 Lucas P. P. Braga
Lucas P. P. Braga Shannon M. Soucy
Shannon M. Soucy Deyvid E. Amgarten
Deyvid E. Amgarten Aline M. da Silva
Aline M. da Silva João C. Setubal
João C. Setubal- 1Departmento de Bioquímica, Instituto de Química, Universidade de São Paulo, São Paulo, Brazil
- 2UMR1347 Agroécologie, Institut National de la Recherche Agronomique, Dijon, France
- 3Evolutionary Computational Genomics Laboratory, Dartmouth College, Hanover, NH, United States
Phages have a major impact on microbial populations. In this work, we discuss how predation, transduction, lysogeny, and phage domestication lead to symbio-centric genomic interactions between bacteria and phages, ranging from antagonistic to mutualistic. Furthermore, these interactions influence bacterial diversification and ecotype formation. We then propose an additional consideration in the form of a symbio-centric ecological speciation framework for bacteria. Our framework builds upon classical morphological and molecular taxonomy by also considering bacteria and their phages as a unit of evolutionary selection. This framework acknowledges the considerable effect that phage interaction has on bacterial genomic content, regulation, and evolution, and will advance our understanding of bacterial evolution.
Introduction
The work of microbiology pioneers Anton van Leeuwenhoek, Ferdinand Cohn, Albert Koch, Julius Petri, and Martinus Beijerinck, revealed the immense diversity of the microbial world and enabled the formulation of the first hypotheses about bacterial ubiquity and diversification. Early hypotheses about forces that regulated microbial diversity acknowledged the importance of environmental factors; that is, given the same conditions, the same microorganisms would grow, thus in similar environments, the same species would be present (Pasteur, 1861; Van Niel, 1949). In other words, “everything is everywhere,” the environment selects. This hypothesis has been generally accepted in the microbial ecology field until recently (see detailed discussion in O'Malley, 2008).
Although it is true that some bacterial species have been detected in various locations across the planet, the hypothesis that “everything is everywhere” may obscure our perception of the evolutionary processes modulating bacterial diversification. For example, Salinibacter ruber strains were isolated from 14 different locations across Mediterranean, Atlantic, and Peruvian sites. However, these strains were found to be considerably different based on metabolic characteristics, and this difference was correlated with geography (Rosselló-Mora et al., 2008). Should all S. ruber isolates be considered the same species? They undoubtedly share common genomic features, but considering the all different strains of S. ruber as a single species would hinder our understanding of the evolutionary processes that contributed to the forces that promoted the contemporary differences in metabolic potential.
Considerable effort has been devoted to determine genomic delineations used to demarcate microbial species as well as provide information on how bacterial species are generated and maintained over time, colloquially referred to as “the bacterial species problem,” but no consensus has been achieved so far (Konstantinidis and Tiedje, 2005; Achtman and Wagner, 2008; Doolittle and Zhaxybayeva, 2009; Shapiro et al., 2012; Cordero and Polz, 2014; Booth et al., 2016). Horizontal gene transfer (HGT) is recognized as a powerful phenomenon capable of generating variation in microbial populations (Soucy et al., 2015; Box 1), however current species delineations fail to consider the impact of HGT during classification of organisms into clusters of species. It seems that all gene families in prokaryotes have been affected by HGT, and generally, no two gene families, including 16S rDNA, share the same exact evolutionary history (Doolittle, 1999; Koonin et al., 2001; Gogarten and Townsend, 2005; Gribaldo and Brochier, 2009; Zhaxybayeva, 2009; Boto, 2010; Andam and Gogarten, 2011; Zhaxybayeva and Doolittle, 2011).
Box 1. Horizontal gene transfer—metabolic expansion and trait diversification.
Horizontal gene transfer (HGT) plays a major role in the evolution of metabolic pathways, and is a major driver of shared physiological traits allowing rapid adaptive evolution in the presence of environmental changes (Koonin et al., 2001; Pál et al., 2005; Treangen and Rocha, 2011; Puigbò et al., 2014). One example is the evolution of photosynthesis. Well-supported events of horizontal transfer of photosystem genes inter and intra phylum (Acidobacteria, Chlorobi, Choroflexi, Cyanobacteria, Firmicutes, and Proteobacteria) suggest that HGT played a considerable role in the evolution of this trait (Raymond, 2009). More specifically, for example, anoxygenic photosynthesis in Rhodopseudomonas palustris (Alpha-proteobacteria), Rhodocyclus purpureus (Beta-proteobacteria), and Ectothiorhodospira marina (Gamma-proteobacteria) rely on the same genes (Bapteste and Boucher, 2009). Other examples about the influence of HGT in the evolution of other traits, such as methylaspartate cycling and methanogenesis, have been described (Fournier, 2009; Maslov et al., 2009; Khomyakova et al., 2011).
Besides the acquisition of novel genes, HGT also provides means for recombination and helps avoid collapse of the population due to mutational meltdown (Muller's ratchet process) (Lynch et al., 1993; Gordo et al., 2002; Iranzo et al., 2016). Koonin and Novozhilov (2009) have pointed out the relation between HGT and the universality of the genetic code, allowing the flow of information necessary for evolution among biological entities in all directions (i.e., vertically and horizontally). HGT promotes diversification through genetic mixing between gigantic evolutionary distances (such as viruses and eukaryotes) thanks to the universality of the genetic code.
Despite significant breakthroughs in our understanding of neutral processes (Zeng et al., 2015), a large and diverse collection of microbial genomes for comparison (National Center for Biotechnology Information—NCBI; Nordberg et al., 2014), a growing appreciation for integrative methods (Edwards and Knowles, 2014), and accepted impact of HGT, we currently lack a detailed understanding of how microbial diversity eventually leads to speciation. Additional consideration of the environmental parameters at the time the samples are taken, though often not feasible, would improve the understanding of contemporary forces of selection that are maintaining some of the metabolic differentiation between strains. However, environmental data represents only a portion of the selective pressures that microbial populations experience. Coevolutionary forces are responsible for promoting considerable changes in the interacting players (Thompson, 2013). Perhaps then, a more significant source of selective pressure imparted on microbial populations is phage predation. Some estimates hypothesize that phages are ten times more abundant than their bacterial hosts, and thus far everywhere bacteria have been found there are also phages. Bacteria-phage interactions often influence genome reticulations in both parties. In particular, infection with lysogenic phages significantly expand the genic repertoire of the host, usually expressing many accessory genes while the phage genome remains dormant in the host genome (Rohwer et al., 2014). In accordance with this, it is relevant to consider environmental selection acting on microbial populations to include the contributions of phage coevolution for microbial selection.
In the following sections, we will discuss how microbial genomic diversity eventually leads to ecological speciation, and we propose that considering the interaction of bacteria and their phage symbionts (Koonin, 2011) will lead to a deeper understanding of the mechanisms driving microbial evolution. We demonstrate how phages may modulate bacterial evolution by influencing fluctuating selection and evolutionary processes driven by HGT, and thus play a relevant role in bacterial ecological speciation.
Ecological Speciation without Phages
Ecological speciation is becoming the current paradigm for studying species evolution (Thompson, 2013). An understanding of bacterial diversification may be easier to achieve in light of ecological speciation, which is the formation of new ecologically distinct populations (i.e., ecotypes). However, before connecting ecological speciation with the context of bacteria-phage coevolution, it is necessary to disentangle the traditional ecological speciation framework that has been proposed for bacteria. The traditional ecological speciation model was proposed by Cohan (2001), and it relies on the assumption that cohesive forces in microbial populations are driven by a diversity-purging process in which genetic variability within the population is periodically reset by environmental changes, and the most adapted genotype prevails through a selective sweep. That is, in a heterogeneous population, a subgroup which evolves an advantageous trait for its specific niche will outcompete the other subgroups, driving the genotypes with lower fitness to extinction (i.e., a genome-wide sweep mechanism). Speciation, based on this model, happens when a subgroup evolves an advantageous trait that allows colonization of a new niche, and this subgroup becomes a new ecotype. Ecotype formation creates two separate but related populations that are no longer subject to the same periodic selection and cohesive forces (Cohan and Perry, 2007).
According to this traditional perspective, the ecological speciation framework resembles directional selection, because it favors the most adapted genotype. However, Thompson (2013) points out that focusing only on directional selection processes obscures the ecology and the whole dynamics of evolutionary changes. Ultimately, selective sweeps with a single genotype would lead to the erosion of the genotypic diversity in the population. Furthermore, a newly formed ecotype dominated by individuals from clonal expansion would be exceptionally prone to extinction given the selective pressure imparted by predators and parasites. Directional selection is undoubtedly important in ecotype formation, while a bacterial population is becoming acclimated to environmental conditions. However, there is a significant genetic diversity at the intra population level, which implies that processes such as predation or HGT are promoting genetic diversity. Therefore, these processes should be considered for understanding the process of ecological speciation and new ecotype formation. Hence they should be included in the ecological speciation framework.
Natural selection acts to preserve diversity within local populations. Fluctuating selection is a particularly efficient process that maintains diversity in populations through ecological interactions such as the host-parasite dynamics (Thompson, 2013); phages are one possible agent of fluctuating selection (see below). Over time ever-present fluctuations in the intensity, direction, and targets of selection, varying from generation to generation (Koskella and Brockhurst, 2014) create a negative frequency-dependent selection (i.e., the more common a host is the less fit it becomes) that promotes diversity in the population (Tellier et al., 2014). A fluctuating selection imposes pressure for constant diversification within the population, and no single genotype becomes completely dominant. However, fluctuating selection only provides pressure for diversification; the extent and direction of diversification will depend on the mechanisms present in the population to generate diversity, the genic potential of a population, and the magnitude of selective pressure imparted through fluctuating selection.
A new ecotype can emerge through the acquisition of novel niche-specific traits. Trait acquisition can occur through an accumulation of mutations (genetic potential), which can be directed by adaptive evolution (i.e., evolvability). Additionally, increased mutation rates or genomic reticulations could lead to the exaptation of genes through duplication, domain shuffling, and altered regulation (True and Carroll, 2002; Zhang, 2003; Taylor and Raes, 2004; Bergthorsson et al., 2007; Pigliucci, 2008). One example of evolution by increased mutation rates is provided by the Escherichia coli long-term experiments by Richard Lenski and co-workers (Blount et al., 2008). Twelve cultures of E. coli from one single population were grown in the absence of phages and serially diluted daily in media containing glucose as a carbon source as well as citrate. After 31,500 generations, a single population evolved the ability to utilize citrate (Blount et al., 2008). Clades containing these mutations coexisted with strains unable to utilize citrate as citrate utilization was optimized in these clades. Eventually the clades that had not developed the ability to utilize both substrates were outcompeted and went extinct (Blount et al., 2012; Turner et al., 2015; Lenski, 2017). These observations are a useful starting point, but are essentially axenic. They show us that without phage interaction bacteria evolution over time resembles periodic selection (Cohan and Perry, 2007) and therefore the traditional ecological speciation model (Figure 1). As discussed above, when considering environmental populations, the effects of fluctuating selection and HGT must be layered on top of the underlying process of periodic selection. Phages in particular represent a link between fluctuating selection, as they are a powerful predatory element in the environment, and HGT, as they are also one of the mediators of HGT. This suggests that a closer look at bacteria-phage interactions may help throw light on the bacterial diversification process.
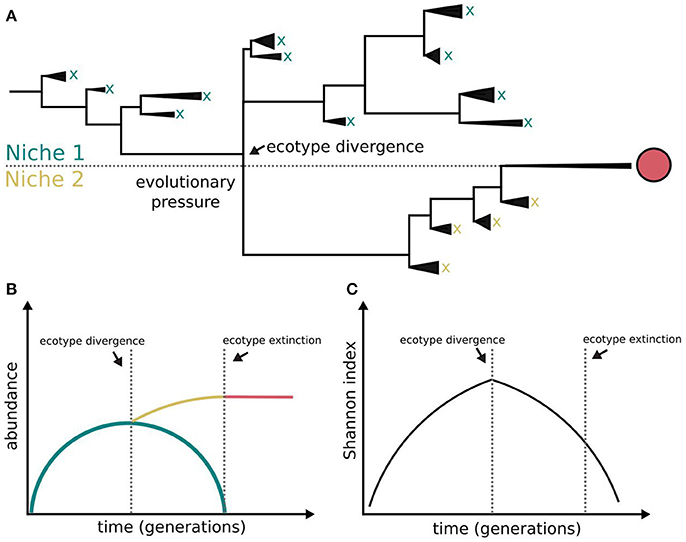
Figure 1. Bacterial diversification in the absence of phage interactions. Different clades within a population colonizing a challenging environment can evolve differently. Some of them may be able to evolve and colonize a different niche, i.e., ecotype differentiation. The capacity for coexistence based on independence of resource consumption may be limited and the evolved clade outcompetes the other clades and dominates both the novel and the ancestral niches (A). The symbols x and red circle represent clade extinction and dominance, respectively. The clades evolving outside evolutionary pressure (lower fitness) become extinct (B) following a diversity-purging process (C), i.e., periodic selection. According to this model ecological speciation in the absence of phages is constrained. This figure is based on Rodriguez-Valera et al. (2009), Blount et al. (2012), Turner et al. (2015), and Lenski (2017).
Ecological Speciation with Phages
Phage-bacteria interactions are unique in that phage interactions with bacteria can range from predatory, to parasitic, to commensal, and even to seemingly mutualistic. Different types of interactions lead to different evolutionary scenarios for the host population, and different genomic signatures. For example, Clustered Regularly Interspersed Short Palindromic Repeat (CRISPR) arrays provide a record of predatory attacks by phages. Considering the number of phage interactions and the type of phage interactions should provide more detail about the process of ecological speciation. The null expectation derives from experiments like the E. coli experiment mentioned above, in which no interaction with phages may result in directional selection and one dominant phenotype with relatively limited genotypic diversity. Below we discuss the implications for genetic diversity in a microbial population that result from each of the categories of phage-bacterial symbioses.
Antagonistic coevolution such as predator-prey dynamics is considered one of the major ecological drivers of genetic variation through fluctuating selection (Thompson, 2013). According to the model proposed by Rodriguez-Valera et al. (2009), the increase in abundance of a successful bacterial strain increases the chance of finding a phage specific to that strain (Lotka-Volterra dynamics). In this model, rare genotypes have a fitness advantage over abundant genotypes (i.e., kill-the-winner dynamics). Therefore, instead of periodic selection (Cohan, 2001), antagonistic interactions between bacteria and phages result in fluctuating selection. In this scenario, bacteria clades of a local population fluctuate around stable levels in a constant-diversity mode of selection, and diversity within the population remains high (Rodriguez-Valera et al., 2009).
Besides selecting for a more diverse population, phage predation can increase mutation rates in their hosts (Pal et al., 2007; Paterson et al., 2010), driving genome-wide evolution (i.e., not only in loci associated with resistance to phages), and increase the genetic divergence in an already diverse population (Scanlan et al., 2015; Cairns et al., 2017). This increase in diversity together with niche selection pressure promotes evolvability and the development of novel niche-specific traits (Box 2). It has been recently predicted that the more violent the perturbations by phage predation (i.e., lytic interactions), the more diverse the community is (Maslov and Sneppen, 2017). Phages contribute to taxonomic variability and functional stability in microbial communities (Louca and Doebeli, 2017).
Box 2. Phage predation favors evolvability of bacterial population.
Rapid and constant evolution of phages and their hosts imply that this specific evolutionary dynamic is likely to be an important factor in many natural scenarios. An empirical example was provided by the Pseudomonas fluorescenses/phi2 model (Buckling and Rainey, 2002), in which genetic divergence in host populations is influenced by phage antagonistic interactions. The authors found that bacterial populations were better able to resist phages from their own microcosm; symmetrically, phages were less able to infect populations from their own microcosm when compared to others. The subgroups of P. fluorescens separated by the microcosms became different from their ancestral population, at least in terms of traits associated with defense mechanisms.
Pal et al. (2007) could verify that with <200 bacterial generations, 25% of the populations coevolving with phages had evolved 10- to 100-fold increases in mutation rates owing to mutations in mismatch-repair genes, while bacteria population evolving without phages showed no significant change in mutation rate. Another example was provided by a study performing three weeks' incubation in chemostat cultures of a marine flavobacterium and virulent phages (Middelboe et al., 2009). The authors observed that a single-strain host population diversified to different multi-strain genotypes; remarkably, they also observed a diversification of metabolic properties in the new strains. Many of these strains have a reduced capacity to metabolize various carbon sources, as demonstrated by BIOLOG assays. It could be argued in this case that the authors have provided a demonstration of the cost associated with the development of phage defenses by the host. This is an important evidence of diversification in traits not directly related with mechanisms of infectivity. In a general way, phage-host coevolution is a decisive factor in the movement of a bacterial population through an adaptive landscape. According to Williams's evolutionary model (Williams, 2013), diversification in response to phage action allows the host community to sample a larger region of the fitness landscape than mutation alone. Thereby, phage action increases the likelihood of intervening fitness valleys and discovering higher fitness peaks. This scenario is similar to heuristics in mathematical optimization, where non-exhaustive search may place solutions at local maximum hills. Similar to the phage influence in the movement of a bacterial population through greater fitness, heuristics start from random points and apply perturbations to increase the likelihood of reaching the global maximum. In this case, the diversification caused by phage predation works as a bridge to higher absolute growth rates, which would otherwise not be reached.
Phages are not only predators but also parasites. Parasites are intrinsic to all life, and are considered major drivers of important innovations and evolutionary transitions (Koonin, 2016). Lysogeny is a parasitic symbiosis where phages delay cell lysis, integrate into the bacterial genome, and lay dormant. Lysogeny is more common when the multiplicity of infection is high, cells are abundant, cells are slower growing, or cells are smaller in size (Touchon et al., 2017). Lysogeny leads to a temporary alignment between the welfare of the bacteria and the phage. Active lysogeny regulates bacterial genes (Feiner et al., 2015). Most microbial genomes sequenced to date have at least one lysogen, and some have more than 20 lysogens, like Sodalis glossinidius which has 29 (Kang et al., in review). These prophages, another term for lysogenic phage, can reside in the host genome for generations, leading to orthologous but divergent prophages among related subgroups of bacteria (Bobay et al., 2014). Over time prophages will evolve through the accumulation of small deletion events, recombination with other phages the host is exposed to, and recombination with other regions of the host genome (Bobay et al., 2014). The accumulation of new genetic information or combinations of genes through recombination can lead to the emergence of diversity in the host (Lang et al., 2012; Gama et al., 2013; Bobay et al., 2014; Davies et al., 2016; Touchon et al., 2017).
Though lysogeny has the potential to lead to cell lysis there can be many potential benefits. For example, in some cases the bacteria has protection from related phage predation through superinfection exclusion, which prevents related phages from injecting their DNA into the host (Seed, 2015). Importantly, lysogens can enable niche invasion through lytic induction of a small portion of the population upon exposure to a new niche. If the established population is not immune it will succumb to phage predation, and the invading population, protected through superinfection exclusion, is exposed to new genic material released from the established population by phage predation (Touchon et al., 2017). Any genes the previous population had acquired enabling them to adapt to the niche are made available to the invading population as a direct consequence of targeted phage predation combined with immunity due to the lysogenic phage. Or, it may happen the other way around, as lysogens can also enable niche protection. Prophage induction in part of the population can confer a phage-mediated form of immunity, thus protecting that population against other bacteria (Silveira and Rohwer, 2016).
Additionally, lysogenic phages often carry genes from previous infections that can be adaptive for their new host. Some genes that are found associated with phage genomes are: virulence factors, tRNAs (Kang et al., in review), toxin-antitoxin addiction modules, glycoproteins, metabolic genes, and antibiotic resistance genes (Popa et al., 2017). One study of 47 strains of E. coli showed that 41% of the genic diversity in these E. coli strains were attributed to genes carried by prophages (Touchon et al., 2017). This is reminiscent of the relationship between humans and their microbiome, which is responsible for the majority of genic diversity in humans (Soucy et al., 2013).
Sometimes during lysis phage error can lead to packaging of bacterial genes rather than phage genes, leading to a form of HGT called transduction. There are two main forms of transduction: specialized transduction, where an error in viral genome excision leads to the packaging of a small piece of host chromosome in addition to the viral genome, and generalized transduction, where a packaging error leads to a segment of host chromosome being packaged in lieu of the viral genome. Transduction frequency varies between environments (Popa et al., 2017), but overall generalized transduction is far more common than specialized transduction (Touchon et al., 2017). However, some Mu-like phages regularly package a few kilobases of neighboring DNA, and thus specialized transduction seems to have become part of the life cycle of these phages. Generalized transduction rates are lowest when the multiplicity of infection is high, probably because cells that are transduced are still susceptible to infection by the surrounding viruses, while cells that are infected with viruses cannot be re-infected due to superinfection exclusion (Casjens and Hendrix, 2015; Howard-Varona et al., 2017).
The longer that a lysogenic phage remains in the bacterial genome, the more potential there is that accumulating deletions and mutations will abolish cell lysis. Once a phage genome cannot establish an infective cycle, the reproductive fate of all of the phage genes are permanently linked to the fate of the host cell. In bacteria, genome streamlining will lead to deterioration of most phage-transferred genes. However, there are several examples of deteriorating phages evolving a more mutualistic phenotype through domestication. For example, gene transfer agents (GTAs) are domesticated phages that promote HGT between microbial cells. GTAs have evolved independently from various types of viruses at least six times (Lang et al., 2012). The best studied GTA is RcGTA in Rhodobacter capsulatus and is regulated by highly conserved gene networks that ensure coordinated release and uptake of the GTAs in the population expressing them (Westbye et al., 2017). Importantly, GTAs cannot package enough DNA to enable the recipient to encode a GTA (Lang et al., 2012). This permanently couples the fate of the GTA to its host. Another example of phage genes being repurposed with a more mutualistic role are tailocins, in which tail subunits from a phage are used in bacterial warfare (Ghequire and De Mot, 2015). Similar to GTAs, tailocins have evolved independently in several microbial groups, and in both GTAs and tailocins the organization of genes is highly similar to the organization in phage genomes, and genes are recognizably viral in origin (Ghequire and De Mot, 2015; Hynes et al., 2016). The fate of any gene lies in its replicative potential. In viral infections hundreds of replicates are produced in a matter of minutes; in contrast lysogens replicate only when the host replicates. This means that viral genes that become permanently linked to the fate of their host are hedging their bets on the replicative potential of their host; prophages often do this with acquisition of new genes, superinfection exclusion, or by enabling niche expansion. Domestication as in GTAs and tailocins are formed through selection that leads to retention of genes that promote the replicative potential of the host.
A Symbio-Centric Ecological Speciation Framework
Once the infection is established, and phages overcome host immunity (i.e., CRISPR defenses), the cell may suffer the lytic or lysogenic cycle. Generally, the phage is likely to take the path that leads to the most convenient mode to produce its progeny. When environmental conditions are favorable, e.g., abundant host cells living in copiotrophic conditions, the lytic cycle is preferred, but when hosts are scarce, the infecting phages may decide for the lysogenic cycle (Oppenheim et al., 2005; Zeng et al., 2010; Koskella and Brockhurst, 2014; Obeng et al., 2016). If phage concentration inside the cell is high enough phages will decide for the lysogenic cycle (Zeng et al., 2010; Shao et al., 2017). As shown recently (Erez et al., 2017), phages may have sophisticated mechanisms for sensing host abundance. A PBdelta group infecting Bacillus produces a peptide under lytic infections, which is released in the medium. With that, during subsequent infections, the phage descendants can measure the amount of lytic infections that the host population has suffered, and then lysogenize if the peptide concentration is high. However, and curiously, bacteria able to grow faster under optimal conditions have more prophages (Touchon et al., 2016).
The extensive research developed in the last 20 years suggests that bacteria-phage interactions can be both antagonistic and mutualistic (Koskella and Brockhurst, 2014; Obeng et al., 2016). The most prominent ecological outcome of this interaction is ecological speciation, which is influenced by phage predation and phage-mediated HGT (Rodriguez-Valera et al., 2009; Koskella and Brockhurst, 2014; Obeng et al., 2016). From both angles, these processes can maintain the ecotype and sustain the emergence of new ecotypes while environmental conditions are changing (Figure 2). With that, bacteria-phage interactions modulate the biogeochemical cycles and ecosystem processes (e.g., photosynthesis; Fridman et al., 2017), thus ensuring that there is always a population that is most suited to use the available resources, even as resource abundance fluctuates.
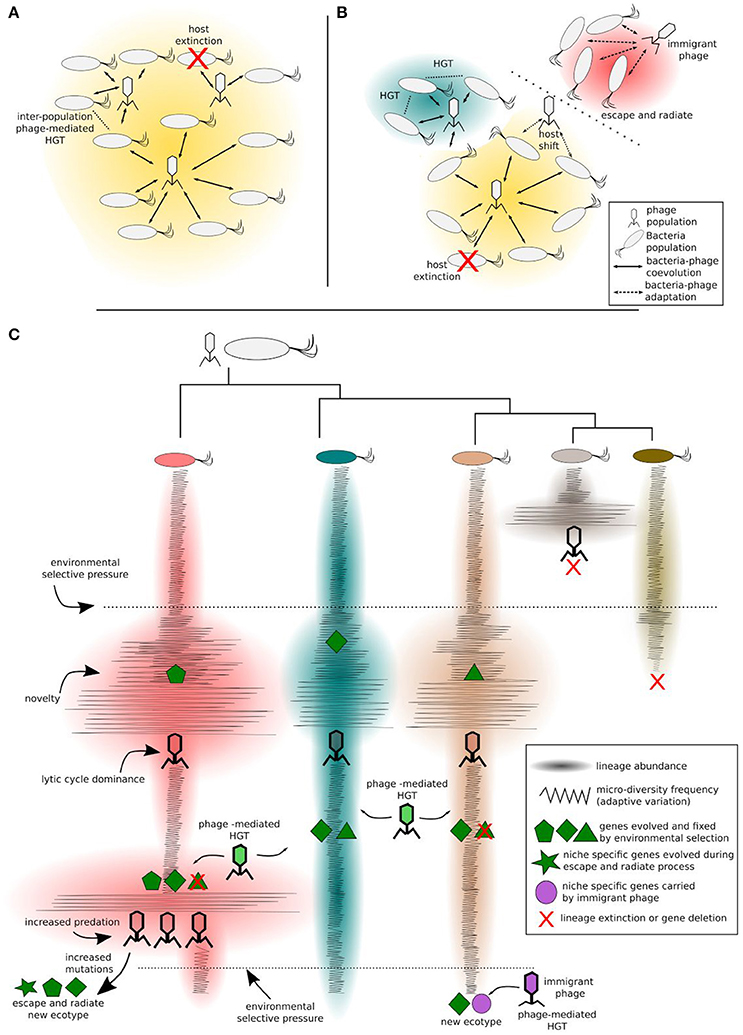
Figure 2. Bacteria-phage coevolution. Different clades of a bacteria populations coevolving with phages in a given ecotype (A), challenged by environmental factors, can diverge and form new ecotypes (B). This process can be modulated either by antagonistic or by mutualistic interactions. Increased predation pressure may induce the escape and radiate process in some clades, while other clades may benefit from phage-mediated HGT. Different ecotypes are represented by different background colors in (A,B). More specifically (C), considering an intra-population scale, the bacteria clade and their phages diversify in a constant-diversity mode of selection (i.e., fluctuating selection). Phage predation selects against the most common phenotype. Upon high selective pressure imposed by phage predation, the host clade can increase evolvability rates and increase its chance to evolve to a new ecotype, e.g., ecological speciation by the escape and radiate process. Mutualistic interactions (i.e., transduction, lysogeny, and phage domestication) mediated by phage transition within clades or by immigrant phages (from different ecotypes), carrying advantageous niche-specific genes, contribute to ecological speciation.
The blurred distinction between the two kinds of ecological interactions suggests that phages are conditionally helpful parasites (i.e., parasites that are beneficial to their host in specific environments but have a negative impact in others; see (Fellous and Salvaudon, 2009)). From this perspective, phages can be seen as bacterial symbionts (the bacterial bona fide symbionts; Koonin, 2011). For the sake of clarity, the coexistence of bacteria and phages, along a continuum, at the ecotype scale (Figure 2), may be seen as a holobiont. The term holobiont is generally used for describing the host and its associated microbes (Soucy et al., 2015). It is worth noting that here, the host (i.e., bacteria ecotype) and its symbiont (i.e., the interacting phages) are more tightly connected since phages can be genetic symbionts. So that the (a) genetic hybridization between the symbiont and host is possible, and (b) the changes that arise from genetic hybridization are passed on to future generations. Then the holobiont formed by bacteria and their phages is the vehicle of evolution, able to diversify and to accommodate environmental changes by ecological speciation (Figure 3).
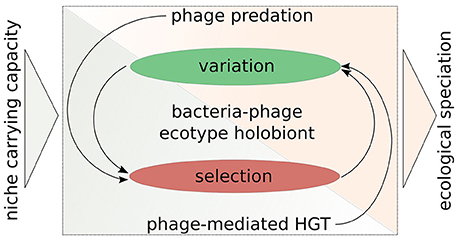
Figure 3. A symbio-centric framework for bacterial ecological speciation in the light of interactions with phages. There are two main phases of evolution: (1) Variation generation; (2) Selection acting on variation. Phage predation acts directly on the selection part and indirectly on the variation part, whereas phage-mediated HGT (as a consequence of transduction, lysogeny, and phage domestication) increases variation directly and indirectly affects selection. These interactions then may facilitate ecological speciation, when niche carrying capacity is not sufficient enough to maintain a given population. This framework outlines the importance of considering bacteria and their phages as an evolutionary unit.
Bacterial genomes show signs of phage-derived interactions; these genomically-fossilized records of bacteria-phage interactions can add up to 30% of a bacterial genome (Obeng et al., 2016). Experiments to remove these prophage-like sequences revealed remarkable changes in host physiological traits (Zeng et al., 2016; Argov et al., 2017; De Smet et al., 2017). These records of interactions between the genomic parasite and its host may encode valuable ecological information (see Box 3). For instance, Vale et al. (2017) have identified a robust phylogeographic pattern in genomic sequences associated with prophages from Helicobacter pylori. According to the authors, four distinct clusters were found: one African, one Asian and two European prophage populations. Therefore, observations of such underlying patterns may be facilitated considering the perspective of a symbio-centric ecological speciation framework.
Box 3. How the “genomically-fossilized” records of bacteria-phage interactions may contribute to a better comprehension of bacterial ecological speciation?
This is one important question for guiding future research. Here we briefly explore the topic by bringing into this context the Enterobacter cloacae complex. These are ubiquitous bacteria considered a threat to human and plant health (Chavda et al., 2016). This complex clusterizes into 18 phylogenomic groups. However these clusters are not informative with respect to the different ecological patterns of these strains. That is, members from the same phylogenetic group can be found in both human and plant microbiomes (Chavda et al., 2016).
We have analyzed the genomic signs of bacteria-phage interactions in two Enterobacter genomes from the same phylogenomic group (Hoffman cluster V) (Chavda et al., 2016), which are closely related by their shared geographic location (U.S.A.); one was isolated from a human microbiome (Chavda et al., 2016) (JVAG00000000) and the other from the rhizosphere (Humann et al., 2011) (CP002886.1). Both are described as pathogenic bacteria. We then identified important differences between them, by comparing the prophage sequences annotated using PHASTER (e < 10−4) (Arndt et al., 2016). Two prophage regions were annotated in the genome of the human microbiome isolate. They are identified as bacteriophages 186 (NC_001317) and mEp237 (NC_019704). The plant Enterobacter genome was also found to encode bacteriophages 186 and mEp237. Besides that, this genome also encodes two other prophage regions. One was identified as PsP3 phage (NC_005340), and the other as phage phiARI0923 (NC_030946).
A previous report found PsP3 phages to be abundant in the rhizosphere (~104 pfu g−1 soil) (Campbell et al., 1995). In that study this phage was reported to have a broad host range infecting several rhizosphere Pseudomonas species (P. aeruginosa, P. fluorescens, P. putida and P. chlororaphis, P. stutzeri, and P. spp.) (Campbell et al., 1995). Phage phiARI0923 has been characterized as a Streptococcus phage. For this phage we found no previous information about its presence in the rhizosphere. Streptococcus is considered a human pathogen and can also be found in the rhizosphere (Zarraonaindia et al., 2015). It is therefore tempting to propose the hypothesis that these two prophages (PsP3 and phiARI0923) are playing important roles in rhizosphere niche bacterial colonization.
Whatever the origins of these Enterobacter strains, plant, or human, from the point of view of an invader the alien bacteria must be able to exchange genetic material with the native microbiome (Mallon et al., 2015). Perhaps interactions with native phages may facilitate this process, especially if one considers the importance of phage-mediated HGT and the high rates of HGT that are likely to be more frequent (25-fold) between members of same microbiome than between members from different microbiomes (Soucy et al., 2015). For the case exemplified here, although the analysis was able to identify the phages taxonomically, the prophage genome showed a “mosaic-like” pattern. Their coding sequences (CDSs), in each one of the prophage regions that were detected, were highly similar with CDSs of phages infecting different Gammaproteobacteria genera (e.g., Pseudomonas, Salmonella, and Enterobacter). Phage-bacteria interactions are likely to occur within the context of host specificity (Popa et al., 2017), but broad host range interactions have been reported in certain conditions (Jensen et al., 1998; Beumer and Robinson, 2005; Koskella and Brockhurst, 2014).
With this simple example we sought to illustrate how future research studying phage records in bacterial genomes may contribute to a better comprehension of bacterial ecological speciation. In this case, these preliminary findings may suggest that different genomes within the same phylogenomic cluster, but with different ecological features, may exhibit different “phage footprints.”
From the point of view of phage evolution, such framework is equally required. The origin of viruses is still obscure (Nasir and Caetano-Anollés, 2015). So far, no common ancestor has been found. Recent findings on virus phylogenomics posit that capsid genes may encode sufficient signal for reconstructing the evolutionary divergences between different phage families (Comeau and Krisch, 2008; Laanto et al., 2017). Interestingly, capsid genes might have evolved from cellular organisms (Krupovic and Koonin, 2017), reinforcing the relevance of a symbio-centric framework.
Conclusion
As stated previously the bacterial species problem is a complex issue that is consistently being re-evaluated considering new experimental data types and evolutionary theories. A core component of the bacterial species problem and microbial ecology is the question of how bacteria diversify and form distinct ecologically coherent clades (Philippot et al., 2010). The evolutionary mechanisms underlying this process are still poorly understood. The discussion presented here emphasizes that the ecology and evolution of bacteria cannot be fully conceptualized without considering the role of phages. A better comprehension about bacterial ecological speciation may be achieved by considering bacteria and their phages as a single unit of selection (Figure 3). This perspective opens a new avenue for further research. Future experiments addressing the symbio-centric ecological speciation framework may accelerate our understanding of the mechanisms underlying microbial evolutionary processes.
The underestimation of the role that viruses play in driving biological diversification extends beyond the microbial sphere. Roossinck (2015) has discussed the importance of viruses as microbial symbionts for all kinds of life. There are plants that owe the successful colonization of geothermal soils (>50°C) to endophytic fungi infected with viruses. There are insects (endoparasitoid braconid wasps) which rely on viruses for genes that suppress the host immune system, so their eggs deposited inside the host body can develop. Even mammalian evolution has been affected by interactions with viruses: a fundamental protein for placental biology may have been domesticated from a virus (Mi et al., 2000). Viral-like sequences are found in the genomes of every organism. Arguably the comparative simplicity of microbial populations is a model system for analyzing the contribution and importance of host-virus interactions for ecological speciation.
Author Contributions
LB: conceived the project; LB, SS, DA, AdS, JS: discussed the ideas; LB, SS, JS: wrote the manuscript; All authors read and approved the final draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Laurent Philippot (INRA, University of Burgundy) for providing helpful discussions and useful suggestions that contributed to this work. We also thank Dr. Luiz Thibério Rangel (Laboratory of Bioinformatics, USP) for contributing in the initial discussions. LB and DA were supported by a fellowship from the Coordination for the Improvement of Higher Education Personnel (CAPES). JS has a research fellowship from CNPq. This work was supported in part by FAPESP grant 2011/50870-6.
References
Achtman, M., and Wagner, M. (2008). Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 6, 431–440. doi: 10.1038/nrmicro1872
Andam, C. P., and Gogarten, J. P. (2011). Biased gene transfer in microbial evolution. Nat. Rev. Microbiol. 9, 543–555. doi: 10.1038/nrmicro2593
Argov, T., Azulay, G., Pasechnek, A., Stadnyuk, O., Ran-Sapir, S., Borovok, I., et al. (2017). Temperate bacteriophages as regulators of host behavior. Curr. Opin. Microbiol. 38, 81–87. doi: 10.1016/j.mib.2017.05.002
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bapteste, E., and Boucher, Y. (2009). Epistemological impacts of horizontal gene transfer on classification in microbiology. Methods Mol. Biol. 532, 55–72. doi: 10.1007/978-1-60327-853-9_4
Bergthorsson, U., Andersson, D. I., and Roth, J. R. (2007). Ohno's dilemma: evolution of new genes under continuous selection. Proc. Natl. Acad. Sci. U.S.A. 104, 17004–17009. doi: 10.1073/pnas.0707158104
Beumer, A., and Robinson, J. B. (2005). A broad-host-range, generalized transducing phage (SN-T) acquires 16S rRNA genes from different genera of bacteria. Appl. Environ. Microbiol. 71, 8301–8304. doi: 10.1128/AEM.71.12.8301-8304.2005
Blount, Z. D., Barrick, J. E., Davidson, C. J., and Lenski, R. E. (2012). Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature 489, 513–518. doi: 10.1038/nature11514
Blount, Z. D., Borland, C. Z., and Lenski, R. E. (2008). Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105, 7899–7906. doi: 10.1073/pnas.0803151105
Bobay, L.-M., Touchon, M., and Rocha, E. P. C. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. U.S.A. 111, 12127–12132. doi: 10.1073/pnas.1405336111
Booth, A., Mariscal, C., and Doolittle, W. F. (2016). The modern synthesis in the light of microbial genomics. Annu. Rev. Microbiol. 70, 279–297. doi: 10.1146/annurev-micro-102215-095456
Boto, L. (2010). Horizontal gene transfer in evolution: facts and challenges. Proc. Biol. Sci. 277, 819–827. doi: 10.1098/rspb.2009.1679
Buckling, A., and Rainey, P. B. (2002). Antagonistic coevolution between a bacterium and a bacteriophage. Proc. Biol. Sci. 269, 931–936. doi: 10.1098/rspb.2001.1945
Cairns, J., Frickel, J., Jalasvuori, M., Hiltunen, T., and Becks, L. (2017). Genomic evolution of bacterial populations under coselection by antibiotics and phage. Mol. Ecol. 26, 1848–1859. doi: 10.1111/mec.13950
Campbell, J. I. A., Albrechtsen, M., and Sørensen, J. (1995). Large Pseudomonas phages isolated from barley rhizosphere. FEMS Microbiol. Ecol. 18, 63–74. doi: 10.1111/j.1574-6941.1995.tb00164.x
Casjens, S. R., and Hendrix, R. W. (2015). Bacteriophage lambda: early pioneer and still relevant. Virology 479–480, 310–330. doi: 10.1016/j.virol.2015.02.010
Chavda, K. D., Chen, L., Fouts, D. E., Sutton, G., Brinkac, L., Jenkins, S. G., et al. (2016). Comprehensive genome analysis of carbapenemase-producing Enterobacter spp.: new insights into phylogeny, population structure, and resistance mechanisms. MBio 7:e02093-16. doi: 10.1128/mBio.02093-16
Cohan, F. M. (2001). Bacterial species and speciation. Syst. Biol. 50, 513–524. doi: 10.1080/10635150118398
Cohan, F. M., and Perry, E. B. (2007). A systematics for discovering the fundamental units of bacterial diversity. Curr. Biol. 17, R373–R386. doi: 10.1016/j.cub.2007.03.032
Comeau, A. M., and Krisch, H. M. (2008). The capsid of the T4 phage superfamily: the evolution, diversity, and structure of some of the most prevalent proteins in the biosphere. Mol. Biol. Evol. 25, 1321–1332. doi: 10.1093/molbev/msn080
Cordero, O. X., and Polz, M. F. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 12, 263–273. doi: 10.1038/nrmicro3218
Davies, E. V., James, C. E., Williams, D., O'Brien, S., Fothergill, J. L., Haldenby, S., et al. (2016). Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc. Natl. Acad. Sci. U.S.A. 113, 8266–8271. doi: 10.1073/pnas.1520056113
De Smet, J., Hendrix, H., Blasdel, B. G., Danis-Wlodarczyk, K., and Lavigne, R. (2017). Pseudomonas predators: understanding and exploiting phage-host interactions. Nat. Rev. Microbiol. 15, 517–530. doi: 10.1038/nrmicro.2017.61
Doolittle, W. F. (1999). Lateral genomics. Trends Cell Biol. 9, M5–M8. doi: 10.1016/S0962-8924(99)01664-5
Doolittle, W. F., and Zhaxybayeva, O. (2009). On the origin of prokaryotic species. Genome Res. 19, 744–756. doi: 10.1101/gr.086645.108
Edwards, D. L., and Knowles, L. L. (2014). Species detection and individual assignment in species delimitation: can integrative data increase efficacy? Proc. Biol. Sci. 281:20132765. doi: 10.1098/rspb.2013.2765
Erez, Z., Steinberger-Levy, I., Shamir, M., Doron, S., Stokar-Avihail, A., Peleg, Y., et al. (2017). Communication between viruses guides lysis–lysogeny decisions. Nature 541, 488–493. doi: 10.1038/nature21049
Feiner, R., Argov, T., Rabinovich, L., Sigal, N., Borovok, I., and Herskovits, A. A. (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 13, 641–650. doi: 10.1038/nrmicro3527
Fellous, S., and Salvaudon, L. (2009). How can your parasites become your allies? Trends Parasitol. 25, 62–66. doi: 10.1016/j.pt.2008.11.010
Fournier, G. (2009). Horizontal gene transfer and the evolution of methanogenic pathways. Methods Mol. Biol. 532, 163–179. doi: 10.1007/978-1-60327-853-9_9
Fridman, S., Flores-Uribe, J., Larom, S., Alalouf, O., Liran, O., Yacoby, I., et al. (2017). A myovirus encoding both photosystem I and II proteins enhances cyclic electron flow in infected Prochlorococcus cells. Nat. Microbiol. 2, 1350–1357. doi: 10.1038/s41564-017-0002-9
Gama, J. A., Reis, A. M., Domingues, I., Mendes-Soares, H., Matos, A. M., and Dionisio, F. (2013). Temperate bacterial viruses as double-edged swords in bacterial warfare. PLoS ONE 8:e59043. doi: 10.1371/journal.pone.0059043
Ghequire, M. G. K., and De Mot, R. (2015). The tailocin tale: peeling off phage tails. Trends Microbiol. 23, 587–590. doi: 10.1016/j.tim.2015.07.011
Gogarten, J. P., and Townsend, J. P. (2005). Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3, 679–687. doi: 10.1038/nrmicro1204
Gordo, I., Navarro, A., and Charlesworth, B. (2002). Muller's ratchet and the pattern of variation at a neutral locus. Genetics 161, 835–848.
Gribaldo, S., and Brochier, C. (2009). Phylogeny of prokaryotes: does it exist and why should we care? Res. Microbiol. 160, 513–521. doi: 10.1016/j.resmic.2009.07.006
Howard-Varona, C., Hargreaves, K. R., Abedon, S. T., and Sullivan, M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. doi: 10.1038/ismej.2017.16
Humann, J. L., Wildung, M., Cheng, C.-H., Lee, T., Stewart, J. E., Drew, J. C., et al. (2011). Complete genome of the onion pathogen Enterobacter cloacae EcWSU1. Stand. Genomic Sci. 5, 279–286. doi: 10.4056/sigs.2174950
Hynes, A. P., Shakya, M., Mercer, R. G., Grüll, M. P., Bown, L., Davidson, F., et al. (2016). Functional and evolutionary characterization of a gene transfer agent's Multilocus “Genome.” Mol. Biol. Evol. 33, 2530–2543. doi: 10.1093/molbev/msw125
Iranzo, J., Puigbò, P., Lobkovsky, A. E., Wolf, Y. I., and Koonin, E. V. (2016). Inevitability of genetic parasites. Genome Biol. Evol. 8, 2856–2869. doi: 10.1093/gbe/evw193
Jensen, E. C., Schrader, H. S., Rieland, B., Thompson, T. L., Lee, K. W., Nickerson, K. W., et al. (1998). Prevalence of broad-host-range lytic bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl. Environ. Microbiol. 64, 575–580.
Khomyakova, M., Bükmez, Ö., Thomas, L. K., Erb, T. J., and Berg, I. A. (2011). A methylaspartate cycle in haloarchaea. Science 331, 334–337. doi: 10.1126/science.1196544
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Koonin, E. V. (2011). The Logic of Chance: The Nature and Origin of Biological Evolution. Upper Saddle River, NJ: FT Press.
Koonin, E. V. (2016). Viruses and mobile elements as drivers of evolutionary transitions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, doi: 10.1098/rstb.2015.0442
Koonin, E. V., Makarova, K. S., and Aravind, L. (2001). Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55, 709–742. doi: 10.1146/annurev.micro.55.1.709
Koonin, E. V., and Novozhilov, A. S. (2009). Origin and evolution of the genetic code: the universal enigma. IUBMB Life 61, 99–111. doi: 10.1002/iub.146
Koskella, B., and Brockhurst, M. A. (2014). Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. doi: 10.1111/1574-6976.12072
Krupovic, M., and Koonin, E. (2017). Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. U.S.A. 114, E2401–E2410. doi: 10.1073/pnas.1621061114
Laanto, E., Mäntynen, S., De Colibus, L., Marjakangas, J., Gillum, A., Stuart, D. I., et al. (2017). Virus found in a boreal lake links ssDNA and dsDNA viruses. Proc. Natl. Acad Sci. U. S. A. 114, 8378–8383. doi: 10.1073/pnas.1703834114
Lang, A. S., Zhaxybayeva, O., and Beatty, J. T. (2012). Gene transfer agents: phage-like elements of genetic exchange. Nat. Rev. Microbiol. 10, 472–482. doi: 10.1038/nrmicro2802
Lenski, R. E. (2017). Experimental evolution and the dynamics of adaptation and genome evolution in microbial populations. ISME J. 11, 2181–2194 doi: 10.1038/ismej.2017.69
Louca, S., and Doebeli, M. (2017). Taxonomic variability and functional stability in microbial communities infected by phages. Environ. Microbiol. 19, 3863–3878. doi: 10.1111/1462-2920.13743
Lynch, M., Bürger, R., Butcher, D., and Gabriel, W. (1993). The mutational meltdown in asexual populations. J. Hered. 84, 339–344. doi: 10.1093/oxfordjournals.jhered.a111354
Mallon, C. A., Elsas, J. D., and Salles, J. F. (2015). Microbial invasions: the process, patterns, and mechanisms. Trends Microbiol. 23, 719–729. doi: 10.1016/j.tim.2015.07.013
Maslov, S., Krishna, S., Pang, T. Y., and Sneppen, K. (2009). Toolbox model of evolution of prokaryotic metabolic networks and their regulation. Proc. Natl. Acad. Sci. U.S.A. 106, 9743–9748. doi: 10.1073/pnas.0903206106
Maslov, S., and Sneppen, K. (2017). Population cycles and species diversity in dynamic Kill-the-Winner model of microbial ecosystems. Sci. Rep. 7:39642. doi: 10.1038/srep39642
Mi, S., Lee, X., Li, X., Veldman, G. M., Finnerty, H., Racie, L., et al. (2000). Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789. doi: 10.1038/35001608
Middelboe, M., Holmfeldt, K., Riemann, L., Nybroe, O., and Haaber, J. (2009). Bacteriophages drive strain diversification in a marine Flavobacterium: implications for phage resistance and physiological properties. Environ. Microbiol. 11, 1971–1982. doi: 10.1111/j.1462-2920.2009.01920.x
Nasir, A., and Caetano-Anollés, G. (2015). A phylogenomic data-drive exploration of viral origins and evolution. Sci. Adv. 1:e1500527. doi: 10.1126/sciadv.1500527
Nordberg, H., Cantor, M., Dusheyko, S., Hua, S., Poliakov, A., Shabalov, I., et al. (2014). The genome portal of the department of energy joint genome institute: 2014 updates. Nucleic Acids Res. 42, 26–31. doi: 10.1093/nar/gkt1069
Obeng, N., Pratama, A. A., and Elsas, J. D. (2016). The significance of mutualistic phages for bacterial ecology and evolution. Trends Microbiol. 24, 440–449. doi: 10.1016/j.tim.2015.12.009
O'Malley, M. A. (2008). “Everything is everywhere: but the environment selects”: ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Philos. Biol. Biomed. Sci. 39, 314–325. doi: 10.1016/j.shpsc.2008.06.005
Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., and Adhya, S. (2005). Switches in bacteriophage lambda development. Annu. Rev. Genet. 39, 409–429. doi: 10.1146/annurev.genet.39.073003.113656
Pal, C., Maciá, M. D., Oliver, A., Schachar, I., and Buckling, A. (2007). Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 450, 1079–1081. doi: 10.1038/nature06350
Pál, C., Papp, B., and Lercher, M. J. (2005). Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 37, 1372–1375. doi: 10.1038/ng1686
Pasteur, L. (1861). “On the organized bodies which exist in the atmosphere; examination of the doctrine of spontaneous generation,” in Milestones in Microbiology, ed T. D. Brock (Washington, DC: American Society for Microbiology). 43–48.
Paterson, S., Vogwill, T., Buckling, A., Benmayor, R., Spiers, A. J., Thomson, N. R., et al. (2010). Antagonistic coevolution accelerates molecular evolution. Nature 464, 275–278. doi: 10.1038/nature08798
Philippot, L., Andersson, S. G. E., Battin, T. J., Prosser, J. I., Schimel, J. P., Whitman, W. B., et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8, 523–529. doi: 10.1038/nrmicro2367
Pigliucci, M. (2008). What, if anything, is an evolutionary novelty? Philos. Sci. 75, 887–898. doi: 10.1086/594532
Popa, O., Landan, G., and Dagan, T. (2017). Phylogenomic networks reveal limited phylogenetic range of lateral gene transfer by transduction. ISME J. 11, 543–554. doi: 10.1038/ismej.2016.116
Puigbò, P., Lobkovsky, A. E., Kristensen, D. M., Wolf, Y. I., and Koonin, E. V. (2014). Genomes in turmoil: quantification of genome dynamics in prokaryote supergenomes. BMC Biol. 12:66. doi: 10.1186/s12915-014-0066-4
Raymond, J. (2009). The role of horizontal gene transfer in photosynthesis, oxygen production, and oxygen tolerance. Methods Mol. Biol. 532, 323–338. doi: 10.1007/978-1-60327-853-9_19
Rodriguez-Valera, F., Martin-Cuadrado, A.-B., Rodriguez-Brito, B., Pasić, L., Thingstad, T. F., Rohwer, F., et al. (2009). Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 7, 828–836. doi: 10.1038/nrmicro2235
Rohwer, F., Youle, M., Maughan, H., and Hisakawa, N. (2014). Life in Our Phage World: A Centennial Field Guide to Earth's Most Diverse Inhabitants. San Diego, CA: Wholon.
Roossinck, M. J. (2015). Move over, bacteria! Viruses make their mark as mutualistic microbial symbionts. J. Virol. 89, 6532–6535. doi: 10.1128/JVI.02974-14
Rosselló-Mora, R., Lucio, M., Peña, A., Brito-Echeverría, J., López-López, A., Valens-Vadell, M., et al. (2008). Metabolic evidence for biogeographic isolation of the extremophilic bacterium Salinibacter ruber. ISME J. 2, 242–253. doi: 10.1038/ismej.2007.93
Scanlan, P. D., Hall, A. R., Blackshields, G., Friman, V.-P., Davis, M. R. Jr., Goldberg, J. B., et al. (2015). Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol. Biol. Evol. 32, 1425–1435. doi: 10.1093/molbev/msv032
Seed, K. D. (2015). Battling phages: how bacteria defend against viral attack. PLoS Pathog. 11:e1004847. doi: 10.1371/journal.ppat.1004847
Shao, Q., Trinh, J. T., McIntosh, C. S., Christenson, B., Balázsi, G., and Zeng, L. (2017). Lysis-lysogeny coexistence: prophage integration during lytic development. Microbiologyopen 6:e00395. doi: 10.1002/mbo3.395
Shapiro, B. J., Friedman, J., Cordero, O. X., Preheim, S. P., Timberlake, S. C., Szabó, G., et al. (2012). Population genomics of early events in the ecological differentiation of bacteria. Science 336, 48–51. doi: 10.1126/science.1218198
Silveira, C. B., and Rohwer, F. L. (2016). Piggyback-the-winner in host-associated microbial communities. NPJ Biofilms Microbiomes 2:16010. doi: 10.1038/npjbiofilms.2016.10
Soucy, S. M., Huang, J., and Gogarten, J. P. (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16, 472–482. doi: 10.1038/nrg3962
Soucy, S., Olendzenski, L., and Gogarten, J. P. (2013). “Orthologues, paralogues and horizontal gene transfer in the human holobiont,” in eLS (Chichester: John Wiley and Sons, Ltd.). doi: 10.1002/9780470015902.a0005298.pub3
Taylor, J. S., and Raes, J. (2004). Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38, 615–643. doi: 10.1146/annurev.genet.38.072902.092831
Tellier, A., Moreno-Gámez, S., and Stephan, W. (2014). Speed of adaptation and genomic footprints of host-parasite coevolution under arms race and trench warfare dynamics. Evolution 68, 2211–2224. doi: 10.1111/evo.12427
Touchon, M., Bernheim, A., and Rocha, E. P. (2016). Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 10, 2744–2754. doi: 10.1038/ismej.2016.47
Touchon, M., Moura de Sousa, J. A., and Rocha, E. P. (2017). Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr. Opin. Microbiol. 38, 66–73. doi: 10.1016/j.mib.2017.04.010
Treangen, T. J., and Rocha, E. P. C. (2011). Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 7:e1001284. doi: 10.1371/journal.pgen.1001284
True, J. R., and Carroll, S. B. (2002). Gene co-option in physiological and morphological evolution. Annu. Rev. Cell Dev. Biol. 18, 53–80. doi: 10.1146/annurev.cellbio.18.020402.140619
Turner, C. B., Blount, Z. D., and Lenski, R. E. (2015). Replaying evolution to test the cause of extinction of one ecotype in an experimentally evolved population. PLoS ONE 10:e0142050. doi: 10.1371/journal.pone.0142050
Vale, F. F., Nunes, A., Oleastro, M., Gomes, J. P., Sampaio, D. A., Rocha, R., et al. (2017). Genomic structure and insertion sites of Helicobacter pylori prophages from various geographical origins. Sci. Rep. 7:42471. doi: 10.1038/srep42471
Van Niel, C. B. (1949). THE “DELFT SCHOOL” AND THE RISE OF GENERAL MICROBIOLOGY. Bacteriol. Rev. 13, 161–174.
Westbye, A. B., Beatty, J. T., and Lang, A. S. (2017). Guaranteeing a captive audience: coordinated regulation of gene transfer agent (GTA) production and recipient capability by cellular regulators. Curr. Opin. Microbiol. 38, 122–129. doi: 10.1016/j.mib.2017.05.003
Williams, H. T. P. (2013). Phage-induced diversification improves host evolvability. BMC Evol. Biol. 13:17. doi: 10.1186/1471-2148-13-17
Zarraonaindia, I., Owens, S. M., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. MBio 6:e02527-14. doi: 10.1128/mBio.02527-14
Zeng, L., Skinner, S. O., Zong, C., Sippy, J., Feiss, M., and Golding, I. (2010). Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell 141, 682–691. doi: 10.1016/j.cell.2010.03.034
Zeng, Q., Sukumaran, J., Wu, S., and Rodrigo, A. (2015). Neutral models of microbiome evolution. PLoS Comput. Biol. 11:e1004365. doi: 10.1371/journal.pcbi.1004365
Zeng, Z., Liu, X., Yao, J., Guo, Y., Li, B., Li, Y., et al. (2016). Cold adaptation regulated by cryptic prophage excision in Shewanella oneidensis. ISME J. 10, 2787–2800. doi: 10.1038/ismej.2016.85
Zhang, J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/S0169-5347(03)00033-8
Zhaxybayeva, O. (2009). Detection and quantitative assessment of horizontal gene transfer. Methods Mol. Biol. 532, 195–213. doi: 10.1007/978-1-60327-853-9_11
Zhaxybayeva, O., and Doolittle, W. F. (2011). Lateral gene transfer. Curr. Biol. 21, R242–R246. doi: 10.1016/j.cub.2011.01.045
Glossary
Microbial population: genetically coherent microbial groups. One microbial population may harbor different subgroups that can be distinguished according to their ecotypes.
Ecological speciation: is the origin of a new microbial group (i.e., ecotype) by niche adaptation.
Ecotype: a genetically coherent microbial group within a given population that differ from other subgroups by niche adaptation.
Traditional ecological speciation framework: proposes that an ecotype is maintained by periodic selection events, which select for the most adapted genotype. The adapted genotype outcompete other genotypes and this causes a diversity-purging process in which genetic variability within the local population is periodically reset by environmental changes. The formation of a new ecotype depends on the acquisition of advantageous trait that allows colonization of a new niche. But to form a stable ecotype, the adapted subgroup needs to be no longer subject to the same periodic selection and cohesive forces. Otherwise it will outcompete or be outcompeted by the other subgroups.
Directional selection: selection of one particular genetic variation within a population. Directional evolutionary change erodes the genetic variation over time.
Fluctuating selection: when natural selection varies in the intensity, direction, or target among generations. Natural selection driven by parasites is considered strong ecological drivers of fluctuating selection. Parasites mediate frequency-dependent selection. Parasites become adapted to the most frequent genotypes of a local host population. This mechanism of interaction can be named as negative-frequency dependent selection, because the more common a host is the less fit it becomes (Thompson, 2013).
Niche-specific traits: evolved feature(s) required for colonization of a particular niche.
Clades: a group of microbes that share some genetic coherence. The genetic coherence of a clade requires often a definition. For example, it can be a Firmicutes clade or one particular clade within an ecotype.
Axenic: observation based on culture-dependent experiment where only one single species is present.
Symbio-centric ecological speciation: ecological speciation influenced by symbiotic interactions.
Unit of selection: elementary biological unit mostly subjected to natural selection. Defining an appropriate unit of selection enables more precisely measurements of evolutionary rates and ecological processes.
Keywords: coevolution, phage predation, lysogeny, transduction, GTAs, ecological speciation, ecotype, symbionts
Citation: Braga LPP, Soucy SM, Amgarten DE, da Silva AM and Setubal JC (2018) Bacterial Diversification in the Light of the Interactions with Phages: The Genetic Symbionts and Their Role in Ecological Speciation. Front. Ecol. Evol. 6:6. doi: 10.3389/fevo.2018.00006
Received: 08 September 2017; Accepted: 11 January 2018;
Published: 26 January 2018.
Edited by:
Lutz Becks, Max Planck Institute for Evolutionary Biology (MPG), GermanyReviewed by:
David Alonso, Consejo Superior de Investigaciones Científicas (CSIC), SpainBertrand Schatz, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2018 Braga, Soucy, Amgarten, da Silva and Setubal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucas P. P. Braga, bHBwYnJhZ2FAaXEudXNwLmJy