. peltata, the genome sequence of Salvia miltiorrhiza (ftp://202.203.187.112/genome/danshen/) was chose as reference because among the plant species whose whole genome had been sequenced, the S. miltiorrhiza was shown to be more closely related to N. peltata. Identification of orthologous groups among these three samples was firstly assessed by using the program, OrthoMCL v1.4 (Li et al., 2003), using default settings. Those genes with lengths longer than 50 amino acids and orthologous groups with a single copy gene for each of the three samples were used for further analysis. Then, the sequences of S. miltiorrhiza were removed and each remaining orthologous group was aligned using the program MUSCLE v3.8.31 (Edgar, 2004) with default parameters. The poorly aligned regions, such as alignment length <200 bp or including unexpected stop codon, were excluded and the remaining sequences were used to generate maximum likelihood (ML) tree using the program PAML v4.8 (Yang, 2007) with the general time reversible (GTR) model.
Calculation of Ka/Ks
The ratio of non-synonymous (Ka) and synonymous (Ks) substitution sites between pairs of orthologous genes identified from long- and short-styled flowered plants of N. peltata were estimated using a maximum likelihood (ML) method. Ka/Ks calculation was performed with PAML v4.8 (Yang, 2007) using default settings. High Ka/Ks (>>1) ratio indicated divergent orthologous genes and the positive selection may act on the orthologous gene, while low Ka/Ks (< <0.1) ratio indicated conserved orthologous genes. These divergent orthologous gene sequences can be used for further study on the genetic basis of distyly and adaptive evolution of distylous plant.
Links to the Deposited Data
Raw reads data for this project were deposited at SRA data base with the accession numbers SRR5154775 (http://www.ncbi.nlm.nih.gov/sra/SRR5154775) for long-styled flowered plant transcriptome and SRR5154807 (http://www.ncbi.nlm.nih.gov/sra/SRR5154807) for short-styled flowered plant transcriptome. Assemblies of long- and short-styled flowered plants transcriptomes of N. peltata are publicly available on Figshare at: https://figshare.com/articles/Nymphoides_peltata_transcriptome_assembly_fasta/4555423. The orthologous genes between long- and short-styled flowered plants are publicly available on Figshare at: https://figshare.com/articles/Nymphoides_peltata_ortholog_genes_/4555432. The list of the divergent orthologous genes (including the Ka/Ks-values, the lengths of the genes and the sequence of each gene) between long- and short-styled flowered plants is publicly available on Figshare at: https://doi.org/10.6084/m9.figshare.4555435.
Author Contributions
QW, JC: conceived this project and acquired funding; ZL, SS: conducted the experiment and drafted the manuscript; ZL, JC: performed data analyses; All authors gave approval of this manuscript to be published.
Funding
This research was supported by grants from the National Natural Science Foundation of China (Nos. 31270278 and 31570220) and Wuhan Botanical Garden (CAS) (No. Y655261W03).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Zhi-Yuan Du, Yang-Yang Yuan, Hua Liu, and Zhi-Cheng Long for their assistance in the laboratory.
References
Barrett, S. C. H. (1992). “Heterostylous genetic polymorphisms: model systems for evolutionary analysis,” in Evolution and Function of Heterostyly, ed S. C. H. Barrett (Berlin; Heidelberg: Springer), 1–29. doi: 10.1007/978-3-642-86656-2_1
Barrett, S. C. H. (2002). The evolution of plant sexual diversity. Nat. Rev. Genet. 3, 274–284. doi: 10.1038/nrg776
Barrett, S. C. H., and Shore, J. S. (2008). “New insights on heterostyly: comparative biology, ecology and genetics,” in Self-Incompatibility in Flowering Plants, ed V. E. Franklin-Tong (Berlin; Heidelberg: Springer), 3–32. doi: 10.1007/978-3-540-68486-2_1
Barrett, S. C. H., Jesson, L. K., and Baker, A. M. (2000). The evolution and function of stylar polymorphisms in flowering plants. Ann. Bot. 85, 253–265. doi: 10.1006/anbo.1999.1067
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Li, J., Webster, M. A., Smith, M. C., and Gilmartin, P. M. (2011). Floral heteromorphy in Primula vulgaris: progress towards isolation and characterization of the S locus. Ann. Bot. 108, 715–726. doi: 10.1093/aob/mcr181
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Nowak, M. D., Russo, G., Schlapbach, R., Huu, C. N., Lenhard, M., and Conti, E. (2015). The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol. 16:12. doi: 10.1186/s13059-014-0567-z
Wang, Y. (2005). The Reproductive Ecology of Distylous Clonal Aquatic Plant- Nymphoides Peltata (Gmel.) O. Kuntze. Doctoral Dissertation of Wuhan University, Wuhan.
Keywords: distyly, Nymphoides peltata, ortholog gene, RNA-Seq, transcriptome
Citation: Li Z-Z, Sun S-S, Wang Q-F and Chen J-M (2017) RNA-Seq Analysis of the Distylous Plant Nymphoides peltata Identified Ortholog Genes between Long- and Short-Styled Flowers. Front. Ecol. Evol. 5:59. doi: 10.3389/fevo.2017.00059
Received: 15 January 2017; Accepted: 23 May 2017;
Published: 06 June 2017.
Edited by:
Stefano Tiozzo, Paris-Sorbonne University, FranceReviewed by:
Delany Francisco Rodriguez, University of California, Santa Barbara, United StatesJohn P. Masly, University of Oklahoma, United States
Copyright © 2017 Li, Sun, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing-Feng Wang, cWZ3YW5nQHdiZ2Nhcy5jbg==
Jin-Ming Chen, am1jaGVuQHdiZ2Nhcy5jbg==
†These authors have contributed equally to this work.