 Dennis Hasselquist
Dennis Hasselquist Michael Tobler
Michael Tobler- Department of Biology, Molecular Ecology & Evolution Lab (MEEL), Lund University, Lund, Sweden
Mild diseases and moderate stressors are seemingly harmless and are therefore often assumed to have negligible impact on Darwinian fitness. Here we argue that the effects of “benign” parasites and other moderate stressors may have a greater impact on lifespan and other fitness traits than generally thought. We outline the “accumulating costs” hypothesis which proposes that moderate strains on the body caused by mild diseases and other moderate stressors that occur throughout life will result in small irreversible “somatic lesions” that initially are invisible (i.e., induce “hidden” costs). However, over time these somatic lesions accumulate until their summed effect reaches a critical point when cell senescence and malfunction begin to affect organ functionality and lead to the onset of degenerative diseases and aging. We briefly discuss three potential mechanisms through which the effects of moderate strains (e.g., mild diseases) could accumulate: Accelerated telomere shortening, loss of repetitious cell compartments and other uncorrected DNA damage in the genome. We suggest that telomere shortening may be a key candidate for further research with respect to the accumulating costs hypothesis. Telomeres can acquire lesions from moderate strains without immediate negative effects, lesions can be accumulated over time and lead to a critically short telomere length, which may eventually cause severe somatic malfunctioning, including aging. If effects of mild diseases, benign parasites and moderate stressors accrued throughout life can have severe delayed consequences, this might contribute to our understanding of life history strategies and trade-offs, and have important implications for medicine, including consideration of treatment therapies for mild (chronic) infections such as malaria.
Introduction
Severe stressors and diseases, which directly affect behavior and performance as well as threaten survival (e.g., malign diseases), entail considerable (Darwinian fitness) costs. Moreover, there is good support for a link between severe (chronic) stressors, and accelerated aging and increased mortality (Sapolsky, 1999; Epel et al., 2004; Öhlin et al., 2004; Aldwin et al., 2011; Steptoe and Kivimäki, 2012). Such severe stressors can be contrasted with moderate (and mild) stressors and diseases that have no immediately apparent effects (i.e., the organism performs in more or less the same way as without the stressor). It is well established that stressors during early life can have constitutive effects with long-lasting consequences for fitness (e.g., Gluckman et al., 2007; Monaghan, 2008). However, how moderate stress episodes that occur later in life (e.g., short periods of heat or cold stress, sleep deprivation, heavy physical workload), and seemingly mild diseases (e.g., short episodes of severe illness due to infection that are cleared by the immune system, or “benign” parasites with low virulence that persist as mild chronic infections) may have impact on aging, life span and lifetime reproductive success is less clear. The predominant view has been that mild diseases (“benign parasites”) and moderate (short-term) stress episodes in the adult life stages have negligible costs in terms of survival and Darwinian fitness (Bensch et al., 2007; LaPointe et al., 2012; Asghar et al., 2015; although this view has sometimes been challenged; e.g., Hamilton and Zuk, 1982). It has been argued that wild animals are generally resilient and able to cope with repeated (but limited) short-term stressors without experiencing any substantial remaining effects (e.g., Boonstra, 2013). Similarly, organisms that get infected with a benign parasite may become sick, mount immune responses to combat the pathogen and then recover to, in most instances, apparently become fully healthy again, without any directly measurable negative effects on performance or survival. However, there is increasing evidence that even mild diseases and moderate short-term stress episodes can have long-term consequences for the Darwinian fitness of individuals, in terms of accelerated aging and increased mortality (e.g., Hanssen et al., 2003; Asghar et al., 2015; Froy et al., 2019). We therefore believe that the somatic effects of mild diseases and other moderate stressors that may appear almost negligible in fact deserve more attention, both in terms of theory and empirical studies. A particularly challenging question is how presumably small effects of short, repeated stress episodes and mild diseases are translated into long-term effects on reproduction, aging and lifespan (i.e., impairing Darwinian fitness). Using some examples from the eco-immunology literature, we emphasize the point that small (physiological) costs induced by moderate strains, such as short disease episodes or benign (chronic) parasite infection accrued throughout life, over time can translate into Darwinian fitness costs. We then outline a hypothesis that proposes an explanation for how such, seemingly limited and benign, short-term costs can translate into more severe costs acting over the longer term. We also discuss possible somatic agents that have the ability to translate seemingly negligible short-term costs into more severe long-term costs, as well as some potential mechanisms (i.e., cost currencies) that can be involved in this process.
Mild Diseases and Darwinian Fitness
Our argument that mild diseases might have considerable effects on Darwinian fitness originates from studies conducted in the field of eco-immunology. In a set of studies using vaccination to induce an immune response that mimic an infection in the body (of wild birds), to us unexpected effects were found. The paradigm at the time was that an energetic (or nutritional) trade-off between heavy workload and immune system activation, both assumed to be energetically costly/nutrition demanding, would directly limit immune responses if other energetically costly/nutrition demanding activities were conducted simultaneously (Sheldon and Verhulst, 1996; Lochmiller and Deerenberg, 2000). However, in vaccination studies on songbirds, in which parents were working hard to feed their nestlings (Ilmonen et al., 2000; Råberg et al., 2000), the surprising finding was that the songbirds decreased their feeding workload to such a high extent that it saved seven times more energy than needed to equal out the energetic cost induced by mounting the immune response to the vaccine antigens (Svensson et al., 1998; Hasselquist and Nilsson, 2012). And at the same time this reduction in workload resulted in significantly decreased number and/or quality of their offspring (Ilmonen et al., 2000; Råberg et al., 2000). Thus, a seemingly moderate stressor induced an unexpectedly high (apparently over-compensatory prudent) response.
Moreover, in a study on wild female eider ducks (Somateria mollissima) that were vaccinated during incubation (a period when these birds are fasting and rely entirely on accumulated fat stores for their energy consumption; Parker and Holm, 1990; Hanssen et al., 2003), there were no differences in body mass loss (i.e., a close proxy of energy consumption in incubating eiders; Hanssen et al., 2003) over the 1 month long incubation period, with respect to whether the females produced antibodies or not against the antigens (Hanssen et al., 2004). Thus, there were no short-term costs indicating an energetic or nutritional trade-off in relation to immune system activation. However, in the subset of females that had mounted humoral immune responses against more than one vaccine antigen, severe long-term costs were found. These females had a substantially reduced return rate to the next breeding season indicating reduced survival in this species that generally has very high (≥80%) between-year return rates (Yoccoz et al., 2002; Hanssen et al., 2004).
Along the same lines, in great reed warblers (Acrocephalus arundinaceus), it has been shown that chronic low-level infections with malaria parasites did not have any apparent effects on song production, feeding effort, annual reproductive success or survival to the next year (Bensch et al., 2007; Asghar et al., 2015). However, when this was investigated over the longer term, it was found that malaria-infected individuals in fact experienced long-term costs in terms of shorter life span and lower lifetime fledgling success (Asghar et al., 2015).
The above studies illustrate how different moderate stressors experienced during adulthood (temporary immune system activation, malaria infection) can be latent and, thus, not immediately apparent. That even short-term disease episodes and mild chronic infections have long-term fitness costs may not be entirely surprising, but hitherto it has been difficult to show such effects. Moreover, it is not clear how such “micro-costs” can be stored and become apparent later in life. The findings above have made us start to think along the lines of some mechanistic model for how relatively modest costs could remain invisible in the short-term, but still accumulate over time to become visible as deterioration of the soma and negative Darwinian fitness consequences at a later stage in life.
The Accumulating Costs Hypothesis
We argue that even moderate strains, i.e., seemingly mild diseases and other moderate stressors accrued at any time in life, can induce physiological costs that ultimately will affect Darwinian fitness. We propose the accumulating costs hypothesis, which describes the processes through which delayed (or “hidden” sensu Asghar et al., 2015) costs can come about and ultimately cause somatic malfunction and senescence. The accumulating costs hypothesis proposes that even moderate stress and seemingly harmless diseases, with no direct visible effects, will leave small “somatic lesions” in the body (López-Otín et al., 2013; see also Selye, 1956; Geronimus et al., 2015; Bateson, 2016). The “invisible costs” of these relatively small somatic lesions will then accumulate over time and eventually result in more severe consequences for somatic maintenance and physiological functions. When these small somatic lesions have accumulated to reach a certain critical threshold, they become “visible.” This may be in the form of “ordinary” senescence in old age individuals with relatively slow accumulation of costs (exposure to relatively low frequency and magnitude of moderate stress and diseases; Figures 1, 2). However, these long-term costs could also become visible as malfunctioning organs and uncorrected DNA damage anywhere in the genome (e.g., cancers and malign mutations) in young or middle-aged individuals with more rapid accumulation of costs, for example, due to exposure to higher frequency or magnitude of (moderate) stress and diseases (Figures 1, 2). Under both these scenarios, the important implication is that there may be “hidden” delayed costs associated also with relatively mild diseases and moderate stressors that accumulate up to a certain “tipping point.” We think of this as an “ättestupa” effect (or “falling off the cliff” effect sensu Monaghan, 2014). “Ättestupa” comes from the Nordic Vikings where the legend says that it was the custom when a person of the clan (“ätt” in Nordic languages) reached a (biological) age where the body would no longer function properly, he/she would go to a culturally assigned cliff (“stup” in Nordic languages) and jump off to avoid being a problem for the youngsters of the clan. The somatic costs are hard to directly observe when they are induced. Instead, they act on the long-term only to become clearly visible at a (often much) later stage of life. It should be noted that, although we in this paper emphasize the potential importance of mild diseases and other moderate stressors as inducers of somatic lesions and potential agents of aging, costs of more severe stressors and diseases may accumulate through the same mechanisms. Depending on the severity and the duration of the disease or stress episodes, somatic lesions may be larger and accumulate faster. Thus, in general we envision that somatic lesions will accumulate over time at different paces in different individuals (Figures 1, 2). The pace depends both on the number and length of moderate stress and disease events, but also on the number of more severe disease and stress episodes accrued throughout life.
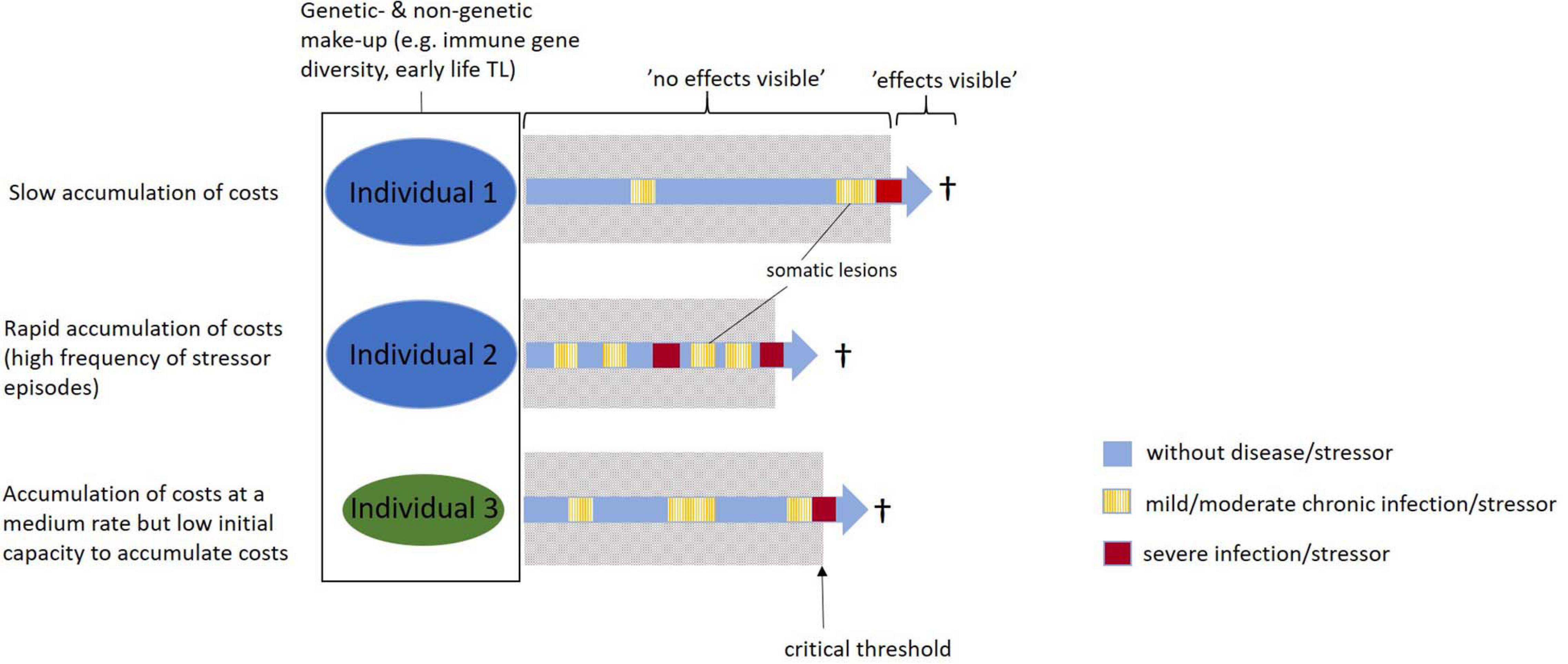
Figure 1. Schematic illustration of how lesions accumulate in different individuals. The accumulating costs hypothesis proposes that lesions accumulate “silently” (i.e., they are “hidden”) until a critical point is reached after which the costs of the accumulated lesions become visible. Individuals that experience more moderate stress/disease events (Ind 2) or have a lower capacity to accumulate lesions (Ind 3) reach the critical point more rapidly than an individual exposed to fewer stress/disease events and a higher capacity to accumulate lesions (Ind 1). Note that for the purpose of illustration we only visualize a few periods during which lesions are accumulated over life for each individual.

Figure 2. Hypothetical example of accumulation of somatic lesions up to a critical threshold (the “critical point”) when (previously hidden) costs become “visible,” as envisioned by the accumulating costs hypothesis. With low effects of diseases and stressors, lesions accumulate slowly over time (blue dashed line). With increasing severity of diseases/stressors (yellow, orange and dark red solid lines) lesions accumulate more rapidly. Note that even though individuals can have different starting points (individual 2 and 3), which may affect their capacity to absorb lesions without any visible effects, individual 2 (with a “better” starting point in early life) may accumulate lesions at a higher rate during adult life and in fact cross-over the trajectory of individual 3 and thus experience a shorter life span due to differences in the magnitudes and frequencies of stress episodes these individuals experience during adult life. Note also that cellular repair mechanisms (e.g., telomerase activity, base or nucleotide excision repair, homologous recombination) might even temporally reverse the slope of accumulating lesions (e.g., third period with no stressor for individual 1).
Potential Processes and Mechanistic Agents Able to Induce Somatic Lesions
Which cellular processes could be affected by mild diseases or moderate stress and leave somatic lesions that can accumulate in the body and eventually cause delayed long-term effects? There are many different physiological processes that could induce strain on the body, and thus result in somatic lesions. We list some rather obvious processes in Table 1. Note that these processes are not independent from each other and, hence, not mutually exclusive. For example, excessive inflammatory response may be a result of immune system over-activation (i.e., immunopathology) and may, in turn, lead to ROS (i.e., reactive oxygen species) damage (von Schantz et al., 1999; Zglinicki, 2000). Similarly, ROS damage may increase the mutation rate (Sakai et al., 2006; Ragu et al., 2007), which in turn could alter performance of cellular functions, e.g., result in less efficient or even malfunctioning mitochondrial processes (López-Otín et al., 2013; Hahn and Zuryn, 2019).

Table 1. Possible cellular processes that could cause the small “somatic lesions” generating small (“hidden”) physiological costs that underlie the cost accumulation process as envisioned in the accumulating costs hypothesis.
Mechanisms Through Which Small Short-Term Costs May Be Translated Into Delayed (Long-Term) Costs
If moderate stressors can induce somatic lesions, for example through the processes listed in Table 1, then the obvious question is “what is the mechanistic basis through which these lesions may accumulate over time and translate into long-term costs”? Or, to put it a bit differently, in what form are the effects of moderate stressors stored in somatic tissue and how can they be converted into more severe syndromes that can be hazardous for the whole soma and, thus, the health and well-being of the entire organism? Here, we briefly discuss three mechanisms through which moderate stress and disease episodes could induce somatic lesions that are stored in somatic tissue and, thus, allow for accumulation of short-term costs and their transfer into severe long-term consequences. We describe these below.
(1) Accelerated telomere shortening. Somatic lesions are manifested as successive telomere shortening (which has been shown to be accelerated through disease and severe stress; e.g., Effros et al., 1996; Epel et al., 2004; Kotrschal et al., 2007; Ilmonen et al., 2008; Asghar et al., 2015). This is consistent with the idea proposed by Bateson (2016) that telomere length is sensitive to the cumulative effects of stress factors in a dose-dependent manner, more negative stress factors being associated with greater loss of telomere repeats, and telomeres then being a biological marker of exposure to stress accrued over time (see also Kotrschal et al., 2007). Here, we take this one step further, arguing that the cumulative loss of telomere repeats may be a key process that has causative negative functional consequences for the organism. Over time, loss of telomere repeats leads to an increasing number of stem cells with too short telomeres (and thus also lower average telomere length), resulting in cell dysfunction, ceased stem cell division, and eventually cell death due to apoptosis (Blackburn, 2005; Shay and Wright, 2019). When cell death increases and stem cell activity decreases, this eventually result in organ dysfunction and physiological impairment.
The above idea contrasts with the hypothesis proposed by Aviv et al. (2015) which suggests that telomeres shorten at more or less similar rates between individuals in somatic tissues after birth. According to Aviv et al. (2015), telomere length after birth essentially reflects early life/birth differences in somatic cell reserves and the ability of stem cells to maintain tissue repair. Hence, in clear contrast to our Accumulating costs hypothesis, Aviv et al. (2015) propose that individuals with shorter early life telomere length will (inevitably) reach the critical threshold at an earlier age, and therefore risk earlier onset of age-related diseases and premature death independent of what happens during the individuals’ adult life stages.
(2) Loss of “repetitous cell compartments.” Gavrilov and Gavrilova (2001) proposed the concept that each cell type compartment contains a (large) set of redundant cells with equal function—more cells of similar function than what is needed for the organism to function properly at any given time point before severe senescence sets in. Moderate stress and diseases will result in cell damage and subsequent cell death. Death of single cells has, however, no measurable negative effects on body organs and physiological performance—not until too many of the redundant cells within a compartment have become dysfunctional or died. At this stage, which may be much later than when most of the cell damage took place, organ failure will set in leading to degenerative disease and lowered physiological capacity/condition (Gavrilov and Gavrilova, 2001; Boonekamp et al., 2013). The hypothesis, as originally proposed, assumes that redundancy elements have a constant failure rate over time and with more elements more are lost per unit of time (Gavrilov and Gavrilova, 2001). Here, however, we assume that the rate of loss of repetitious cell compartments is dependent on the frequency and duration of the disease and stress factors.
(3) Increased uncorrected DNA damage throughout the genome. Accumulation of genetic damage over time is one of the hallmarks of aging (López-Otín et al., 2013; Moskalev et al., 2013). Accumulated uncorrected DNA damage in terms of somatic mutations in nuclear and mitochondrial DNA (sensu López-Otín et al., 2013), is a process that might be equivalent to the accumulation of somatic lesions over time. It has been shown that mutations increase in response to stressors (e.g., ultraviolet radiation, oxidative stress; Hoeijmakers, 2009) and with age across multiple tissues (Martincorena et al., 2018), including stem cells (Rossi et al., 2007, 2008). Hence, accumulation of mutations has been suggested as a potentially important factor driving aging (Freitas and de Magalhães, 2011; López-Otín et al., 2013). Although many mutations do not alter cell function, accumulation of mutations and deletions in the nuclear and mitochondrial genome may lead to progressive loss of functional cells and induce organ dysfunction (López-Otín et al., 2013; Aviv et al., 2015). However, severe mutations can arise at any point during an organism’s lifetime which, in turn, can result in a more erratic pattern of induction of severe costs, such as cancer that can be induced at any chronological age, and therefore not necessarily mirror the processes typical for aging. Instead, if accumulation of mutations is a process underlying the accumulating costs hypothesis, as we envision it, it has to build on the accumulation of “less severe” mutations in a gradual and less punctuated way, in which each somatic lesion only slightly reduces the organism’s ability to be resilient to additional stress and disease factors without any immediately visible effects on organ function. Hence, reduction in cellular function due to accumulated mutations should progress relatively slowly, although with different speed in different individuals, until the soma of an organism is failing in several tissues and organ systems, resulting in senescence pathologies and eventually death.
The three mechanisms described above are not mutually exclusive but may instead act in concert. For example, telomeric repeats that protect coding DNA may accumulate damage (e.g., due to effects of oxidative stress; Reichert et al., 2014; Chatelain et al., 2020), which in turn may result in a less protective cap structure that makes coding DNA vulnerable to degradation. Damage and changes to coding DNA may then lead to a lowered functional performance of cells and, ultimately, cell death. One could also envision a “vicious” loop so that short telomeres in some cells of a redundancy element leads to cell malfunction, which may then send out cell stress signals to neighboring cells thus starting a cascade of cell stress (López-Otín et al., 2013). Stressed cells will experience an accelerated rate of telomere shortening causing more cells to reach a critically short telomere length, inducing even more severe bursts of cell senescence and apoptosis (Nelson et al., 2012). This could result in a “chain reaction” of cell malfunction and apoptosis in the repetitive cell compartment, leading to exhaustion of the repetitive cells, which may result in organ dysfunction, aging and even premature death.
The mechanisms described above illustrate how even relatively minor costs resulting from exposure to mild diseases and moderate stressor could be “stored” and how these may affect organismal fitness at a later stage in life. However, whether for example relatively minor disease episodes or chronic exposure to seemingly benign parasites result in (somatic) lesions will also depend on the cellular repair mechanisms, which in turn, can differ between cell lineages or tissue. As we outline above, we would not expect mild diseases or moderate stressors to induce excessive damage and therefore cell lineages or tissues with limited repair capacity should be most susceptible. The soma, for example, is more likely to accumulate lesions resulting from moderate stressors than the germline given that the latter is known to have a more efficient repair system (Vijg, 2007), a fact that has also given raise to the disposable soma theory (Kirkwood, 1977). Similarly, in mammals mitochondrial DNA repair mechanisms are limited compared to repair mechanisms for nuclear DNA (Druzhyna et al., 2008) and might, thus, be more susceptible to moderate insults. Finally, the literature on humans and other mammalian species also shows that telomeres have restricted DNA repair to avoid exposure of linear telomeric DNA, which would result in telomere repair and chromosome fusion (Fumagalli et al., 2012; Hewitt et al., 2012). Thus, telomeres might be particularly likely as “storage structure” of cumulative micro-costs resulting from mild disease and moderate stressors (see below).
In humans, the consequences of cell and tissue degeneration can become manifested as degenerative diseases (Edkert, 2002). In short-lived and wild animals, however, cell and tissue degeneration may instead rather be manifested in the form of reduced physiological capacity/”condition” (Palacios et al., 2007; Cote et al., 2010; Nussey et al., 2012; Elliott et al., 2015). In wild animals, the latter effects may result in increased predation rate (Murray, 2002; Genovart et al., 2010), reduced competitive ability (Briffa and Sneddon, 2007), impaired attractiveness in mate choice (von Schantz et al., 1999), and lowered immunocompetence (Palacios et al., 2007; Froy et al., 2019), all of which can ultimately reduce survival and reproductive success (e.g., Hanssen et al., 2004; Asghar et al., 2015).
Of the three mechanisms described above, we argue that telomere shortening is a strong candidate for how seemingly negligible (“hidden”) costs are translated into delayed costs with more severe effects on the soma (see also Giraudeau et al., 2019). Telomere shortening is known to be affected by adverse conditions and stressors (Bateson, 2016; Chatelain et al., 2020) and somatic lesions may, thus, be measured as the loss of telomere repeats. It has also been suggested that there is a telomeric brink beyond which senescence will progress rapidly (Aviv et al., 2015). Under this scenario, (severe) telomere shortening would be the causative process behind the need for having repetitious cell compartments in the body, and, thus, be the underlying cause in the repetitious cell compartment mechanism. The observation that the process of telomere shortening in a wild vertebrate behaves the same way as predicted from a model of redundancy element loss (Boonekamp et al., 2013) lends some support to this idea. Note, however, that the time it will take for an organism to reach the critical point in telomere length is also affected by the initial, early life telomere length, which typically shows rather large variation between individuals of a species (Heidinger et al., 2012; Asghar et al., 2015; Aviv et al., 2015; Watson et al., 2015; Lieshout et al., 2019). This large initial difference in early life telomere length will make it harder to predict the time it will take to reach the critical point in telomere length based on an individual’s exposure to disease and stress factors (Figures 1, 2). The same is true if telomerase (a reverse transcriptase, an enzyme that adds telomere repeat sequences at the end of telomeres during cell division; Blackburn, 2005) is being activated in adult individuals of a species, as it allows for elongation and restoring of telomere length thus prolonging the time it would take to reach the critical point in telomere length.
Does Telomere Length and Shortening Rate Reflect Tolerance to Diseases and Stressors?
It has recently been suggested that the ability to limit damage caused by a given parasite burden (tolerance) should be negatively associated with telomere shortening (Giraudeau et al., 2019). Under the accumulating costs hypothesis, tolerant phenotypes might then be those that accumulate somatic lesions at a slower pace than less tolerant ones, because the latter pay the costs of inducing immune responses. However, another possibility is that tolerant phenotypes can cope with higher levels of somatic lesions before experiencing significant Darwinian fitness costs. In this case, higher levels of telomere shortening might also reflect higher tolerance, and somatic maintenance is then traded-off with other traits such as reproductive performance. We can also envision that being tolerant with respect to certain parasites means becoming chronically infected which, in turn, could lead to repeated induction of somatic lesions over time. In this case, the host accepts somatic lesions induced by the parasites that it tolerates and therefore now reside in its body. Somatic lesions may accumulate and become manifested as accelerated telomere shortening with the consequence that tolerant phenotypes are exposed to a faster pace of telomere shortening and thus potentially premature aging. An apparent case of parasitic tolerance was recently reported in the African buffalo (Syncerus caffer) where infection with the gastrointestinal worm Cooperia fuelleborni was positively correlated with condition and possibly reproduction (Budischak et al., 2016). In this case, we can imagine a scenario in which the costs of the “tolerated” infection might be “hidden” as in the great reed warbler example mentioned earlier, and if so, we expect to find an increase in telomere shortening with increased parasite burden.
Discussion
One aim of this paper was to highlight that moderate stressors and so-called mild diseases might have a greater impact on Darwinian fitness than generally thought. This proposition is based on the idea that there might be mechanisms through which seemingly negligible short-term costs can be translated into more severe delayed costs affecting lifespan and lifetime reproductive success. The accumulating costs hypothesis proposed here, suggests that moderate costs accumulate gradually as somatic lesions throughout life until they eventually trigger cellular and organism dysfunction. One of the major challenges for testing the hypothesis is to find the mechanistic pathway through which accumulation of (not directly visible) somatic lesions can occur and be translated into delayed severe long-term costs. We suggest that telomere shortening might be a key mechanism. It reflects the loss of telomeric repeats which can be equaled with accumulation of small somatic lesions that carry no immediate (outwardly observable) costs, but it has the potential to induce negative effects later in life when a critically short telomere length is reached (rather than just being a biomarker). However, other mechanisms may also contribute to accumulation of lesions. If it is possible to measure whether accumulation of somatic lesions vary over time and in association with different types of mild diseases and other moderate stressors, for example through measurement of telomere shortening, this would give us a better idea of whether the accumulating costs hypothesis is valid. The hypothesis predicts that somatic lesions accumulate in a dynamic way because the severity and occurrence of stress episodes and infections are unpredictable. For telomeres, however, it is currently not clear whether telomeres shorten in a dynamic way or at a constant rate over time (Nettle and Bateson, 2017). The best way to address this issue would be to perform longitudinal experimental studies in which organisms are exposed to repeated moderate stressors, mild disease and/or immune challenges. One could then measure how changes in telomere dynamics are associated with the episodes of disease, immune responses and stress. This could be tested, for example, with a 2 × 2 experimental design in which the repair mechanisms for somatic lesions (e.g., manipulation of telomerase activity through supplementation of the root extract TA-65; see Criscuolo et al., 2018) and a moderate stressor (e.g., a parasite of low virulence or one that induces chronic low-level infections) are manipulated concomitantly. If possible, such an experiment should cover a significant part of the organism’s lifetime. This would make it possible to evaluate whether the stressor has a cumulative effect in terms of acquired lesions and whether this effect could be mitigated through repair of the accumulating agent. We also believe that purely observational data can be illuminating as circumstantial evidence, for example for assessing the critical threshold. It might be useful to look for a (lower) critical threshold in telomere length at the population level. An obvious problem with this approach is that telomere length is not measured when the organism dies, but instead at an earlier time point, and that the time period between the last measurement point and the date of death is highly variable (and unknown) between individuals. One way of partly overcoming this problem is to use mean population telomere shortening rate over successive life stages prior to disappearance, to predict the telomere lengths of those individuals which have disappeared from the population.
Another interesting avenue for research would be to study more host-parasite systems in which parasites have no apparent negative effects on host physiology and behavior including mild chronic infections (Brown et al., 2006; Budischak et al., 2016; Bergstrom et al., 2019). Based on the accumulating costs hypothesis, one might expect that even apparently mild parasitic infections are not cost-free. Somatic lesions (e.g., telomere shortening, mutations) may increase with intensity and duration of infection even in these systems and measurable negative effects on physiology and Darwinian fitness factors may only become visible in the long-term (similar to the study by Asghar et al., 2015).
Finally, another aspect that is relevant when testing the importance of telomere dynamics for the accumulating costs hypothesis that could be approached by experimental and observational long-term longitudinal studies is to compare the relative importance of early life telomere length, telomere loss rate and critical point values between individuals with different life spans within a species. Is the critical point value similar for individuals that differ in life span, and if so, is the time it takes to reach the critical point mainly determined by early life telomere length or by telomere loss rate in adult life? In terms of the accumulating costs hypothesis, we predict that the critical point values are similar and that the time it takes to reach the critical point is largely dependent on the telomere loss rate at adult age when comparing individuals with different life spans within a species. In contrast, in between-species studies, we envision that critical points values may differ between species, due to natural selection inflicting very different pressures leading to different evolutionary trajectories of the lower limit of telomere length inducing malfunctioning cells (i.e., critical point values) in different species, e.g., depending on differences in life history strategies.
If the accumulating costs hypothesis can be tested and proven true, this should have important implications for various research fields. In ecology, it is likely to have implications for research fields such as ecoimmunology, ecophysiology and life history theory. For example, in life history theory, it would provide us with a new mechanism for understanding the trade-offs between current and future reproduction, and between reproduction and (somatic) maintenance. These trade-offs are not always easily reconciled based on the current paradigm of energy being the decisive limiting factor underlying them, because even when energy is readily available trade-offs are sometimes found. Moreover, energetic costs are often rather small and, thus, seemingly insufficient to explain the often quite high reproductive success losses taken on by organisms that are exposed to moderate energetic constraints (Hasselquist and Nilsson, 2012). If such unexpectedly prudent outcomes of these trade-offs are the response to accumulating costs that later will accelerate (organ) senescence, shorten lifespan and decrease lifetime reproductive success, the prudency reactions of the organisms to avoid paying the potentially severe delayed costs, are, at least in our view, more matching in scale. In medicine, it would imply that also seemingly harmless diseases and immune responses may entail small costs than will have impact on the rate of aging and degenerative diseases, a surging problem in modern human societies where life span successively increases and thus an increasing proportion of the populations are made up of people of old age. Moreover, it would also influence decisions on strategies and efforts to prevent and cure seemingly benign and low-level chronic diseases and immune reactions, e.g., human malaria in malaria endemic regions (cf. Asghar et al., 2018).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
DH contributed with all the funding. Both authors contributed equally to all parts of this manuscript.
Funding
This study was funded by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (ERC Advanced grant no. 742646 to DH) and the Swedish Research Council (grants nos. 2016-04391 and 2020-03976 to DH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank two reviewers for constructive comments on an earlier version of the manuscript.
References
Aldwin, C. M., Molitor, N.-T., Spiro, A., Levenson, M. R., Molitor, J., and Igarashi, H. (2011). Do stress trajectories predict mortality in older men? longitudinal findings from the VA normative aging study. J. Aging Res. 2011:896109. doi: 10.4061/2011/896109
Angelier, F., Costantini, D., Blévin, P., and Chastel, O. (2018). Do glucocorticoids mediate the link between environmental conditions and telomere dynamics in wild vertebrates? a review. General Comp. Endocrinol. 256, 99–111. doi: 10.1016/j.ygcen.2017.07.007
Asghar, M., Hasselquist, D., Hansson, B., Zehtindjiev, P., Westerdahl, H., and Bensch, S. (2015). Hidden costs of infection: chronic malaria accelerates telomere degradation and senescence in wild birds. Science 347, 436–438. doi: 10.1126/science.1261121
Asghar, M., Yman, V., Homann, M. V., Sondén, K., Hammar, U., Hasselquist, D., et al. (2018). Cellular aging dynamics after acute malaria infection: a 12-month longitudinal study. Aging Cell 17:e12702. doi: 10.1111/acel.12702
Aviv, A., Kark, J. D., and Susser, E. (2015). Telomeres, atherosclerosis, and human longevity: a causal hypothesis. Epidemiology 26, 295–299. doi: 10.1097/EDE.0000000000000280
Bateson, M. (2016). Cumulative stress in research animals: telomere attrition as a biomarker in a welfare context? Bioessays 38, 201–212. doi: 10.1002/bies.201500127
Bensch, S., Waldenström, J., Jonzén, N., Westerdahl, H., Hansson, B., Sejberg, D., et al. (2007). Temporal dynamics and diversity of avian malaria parasites in a single host species. J. Animal Ecol. 76, 112–122. doi: 10.1111/j.1365-2656.2006.01176.x
Bergstrom, B. J., Rose, R. K., and Bellows, A. S. (2019). Stomach nematodes of cotton rats: parasites, commensals, or mutualists? J. Mammal. 100, 1831–1836. doi: 10.1093/jmammal/gyz136
Blackburn, E. H. (2005). Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett. 579, 859–862. doi: 10.1016/j.febslet.2004.11.036
Blasco, M. A. (2002). Immunosenescence phenotypes in the telomerase knockout mouse. Springer Semin. Immunopathol. 24, 75–85. doi: 10.1007/s00281-001-0096-1
Boonekamp, J. J., Simons, M. J. P., Hemerik, L., and Verhulst, S. (2013). Telomere length behaves as biomarker of somatic redundancy rather than biological age. Aging Cell 12, 330–332. doi: 10.1111/acel.12050
Boonstra, R. (2013). The ecology of stress: a marriage of disciplines. Funct. Ecol. 27, 7–10. doi: 10.1111/1365-2435.12048
Briffa, M., and Sneddon, L. U. (2007). Physiological constraints on contest behaviour. Funct. Ecol. 21, 627–637. doi: 10.1111/j.1365-2435.2006.01188.x
Brown, G. P. B. P., Shilton, C. M. S. M., and Shine, R. S. (2006). Do parasites matter? assessing the fitness consequences of haemogregarine infection in snakes. Can. J. Zool. 84, 668–676. doi: 10.1139/z06-044
Budischak, S. A., Hoberg, E. P., Abrams, A., Jolles, A. E., and Ezenwa, V. O. (2016). Experimental insight into the process of parasite community assembly. J. Animal Ecol. 85, 1222–1233. doi: 10.1111/1365-2656.12548
Chatelain, M., Drobniak, S. M., and Szulkin, M. (2020). The association between stressors and telomeres in non-human vertebrates: a meta-analysis. Ecol. Lett. 23, 381–398. doi: 10.1111/ele.13426
Cote, J., Arnoux, E., Sorci, G., Gaillard, M., and Faivre, B. (2010). Age-dependent allocation of carotenoids to coloration versus antioxidant defences. J. Exp. Biol. 213, 271–277. doi: 10.1242/jeb.035188
Criscuolo, F., Smith, S., Zahn, S., Heidinger, B. J., and Haussmann, M. F. (2018). Experimental manipulation of telomere length: does it reveal a corner-stone role for telomerase in the natural variability of individual fitness? Philos. Trans. R. Soc. B. 373:20160440. doi: 10.1098/rstb.2016.0440
Druzhyna, N. M., Wilson, G. L., and LeDoux, S. P. (2008). Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 129, 383–390. doi: 10.1016/j.mad.2008.03.002
Effros, R. B., Allsopp, R., Chiu, C. P., Hausner, M. A., Hirji, K., Wang, L., et al. (1996). Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS 10, F17–F22. doi: 10.1097/00002030-199607000-00001
Elliott, K. H., Hare, J. F., Vaillant, M. L., Gaston, A. J., Ropert-Coudert, Y., and Anderson, W. G. (2015). Ageing gracefully: physiology but not behaviour declines with age in a diving seabird. Funct. Ecol. 29, 219–228. doi: 10.1111/1365-2435.12316
Epel, E. S., Blackburn, E. H., Lin, J., Dhabhar, F. S., Adler, N. E., Morrow, J. D., et al. (2004). Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. U S A. 101, 17312–17315. doi: 10.1073/pnas.0407162101
Freitas, A. A., and de Magalhães, J. P. (2011). A review and appraisal of the DNA damage theory of ageing. Mutat. Res. 728, 12–22. doi: 10.1016/j.mrrev.2011.05.001
Froy, H., Sparks, A. M., Watt, K., Sinclair, R., Bach, F., Pilkington, J. G., et al. (2019). Senescence in immunity against helminth parasites predicts adult mortality in a wild mammal. Science 365, 1296–1298. doi: 10.1126/science.aaw5822
Fumagalli, M., Rossiello, F., Clerici, M., Barozzi, S., Cittaro, D., Kaplunov, J. M., et al. (2012). Telomeric DNA damage is irreparable and causes persistent DNA damage response activation. Nat. Cell Biol. 14, 355–365. doi: 10.1038/ncb2466
Gavrilov, L. A., and Gavrilova, N. S. (2001). The reliability theory of aging and longevity. J. Theor. Biol. 213, 527–545. doi: 10.1006/jtbi.2001.2430
Genovart, M., Negre, N., Tavecchia, G., Bistuer, A., Parpal, L., and Oro, D. (2010). The young, the weak and the sick: evidence of natural selection by predation. PLoS One 5:e9774. doi: 10.1371/journal.pone.0009774
Geronimus, A. T., Pearson, J. A., Linnenbringer, E., Schulz, A. J., Reyes, A. G., Epel, E. S., et al. (2015). Race-Ethnicity, poverty, urban stressors, and telomere length in a detroit community-based sample. J. Health Soc. Behav. 56, 199–224. doi: 10.1177/0022146515582100
Ghosh, K., and Ghosh, K. (2007). Pathogenesis of anemia in malaria: a concise review. Parasitol. Res. 101, 1463–1469. doi: 10.1007/s00436-007-0742-1
Ghosh, S., Padalia, J., and Moonah, S. (2019). Tissue destruction caused by entamoeba histolytica parasite?: cell death, inflammation, invasion, and the gut microbiome. Curr. Clin. Micro Rpt. 6, 51–57. doi: 10.1007/s40588-019-0113-6
Giraudeau, M., Heidinger, B., Bonneaud, C., and Sepp, T. (2019). Telomere shortening as a mechanism of long-term cost of infectious diseases in natural animal populations. Biol. Lett. 15:20190190. doi: 10.1098/rsbl.2019.0190
Gluckman, P. D., Hanson, M. A., and Beedle, A. S. (2007). Early life events and their consequences for later disease: a life history and evolutionary perspective. Am. J. Hum. Biol. 19, 1–19. doi: 10.1002/ajhb.20590
Hahn, A., and Zuryn, S. (2019). The cellular mitochondrial genome landscape in disease. Trends Cell Biol. 29, 227–240. doi: 10.1016/j.tcb.2018.11.004
Hamilton, W. D., and Zuk, M. (1982). Heritable true fitness and bright birds: a role for parasites? Science 218, 384–387. doi: 10.1126/science.7123238
Hanssen, S. A., Folstad, I., Erikstad, K. E., and Oksanen, A. (2003). Costs of parasites in common eiders: effects of antiparasite treatment. Oikos 100, 105–111. doi: 10.1034/j.1600-0706.2003.12162.x
Hanssen, S. A., Hasselquist, D., Folstad, I., and Erikstad, K. E. (2004). Costs of immunity: immune responsiveness reduces survival in a vertebrate. Proc. R. Soc. B. 271, 925–930.
Hasselquist, D., and Nilsson, J. -Å (2012). Physiological mechanisms mediating costs of immune responses: what can we learn from studies of birds? Animal Behav. 83, 1303–1312. doi: 10.1016/j.anbehav.2012.03.025
Heidinger, B. J., Blount, J. D., Boner, W., Griffiths, K., Metcalfe, N. B., and Monaghan, P. (2012). Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. U. S. A. 109, 1743–1748. doi: 10.1073/pnas.1113306109
Hewitt, G., Jurk, D., Marques, F. D. M., Correia-Melo, C., Hardy, T., Gackowska, A., et al. (2012). Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3:708. doi: 10.1038/ncomms1708
Hoeijmakers, J. H. J. (2009). DNA damage, aging, and cancer. New Eng. J. Med. 361, 1475–1485. doi: 10.1056/NEJMra0804615
Ilmonen, P., Kotrschal, A., and Penn, D. J. (2008). Telomere attrition due to infection. PLoS One 3:e2143. doi: 10.1371/journal.pone.0002143
Ilmonen, P., Taarna, T., and Hasselquist, D. (2000). Experimentally activated immune defence in female pied flycatchers results in reduced breeding success. Proc. R. Soc. B. 267, 665–670. doi: 10.1098/rspb.2000.1053
Kang, Y., Zhang, H., Zhao, Y., Wang, Y., Wang, W., He, Y., et al. (2018). Telomere dysfunction disturbs macrophage mitochondrial metabolism and the NLRP3 inflammasome through the PGC-1α/TNFAIP3 Axis. Cell Rep. 22, 3493–3506. doi: 10.1016/j.celrep.2018.02.071
Kotrschal, A., Ilmonen, P., and Penn, D. J. (2007). Stress impacts telomere dynamics. Biol. Lett. 3, 128–130. doi: 10.1098/rsbl.2006.0594
LaPointe, D. A., Atkinson, C. T., and Samuel, M. D. (2012). Ecology and conservation biology of avian malaria. Ann. N. Y. Acad. Sci. 1249, 211–226. doi: 10.1111/j.1749-6632.2011.06431.x
Lieshout, S. H. J., van, Bretman, A., Newman, C., Buesching, C. D., Macdonald, D. W., et al. (2019). Individual variation in early-life telomere length and survival in a wild mammal. Mol. Ecol. 28, 4152–4165. doi: 10.1111/mec.15212
Lochmiller, R. L., and Deerenberg, C. (2000). Trade-offs in evolutionary immunology: just what is the cost of immunity? Oikos 88, 87–98. doi: 10.1034/j.1600-0706.2000.880110.x
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Martincorena, I., Fowler, J. C., Wabik, A., Lawson, A. R. J., Abascal, F., Hall, M. W. J., et al. (2018). Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917. doi: 10.1126/science.aau3879
Monaghan, P. (2008). Early growth conditions, phenotypic development and environmental change. Philos. Trans. R. Soc. B. 363, 1635–1645. doi: 10.1098/rstb.2007.0011
Monaghan, P. (2014). Organismal stress, telomeres and life histories. J. Exp. Biol. 217, 57–66. doi: 10.1242/jeb.090043
Monaghan, P., Metcalfe, N. B., and Torres, R. (2009). Oxidative stress as a mediator of life history trade-offs: mechanisms, measurements and interpretation. Ecol. Lett. 12, 75–92. doi: 10.1111/j.1461-0248.2008.01258.x
Moskalev, A. A., Shaposhnikov, M. V., Plyusnina, E. N., Zhavoronkov, A., Budovsky, A., Yanai, H., et al. (2013). The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 12, 661–684. doi: 10.1016/j.arr.2012.02.001
Murray, D. L. (2002). Differential body condition and vulnerability to predation in snowshoe hares. J. Animal Ecol. 71, 614–625. doi: 10.1046/j.1365-2656.2002.00632.x
Nelson, G., Wordsworth, J., Wang, C., Jurk, D., Lawless, C., Martin-Ruiz, C., et al. (2012). A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349. doi: 10.1111/j.1474-9726.2012.00795.x
Nettle, D., and Bateson, M. (2017). Detecting telomere elongation in longitudinal datasets: analysis of a proposal by Simons, Stulp and Nakagawa. PeerJ 5:e3265. doi: 10.7717/peerj.3265
Nussey, D. H., Watt, K., Pilkington, J. G., Zamoyska, R., and McNeilly, T. N. (2012). Age-related variation in immunity in a wild mammal population. Aging Cell 11, 178–180. doi: 10.1111/j.1474-9726.2011.00771.x
Öhlin, B., Nilsson, P. M., Nilsson, J. -Å, and Berglund, G. (2004). Chronic psychosocial stress predicts long-term cardiovascular morbidity and mortality in middle-aged men. Eur. Heart J. 25, 867–873. doi: 10.1016/j.ehj.2004.03.003
Palacios, M. G., Cunnick, J. E., Winkler, D. W., and Vleck, C. M. (2007). Immunosenescence in some but not all immune components in a free-living vertebrate, the tree swallow. Proc. R. Soc. B. 274, 951–957. doi: 10.1098/rspb.2006.0192
Parker, H., and Holm, H. (1990). Patterns of nutrient and energy expenditure in female common eiders nesting in the high arctic. Auk 107, 660–668. doi: 10.2307/4087996
Råberg, L., Grahn, M., Hasselquist, D., and Svensson, E. (1998). On the adaptive significance of stress-induced immunosuppression. Proc. R. Soc. B. 265, 1637–1641. doi: 10.1098/rspb.1998.0482
Råberg, L., Nilsson, J. -Å, Ilmonen, P., Stjernman, M., and Hasselquist, D. (2000). The cost of an immune response: vaccination reduces parental effort. Ecol. Lett. 3, 382–386. doi: 10.1046/j.1461-0248.2000.00154.x
Ragu, S., Faye, G., Iraqui, I., Masurel-Heneman, A., Kolodner, R. D., and Huang, M.-E. (2007). Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc. Natl. Acad. Sci. U.S.A. 104, 9747–9752. doi: 10.1073/pnas.0703192104
Reichert, S., Bize, P., Arrivé, M., Zahn, S., Massemin, S., and Criscuolo, F. (2014). Experimental increase in telomere length leads to faster feather regeneration. Exp. Gerontol. 52, 36–38. doi: 10.1016/j.exger.2014.01.019
Rossi, D. J., Bryder, D., Seita, J., Nussenzweig, A., Hoeijmakers, J., and Weissman, I. L. (2007). Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729. doi: 10.1038/nature05862
Rossi, D. J., Jamieson, C. H. M., and Weissman, I. L. (2008). Stems cells and the pathways to aging and cancer. Cell 132, 681–696. doi: 10.1016/j.cell.2008.01.036
Sakai, A., Nakanishi, M., Yoshiyama, K., and Maki, H. (2006). Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli. Genes Cells 11, 767–778. doi: 10.1111/j.1365-2443.2006.00982.x
Sapolsky, R. M. (1999). Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp. Gerontol. 34, 721–732. doi: 10.1016/S0531-5565(99)00047-9
Shay, J. W., and Wright, W. E. (2019). Telomeres and telomerase: three decades of progress. Nat. Rev. Genet. 20, 299–309. doi: 10.1038/s41576-019-0099-1
Sheldon, B. C., and Verhulst, S. (1996). Ecological immunology: costly parasite defences and trade-offs in evolutionary ecology. Trends Ecol. Evol. 11, 317–321. doi: 10.1016/0169-5347(96)10039-2
Steptoe, A., and Kivimäki, M. (2012). Stress and cardiovascular disease. Nat. Rev. Cardiol. 9, 360–370. doi: 10.1038/nrcardio.2012.45
Svensson, E., Råberg, L., Koch, C., and Hasselquist, D. (1998). Energetic stress, immunosuppression and the costs of an antibody response. Funct. Ecol. 12, 912–919. doi: 10.1046/j.1365-2435.1998.00271.x
Vijg, J. (2007). Aging of the Genome: The Dual Role of DNA in Life and Death, Aging of the Genome. Oxford: Oxford University Press.
von Schantz, T., Bensch, S., Grahn, M., Hasselquist, D., and Wittzell, H. (1999). Good genes, oxidative stress and condition-dependent sexual signals. Proc. R. Soc. B. 266, 1–12.
von Zglinicki, T. (2001). Stress, DNA damage and ageing — an integrative approach. Exp. Gerontol. 36, 1049–1062. doi: 10.1016/S0531-5565(01)00111-5
von Zglinicki, T. (2002). Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344. doi: 10.1016/s0968-0004(02)02110-2
Watson, H., Bolton, M., and Monaghan, P. (2015). Variation in early-life telomere dynamics in a long-lived bird: links to environmental conditions and survival. J. Exp. Biol. 218, 668–674. doi: 10.1242/jeb.104265
Yoccoz, N., Erikstad, K., Bustnes, J., Hanssen, S., and Tveraa, T. (2002). Costs of reproduction in common eiders (Somateria mollissima): an assessment of relationships between reproductive effort and future survival and reproduction based on observational and experimental studies. J. Appl. Statist. 29, 57–64.
Keywords: hidden costs, benign parasites, mild stressors and diseases, premature aging, delayed long-term effects, telomeres, critical point in telomere length
Citation: Hasselquist D and Tobler M (2021) The Accumulating Costs Hypothesis—to Better Understand Delayed “Hidden” Costs of Seemingly Mild Disease and Other Moderate Stressors. Front. Ecol. Evol. 9:685057. doi: 10.3389/fevo.2021.685057
Received: 24 March 2021; Accepted: 05 July 2021;
Published: 23 July 2021.
Edited by:
Andrew James Jonathan MacIntosh, Kyoto University, JapanReviewed by:
Steve Smith, University of Veterinary Medicine Vienna, AustriaMichaël Beaulieu, German Oceanographic Museum, Germany
Copyright © 2021 Hasselquist and Tobler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dennis Hasselquist, RGVubmlzLmhhc3NlbHF1aXN0QGJpb2wubHUuc2U=