 Vítor G. Faria
Vítor G. Faria Élio Sucena
Élio Sucena- 1Instituto Gulbenkian de Ciência, Oeiras, Portugal
- 2Departamento de Biologia Animal, Faculdade de Ciências, Universidade de Lisboa, Lisboa, Portugal
In recent years important tools have been developed in Drosophila to capture with the greatest possible accuracy the variation found in nature. Efforts, such as the Drosophila melanogaster Genetic Reference Panel (DGRP) or the Drosophila Synthetic Population Resource (DSPR) allied to the advances in whole-genome sequencing and analysis have propelled to unprecedented level our capacity to dissect the genotype-phenotype map. However, several practical problems arise upstream of these analyses starting with the collection and identification of wild specimens. These problems are dealt with in different ways by each researcher generating solutions not necessarily compatible across laboratories. Here, we provide a systematic coverage of every phase of this process based on our experience, and suggest procedures to maximize and share the generated resources potentiating future applications. We propose a detailed pipeline to guide researchers from collection in the wild to the development of a large array of molecular and genetic resources. We designed a multiplex-PCR that distinguishes sister species D. melanogaster and D. simulans and is diagnostic of the presence/absence of Wolbachia infection. These procedures may extend to other cryptic species pairs and endosymbionts. We developed a standardized protocol to create, replicate and maintain isofemale lines and outbred populations. Finally, we explore the potential of outbred populations across several applications from experimental evolution, to introgression of transgenic constructs or mutant alleles, and genomic analyses. We hope to contribute to the success in developing Drosophila resources for evolutionary genetics studies and facilitate exchanges across laboratories based on a common set of procedures.
Introduction
To study the evolution of species in a controlled and accurate way is undoubtedly a challenge. The complexity of factors acting simultaneously on individuals inevitably blurs the conclusions that can be drawn. To minimize this risk by facilitating the control of experimental conditions, we must bring populations from nature to the laboratory. Maintaining the original populations variation is challenging and can be achieved through two different methods, using parental lineages (isolines) or recreating a new outbred population, the central element to experimental evolution studies.
Experimental evolution can establish direct causation between selection in a given environment and the genetic and phenotypic changes observed in a population. This powerful approach departs from the comparative method in three fundamental aspects: (i) knowledge of the ancestral state, (ii) knowledge of the adaptive trajectories in real-time, (iii) high degree of replication under controlled selection and control regimes (Gibbs et al., 1997; Magalhães and Matos, 2012; Kawecki et al., 2012).
At a different plane, recent years have witnessed the rise of genomic studies, which have provided significant insights into the genetic basis of adaptation for a variety of complex traits. Examples cross all biological organization levels and include studies on transposable element population dynamics (Barrón et al., 2014), developmental time (Burke et al., 2010), immune response (Martins et al., 2013, 2014), hypoxia tolerance (Zhou et al., 2011), body size (Turner et al., 2011), climatic adaptation (Orozco-terWengel et al., 2012; Tobler et al., 2014; Zhao et al., 2016), courtship behavior (Turner et al., 2013) and life span (Remolina et al., 2012; Durham et al., 2014).
The combination of genomics with experimental evolution can provide a nearly unbiased estimate of the genetic changes that underlie the adaptation of populations to a given selection pressure, a central issue in evolutionary biology (Kawecki et al., 2012). The success of this methodology lies in the choice of the model species (with solid genomic tools, such as Drosophila) and the availability of outbred populations (with high levels of genetic variability) in which adaptation relies mostly on standing genetic variation (SGV) (Barrett and Schluter, 2008; Teotónio et al., 2009). The potential of this methodology has been confirmed in a number of recent studies (Turner and Miller, 2012; Abbott et al., 2013; Klepsatel et al., 2013; Martins et al., 2013; Durham et al., 2014; Tobler et al., 2014), and expectedly this approach will gain more and more followers in the coming years (Kawecki et al., 2012; Kofler and Schlötterer, 2014).
Other important, and complementary approaches, underscore the importance of describing and understanding the nature of SGV in natural and laboratory populations. For example, the increased use of isogenic lines propelled by the DGRP-Drosophila melanogaster Genetic Reference Panel (Mackay et al., 2012), has inaugurated an era of unprecedented studies in Drosophila GWAS studies (Huang et al., 2012; Jordan et al., 2012; Magwire et al., 2012; Weber et al., 2012; Kislukhin et al., 2013; Swarup et al., 2013). In parallel, the development of recombinant inbred lines that constitute the Drosophila Synthetic Population Resource (DSPR) have provided another important resource for the dissection of the genetic basis of complex traits (King et al., 2012; Burke et al., 2014; Marriage et al., 2014; Cogni et al., 2016). Before this, isofemale lines had been at the core of fecund research programs aiming at describing and comparing genomic variation between D. melanogaster and its sister species (Begun et al., 2007), and comprehending their genotype-phenotype map (Hoffmann et al., 1990; Turelli and Hoffmann, 1995; Lazzaro et al., 2004, 2006; Scott et al., 2011; Zhu et al., 2012; Ventura et al., 2013). Finally, individually wild-collected flies have provided valuable information in describing and quantifying natural variants (Clark et al., 1994; Nunes et al., 2008), characterizing ecological and evolutionary dynamics of natural populations (Fabian et al., 2012; Bergland et al., 2014; Rajpurohit et al., 2017), estimating the spread dynamics of endosymbionts in natural populations (Kriesner et al., 2013), contrasting or validating laboratory results (Macdonald, 2004; Mathur and Schmidt, 2017) and testing high throughput re-sequencing techniques (Zhu et al., 2012).
Though these different approaches differ in the nature and level at which they ask their questions, they share a common founding feature: the collection of material from nature and its subsequent establishment as a laboratory resource. In the case of Drosophila melanogaster, one seemingly trivial yet important question that must be resolved consists of its co-existence with the cryptic species D. simulans. Here, we present a protocol to streamline the collection, identification and establishment of D. melanogaster in the laboratory. We provide a high-throughput method by multiplex-PCR to identify the species and the individual status of Wolbachia infection that can be easily extended to other species. We provide a pipeline to maximize the resources generated, namely the establishment of outbred populations, isofemale lines, and DNA/RNA banks for genomic studies.
Materials and Methods
Collection
Using a portable vacuum cleaner coupled to a simple acrylic tube custom made with a soft net on one end (Figure 1B), we collected large numbers of flies from a vineyard dump site (Figure 1C). After collection, flies were transferred directly to bottles containing standard cornmeal-agar medium (Figure 1D). Around 1000 females were separated and individually distributed to vials with standard food. These females were transferred twice to new vials, laying eggs during 3 days in each vial.

Figure 1. Vineyards are large-scale collection sites for wild Drosophila specimens. (A) Winery dump site in Southern Portugal (B) portable vacuum cleaner coupled to a acrylic tube with a soft net (C) collecting a large number of individuals (D) flies are transferred directly to bottles containing standard fly food.
Nucleic Acid Extraction
To perform nucleic acid extraction, after the second round of egg laying, each vial with fertile progeny (checking the presence of F1 larvae in the food) was numbered and the respective female was anesthetized with CO2. A 96-well plate was previously prepared for DNA extraction, cooled over dry ice to facilitate the placing of the anesthetized females in the respective wells.
Nucleic acid extraction was performed according to (http://www.drosdel.org.uk/molecular_methods.php) with minor modifications. Briefly, each biological sample was homogenized with metallic beads and detergent for cell lysis. After removing the cellular waste by centrifugation, the supernatant was transferred to a new 96-well plate and nucleic acids were separated and precipitated with KCl and isopropanol. Samples were further washed in EtOH 70% and afterwards ressuspended in milliQ H2O. All steps were alternated with centrifugation steps. DNA or RNA were isolated after incubation with either RNase or Dnase, respectively.
Diagnostic PCR
After DNA extraction, a multiplex PCR reaction was performed in 96-well plates using 1 μL of diluted DNA, GoTaq DNA polymerase (Promega) in a 20 μL total reaction volume per well, using the primers Slif (Fwd–5′GTTAGCGCCTATTAGCACAT; Rev–5′CGGGACAACTCAGTCTGTAA) to distinguish between D. melanogaster and D. simulans, and wsp (Fwd8 1-5′-TGGTCCAATAAGTGATGAAGAAAC-3′; Rev691-5′-AAAAATTAAACGCTACTCCA-3′), to diagnose for the presence or absence of Wolbachia. The PCR program was used as follows: 95°C for 10 min; 30 cycles at 95°C for 30 s (denaturation), 60°C for 1 min (annealing), 72°C for 1 min (elongation) and a final extension step at 72°C for 10 min. PCR amplification products were visualized after electrophoresis in agarose gel (1.5% in TAE supplied with 0.5% RedSafe).
After analysis, the F1 vials of each tested-isofemale line could be separated into four groups, D. simulans or D. melanogaster, and Wolbachia positive or negative.
Tetracycline Treatment
To create a Wolbachia-negative population from the founded outbred infected population, an egg lay with controlled density was allowed in standard food containing 0.05 mg/ml tetracycline hydrochloride (Sigma). Throughout 3 generations, the full life cycle was carried out in food containing tetracycline at the same concentration, after which the population returned to regular maintenance in standard food for two generations for mitochondrial recovery. Finally, we standardized the microbiota by transferring the gut microbiome from the original population (Wolbachia-positive), as described in Faria et al. (2016). In the case of isofemale lines, the same procedure can be applied and adjusted to tubes.
Outbred Populations Foundation
From 160 isofemale lines of each group previously diagnosed, 10 F1-flies, 3–5 virgin females and 3–5 males were separated. Groups of males and virgin females were placed simultaneously in population acrylic boxes (50 × 30 × 25 cm), thus minimizing sib-mating. Around 1,500 individuals founded each population.
Populations were kept on a 3-week non-overlapping generation cycle. Treatments were always performed 3–5 days after eclosion and reproduction occurred 5–7 days after treatment. Reproduction was performed in 10 plastic cups (5 per day) with standard food. Egg density was limited to 400 per cup, a density determined experimentally to enable optimal larval development and population effective numbers. Flies were maintained under constant temperature (25°C), humidity (60–70%) and light-darkness cycle (12:12 h), and fed with standard cornmeal-agar medium.
Each population was kept in laboratory cages with high census (between 1,500 and 2,000 individuals). Census above 2,000 flies will lead to excess moisture inside the boxes that compromises egglaying, mobility and viability of flies and promotes bacterial and fungal proliferation.
Heterozygosity and Fst Analyses
Average expected heterozygosity in Initial population (2007) and outbred population adapted to the laboratorial conditions (2010), were estimated using 103 SNP markers described in Teotónio et al. (2009) and were analyzed as described in (Martins et al., 2013; Supplementary Information from Martins et al., 2014). We used samples of 52 individuals from initial founding females and 48 individuals in 2010.
Starting Experimental Evolution
Before the initiation of experimental evolution experiments, populations were maintained under the laboratory previously described conditions for a minimum of 15 generations and then serially expanded for two generations to allow the establishment of the new replicate populations. In our case, all lines of all treatments were derived from the same base population (from 1 to 6 in first generations and from 6 to 36 in the second). It enables that all populations under different experimental treatments will start with the same genetic pool, even losing some initial variability due to adaptation to the laboratory or to an unequal representation of founder lines. The egg laying for the foundation should be randomly distributed across the replicates to avoid any selection for fertility. In each generation, pools of 200–300 flies of each replicate were frozen in liquid nitrogen and kept at −80°C.
Isofemale Lines
Isofemale lines were established using the vials of each original line used to extract the virgins and males which have founded the outbred population. In this case the lines were maintained, as is customary, with overlapping generations.
Starting from the isofemale lines, one can also establish isogenic lines. To do so, each isofemale line can be taken through 20 generations of full-sib mating as done by Mackay et al. (2012). This procedure should purge deleterious alleles and provide, at a reasonable frequency, fertile and viable genetically-homogeneous lines.
Introgression
We introgressed the white mutant allele (w1118) into the outbred population. Using 80–100 single female crosses in the first two generations, we replaced all second and third chromosomes from the w1118 stock by “wild” chromosomes of the outbred population. In each odd generation (F1, F3, F5, F7, F9, F11) recombination in females reduces the contribution of the w1118 stock. From these crosses, 2 white-eyed males were used to establish at least 100 single female crosses with virgins from the outbred stock (all even generation F2, F4, F6, F8, and F10). In the F12 generation, 3–5 virgin females and 3–5 males from 140 F11 single-female crosses were released into a population cage to establish the white introgressed population.
Crossing scheme:
(P) Outbred females (O;O;O) × w; If/CyO; MKRS/TM6b males,
(F1) O/w; O/CyO; O/TM6b × Outbred males (O; O; O),
(F2) Outbred females (O;O;O) × w; O; O,
(F3) O/w; O; O × Outbred males (O; O; O),
F2 and F3 crosses were repeated 4 more rounds,
(F12) O/w; O; O × O/w; O; O,
Number of individuals used in each single-female cross throughout the introgression procedure.

Results
Collection
Generic methods to collect Drosophila species have been described in Markow and O'Grady (Markow, 2006). For the specific collection of Drosophila melanogaster, our own experience favors the choice of a vineyard as the collection site, given the advantages that collecting from large populations provides (particularly during harvest, which corresponds roughly to the period from August to October in the northern hemisphere) (Figure 1). Using a hand vacuum cleaner adapted to this purpose (see M&M), in 2007 and 2013, we collected in 1h around 5000 females from the José Maria da Fonseca winery in Southern Portugal (Azeitão, Portugal GPS: 38° 31′ 04.91″ N9° 00′ 56.24″).
Back to the lab, we followed systematically the steps schematically represented in Figure 2 in order to maximize the collected resources. First, single females were separated in vials to lay eggs and ensure the next generation. Then, the progenitor females were sacrificed and used as starting material for individual nucleic acids extraction (both DNA and RNA) in 96-well plates.
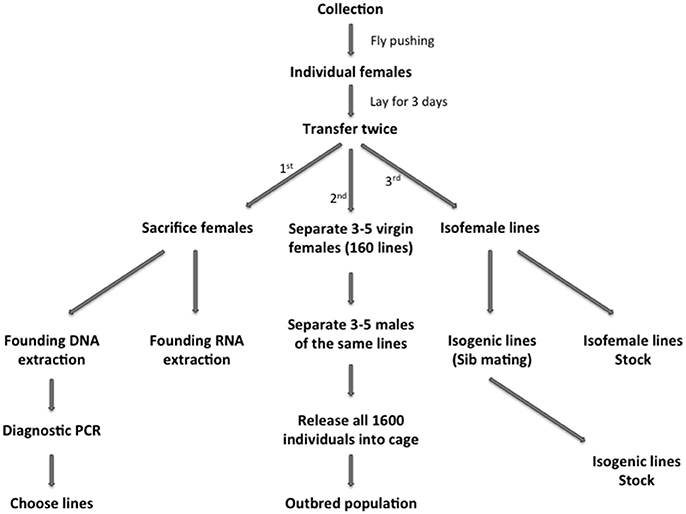
Figure 2. Pipeline of how to establish Drosophila laboratory resources upon collection from nature.
Screening Isofemale Lines
Guided by Alberto Civetta (Minuk and Civetta, 2011), we have scanned the genomes of D. melanogaster and D. simulans for large indels that would be diagnostic of species through a simple PCR followed by electrophoresis. We chose Slif (CG11128) for which we designed a primer pair (see M&M) that amplifies fragments of 939 bp in D. melanogaster and 1058 bp in D. simulans. To detect Wolbachia, we used primers designed for wsp (Wolbachia surface protein) by Zhou et al. (1998) (see M&M), which produce two differently sized fragments of 610 and 590 bp, for D. melanogaster and D. simulans, respectively. The Wolbachia wsp gene amplification product serves therefore two purposes: the characterization of the individual's infection status and a second (indirect) confirmation of the Drosophila species itself, conveyed by the different amplicon sizes generated by respective Wolbachia strains.
Figure 3 shows how this combination of primers reveals simultaneously, in a multiplex PCR (see M&M), the status of Wolbachia infection and which of the cryptic species it is. The PCR program used permits the simultaneous amplification of both fragments without any primer incompatibility or confounding effects from nonspecific bands. We have performed this method at high throughput rate using 96-well plates.
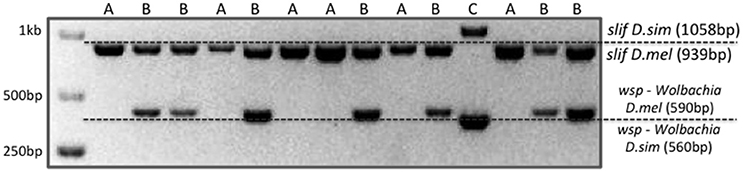
Figure 3. Rapid Drosophila species identification and infection status by multiplex PCR. Each lane contains the PCR amplification products using Wolbachia specific primers and primers for the gene slif. In both cases, amplification products have distinctive sizes that allow identification of the species and determination of Wolbachia infection status. Letters A, B, or C refer to D. melanogaster/ Wolbachia-negative, D. melanogaster/ Wolbachia-positive and D. simulans/ Wolbachia-positive samples, respectively.
To validate the species diagnosis, we performed in parallel a blind test running our method and performing the classical male genitalia classification based on morphological analysis. For this we took 50 males from one collection and mounted their genitalia (Figure 4) as described in Ashburner (Ashburner et al., 2011) and used the carcass to prepare genomic DNA for each individual. We found a 100% match (50 in 50) fit between the two classification methods, leading to the conclusion that the method we developed is at least as good as the more time-consuming (and more subjective) classical method.

Figure 4. Species confirmation using male genitalia. As described in (Ashburner et al., 2011) we show the distinctive male genital arch (arrows) of D. simulans (left) and D. melanogaster (right).
As an illustration of the proportions found in our specific case, in the 2013 collection we genotyped 576 (96 × 6) fertile females (progenitors of isofemale lines), being 341 (D.mel/Wol+), 189 (D.mel/Wol−), 36 (D.sim/Wol+), 1 (D.sim/Wol+), and 9 failed extractions/amplifications.
Isofemale Lines and Outbred Population Establishment
From the collection described above, we established an outbred population from 160 fertilized Wolbachia-positive females. We used 3–5 virgin females and 3–5 males from the F1 of each previously screened line. In parallel, we started 160 isofemale lines, kept in similar maintenance conditions but under an inbreeding regime, in vials with uncontrolled census and overlapping generations. Further, after 3 years we re-derived an outbred population without Wolbachia through tetracycline treatment (see M&M). After the full procedure, the new population (Wolbachia-free) was ready to be expanded and tested.
We performed extensive analyses for quality-control purposes of these established populations. Specifically, we quantified heterozygosity (H) and Fst for a subset of the starting foundation lines (using the extracted and stored DNA as described above) and in the two established outbred populations. We used the genomic DNA extracted from the founding females in 2007 to genotype 48 individuals and estimated an Expected Heterozygosities (Hexp) of 0.344 (±0.016 SE). In 2010 we repeated the procedure on the established outbred population to find a Hexp of 0.320 (±0.017 SE) (Martins et al., 2014). We estimated the Fst at 0.038.
Experimental Evolution
After the establishment of laboratory-controlled outbred populations, namely of Drosophila melanogaster infected with Wolbachia, we waited several generations for adaptation to the lab and consecutive stabilization of genetic pool. After that, we sequentially expanded the population in order to generate several identical replicas (see M&M). In our case, we used those populations to follow their response and genetic configuration upon different pathogenic challenges and different infection routes (Martins et al., 2013, 2014). Additionally, we also approach the eventual costs of adaptation and the influence of the increase of immunocompetence on Wolbachia population (Faria et al., 2015) and the evolutionary significance of its presence (Faria et al., 2016).
In each generation of experimental evolution, 200–400 flies (males and females) were frozen and posteriorly used for different analyses, such as PoolSeq and genotyping (Martins et al., 2014; Faria et al., 2016).
Introgression
After the foundation of outbred populations, the introgression of specific alleles in the populations can be performed. In the case of visible markers this procedure is rather simple, though laborious, as exemplified by the scheme below used to introgress the white mutant allele (w1118) into the outbred population. Using over 100 single female crosses per generation, we recombined the white mutant allele into “wild” chromossomes 6 times. Also, all other chromosomes were outbred as they were replaced in full (using balancer chromosomes) from the initial generations of the crossing scheme (see M&M). With this number of recombination rounds the estimated proportion of the white allele-carrying X chromossome in the final population is theoretically inferior to 2% (2∧6 = 1/64). Except for the fragment of the X in linkage with the white locus, every other fragment from the w1118 stock was randomized and should have virtually no impact on the phenotypes observed at the population level. Effectively, we have generated an outbred population genetically indistinguishable from the wild-derived initial outbred population except for the fact it carries the w1118 allele.
Additionally, we have started a systematic introgression of transgenic lines into the outbred background using the same crossing scheme.
Discussion
We have attempted to propose a systematic and normalized set of procedures when establishing Drosophila tools upon collection of flies from nature. This approach can be extended to any cryptic species pair, including both intra and extracellular symbionts. In our case, a collection in vineyard in Portugal (Figure 1) potentially allows the establishment a total of 4 different populations: D. melanogaster and D. simulans, with or without Wolbachia. Interestingly, we also observed in our collections the sporadic presence of parasitoid wasps, in particular species belonging to the genus Leptopilina. Although it is possible to start some laboratorial isolines with captured wasps, the low frequency indicates that the open-air method (or even the location) used to flies is not the most suitable to catch high number of wasps to, for example, initiate an outbred population of wasps. Other relevant studies could be performed with these flies concerning the gut microbiome of different capture regions and seasons, which probably reflects the difference in locations and diets.
The generic pipeline here presented (Figure 2) can be applied to any collection regardless of the fly species and initial object of study as it preserves to the fullest the potential of samples for future analyses. Inbred lines and the outbred population have, in principle, retained to a great extent the same qualitative variation of the sampled population. Yet, these different methods of maintaining specimens impact differently in genetic variation, namely in what regards the frequencies of deleterious recessive alleles and epistatic complexes (Rose, 1984; Mackay, 2013). For example, this has been approached by Huang and co-workers that found distinct genetic bases for the same traits analyzed by GWAS on the DGRP panel or on a population reconstituted from the same DGRP panel lines (Huang et al., 2012). Instead, a recent study shows that no significant allele differences are found between an ancestral population and a reconstituted counterpart generated by isofemale lines derived from the same original population (Nouhaud et al., 2016). Moreover, these approaches may reveal comparable outcomes and corroborate one another as illustrated by resistance against DCV infection, for example (Magwire et al., 2012; Martins et al., 2014). Thus, since there is not always compatibility in the results revealed by both methods, it is essential to deeply understand where exactly lies the difference of power: (i) in signal detection or statistical constraint or (ii) in biological differences, such as the absence of new epistasis in isolines (GWAS) or costs under-selection (Evolve&resequence). Be that as it may, isofemale lines or isogenic lines, on one hand, and outbred populations, on another, are best suited for different purposes and questions.
The sequential 96 well-plate protocols of DNA extraction, multiplex PCR and agarose gel electrophoresis allows a quick analysis of a large number of specimens. Moreover, the method is also very reliable to distinguish between D. melanogaster and D. simulans, as evidenced by the comparison with the male genitalia method (Figure 4). However, while the preparation of the genitalia is a time consuming procedure that may need an experienced manipulator to dissect, assemble and efficiently distinguish both species, the PCR here described easily allows a high-throughput scale. This method tolerates the testing of large numbers of individuals necessary for the foundation of outbred populations. In addition, after this first nucleic acids extraction, the resulting material may be used for further genetic tests. In addition, in this setup, other primers may be included or changed to quickly diagnose different species and/or strains.
Prior to starting selection experiments, it is essential to adapt the outbred population to the lab, in itself a novel environment to which the population is exposed (Simões et al., 2007; Santos et al., 2010). Major changes may occur in the population structure during this period of adaptation to laboratorial conditions, both in flies and associated microbiome. After this step, performing pilot tests is essential to confirm that replicate populations respond similarly against the chosen selective pressure. Another complementary and important verification in each foundation is to investigate the linkage disequilibrium in both approaches, isolines and outbred populations, to then understand the real effect of bringing specimens to the lab and of the consequent associated bottleneck. This information helps to characterize and validate the populations before testing and provides a clear idea of the potential of this tool in future studies. In our case, and based in the markers used, the estimation of heterozygosity reveals that the populations do not loose significant variability over time. The estimation of the effective population size based on linkage disequilibrium revealed that we had enough mapping resolution to work with the chosen markers.
Population replicates are therefore ready to be submitted against a vast range of selective pressures. In our case, as already mentioned, we evolved those populations in the presence or absence of pathogens, namely Pseudomonas entomophila and Drosophila C Virus (DCV). Throughout experimental evolution, we have frozen adult flies in each generation, creating a bank to explore genetic questions about the adaptive processes.
The posterior introgression of genetic markers could also create very useful tools to evolutionary and genetic questions. The outbred w1118 population can be compared and/or used as control as it is easily distinguishable from the outbred population, though virtually identical from a genetic perspective. Behavioral and competition experiments are also important applications of this tool. However, caution is advised in this case, as the white mutation is far from being a fully innocuous marker. Indeed, white codes for an ABC transporter subunit (Sullivan and Sullivan, 1975) described to play a role in a number of homeostatic functions, namely in the nervous system. This important role may impinge functionally on a number of traits (Diegelmann, 2006; Sitaraman et al., 2008).
Finally, this population can be used to introgress transgenic constructs into the outbred background. Following the same crossing scheme (see M&M) it is straightforward to introgress into the outbred background mini-white containing transgenics, namely of the vast available collection of UAS and GAL4 lines. In this case, upon recombination in heterozygous females, non-white males can be selected to cross against outbred white females. This may prove to be an interesting tool to test the effects of such transgenes in a properly controlled genetic background.
In pace with the growing technological and conceptual advances developed for the dissection of the genotype-phenotype map, rises the interest in studying SGV in natural populations. Here, we have attempted to provide the community with a comprehensive guide for the establishment and development of the necessary laboratory resources stemming from the initial collection of wild Drosophila specimens. With this we hope also to contribute to the standardization of procedures permitting an easier exchange of resources across researchers engaging in the study of natural variation in laboratory conditions.
Author Contributions
VF and ES designed and performed the work here presented. VF and ES wrote the paper.
Funding
Fundação para a Ciência e a Tecnologia (FCT, Portugal) supported VF (#SFRH/BD/82299/2011). This work was supported by Instituto Gulbenkian de Ciência/Fundação Calouste Gulbenkian.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the ES, Beldade, and Mirth Labs for discussion. We are indebted to Alberto Civetta for sharing unpublished data and for advice on the choice of the species-diagnostic polymorphisms. We thank Henrique Teotónio for his help in setting up the outbred population in 2007. We thank Ivo Chelo, Henrique Teotónio, Nelson Martins, and Tânia Paulo for their help with the genomic analyses.
References
Abbott, J. K., Innocenti, P., Chippindale, A. K., and Morrow, E. H. (2013). Epigenetics and sex-specific fitness: an experimental test using male-limited evolution in Drosophila melanogaster. PLoS ONE 8:e70493. doi: 10.1371/journal.pone.0070493
Ashburner, M., Golic, K., and Hawley, R. S. (2011). Drosophila: A Laboratory Handbook. 2nd Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Barrett, R. D. H., and Schluter, D. (2008). Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44. doi: 10.1016/j.tree.2007.09.008
Barrón, M. G., Fiston-Lavier, A.-S., Petrov, D. A., and González, J. (2014). Population genomics of transposable elements in Drosophila. Annu. Rev. Genet. 48, 561–581. doi: 10.1146/annurev-genet-120213-092359
Begun, D. J., Holloway, A. K., Stevens, K., Hillier, L. W., Poh, Y.-P., Hahn, M. W., et al. (2007). Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 5:e310. doi: 10.1371/journal.pbio.0050310
Bergland, A. O., Behrman, E. L., O'Brien, K. R., Schmidt, P. S., and Petrov, D. A. (2014). Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genet. 10:e1004775. doi: 10.1371/journal.pgen.1004775
Burke, M. K., Dunham, J. P., Shahrestani, P., Thornton, K. R., Rose, M. R., and Long, A. D. (2010). Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature 467, 587–590. doi: 10.1038/nature09352
Burke, M. K., King, E. G., Shahrestani, P., Rose, M. R., and Long, A. D. (2014). Genome-wide association study of extreme longevity in Drosophila melanogaster. Genome Biol. Evol. 6, 1–11. doi: 10.1093/gbe/evt180
Clark, A. G., Silveria, S., Meyers, W., and Langley, C. H. (1994). Nature screen: an efficient method for screening natural populations of Drosophila for targeted P-element insertions. Proc. Natl. Acad. Sci. U.S.A. 91, 719–722. doi: 10.1073/pnas.91.2.719
Cogni, R., Cao, C., Day, J. P., Bridson, C., and Jiggins, F. M. (2016). The genetic architecture of resistance to virus infection in Drosophila. Mol. Ecol. 25, 5228–5241. doi: 10.1111/mec.13769
Diegelmann, S. (2006). Genetic dissociation of acquisition and memory strength in the heat-box spatial learning paradigm in Drosophila. Learn. Mem. 13, 72–83. doi: 10.1101/lm.45506
Durham, M. F., Magwire, M. M., Stone, E. A., and Leips, J. (2014). Genome-wide analysis in Drosophila reveals age-specific effects of SNPs on fitness traits. Nat. Commun. 5, 4338. doi: 10.1038/ncomms5338
Fabian, D. K., Kapun, M., Nolte, V., Kofler, R., Schmidt, P. S., Schlötterer, C., et al. (2012). Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol. Ecol. 21, 4748–4769. doi: 10.1111/j.1365-294X.2012.05731.x
Faria, V. G., Martins, N. E., Magalhães, S., Paulo, T. F., Nolte, V., Schlötterer, C., et al. (2016). Drosophila adaptation to viral infection through defensive symbiont evolution. PLoS Genet. 12:e1006297. doi: 10.1371/journal.pgen.1006297
Faria, V. G., Martins, N. E., Paulo, T., Teixeira, L., Sucena, É., and Magalhães, S. (2015). Evolution of Drosophila resistance against different pathogens and infection routes entails no detectable maintenance costs. Evolution 69, 2799–2809. doi: 10.1111/evo.12782
Gibbs, A. G., Chippindale, A. K., and Rose, M. R. (1997). Physiological mechanisms of evolved desiccation resistance in Drosophila melanogaster. J. Exp. Biol. 200, 1821–1832.
Hoffmann, A. A., Turelli, M., and Harshman, L. G. (1990). Factors affecting the distribution of cytoplasmic incompatibility in Drosophila simulans. Genetics 126, 933–948.
Huang, W., Richards, S., Carbone, M. A., Zhu, D., Anholt, R. R., Ayroles, J. F., et al. (2012). Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proc. Natl. Acad. Sci. U.S.A. 109, 15553–15559. doi: 10.1073/pnas.1213423109
Jordan, K. W., Craver, K. L., Magwire, M. M., Cubilla, C. E., Mackay, T. F., and Anholt, R. R. (2012). Genome-wide association for sensitivity to chronic oxidative stress in Drosophila melanogaster. PLoS ONE 7:e38722. doi: 10.1371/journal.pone.0038722
Kawecki, T. J., Lenski, R. E., Ebert, D., Hollis, B., Olivieri, I., and Whitlock, M. C. (2012). Experimental evolution. Trends Ecol. Evol. 27, 547–560. doi: 10.1016/j.tree.2012.06.001
King, E. G., Macdonald, S. J., and Long, A. D. (2012). Properties and power of the Drosophila synthetic population resource for the routine dissection of complex traits. Genetics 191, 935–949. doi: 10.1534/genetics.112.138537
Kislukhin, G., King, E. G., Walters, K. N., Macdonald, S. J., and Long, A. D. (2013). The genetic architecture of methotrexate toxicity is similar in Drosophila melanogaster and humans. G3 (Bethesda) 3, 1301–1310. doi: 10.1534/g3.113.006619
Klepsatel, P., Gáliková, M., De Maio, N., Huber, C. D., Schlötterer, C., and Flatt, T. (2013). Variation in thermal performance and reaction norms among populations of Drosophila melanogaster: thermal performance in Drosophila. Evolution 67, 3573–3587. doi: 10.1111/evo.12221
Kofler, R., and Schlötterer, C. (2014). A guide for the design of evolve and resequencing studies. Mol. Biol. Evol. 31, 474–483. doi: 10.1093/molbev/mst221
Kriesner, P., Hoffmann, A. A., Lee, S. F., Turelli, M., and Weeks, A. R. (2013). Rapid sequential spread of two wolbachia variants in Drosophila simulans. PLoS Pathog. 9:e1003607. doi: 10.1371/journal.ppat.1003607
Lazzaro, B. P., Sackton, T. B., and Clark, A. G. (2006). Genetic variation in Drosophila melanogaster resistance to infection: a comparison across bacteria. Genetics 174, 1539–1554. doi: 10.1534/genetics.105.054593
Lazzaro, B. P., Sceurman, B. K., and Clark, A. G. (2004). Genetic basis of natural variation in D. melanogaster antibacterial immunity. Science 303, 1873–1876. doi: 10.1126/science.1092447
Macdonald, S. J. (2004). A Potential Regulatory polymorphism upstream of hairy is not associated with bristle number variation in wild-caught Drosophila. Genetics 167, 2127–2131. doi: 10.1534/genetics.104.026732
Mackay, T. F. (2013). Epistasis and quantitative traits: using model organisms to study gene–gene interactions. Nat. Rev. Genet. 15, 22–33. doi: 10.1038/nrg3627
Mackay, T. F., Richards, S., Stone, E. A., Barbadilla, A., Ayroles, J. F., Zhu, D., et al. (2012). The Drosophila melanogaster genetic reference panel. Nature 482, 173–178. doi: 10.1038/nature10811
Magalhães, S., and Matos, M. (2012). Strengths and weaknesses of experimental evolution. Trends Ecol. Evol. 27, 649–650. doi: 10.1016/j.tree.2012.08.004
Magwire, M. M., Fabian, D. K., Schweyen, H., Cao, C., Longdon, B., Bayer, F., et al. (2012). Genome-wide association studies reveal a simple genetic basis of resistance to naturally coevolving viruses in Drosophila melanogaster. PLoS Genet. 8:e1003057. doi: 10.1371/journal.pgen.1003057
Markow, T. A. (2006). Drosophila: A Guide to Species Identification and Use. Amsterdam; Boston, MA: Elsevier/AP.
Marriage, T. N., King, E. G., Long, A. D., and Macdonald, S. J. (2014). Fine-mapping nicotine resistance loci in Drosophila using a multiparent advanced generation inter-cross population. Genetics 198, 45–57. doi: 10.1534/genetics.114.162107
Martins, N. E., Faria, V. G., Nolte, V., Schlötterer, C., Teixeira, L., Sucena, É., et al. (2014). Host adaptation to viruses relies on few genes with different cross-resistance properties. Proc. Natl. Acad. Sci. U.S.A. 111, 5938–5943. doi: 10.1073/pnas.1400378111
Martins, N. E., Faria, V. G., Teixeira, L., Magalhães, S., and Sucena, É. (2013). Host adaptation is contingent upon the infection route taken by pathogens. PLoS Pathog. 9:e1003601. doi: 10.1371/journal.ppat.1003601
Mathur, V., and Schmidt, P. S. (2017). Adaptive patterns of phenotypic plasticity in laboratory and field environments in Drosophila melanogaster. Evolution 71, 465–474. doi: 10.1111/evo.13144
Minuk, L., and Civetta, A. (2011). Drosophila genomes and the development of affordable molecular markers for species genotyping. Genome 54, 341–347. doi: 10.1139/g10-120
Nouhaud, P., Tobler, R., Nolte, V., and Schlötterer, C. (2016). Ancestral population reconstitution from isofemale lines as a tool for experimental evolution. Ecol. Evol. 6, 7169–7175. doi: 10.1002/ece3.2402
Nunes, M. D., Neumeier, H., and Schlötterer, C. (2008). Contrasting patterns of natural variation in global Drosophila melanogaster populations. Mol. Ecol. 17, 4470–4479. doi: 10.1111/j.1365-294X.2008.03944.x
Orozco-terWengel, P., Kapun, M., Nolte, V., Kofler, R., Flatt, T., and SchlöTterer, C. (2012). Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles: genomic signatures of adaptation to new environment. Mol. Ecol. 21, 4931–4941. doi: 10.1111/j.1365-294X.2012.05673.x
Rajpurohit, S., Hanus, R., Vrkoslav, V., Behrman, E. L., Bergland, A. O., Petrov, D., et al. (2017). Adaptive dynamics of cuticular hydrocarbons in Drosophila. J. Evol. Biol. 30, 66–80. doi: 10.1111/jeb.12988
Remolina, S. C., Chang, P. L., Leips, J., Nuzhdin, S. V., and Hughes, K. A. (2012). Genomic basis of aging and life-history evolution in Drosophila melanogaster. Evolution 66, 3390–3403. doi: 10.1111/j.1558-5646.2012.01710.x
Rose, M. R. (1984). Genetic covariation in Drosophila life-history: untangling the data. Am. Nat. 123, 565–569. doi: 10.1086/284222
Santos, M., Fragata, I., Santos, J., Simoes, P., Marques, A., Lima, M., et al. (2010). Playing darwin. part b. 20 years of domestication in Drosophila subobscura. Theory Biosci. 129, 97–102. doi: 10.1007/s12064-010-0086-8
Scott, D., Shields, A., Straker, M., Dalrymple, H., Dhillon, P. K., and Harbinder, S. (2011). Variation in the male pheromones and mating success of wild caught Drosophila melanogaster. PLoS ONE 6:e23645. doi: 10.1371/journal.pone.0023645
Simões, P., Rose, M. R., Duarte, A., GonçAlves, R., and Matos, M. (2007). Evolutionary domestication in Drosophila subobscura. J. Evol. Biol. 20, 758–766. doi: 10.1111/j.1420-9101.2006.01244.x
Sitaraman, D., Zars, M., LaFerriere, H., Chen, Y.-C., Sable-Smith, A., Kitamoto, T., et al. (2008). Serotonin is necessary for place memory in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 105, 5579–5584. doi: 10.1073/pnas.0710168105
Sullivan, D. T., and Sullivan, M. C. (1975). Transport defects as the physiological basis for eye color mutants of Drosophila melanogaster. Biochem. Genet. 13, 603–613. doi: 10.1007/BF00484918
Swarup, S., Huang, W., Mackay, T. F., and Anholt, R. R. (2013). Analysis of natural variation reveals neurogenetic networks for Drosophila olfactory behavior. Proc. Natl. Acad. Sci. U.S.A. 110, 1017–1022. doi: 10.1073/pnas.1220168110
Teotónio, H., Chelo, I. M., Bradić, M., Rose, M. R., and Long, A. D. (2009). Experimental evolution reveals natural selection on standing genetic variation. Nat. Genet. 41, 251–257. doi: 10.1038/ng.289
Tobler, R., Franssen, S. U., Kofler, R., Orozco-Terwengel, P., Nolte, V., Hermisson, J., et al. (2014). Massive habitat-specific genomic response in D. melanogaster populations during experimental evolution in hot and cold environments. Mol. Biol. Evol. 31, 364–375. doi: 10.1093/molbev/mst205
Turelli, M., and Hoffmann, A. A. (1995). Cytoplasmic incompatibility in Drosophila simulans: dynamics and parameter estimates from natural populations. Genetics 140, 1319–1338.
Turner, T. L., and Miller, P. M. (2012). Investigating natural variation in Drosophila courtship song by the evolve and resequence approach. Genetics 191, 633–642. doi: 10.1534/genetics.112.139337
Turner, T. L., Miller, P. M., and Cochrane, V. A. (2013). Combining genome-wide methods to investigate the genetic complexity of courtship song variation in Drosophila melanogaster. Mol. Biol. Evol. 30, 2113–2120. doi: 10.1093/molbev/mst111
Turner, T. L., Stewart, A. D., Fields, A. T., Rice, W. R., and Tarone, A. M. (2011). Population-based resequencing of experimentally evolved populations reveals the genetic basis of body size variation in Drosophila melanogaster. PLoS Genet. 7:e1001336. doi: 10.1371/journal.pgen.1001336
Ventura, I. M., Costa, T., and Klaczko, L. B. (2013). Low temperature reveals genetic variability against male-killing spiroplasma in Drosophila melanogaster natural populations. Microb. Ecol. 67, 229–235. doi: 10.1007/s00248-013-0295-z
Weber, A. L., Khan, G. F., Magwire, M. M., Tabor, C. L., Mackay, T. F., and Anholt, R. R. (2012). Genome-wide association analysis of oxidative stress resistance in Drosophila melanogaster. PLoS ONE 7:e34745. doi: 10.1371/journal.pone.0034745
Zhao, X., Bergland, A. O., Behrman, E. L., Gregory, B. D., Petrov, D. A., and Schmidt, P. S. (2016). Global transcriptional profiling of diapause and climatic adaptation in Drosophila melanogaster. Mol. Biol. Evol. 33, 707–720. doi: 10.1093/molbev/msv263
Zhou, D., Udpa, N., Gersten, M., Visk, D. W., Bashir, A., Xue, J., et al. (2011). Experimental selection of hypoxia-tolerant Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 108, 2349–2354. doi: 10.1073/pnas.1010643108
Zhou, W., Rousset, F., and O'Neil, S. (1998). Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc. Biol. Sci. 265, 509–515. doi: 10.1098/rspb.1998.0324
Keywords: experimental evolution, Drosophila outbred populations, cryptic species multiplex PCR, isofemale lines, introgression
Citation: Faria VG and Sucena É (2017) From Nature to the Lab: Establishing Drosophila Resources for Evolutionary Genetics. Front. Ecol. Evol. 5:61. doi: 10.3389/fevo.2017.00061
Received: 11 October 2016; Accepted: 26 May 2017;
Published: 08 June 2017.
Edited by:
Brian P. Lazzaro, Pennsylvania State University, United StatesReviewed by:
Paul Schmidt, University of Pennsylvania, United StatesRobert Unckless, University of Kansas, United States
Copyright © 2017 Faria and Sucena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vitor G. Faria, dmZhcmlhQGlnYy5ndWxiZW5raWFuLnB0
Élio Sucena, ZXN1Y2VuYUBpZ2MuZ3VsYmVua2lhbi5wdA==