 Chauncy Hinshaw
Chauncy Hinshaw Kathleen C. Evans
Kathleen C. Evans Cristina Rosa1
Cristina Rosa1 Margarita M. López-Uribe
Margarita M. López-Uribe- 1Department of Plant Pathology and Environmental Microbiology, The Pennsylvania State University, University Park, PA, United States
- 2Department of Entomology, University of Maryland, College Park, College Park, MD, United States
- 3Department of Entomology, Center for Pollinator Research, The Pennsylvania State University, University Park, PA, United States
Studies of the ecoimmunology of feral organisms can provide valuable insight into how host–pathogen dynamics change as organisms transition from human-managed conditions back into the wild. Honey bees (Apis mellifera Linnaeus) offer an ideal system to investigate these questions as colonies of these social insects often escape management and establish in the wild. While managed honey bee colonies have low probability of survival in the absence of disease treatments, feral colonies commonly survive in the wild, where pathogen pressures are expected to be higher due to the absence of disease treatments. Here, we investigate the role of pathogen infections [Deformed wing virus (DWV), Black queen cell virus (BQCV), and Nosema ceranae] and immune gene expression (defensin-1, hymenoptaecin, pgrp-lc, pgrp-s2, argonaute-2, vago) in the survival of feral and managed honey bee colonies. We surveyed a total of 25 pairs of feral and managed colonies over a 2-year period (2017–2018), recorded overwintering survival, and measured pathogen levels and immune gene expression using quantitative polymerase chain reaction (qPCR). Our results showed that feral colonies had higher levels of DWV but it was variable over time compared to managed colonies. Higher pathogen levels were associated with increased immune gene expression, with feral colonies showing higher expression in five out of the six examined immune genes for at least one sampling period. Further analysis revealed that differential expression of the genes hymenoptaecin and vago increased the odds of overwintering survival in managed and feral colonies. Our results revealed that feral colonies express immune genes at higher levels in response to high pathogen burdens, providing evidence for the role of feralization in altering pathogen landscapes and host immune responses.
Introduction
Feralization is the process by which previously domesticated organisms establish populations in the wild in the absence of anthropogenic influence (Gering et al., 2019a). The outcomes of feralization are typically studied in an evolutionary context, examining how environmental and genetic factors affect the fitness of feral organisms compared to their domesticated sources. It has been suggested that because of genetic bottlenecks and artificial selection during the domestication process, fitness decreases outside of captivity, making feral organisms exhibit reduced fitness upon reintroduction to the wild (Araki et al., 2009; Baskett and Waples, 2013; Meyer and Purugganan, 2013). However, feral organisms often thrive (e.g., cats, dogs, pigs) and do not always revert back to the ‘wild-type’ (Taylor et al., 1998; Bellard et al., 2017). Indeed, feral organisms often outnumber their wild counterparts, and can lead to shifts in community composition at the ecosystem level by increasing predation pressure on available prey and potentially spilling over pathogens to wild species (Leiser et al., 2013; Bevins et al., 2014; Maeda et al., 2019; Lepczyk et al., 2020). These ecological effects of feralization are gaining more attention due to their implications for conservation biology and ecosystem management.
Feral organisms frequently interact with both domesticated and wild species, and play a critical ecological role in the dynamics of pathogens shared among these closely related groups. The increased fecundity and expanded geographic ranges that result from the domestication processes, often result in large uncontrolled populations of feral organisms harboring infections and serving as a bridge between domesticated and wild hosts (Bevins et al., 2014). This is the case of the feral swine (Sus scrofa domestica), a species that has high rates of reproduction, high pathogen loads, and overlaps in range with domestic pigs (S. scrofa domestica) and wild boars (S. scrofa) (Taylor et al., 1998; Hill et al., 2014). Transmission of several pathogens, including viruses and bacteria, have been documented from feral swine to domestic pigs and wild boars, posing concerns for the role of feral organisms as reservoirs of pathogen transmission to both wild and domesticated populations (Cvetnic et al., 2003; Le Potier et al., 2006; Meng et al., 2009).
The ecological conditions of feralization may facilitate tolerance or resistance to disease. Pathogens of domesticated species are usually managed by humans to avoid the rapid spread of diseases among domesticated animals. On the contrary, pathogen transmission in feral populations is uncontrolled. Under these conditions, host–pathogen interactions in feral populations may therefore facilitate the rapid evolution of natural mechanisms of disease tolerance or resistance (LeConte et al., 2007; Locke, 2016). Thus, the maintenance of traits associated with disease resilience may be relaxed in domesticated species, while greater ability to mitigate the negative effects of pathogens is critical for the survival of feral organisms (Moreira et al., 2018).
Feral colonies of the honey bee (Apis mellifera L.) provide an ideal model to investigate the hypothesis that host–pathogen dynamics during feralization favor higher expression of defenses and disease tolerance in feral organisms. Apis mellifera is a eusocial bee species that has undergone extensive domestication efforts for traits such as increased honey production, decreased aggressive behavior and reduced frequency of swarming (Lecocq, 2018). Managed honey bee colonies frequently colonize wild environments and become feral because colonies reproduce through swarming (Winston, 1991). Both domesticated and feral honey bees face serious challenges due to a large number of pests and pathogens (Calderone, 2012; Mcmahon et al., 2016). One of the major drivers of disease and colony losses among honey bees is the ectoparasitic mite Varroa destructor, which acts as a vector for multiple bee-infecting RNA viruses that significantly weaken colonies and decrease their overwintering survival (Gisder et al., 2009; Martin et al., 2012). Varroa mites and associated viruses are considered major antagonists of honey bee health and, because of their strong effects on honey bee survival, managed colonies are often treated with chemical acaricides multiple times per year to decrease mite numbers. If untreated, most managed honey bee colonies die within the first year (Kraus and Page, 1995; LeConte et al., 2010). Nevertheless, feral honey bee colonies have been documented as surviving long-term in the wild in the absence of beekeeper management, where Varroa mites and viruses are not artificially controlled, and can therefore pose high selective pressure on colonies (Locke, 2016).
Previous studies have indicated that feral honey bee colonies may exhibit higher immune responses than managed colonies (Youngsteadt et al., 2015, but see Lowe et al., 2011). However, it is unclear how the expression of different immune phenotypes in managed and feral conditions is associated with colony survival and resistance or tolerance to parasites. Honey bees rely on both individual and social mechanisms of immunity to protect the colony from pests and pathogens, and management likely has an influence on both types of defenses (Neumann and Blacquière, 2017; Taric et al., 2020). While behavioral responses, such as hygienic behavior, play key roles in protection against pathogens (Simone-Finstrom, 2017), humoral immune responses in individual bees are also critical for pathogen defense and control of infections (Di Prisco et al., 2016; McMenamin et al., 2018). Several immune pathways are involved in the immune response against viruses, bacteria and fungi. For example, antimicrobial peptides (AMPs) produced from several immune pathways (e.g., Toll and Imd) have key general roles in insect immune systems (Yi et al., 2014; Brutscher et al., 2015). The RNA interference (RNAi) pathway is an antiviral defense pathway in insects that targets sequence-specific double-stranded RNA produced during RNA virus replication (Gammon and Mello, 2015).
Here, we investigate the role of pathogen infections and immune gene expression in the survival of feral and managed honey bees to answer the following questions: (1) are feral colonies reservoirs of pathogens with increased levels of pathogens compared to managed colonies?; (2) do increased pathogen levels lead to higher expression of immune genes in feral colonies than in managed colonies?; (3) is immune gene expression correlated with survival of honey bee colonies? Over a 2-year period, we sampled feral and managed colonies in the same landscapes with the participation of beekeepers who reported the location of colonies. We collected individuals from a total of 44 colonies during the spring and fall of the years 2017 and 2018 to compare the ecoimmunology and survival of colonies in apiaries and in the wild. The virus most directly linked to Varroa mite infestations (DWV) was found at significantly higher levels in feral than managed colonies, and the expression of most immune genes was also higher in feral colonies. We also identified two immune genes that are associated with colony survival and that can potentially be used as biomarkers of health in honey bee colonies.
Materials and Methods
Sample Collection
We established a collaborative community science project to locate feral honey bee colonies across the state of Pennsylvania, United States. Participants reported the locations of feral colonies by submitting GPS locations of colonies into a web portal or via email. To be included in the study, all feral colonies needed to survive at least one winter in wild, unmanaged conditions. We checked each reported colony in early spring to corroborate activity and record overwintering survival (Figure 1). We paired each feral colony with one managed colony located within a seven-mile radius to control for site variation between colonies located in geographic areas with different landscapes and climates. A total of eight pairs of feral and managed colonies (n = 16 colonies) in 2017 and 17 pairs (n = 34) in 2018 were included in the laboratory analyses (Figure 2). In the case where a managed colony was not able to be sampled a second time due to death or other reasons, the feral colony was paired with a different managed colony in the same location (between 2017 and 2018, n = 3; between spring and fall 2018, n = 1). This resulted in 20 unique feral colonies and 24 unique managed colonies being sampled over the course of this study (Supplementary Data Sheet 1). Additionally, we were unable to obtain data on the overwintering status of one managed colony in 2018, therefore it was omitted from the analyses of overwintering survival. Due to the death of either a managed or feral colony in a pair, only two pairs of colonies were sampled in both 2017 and 2018.

Figure 1. Cavities used by feral honey bee colonies included in the study. (A) Colony inhabiting a tree cavity with abundant propolis at the entrance of the nest in Saxonburg (PA, United States). (B) Feral colony nesting inside the wall of a house in New Bethlehem (PA, United States) (C) Entrance of a feral colony nesting in an abandoned shed in Harrison Valley (PA, United States).
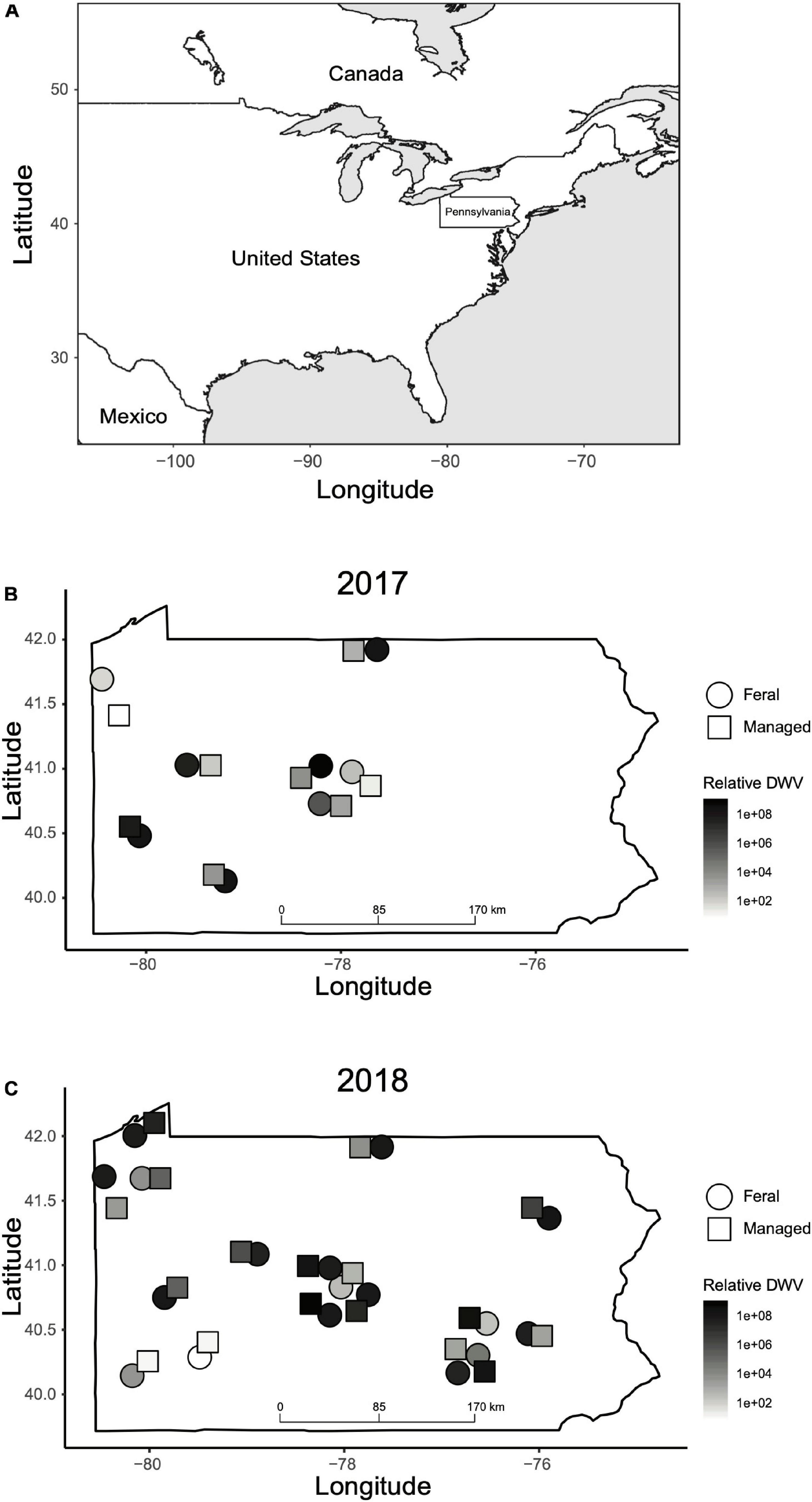
Figure 2. Geographic locations of feral and managed colonies sampled in this study. (A) Map of North America indicating the location of Pennsylvania in the northeast of the United States. (B) Location of feral (circles) and managed (squares) colonies sampled in 2017. (C) Location of feral (circles) and managed (squares) colonies sampled in 2018. The color of each shape corresponds to relative DWV levels (2–ΔΔCT values) recorded in the fall for that colony. Colony locations are approximate and have been adjusted slightly due to overlapping points. GPS coordinates of each colony can be found in Supplementary Data Sheet 1.
We sampled approximately 75 forager bees from the entrance of each colony in the spring (March–June) and fall (August–October) (dates in Supplementary Data Sheet 1). Individuals were sampled with aerial insect nets and transferred to 50 ml conical tubes that were placed on dry ice to preserve RNA quality before long-term storage at −80°C. All sampling sites were on private property and permission was obtained from the land owners. The specific locations and contact information of participants was kept confidential. No protected or endangered species were involved in these studies.
Selection of Pathogens and Immune Genes
To characterize disease dynamics in honey bee colonies, we quantified three pathogens that commonly infect honey bees and negatively impact colony health. Deformed wing virus (DWV) is an RNA virus that is considered the most detrimental honey bee pathogen for its ubiquity, global distribution, and role in overwintering losses of honey bees (Martin et al., 2012; Brutscher et al., 2016). It can be transmitted horizontally and vertically within a colony, but is also efficiently transmitted by Varroa mites which can increase the titer of this virus leading to overt, clinically symptomatic infections (Gisder et al., 2009; Möckel et al., 2011). We also quantified Black queen cell virus (BQCV), another RNA virus that is highly prevalent in adult honey bees globally, but predominantly affects immature bee stages (prepupae and pupae) (Mondet et al., 2014). BQCV is transmitted vertically and horizontally between adults and from adult bees to developing bees, but has not been shown to be transmitted by Varroa mites (Chen et al., 2006). BQCV may also have synergistic interactions with other pathogens due to its correlation with viruses and the fungal parasite Nosema ceranae Fries (D’Alvise et al., 2019). Nosema ceranae is a common microsporidian gut parasite of honey bees, contributing to decreased lifespans in infected bees (Higes et al., 2008; Goblirsch, 2018). We quantified this pathogen in all colonies sampled in 2018.
To characterize immune gene expression in feral and managed colonies, we quantified transcript expression of six genes (argonaute-2, vago, pgrp-s2, pgrp-lc, defensin-1 and hymenoptaecin) from several immune pathways. The genes argonaute-2 (ago2) and vago, from the RNAi pathway, have been shown to be upregulated after viral infection (Brutscher et al., 2015). The gene pgrp-s2 encodes for an upstream recognition receptor involved in activation of the Toll immune pathway, and pgrp-lc encodes a transmembrane protein activator of the Imd (Immune Deficiency) pathway. Both of these genes are upregulated in pathogen-infected honey bees (Evans et al., 2006; Brutscher et al., 2017). Additionally, we quantified genes corresponding to two antimicrobial peptides (AMPs), defensin-1 (Def1) and hymenoptaecin (Hym), produced by the Toll and Imd pathways. These AMPs have key roles in honey bee immune responses to viruses, bacterial and fungal pathogens (Yi et al., 2014; Brutscher et al., 2015).
RNA Extraction and Quantitative PCR
For total RNA extraction, we dissected abdomens from thirty bees per colony by removing the stingers, hindguts, and midguts. Ten abdomens per sample (three samples per colony) were pooled into 2.0 ml tubes with 2.0 mm BashingBead Lysis Tubes (Zymo Research, Irvine, CA, United States) and homogenized using a BeadBlasterTM24 (Benchmark Scientific, Edison, NJ, United States) at 6.0 m/s for three 30 s intervals. We extracted RNA from homogenate using RNeasy spin columns (QIAGEN, Hilden, Germany), according to the manufacturer’s protocol and eluted into nuclease-free water. We assessed the quantity and quality of RNA using a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, United States).
We quantified the three pathogens and the expression of six immune genes through quantitative reverse-transcriptase PCR (qRT-PCR) using previously developed primer sequences (Table 1). The three RNA extracts from pooled individuals per colony were individually used as templates to produce cDNA using random primers and MultiScribe RT, according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA, United States). A total of 2 μg RNA was used for each cDNA synthesis. A total of 40 ng of cDNA was used for each qPCR reaction. We carried out reactions in 384-well plates using a QuantStudio 5 Real-Time PCR System (Applied Biosystems). Each well contained 5 μl Luna Universal qPCR Master Mix (New England Biolabs, Ipswich, MA, United States), 0.25 μl of each of the forward and reverse primers (10 μM), 2.5 μl nuclease-free H2O, and 2 μl cDNA template. The following reaction conditions were used: 60 s at 95°C for initial denaturation, then 40 cycles of 15 s at 95°C for denaturation, and 30 s at 60°C for annealing, extension, and data collection followed by a melting curve analysis of 15 s at 95°C, 60 s at 60°C, and 1 s at 95°C to determine the specificity of amplification products. In each plate, we ran all reactions in triplicate and included negative controls of nuclease-free water for each set of primers. After surveying three reference genes (ef1-alpha, eIFS8, and GAPDH1), we determined that elongation factor 1-alpha (ef1-alpha) was a suitable reference gene due to its similar level of expression in all samples, and we used it as the reference gene for these experiments (Table 1).
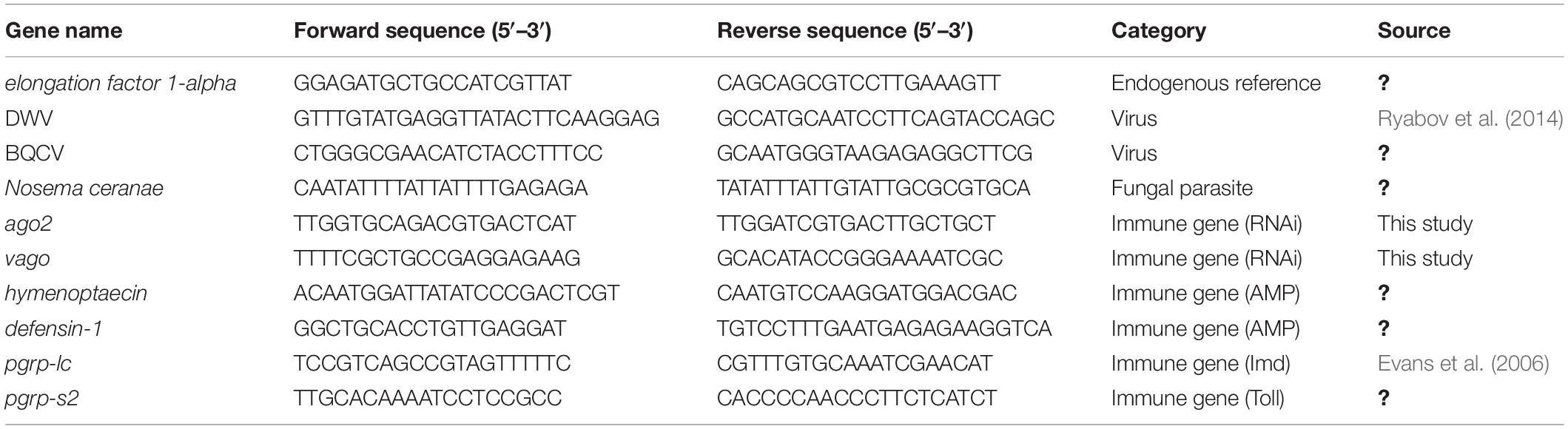
Table 1. Sequences of forward and reverse primers used for the quantification of pathogens and immune genes through quantitative PCR.
We determined the Ct value for each sample by taking the mean of the three technical replicates. We used the Ct value for the reference gene and subtracted this from the Ct value for the target to generate ΔCt values for each sample. These ΔCt values were then normalized to the managed colony with the lowest relative abundance (highest ΔCt) of the target for that sampling period (spring or fall of the year sampled), generating ΔΔCt values. We then calculated the relative amounts of transcripts using the 2–ΔΔCT method (Livak and Schmittgen, 2001). As each colony had three samples consisting of ten bees each, we took the average 2–ΔΔCT value of the three biological replicates and used this for subsequent analyses.
For absolute quantification of DWV copy numbers in samples collected in the fall of each year, we used synthetic DNA corresponding to the sequence of DWV amplified by our primers, in the form of gBlocks gene fragments (Integrated DNA Technologies, Coralville, IA, United States), producing ten-fold dilutions (101–107 copies of synthetic DNA) run in each qPCR plate. We calculated the copy number using the formula: copy number = DNA concentration (ng/mL) × 6.02 × 1023 (copies/mol)/length (140 bp) × 6.6 × 1011 (Wu et al., 2017). The log DNA copy numbers were then plotted with Ct values, producing linear standard curves for each qPCR plate, allowing for the estimation of DWV copy number in each sample.
Statistical Analysis
Due to non-normality, 2–ΔΔCT values were log-transformed and analyzed through generalized linear models (GLM) with a Gaussian distribution using the glm function in R package ‘stats’ (R Core Team, 2019). The relative expression of each target was analyzed separately using management (feral or managed), sampling time (spring or fall of 2017 or 2018), and their interactions as fixed effects. Two-way ANOVA was used to test for overall effects of management and sampling time on target expression. The estimated marginal means were then calculated with the emmeans function in R package ‘emmeans’ (Lenth, 2020). These values were then used for post hoc tests with a Tukey adjustment for multiple comparisons to determine differences in relative target abundance between feral and managed colonies at each timepoint. To evaluate associations between pathogen levels and immune gene expression among all samples, we calculated the Spearman’s rank correlation coefficients using the cor.test function in R package ‘stats.’
To assess the role of immune gene expression, pathogen levels, and management in overwintering survival, we used a log linear generalized linear mixed model (GLMM) with a binomial distribution using the glmer function in R package ‘lme4’ (Bates et al., 2015). The original full model included data from the fall of both years with overwintering survival as the response variable, and the relative levels of DWV and BQCV, relative expression of all six immune genes, and management as fixed effects, and year of sampling as a random effect. We used a backward model selection to identify the fixed effects that contributed significantly to the model. We calculated the variance inflation factors (VIF) for the model using the vif function in R package ‘car’ (Fox and Weisberg, 2019). All VIF values were less than 3, and thus all fixed effects were kept in the final model. Ultimately, the model with the lowest AIC value was chosen. All analyses were conducted in R version 3.6.2.
Results
Pathogen Levels
Our results indicate that mean DWV levels were significantly higher in feral colonies compared to managed colonies in fall of 2017 (P < 0.05, z = −2.470), but not at other timepoints (P > 0.05) (Table 2 and Figure 3). Levels of BQCV and N. ceranae did not differ between groups (P > 0.05). All 44 colonies tested positive for the presence of DWV and BQCV at all timepoints, while N. ceranae presence was more variable over time (Supplementary Figure 1). Out of the 34 colonies tested, N. ceranae was detected in eight feral and 11 managed colonies in the spring (47.1% and 65%, respectively), and 12 feral and 10 managed colonies in the fall (76% and 59%, respectively).
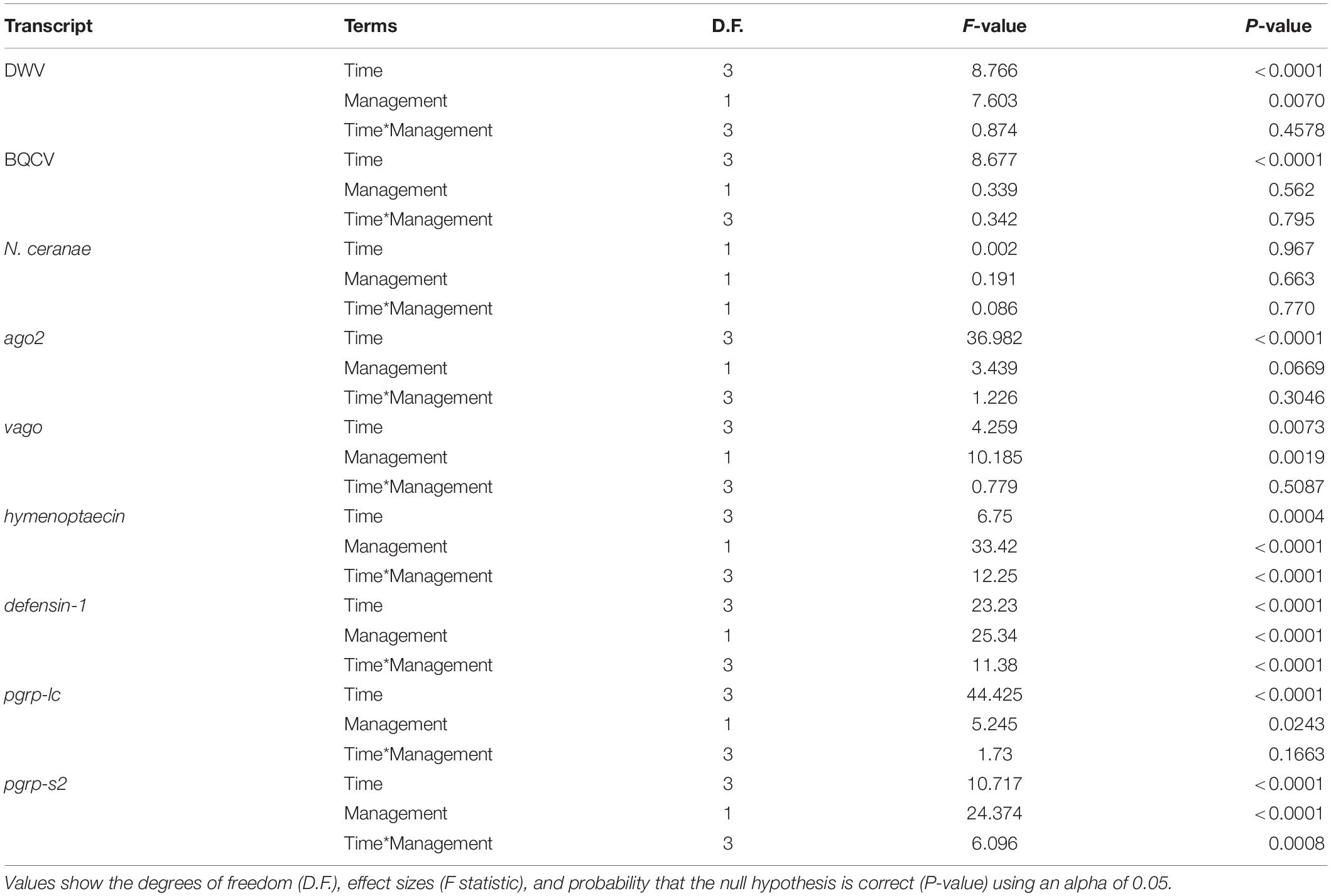
Table 2. Results of two-way ANOVA on generalized linear models (GLM) assessing the effects of time, management, and their interaction on pathogen levels and immune gene expression.
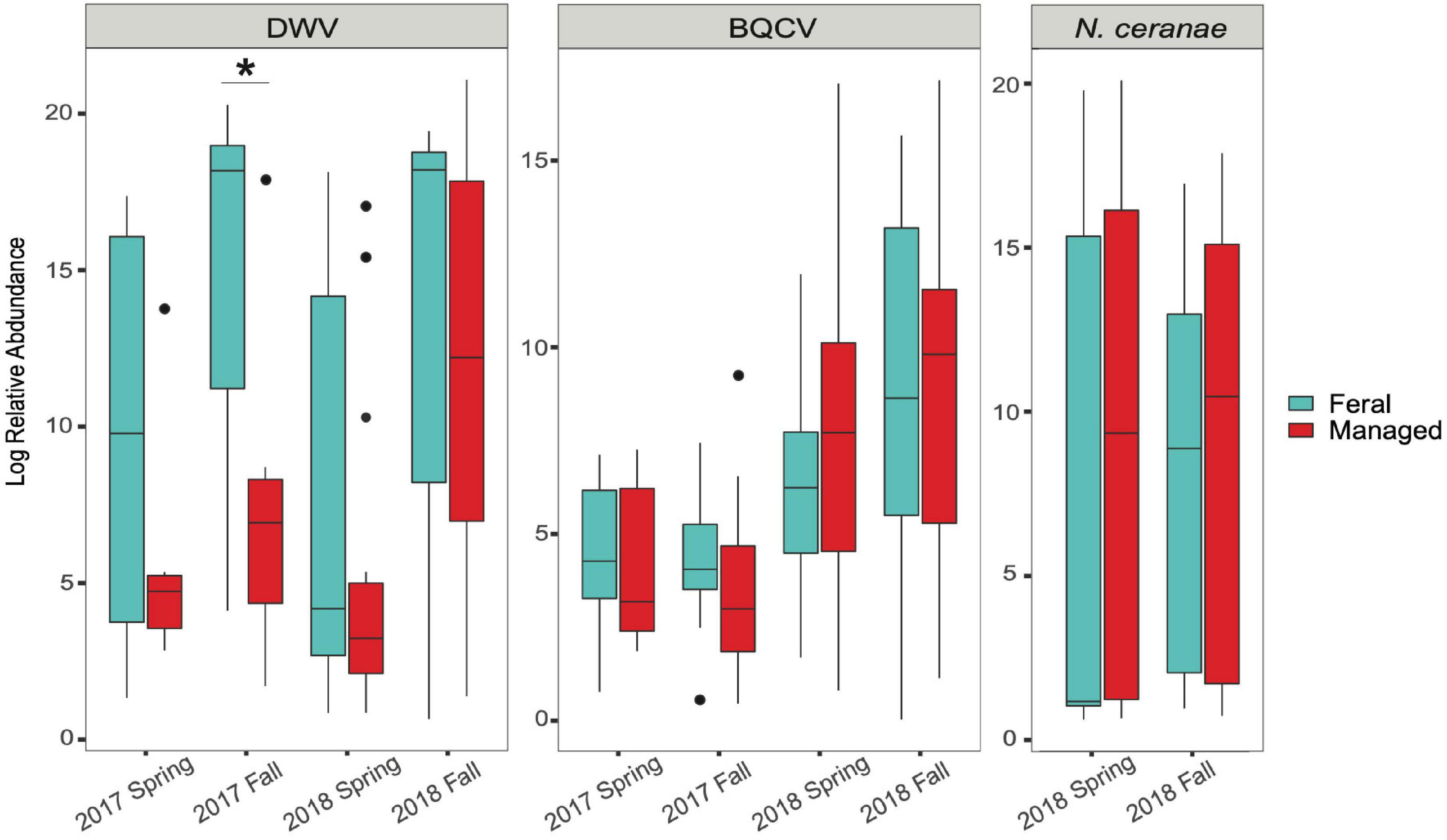
Figure 3. Boxplots showing relative abundance of pathogens in feral (blue) and managed (red) honey bee colonies for each sampling period. Relative abundance = 2–ΔΔCT values for each colony (calculations described in main text). In 2017, n = 8 pairs of colonies. In 2018, n = 17 pairs of colonies. Horizontal line of each box represents the median and circles represent outliers. Asterisk denotes statistical significance (*P < 0.05).
Immune Gene Expression
In the spring of 2017, feral colonies exhibited higher average expression of five out of the six immune genes tested (defensin-1, hymenoptaecin, pgrp-lc, pgrp-s2, and ago2), although pathogen levels were not significantly different between managed and feral colonies (Table 2 and Figure 4). In the fall of 2017, expression of defensin-1, hymenoptaecin, pgrp-s2 remained higher in feral colonies, while the gene vago had higher average expression in managed colonies. In 2018, immune gene expression was similar between feral and managed colonies, yet feral colonies had higher average expression of hymenoptaecin and pgrp-s2 in the spring, regardless of the similar levels of pathogens in feral and managed colonies. No significant differences in average gene expression between feral and managed colonies were observed in the fall of 2018. Interestingly, pathogen pressures were also similar at this time point.
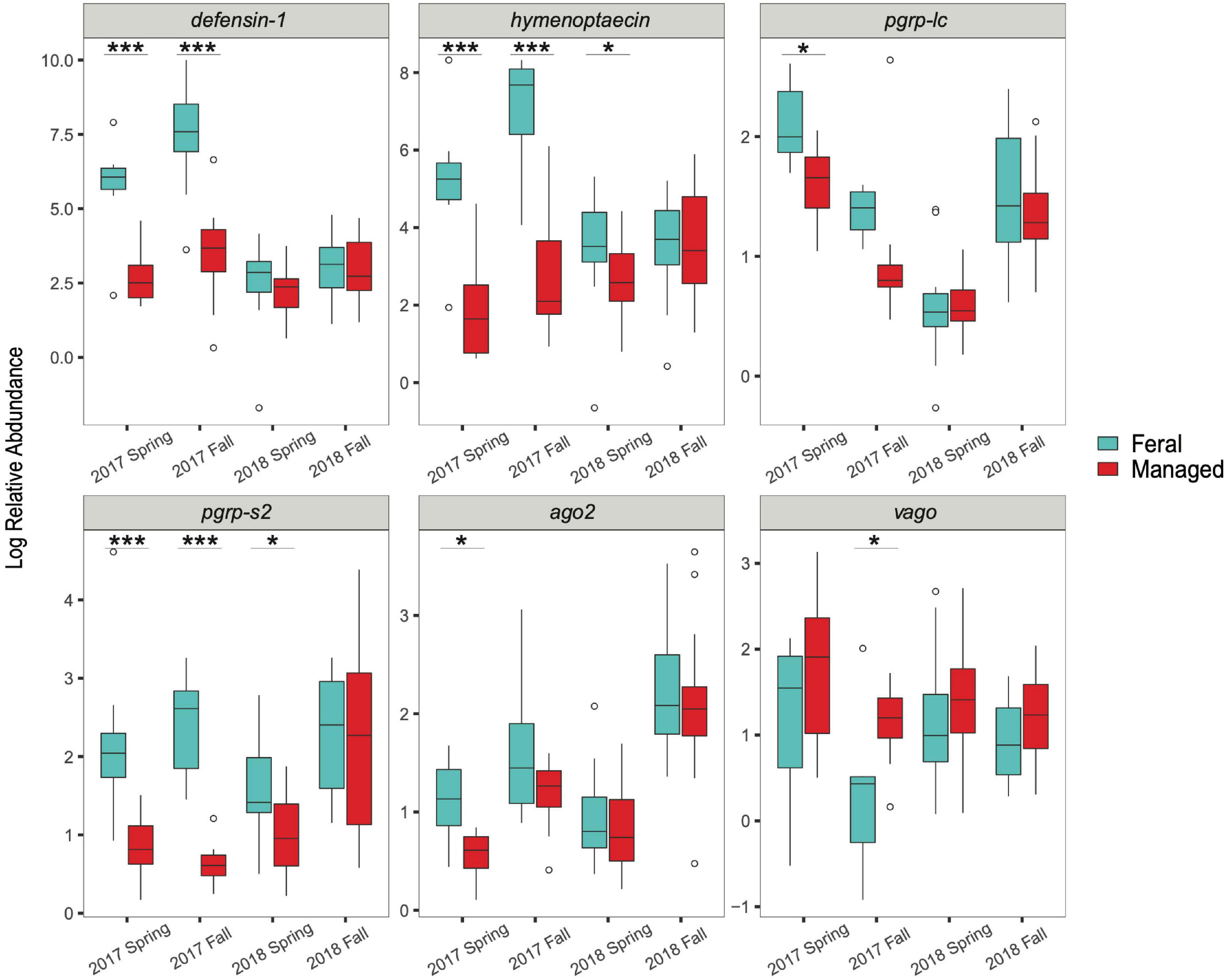
Figure 4. Boxplots showing immune gene expression of feral (blue) and managed (red) honey bee colonies for 2017 (n = 8 pairs of colonies) and 2018 (n = 17 pairs of colonies). Relative abundance = 2–ΔΔCT values for each colony (calculations described in main text). Horizontal line of each box represents the median and circles represent outliers. Asterisks denote statistical significance (*P < 0.05, ***P < 0.001).
Spearman’s correlations (ρ) revealed that DWV levels were positively correlated with the expression of hymenoptaecin in the spring (ρ = 0.32), but not significantly correlated with any other pathogen levels or immune genes, while BQCV levels were positively correlated with N. ceranae levels in the spring (ρ = 0.48). Nosema ceranae levels were also positively correlated with the expression of defensin-1 and pgrp-s2 in the fall (ρ = 0.39; ρ = 0.35, respectively). However, all correlation coefficients between pathogen levels and immune gene expression were low (ρ < 0.4) (Mukaka, 2012), suggesting factors other than pathogen levels likely contribute to immune gene expression (Supplementary Table 1).
Overwintering Survival
Total survival of colonies over the 2017–2018 winter was 63% for both feral and managed colonies. For the 2018–2019 winter, survival was 47% and 38% for feral and managed colonies, respectively. Of the five feral colonies that survived the 2017–2018 winter, two also survived the 2018–2019 winter. Two managed colonies were sampled in both years, and one of these also survived the 2018–2019 winter. Despite the similar overall survival between managed and feral colonies, more feral than managed colonies survived with high copy numbers of DWV (>107) (Supplementary Figure 2).
The best predictive model for overwintering survival included management, DWV levels, and expression of hymenoptaecin, vago, and pgrp-s2 as fixed effects (AIC = 65.5). The level of DWV in a colony was negatively correlated with overwintering survival (DWV: P < 0.05, standard estimation error of coefficient = 0.06). Management and pgrp-s2 expression were also negatively correlated with overwintering survival, although neither were significant predictors (Management: P = 0.105, s.e. = 0.877; pgrp-s2: P = 0.062, s.e. = 0.51) (Figure 5). The expression of hymenoptaecin and vago were both significantly positively correlated with survival (hymenoptaecin: P < 0.05, s.e. = 0.265; vago: P < 0.05, s.e. = 0.757) (Figure 5).
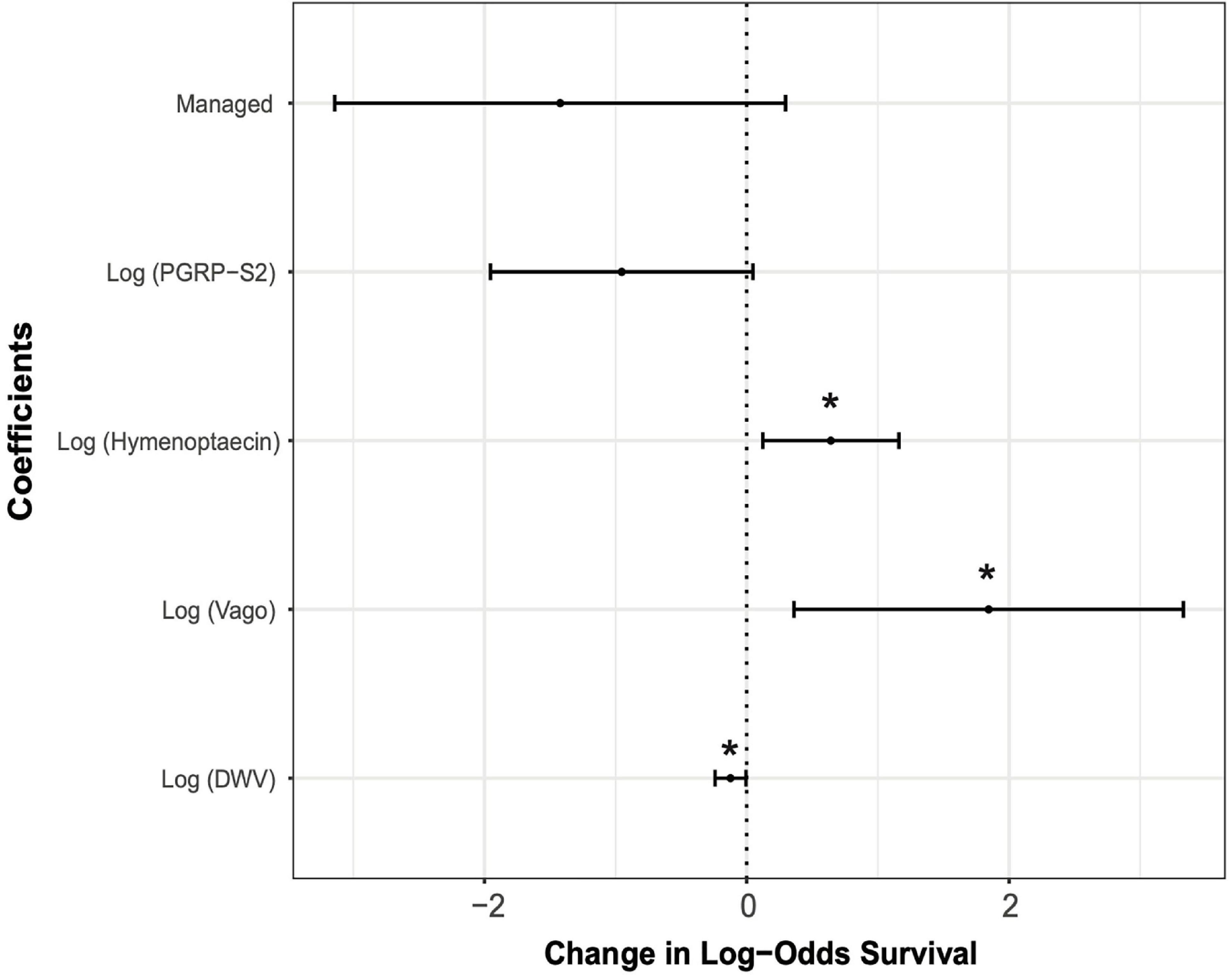
Figure 5. Effect sizes of the generalized linear mixed model (GLMM) showing estimates of each variable along with 95% confidence intervals. Asterisks denote variables that were statistically significant predictors of overwintering survival (*P < 0.05). Estimates of fixed effects are: Management = –1.422; Log (pgrp-s2) = –0.953; Log (hymenoptaecin) = 0.641; Log (vago) = 1.845; Log (DWV) = –0.124.
Discussion
In this study, we investigated the ecoimmunology of feral and managed bees over a 2-year period. Our results show that overall feral honey bee colonies have higher levels of DWV despite yearly and seasonal variation. Alternatively, BQCV and N. ceranae, which were constant across seasons, did not differ with management and were not linked to decreased survival in this study. While we did not investigate the transmission efficiencies of pathogens from feral to managed honey bees, our results provide support for the ability of feral colonies to serve as reservoirs of DWV. We also found evidence of higher immune gene expression in feral colonies, even at timepoints when DWV levels were similar between managed and feral colonies. Further analysis of all colonies revealed that levels of DWV infection were positively correlated with the expression of hymenoptaecin in the spring and N. ceranae levels were correlated with defensin-1 and pgrp-s2 in the fall. The strength of correlations was low suggesting additional factors such as genetic background and environmental conditions play an important role in immune phenotypes. Last, we found significant associations between the expression of two immune genes (hymenoptaecin and vago) and survival in both feral and managed colonies. These genes have been previously identified as differentially expressed in virus-infected honey bees, but this is the first report of expression being correlated with reduced host mortality (Kuster et al., 2014; Ryabov et al., 2014). Feral and managed colonies also had similar probabilities of survival, despite higher DWV titers in feral than managed colonies.
The survival of feral colonies in the presence of high viral titers suggests that feralization may facilitate the expression of traits that confer virus tolerance. Virus tolerance to DWV has previously been observed among mite-resistant honey bees in several locations across many years suggesting that it is heritable (Locke et al., 2014; Russo et al., 2020). Additionally, even without miticidal treatment, several feral colonies had low levels of DWV, suggesting these colonies may exhibit additional traits such as hygienic behavior to reduce the number of Varroa mites, and thus viruses present (Supplementary Figure 2) (Wagoner et al., 2019). Natural selection is a primary driver of evolution in host–pathogen dynamics and allows organisms to potentially respond to many stressors (Papkou et al., 2019). Management practices of domesticated organisms change the fitness landscape and can alter their evolutionary trajectories (Wilkins et al., 2014; Nygren et al., 2015; Milla et al., 2018). Managed honey bees are typically under different environmental conditions compared to their feral counterparts, and while beekeeping practices often aim to limit pathogen abundance, these practices may relax the selective pressures that pathogens exert on managed colonies, potentially delaying host-parasite coevolution (Neumann and Blacquière, 2017). While low input beekeeping management reduces pathogen burdens and oxidative stress compared to commercially managed colonies, highly invasive beekeeping practices may cause added stresses (Taric et al., 2019). Even though the degree of disease management varies greatly among beekeepers (Underwood et al., 2019), Varroa mite management may inhibit adaptations for increased resilience to mites and viruses in managed honey bee colonies (Blacquière et al., 2019). In addition to virus tolerance, other traits such as small colony sizes, frequent swarming, and increased hygienic and grooming behavior appear to be critical for the survival of feral colonies in the presence of high pathogen pressure (Gramacho and Spivak, 2003; Seeley, 2007; Locke, 2016; Loftus et al., 2016; Russo et al., 2020). Future studies should focus on providing information about the molecular mechanisms of disease tolerance in these feral colonies to directly test for the role of feralization in altering selective pressures and leading to different immune phenotypes in honey bees.
Although mechanisms of virus tolerance are still unknown for insects, one possible explanation is the ability of highly infected bees to limit the overreaction of immune responses. Immune effectors, such as pro-phenoloxidase, produced by insects can have cytotoxic effects that work to limit infections, but also cause damage to host tissues (Sadd and Siva-Jothy, 2006; Hillyer, 2016). This self-damage can lead to increased aging and higher mortality as a result of increased inflammation (Alaux et al., 2010; Khan et al., 2017). Virus-tolerant colonies may therefore have mechanisms to limit inflammation-induced damage. A second possible mechanism includes transgenerational immune priming, the development of immune memory via vertical transmission of immunological experiences, and its effects have been demonstrated in several invertebrates (Tetreau et al., 2019). Vertical transmission of DWV and the fact that queens may preside over a colony for several years would favor immune-primed offspring in feral colonies, in contrast to beekeeping operations where queens are replaced frequently. Another potential mechanism includes changes in the virulence of DWV and Varroa mites in feral colonies. Genotypes of DWV are known to differ in virulence and Varroa mite transmission can favor certain strains of the virus (Ryabov et al., 2019). While our study did not assess Varroa mite levels or the genetic diversity of DWV, previous work showed that Varroa mites from managed colonies had increased population growth compared to mites from feral colonies (Dynes et al., 2020), providing evidence for the role of management in selecting for mites with greater reproductive rates. While the specific mechanisms are unclear, the absence of human management and the process of feralization may lead to changes in virus virulence and host tolerance in honey bees.
In addition to different selective pressures experienced by feral and managed colonies, feral colonies can also have different genetic backgrounds, contributing to differences in immune phenotypes and disease outcomes. Previously, López-Uribe et al. (2017) showed that feral and managed honey bees exhibit some genetic differentiation even at short geographic scales. Although we provide evidence for differential immune gene expression between feral and managed colonies, the role of ancestry and genetic diversity in this difference remains unclear. Immune gene expression is heritable in honey bees and virus tolerance may have a heritable basis as well, making it plausible that different genetic backgrounds play an important role in the different immune phenotypes observed between feral and managed colonies (Decanini et al., 2007; Thaduri et al., 2019). The underlying variability and heritability of immunological traits combined with different selection regimes may lead to different evolutionary trajectories for feral and managed honey bees.
We found higher expression of several immune genes in feral colonies compared to managed colonies, suggesting that feralization has led to increased pathogen defenses, although this was not true for all genes and differed with time of sampling. Specifically, we found that the expression of hymenoptaecin was higher in feral colonies, while the expression of vago was higher in managed colonies. However, both genes were associated with increased colony survival. Hymenoptaecin is a general AMP involved in responses to many pathogens including DWV and Varroa mites (Evans et al., 2006; Kuster et al., 2014; Brutscher et al., 2015; Wu et al., 2020). Other studies have shown that this gene is consistently upregulated in response to pathogens and during wounding events in honey bees and may be a potential biomarker to quantify honey bee health (Galbraith et al., 2015; Brutscher et al., 2017; Doublet et al., 2017; Zanni et al., 2017). We also identified the expression of vago as important for the overall survival of colonies. This transcript is expressed upon activation of the RNAi pathway, which recognizes the dsRNA of viruses and leads to the increased expression of vago or its orthologs in mosquitoes, fruit flies, bumble bees, and honey bees (Deddouche et al., 2008; Paradkar et al., 2012; Ryabov et al., 2014; Niu et al., 2016). In DWV-infected honey bees, vago expression has been shown to be significantly increased, providing evidence for its role in antiviral responses (Ryabov et al., 2014). To our knowledge this is the first report of vago expression being directly linked to increased survival in honey bees. These two genes could be considered biomarkers of honey bee health that can be used to predict the ability of a colony to survive the winter (López-Uribe et al., 2020). Additionally, genome-level studies looking for signatures of selection at regulatory regions of immunity in honey bees may also provide important information on mechanisms of pathogen resilience.
Feral organisms offer valuable systems to study potential negative consequences of domestication and anthropogenic influence by examining host–pathogen interactions of organisms that recently escaped managed conditions (Burdon and Thrall, 2008; Gering et al., 2019b). Previous studies of feral honey bees have examined levels of mite infestation, pathogen pressures, or the combination of pathogen pressures and immune gene expression, but the association of host–pathogen dynamics with colony survival was not previously investigated (Seeley, 2007; Thompson et al., 2014; Youngsteadt et al., 2015). Here, we quantified pathogen levels, immune gene expression, and linked this to overwintering survival in managed and feral honey bee colonies. This allowed for the identification of specific genes associated with overwintering survival of honey bees and evidence of virus tolerance in feral colonies, linking immunity, infection, and survival under natural conditions. Further identification of the genetic mechanisms of virus tolerance and biomarkers of bee health can help breeding efforts to focus on increasing these traits in selected honey bee stocks (e.g., Robertson et al., 2020), thus decreasing overall colony losses for the beekeeping industry. Future studies should assess the role of feralization on pathogen dynamics and ecoimmunology in other domesticated species.
Data Availability Statement
The raw data and R script to reproduce the statistical analyses of this study can be found online at the ScholarSphere repository: https://doi.org/10.26207/w2wv-ax07.
Author Contributions
CH and MML-U conceived the study. CH and KCE led data collection. KCE was responsible for correspondence with community members and locating honey bee colonies. CH conducted the data analysis with insight from CR and MML-U. CH and MML-U drafted the manuscript. All authors critically revised the manuscript and gave final approval for publication.
Funding
CH was supported by the Pennsylvania State University Graduate Training Program in Integrative Pollinator Ecology, funded by the Penn State College of Agricultural Sciences’ Strategic Networking Initiative Program. CR was funded through the USDA NIFA Hatch Appropriations under Project PEN04652 and Accession No. 1016243. MML-U was funded through the USDA NIFA Appropriations under Projects PEN04716 Accession No. 1020527 and PEN04620 Accession No. 1011873.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank John Albright, Ross and Peggy Bell, Ken Bowman, Charles Brudowsky, Carolyn Burgh, Lou Caputzal, Fran and Bob Cooley, Laurie Emerson, Stephen Finke, Bill Fisher, Mardi Frye, Daryln Hoffstot, Robert Hoppe, Rita and Roper Houston, Justin Jacobeen, Bonnie Kaushik, Jackie Kimmel, Thomas Kretchik, Bob Landis, Daryl Martin, Hank and Patty Jo McClenahan, Charles McGee, Josh Nearhoof, Stephen Repasky, Bruce Rodriguez, Amy Sharp, Avi Sommerville, Joshua Tyrone, Lynn Urban, Charlie Vorisek, John Wenzel, and the beekeepers of Pennsylvania (United States) for their participation in this study and the Pennsylvania State Beekeepers Association for their support throughout the project. We are also thankful to Ryan Ford for help with laboratory analyses, Kristen Brochu for help with statistical analyses, Catalina Mejia for help with figure design, Romina Russo for discussions about experimental design, and Brock Harpur, the López-Uribe lab and the reviewers for comments on previous versions of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.594263/full#supplementary-material
Supplementary Figure 1 | Prevalence of pathogens in feral and managed colonies. Deformed wing virus (DWV) and Black queen cell virus (BQCV) were detected in all colonies at all times of sampling. Presence of Nosema ceranae was only tested in colonies sampled in 2018. Nosema ceranae was found in 47.1% and 76.47% of feral colonies in spring and fall, respectively, while 64.7% and 58.8% of managed colonies tested positive for N. ceranae in spring and fall, respectively.
Supplementary Figure 2 | DWV copy number in feral and managed honey bee colonies that died (No) and survived (Yes). The effects of management, overwintering survival, and their interaction were analyzed using GLM, followed by ANOVA to assess their influence on DWV copy number (management: df = 1, F = 2.895, P = 0.096; overwintering survival: df = 1, F-value = 4.281, P = 0.045).
Supplementary Table 1 | Spearman’s correlations of pathogens and immune genes for combined data from feral and managed colonies sampled in 2017 and 2018, separated by season of sampling. Asterisks denote statistical significance (∗P < 0.1, ∗∗P < 0.05, ∗∗∗P < 0.001).
Supplementary Data Sheet 1 | Summary of data from honey bee colonies including: date of sampling, calculated 2–ΔΔCT gene and pathogen expression values, overwintering survival, abbreviated GPS coordinates, and distance from paired colony.
References
Alaux, C., Ducloz, F., Crauser, D., and Le Conte, Y. (2010). Diet effects on honeybee immunocompetence. Biol. Lett. 6, 562–565. doi: 10.1098/rsbl.2009.0986
Araki, H., Cooper, B., and Blouin, M. S. (2009). Carry-over effect of captive breeding reduces reproductive fitness of wild-born descendants in the wild. Biol. Lett. 5, 621–624. doi: 10.1098/rsbl.2009.0315
Baskett, M. L., and Waples, R. S. (2013). Evaluating alternative strategies for minimizing unintended fitness consequences of cultured individuals on wild populations. Conserv. Biol. 27, 83–94. doi: 10.1111/j.1523-1739.2012.01949.x
Bates, D., Mächler, M., Bolker, B. M., and Walker, S. C. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. doi: 10.18637/jss.v067.i01
Bellard, C., Rysman, J. F., Leroy, B., Claud, C., and Mace, G. M. (2017). A global picture of biological invasion threat on islands. Nat. Ecol. Evol. 1, 1862–1869. doi: 10.1038/s41559-017-0365-6
Bevins, S. N., Pedersen, K., Lutman, M. W., Gidlewski, T., and Deliberto, T. J. (2014). Consequences associated with the recent range expansion of nonnative feral swine. Bioscience 64, 291–299. doi: 10.1093/biosci/biu015
Blacquière, T., Boot, W., Calis, J., Moro, A., Neumann, P., and Panziera, D. (2019). Darwinian black box selection for resistance to settled invasive Varroa destructor parasites in honey bees. Biol. Invasions 21, 2519–2528. doi: 10.1007/s10530-019-020010
Brutscher, L. M., Daughenbaugh, K. F., and Flenniken, M. L. (2015). Antiviral defense mechanisms in honey bees. Curr. Opin. Insect Sci. 10, 71–82. doi: 10.1016/j.cois.2015.04.016
Brutscher, L. M., Daughenbaugh, K. F., and Flenniken, M. L. (2017). Virus and dsRNA-triggered transcriptional responses reveal key components of honey bee antiviral defense. Sci. Rep. 7:6448. doi: 10.1038/s41598-017-06623-z
Brutscher, L. M., McMenamin, A. J., and Flenniken, M. L. (2016). The buzz about honey bee viruses. PLoS Pathog. 12:e1005757. doi: 10.1371/journal.ppat.1005757
Burdon, J. J., and Thrall, P. H. (2008). Pathogen evolution across the agro-ecological interface: implications for disease management. Evol. Appl. 1, 57–65. doi: 10.1111/j.1752-4571.2007.00005.x
Calderone, N. W. (2012). Insect pollinated crops, insect pollinators and US agriculture: trend analysis of aggregate data for the period 1992–2009. PLoS One 7:e37235. doi: 10.1371/journal.pone.0037235
Chen, Y. P., Pettis, J. S., Collins, A., and Feldlaufer, M. F. (2006). Prevalence and transmission of honeybee viruses. Appl. Environ. Microbiol. 72, 606–611. doi: 10.1128/AEM.72.1.606-611.2006
Cvetnic, Z., Mitak, M., Ocepek, M., Lojkic, M., Terzic, S., Jemersic, L., et al. (2003). Wild boars (Sus scrofa) as reservoirs of Brucella suis biovar 2 in Croatia. Acta Vet. Hung. 51, 465–473. doi: 10.1556/AVet.51.2003.4.4
D’Alvise, P., Seeburger, V., Gihring, K., Kieboom, M., and Hasselmann, M. (2019). Seasonal dynamics and co-occurrence patterns of honey bee pathogens revealed by high-throughput RT-qPCR analysis. Ecol. Evol. 9, 10241–10252. doi: 10.1002/ece3.5544
Decanini, L. I., Collins, A. M., and Evans, J. D. (2007). Variation and heritability in immune gene expression by diseased honeybees. J. Hered. 98, 195–201. doi: 10.1093/JHERED
Deddouche, S., Matt, N., Budd, A., Mueller, S., Kemp, C., Galiana-Arnoux, D., et al. (2008). The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in drosophila. Nat. Immunol. 9, 1425–1432. doi: 10.1038/ni.1664
Di Prisco, G., Annoscia, D., Margiotta, M., Ferrara, R., Varricchio, P., Zanni, V., et al. (2016). A mutualistic symbiosis between a parasitic mite and a pathogenic virus undermines honey bee immunity and health. Proc. Natl. Acad. Sci. U.S.A. 113, 3203–3208. doi: 10.1073/pnas.1523515113
Doublet, V., Poeschl, Y., Gogol-Döring, A., Alaux, C., Annoscia, D., Aurori, C., et al. (2017). Unity in defence: honeybee workers exhibit conserved molecular responses to diverse pathogens. BMC Genomics 18:207. doi: 10.1186/s12864-017-3597-6
Dynes, T. L., Berry, J. A., Delaplane, K. S., de Roode, J. C., and Brosi, B. J. (2020). Assessing virulence of Varroa destructor mites from different honey bee management regimes. Apidologie 51, 276–289. doi: 10.1007/s13592-019-00716-6
Evans, J. D., Aronstein, K., Chen, Y. P., Hetru, C., Imler, J.-L., Jiang, H., et al. (2006). Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol. Biol. 15, 645–656. doi: 10.1111/j.1365-2583.2006.00682.x
Fox, J., and Weisberg, S. (2019). An {R} Companion to Applied Regression, 3rd Edn. Thousand Oaks CA: Sage.
Galbraith, D. A., Yang, X., Niño, E. L., Yi, S., and Grozinger, C. (2015). Parallel epigenomic and transcriptomic responses to viral infection in honey bees (Apis mellifera). PLoS Pathog. 11:e1004713. doi: 10.1371/journal.ppat.1004713
Gammon, D., and Mello, C. (2015). RNA interference-mediated antiviral defense in insects. Curr. Opin. Insect Sci. 8, 111–120. doi: 10.1016/j.cois.2015.01.006.RNA
Gering, E., Incorvaia, D., Henriksen, R., Conner, J., Getty, T., and Wright, D. (2019a). Getting back to nature: feralization in animals and plants. Trends Ecol. Evol. 34, 1137–1151. doi: 10.1016/j.tree.2019.07.018
Gering, E., Incorvaia, D., Henriksen, R., Wright, D., and Getty, T. (2019b). Maladaptation in feral and domesticated animals. Evol. Appl. 12, 1274–1286. doi: 10.1111/eva.12784
Gisder, S., Aumeier, P., and Genersch, E. (2009). Deformed wing virus: replication and viral load in mites (Varroa destructor). J. Gen. Virol. 90, 463–467. doi: 10.1099/vir.0.005579-0
Goblirsch, M. (2018). Nosema ceranae disease of the honey bee (Apis mellifera). Apidologie 49, 131–150. doi: 10.1007/s13592-017-0535-1
Gramacho, K. P., and Spivak, M. (2003). Differences in olfactory sensitivity and behavioral responses among honey bees bred for hygienic behavior. Behav. Ecol. Sociobiol. 54, 472–479. doi: 10.1007/s00265-003-0643-y
Higes, M., Martín-Hernández, R., Botías, C., Bailón, E. G., González-Porto, A. V., Barrios, L., et al. (2008). How natural infection by Nosema ceranae causes honeybee colony collapse. Environ. Microbiol. 10, 2659–2669. doi: 10.1111/j.1462-2920.2008.01687.x
Hill, D. E., Dubey, J. P., Baroch, J. A., Swafford, S. R., Fournet, V. F., Hawkins-Cooper, D., et al. (2014). Surveillance of feral swine for Trichinella spp. and Toxoplasma gondii in the USA and host-related factors associated with infection. Vet. Parasitol. 205, 653–665. doi: 10.1016/j.vetpar.2014.07.026
Hillyer, J. F. (2016). Insect immunology and hematopoiesis. Dev. Comp. Immunol. 58, 102–118. doi: 10.1016/j.dci.2015.12.006
Khan, I., Agashe, D., and Rolff, J. (2017). Early-life inflammation, immune response and ageing. Proc. R. Soc. B Biol. Sci. 284:20170125. doi: 10.1098/rspb.2017.0125
Kraus, B., and Page, R. E. (1995). Effect of Varroa jacobsoni (Mesostigmata: Varroidae) on feral Apis mellifera (Hymenoptera: Apidae) in California. Environ. Entomol. 24, 1473–1480. doi: 10.1093/EE/24.6.1473
Kuster, R. D., Boncristiani, H. F., and Rueppell, O. (2014). Immunogene and viral transcript dynamics during parasitic Varroa destructor mite infection of developing honey bee (Apis mellifera) pupae. J. Exp. Biol. 217, 1710–1718. doi: 10.1242/jeb.097766
Le Potier, M. F., Mesplede, A., and Vannier, P. (2006). “Classical swine fever and other pestiviruses,” in Diseases of Swine, 9th Edn, eds B. E. Straw, J. J. Zimmerman, S. D’Allaire, and D. J. Taylor (Oxford: Blackwell Publishing Ltd), 309–322.
Lecocq, T. (2018). “Insects: the disregarded domestication histories,” in Animal Domestication, ed. F. Teletchea (London: IntechOpen). doi: 10.5772/intechopen.81834
LeConte, Y., de Vaublanc, G., Crauser, D., Jeanne, F., Rousselle, J.-C., and Bécard, J.-M. (2007). Honey bee colonies that have survived Varroa destructor. Apidologie 38, 566–572. doi: 10.1051/apido:2007040
LeConte, Y., Ellis, M., and Ritter, W. (2010). Varroa mites and honey bee health: can Varroa explain part of the colony losses? Apidologie 41, 353–363. doi: 10.1051/apido/2010017
Leiser, O. P., Corn, J. L., Schmit, B. S., Keim, P. S., and Foster, J. T. (2013). Feral swine brucellosis in the United States and prospective genomic techniques for disease epidemiology. Vet. Microbiol. 166, 1–10. doi: 10.1016/j.vetmic.2013.02.025
Lenth, R. (2020). emmeans: Estimated Marginal Means, aka Least-Squares Means. R package version 1.4.4. Available online at: https://cran.r-project.org/package=emmeans (accessed August 8, 2020).
Lepczyk, C. A., Haman, K. H., Sizemore, G. C., and Farmer, C. (2020). Quantifying the presence of feral cat colonies and Toxoplasma gondii in relation to bird conservation areas on O’ahu, Hawai’i. Conserv. Sci. Pract. 2:e179. doi: 10.1111/csp2.179
Li, W., Evans, J. D., Huang, Q., Rodríguez-García, C., Liu, J., Hamilton, M., et al. (2016). Silencing the honey bee (Apis mellifera) naked cuticle gene (nkd) improves host immune function and reduces Nosema ceranae infections. Appl. Environ. Microbiol. 82, 6779–6787. doi: 10.1128/AEM.02105-16
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Locke, B. (2016). Natural Varroa mite-surviving Apis mellifera honeybee populations. Apidologie 47, 467–482. doi: 10.1007/s13592-015-0412-8
Locke, B., Forsgren, E., and De Miranda, J. R. (2014). Increased tolerance and resistance to virus infections: a possible factor in the survival of Varroa destructor-resistant honey bees (Apis mellifera). PLoS One 9:e0099998. doi: 10.1371/journal.pone.0099998
Loftus, J. C., Smith, M. L., and Seeley, T. D. (2016). How honey bee colonies survive in the wild: testing the importance of small nests and frequent swarming. PLoS One 11:e0150362. doi: 10.1371/journal.pone.0150362
López-Uribe, M. M., Appler, R. H., Youngsteadt, E., Dunn, R. R., Frank, S. D., and Tarpy, D. R. (2017). Higher immunocompetence is associated with higher genetic diversity in feral honey bee colonies (Apis mellifera). Conserv. Genet. 18, 659–666. doi: 10.1007/s10592-017-0942-x
López-Uribe, M. M., Ricigliano, V. A., and Simone-Finstrom, M. (2020). Defining pollinator health: a holistic approach based on ecological, genetic, and physiological factors. Annu. Rev. Anim. Biosci. 8, 269–294. doi: 10.1146/annurev-animal-020518-115045
Lourenço, A. P., Mackert, A., dos Santos Cristino, A., and Simões, Z. L. P. (2008). Validation of reference genes for gene expression studies in the honey bee, Apis mellifera, by quantitative real-time RT-PCR. Apidologie 39, 372–385. doi: 10.1051/apido:2008015
Lowe, E. C., Simmons, L. W., and Baer, B. (2011). Worker heterozygosity and immune response in feral and managed honeybees (Apis mellifera). Aust. J. Zool. 59, 73–78. doi: 10.1071/ZO11041
Luong, G. T. H., Lee, J.-S., Yong, S.-J., and Yoon, B.-S. (2015). Development of ultra-rapid reverse transcription real-time PCR for detection against black queen cell virus in honeybee. J. Apiculture 30:171. doi: 10.17519/apiculture.2015.09.30.3.171
Maeda, T., Nakashita, R., Shionosaki, K., Yamada, F., and Watari, Y. (2019). Predation on endangered species by human-subsidized domestic cats on Tokunoshima Island. Sci. Rep. 9:16200. doi: 10.1038/s41598-019-52472-3
Martin, S. J., Highfield, A. C., Brettell, L., Villalobos, E. M., Budge, G. E., Powell, M., et al. (2012). Global honey bee viral landscape altered by a parasitic mite. Science 336, 1304–1306. doi: 10.1126/science.1220941
Mcmahon, D. P., Paxton, R. J., Natsopoulou, M. E., Doublet, V., Fürst, M., Weging, S., et al. (2016). Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R. Soc. B Biol. Sci. 283:20160811. doi: 10.1098/rspb.2016.0811
McMenamin, A. J., Daughenbaugh, K. F., Parekh, F., Pizzorno, M. C., and Flenniken, M. L. (2018). Honey bee and bumble bee antiviral defense. Viruses 10, 1–22. doi: 10.3390/v10080395
Meng, X. J., Lindsay, D. S., and Sriranganathan, N. (2009). Wild boars as sources for infectious diseases in livestock and humans. Philos. Trans. R. Soc. B Biol. Sci. 364, 2697–2707. doi: 10.1098/rstb.2009.0086
Meyer, R. S., and Purugganan, M. D. (2013). Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14, 840–852. doi: 10.1038/nrg3605
Milla, R., Bastida, J. M., Turcotte, M. M., Jones, G., Violle, C., Osborne, C. P., et al. (2018). Phylogenetic patterns and phenotypic profiles of the species of plants and mammals farmed for food. Nat. Ecol. Evol. 2, 1808–1817. doi: 10.1038/s41559-018-0690-4
Möckel, N., Gisder, S., and Genersch, E. (2011). Horizontal transmission of deformed wing virus: pathological consequences in adult bees (Apis mellifera) depend on the transmission route. J. Gen. Virol. 92, 370–377. doi: 10.1099/vir.0.025940-0
Mondet, F., de Miranda, J. R., Kretzschmar, A., Le Conte, Y., and Mercer, A. R. (2014). On the front line: quantitative virus dynamics in honeybee (Apis mellifera L.) colonies along a new expansion front of the parasite Varroa destructor. PLoS Pathog. 10:e1004323. doi: 10.1371/journal.ppat.1004323
Moreira, X., Abdala-Roberts, L., Gols, R., and Francisco, M. (2018). Plant domestication decreases both constitutive and induced chemical defences by direct selection against defensive traits. Sci. Rep. 8:12678. doi: 10.1038/s41598-018-31041-0
Mukaka, M. M. (2012). Statistics corner: a guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 24, 69–71.
Neumann, P., and Blacquière, T. (2017). The Darwin cure for apiculture? Natural selection and managed honeybee health. Evol. Appl. 10, 226–230. doi: 10.1111/eva.12448
Niu, J., Meeus, I., and Smagghe, G. (2016). Differential expression pattern of Vago in bumblebee (Bombus terrestris), induced by virulent and avirulent virus infections. Sci. Rep. 6:34200. doi: 10.1038/srep34200
Nygren, J., Shad, N., Kvarnheden, A., and Westerbergh, A. (2015). Variation in susceptibility to wheat dwarf virus among wild and domesticated wheat. PLoS One 10:e0121580. doi: 10.1371/journal.pone.0121580
Papkou, A., Guzella, T., Yang, W., Koepper, S., Pees, B., Schalkowski, R., et al. (2019). The genomic basis of red queen dynamics during rapid reciprocal host–pathogen coevolution. Proc. Natl. Acad. Sci. U.S.A. 116, 923–928. doi: 10.1073/pnas.1810402116
Paradkar, P. N., Trinidad, L., Voysey, R., Duchemin, J. B., and Walker, P. J. (2012). Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc. Natl. Acad. Sci. U.S.A. 109, 18915–18920. doi: 10.1073/pnas.1205231109
R Core Team (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Robertson, A. J., Scruten, E., Mostajeran, M., Robertson, T., Denomy, C., Hogan, D., et al. (2020). Kinome analysis of honeybee (Apis mellifera L.) dark-eyed pupae identifies biomarkers and mechanisms of tolerance to Varroa mite infestation. Sci. Rep. 10:2117. doi: 10.1038/s41598-020-58927-2
Russo, R. M., Liendo, M. C., Landi, L., Pietronave, H., Merke, J., Fain, H., et al. (2020). Grooming behavior in naturally Varroa-resistant Apis mellifera colonies from north-central Argentina. Front. Ecol. Evol. 8:590281. doi: 10.3389/fevo.2020.590281
Ryabov, E. V., Childers, A. K., Lopez, D., Grubbs, K., Posada-Florez, F., Weaver, D., et al. (2019). Dynamic evolution in the key honey bee pathogen deformed wing virus: novel insights into virulence and competition using reverse genetics. PLoS Biol. 17:e3000502. doi: 10.1371/journal.pbio.3000502
Ryabov, E. V., Wood, G. R., Fannon, J. M., Moore, J. D., Bull, J. C., Chandler, D., et al. (2014). A virulent strain of deformed wing virus (DWV) of honeybees (Apis mellifera) prevails after Varroa destructor-mediated, or in vitro, transmission. PLoS Pathog. 10:e1004230. doi: 10.1371/journal.ppat.1004230
Sadd, B. M., and Siva-Jothy, M. T. (2006). Self-harm caused by an insect’s innate immunity. Proc. R. Soc. B Biol. Sci. 273, 2571–2574. doi: 10.1098/rspb.2006.3574
Seeley, T. D. (2007). Honey bees of the Arnot Forest: a population of feral colonies persisting with Varroa destructor in the northeastern United States. Apidologie 38, 19–29. doi: 10.1051/apido:2006055
Simone-Finstrom, M. (2017). Social immunity and the superorganism: behavioral defenses protecting honey bee colonies from pathogens and parasites. Bee World 94, 21–29. doi: 10.1080/0005772x.2017.1307800
Taric, E., Glavinic, U., Stevanovic, J., Vejnovic, B., Aleksic, N., Dimitrijevic, V., et al. (2019). Occurrence of honey bee (Apis mellifera L.) pathogens in commercial and traditional hives. J. Apicult. Res. 58, 433–443. doi: 10.1080/00218839.2018.1554231
Taric, E., Glavinic, U., Vejnovic, B., Stanojkovic, A., Aleksic, N., Dimitrijevic, V., et al. (2020). Oxidative stress, endoparasite prevalence and social immunity in bee colonies kept traditionally vs. those kept for commercial purposes. Insects 11:266. doi: 10.3390/insects11050266
Taylor, R. B., Hellgren, E. C., Gabor, T. M., and Ilse, L. M. (1998). Reproduction of feral pigs in southern Texas. J. Mammal. 79, 1325–1331. doi: 10.2307/1383024
Tetreau, G., Dhinaut, J., Gourbal, B., and Moret, Y. (2019). Trans-generational immune priming in invertebrates: current knowledge and future prospects. Front. Immunol. 10:1938. doi: 10.3389/fimmu.2019.01938
Thaduri, S., Stephan, J. G., de Miranda, J. R., and Locke, B. (2019). Disentangling host-parasite-pathogen interactions in a varroa-resistant honeybee population reveals virus tolerance as an independent, naturally adapted survival mechanism. Sci. Rep. 9:6221. doi: 10.1038/s41598-019-42741-6
Thompson, C. E., Biesmeijer, J. C., Allnutt, T. R., Pietravalle, S., and Budge, G. E. (2014). Parasite pressures on feral honey bees (Apis mellifera sp.). PLoS One 9:e0105164. doi: 10.1371/journal.pone.0105164
Underwood, R. M., Traver, B. E., and López-Uribe, M. M. (2019). Beekeeping management practices are associated with operation size and beekeepers’ philosophy towards in-hive chemicals. Insects 10:10. doi: 10.3390/insects10010010
vanEngelsdorp, D., Evans, J. D., Saegerman, C., Mullin, C., Haubruge, E., Nguyen, B. K., et al. (2009). Colony collapse disorder: a descriptive study. PLoS One 4:e6481. doi: 10.1371/journal.pone.0006481
Vannette, R. L., Mohamed, A., and Johnson, B. R. (2015). Forager bees (Apis mellifera) highly express immune and detoxification genes in tissues associated with nectar processing. Sci. Rep. 5. doi: 10.1038/srep16224
Wagoner, K., Spivak, M., Hefetz, A., Reams, T., and Rueppell, O. (2019). Stock-specific chemical brood signals are induced by Varroa and Deformed Wing Virus, and elicit hygienic response in the honey bee. Sci. Rep. 9:8753. doi: 10.1038/s41598-019-45008-2
Wilkins, A. S., Wrangham, R. W., and Tecumseh Fitch, W. (2014). The “domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics. Genetics 197, 795–808. doi: 10.1534/genetics.114.165423
Wu, Y., Dong, X., and Kadowaki, T. (2017). Characterization of the copy number and variants of deformed wing virus (DWV) in the pairs of honey bee pupa and infesting Varroa destructor or Tropilaelaps mercedesae. Front. Microbiol. 8:1558. doi: 10.3389/fmicb.2017.01558
Wu, Y., Liu, Q., Weiss, B., Kaltenpoth, M., and Kadowaki, T. (2020). Honey bee suppresses the parasitic mite Vitellogenin by antimicrobial peptide. Front. Microbiol. 11:1037. doi: 10.3389/fmicb.2020.01037
Yi, H.-Y., Chowdhury, M., Huang, Y.-D., and Yu, X.-Q. (2014). Insect antimicrobial peptides and their applications. Appl. Microbiol. Biotechnol. 98, 5807–5822. doi: 10.1007/s00253-014-5792-6
Youngsteadt, E., Appler, R. H., López-Uribe, M. M., Tarpy, D. R., and Frank, S. D. (2015). Urbanization increases pathogen pressure on feral and managed honey bees. PLoS One 10:e0142031. doi: 10.1371/journal.pone.0142031
Keywords: Apis mellifera, ecoimmunology, Deformed wing virus (DWV), hymenoptaecin, vago
Citation: Hinshaw C, Evans KC, Rosa C and López-Uribe MM (2021) The Role of Pathogen Dynamics and Immune Gene Expression in the Survival of Feral Honey Bees. Front. Ecol. Evol. 8:594263. doi: 10.3389/fevo.2020.594263
Received: 12 August 2020; Accepted: 14 December 2020;
Published: 13 January 2021.
Edited by:
Nikolaos Papadopoulos, University of Thessaly, GreeceReviewed by:
Jevrosima Stevanovic, University of Belgrade, SerbiaBin Tang, Hangzhou Normal University, China
Copyright © 2021 Hinshaw, Evans, Rosa and López-Uribe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chauncy Hinshaw, Y3JoNTRAcHN1LmVkdQ==; Margarita M. López-Uribe, bW1sNjRAcHN1LmVkdQ==