Abstract
Animal domestication is considered a complex and multistage process that altered behaviorally, morphologically, and physiologically the domesticates relative to their wild ancestors. Ever since Darwin, scientists have been concerned about the history of domestication. To determine the domestication origins of the species, it is crucial to discover their ancestors and identify the approximate local domestication. Domestication has been the focus of several studies from different specialties. Studying when, where, and how domestication happened is essential to understand the origins of civilizations and the evolution of domesticated species. The development of both humans and domestic animals is hard to justify, and the genetic variations that occurred during the early animal domestication process remain vague. The recent and potential applications of evolutionary biology may deliver answers for main social challenges. It is important to examine the relationship among the environment and the traits of organisms that have been influenced through the adaptation to modern environments and the patterns of selection triggered by their environments during domestication period. Once domestication occurred, several events such as gene flow and selective pressures occurred, leading to genomic and phenotypic alterations. In this review, we discuss the current knowledge about the spatiotemporal outlines of domestication and debates surrounding the intent, speed, and evolutionary landscapes of this event. We also focus on the core challenges for future research. In conclusion, we argue that although the current growth in domestication information has been remarkable, the next era will produce even more significant insights into not only how domestication occurred but also where and when it did so.
Introduction
Domestication is an evolutionary process by which animals are artificially selected and undergo huge phenotypic behavioral and physiological alterations (Trut et al., 2009). These transformations occurred at the same time in several regions with a tremendous impact on human societies (Neolithization) (Vigne, 2011). Domestication is well known not only for its slow course but also for its extreme techno-economic alterations, from hunting-gathering to food production (Price, 1999). It depends on many important factors, such as strong demographic transition, cultivation, and husbandry of valuable domesticates (Bocquet-Appel, 2008), along with profound social and spiritual changes (Price, 2002).
Dogs (Canis lupus familiaris) were the earliest species domesticated by Asian and European gatherers during the late glacial period approximately 17–15 thousand years (kyrs) before the present (BP) (Pionnier-Capitan et al., 2011; Frantz et al., 2016) followed later by the domestication of livestock and crops (Mignon-Grasteau et al., 2005). Interestingly, while in some species (dogs and cattle), domestication was a human-driven process, for other species (cat, rat, and house sparrow), it occurred naturally (Driscoll et al., 2009). Nowadays, several authors agree that there are approximately 40 animal species domesticated in different geographic areas (Scherf and Pilling, 2015; Leroy et al., 2018).
Differences in morphological features and behavior of most domestic animals from their wild counterparts came about by controlling breeding and persistent animal husbandry. These practices also designed and shaped the diverse genetic makeup among different breeding populations. The variation in the phenotypes of the domesticated animal led to the basis of the Darwinian evolutionary study, which highlighted several queries for further studies, namely: when, where, and how did the domestication of these animals start, and what are the genetic origins of domestication development (Darwin, 1859). Darwin’s initial observations in “On the Origin of Species,” and his variation under domestication began an essential debate for future works (Darwin, 2010). Commonly, for different domestic animals, there are two evolutionary phases: an ancient domestication event where a wild ancestor become a domesticated species followed by a modern breeding process (Driscoll et al., 2009). Ancient domestication occurred approximately 12,000–14,000 years ago, during the agricultural surge of the initial Neolithic period, along with the domestication of major crops (Wang et al., 2014b). On the other hand, modern breeding occurred only in the past 300 years (since the eighteenth century) and started with the selection of breeding animals based on their demand by human societies (Crowley and Adelman, 1998).
In the current review, we try to summarize the existing information on domestication from the literature. We start by addressing the evolution of animal domestication through an evolutionary perspective. We address the roots, demographic history, and impacts on domestication, genetic architecture, and methodologies taken to monitor domestication procedures by selective pressures. We also conclude presenting the challenges to the research and a preview of future directions for this thematic.
Animal Domestication
Animal domestication is the process of a prompt, artificial, and intensive selection of wild animals that, over the last 11,500 years, has altered the Earth’s biosphere, shaped human evolution, and influenced the size of the human population. The foundation of domestication is linked to the cultural progression from hunting to farming in ancient civilizations during the Neolithic period, possibly with the exclusion of dogs, which were the earliest domesticated animals (Savolainen et al., 2002) and diverge from other species regarding both location and timing of domestication (Beja-Pereira et al., 2006; Liu et al., 2006; Driscoll et al., 2009). The periods where animal domestication occurred were determined through archeological clues and reflected selection forces generated by human activities and by human-adapted environments. During the last years, several studies (Chen et al., 2007; Wright, 2015; Hunter, 2018) have focused on animal domestication, and nowadays, it is possible to establish a timeline for the domestication of several animals (Figure 1). Despite this, there are still several questions regarding the timing, location, and the evolutionary domestication process (Larson and Fuller, 2014).
FIGURE 1
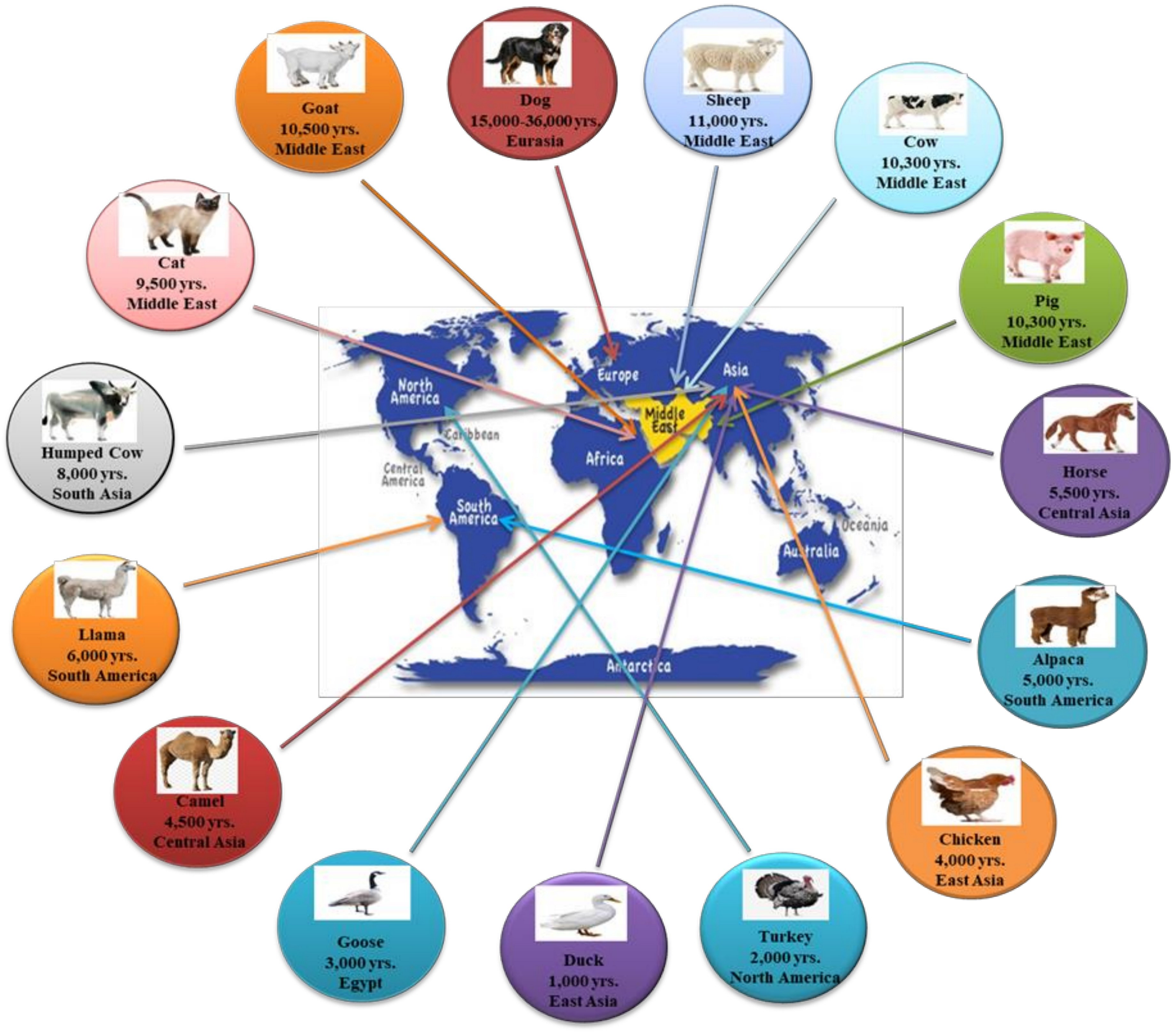
The timeline of animal domestication (adapted from Zeder, 2008).
Since the Neolithic period, humans struggled to domesticate wild animals and use them as food sources (milk and meat), commodity manufacturers (silk and wool), protection, and transportation. There are three pathways described for domestication: commensal, prey, and direct pathways (Zeder, 2012; Figure 2). In the commensal pathway, the wild animals were attracted to anthropogenic habitats, mainly for human food waste or small prey, establishing a commensal relationship with humans. Dogs, cats, or chickens are some of the species that followed this pathway. In the prey pathway, humans start hunting some species like pigs and cattle for their meat in response to depletion of the local stock of these animals. Over time, these game management strategies developed into controlled breeding of these species. In the direct pathway, humans captured wild animals (horses, donkeys, and camelids) to obtain some resources by controlling their movements, their nutrition, and reproduction, which lead to a dramatic bottleneck (Zeder, 2012).
FIGURE 2
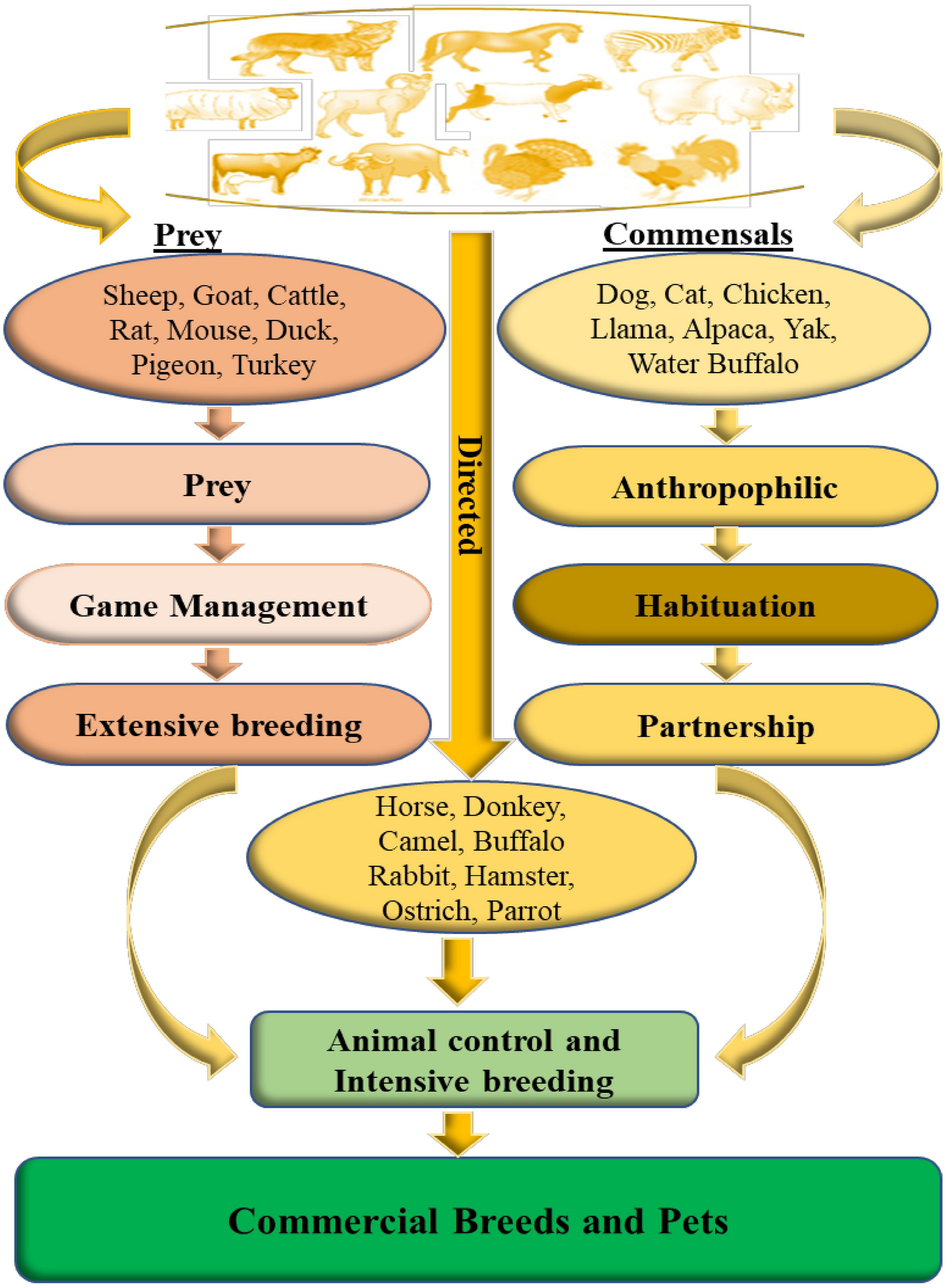
Different pathways from wild to domestic animals (adapted from Larson and Burger, 2013).
Dogs
Dogs were the first animal to be domesticated by humans more than 15,000 years ago. Their wild ancestor is the extinct gray wolf, and despite being intensively studied, there are still questions regarding their geographical and temporal origins and events of domestication. In the literature, there are several places of origin of dogs, including Europe (Thalmann et al., 2013), the Central, Middle East, and East Asia (Pollinger et al., 2010; Shannon et al., 2015; Wang et al., 2016). More recently, a study using mtDNA states that dogs may have been domesticated independently in Eastern and Western Eurasia from different wolf populations. Later the eastern dogs accompanied humans through their dispersion to Western Europe, where they replaced the Western Eurasian and European Paleolithic dogs (Frantz et al., 2016). The same event occurred for American (Leathlobhair et al., 2018) and African (Liu et al., 2018) dogs that arrived with human expansions. These expansions through the world, and the evolutionary dog history, involve bottlenecks and gene flow.
Cattle
Since ancient times, wild cattle and humans are interconnected, and during the last 10,500 years, the number of domesticated bovine species is approximately five (Helmer et al., 2005), resulted in human benefits of meat and milk to drought animals. Recently wild cattle are the source of the genetic pool for domestic breeds’ adaptation to changing pressures of climate and infectious diseases (Melletti and Burton, 2014). Current knowledge of cattle domestication is predominantly based on mitochondrial DNA analysis (Groeneveld et al., 2010). Taurine cattle domestication reported back to the wild and now extinct in the near east, about 8500 years ago, after sheep and goat domestication (Helmer et al., 2005; Bollongino et al., 2012).
Cattle with Longhorn phenotype were the first one to be domesticated; still, this phenotype is common in the number of British, French, Mediterranean, and African breeds to date (Schafberg and Swalve, 2015). First cattle with short horns reported back to 3000 years BC; the phenotype was fit to these habitats and switched by next wave of migrants (Bradley and Magee, 2006). Britain took the most long-horn forms from Asia and neighboring continents about 1000–2000 years BC (Epstein, 1984). In Europe, the most common type of cattle was short-horn until around 1000 BC (Lenstra et al., 2014). Several ecosystems in the various regions of the world are attributed to the domestication and distribution of cattle to and their adaptation to local environments (Lenstra et al., 2014). Moreover, several “agrotypes,” generated by human selection, let the breeds differ in coat color, development of horns, and docility (Ajmone-Marsan et al., 2010). Systematics has accelerated the cattle diversity in the last 200 years and stretched the castles to the main breeds, like with dairy production (Barker et al., 1991) cattle acquired the large udder. The process of the domestication resulted in a continuous decrease of size until the Middle Ages, but it was less pronounced in long-horned Italian forms and draught cattle (Lenstra et al., 2014).
Sheep
Primarily sheep were raised for meat around 4000–5000 years ago but later specified for other foodstuffs. Developments in animal husbandry and application of direct mating systems have evolved a variety of current sheep breeds, not only the most adaptable to the range of climates but also specific to the production of milk, meat, and wool (Chessa et al., 2009; Kijas et al., 2012). Mongolian sheep origin is the wild Argali sheep from highland areas of Central Asia. More than 2,000 years ago, many populations had moved to the south of the Great wall for several reasons; hence the most present Chinese breeds are linked to Mongolian sheep (Huang et al., 2017). However, with exposure to a changing environment, and feeding situations to various eco-regions across the country than normal habitat, Mongolian Sheep faced extensive artificial selection in diverse orders (Liu et al., 2016).
The subspecies of Mongolian sheep display substantial changes in several traits, particularly associated with reproduction, but how species diverge natively relative to these characters is not well interpreted (Huang et al., 2017). These studies identified the genes of significant importance for domestication process (Petersen et al., 2013b; Carneiro et al., 2014), capability to withstand the harsh climates (Gou et al., 2014; Wang et al., 2014a), or conspicuous economic characters (Choi et al., 2014; Yang et al., 2014; Wang et al., 2015). Tibet wild pig (Li et al., 2013) and Tibet mastiffs (Gou et al., 2014; Wang et al., 2014a) have been a subject for popular studies from Tibetan Plate, and linked many genes for adoption to high altitude and hypoxia. Several studies have explored the genome-wide difference among various indigenous breeds to fix the molecular basis of various physical traits of vital importance in the livestock, such as chicken (Fan W.-L. et al., 2013), pig (Yang et al., 2014; Wang et al., 2015), and cattle (Choi et al., 2014). However, the studies for Chinese short fat tail sheep are limited.
Goat
Fertile Crescent Land has reported as the first domestication habitat for goat (Capra hircus) in Near East almost < 10,000 years ago by various genetic and archeological studies (Pringle, 1998). The domestication of goats has a significant role in human society by providing valued products such as milk, meat, furs, and fiber, predominantly in China and other developing countries (Joshi et al., 2004). During the epoch, these domestic breeds rich in genetic assets incited us to pay more consideration; consequently, keeping the domestic animal variety is imperative to accomplish the forthcoming necessities.
Concerning precise economic and environmental features, China has started focusing on conservation strategies for these native breeds, including specified conservation zone, conservation farms, and the gene bank of the genetic reserve for distinctive breeds (Wei et al., 2014). These highlands had allocated West China into Southwest and Northwest China, and consequence to the diverse climatic regions and ecological structure. China is an extensive subcontinent of merged topographical locations, so Chinese goat breeds exhibit a great range of variation in productivity, milk production, meat, and fiber; draught ability; heat tolerance; and disease resistance. Besides, several preceding studies on Chinese goats were conducted in the restricted number of trials within fewer breeds and counties (Di et al., 2011; Wei et al., 2014).
Cat
A recent evolutionary study of domesticated cats using DNA analysis suggested thousand years of interaction between the cats and humans before their domestication, and this interaction let the cats’ genes changed pretentiously from those of wild cats except the development of unique stripes and spots of the tabby (O’Brien and Johnson, 2007). Evaluation of the DNA of cats bridging the last 9,000 years – counting the remains, mummies, and specimens of cats from ancient Roman, Egypt, and modern African wildcat – suggested the current domestic feline have a great contribution of two major cat lines (Ottoni et al., 2017).
Thus early 4400 B.C., earlier ancestors of our domestic cats spread from Southwest Asia and into Europe (Ottoni et al., 2017). Farming communities settled the cats to control the rodent patrol. Mice and rats were drawn to the crops, and the rodent populations were possibly pursued by cats (Montague et al., 2014). In turn, both often came close to the human settlement. Hence, the domestication changed the wild cats to a domesticated human companion without changing much, says Eva-Maria Geigl, the evolutionary geneticist and article co-author (Ottoni et al., 2017). Domesticated cats look like wildcats, but they are not lonely and capable of tolerating both humans and other animals (O’Brien and Johnson, 2007).
Horses
The date of horse domestication is conceived from the definition of domestications (Cieslak et al., 2010). Two theories of domestication are as follows: first, the human control over breeding depicted by changes in the size and variability of ancient skeletal samples from ancient horse populations, and second includes broader evidence of skeletal and dental activity weapons, arts, and spiritual artifacts; and patterns of human lifestyles (Ross-Ibarra, 2004). Evidence claim horses were kept as meat animals before trained as working animals (Hausberger et al., 2008). The concept of isolated genotypes between domesticated and wild populations leads to exploring the domestication attempts using genetics or physical traits examination (Rollin, 2011). But these methods could only find the latest footprints of domestication but failed to determine uncertain primitive gene flow between the two groups (which occurs naturally as long as the domesticated population remains within wildlife habitat) (Vilà et al., 2001). Furthermore, being descendent of captive-fledged ancestors, all the domesticated horses, and feral horses are capable of retaining either characteristic (Dobbie and Braysher, 1993). Time frames chosen for the horses’ domestication are influenced equally, either following the narrower zoological concept of domestication or the broader cultural definition, i.e., the combination of zoological and archeological facts (Aberle and Distl, 2004). The date of 4000 BCE is based on evidence that includes the appearance of bite-related dental pathologies, changes in butchering practices, changes in humans’ economies and patterns of settlement, the depiction of horses as symbols of power in artifacts, and the appearance of horse bones in human graves (Larson and Burger, 2013).
Buffalo
The domestication process led to the adaptation of various bovine species to an agricultural environment, and the most important species are indicated and taurine cattle followed by swamp and riverine and buffalo (Barker, 2014). These species have ranged to several regions, while the domestic forms of gaur and yak are grouped near their wild ancestors’ sharing areas (Groves, 1981). The genus Bubalus distribution was initially started in the Pleistocene, Europe, and South Asia, but was later constrained to southeast Asia and the Indian subcontinent (Flamand et al., 2003). In ancient times, the wild Asian buffalo (Choudhury and Barker, 2014) spread across southeast Asia to Indo-China. It is presently listed as an endangered species, with a world population lesser than 4,000, possibly fewer than 2001. Domestic water buffalo (B. bubalis) has been grouped into two types: river and swamp based on behavioral and morphological criteria (Barker, 2014). Some have referred to these as distinct subspecies, naming swamp buffalo B. bubalis carabenesis and riverine B. bubalis bubalis. However, because they resulted from different domestications, here they are designated distinctly. The same applies to the swamp and river types of water buffalo, the cross-fertile subspecies of the wild Bubalus arnee (Groeneveld et al., 2010; Yindee et al., 2010).
Chicken
The history of domestic chicken species can be discussed in three different periods of time. The longest period is the evolutionary history, shared with other species before speciation, and along with various courses after speciation. The currently shared ancestor of birds and mammals existed 300 my ago (Tixier-Boichard et al., 2011). The most common ancestor between chicken and quail lived 40 my ago (Mwacharo et al., 2013). The second period of species domestication starts several thousand years ago and led to domestic species diversification. The recent period of rigorous production trait selection applied to a subset of these breeds is shorter, with just a few decades (Kanginakudru et al., 2008). Therefore, on an evolutionary scale, domestication and, more significantly, a good selection for higher levels of production are very important (MacDonald, 1992). Current domestic chickens’ genome diversity is a result of the founding effects at the time of domestication, the long-term domestication process, subsequent breed differentiation, and recent strong production selection (Nishibori et al., 2005). The cumulative effect of human-made domestication and subsequent selection has given rise to a remarkable phenotypic diversification of the chicken, both at the molecular level (MacDonald, 1992).
Domestication and Evolution
Domestication has been a core question of interdisciplinary scientific research, and those traits that enhance the survival or the reproductive competence of the organisms are subject to selection and transferred to the next generation to increase the population prevalence (Vitti et al., 2013). Since 2006, the study the domestication through complete genome sequence has become possible, and it has been associated with the detection of selection in a large number of genomic loci that have likely evolved by selective pressures (Carneiro et al., 2014; Larson et al., 2014). Carneiro et al. (2014) studied genes involved in brain and neuronal development and proposed that domestic animals evolved by accumulating several mutations with small effects instead of critical alterations in a few loci. Later in 2016, Messer et al. (2016) reviewed some limitations to this model and provided some examples of studies using fishes, bugs, and reptiles where the phenotypic traits altered in a few generations. Strong animal husbandry practices and controlled breeding have shaped the behavior, the morphological features, and the genetic diversity of domestic animals when compared to their wild ancestors (Wang et al., 2014b). Several alterations were observed across different domestic species and captivated researchers at least since Darwin’ findings, namely lack of fear; enhanced reproductive system (able to reproduce in any season); disparities in the coat length, texture, and color; modifications in skull form, tooth crowding, and corporal sizes; floppy ears, and rolled tails (Driscoll et al., 2009; Trut et al., 2009; Larson and Fuller, 2014; Wilkins et al., 2014). These characteristics make domesticated animals valued models for different areas with highlights in genetics and biomedical research (Andersson, 2016; Wolf et al., 2018): descendants are commonly known, samples are usually unlimited, many breeds are inbred, gene variants responsible for particular phenotypic characters are fixed, and the genetic differences (a requirement for selection) are limited (Andersson, 2001; Gentry et al., 2004; Lyons et al., 2016). All these differences are associated with genetic events as inbreeding, gene flow, and selection pressures. Inbreeding leads to a decrease in genetic diversity and is associated with the isolation of small populations at the beginning of the domestication process (Cieslak et al., 2011). There is evidence of long-term gene flow between wild and domestic animals such as donkeys, horses, camelids, pigs, wolves, cats, and the reindeer (Marshall et al., 2014; Frantz et al., 2015; Bolstad et al., 2017). Once domestication became established, a relaxation in the natural selective pressures (both environmental and induced by humans) enabled the increase of new mutations (mostly non-synonymous mutations), leading to even more differentiated species (Petersen et al., 2013a). Despite this, and due to the multitude of selective pressures involved, it is difficult to isolate any causal factors that result in specific genetic differences (Zeder, 2015). Over the past 40 years, several species (mostly livestock) have been intensively selected, and notable phenotypic variations have been observed. Since many of these genetic mutations lead to phenotypic alterations, identification of the signatures of positive selection is considered a valuable tool to recognize genes that might underlie important traits allowing to link genetic variants to a particular phenotype (Consortium, 2009).
As some authors suggest, domestication started unconsciously (Tchernov and Horwitz, 1991), and later a conscious selection of human-defined traits led to a high level of diversity (Trut et al., 2009). Indeed, during the initial period of their domestication, horses were mainly used for meat and milk (Outram et al., 2009), and later they became important for transportation, warfare, and sport horseracing. Another example is the dog domestication process that focused not only on preferred physical characteristics, such as body thickness and body length, texture, skull shape, tail size and shape (Wayne, 1986), but also on improved behavioral patterns, with advanced features of guarding, herding, speed, agility, and companionship (Ostrander et al., 2000). Indeed, all over the domestication process, studies suggest that humans did not maintain constant selective pressures. Most likely, they selected different traits in different places at different times. Thus, extra care must be taken since the discovery of certain traits in current breeds does not necessarily mean that the trait was a target during the early domestication (MacHugh et al., 2017).
Domestication Studies
Domestication has fascinated scientists from different fields through its importance as a model of evolutionary and demographic change (Zeder et al., 2006). Ever since Darwin, scientists have been concerned about the history of domestication. To determine the domestication origins of the species, it is crucial to discover their ancestors and identify the approximate local domestication. For example, dogs were domesticated before the start of agriculture from gray wolves (Larson and Fuller, 2014). However, gray wolves were disseminated across the Northern hemisphere, hampering the findings on how, why, when, and where dogs were domesticated and if this process occurred just once or independently at different times (Larson, 2017). Over the years, different genetic methodologies have been used to explore these questions (Larson, 2011).
In the first studies, DNAs were extracted from samples of a given species (different locations, breeds, and populations) and used to amplify and sequence control-regions of the mitochondrial genome. Additionally, phylogenetic trees and haplotypes networks were generated (Larson, 2011). Mitochondrial DNA (mtDNA) is considered an ideal marker, being extremely mutable within species and has been used to study demographic expansion, genetic diversity, and phylogenetic structure (Bruford et al., 2003). However, these sequences are derived from the maternally inherited genome and have a limited power to identify and quantify hybridization between different populations (Larson, 2011). This approach was intensively used for several species as dogs (Verginelli et al., 2005), pigs (Giuffra et al., 2000), horses (Jansen et al., 2002), and cattle (Loftus et al., 1994), however with no deep insights into their domestication process. Some authors suggest that recent selective breeding may contribute to undermining the signatures of mtDNA between domesticates and their ancestors (Librado et al., 2016). Additionally, quantitative trait loci (QTL) mapping has been used to identify candidate genes associated with domestication traits. With these analyses, there have been some improvements in the domestication process, namely for fox (Kukekova et al., 2011), chickens (Fallahsharoudi et al., 2017), pigs (Rodriguez et al., 2005), and cattle (Khatkar et al., 2004).
GWAS, NGS, Microarray, and mtDNA
In domestic animals, the genome-wide association studies (GWAS) has become an important method to study the genomic regions involved in traits of concern with the sequencing of the pig, cow, and dog genomes (Lindblad-Toh et al., 2005; Groenen et al., 2012; Hekman et al., 2015). This method investigates the likelihood of the association of the genetic markers with a specific trait in domestic animals (Cadieu et al., 2009). The variation in these traits is due to intensive selective pressure elucidated by a small number of loci in these species (Boyko et al., 2010). Consequently, in several domesticated species, GWAS have effectively recognized contributing genes both for complex traits and Mendelian traits controlled by loci with big influence size (Hekman et al., 2015). The use of genome-wide methods initiated the microarray gene expression studies in the search for a group of genes or gene linkages involved in composite phenotypes in domesticated animals (Everts et al., 2005). However, the use of microarray techniques is narrow due to their dependencies on the use of identified probes, needing species-specific markers for the most precise results (Hekman et al., 2015). The introduction of next-generation sequencing (NGS) technologies has revolutionized the gene expression research by removing the need for prior probes for transcripts. RNA-seq or RNA sequencing requires the high-throughput reads generated by NGS to characterize the whole transcriptome: in other words, all transcripts generated in a tissue sample plus novel isoforms and formerly uncharacterized recorded sequences (Allen et al., 2010). RNA-seq is generally used for a range of applications, such as to compare the group of differentially expressed genes in tissue samples or samples from different experimental groups or populations (Bottomly et al., 2011; Roy et al., 2013). Based on NGS or RNA-seq data, the individuals can be grouped into healthy or diseased by identifying the gene linkages associated with hereditary diseases or other genetic traits or by mapping various genetic loci (Gautier et al., 2012; Tonomura et al., 2015). Furthermore, mtDNA technology was used to recognize the genomic regions associated with important phenotypic traits as well as to identify the evolutionary history and the origin of domestication in animal species as compared with nuclear markers (MacHugh and Bradley, 2001; Akey et al., 2010). Since 2000, sequences of mtDNA fragments such as D-loop and cytochrome b regions have been used to study distribution of different domesticated animals, including dogs (Savolainen et al., 2002), sheep (Hiendleder et al., 2002), pigs (Giuffra et al., 2000), cattle (Troy et al., 2001), goats (Luikart et al., 2001), horses (Jansen et al., 2002), chickens (Liu et al., 2006), donkeys (Beja-Pereira et al., 2004), and cats (Driscoll et al., 2007), as well as those with more restricted dispersals, such as water buffalo (Kierstein et al., 2004), and zebu cattle (Chen et al., 2010). mtDNA research has provided a viewpoint, at least from the parental side, as to the probable ancestors and candidate pedigrees involved in the domestication of species (Wang et al., 2014b).
Understanding the genetic basis of phenotypic variations is the major aim of genetics. Domestic animals offer a subjective opening for making significant improvement toward the goal of minimizing the gap between human biology and traditional model species (Qanbari and Simianer, 2014). The continuous progress of high throughput sequencing technologies and modern bioinformatics approaches provide the complete genetic variation map of an individual, and it is now likely to test for phenotypic variation caused by single nucleotide polymorphisms (SNPs). The genome-wide association studies (GWAS) and candidate gene approach are two primary methods that are currently followed to recognize genomic regions or genes affecting the particular trait. Population genomics has presented a new model for linking DNA with a phenotype that has been revealed as a selection signature analysis. This is a genome to phenotype method that includes the statistical assessment of population genomic data irrespective of phenotype to find out targets of the previous selection. Selection analysis can be employed in the natural population so numerous species (Akey et al., 2010) for which a high-density genetic record is available. An additional benefit is that it can detect selection if the preferred allele is previously fixed, while GWAS fails in such a condition (Qanbari and Simianer, 2014).
SNP Chip
In modern researches, through using recent genomics technologies, it has become likely to explore the micro-evolutionary developments underlying animal domestication at the molecular level. In this regard, various studies that have been done to produce domestic red fox, silver foxes (Vulpes vulpes), and rats (Rattus norvegicus) have provided valuable understandings that were introduced during the middle of the twentieth century (Albert et al., 2009, 2011; Trut et al., 2009). Furthermore, functional genomics, quantitative real-time PCR, microarray and reverse transcription studies of brain tissues from domestic dogs and wolves, transcriptional profiling through RNA-sequencing of rat brains combining genome mapping studies have identified numerous candidate genes and putative regulatory regions that have influenced docility and viciousness in animals (Heyne et al., 2014). It is necessary to annotate that the acute changes in gene expression linked to domestication possibly affect the growing stages in a specific tissue and will require extensive work to be conclusive (Carneiro et al., 2014). Gene enrichment analysis recognized that neurobiology is affected by the loci of those genes that were over-represented and targeted by directional selection, and principle functional analyses exposed that derived single nucleotide polymorphisms in developmental genes (PAX2 and SOX2) were possible to be fixed within, or close to, regulatory sequences. Most remarkably, it was concluded that domestication was predominantly associated with selective sweeps causing genetic variations on regulatory regions throughout the animal genome, therefore indicating micro-evolutionary developments during the initial periods of domestication of vertebrate species (Carneiro et al., 2014). Understanding the significance of single disease-associated SNP alleles itself is neither necessary nor enough in causal of a disease. Rather, it is possibly the collective consequence of a set of SNP alleles enclosed by key genes, plus environmental factors that jointly conclude whether an individual experiences a certain disease. The association study process includes the frequency determination of test factor (e.g., an SNP allele) among several patients and in the race and age-matched controls. The determination of the validity of this test crucially depends on appropriate patient-to-control matching (population stratification). Another way of performing association studies more efficiently is by limiting the SNPs through pre-selection by testing the pathogenic effects of SNPs. For the betterment of association studies design, one way is to exploit pathogenic allele’s linkage disequilibrium. When there is strong linkage disequilibrium among the SNP marker and an unknown pathogenic allele, both can show a parallel association with the disease (Pruvost et al., 2011; Corbett-Detig et al., 2015).
Nuclear Genes Epigenetics
More recently (since 2006), “next-generation sequencing” (NGS) technologies allowed access to the whole-genome sequences and gained new information about timing and location of domestication (Wang et al., 2014b). These approaches expanded the scope of comparative genomics from single genes to gene families and entire genomes. Mutations in DNA can vary from a single polymorphism to gene duplication or even complete genome duplication leading to many consequences, including phenotypic alterations. Additionally, a single gene can be involved in multiple, unrelated phenotypes (Hodgkin, 2002; Hartl, 2009), and genes of polygenic traits can act in combination to produce a single phenotype. The variations within the gene could be triggered by nucleotide exchange (non-synonymous or synonymous) or indels, which can generate adaptive, negative, or neutral alterations in the gene. More recently, these approaches allowed the availability of genomes for several species and permitted the association of genes to specific traits of domestication in cattle (Yurchenko et al., 2018), rabbits (Carneiro et al., 2014), sheep (Zamani et al., 2018), pigs (Rubin et al., 2012), horses (Zhang et al., 2018), dogs (Pendleton et al., 2018), and ducks (Zhou et al., 2018). Several other studies focus on the search for signatures of selection in those genes already described as important for the domestication process (Neves et al., 2014; Ahmad et al., 2017a, b).
Paleogenomics, also known as genome-wide ancient DNA (aDNA) analysis, gives valuable information and has been crucial to investigate when, where, and how rapidly adaptive alleles spread in the populations (Irving-Pease et al., 2018; Brunson and Reich, 2019). Indeed, recent studies using this approach provide new insights into the evolution and history of the cave bear (Barlow et al., 2018) and horses (Gaunitz et al., 2018). In livestock, biotechnology and conservative tools have contributed considerably to improve productivity, preserve genetic diversity, and enhance the adaptation to the environment (Ko and Takahashi, 2006). Furthermore, functional genomics, quantitative real-time PCR, microarray, and reverse transcription studies identified many candidate genes and putative regulatory regions that have influenced docility and viciousness in animals (Heyne et al., 2014).
With these advances in technology, the genetic architecture of domestication and the domestication process in some species became clearer (Caliebe et al., 2017; Kemp et al., 2017; Pitt et al., 2019). For example, it was possible to discover that pig populations were domesticated in one place and then moved to new areas successively gained the mitochondrial signature of native wild populations (Ottoni et al., 2012). The same applies to other taxa. These new approaches also allowed to verify that African cattle are mixtures of “taurine” and “indicine” that possess both Y-chromosome signature and mitochondrial signals (Larson and Fuller, 2014).
Genomics and the Domestication Process
The development of genome technologies such as genome assembly by sequencing, whole-genome shotgun (WGS) method (Lindblad-Toh et al., 2005), NGS (Dong et al., 2013), and third-generation single-molecule sequencing tools (Koren et al., 2012) are outstanding approaches are advancing our understanding to study animal domestication. Genome sequencing of domesticated species not only provides important resources to answer the queries raised by Darwin but also provides prospects to discover the genetic origin of profitable traits in domesticated species (Hillier et al., 2014). The current information on genome projects highlights the demographic history, origins, and the artificial selection of domesticated animal species (Anthony et al., 1986). Additionally, it was determined with a preview of future guidelines for animal domestication. Various techniques were established to allow the de novo assembly of genomes during the human genome project (Venter et al., 2015). The most effective technique was WGS sequencing, along with the building of physical maps. By 2009, four domesticated animals (horse, cat, dog, and taurine cattle) genomes and one wild species genome (the red jungle fowl) were sequenced and assembled based on this method (Wallis et al., 2004).
The domestication process is demonstrated through the detection of selection at a very large number of genomic loci that have likely evolved by natural and artificial selection (Carneiro et al., 2014; Larson et al., 2014). Recently, it has become possible to explore the domestication process by complete genome sequences analyses at high resolution to define how it has formed modern domestic animal. Several genomic loci such as the DGAT1 gene that is associated with lactation traits comprise a major quantitative trait nucleotide has been identified putatively under the selection through genome-wide comparisons of data from modern taurine and indicine cattle (Park et al., 2015). Furthermore, 106 candidate genes were found under selection using population genomics approaches that were particularly involved in muscle development, growth, function, and immunity. This first genome-wide selection analyses detected genes that are considered as important candidates of domestication (Ludwig et al., 2015). However, many methods are available to retrieve and sequence genomes routinely and recognize hundreds of genomic loci under selection through genotyping. It is revealed that paleogenomics techniques will be used to investigate when, where, and how rapidly adaptive alleles spread at the population in domestic animals (MacHugh et al., 2017). Several arithmetical methods have been established by scientists to reveal diverse features of how to accomplish variations from and what is anticipated with respect to genetic differences in the neutral model (Voight et al., 2006). While all statistical methods are based on neutral genomic differences, not all of them are based on similar information. Most of the methods were established for complete sequence data and not for genome-wide pools of predetermined SNPs, which are presently accessible in few livestock species (Corbett-Detig et al., 2015).
Epigenetics in the Evolution of the Domestic Traits
Evidence illustrates that in addition to genetic factors, epigenetic factors can affect the behavioral phenotypes as well as other traits within breed or species (Jensen, 2015; Bélteky et al., 2018). For example, the difference in the behavior of the great tit is statistically linked to the DNA methylation at dopamine receptor genes (Verhulst et al., 2016). Similarly, maternal behavior affects the DNA methylation of the hippocampal glucocorticoid receptor gene in rats (Weaver et al., 2004). In domestic chickens, variances in DNA methylation are linked to disease exposure (Tian et al., 2013), immune reactions (Berghof et al., 2013), metabolism, and growth (Hu et al., 2013).
Furthermore, epigenetic changes can occur just after individuals are exposed to various rearing environments (Pértille et al., 2017). Although cell division maintains the DNA methylation patterns, sometimes these could be regulated by external stimuli (Raynal et al., 2012). The DNA methylation changes that are controlled by the environment can be transferred through the germline (Guerrero-Bosagna et al., 2010) and remain unchanged over generations in somatic tissues (Franklin et al., 2010; Goerlich et al., 2012). Somatic epigenetic changes can affect phenotypic traits, whether selected deliberately or involuntarily or formed by the environment. Therefore, mechanisms of epigenetic changes could be an essential factor in the development of prompt phenotypic variations that arise during domestication. For example, these methylation changes are controlled by substantial hyper-methylation in domestic white Leghorn chickens as compared to the red jungle fowl (Nätt et al., 2012), in pedigree dogs compared to wolves (Janowitz Koch et al., 2016), and in domestic compared to wild worms (Xiang et al., 2013).
Domestication Genes
As described above, selection played a crucial role during domestication accelerating phenotypic variations. Skin and coat color are considered the only domestic traits subject to early selection by humans, thus becoming an essential genetic marker. Coat color can have patterned (spotted, striped) and non-patterned (solid colors) phenotypes that are defined by the proportion of two pigments: eumelanin (black/brown) and pheomelanin (red/yellow) (Cieslak et al., 2011; Koseniuk et al., 2018). During the past years, this has been a central question in domestication studies, and MC1R, ASIP, TYRP1, CBD103, KIT, and PMEL17 genes were associated with different traits in several species (Table 1). A variety of mutations, including single nucleotide polymorphisms (SNPs), insertions, deletions, and duplications are responsible for coat color discrepancies in both domestic and wild populations (Cui et al., 2018).
TABLE 1
| Related traits | Gene | Gene name | Species | References |
| Coat/skin/fur color | MC1R | Melanocortin-1 receptor | Rabbit, dogs, cat, pigs, horses, dromedary, sheep, goat, chicken, cattle, mice | Schmutz et al., 2002; Andersson, 2003; Fontanesi et al., 2006; Almathen et al., 2018 |
| ASIP | Agouti signaling protein | Horses, dromedary, dogs, pigs, sheep, goat, chicken, cattle, donkey | Rieder et al., 2001; Royo et al., 2005; Schmutz and Berryere, 2007a; Norris and Whan, 2008; Mao et al., 2010; Ahmad et al., 2017b; Almathen et al., 2018 | |
| TYRP1 | Tyrosinase-related protein 1 | Dogs, horses, ferret, hamster, sheep, goat, chicken, cattle, mice, donkey, cat | Rieder et al., 2001; Guibert et al., 2004; Schmutz and Berryere, 2007a | |
| CDB103 | Beta-defensin | Dog, cat, chicken, turkey | Ollivier et al., 2013; Cheng et al., 2015; Galov et al., 2015 | |
| KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | Rabbit, fox, mice, pigs, horses, dog, cat, sheep, goat | Haase et al., 2007; Haase et al., 2009; Reissmann and Ludwig, 2013; Wong et al., 2013; Fontanesi et al., 2014b; Johnson et al., 2015 | |
| PMEL17 | Premelanosome Protein 17 | Dogs, horses, ferret, hamster, chicken, cattle, mice, donkey, cat | Kerje et al., 2004; Brunberg et al., 2006; Schmutz and Berryere, 2007a; Karlsson et al., 2009; Komáromy et al., 2011; Reissmann and Ludwig, 2013; Schmutz and Dreger, 2013 | |
| MLPH | Melanophilin | Rabbit, dogs, horses, ferret, hamster, sheep, goat, chicken, cattle, mice, donkey, cat | Philipp et al., 2005; Schmutz and Berryere, 2007a; Vaez et al., 2008; Hauswirth et al., 2012; Fan R. et al., 2013; Fontanesi et al., 2014a; Demars et al., 2018 | |
| LYST | Lysosomal trafficking regulator | Dogs, ferret, hamster, chicken, cattle, mice, donkey, cat | Kunieda et al., 1999; Runkel et al., 2006; Tryon et al., 2007; Anistoroaei et al., 2013 | |
| SLC45A2 | Solute carrier family 45 member 2 | Rabbit, dogs, ferret, hamster, sheep, goat, chicken, cattle, mice, donkey, cat | Steingrímsson et al., 2006; Gunnarsson et al., 2007; Schmutz and Berryere, 2007b; Manceau et al., 2010; Wijesena and Schmutz, 2015 | |
| MITF | Microphthalmia-associated transcription factor | Dogs, cat, pigs, chicken, cattle, mice | Rieder et al., 2001; Guibert et al., 2004; Manceau et al., 2010; Hauswirth et al., 2012; Haase et al., 2013; Körberg et al., 2014; Han et al., 2015 | |
| TRPM1 | Transient receptor potential cation channel subfamily member 1 | Horses, dromedary, dogs, pigs, sheep, goat, chicken, cattle, mice, donkey, cat | Bellone et al., 2010; Webb and Cullen, 2010; Cieslak et al., 2011; Reissmann and Ludwig, 2013 | |
| Horn type | RXFP2 | Relaxin family peptide receptor 2 | Sheep, goat, cattle | Lühken, 2012; Wang X. et al., 2014; Pan et al., 2018 |
| FOXL2 | Forkhead box L2 | Cattle | ||
| Gait type | DMRT3 | Doublesex and Mab-3 related transcription factor 3 | Horses | Kristjansson et al., 2014; Promerová et al., 2014; Pereira et al., 2016; Staiger et al., 2017 |
| Ear size | PPARD | Peroxisome proliferator activated receptor delta | Pigs | Ren et al., 2011; Wilkinson et al., 2013 |
Genes involved in domestication phenotypes in animals.
With the advances in technology and the knowledge on domestication, recent studies were able to associate Tph1 and Gabra5 genes to tameness and to produce tamed animals, namely foxes (Vulpes vulpes) and rats (Rattus norvegicus) (Albert et al., 2009, 2011; Trut et al., 2009). Besides, several other genes have been associated with different phenotypes in different animals. In dogs, genes associated with wrinkled skin (HAS2), body size (IGF1), leg length (FGF4), and fur growth and texture (FGF5, RSPO2, and KRT71) were identified and reported (Sutter et al., 2007; Cadieu et al., 2009; Parker et al., 2009; Hellström et al., 2010). In cattle (Grobet et al., 1997), sheep (Clop et al., 2006), pigs (Stinckens et al., 2008), goat (Zhang et al., 2012), horses (Dall’olio et al., 2010), and dogs (Mosher et al., 2007), a mutation in the MSTN gene is related to increased muscular development (Ahad et al., 2017). Also, in cattle, the DGAT1 and ABCG2 genes are responsible for variations in milk production and composition, respectively (Ogorevc et al., 2009). The HMGA2 and LCORL-NCAPG genes are associated with stature and body size in cattle (Pryce et al., 2011), rabbits (Carneiro et al., 2017), pigs (Rubin et al., 2012), horses (Frischknecht et al., 2015), and dogs (Jones et al., 2008). Another trait that has been the focus of several studies is the meat tenderness with genes associated with cattle (CAST2, HSP90AA1, DNAJA1, and HSPB1) (Malheiros et al., 2018) and pigs (CAST, HAL, RYR1, and RN).
Challenges and Prospects
Ancient information-rich biomolecules such as DNA and proteins have long been under discussion and systematically analyzed by the scientists (Pääbo et al., 1989). In the early 1980s, the DNA cloning was made possible through a cumbersome molecular cloning approach (Higuchi et al., 1984), which eventually proved unreliable, particularly making fake DNA sequences from a 2400-year-old Egyptian mummy (Pääbo, 1985). In 1980s, the amplification of a DNA from archeological material and museum samples using polymerase chain reaction (PCR) method brought an important revolution (Pääbo et al., 1988). However, it was a big challenge that got attention in scientific community to recover reliable and reproducible DNA (Thomas et al., 1989). Therefore, the DNA field has been affected with major technical hurdles such as the presence of inhibitors of enzymatic reactions, post-mortem loss, contamination of preserved samples, and all the influences that can irreversibly contain the validity and reproducibility of a DNA amplified from archeological specimens (Lindahl, 1997; Cooper and Poinar, 2000). However, the field of archaeo-genetics has been developed over the last four decades, scientists have scientifically tackled the methodological challenges related to DNA recovery from long-dead materials, and it is now well-known that an authentic and reproducible genetics information can be produced from the fossils of vertebrates. Thus a DNA research has had an ancient concern in understanding the biology and evolution of domestic animals and their wild ancestors (Troy et al., 2001; Leonard et al., 2002).
Domestication research has challenged scientists since Darwin, and despite the amount of new literature published every year, there are still many questions regarding this thematic. This endless process began several million years ago and included diverse pressures that shaped animals in different ways and differentiate them from their ancestors. Genes linked with coat color were the ones associated with early domestication, being widely studied. With this exception, no other genes were associated with the early stages of animal domestication. Recent studies described that animals were firstly selected based on behavioral characteristics, making hard the research when compared to morphological traits. Indeed, if we look at the crop and plant domestication where the insights obtained in the past years, with the same approaches, are significant when comparing to animals.
With the advances in sequencing and assembly technologies, genomes from different domestic and wild animals are becoming accessible. In addition to these, genomes from ancient populations are becoming available. Genetic research has a wide range of toolkits to explain not only the relationships between the domesticated animals and their wild ancestors but also the domestication traits and their genetic architecture. These results are essential to compare the patterns in the modern populations with those of the previous generations and data from phylogeography and also to identify new genes and associate them with specific traits. Methylomics and transcriptomic analyses are essential to study the epigenetic factors and expression present in wild and domesticated animals to support the variations linked to domestication.
The recent and potential applications of evolutionary biology may deliver answers for main social challenges. It is important to examine the relationship among the environment and the traits of organisms that have been influenced through the adaptation to modern environments and the patterns of selection triggered by their environments during domestication period. A conceptual perspective connecting all of these environmental, genetic, and developmental manipulations is expected to lead to better application and cross-disciplinary incorporation of functional evolutionary approaches to study domestication of animals and their relationship to wild ancestors. It is important to highlight the evolutionary plans/policies that may be used to accomplish required targets of sustainable development for better health, use of natural resource and biodiversity conservation, including how domestication conflicts have been reduced to accomplish preferred outcomes. Therefore, the facts of building a more integrated field of evolutionary biology must be underscored to address global challenges of domestication. Human impact on the biosphere has deep consequences for both direction and the rate of evolution during evolution. At the same time, animals and other organisms that are worthy for ecological, aesthetic or economic reasons are often not able to adapt rapidly enough to keep pace with changes of the environment impacted by human activities. These modern dilemmas progressively threaten animal health, biological diversity, and domestication history. Meanwhile, the problem of earth’s sixth mass extinction of species becomes impending as species are inept to adapt quickly to environmental variations. A developing interest of evolutionary biology may help us to improve our skills to cope with challenges to solve most of pressing problems of domestication of animal species during the twenty-firstcentury.
Future works should apply these technologies and obtain genomes of a large number of individuals inside different species worldwide in order to better comprehend the genetic, morphological, and behavioral characteristics of different species. With these achievements, we hope to fully understand when, where, and how animals where domesticated and consequently understand the human civilizations.
Conclusion
Although the genetic and geographic pattern of early animal domestication is poorly understood, a clear background for understanding the evolutionary routes of domesticated animals is progressing. The early stages of animal domestication show an extended coevolutionary progression with various phases along diverse trajectories that enhanced the reproduction and survival of domesticates. Natural selective pressures relaxed, and new mutations arose and allowed for unique traits.
Our understanding of the genetic basis of animal domestication facilitates improvements through breeding using new techniques. Defining important events of domestication delivers a unique aspect in studying the linkage between humans and the natural world and determines the events that drive human cultural evolution to interact with that leading biological evolution.
Statements
Author contributions
JC, NA, AE, SA, and MA: conception and design of the work. HA: data acquisition. HA, MA, and FJ: analysis. SA: writing of the manuscript. HA, NA, and FJ: critical review.
Funding
This work was funded by the GDAS project of Science and Technology Development (2019-GDASYL-0103059 and 2018GDASCX-0107).
Acknowledgments
We thankful to reviewers for their comments and critical reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Aberle K. S. Distl O. (2004). Domestication of the horse: results based on microsatellite and mitochondrial DNA markers.Archiv. Fur. Tierzucht.47517–536.
2
Ahad W. A. Andrabi M. Beigh S. A. Bhat R. A. Shah R. A. (2017). Applications of myostatin (MSTN) gene in the livestock animals and humans: a review.Int. J. Curr. Microbiol. App. Sci.61807–1811.
3
Ahmad H. I. Liu G. Jiang X. Liu C. Chong Y. Huarong H. (2017a). Adaptive molecular evolution of MC 1R gene reveals the evidence for positive diversifying selection in indigenous goat populations.Ecol. Evol.75170–5180. 10.1002/ece3.2919
4
Ahmad H. I. Liu G. Jiang X. Liu C. Fangzheng X. Chong Y. et al (2017b). Adaptive selection at agouti gene inferred breed specific selection signature within the indigenous goat populations.Asian Austr. J. Anim. Sci.10.5713/ajas.16.0994
5
Ajmone-Marsan P. Garcia J. F. Lenstra J. A. (2010). On the origin of cattle: how aurochs became cattle and colonized the world.Evol. Anthropol.19148–157.
6
Akey J. M. Ruhe A. L. Akey D. T. Wong A. K. Connelly C. F. Madeoy J. et al (2010). Tracking footprints of artificial selection in the dog genome.Proc. Natl. Acad. Sci. U.S.A.1071160–1165. 10.1073/pnas.0909918107
7
Albert F. W. Carlborg O. Plyusnina I. Besnier F. Hedwig D. Lautenschläger S. et al (2009). Genetic architecture of tameness in a rat model of animal domestication.Genetics182541–554. 10.1534/genetics.109.102186
8
Albert F. W. Hodges E. Jensen J. Besnier F. Xuan Z. Rooks M. et al (2011). Targeted resequencing of a genomic region influencing tameness and aggression reveals multiple signals of positive selection.Heredity107205–214. 10.1038/hdy.2011.4
9
Allen H. L. Estrada K. Lettre G. Berndt S. I. Weedon M. N. Rivadeneira F. et al (2010). Hundreds of variants clustered in genomic loci and biological pathways affect human height.Nature467832–838. 10.1038/nature09410
10
Almathen F. Elbir H. Bahbahani H. Mwacharo J. Hanotte O. (2018). Polymorphisms in MC1R and ASIP genes are associated with coat color variation in the arabian camel.J. Hered.109700–706. 10.1093/jhered/esy024
11
Andersson L. (2001). Genetic dissection of phenotypic diversity in farm animals.Nat. Rev. Genet.2:130. 10.1038/35052563
12
Andersson L. (2003). Melanocortin receptor variants with phenotypic effects in horse, pig, and chicken.Ann. N. Y. Acad. Sci.994313–318. 10.1111/j.1749-6632.2003.tb03195.x
13
Andersson L. (2016). Domestic animals as models for biomedical research.UPSALA J. Med. Sci.1211–11. 10.3109/03009734.2015.1091522
14
Anistoroaei R. Krogh A. Christensen K. (2013). A frameshift mutation in the LYST gene is responsible for the Aleutian color and the associated Chédiak–Higashi syndrome in American mink.Anim. Genet.44178–183. 10.1111/j.1365-2052.2012.02391.x
15
Anthony D. W. Bogucki P. Comşa E. Gimbutas M. Jovanović B. Mallory J. et al (1986). The Kurgan culture, indo-european origins, and the domestication of the horse: a reconsideration and comments and replies.Curr. Anthropol.27291–313.
16
Barker J. (2014). Water buffalo: domestication.L’Anthropologie94619–642.
17
Barker J. T. Tan S. G. Mukherjee T. (1991). Future studies of genetic differentiation among swamp buffalo and native goat populations. Buffalo and goats in Asia: genetic diversity and its application. 34:144.
18
Barlow A. Cahill J. A. Hartmann S. Theunert C. Xenikoudakis G. Fortes G. G. et al (2018). Partial genomic survival of cave bears in living brown bears.Nat. Ecol. Evol.2:1563. 10.1038/s41559-018-0654-8
19
Beja-Pereira A. Caramelli D. Lalueza-Fox C. Vernesi C. Ferrand N. Casoli A. et al (2006). The origin of European cattle: evidence from modern and ancient DNA.Proc. Natl. Acad. Sci. U.S.A.1038113–8118. 10.1073/pnas.0509210103
20
Beja-Pereira A. England P. R. Ferrand N. Jordan S. Bakhiet A. O. Abdalla M. A. et al (2004). African origins of the domestic donkey.Science3041781–1781.
21
Bellone R. R. Forsyth G. Leeb T. Archer S. Sigurdsson S. Imsland F. et al (2010). Fine-mapping and mutation analysis of TRPM1: a candidate gene for leopard complex (LP) spotting and congenital stationary night blindness in horses.Briefings Funct. Genom.9193–207. 10.1093/bfgp/elq002
22
Bélteky J. Agnvall B. Bektic L. Höglund A. Jensen P. Guerrero-Bosagna C. (2018). Epigenetics and early domestication: differences in hypothalamic DNA methylation between red junglefowl divergently selected for high or low fear of humans.Genet. Select. Evol.50:13. 10.1186/s12711-018-0384-z
23
Berghof T. Parmentier H. Lammers A. (2013). Transgenerational epigenetic effects on innate immunity in broilers: An underestimated field to be explored?Poul. Sci.922904–2913. 10.3382/ps.2013-03177
24
Bocquet-Appel J.-P. (2008). “Explaining the Neolithic demographic transition,” in The Neolithic Demographic Transition And Its Consequences, edsBar-YosefJ. P.Bocquet-AppelO. (Dordrecht: Springer), 35–55.
25
Bollongino R. Burger J. Powell A. Mashkour M. Vigne J.-D. Thomas M. G. (2012). Modern taurine cattle descended from small number of Near-Eastern founders.Mol. Biol. Evol.292101–2104. 10.1093/molbev/mss092
26
Bolstad G. H. Hindar K. Robertsen G. Jonsson B. Saegrov H. Diserud O. H. et al (2017). Gene flow from domesticated escapes alters the life history of wild Atlantic salmon.Nat. Ecol. Evol.1:0124. 10.1038/s41559-017-0124
27
Bottomly D. Walter N. A. Hunter J. E. Darakjian P. Kawane S. Buck K. J. et al (2011). Evaluating gene expression in C57BL/6J and DBA/2J mouse striatum using RNA-Seq and microarrays.PLoS One6:e17820. 10.1371/journal.pone.0017820
28
Boyko A. R. Quignon P. Li L. Schoenebeck J. J. Degenhardt J. D. Lohmueller K. E. et al (2010). A simple genetic architecture underlies morphological variation in dogs.PLoS Biol.8:e1000451. 10.1371/journal.pbio.1000451
29
Bradley D. G. Magee D. A. (2006). Genetics and the origins of domestic cattle.Document. Domest.22317–328.
30
Bruford M. W. Bradley D. G. Luikart G. (2003). DNA markers reveal the complexity of livestock domestication.Nat. Rev. Genet.4:900. 10.1038/nrg1203
31
Brunberg E. Andersson L. Cothran G. Sandberg K. Mikko S. Lindgren G. (2006). A missense mutation in PMEL17 is associated with the Silver coat color in the horse.BMC Genet.7:46. 10.1186/1471-2156-7-46
32
Brunson K. Reich D. (2019). The promise of paleogenomics beyond our own species.Trends Genet.35319–329. 10.1016/j.tig.2019.02.006
33
Cadieu E. Neff M. W. Quignon P. Walsh K. Chase K. Parker H. G. et al (2009). Coat variation in the domestic dog is governed by variants in three genes.Science326150–153. 10.1126/science.1177808
34
Caliebe A. Nebel A. Makarewicz C. Krawczak M. Krause-Kyora B. (2017). Insights into early pig domestication provided by ancient DNA analysis.Sci. Rep.7:44550. 10.1038/srep44550
35
Carneiro M. Hu D. Archer J. Feng C. Afonso S. Chen C. et al (2017). Dwarfism and altered craniofacial development in rabbits is caused by a 12.1 kb deletion at the HMGA2 locus.Genetics205955–965. 10.1534/genetics.116.196667
36
Carneiro M. Rubin C.-J. Di Palma F. Albert F. W. Alföldi J. Barrio A. M. et al (2014). Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication.Science3451074–1079. 10.1126/science.1253714
37
Chen K. Baxter T. Muir W. M. Groenen M. A. Schook L. B. (2007). Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa).Intern. J. Biol. Sci.3:153. 10.7150/ijbs.3.153
38
Chen S. Lin B.-Z. Baig M. Mitra B. Lopes R. J. Santos A. M. et al (2010). Zebu cattle are an exclusive legacy of the South Asia Neolithic.Mol. Biol. Evol.271–6. 10.1093/molbev/msp213
39
Cheng Y. Prickett M. D. Gutowska W. Kuo R. Belov K. Burt D. W. (2015). Evolution of the avian β-defensin and cathelicidin genes.BMC Evol. Biol.15:188. 10.1186/s12862-015-0465-3
40
Chessa B. Pereira F. Arnaud F. Amorim A. Goyache F. Mainland I. et al (2009). Revealing the history of sheep domestication using retrovirus integrations.Science324532–536. 10.1126/science.1170587
41
Choi J.-W. Liao X. Stothard P. Chung W.-H. Jeon H.-J. Miller S. P. et al (2014). Whole-genome analyses of Korean native and Holstein cattle breeds by massively parallel sequencing.PLoS One9:e101127. 10.1371/journal.pone.0101127
42
Choudhury A. Barker J. (2014). Wild water buffalo Bubalus arnee (Kerr, 1792).Ecol. Evol. Behav. Wild Cattle255–301.
43
Cieslak M. Pruvost M. Benecke N. Hofreiter M. Morales A. Reissmann M. et al (2010). Origin and history of mitochondrial DNA lineages in domestic horses.PLoS One5:e15311. 10.1371/journal.pone.0015311
44
Cieslak M. Reissmann M. Hofreiter M. Ludwig A. (2011). Colours of domestication.Biol. Rev.86885–899.
45
Clop A. Marcq F. Takeda H. Pirottin D. Tordoir X. Bibé B. et al (2006). A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep.Nat. Genet.38:813. 10.1038/ng1810
46
Consortium B. H. (2009). Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds.Science324528–532. 10.1126/science.1167936
47
Cooper A. Poinar H. N. (2000). Ancient DNA: do it right or not at all.Science2891139–1139. 10.1126/science.289.5482.1139b
48
Corbett-Detig R. B. Hartl D. L. Sackton T. B. (2015). Natural selection constrains neutral diversity across a wide range of species.PLoS Biol.13:e1002112. 10.1371/journal.pbio.1002112
49
Crowley J. Adelman B. (1998). The Complete Dog Book: Official Publication Of The American Kennel Club.New York, NY: Howell House.
50
Cui Y. Yan H. Wang K. Xu H. Zhang X. Zhu H. et al (2018). Insertion/deletion within the KDM6A gene is significantly associated with litter size in goat.Front. Genet.9:91. 10.3389/fgene.2018.00091
51
Dall’olio S. Fontanesi L. Nanni Costa L. Tassinari M. Minieri L. Falaschini A. (2010). Analysis of horse myostatin gene and identification of single nucleotide polymorphisms in breeds of different morphological types.Biomed. Res. Intern.2010:542945. 10.1155/2010/542945
52
Darwin C. (1859). On the Origin of Species by Means of Natural Selection.London: J. Murray.
53
Darwin C. (2010). The Variation Of Animals And Plants Under Domestication.Cambridge: Cambridge University Press.
54
Demars J. Iannuccelli N. Utzeri V. Auvinet G. Riquet J. Fontanesi L. et al (2018). New insights into the melanophilin (MLPH) gene affecting coat color dilution in rabbits.Genes9:430. 10.3390/genes9090430
55
Di R. Farhad Vahidi S. Ma Y. He X. Zhao Q. Han J. et al (2011). Microsatellite analysis revealed genetic diversity and population structure among Chinese cashmere goats.Anim. Genet.42428–431. 10.1111/j.1365-2052.2010.02072.x
56
Dobbie W. R. Braysher M. (1993). Managing Vertebrate Pests: Feral Horses.Canberra: Australian Government Publishing Service.
57
Dong Y. Xie M. Jiang Y. Xiao N. Du X. Zhang W. et al (2013). Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus).Nat. Biotechnol.31135–141. 10.1038/nbt.2478
58
Driscoll C. A. Macdonald D. W. O’brien S. J. (2009). From wild animals to domestic pets, an evolutionary view of domestication.Proc. Natl. Acad. Sci. U.S.A.1069971–9978. 10.1073/pnas.0901586106
59
Driscoll C. A. Menotti-Raymond M. Roca A. L. Hupe K. Johnson W. E. Geffen E. et al (2007). The Near Eastern origin of cat domestication.Science317519–523. 10.1126/science.1139518
60
Epstein H. (1984). “Cattle,” in Evolution of Domesticated Animals, ed.MasonI. L. (New York, NY: Longman).
61
Everts R. E. Band M. R. Liu Z. L. Kumar C. G. Liu L. Loor J. J. et al (2005). A 7872 cDNA microarray and its use in bovine functional genomics.Vet. Immunol. Immunopathol.105235–245. 10.1016/j.vetimm.2005.02.003
62
Fallahsharoudi A. De Kock N. Johnsson M. Bektic L. Ubhayasekera S. K. A. Bergquist J. et al (2017). Genetic and targeted eQTL mapping reveals strong candidate genes modulating the stress response during chicken domestication.G37497–504. 10.1534/g3.116.037721
63
Fan R. Xie J. Bai J. Wang H. Tian X. Bai R. et al (2013). Skin transcriptome profiles associated with coat color in sheep.BMC Genomics14:389. 10.1186/1471-2164-14-389
64
Fan W.-L. Ng C. S. Chen C.-F. Lu M.-Y. J. Chen Y.-H. Liu C.-J. et al (2013). Genome-wide patterns of genetic variation in two domestic chickens.Genome Biol. Evol.51376–1392. 10.1093/gbe/evt097
65
Flamand J. Vankan D. Gairhe K. Duong H. Barker J. (2003). Genetic identification of wild Asian water buffalo in Nepal.Anim. Conserv. Forum6265–270.
66
Fontanesi L. Scotti E. Allain D. Dall’olio S. (2014a). A frameshift mutation in the melanophilin gene causes the dilute coat colour in rabbit (Oryctolagus cuniculus) breeds.Anim. Genet.45248–255. 10.1111/age.12104
67
Fontanesi L. Vargiolu M. Scotti E. Latorre R. Pellegrini M. S. F. Mazzoni M. et al (2014b). The KIT gene is associated with the English spotting coat color locus and congenital megacolon in Checkered Giant rabbits (Oryctolagus cuniculus).PLoS One9:e93750. 10.1371/journal.pone.0093750
68
Fontanesi L. Tazzoli M. Beretti F. Russo V. (2006). Mutations in the melanocortin 1 receptor (MC1R) gene are associated with coat colours in the domestic rabbit (Oryctolagus cuniculus).Anim. Genet.37489–493. 10.1111/j.1365-2052.2006.01494.x
69
Franklin T. B. Russig H. Weiss I. C. Gräff J. Linder N. Michalon A. et al (2010). Epigenetic transmission of the impact of early stress across generations.Biol. Psychiatry68408–415. 10.1016/j.biopsych.2010.05.036
70
Frantz L. A. Mullin V. E. Pionnier-Capitan M. Lebrasseur O. Ollivier M. Perri A. et al (2016). Genomic and archaeological evidence suggest a dual origin of domestic dogs.Science3521228–1231. 10.1126/science.aaf3161
71
Frantz L. A. Schraiber J. G. Madsen O. Megens H.-J. Cagan A. Bosse M. et al (2015). Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes.Nat. Genet.47:1141. 10.1038/ng.3394
72
Frischknecht M. Jagannathan V. Plattet P. Neuditschko M. Signer-Hasler H. Bachmann I. et al (2015). A non-synonymous HMGA2 variant decreases height in Shetland ponies and other small horses.PLoS One10:e0140749. 10.1371/journal.pone.0140749
73
Galov A. Fabbri E. Caniglia R. Arbanasiæ H. Lapalombella S. Florijanèiæ T. et al (2015). First evidence of hybridization between golden jackal (Canis aureus) and domestic dog (Canis familiaris) as revealed by genetic markers.R. Soc. Open Sci.2:150450. 10.1098/rsos.150450
74
Gaunitz C. Fages A. Hanghøj K. Albrechtsen A. Khan N. Schubert M. et al (2018). Ancient genomes revisit the ancestry of domestic and Przewalski’s horses.Science360111–114. 10.1126/science.aao3297
75
Gautier E. L. Shay T. Miller J. Greter M. Jakubzick C. Ivanov S. et al (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages.Nat. Immunol.13:1118. 10.1038/ni.2419
76
Gentry A. Clutton-Brock J. Groves C. P. (2004). The naming of wild animal species and their domestic derivatives.J. Archaeol. Sci.31645–651.
77
Giuffra E. Kijas J. Amarger V. Carlborg O. Jeon J.-T. Andersson L. (2000). The origin of the domestic pig: independent domestication and subsequent introgression.Genetics1541785–1791.
78
Goerlich V. C. Nätt D. Elfwing M. Macdonald B. Jensen P. (2012). Transgenerational effects of early experience on behavioral, hormonal and gene expression responses to acute stress in the precocial chicken.Horm. Behav.61711–718. 10.1016/j.yhbeh.2012.03.006
79
Gou X. Wang Z. Li N. Qiu F. Xu Z. Yan D. et al (2014). Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia.Genome Res.241308–1315. 10.1101/gr.171876.113
80
Grobet L. Martin L. J. R. Poncelet D. Pirottin D. Brouwers B. Riquet J. et al (1997). A deletion in the bovine myostatin gene causes the double–muscled phenotype in cattle.Nat. Genet.17:71. 10.1038/ng0997-71
81
Groenen M. A. Archibald A. L. Uenishi H. Tuggle C. K. Takeuchi Y. Rothschild M. F. et al (2012). Analyses of pig genomes provide insight into porcine demography and evolution.Nature491393–398. 10.1038/nature11622
82
Groeneveld L. Lenstra J. Eding H. Toro M. Scherf B. Pilling D. et al (2010). Genetic diversity in farm animals–a review.Anim. Genet.416–31. 10.1111/j.1365-2052.2010.02038.x
83
Groves C. (1981). Systematic relationships in the bovini (Artiodactyla, Bovidae).J. Zool. Syst. Evol. Res.19264–278.
84
Guerrero-Bosagna C. Settles M. Lucker B. Skinner M. K. (2010). Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome.PLoS One5:e13100. 10.1371/journal.pone.0013100
85
Guibert S. Girardot M. Leveziel H. Julien R. Oulmouden A. (2004). Pheomelanin coat colour dilution in French cattle breeds is not correlated with the TYR, TYRP1 and DCT transcription levels.Pigment. Cell Res.17337–345. 10.1111/j.1600-0749.2004.00152.x
86
Gunnarsson U. Hellström A. R. Tixier-Boichard M. Minvielle F. Bed’hom B. Ito S. I. et al (2007). Mutations in SLC45A2 cause plumage color variation in chicken and Japanese quail.Genetics175867–877. 10.1534/genetics.106.063107
87
Haase B. Brooks S. Tozaki T. Burger D. Poncet P. A. Rieder S. et al (2009). Seven novel KIT mutations in horses with white coat colour phenotypes.Anim. Genet.40623–629. 10.1111/j.1365-2052.2009.01893.x
88
Haase B. Brooks S. A. Schlumbaum A. Azor P. J. Bailey E. Alaeddine F. et al (2007). Allelic heterogeneity at the equine KIT locus in dominant white (W) horses.PLoS Genet.3:e195. 10.1371/journal.pgen.0030195
89
Haase B. Signer-Hasler H. Binns M. M. Obexer-Ruff G. Hauswirth R. Bellone R. R. et al (2013). Accumulating mutations in series of haplotypes at the KIT and MITF loci are major determinants of white markings in Franches-Montagnes horses.PLoS One8:e75071. 10.1371/journal.pone.0075071
90
Han J.-L. Min Y. Guo T.-T. Yue Y.-J. Liu J.-B. Niu C.-E. et al (2015). Molecular characterization of two candidate genes associated with coat color in Tibetan sheep (Ovis arise).J. Integr. Agric.141390–1397. 10.4238/2015.February.6.22
91
Hartl D. L. (2009). Essential Genetics: A Genomics Perspective: A Genomics Perspective.Burlington, MA: Jones & Bartlett Publishers.
92
Hausberger M. Roche H. Henry S. Visser E. K. (2008). A review of the human–horse relationship.Appl. Anim. Behav. Sci.1091–24.
93
Hauswirth R. Haase B. Blatter M. Brooks S. A. Burger D. Drögemüller C. et al (2012). Mutations in MITF and PAX3 cause “splashed white” and other white spotting phenotypes in horses.PLoS Genet.8:e1002653. 10.1371/journal.pgen.1008321
94
Hekman J. P. Johnson J. L. Kukekova A. V. (2015). Transcriptome analysis in domesticated species: challenges and strategies.Bioinform. Biol. Insights9:S29334. 10.4137/BBI.S29334
95
Hellström A. R. Sundstrom E. Gunnarsson U. Bed’hom B. Tixier-Boichard M. Honaker C. F. et al (2010). Sex-linked barring in chickens is controlled by the CDKN2A/B tumour suppressor locus.Pigment Cell Melanom. Res.23521–530. 10.1111/j.1755-148X.2010.00700.x
96
Helmer D. Gourichon L. Monchot H. Peters J. Segui M. S. (2005). Identifying Early Domestic Cattle From Pre-Pottery Neolithic Sites On The Middle Euphrates Using Sexual Dimorphism.Oxford: Oxbow Books.
97
Heyne H. O. Lautenschläger S. Nelson R. Besnier F. Rotival M. Cagan A. et al (2014). Genetic influences on brain gene expression in rats selected for tameness and aggression.Genetics1981277–1290. 10.1534/genetics.114.168948
98
Hiendleder S. Kaupe B. Wassmuth R. Janke A. (2002). Molecular analysis of wild and domestic sheep questions current nomenclature and provides evidence for domestication from two different subspecies.Proc. R. Soc. Lond. Ser. B Biol. Sci.269893–904. 10.1098/rspb.2002.1975
99
Higuchi R. Bowman B. Freiberger M. Ryder O. A. Wilson A. C. (1984). DNA sequences from the quagga, an extinct member of the horse family.Nature312282–284. 10.1038/312282a0
100
Hillier L. W. Miller W. Birney E. Warren W. Hardison R. C. Ponting C. P. et al (2014). Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution.Nature423695–777. 10.1038/nature03154
101
Hodgkin J. (2002). Seven types of pleiotropy.Intern. J. Dev. Biol.42501–505.
102
Hu Y. Xu H. Li Z. Zheng X. Jia X. Nie Q. et al (2013). Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers.PLoS One8:e56411. 10.1371/journal.pone.0056411
103
Huang Y.-F. Zhao Y.-J. He J.-N. (2017). Genetic diversity of three Chinese native sheep breeds.Russia. J. Genet.53118–127.
104
Hunter P. (2018). The genetics of domestication.EMBO Rep.19201–205.
105
Irving-Pease E. K. Ryan H. Jamieson A. Dimopoulos E. A. Larson G. Frantz L. A. (2018). “Paleogenomics of animal domestication,” in Paleogenomics Aleogenomics Population Genomics, edsRajoraC.LindqvistO. (Cham: Springer), 225–272.
106
Janowitz Koch I. Clark M. M. Thompson M. J. Deere-Machemer K. A. Wang J. Duarte L. et al (2016). The concerted impact of domestication and transposon insertions on methylation patterns between dogs and grey wolves.Mol. Ecol.251838–1855. 10.1111/mec.13480
107
Jansen T. Forster P. Levine M. A. Oelke H. Hurles M. Renfrew C. et al (2002). Mitochondrial DNA and the origins of the domestic horse.Proc. Natl. Acad. Sci. U.S.A.9910905–10910. 10.1073/pnas.152330099
108
Jensen P. (2015). Adding ‘epi-’to behaviour genetics: implications for animal domestication.J. Exp. Biol.21832–40. 10.1242/jeb.106799
109
Johnson J. Kozysa A. Kharlamova A. Gulevich R. Perelman P. Fong H. et al (2015). Platinum coat color in red fox (V ulpes vulpes) is caused by a mutation in an autosomal copy of KIT.Anim. Genet.46190–199. 10.1111/age.12270
110
Jones P. Chase K. Martin A. Davern P. Ostrander E. A. Lark K. G. (2008). Single-nucleotide-polymorphism-based association mapping of dog stereotypes.Genetics1791033–1044. 10.1534/genetics.108.087866
111
Joshi M. B. Rout P. K. Mandal A. K. Tyler-Smith C. Singh L. Thangaraj K. (2004). Phylogeography and origin of Indian domestic goats.Mol. Biol. Evol.21454–462. 10.1093/molbev/msh038
112
Kanginakudru S. Metta M. Jakati R. Nagaraju J. (2008). Genetic evidence from Indian red jungle fowl corroborates multiple domestication of modern day chicken.BMC Evol. Biol.8:174. 10.1186/1471-2148-8-174
113
Karlsson A. C. Kerje S. Hallböök F. Jensen P. (2009). The Dominant white mutation in the PMEL17 gene does not cause visual impairment in chickens.Vet. Ophthalmol.12292–298. 10.1111/j.1463-5224.2009.00714.x
114
Kemp B. M. Judd K. Monroe C. Eerkens J. W. Hilldorfer L. Cordray C. et al (2017). Prehistoric mitochondrial DNA of domesticate animals supports a 13th century exodus from the northern US southwest.PLoS One12:e0178882. 10.1371/journal.pone.0178882
115
Kerje S. Sharma P. Gunnarsson U. Kim H. Bagchi S. Fredriksson R. et al (2004). The dominant white, dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene.Genetics1681507–1518. 10.1534/genetics.104.027995
116
Khatkar M. S. Thomson P. C. Tammen I. Raadsma H. W. (2004). Quantitative trait loci mapping in dairy cattle: review and meta-analysis.Genet. Select. Evol.36:163. 10.1186/1297-9686-36-2-163
117
Kierstein G. Vallinoto M. Silva A. Schneider M. P. Iannuzzi L. Brenig B. (2004). Analysis of mitochondrial D-loop region casts new light on domestic water buffalo (Bubalus bubalis) phylogeny.Mol. Phylogenet. Evol.30308–324. 10.1016/s1055-7903(03)00221-5
118
Kijas J. W. Lenstra J. A. Hayes B. Boitard S. Neto L. R. P. San Cristobal M. et al (2012). Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection.PLoS Biol.10:e1001258. 10.1371/journal.pbio.1001258
119
Ko C. H. Takahashi J. S. (2006). Molecular components of the mammalian circadian clock.Hum. Mol. Genet.15R271–R277. 10.1007/978-3-642-25950-0_1
120
Komáromy A. M. Rowlan J. S. La Croix N. C. Mangan B. G. (2011). Equine Multiple Congenital Ocular Anomalies (MCOA) syndrome in PMEL17 (Silver) mutant ponies: five cases.Vet. Ophthalmol.14313–320. 10.1111/j.1463-5224.2011.00878.x
121
Körberg I. B. Sundström E. Meadows J. Pielberg G. R. Gustafson U. Hedhammar A. et al (2014). A simple repeat polymorphism in the MITF-M promoter is a key regulator of white spotting in dogs.PLoS One9:e104363. 10.1371/journal.pone.0104363
122
Koren S. Schatz M. C. Walenz B. P. Martin J. Howard J. T. Ganapathy G. et al (2012). Hybrid error correction and de novo assembly of single-molecule sequencing reads.Nat. Biotechnol.30:693. 10.1038/nbt.2280
123
Koseniuk A. Ropka-Molik K. Rubiś D. Smołucha G. (2018). Genetic background of coat colour in sheep.Archiv. Anim. Breed.61173–178.
124
Kristjansson T. Bjornsdottir S. Sigurdsson A. Andersson L. Lindgren G. Helyar S. et al (2014). The effect of the ‘Gait keeper’mutation in the DMRT3 gene on gaiting ability in Icelandic horses.J. Anim. Breed. Genet.131415–425. 10.1111/jbg.12112
125
Kukekova A. V. Trut L. N. Chase K. Kharlamova A. V. Johnson J. L. Temnykh S. V. et al (2011). Mapping loci for fox domestication: deconstruction/reconstruction of a behavioral phenotype.Behav. Genet.41593–606. 10.1007/s10519-010-9418-1
126
Kunieda T. Nakagiri M. Takami M. Ide H. Ogawa H. (1999). Cloning of bovine LYST gene and identification of a missense mutation associated with Chediak-higashi syndrome of cattle.Mamm. Genome101146–1149. 10.1007/s003359901181
127
Larson G. (2011). Genetics and domestication: important questions for new answers.Curr. Anthropol.52S485–S495.
128
Larson G. (2017). Reconsidering the distribution of gray wolves.Zool. Res.38:115. 10.24272/j.issn.2095-8137.2017.021
129
Larson G. Burger J. (2013). A population genetics view of animal domestication.Trends Genet.29197–205. 10.1016/j.tig.2013.01.003
130
Larson G. Fuller D. Q. (2014). The evolution of animal domestication.Ann. Rev. Ecol. Evol. Syst.45115–136.
131
Larson G. Piperno D. R. Allaby R. G. Purugganan M. D. Andersson L. Arroyo-Kalin M. et al (2014). Current perspectives and the future of domestication studies.Proc. Natl. Acad. Sci. U.S.A.1116139–6146. 10.1073/pnas.1323964111
132
Leathlobhair M. N. Perri A. R. Irving-Pease E. K. Witt K. E. Linderholm A. Haile J. et al (2018). The evolutionary history of dogs in the Americas.Science36181–85. 10.1126/science.aao4776
133
Lenstra J. A. Ajmone-Marsan P. Beja-Pereira A. Bollongino R. Bradley D. G. Colli L. et al (2014). Meta-analysis of mitochondrial DNA reveals several population bottlenecks during worldwide migrations of cattle.Diversity6178–187.
134
Leonard J. A. Wayne R. K. Wheeler J. Valadez R. Guillén S. Vila C. (2002). Ancient DNA evidence for old world origin of new world dogs.Science2981613–1616. 10.1126/science.1076980
135
Leroy G. Baumung R. Boettcher P. Besbes B. From T. Hoffmann I. (2018). Animal genetic resources diversity and ecosystem services.Glob. Food Securi.1784–91.
136
Li M. Tian S. Jin L. Zhou G. Li Y. Zhang Y. et al (2013). Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars.Nat. Genet.451431–1438. 10.1038/ng.2811
137
Librado P. Fages A. Gaunitz C. Leonardi M. Wagner S. Khan N. et al (2016). The evolutionary origin and genetic makeup of domestic horses.Genetics204423–434. 10.1534/genetics.116.194860
138
Lindahl T. (1997). Facts and artifacts of ancient DNA.Cell901–3. 10.1016/s0092-8674(00)80306-2
139
Lindblad-Toh K. Wade C. M. Mikkelsen T. S. Karlsson E. K. Jaffe D. B. Kamal M. et al (2005). Genome sequence, comparative analysis and haplotype structure of the domestic dog.Nature438803–819. 10.1038/nature04338
140
Liu Y.-H. Wang L. Xu T. Guo X. Li Y. Yin T.-T. et al (2018). Whole-genome sequencing of African dogs provides insights into adaptations against tropical parasites.Mol. Biol. Evol.35287–298. 10.1093/molbev/msx258
141
Liu Y.-P. Wu G.-S. Yao Y.-G. Miao Y.-W. Luikart G. Baig M. et al (2006). Multiple maternal origins of chickens: out of the Asian jungles.Mol. Phylogenet. Evol.3812–19. 10.1016/j.ympev.2005.09.014
142
Liu Z. Ji Z. Wang G. Chao T. Hou L. Wang J. (2016). Genome-wide analysis reveals signatures of selection for important traits in domestic sheep from different ecoregions.BMC Genomics17:863. 10.1186/s12864-016-3212-2
143
Loftus R. T. Machugh D. E. Bradley D. G. Sharp P. M. Cunningham P. (1994). Evidence for two independent domestications of cattle.Proc. Natl. Acad. Sci. U.S.A.912757–2761. 10.1073/pnas.91.7.2757
144
Ludwig A. Reissmann M. Benecke N. Bellone R. Sandoval-Castellanos E. Cieslak M. et al (2015). Twenty-five thousand years of fluctuating selection on leopard complex spotting and congenital night blindness in horses.Philos. Trans. R. Soc. B370:20130386. 10.1098/rstb.2013.0386
145
Lühken G. (2012). Genetic testing for phenotype-causing variants in sheep and goats.Mol. Cell. Probes26231–237. 10.1016/j.mcp.2012.04.005
146
Luikart G. Gielly L. Excoffier L. Vigne J.-D. Bouvet J. Taberlet P. (2001). Multiple maternal origins and weak phylogeographic structure in domestic goats.Proc. Natl. Acad. Sci. U.S.A.985927–5932. 10.1073/pnas.091591198
147
Lyons S. K. Amatangelo K. L. Behrensmeyer A. K. Bercovici A. Blois J. L. Davis M. et al (2016). Holocene shifts in the assembly of plant and animal communities implicate human impacts.Nature529:80. 10.1038/nature19104
148
MacDonald K. C. (1992). The domestic chicken (Gallus gallus) in sub-Saharan Africa: a background to its introduction and its osteological differentiation from indigenous fowls (Numidinae and Francolinus sp.).J. Archaeol. Sci.19303–318.
149
MacHugh D. E. Bradley D. G. (2001). Livestock genetic origins: goats buck the trend.Proc. Natl. Acad. Sci. U.S.A.985382–5384. 10.1073/pnas.111163198
150
MacHugh D. E. Larson G. Orlando L. (2017). Taming the past: ancient DNA and the study of animal domestication.Ann. Rev. Anim. Biosci.5329–351. 10.1146/annurev-animal-022516-022747
151
Malheiros J. M. Enríquez-Valencia C. E. Da Silva Duran B. O. De Paula T. G. Curi R. A. De Vasconcelos Silva J. et al (2018). Association of CAST2, HSP90AA1, DNAJA1 and HSPB1 genes with meat tenderness in Nellore cattle.Meat Sci.13849–52. 10.1016/j.meatsci.2018.01.003
152
Manceau M. Domingues V. S. Linnen C. R. Rosenblum E. B. Hoekstra H. E. (2010). Convergence in pigmentation at multiple levels: mutations, genes and function.Philos. Trans. R. Soc. B Biol. Sci.3652439–2450. 10.1098/rstb.2010.0104
153
Mao H. Ren J. Ding N. Xiao S. Huang L. (2010). Genetic variation within coat color genes of MC1R and ASIP in Chinese brownish red Tibetan pigs.Anim. Sci. J.81630–634. 10.1111/j.1740-0929.2010.00789.x
154
Marshall F. B. Dobney K. Denham T. Capriles J. M. (2014). Evaluating the roles of directed breeding and gene flow in animal domestication.Proc. Natl. Acad. Sci. U.S.A.1116153–6158. 10.1073/pnas.1312984110
155
Melletti M. Burton J. (2014). Ecology, Evolution And Behaviour Of Wild Cattle: Implications For Conservation.Cambridge: Cambridge University Press.
156
Messer P. W. Ellner S. P. Hairston N. G. Jr. (2016). Can population genetics adapt to rapid evolution?Trends Genet.32408–418. 10.1016/j.tig.2016.04.005
157
Mignon-Grasteau S. Boissy A. Bouix J. Faure J.-M. Fisher A. D. Hinch G. N. et al (2005). Genetics of adaptation and domestication in livestock.Livestock Product. Sci.933–14.
158
Montague M. J. Li G. Gandolfi B. Khan R. Aken B. L. Searle S. M. et al (2014). Comparative analysis of the domestic cat genome reveals genetic signatures underlying feline biology and domestication.Proc. Natl. Acad. Sci. U.S.A.11117230–17235. 10.1073/pnas.1410083111
159
Mosher D. S. Quignon P. Bustamante C. D. Sutter N. B. Mellersh C. S. Parker H. G. et al (2007). A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs.PLoS Genet.3:e79. 10.1371/journal.pgen.0030079
160
Mwacharo J. M. Bjørnstad G. Han J. Hanotte O. (2013). The history of African village chickens: an archaeological and molecular perspective.Afr. Archaeol. Rev.3097–114. 10.1007/s10437-013-9128-1
161
Nätt D. Rubin C.-J. Wright D. Johnsson M. Beltéky J. Andersson L. et al (2012). Heritable genome-wide variation of gene expression and promoter methylation between wild and domesticated chickens.BMC Genomics13:59. 10.1186/1471-2164-13-59
162
Neves F. Abrantes J. Steinke J. W. Esteves P. J. (2014). Maximum-likelihood approaches reveal signatures of positive selection in IL genes in mammals.Innate Immun.20184–191. 10.1177/1753425913486687
163
Nishibori M. Shimogiri T. Hayashi T. Yasue H. (2005). Molecular evidence for hybridization of species in the genus Gallus except for Gallus varius.Anim. Genet.36367–375. 10.1111/j.1365-2052.2005.01318.x
164
Norris B. J. Whan V. A. (2008). A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep.Genome Res.181282–1293. 10.1101/gr.072090.107
165
O’Brien S. J. Johnson W. E. (2007). The evolution of cats.Sci. Am.29768–75.
166
Ogorevc J. Kunej T. Razpet A. Dovc P. (2009). Database of cattle candidate genes and genetic markers for milk production and mastitis.Anim. Genet.40832–851. 10.1111/j.1365-2052.2009.01921.x
167
Ollivier M. Tresset A. Hitte C. Petit C. Hughes S. Gillet B. et al (2013). Evidence of coat color variation sheds new light on ancient canids.PLoS One8:e75110. 10.1371/journal.pone.0075110
168
Ostrander E. A. Galibert F. Patterson D. F. (2000). Canine genetics comes of age.Trends Genet.16117–124. 10.1016/s0168-9525(99)01958-7
169
Ottoni C. Girdland Flink L. Evin A. Geörg C. De Cupere B. Van Neer W. et al (2012). Pig domestication and human-mediated dispersal in western Eurasia revealed through ancient DNA and geometric morphometrics.Mol. Biol. Evol.30824–832. 10.1093/molbev/mss261
170
Ottoni C. Van Neer W. De Cupere B. Daligault J. Guimaraes S. Peters J. et al (2017). The palaeogenetics of cat dispersal in the ancient world.Nat. Ecol. Evol.11–7.
171
Outram A. K. Stear N. A. Bendrey R. Olsen S. Kasparov A. Zaibert V. et al (2009). The earliest horse harnessing and milking.Science3231332–1335. 10.1126/science.1168594
172
Pääbo S. (1985). Molecular cloning of ancient Egyptian mummy DNA.Nature314644–645. 10.1038/314644a0
173
Pääbo S. Gifford J. A. Wilson A. C. (1988). Mitochondrial DNA sequences from a 7000-year old brain.Nucleic Acids Res.169775–9787. 10.1093/nar/16.20.9775
174
Pääbo S. Higuchi R. G. Wilson A. C. (1989). Ancient DNA and the polymerase chain reaction: the emerging field of molecular archaeology (Minireview).J. Biol. Chem.2649709–9712.
175
Pan Z. Li S. Liu Q. Wang Z. Zhou Z. Di R. et al (2018). Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization.Gigascience7:giy019. 10.1093/gigascience/giy019
176
Park S. D. Magee D. A. Mcgettigan P. A. Teasdale M. D. Edwards C. J. Lohan A. J. et al (2015). Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle.Genome Biol.16:234. 10.1186/s13059-015-0790-2
177
Parker H. G. Vonholdt B. M. Quignon P. Margulies E. H. Shao S. Mosher D. S. et al (2009). An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs.Science325995–998. 10.1126/science.1173275
178
Pendleton A. L. Shen F. Taravella A. M. Emery S. Veeramah K. R. Boyko A. R. et al (2018). Comparison of village dog and wolf genomes highlights the role of the neural crest in dog domestication.BMC Biol.16:64. 10.1186/s12915-018-0535-2
179
Pereira G. L. De Matteis R. Regitano L. C. Chardulo L. A. L. Curi R. A. (2016). MSTN, CKM, and DMRT3 gene variants in different lines of quarter horses.J. Equine Vet. Sci.3933–37.
180
Pértille F. Brantsaeter M. Nordgreen J. Coutinho L. L. Janczak A. M. Jensen P. et al (2017). DNA methylation profiles in red blood cells of adult hens correlate with their rearing conditions.J. Exp. Biol.2203579–3587. 10.1242/jeb.157891
181
Petersen J. L. Mickelson J. R. Cothran E. G. Andersson L. S. Axelsson J. Bailey E. et al (2013a). Genetic diversity in the modern horse illustrated from genome-wide SNP data.PLoS One8:e54997. 10.1371/journal.pone.0054997
182
Petersen J. L. Mickelson J. R. Rendahl A. K. Valberg S. J. Andersson L. S. Axelsson J. et al (2013b). Genome-wide analysis reveals selection for important traits in domestic horse breeds.PLoS Genet.9:e1003211. 10.1371/journal.pgen.1003211
183
Philipp U. Hamann H. Mecklenburg L. Nishino S. Mignot E. Günzel-Apel A.-R. et al (2005). Polymorphisms within the canine MLPH gene are associated with dilute coat color in dogs.BMC Genet.6:34. 10.1186/1471-2156-6-34
184
Pionnier-Capitan M. Bemilli C. Bodu P. Célérier G. Ferrié J.-G. Fosse P. et al (2011). New evidence for upper palaeolithic small domestic dogs in south-western Europe.J. Archaeol. Sci.382123–2140.
185
Pitt D. Sevane N. Nicolazzi E. L. Machugh D. E. Park S. D. Colli L. et al (2019). Domestication of cattle: Two or three events?Evol. Appl.12123–136. 10.1111/eva.12674
186
Pollinger J. P. Lohmueller K. E. Han E. Parker H. G. Quignon P. Degenhardt J. D. et al (2010). Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication.Nature464898–902. 10.1038/nature08837
187
Price E. O. (1999). Behavioral development in animals undergoing domestication.Appl. Anim. Behav. Sci.65245–271.
188
Price E. O. (2002). Animal Domestication and Behavior.Wallingford: CABI.
189
Pringle H. (1998). Reading the signs of ancient animal domestication.Science2821827–1827.
190
Promerová M. Andersson L. Juras R. Penedo M. Reissmann M. Tozaki T. et al (2014). Worldwide frequency distribution of the ‘G ait keeper’mutation in the DMRT 3 gene.Anim. Genet.45274–282. 10.1111/age.12120
191
Pruvost M. Bellone R. Benecke N. Sandoval-Castellanos E. Cieslak M. Kuznetsova T. et al (2011). Genotypes of predomestic horses match phenotypes painted in Paleolithic works of cave art.Proc. Natl. Acad. Sci. U.S.A.10818626–18630. 10.1073/pnas.1108982108
192
Pryce J. E. Hayes B. J. Bolormaa S. Goddard M. E. (2011). Polymorphic regions affecting human height also control stature in cattle.Genetics187981–984. 10.1534/genetics.110.123943
193
Qanbari S. Simianer H. (2014). Mapping signatures of positive selection in the genome of livestock.Livestock Sci.166133–143.
194
Raynal N. J.-M. Si J. Taby R. F. Gharibyan V. Ahmed S. Jelinek J. et al (2012). DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory.Cancer Res.721170–1181. 10.1158/0008-5472.CAN-11-3248
195
Reissmann M. Ludwig A. (2013). Pleiotropic effects of coat colour-associated mutations in humans, mice and other mammals.Semin. Cell Dev. Biol.24576–586. 10.1016/j.semcdb.2013.03.014
196
Ren J. Duan Y. Qiao R. Yao F. Zhang Z. Yang B. et al (2011). A missense mutation in PPARD causes a major QTL effect on ear size in pigs.PLoS Genet.7:e1002043. 10.1371/journal.pgen.1002043
197
Rieder S. Taourit S. Mariat D. Langlois B. Guérin G. (2001). Mutations in the agouti (ASIP), the extension (MC1R), and the brown (TYRP1) loci and their association to coat color phenotypes in horses (Equus caballus).Mamm. Genome12450–455. 10.1007/s003350020017
198
Rodriguez C. Tomas A. Alves E. Ramirez O. Arque M. Munoz G. et al (2005). QTL mapping for teat number in an Iberian-by-Meishan pig intercross.Anim. Genet.36490–496. 10.1111/j.1365-2052.2005.01358.x
199
Rollin B. (2011). Putting the Horse Before Descartes: My Life’s Work On Behalf Of Animals.Philadelphia: Temple University Press.
200
Ross-Ibarra J. (2004). The evolution of recombination under domestication: a test of two hypotheses.Am. Nat.163105–112. 10.1086/380606
201
Roy M. Kim N. Kim K. Chung W.-H. Achawanantakun R. Sun Y. et al (2013). Analysis of the canine brain transcriptome with an emphasis on the hypothalamus and cerebral cortex.Mamm. Genome24484–499. 10.1007/s00335-013-9480-0
202
Royo L. Álvarez I. Fernandez I. Arranz J. Gomez E. Goyache F. (2005). The coding sequence of the ASIP gene is identical in nine wild-type coloured cattle breeds.J. Anim. Breed. Genet.122357–360. 10.1111/j.1439-0388.2005.00541.x
203
Rubin C.-J. Megens H.-J. Barrio A. M. Maqbool K. Sayyab S. Schwochow D. et al (2012). Strong signatures of selection in the domestic pig genome.Proc. Natl. Acad. Sci. U.S.A.10919529–19536. 10.1073/pnas.1217149109
204
Runkel F. Büssow H. Seburn K. L. Cox G. A. Ward D. M. Kaplan J. et al (2006). Grey, a novel mutation in the murine Lyst gene, causes the beige phenotype by skipping of exon 25.Mamm. Genome17203–210. 10.1007/s00335-005-0015-1
205
Savolainen P. Zhang Y.-P. Luo J. Lundeberg J. Leitner T. (2002). Genetic evidence for an East Asian origin of domestic dogs.Science2981610–1613. 10.1126/science.1073906
206
Schafberg R. Swalve H. (2015). The history of breeding for polled cattle.Livestock Sci.17954–70.
207
Scherf B. D. Pilling D. (2015). The Second Report On The State Of The World’s Animal Genetic Resources For Food And Agriculture.Rome: FAO.
208
Schmutz S. M. Berryere T. G. (2007a). Genes affecting coat colour and pattern in domestic dogs: a review.Anim. Genet.38539–549. 10.1111/j.1365-2052.2007.01664.x
209
Schmutz S. M. Berryere T. G. (2007b). The genetics of cream coat color in dogs.J. Hered.98544–548. 10.1093/jhered/esm018
210
Schmutz S. M. Berryere T. G. Goldfinch A. D. (2002). TYRP1 and MC1R genotypes and their effects on coat color in dogs.Mamm. Genome13380–387. 10.1007/s00335-001-2147-2
211
Schmutz S. M. Dreger D. L. (2013). Interaction of MC1R and PMEL alleles on solid coat colors in H ighland cattle.Anim. Genet.449–13. 10.1111/j.1365-2052.2012.02361.x
212
Shannon L. M. Boyko R. H. Castelhano M. Corey E. Hayward J. J. Mclean C. et al (2015). Genetic structure in village dogs reveals a Central Asian domestication origin.Proc. Natl. Acad. Sci. U.S.A.11213639–13644. 10.1073/pnas.1516215112
213
Staiger E. A. Almén M. S. Promerová M. Brooks S. Cothran E. G. Imsland F. et al (2017). The evolutionary history of the DMRT3 ‘Gait keeper’haplotype.Anim. Genet.48551–559. 10.1111/age.12580
214
Steingrímsson E. Copeland N. G. Jenkins N. A. (2006). Mouse coat color mutations: from fancy mice to functional genomics.Dev. Dyn.2352401–2411. 10.1002/dvdy.20840
215
Stinckens A. Luyten T. Bijttebier J. Van Den Maagdenberg K. Dieltiens D. Janssens S. et al (2008). Characterization of the complete porcine MSTN gene and expression levels in pig breeds differing in muscularity.Anim. Genet.39586–596. 10.1111/j.1365-2052.2008.01774.x
216
Sutter N. B. Bustamante C. D. Chase K. Gray M. M. Zhao K. Zhu L. et al (2007). A single IGF1 allele is a major determinant of small size in dogs.Science316112–115. 10.1126/science.1137045
217
Tchernov E. Horwitz L. K. (1991). Body size diminution under domestication: unconscious selection in primeval domesticates.J. Anthropol. Archaeol.1054–75.
218
Thalmann O. Shapiro B. Cui P. Schuenemann V. J. Sawyer S. K. Greenfield D. et al (2013). Complete mitochondrial genomes of ancient canids suggest a European origin of domestic dogs.Science342871–874. 10.1126/science.1243650
219
Thomas R. H. Schaffner W. Wilson A. C. Pääbo S. (1989). DNA phylogeny of the extinct marsupial wolf.Nature340465–467. 10.1038/340465a0
220
Tian F. Zhan F. Vanderkraats N. D. Hiken J. F. Edwards J. R. Zhang H. et al (2013). DNMT gene expression and methylome in Marek’s disease resistant and susceptible chickens prior to and following infection by MDV.Epigenetics8431–444. 10.4161/epi.24361
221
Tixier-Boichard M. Bed’hom B. Rognon X. (2011). Chicken domestication: from archeology to genomics.C. R. Biol.334197–204. 10.1016/j.crvi.2010.12.012
222
Tonomura N. Elvers I. Thomas R. Megquier K. Turner-Maier J. Howald C. et al (2015). Genome-wide association study identifies shared risk loci common to two malignancies in golden retrievers.PLoS Genet.11:e1004922. 10.1371/journal.pgen.1005339
223
Troy C. S. Machugh D. E. Bailey J. F. Magee D. A. Loftus R. T. Cunningham P. et al (2001). Genetic evidence for Near-Eastern origins of European cattle.Nature4101088–1091. 10.1038/35074088
224
Trut L. Oskina I. Kharlamova A. (2009). Animal evolution during domestication: the domesticated fox as a model.Bioessays31349–360. 10.1002/bies.200800070
225
Tryon R. C. White S. D. Bannasch D. L. (2007). Homozygosity mapping approach identifies a missense mutation in equine cyclophilin B (PPIB) associated with HERDA in the American Quarter Horse.Genomics9093–102. 10.1016/j.ygeno.2007.03.009
226
Vaez M. Follett S. A. Bed’hom B. Gourichon D. Tixier-Boichard M. Burke T. (2008). A single point-mutation within the melanophilin gene causes the lavender plumage colour dilution phenotype in the chicken.BMC Genet.9:7. 10.1186/1471-2156-9-7
227
Venter J. C. Smith H. O. Adams M. D. (2015). The sequence of the human genome.Clin. Chem.611207–1208.
228
Verginelli F. Capelli C. Coia V. Musiani M. Falchetti M. Ottini L. et al (2005). Mitochondrial DNA from prehistoric canids highlights relationships between dogs and South-East European wolves.Mol. Biol. Evol.222541–2551. 10.1093/molbev/msi248
229
Verhulst E. C. Mateman A. C. Zwier M. V. Caro S. P. Verhoeven K. J. Van Oers K. (2016). Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD 4 DNA methylation.Mol. Ecol.251801–1811. 10.1111/mec.13519
230
Vigne J.-D. (2011). The origins of animal domestication and husbandry: a major change in the history of humanity and the biosphere.C. R. Biol.334171–181. 10.1016/j.crvi.2010.12.009
231
Vilà C. Leonard J. A. Götherström A. Marklund S. Sandberg K. Lidén K. et al (2001). Widespread origins of domestic horse lineages.Science291474–477. 10.1126/science.291.5503.474
232
Vitti J. J. Grossman S. R. Sabeti P. C. (2013). Detecting natural selection in genomic data.Annu. Rev. Genet.4797–120. 10.1093/molbev/msv334
233
Voight B. F. Kudaravalli S. Wen X. Pritchard J. K. (2006). A map of recent positive selection in the human genome.PLoS Biol.4:e72. 10.1371/journal.pbio.0040072
234
Wallis J. W. Aerts J. Groenen M. A. Crooijmans R. P. Layman D. Graves T. A. et al (2004). A physical map of the chicken genome.Nature432761–764.
235
Wang C. Wang H. Zhang Y. Tang Z. Li K. Liu B. (2015). Genome-wide analysis reveals artificial selection on coat colour and reproductive traits in Chinese domestic pigs.Mol. Ecol. Resour.15414–424. 10.1111/1755-0998.12311
236
Wang G.-D. Fan R.-X. Zhai W. Liu F. Wang L. Zhong L. et al (2014a). Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the tibetan plateau.Genome Biol. Evol.62122–2128. 10.1093/gbe/evu162
237
Wang G.-D. Xie H.-B. Peng M.-S. Irwin D. Zhang Y.-P. (2014b). Domestication genomics: evidence from animals.Annu. Rev. Anim. Biosci.265–84. 10.1146/annurev-animal-022513-114129
238
Wang X. Zhou G. Li Q. Zhao D. Chen Y. (2014). Discovery of SNPs in RXFP2 related to horn types in sheep.Small Rum. Res.116133–136.
239
Wang G.-D. Zhai W. Yang H.-C. Wang L. Zhong L. Liu Y.-H. et al (2016). Out of southern East Asia: the natural history of domestic dogs across the world.Cell Res.2621–33. 10.1038/cr.2015.147
240
Wayne R. K. (1986). Limb morphology of domestic and wild canids: the influence of development on morphologic change.J. Morphol.187301–319. 10.1002/jmor.1051870304
241
Weaver I. C. Cervoni N. Champagne F. A. D’alessio A. C. Sharma S. Seckl J. R. et al (2004). Epigenetic programming by maternal behavior.Nat. Neurosci.7847–854.
242
Webb A. A. Cullen C. L. (2010). Coat color and coat color pattern-related neurologic and neuro-ophthalmic diseases.Can. Vet. J.51:653.
243
Wei C. Lu J. Xu L. Liu G. Wang Z. Zhao F. et al (2014). Genetic structure of Chinese indigenous goats and the special geographical structure in the Southwest China as a geographic barrier driving the fragmentation of a large population.PLoS One9:e94435. 10.1371/journal.pone.0094435
244
Wijesena H. R. Schmutz S. M. (2015). A missense mutation in SLC45A2 is associated with albinism in several small long haired dog breeds.J. Hered.106285–288. 10.1093/jhered/esv008
245
Wilkins A. S. Wrangham R. W. Fitch W. T. (2014). The “domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics.Genetics197795–808. 10.1534/genetics.114.165423
246
Wilkinson S. Lu Z. H. Megens H.-J. Archibald A. L. Haley C. Jackson I. J. et al (2013). Signatures of diversifying selection in European pig breeds.PLoS Genet.9:e1003453. 10.1371/journal.pgen.1003453
247
Wolf E. Kind A. Aigner B. Schnieke A. (2018). Genetically engineered large animals in biomedicine.Anim. Biotechnol.2169–214.
248
Wong A. Ruhe A. Robertson K. Loew E. Williams D. Neff M. (2013). A de novo mutation in KIT causes white spotting in a subpopulation of G erman S hepherd dogs.Anim. Genet.44305–310. 10.1111/age.12006
249
Wright D. (2015). Article commentary: the genetic architecture of domestication in animals.Bioinform. Biol. Insights9:S28902.
250
Xiang H. Li X. Dai F. Xu X. Tan A. Chen L. et al (2013). Comparative methylomics between domesticated and wild silkworms implies possible epigenetic influences on silkworm domestication.BMC Genomics14:646. 10.1186/1471-2164-14-646
251
Yang S. Li X. Li K. Fan B. Tang Z. (2014). A genome-wide scan for signatures of selection in Chinese indigenous and commercial pig breeds.BMC Genet.15:7. 10.1186/1471-2156-15-7
252
Yindee M. Vlamings B. Wajjwalku W. Techakumphu M. Lohachit C. Sirivaidyapong S. et al (2010). Y-chromosomal variation confirms independent domestications of swamp and river buffalo.Anim. Genet.41433–435. 10.1111/j.1365-2052.2010.02020.x
253
Yurchenko A. A. Daetwyler H. D. Yudin N. Schnabel R. D. Vander Jagt C. J. Soloshenko V. et al (2018). Scans for signatures of selection in Russian cattle breed genomes reveal new candidate genes for environmental adaptation and acclimation.Sci. Rep.8:12984. 10.1038/s41598-018-31304-w
254
Zamani W. Ghasempouri S. M. Rezaei H. R. Naderi S. Hesari A. R. E. Ouhrouch A. (2018). Comparing polymorphism of 86 candidate genes putatively involved in domestication of sheep, between wild and domestic Iranian sheep.Meta Gene17223–231.
255
Zeder M. A. (2008). Domestication and early agriculture in the mediterranean basin: origins, diffusion, and impact.Proc. Natl. Acad. Sci. U.S.A.10511597–11604. 10.1073/pnas.0801317105
256
Zeder M. A. (2012). Pathways to Animal Domestication.Cambridge: Cambridge University Press.
257
Zeder M. A. (2015). Core questions in domestication research.Proc. Natl. Acad. Sci. U.S.A.1123191–3198.
258
Zeder M. A. Emshwiller E. Smith B. D. Bradley D. G. (2006). Documenting domestication: the intersection of genetics and archaeology.Trends Genet.22139–155. 10.1016/j.tig.2006.01.007
259
Zhang C. Liu Y. Xu D. Wen Q. Li X. Zhang W. et al (2012). Polymorphisms of myostatin gene (MSTN) in four goat breeds and their effects on Boer goat growth performance.Mol. Biol. Rep.393081–3087. 10.1007/s11033-011-1071-0
260
Zhang C. Ni P. Ahmad H. I. Gemingguli M. Baizilaitibei A. Gulibaheti D. et al (2018). Detecting the population structure and scanning for signatures of selection in horses (Equus caballus) from whole-genome sequencing data.Evol. Bioinform.14:1176934318775106. 10.1177/1176934318775106
261
Zhou Z. Li M. Cheng H. Fan W. Yuan Z. Gao Q. et al (2018). An intercross population study reveals genes associated with body size and plumage color in ducks.Nat. Commun.9:2648. 10.1038/s41467-018-06521-6
Summary
Keywords
animal domestication, evolution, genes, adaptation, diversification
Citation
Ahmad HI, Ahmad MJ, Jabbir F, Ahmar S, Ahmad N, Elokil AA and Chen J (2020) The Domestication Makeup: Evolution, Survival, and Challenges. Front. Ecol. Evol. 8:103. doi: 10.3389/fevo.2020.00103
Received
15 October 2019
Accepted
30 March 2020
Published
08 May 2020
Volume
8 - 2020
Edited by
Fulvio Cruciani, Sapienza University of Rome, Italy
Reviewed by
Muniyandi Nagarajan, Central University of Kerala, India; Paul Gepts, University of California, Davis, United States
Updates
Copyright
© 2020 Ahmad, Ahmad, Jabbir, Ahmar, Ahmad, Elokil and Chen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinping Chen, chenjp@giabr.gd.cn
This article was submitted to Evolutionary and Population Genetics, a section of the journal Frontiers in Ecology and Evolution
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.