 Madeleine E. Aase-Remedios
Madeleine E. Aase-Remedios David E. K. Ferrier
David E. K. Ferrier- The Scottish Oceans Institute, Gatty Marine Laboratory, School of Biology, University of St Andrews, St Andrews, United Kingdom
Comparative approaches to understanding chordate genomes have uncovered a significant role for gene duplications, including whole genome duplications (WGDs), giving rise to and expanding gene families. In developmental biology, gene families created and expanded by both tandem and WGDs are paramount. These genes, often involved in transcription and signalling, are candidates for underpinning major evolutionary transitions because they are particularly prone to retention and subfunctionalisation, neofunctionalisation, or specialisation following duplication. Under the subfunctionalisation model, duplication lays the foundation for the diversification of paralogues, especially in the context of gene regulation. Tandemly duplicated paralogues reside in the same regulatory environment, which may constrain them and result in a gene cluster with closely linked but subtly different expression patterns and functions. Ohnologues (WGD paralogues) often diversify by partitioning their expression domains between retained paralogues, amidst the many changes in the genome during rediploidisation, including chromosomal rearrangements and extensive gene losses. The patterns of these retentions and losses are still not fully understood, nor is the full extent of the impact of gene duplication on chordate evolution. The growing number of sequencing projects, genomic resources, transcriptomics, and improvements to genome assemblies for diverse chordates from non-model and under-sampled lineages like the coelacanth, as well as key lineages, such as amphioxus and lamprey, has allowed more informative comparisons within developmental gene families as well as revealing the extent of conserved synteny across whole genomes. This influx of data provides the tools necessary for phylogenetically informed comparative genomics, which will bring us closer to understanding the evolution of chordate body plan diversity and the changes underpinning the origin and diversification of vertebrates.
Introduction
The genetic basis of development is of fundamental interest to understanding animal evolution. In the course of the evolution of chordates, gene and genome duplications have played important roles in shaping gene families by providing the means with which genetic novelties can arise (Ohno, 1970). Whole genome duplications (WGDs) have occurred at the base of speciose and diverse lineages, including the vertebrate two rounds of whole genome duplication (2R WGD) (Meyer and Schartl, 1999; Dehal and Boore, 2005), or the teleost-specific 3R WGD (Hoegg et al., 2004; Meyer and Van de Peer, 2005), and have been implicated in the evolution of more complex, novel, or diverse clades. The 2R WGD duplicated the genome of the vertebrate ancestor twice, such that many genes were retained in as many as four copies, though the majority of paralogues were lost (Dehal and Boore, 2005). The paralogues that were retained in duplicate, ohnologues, are of particular importance to understanding chordate evolution because they may be candidate genes underpinning the evolutionary transition to the vertebrates, and many of these genes retained in duplicate have roles in development (Brunet et al., 2006). While the 2R WGD certainly affected the evolution and subsequent radiation of the vertebrates, duplications also occur frequently on much smaller scales than polyploidisation, and small-scale duplications of developmental genes have also played a role in chordate evolution. Our current understanding of how gene and genome duplications have shaped chordate lineages has progressed rapidly due to the advancements in comparative genomics and with the availability of new genome and transcriptome sequences from phylogenetically informative lineages like amphioxus, lampreys, coelacanth, and monotremes.
Addressing the Origin of Vertebrates
Within the chordates, there are three extant subphyla, the cephalochordates, the tunicates/urochordates, and vertebrates, the vertebrates being the most familiar and speciose of the three, which in turn includes cyclostomes (lampreys and hagfish), actinopterygians (ray-finned fish), amphibians, sauropsids (birds and reptiles), and mammals (Figure 1). The three chordate lineages share common traits including a hollow neural tube located dorsal to a notochord, pharyngeal slits, and a post-anal tail, but the vertebrate lineage is far more speciose and diverse than either invertebrate chordate sub-phylum. The field of developmental biology has spent decades determining the genetic basis for the construction of the morphologies of a select few model species, including a handful of vertebrates (like mouse, frog, chick, and zebrafish), but in our current age of genomics, our understanding is growing at an unprecedented pace, with much broader taxon-sampling now making possible a more “phylogenetically informed” and hence evolutionarily robust era of comparative biology and evolutionary developmental biology.
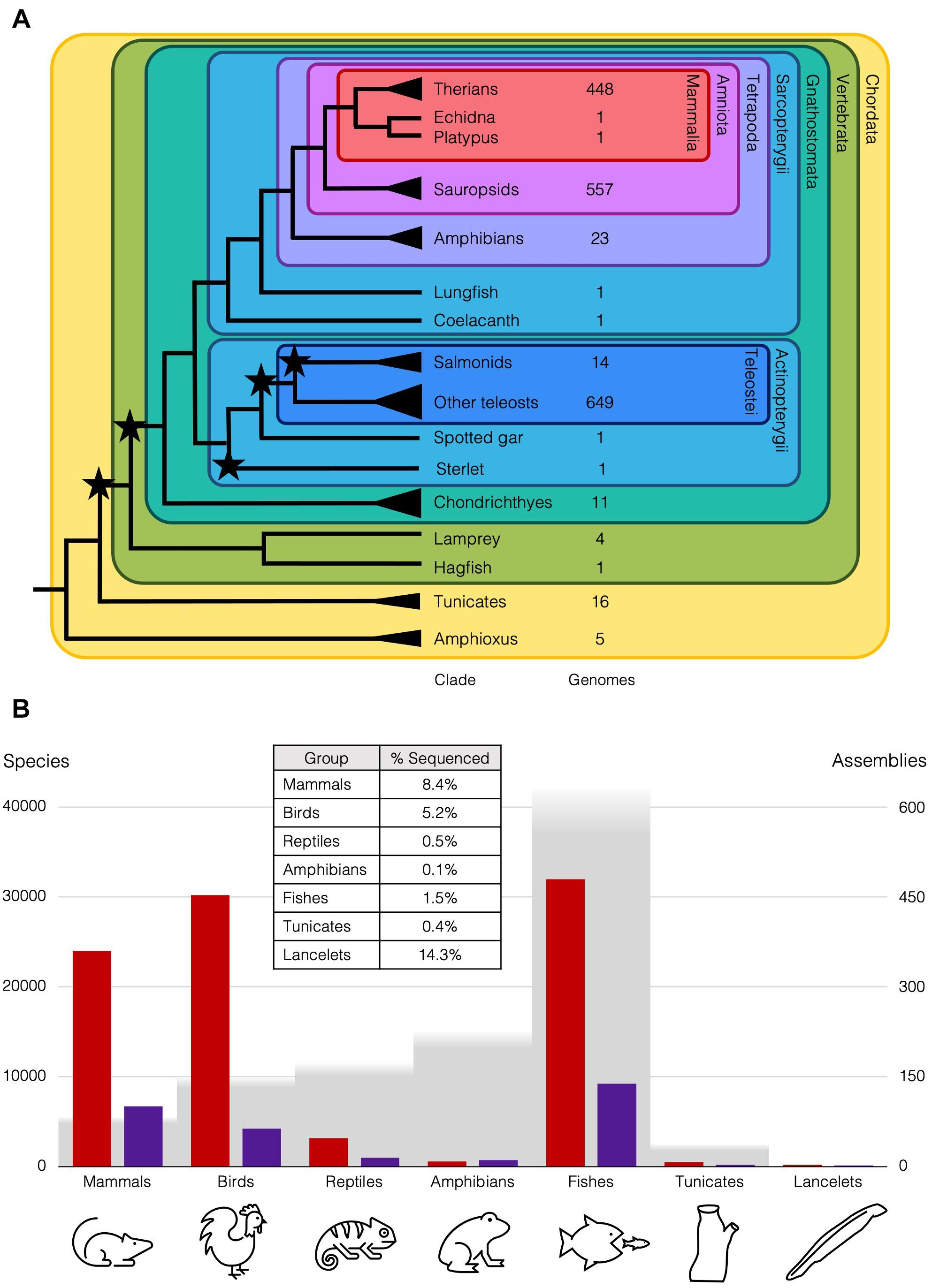
Figure 1. Diversity of chordates and available genome assemblies. (A) Cladogram of chordate lineages and count of representative genome assemblies available on NCBI (https://www.ncbi.nlm.nih.gov/assembly/organism; Accessed 25 April 2021). Branches with single species mentioned in the text end with a line, but lineages with multiple species end with a triangle, e.g., there are ∼35 amphioxus species (Bertrand and Escriva, 2011). Stars represent WGDs. NCBI Taxonomic codes used to count genomes for each lineage: Therians (taxid:32525); Sauropsids (taxid:8457); Amphibians (taxid:8292); Salmonids (taxid:8015); Teleosts (taxid:32443); Cartilaginous fishes (taxid:7777); Tunicates (taxid:7712); Cephalochordata (taxid:7735). (B) Genome assemblies at scaffold (red) and chromosome (purple) level available on NCBI for animal subgroups (https://www.ncbi.nlm.nih.gov/genome/browse#!/eukaryotes/; Accessed 25th April 2021), right axis, relative to approximate number of species (grey), left axis. Table inset is approximate percentage of species in each group that has been sequenced at least to the draft level.
Invertebrate Chordate Models
One of the major questions facing evolutionary developmental biology is the origin of the vertebrates. For a comparison, sister to the vertebrates is the tunicate lineage, the two lineages together making up the clade Olfactores (Delsuc et al., 2006). Tunicates generally have two life stages, comprising the tadpole larval stage and the sessile filter-feeding adult in which chordate-like larval traits such as the notochord, tail, and head are absent or highly reorganised (Lemaire, 2011). Tunicate models such as Ciona spp. have been used for decades, representing a growing basis of comparative genomics and transcriptomics studies in chordate developmental biology. Tunicate embryos have been used as a model for the chordate nervous system, as well as for key chordate traits such as the notochord and the precursors of the vertebrate neural crest cells (Schubert et al., 2006; Kugler et al., 2011; Holland, 2016). Neural crest cells are characteristic of vertebrates and constitute a population which migrates and differentiates to contribute to the vertebrate peripheral nervous system, pigmented cells, and elements of the skeletal system, thus the origin of these cells is of particular importance to understanding the invertebrate–vertebrate transition (Gans and Northcutt, 1983). While invertebrate chordates are considered to lack a true neural crest, they do have cells with some similar properties. These include migratory cell types emerging from the neural plate boundary and differentiating into neurons or pigment cells but, critically, the skeletogenic role is lacking from these tunicate cells (Jeffery et al., 2004; Jeffery, 2006; Stolfi et al., 2015). Tunicates have also been instrumental in understanding the control of gene expression because of their relatively simple body plan and a small number of transcription factors compared to vertebrate models, and they have greater extents of homology to vertebrates than other comparisons outside of the phylum Chordata (Lemaire, 2011). There is a growing database of genomic resources for ascidians (ANISEED), which in recent years has acquired datasets focussing on gene expression, including RNA-seq and epigenetic datasets, as well as many new genome annotations (Brozovic et al., 2018), reflecting the utility of tunicate models for molecular biology. The most recent update to this database includes the addition of a larvacean, Oikopleura dioica, and the expansion of tools to explore gene expression data in larvae (Dardaillon et al., 2019).
Tunicate genomes, while small, evolve very quickly and have undergone extensive rearrangements relative to the other chordate subphyla (Denoeud et al., 2010; Holland, 2016). At first, this made genomic comparisons between tunicates and vertebrates confusing, and more than a fifth of Ciona robusta (previously C. intestinalis type A) genes had no clear homology to known bilaterian genes when the genome was first sequenced (Cañestro et al., 2003). Gene families typically conserved in clusters in the genomes of other organisms are often disorganised in tunicate genomes, e.g., the Hox cluster (Sekigami et al., 2017). There is also an issue with long-branch attraction in molecular phylogenies with the highly divergent C. robusta sequences, but the sequencing of a second clade of tunicates, the larvacean O. dioica to complement the ascidian C. robusta, helped resolve this (Delsuc et al., 2006). Now genomes are available for several different tunicate lineages, including a chromosomal level assembly of O. dioica (Bliznina et al., 2021). Sequencing of two C. intestinalis Type B (i.e., C. intestinalis rather than C. robusta) genomes revealed that between and even within Ciona species, there have been many chromosomal inversions and there are high levels of polymorphism (Satou et al., 2021). Because of their fast rates of evolution, between tunicate and vertebrate genomes there is little conserved synteny remaining, making inferences about the genomic changes at the origin of the vertebrates difficult from these comparisons.
2R and Amphioxus
It is now widely understood that 2R WGD occurred in the ancestors leading to vertebrates, and these events have been implicated as playing a permissive role in the evolution of vertebrate novelties. Still, the specifics of how the 2R WGD provided the genetic basis for these traits is not yet fully understood. Much of the evidence comes from comparative studies with amphioxus, the basal chordate lineage. Considered the “archetypal” chordate, both its genome and morphology are thought to have not changed much in the nearly 600 million years since it diverged, in contrast to the fast-evolving tunicates (Holland et al., 2008; Putnam et al., 2008; Zhang et al., 2018), making it an ideal model for inferring features of the chordate ancestor (Dehal and Boore, 2005; Schubert et al., 2006; Bertrand and Escriva, 2011). While amphioxus lacks a true neural crest, as do tunicates (Holland and Holland, 2001), the draft genome of Branchiostoma floridae contains most pro-orthologues of the genes that constitute the regulatory network required for vertebrate neural crest formation (Yu et al., 2008). This suggests that the ancestral chordate may have possessed the components of a neural crest rudiment, but it was the 2R WGD at the origin of the vertebrates that may have facilitated the evolution of the true neural crest.
With the sequencing of an amphioxus genome came the most solid evidence for the 2R WGD, revealing the prevalent one-to-four syntenic relationship between the scaffolds of B. floridae and the human genome (Putnam et al., 2008). This amphioxus genome was highly polymorphic, thus each haplotype assembled separately, which complicated these analyses. More recently, the application of newer techniques like long-read sequencing and Hi-C has allowed the genome to be assembled to the chromosome level, revealing the depth of synteny conserved amongst chordate lineages (Simakov et al., 2020). Advancements in long-read sequencing reduce the assembly errors that can arise in highly polymorphic genomes that can cause false gene gains to be inferred for alleles rather than paralogues (Denton et al., 2014). With this chromosome-level assembly, the authors were able to precisely determine the sequence and timing of polyploidisations, and the chromosomal rearrangements that occurred after each one in the process of rediploidisation characterised by chromosomal fusions and rampant gene losses (Simakov et al., 2020).
Cyclostomes—the Agnathan Lampreys and Hagfish
This recent study also took a comparative approach to determine the timing of 2R WGD in vertebrate ancestors, with particular focus on the placement of the divergence of the cyclostome lineage. The cyclostomes (lampreys and hagfish) diverged from the gnathostomes 500 Mya, and provide a phylogenetically well-placed point of comparison to the jawed vertebrates (Janvier, 2007). They share a cartilaginous skeleton including a cranium, paired sensory organs, and true neural crest, but lack key gnathostome structures like a jaw and paired fins (Green and Bronner, 2014). An initial lamprey genome assembly from a sample of liver tissue was later updated with a germline assembly, since hundreds of genes are eliminated from somatic tissues, though both assemblies revealed a high number of rearrangements and repeats (Smith et al., 2013, 2018). These complicated genomic characteristics have hindered the genome assembly and contributed to uncertainty about the number of duplications in the cyclostome lineage, and how many, if any, are shared with the gnathostomes.
The Timing of 2R
One potential marker of genome duplications is the Hox cluster. This cluster of developmental transcription factor genes is highly conserved across animals, so that comparison of the single cluster in amphioxus to the four in mammals gave an early line of evidence for 2R (Garcia-Fernàndez and Holland, 1994) and was complemented by the discovery of the teleost-specific 3R and the seven (i.e., eight minus one) Hox clusters in zebrafish (Amores, 1998). Lampreys were reported to have at least six Hox clusters, and the updated germline assembly of Petromyzon marinus revealed the synteny of these regions shared with other vertebrates’ Hox chromosomes (Mehta et al., 2013; Smith et al., 2018). Amongst these six, two pairs appear to be the product of more recent, chromosome-level duplications, suggesting there may be a high prevalence of large-scale duplications occurring more recently besides the WGDs in lampreys. Smith and Keinath (2015) had previously constructed the lamprey meiotic map using RAD-seq, and with these syntenic comparisons, concluded that fewer chromosomal rearrangements were required if lampreys diverged after 1R WGD, but before 2R.
These data are intriguing, and indeed several different scenarios of duplications could result in the six Hox clusters, among other unexpected or unique patterns of synteny. Lampreys could have undergone the 2R WGD and several independent chromosomal duplications and losses or diverged after 1R then undergone their own WGD or several chromosome-level duplications (Figure 2). If lampreys and gnathostomes share 2R, the lack of direct orthology between lamprey Hox clusters and those of gnathostomes can be explained by cyclostome divergence during rediploidisation, before the ancestral gnathostome karyotype was established. This pattern, where orthology is obscured because lineages retained different paralogues when they diverged, termed tetralogy, has been described for early-branching 3R teleosts, and may apply here as well (Martin and Holland, 2014). Many of the paralogues in lamprey are younger than 2R ohnologues, suggesting some independent duplications have occurred more recently than either of the 2R WGD (Mehta et al., 2013). Some phylogenies suggest that cyclostomes and gnathostomes share both WGDs (Kuraku et al., 2009), while others support independent duplications (Force et al., 2002) or that cyclostomes diverged before either WGD (Fried et al., 2003), though these early conclusions may have been misled by long-branch attraction and hidden paralogy due to the divergent sequences and independent gene gains and losses in cyclostome genomes. Furthermore, with only one lamprey genome assembled as of 2018, the rigour of any of these conclusions was tenuous (Holland and Ocampo Daza, 2018). Because of this, the extent to which lamprey duplications may be shared with the other cyclostome lineage or if one or both of the 2R WGD occurred in the vertebrate ancestor has been contentious. Previous synteny evidence suggested that all vertebrates share at least 1R, and likely the 2R as well (Holland and Ocampo Daza, 2018; Sacerdot et al., 2018). However, with the new chromosome-level assembly of B. floridae, the distinct chromosomal fusion events that occurred between the first and second round of WGD could be discerned. Simakov et al. (2020) provide evidence that lampreys diverged after only 1R, as certain genomic rearrangements predating the 2R are unique to jawed vertebrates and lacking from lampreys, in particular the gnathostome-specific CLGE-CLGO fusion, and the observation that lamprey syntenic blocks are generally unmixed. This new assembly improves upon the extensive synteny work aimed at determining the origin of chordate karyotypes from previous studies (Nakatani et al., 2007; Putnam et al., 2008; Sacerdot et al., 2018) and provides the latest working hypothesis for the timing of the 2R WGD and their impact on the evolution of vertebrates.
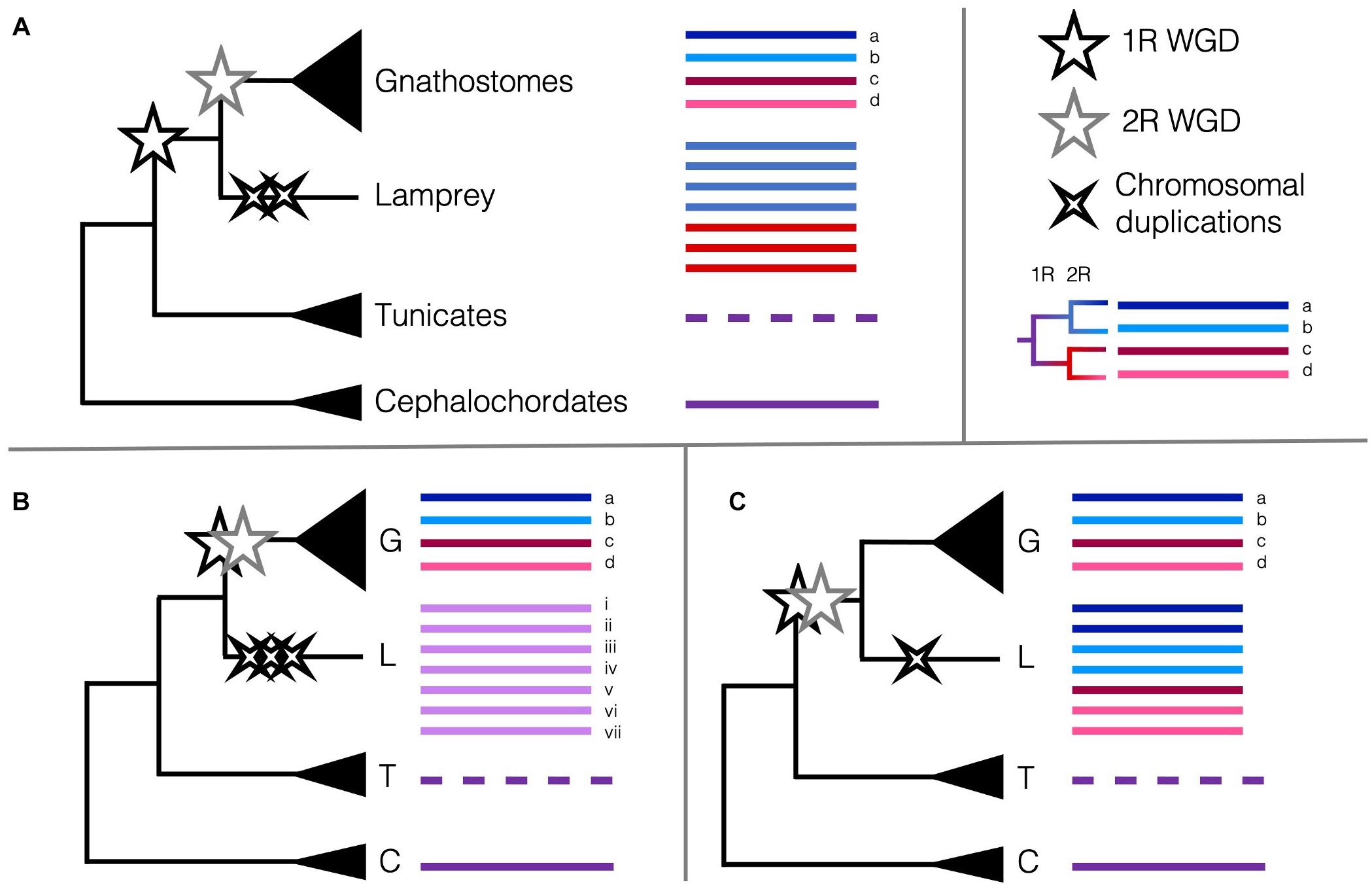
Figure 2. Scenarios of the timing of the 2R WGD relative to the divergence of cyclostomes and the Hox cluster as a marker of WGDs. Pre-duplicate chordate phyla with single Hox clusters are coloured to represent the ancestral state (purple lines). The reorganised Hox cluster in tunicates is represented by a dashed line. (A) The current consensus that cyclostomes diverged from the gnathostomes after 1R (black star) but before 2R (grey star) and underwent some independent chromosome-level duplications (black crosses) (Smith et al., 2018; Simakov et al., 2020). Remnants of 1R paralogy (red versus blue) may distinguish Hox clusters in the cyclostomes. (B) Alternate hypothesis that cyclostomes diverged before 1R and only underwent several chromosome-level duplications (Fried et al., 2003; Furlong et al., 2007) (or perhaps an independent WGD). (C) Alternate hypothesis that cyclostomes diverged after 2R and underwent few independent chromosome-level duplications (Caputo Barucchi et al., 2013; Sacerdot et al., 2018). Remnants of 1R paralogy and 2R paralogy (dark red versus pink, dark blue versus light blue) may distinguish Hox clusters in the cyclostomes. Cyclostome Hox cluster numbering and colouring does not represent actual homology [(Smith et al., 2018) found homology between lamprey Hox clusters i. and iv., and ii. and v.]; no conclusive affinities have been found between cyclostome Hox clusters and any particular gnathostome Hox cluster.
Cyclostome Peculiarities
Genomes of amphioxus and lamprey have been instrumental in understanding the origin of the vertebrates as these lineages are phylogenetic outgroups to larger chordate lineages. While amphioxus is remarkably conserved, making inferences about the chordate ancestor fairly straightforward, the lamprey genome is much more complicated. These complications, however, are interesting themselves, especially the programmed genomic rearrangements (PGRs) in the somatic tissues, presumably an adaptation reflecting the genetic conflict between germline and soma (Smith et al., 2012; Timoshevskiy et al., 2017). The hagfish genome also undergoes PGR (Nakai et al., 1991, 1995), and further resolution of genomes in this cyclostome clade should be instrumental in deciphering the mechanism of PGR. In addition, comparisons to determine homology of structures and their genetic underpinnings depend on reliable gene annotations. For instance, while amphioxus lacks myelin, lampreys were reported to have it (Smith et al., 2013), though lamprey neurons are not myelinated. In fact, the genes annotated as myelin in lamprey are likely to be in another gene family altogether (Werner, 2013). This highlights the pitfalls of automated gene annotations and the need for careful gene curation and annotation with a focus on phylogenetically informed and robust gene nomenclature.
More recently, the genome sequence of a Reissner’s lamprey (Lethenteron reissneri) was assembled to higher completeness than any previous lamprey assemblies, aided by the combinatorial use of short- and long-read sequencing, and Hi-C (Zhu et al., 2021). The protein-coding genes of this genome were annotated using a wider and more phylogenetically informed array of species, including several genomes only recently sequenced from both invertebrates and vertebrates, revealing a total of seven Hox clusters (Figure 2). This new assembly, as well as the improved sea lamprey genome (Smith et al., 2018) show that the independent duplications indicated by the seven Hox clusters are likely synapomorphic to at least the lampreys, but maybe to the cyclostomes as a whole. Comparisons of lamprey and hagfish Hox clusters revealed that cyclostomes share the loss of Hox genes from paralogy group 12 and likely independent duplications, as hagfish may have six or more Hox clusters as well (Pascual-Anaya et al., 2018). These data are taking us closer to finally determining the cyclostomes’ placement in relation to the 2R WGD. The sequencing of multiple cyclostomes, particularly from hagfishes, would build on current data in order to enable further understanding of the role 2R WGD played in chordate evolution, and particularly, the evolution of vertebrate- or gnathostome-specific developmental novelties.
Duplications in the Hox Cluster
As mentioned above, the Hox gene cluster has been a fruitful system for determining how gene duplication has affected animal development. Not only have multiple ohnologous clusters been maintained following WGDs, including 2R as well as the teleost-specific 3R and others (Hoegg and Meyer, 2005), but the cluster (and others) arose via extensive tandem duplications of a single ANTP-class transcription factor gene early in animal evolution (Butts et al., 2008; Holland, 2013; Ferrier, 2016). Furthermore, there tends to be a link between the expression of the Hox genes and their clustered arrangement, as genes at one end of the cluster are expressed anteriorly, and expression follows successively toward the other end of the cluster, where those genes are expressed in the posterior of the embryo (Duboule and Dollé, 1989; Graham et al., 1989; Duboule, 2007; Tschopp et al., 2009). This spatial collinearity is frequently observed alongside temporal collinearity, where the activation of the genes follows the same sequence of early to late as anterior to posterior in many animals (Duboule, 1994; Kmita and Duboule, 2003; Iimura and Pourquié, 2006; Monteiro and Ferrier, 2006; Gaunt, 2018; Krumlauf, 2018; Ferrier, 2019). Hox genes are transcription factors, and their expression in the developing embryo is strictly regulated so as to provide positional identity along the anterior–posterior axis of the animal. Changes to the expression of Hox genes therefore changes rostro-caudal identity, most obviously manifested in vertebrates in the formation of the axial skeleton (Casaca et al., 2014). Between different vertebrate groups, differential expression of Hox genes drives the different identities of vertebral segments, illustrating how the Hox gene diversification directly underpins the diversity of vertebrate body-plans (Krumlauf, 1994; Burke et al., 1995). The Hox cluster perfectly illustrates the interplay between gene regulation, expression, duplication, and subfunctionalisation, and the effects of these processes on development and animal evolution.
Hox Clusters Through WGDs
Other studies have turned to the Hox cluster in order to understand the impact of the 3R WGD. Early teleost genome projects revealed excess Hox clusters in zebrafish, medaka, and two pufferfish (Hoegg and Meyer, 2005). In these, as above, the genome sequence and transcriptomics of an appropriate outgroup was essential. PCR screening of the birchir, a non-teleost ray-finned fish, revealed only four Hox clusters, which allowed the timing of the 3R WGD to be determined (Ledje et al., 2002). Another more basally branching fish lineage, the spotted gar, also has four Hox clusters (Braasch et al., 2016) while zebrafish has seven (Amores, 1998). As many as eight Hox clusters can be found in different teleost lineages, though some have been lost from different lineages (Sato and Nishida, 2010; Martin and Holland, 2017). Detection of the 3R WGD has led to speculation that WGDs may play a permissive role in the evolution of diverse lineages, as within the more than 30,000 actinopterygian species, only 50 are non-teleosts which did not undergo 3R (Near et al., 2012). The link of WGDs and subsequent species diversity has been posited for vertebrates as well (Cañestro et al., 2013), but evidence for this link is still lacking (Glasauer and Neuhauss, 2014).
A more recent WGD allows the study of rediploidisation as it is occurring. Salmonids have thirteen Hox clusters (Mungpakdee et al., 2008), indicative that this lineage has undergone a fourth whole genome duplication (4R WGD) more recently, around 88 Mya (Figure 1; Macqueen and Johnston, 2014; Lien et al., 2016). The Atlantic salmon (Salmo salar) genome was particularly difficult to assemble because of the repetitiveness created by the fourth more recent WGD, but its assembly enables our understanding of the processes of rediploidisation (Lien et al., 2016). Many of the paralogues created in the 4R WGD were able to be identified, even those that were recently pseudogenised (Lien et al., 2016). In another salmonid, the rainbow trout (Oncorhynchus mykiss), nearly half of the 4R ohnologues remain in duplicate (Berthelot et al., 2014). Many of the 4R ohnologue pairs in salmon consist of one gene that maintained the ancestral function, as conserved between the salmon and pike, while the second had a different expression pattern (Lien et al., 2016; Robertson et al., 2017). Genomes of 4R salmonids also enable detection of a category of genes retained following successive WGDs, which is enriched in genes involved in development and transcription factors (Berthelot et al., 2014). This was associated with changes to the ecology of salmonids, but did not directly cause rapid species diversification as was hypothesised for the 3R in teleosts, rather, it is correlated with the adaptation of anadromy (Macqueen and Johnston, 2014). These observations suggest that environmental adaptation may have had a larger impact on teleost diversity than the 3R WGD because there are significant discrepancies between the timing of 3R and the divergence of major teleost lineages (Donoghue and Purnell, 2005; Santini et al., 2009; Near et al., 2012), as well as 40–50 million years between the salmonid 4R WGD and their subsequent radiation (Macqueen and Johnston, 2014). The salmonid clade is still within the process of rediploidisation, and comparisons between lineages reveals that a quarter of the Atlantic salmon genome underwent rediploidisation independently of the trout, suggesting that rediploidisation occurred before and after speciation (Robertson et al., 2017). These differently resolved regions may underpin the differences in ecology amongst salmonid lineages and also help to explain the (perhaps surprising) length of time that is required post-WGD before species diversification might be observable (Robertson et al., 2017). The differences between salmon and trout karyotypes also provide support for tetralogy, the pattern of homology between genes in lineages that diverged during rediploidisation (Martin and Holland, 2014). These assemblies of salmon and trout revealed the processes of rediploidisation and provided a case study for examining the fate of recently duplicated genes in the context of a WGD.
The Fate of Duplicated Genes
Several mechanisms have been proposed for the maintenance of duplicated genes, despite the likelihood of non-functionalisation of redundant paralogues (Nakatani et al., 2007). For some gene types, increased copy number is advantageous, despite their redundancy, such as ribosomal RNAs (Zhang, 2003). Alternatively, once a dosage-sensitive network of genes is duplicated in entirety, perhaps along with the entire genome following a WGD, to preserve the balance of the network components, no loss can occur, therefore entire duplicate networks are retained (Lynch and Conery, 2000). This dosage compensation theory, however, does not result in as high an incidence of retained paralogues being related to dosage-sensitive complexes and pathways as one might expect; instead, this model might better explain a stop-gap mechanism that prevents initial loss and allows time for sub- or neo-functionalisation processes to occur (Hughes et al., 2007).
While duplicated genes are likely to be redundant initially, this redundancy presumably reduces selective constraint and allows one or both daughter genes to evolve, which explains how duplication can facilitate evolutionary novelties. Under the duplication-degeneration-complementation (DDC) model, genes undergo degenerative mutations following their duplication, resulting in the complementary partitioning of the ancestral gene’s functions among the daughters (Force et al., 1999). This mechanism of subfunctionalisation is particularly relevant to developmental genes, which have complex cis-regulatory regions, since degenerative mutations between the regulatory regions of the daughter genes can quickly partition their expression profiles and therefore their functions. One step further is neofunctionalisation, which suggests that the reduced constraint on paralogues allows for novel functions to evolve in one or both of the paralogues, including new upstream transcription binding sites or even protein domains [sometimes referred to as the duplication-degeneration-innovation (DDI) model] (Jimenez-Delgado et al., 2009). The role of DDC or DDI in the evolution of developmental genes is supported by the overrepresentation of transcription factors among retained paralogues, the often complicated cis-regulatory regions these transcription factors have, and the fact that expression rather than sequence changes are the primary observed differences between different animals’ developmental genes.
Evidence of Subfunctionalisation of 3R Paralogues
Evidence supporting the role of DDC, or DDI, in chordate evolution is growing. Large RNA-seq datasets have been instrumental in detecting subfunctionalised ohnologues, for instance the reduced expression of genes retained in duplicate compared to single orthologues in mammals (Qian et al., 2010). One of the largest datasets of expression data of paralogues and their pro-orthologues in another species comes from the gar genome project. The spotted gar is a ray-finned fish whose lineage diverged from the teleosts before the 3R WGD that characterises this latter clade (Figure 1; Braasch et al., 2016). This makes it an ideal outgroup to understand the impact of the 3R WGD on teleost evolution that has been implicated in possibly permitting the huge diversity of fishes (Hoegg et al., 2004; Sato and Nishida, 2010). This model suggests that speciation occurs following WGD, but before the genomes undergo rediploidisation, so that changes to genome architecture like chromosomal fusions reinforce speciation (Taylor et al., 2001). Because it has not undergone the 3R, but still lies within Actinopterygii, the spotted gar possesses pro-orthologues of the duplicated 3R ohnologues. Thus, expression of gar genes is expected to be similar to that of the ancestral gene. Between the gar and teleost transcriptomes, each of the two teleost 3R ohnologues were expressed at lower levels than the gar pro-orthologue, but the pooled expression of the ohnologue pair was similar to the gar gene’s expression (Braasch et al., 2016). This pattern suggests that following 3R, many teleost ohnologues underwent subfunctionalisation in accordance with the predictions of the DDC model, and for certain gene families, subfunctionalisation amongst paralogues has been traced to specific regulatory regions.
These inter-species comparisons are giving us concrete examples of the DDC process. For example, comparisons between mammals and zebrafish revealed that in the gene-poor region around the Pax6 locus, there are cross-functional conserved non-coding elements (CNEs) that have been retained in a patchwork pattern between the zebrafish Pax6 paralogues relative to the single mouse Pax6 (Navratilova et al., 2009). The zebrafish, a 3R teleost, has two Pax6 paralogues: pax6a and pax6b, which are expressed in overlapping yet complementarily constricted patterns relative to tetrapod Pax6; pax6a is expressed widely in the eye and brain, while pax6b is found in the eye, a restricted section of the developing brain, and the pancreas (Kleinjan et al., 2008). The changes to expression of the two ohnologues was traced to the differential loss of certain cis-regulatory regions around the two genes; brain-specific elements from pax6b and the pancreas control element from pax6a (Kleinjan et al., 2008). This pattern of retention of the two Pax6 ohnologues in zebrafish is consistent with the DDC framework of subfunctionalisation, illustrating the role of cis-regulatory elements in this process.
Specialisation of Paralogues
Among daughter paralogues, subfunctionalisation processes are not necessarily symmetrical. Specialisation results in one paralogue with a function more similar to that of the ancestral gene, while the other paralogue’s function diverges, either to a small subset of the ancestral function, or even the adaptation of new functions (Farrè and Albà, 2010; Marlétaz et al., 2018). Specialisation of ohnologues has been detected often in the past but has been described by several different mechanisms, corresponding to the different mechanisms of paralogue retention following duplication. Previous studies focusing on protein sequence evolution and detecting neofunctionalisation have found examples of specialisation in protein function (Chain and Evans, 2006; Steinke et al., 2006; Sémon and Wolfe, 2008). This takes place in accordance with the process described as qualitative subfunctionalisation, driven by adaptive evolution of the paralogue’s protein sequence (Espinosa-Cantú et al., 2015). For many developmental genes, however, the DDC hypothesis, which falls under quantitative subfunctionalisation and occurs by neutral processes affecting gene regulation, is more relevant (Espinosa-Cantú et al., 2015; Braasch et al., 2018). Following duplication, for some gene families, one paralogue retains widespread expression across several domains consistent with the inferred ancestral function, while the other’s function is reduced to a specialised domain. This process is linked to the changes to regulatory domains occurring primarily on one paralogue instead of symmetrically across both (during the degeneration and complementation aspects of the DDC), though this was detected for the most part in genes with complex or widespread ancestral expression patterns (Marlétaz et al., 2018). Some studies of expressed sequence tag (EST) data between the polyploid frog Xenopus laevis and the non-duplicate species X. tropicalis found that many paralogues exhibited subfunctionalisation, but others had patterns where only one of the two paralogues was greatly reduced in expression (Hellsten et al., 2007). Also, for many pairs of 4R ohnologues in the salmon, one retained a broader more ancestral expression pattern, while only the other ohnologue changed, often even neofunctionalising (Lien et al., 2016). These observations of specialisation are consistent with an asymmetric mechanism of the DDC, where one ohnologue’s regulatory region degenerates much more than the other.
A more recent detection of this pattern extends more broadly across the amphioxus transcriptome, with comparisons to multiple vertebrates (Marlétaz et al., 2018). For certain ohnologues, new regulatory elements were found near the specialised gene, indicating a role for neofunctionalisation as well as the asymmetrical loss of one ohnologue’s “ancestral” regulatory elements (Marlétaz et al., 2018). These studies rely on broad overviews of gene expression from extensive transcriptome datasets, so may be unable to detect more focused and specific changes to gene function and expression between particular ohnologues, such as qualitative changes to protein sequences described above. Furthermore, specialisation may only be detectable for genes whose ancestral function or expression was widespread and complex (pleiotropic), while other studies detecting asymmetrical changes in expression amongst paralogues with more specialised or localised functions ascribe the process to sub- or neo-functionalisation. The mechanisms resulting in more symmetrical patterns of subfunctionalisation, namely the relaxed constraint provided by redundancy allowing degenerative mutations to accrue in regulatory regions, are not required to affect each paralogue equally, so patterns described as specialisation or neofunctionalisation can also occur by the same logic, if for some reason one paralogue is affected more than the other. This asymmetry amongst paralogues can also explain how novelties arose via co-option of redundant ohnologues to novel functions, through changes in gene regulation.
Regulatory Elements and Subfunctionalisation
Advancements in bioinformatic approaches to comparative genomics have facilitated the detection of CNEs, aiding in the identification of the regulatory changes between paralogues. The patterns of retention of these CNEs, many of which are situated within the introns of neighbouring genes, may have important consequences for the evolutionary maintenance of the synteny of gene clusters or highly conserved genomic neighbourhoods, in which many developmental genes can be found. For example, VISTA plots show that the conserved regulatory elements for Pax6 are situated in the introns of several gene neighbours across vertebrates (Kleinjan et al., 2008; Navratilova et al., 2009), providing the selective pressure to maintain these gene clusters. This can be seen in many other developmental gene clusters, including the MyoD-related genes that are key for muscle development. In most non-teleost 2R vertebrates, there are four myogenic regulatory factors (MRFs; MyoD, Myog, Myf5, and Myf6), two of which, Myf5 and Myf6, reside in a closely linked cluster and share many overlapping regulatory elements (Braun et al., 1990; Carvajal et al., 2008). While in zebrafish these genes act early in myoblast determination and later in myoblast differentiation, respectively, so that Myf5 expression is followed by that of its neighbour, Myf6, in mice Myf6 also has an early role in determination and so has a biphasic expression pattern (Kassar-Duchossoy et al., 2004; Schnapp et al., 2009; Moncaut et al., 2013; Hernández-Hernández et al., 2017). This early versus late distinction is exemplified by MyoD binding the Myog promoter (Tapscott, 2005). Deletion of the minimal Myf5 promoter results in misexpression of Myf6 in Myf5 regions, thus it is clear that the regulatory domains of these two genes are intrinsically linked (Carvajal et al., 2008).
Mirroring this cluster, a cryptic fifth MRF ohnologue is located adjacent to MyoD in the genomes of the coelacanth, sterlet, and spotted gar (Aase-Remedios et al., 2020). Only with the recent availability of the genomes of these “non-model” organisms could this fifth ohnologue, Myf7, be found, since it has been lost from most other vertebrate lineages to which common model organisms belong, including tetrapods, cartilaginous fish, and teleosts. The finding of this fifth vertebrate MRF upended previous interpretations of the evolution of the four MRFs shared amongst all vertebrates. Instead of a single ancestral gene, duplicated in 2R into four ohnologues, and a highly unusual translocation event, it is much more likely that there was an ancestral two-gene state resulting from a tandem duplication that generated the two types of MRF, early and late (Aase-Remedios et al., 2020). This cluster nevertheless shares synteny with the MyoD locus across the vertebrates, potentially as a result of constraint on regulatory elements that overlap with the MRFs and their gene neighbours. Other developmental gene families also have so-called “cryptic” vertebrate paralogues analogous to Myf7, that have been lost from common model system lineages and found only with the availability of genomes from more basally branching species (Kuraku et al., 2016), such as Bmp16 (Feiner et al., 2019) and Foxl2B (Geraldo et al., 2013). The wider availability of sequencing has allowed more phylogenetically informative outgroups and lineages to be studied, changing our understanding of chordate evolution.
Independent Duplications
Small-Scale Independent Duplications in Chordate Lineages
Wider taxon-sampling has enabled the detection of independent duplications in certain lineages overlaid on the larger general trend of various WGDs. In many important developmental gene families, amphioxus has undergone its own expansions of gene clusters, formed by tandem duplications (Minguillón et al., 2002). This ranges from expansions within existing clusters, possibly like the Posterior Hox gene AmphiHox15 within the Hox cluster (Holland et al., 2008), to the advent of new clusters, like the expansion of the amphioxus MRFs (Schubert et al., 2003; Urano et al., 2003; Yuan et al., 2003; Somorjai et al., 2008; Bertrand et al., 2011; Tan et al., 2014; Aase-Remedios et al., 2020). The five genes in this cluster are expressed in different temporal and spatial patterns during amphioxus muscle development, indicating these genes have subfunctionalised as well, providing an independent case of MRF subfunctionalisation from that seen with the vertebrate MRFs (Aase-Remedios et al., 2020). In the Pax gene family of developmental transcription factors, we see a comparable case of independent subfunctionalisation between amphioxus and vertebrates. Amphioxus has, independently from vertebrates, duplicated the ancestral chordate pro-orthologue of Pax3/7 to produce two Pax3/7 trans-homologues of vertebrate Pax3 and Pax7, which have again evolved distinct expression patterns (Barton-Owen et al., 2018). Being that in vertebrates, Pax3 and Pax7 are upstream regulators of the MRFs, perhaps this duplication is acting in concert with the MRF expansion, as an expanding network of muscle regulators. These observations complicate the overall relationship between the pre-duplicate amphioxus genome and vertebrate genomes shaped by 2R WGD and require careful “mining” of the latest genome sequence data and close consideration of gene phylogenies. Overall, these cases show that although amphioxus does indeed provide us with an excellent proxy for much of the chordate ancestor, this animal also has its own unique evolutionary history. This needs to be carefully considered in order to properly infer the features of the chordate ancestor.
Lineage-Specific WGDs
Wider taxon-sampling allows us to study not only phylogenetically informative lineages, but also species that serve as case studies for interesting phenomena like more recent lineage-specific WGDs. Recent sequencing efforts have provided a characterisation of the WGD in the sterlet lineage (Cheng et al., 2019; Du et al., 2020). Not only does this genome provide an outgroup comparison to 3R teleosts as a basal osteichthyan (Figure 1), but because sturgeons are slow-evolving, they have retained ancestral characteristics possibly remnant of the bony fish ancestor. Evidence of several levels of polyploidy has been observed in different sturgeon groups (Vasil’yev et al., 1980; Fontana et al., 2008), but the WGD identified at the base of the sturgeon lineage provides a case study to examine the process of rediploidisation. In the sturgeon, while large chromosomes were retained in duplicate, many of the second copies of the smaller chromosomes were lost, meaning some chromosomes are tetraploid, while others are diploid (Romanenko et al., 2015; Du et al., 2020). In contrast, in 4R salmonids, ohnologues were lost gene by gene (Lien et al., 2016). The sterlet genome’s mobilome shows identical transposable element content between each haplotype, suggesting an origin by autopolyploidy (Du et al., 2020). Comparisons to allopolyploid genomes could reveal the different mechanisms of genomic rediploidisation following the different modes of duplication; perhaps an autopolyploid genome allows the loss of entire chromosomes, while allopolyploid genomes are constrained by differences between haplotypes, and therefore losses occur on smaller scales.
An important allopolyploid is the African clawed frog (Xenopus laevis) (Kobel and Du Pasquier, 1986; Session et al., 2016). This frog has a diploid chromosome number of 36, almost twice the number of its congeneric X. tropicalis, with 20, and arose via hybridisation of two 18n frogs that had undergone chromosomal fusion between homologues of X. tropicalis chromosomes 9 and 10 (Evans, 2008). There is extensive conserved homology between the two sub-genomes in X. laevis and the diploid X. tropicalis, reflecting the recentness of this duplication, and only 17% of duplicated genes have been lost (Uno et al., 2013), in contrast to ∼80% of teleost 3R ohnologues for example (Brunet et al., 2006). The mechanism for retention in the frog may be dosage-based, though some paralogues may also have subfunctionalised (Charbonnier et al., 2002; Hellsten et al., 2007; Sémon and Wolfe, 2008), and different types of genes are lost at different rates (Session et al., 2016). Notably, the karyotype is stable, unlike in the sterlet, probably due to the lack of recombination between the two X. laevis sub-genomes (S and L), though more genes are lost from sub-genome S, which has also undergone more intra-chromosomal rearrangements (Session et al., 2016). Altogether, this sheds light on the patterns of rediploidisation in an allopolyploid, where each sub-genome is more intact and independent, as opposed to the sterlet example of an autopolyploid, where the polyploid genome is truly redundant and entire chromosomes can be lost.
These differences in rediploidisation are relevant more widely, as the 2R WGD at the base of the vertebrates is thought to have taken place with an initial autopolyploidy, then subsequent allopolyploidy of two descendants of that first polyploid (Simakov et al., 2020). This is supported by the genomic rearrangements and symmetrical gene losses that follow 1R, also seen in the rediploidisation process in the sterlet. These rearrangements are shared with lampreys, but the asymmetric gene losses and independence of haplotypes characteristic of allopolyploids are not. Ancient duplications can now be understood in more detail with our better understanding of the different rediploidisation processes, only now possible with the wider taxon-sampling of case-study species with lineage-specific WGDs. For instance, only 0.2% of the diversity of amphibian genomes has been sequenced, while over 8% of mammal genomes have been sequenced, and roughly the same number of genomes are available for teleosts as sauropsids, though there are twice as many teleost species (Figure 1B). This has changed remarkably over the past few decades with the rapid improvements in genome sequencing, and is set to improve still further with initiatives like the Earth Biogenome Project (Lewin et al., 2018), which should release us from our anthropocentric concentration on only biomedically and economically relevant species and allow us instead to obtain an unbiased overview of genomic biodiversity and the processes that generated it.
New Genomes and Transitions Within Vertebrates
The Origin of Tetrapods
Of these more recently sequenced species, the coelacanth genome has been particularly instrumental in addressing the origin of tetrapods and the transition to land, as it is the most basal-branching sarcopterygian, to which tetrapods also belong (Figure 1). Likewise, the lungfish genome has been instrumental in revealing the genomic changes underpinning this transition, as it is the closest living relative to tetrapods (Figure 1; Meyer et al., 2021). These groups make more suitable comparisons to tetrapods than commonly used teleost outgroups, since they have not undergone the teleost-specific 3R WGD and have the fourfold paralogy of a 2R vertebrate (Koh et al., 2003; Noonan et al., 2004; Amemiya et al., 2010). They also allow observed differences between tetrapods and teleosts to be timed specifically to the tetrapod or sarcopterygian lineages (Figure 1). Many cryptic vertebrate paralogues have been found in such lineages, and indeed more than fifty developmental genes were found in the coelacanth that had been lost in tetrapods (Amemiya et al., 2013), including HoxA14 (Amemiya et al., 2010). While these gene losses corresponded to the transition to land, several thousand CNEs near genes involved in morphogenesis and development were identified that evolved anew in the tetrapod lineage (Amemiya et al., 2013).
Looking more closely at developmental genes to address the morphological evolution of limbs in the transition to land, the genomes of coelacanth and lungfish reveal changes to the Hox clusters that may underpin these morphological changes. The coelacanth genome study identified a limb development enhancer in the gene desert upstream of the HoxD cluster shared between tetrapods and the coelacanth but absent in the teleost comparison, suggesting this sequence could have been co-opted into lower limb development in tetrapods from an element present in the aquatic ancestral sarcopterygian (Amemiya et al., 2013). In lungfish, HoxA14 is also present, indicating its loss occurred in tetrapods, and HoxC13 expression was detected in distal fins, showing the early evidence of its co-option to tetrapod limb development (Meyer et al., 2021). Also implicated in the adaptation to land is an enhancer near HoxA14 in coelacanth that controls the expression of Posterior Hox genes, though HoxA14 itself is lost in tetrapods. This enhancer has evolved to be essential for the formation of the placenta (Amemiya et al., 2013). This analysis revealed the impact of new regulatory elements in major transitions in the vertebrates, which are key players in the differentiation among paralogous loci with the potential to evolve different or new functions because of genetic redundancy.
Gene duplications also characterise this transition, particularly in gene families involved in adaptation to land. The lungfish genome contains a gene encoding a respiratory surfactant, Sftpc, which originated via duplication earlier in the sarcopterygian lineage, and further duplicated in lungfish and tetrapods (Wang et al., 2021). Also expanded in the lungfish genome are several gene families for olfactory receptors to detect airborne chemicals as well as the vomeronasal receptor family, showing the timing of this adaptation coincides with the adaptation to land (Meyer et al., 2021). Likewise, in comparison with the coelacanth, many tetrapod-specific CNEs were identified near genes involved in olfaction (Amemiya et al., 2013), showing that for the adaptation to land, both gene duplications and changes to gene regulation likely contributed to some extent. These candidate genes and potential regulatory elements may represent some of the genomic changes underpinning the evolution of land vertebrates and were only found with new comparative genomics approaches and new genome sequences. In particular, the lungfish genome is the largest of any animal, so only with new sequencing techniques, in this case ultra-long-read Nanopore technology, could a genome with this quantity of repetitive elements be sequenced (Meyer et al., 2021), enabling a better understanding of the origin of tetrapods.
The Origin of Mammals
A major transition within the tetrapods was the evolution of mammals, most of which are warm-blooded, viviparous, milk-producing, and have fur, distinct from the reptilian ancestor. Of particular relevance to this transition are the monotremes, the basal lineage of mammals who simultaneously exhibit both mammalian and reptilian traits, including milk production and fur, but also have oviparity and produce venom like reptiles, thus providing an excellent point of comparison to address the genetics supporting mammalian adaptations (Warren et al., 2008). There are two extant monotreme lineages, the platypuses and echidnas (Figure 1). When the draft genome of the platypus was sequenced, platypuses were found to have many microchromosomes like in reptiles (Warren et al., 2008). Nearly three quarters of the first genome sequence was unmapped to chromosomes, but now, with advancements like long-read sequencing and Hi-C, the platypus genome is assembled to the chromosomal level and the echidna genome was much improved as well (Zhou et al., 2021). These new assemblies allow us to address monotreme traits like their many sex chromosomes, electromagnetic sensory organs, and venom. Mammalian venom evolved convergently to reptilian venoms, originating in monotremes by tandem duplication of defensin family genes in glands that were originally sweat glands, similarly to the evolution of shrew venom and in contrast to snake venom, which originated by duplication of other defensins in salivary glands (Whittington et al., 2008). The many sex chromosomes in monotremes evolved from the same autosomes as bird sex chromosomes (ZW female and ZZ male) rather than the SRY (XX female, XY male) sex determination genes of therian mammals, which remain autosomal in monotreme genomes (Veyrunes et al., 2008). The new assembly recharacterised the 10 platypus sex chromosomes and found female-biassed expression of sexually differentiated regions of these chromosomes, consistent with a heterozygous female, and allowed the reconstruction of the series of recombinations and fusions that occurred in their formation (Zhou et al., 2021). This showed that the therian sex determination system evolved independently from the more reptile-like system in monotremes.
A major advancement within mammals was the evolution of viviparity. Monotremes lay small eggs that hatch early in development, as young rely primarily on milk for nutrition, as in therians. The first platypus genome enabled the identification of the genes involved in the transition from oviparity to viviparity, including the retention of reptile-like ZPAX genes and nothepsin, but paired with the loss of vitellogenin genes, leaving only one in the platypus and none in therians, compared to three in birds (Warren et al., 2008). Casein genes are integral to milk production in mammals and evolved via tandem gene duplications of genes in the secretory calcium-binding phosphoprotein family (Kawasaki et al., 2011). Monotremes not only have the full complement of therian caseins, but have a second CSN2 paralogue (Lefèvre et al., 2009). The new assemblies of platypus and echidna genomes have built on these previous findings, including the identification of a CSN3 paralogue and determining the origin of these genes from teeth-related genes based on their syntenic locations (Zhou et al., 2021). A comparative genomics study of the new platypus assembly also revealed that genes involved in the development of the therian placenta were co-opted from genes with other functions, and this transition was underpinned by changes to gene regulation and expression in old genes (Hao et al., 2020). Taken together, these studies show that only with new, chromosome-level assemblies of phylogenetic outgroups can the genetic basis of transitions in chordate evolution be studied.
Conclusion
The age of genomics has made sequencing easier and cheaper than ever before, and important advancements such as long-molecule technologies and Hi-C are enabling chromosome-level assemblies for not just traditional model organisms, but also those of particular evolutionary importance. These advances have allowed us to more accurately understand transitions in evolution, for instance using the chromosome-level assembly of B. floridae to determine the specific chromosomal rearrangements in and around the 2R WGD. Comparative genomics is now equipped with the material with which to determine the genetic changes that underpin these major evolutionary transitions. It is clear that both gene and genome duplications have been instrumental in the evolution of chordate lineages, from the revolutionary 2R WGD around the origin of the vertebrates, to smaller scale expansions of developmental gene clusters, and tandem duplications underpinning changes to particular lineages or in specific gene families. These duplications can be linked to functional changes as studies of gene expression have revealed the role of regulatory regions, non-coding RNAs, and epigenetics in the evolution of chordate genomes. The continuously improving sequence resources for key outgroup lineages, including amphioxus, cyclostomes, coelacanth and lungfish, and the monotremes, have enabled us to detect gene and genome duplications, pinpoint their timing in the tree of chordates, and to infer the evolutionary impact of these events.
Author Contributions
Both authors wrote the manuscript, are accountable for the content, and approved the submitted version.
Funding
MEA-R is supported by funding from the University of St Andrews School of Biology and St Leonard’s College. Work in the lab of DEKF is funded by the BBSRC (BB/S016856/1) with additional support from the European project Assemble Plus (H2020-INFRAIA-1-2016–2017; grant no. 730984).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge discussions with members of the Ferrier laboratory that helped to shape the content of this manuscript.
References
Aase-Remedios, M. E., Coll-Lladó, C., and Ferrier, D. E. K. (2020). More Than One-to-Four via 2R: Evidence of an Independent Amphioxus Expansion and Two-Gene Ancestral Vertebrate State for MyoD-Related Myogenic Regulatory Factors (MRFs). Mol. Biol. Evol. 37, 2966–2982. doi: 10.1093/molbev/msaa147
Amemiya, C. T., Alfoldi, J., Lee, A. P., Fan, S., Philippe, H., MacCallum, I., et al. (2013). The African coelacanth genome provides insights into tetrapod evolution. Nature 496, 311–316. doi: 10.1038/nature12027
Amemiya, C. T., Powers, T. P., Prohaska, S. J., Grimwood, J., Schmutz, J., Dickson, M., et al. (2010). Complete HOX cluster characterization of the coelacanth provides further evidence for slow evolution of its genome. Proc. Natl. Acad. Sci. U. S. A. 107, 3622–3627. doi: 10.1073/pnas.0914312107
Amores, A. (1998). Zebrafish hox Clusters and Vertebrate Genome Evolution. Science 282, 1711–1714. doi: 10.1126/science.282.5394.1711
Barton-Owen, T. B., Ferrier, D. E. K., and Somorjai, I. M. L. (2018). Pax3/7 duplicated and diverged independently in amphioxus, the basal chordate lineage. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-27700-x
Berthelot, C., Brunet, F., Chalopin, D., Juanchich, A., Bernard, M., Noël, B., et al. (2014). The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 5:3657. doi: 10.1038/ncomms4657
Bertrand, S., Camasses, A., Somorjai, I., Belgacem, M. R., Chabrol, O., Escande, M.-L., et al. (2011). Amphioxus FGF signaling predicts the acquisition of vertebrate morphological traits. Proc. Natl. Acad. Sci. U. S. A. 108, 9160–9165. doi: 10.1073/pnas.1014235108
Bertrand, S., and Escriva, H. (2011). Evolutionary crossroads in developmental biology: amphioxus. Development 138, 4819–4830. doi: 10.1242/dev.066720
Bliznina, A., Masunaga, A., Mansfield, M. J., Tan, Y., Liu, A. W., West, C., et al. (2021). Telomere-to-telomere assembly of the genome of an individual Oikopleura dioica from Okinawa using Nanopore-based sequencing. BMC Genomics 22:222. doi: 10.1186/s12864-021-07512-6
Braasch, I., Bobe, J., Guiguen, Y., and Postlethwait, J. H. (2018). Reply to: “Subfunctionalization versus neofunctionalization after whole-genome duplication.”. Nat. Genet. 50, 910–911. doi: 10.1038/s41588-018-0163-3
Braasch, I., Gehrke, A. R., Smith, J. J., Kawasaki, K., Manousaki, T., Pasquier, J., et al. (2016). The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 48, 427–437. doi: 10.1038/ng.3526
Braun, T., Bober, E., Winter, B., Rosenthal, N., and Arnold, H. H. (1990). Myf-6, a new member of the human gene family of myogenic determination factors: evidence for a gene cluster on chromosome 12. EMBO J. 9, 821–831.
Brozovic, M., Dantec, C., Dardaillon, J., Dauga, D., Faure, E., Gineste, M., et al. (2018). ANISEED 2017: Extending the integrated ascidian database to the exploration and evolutionary comparison of genome-scale datasets. Nucleic Acids Res. 46, D718–D725. doi: 10.1093/nar/gkx1108
Brunet, F., Crollius, H. R., Paris, M., Aury, J. M., Gibert, P., Jaillon, O., et al. (2006). Gene loss and evolutionary rates following whole-genome duplication in teleost fishes. Mol. Biol. Evol. 23, 1808–1816. doi: 10.1093/molbev/msl049
Burke, A. C., Nelson, C. E., Morgan, B. A., and Tabin, C. (1995). Hox genes and the evolution of vertebrate axial morphology. Development 121, 333–346.
Butts, T., Holland, P. W. H., and Ferrier, D. E. K. (2008). The Urbilaterian Super-Hox cluster. Trends Genet. 24, 259–262. doi: 10.1016/j.tig.2007.09.006
Cañestro, C., Bassham, S., and Postlethwait, J. H. (2003). Seeing chordate evolution through the Ciona genome sequence. Genome Biol. 4:208. doi: 10.1186/gb-2003-4-3-208
Cañestro, C., Albalat, R., Irimia, M., and Garcia-Fernàndez, J. (2013). Impact of gene gains, losses and duplication modes on the origin and diversification of vertebrates. Semin. Cell Dev. Biol. 24, 83–94. doi: 10.1016/j.semcdb.2012.12.008
Caputo Barucchi, V., Giovannotti, M., Nisi Cerioni, P., and Splendiani, A. (2013). Genome duplication in early vertebrates: Insights from agnathan cytogenetics. Cytogenet. Genome Res. 141, 80–89. doi: 10.1159/000354098
Carvajal, J. J., Keith, A., and Rigby, P. W. J. (2008). Global transcriptional regulation of the locus encoding the skeletal muscle determination genes Mrf4 and Myf5. Genes Dev. 22, 265–276. doi: 10.1101/gad.442408
Casaca, A., Santos, A. C., and Mallo, M. (2014). Controlling Hox gene expression and activity to build the vertebrate axial skeleton. Dev. Dyn. 243, 24–36. doi: 10.1002/dvdy.24007
Chain, F. J. J., and Evans, B. J. (2006). Multiple Mechanisms Promote the Retained Expression of Gene Duplicates in the Tetraploid Frog Xenopus laevis. PLoS Genet. 2:e56. doi: 10.1371/journal.pgen.0020056
Charbonnier, F., Gaspera, B., Della, Armand, A.-S., Van der Laarse, W. J., Launay, T., et al. (2002). Two myogenin-related genes are differentially expressed in Xenopus laevis myogenesis and differ in their ability to transactivate muscle structural genes. J. Biol. Chem. 277, 1139–1147. doi: 10.1074/jbc.M107018200
Cheng, P., Huang, Y., Du, H., Li, C., Lv, Y., Ruan, R., et al. (2019). Draft Genome and Complete Hox-Cluster Characterization of the Sterlet (Acipenser ruthenus). Front. Genet. 10:776. doi: 10.3389/fgene.2019.00776
Dardaillon, J., Dauga, D., Simion, P., Faure, E., Onuma, T. A., DeBiasse, M. B., et al. (2019). ANISEED 2019: 4D exploration of genetic data for an extended range of tunicates. Nucleic Acids Res. 48, D668–D675. doi: 10.1093/nar/gkz955
Dehal, P., and Boore, J. L. (2005). Two Rounds of Whole Genome Duplication in the Ancestral Vertebrate. PLoS Biol. 3:e314. doi: 10.1371/journal.pbio.0030314
Delsuc, F., Brinkmann, H., Chourrout, D., and Philippe, H. (2006). Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439, 965–968. doi: 10.1038/nature04336
Denoeud, F., Henriet, S., Mungpakdee, S., Aury, J. M., Da Silva, C., Brinkmann, H., et al. (2010). Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science 330, 1381–1385. doi: 10.1126/science.1194167
Denton, J. F., Lugo-Martinez, J., Tucker, A. E., Schrider, D. R., Warren, W. C., and Hahn, M. W. (2014). Extensive Error in the Number of Genes Inferred from Draft Genome Assemblies. PLoS Comput. Biol. 10:e1003998. doi: 10.1371/journal.pcbi.1003998
Donoghue, P. C. J., and Purnell, M. (2005). Genome duplication, extinction and vertebrate evolution. Trends Ecol. Evol. 20, 312–319. doi: 10.1016/j.tree.2005.04.008
Du, K., Stöck, M., Kneitz, S., Klopp, C., Woltering, J. M., Adolfi, M. C., et al. (2020). The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 4, 841–852. doi: 10.1038/s41559-020-1166-x
Duboule, D. (1994). Temporal colinearity and the phylotypic progression: A basis for the stability of a vertebrate Bauplan and the evolution of morphologies through heterochrony. Development 120, 135–142.
Duboule, D. (2007). The rise and fall of Hox gene clusters. Development 134, 2549–2560. doi: 10.1242/dev.001065
Duboule, D., and Dollé, P. (1989). The structural and functional organization of the murine HOX gene family resembles that of Drosophila homeotic genes. EMBO J. 8, 1497–1505. doi: 10.1002/j.1460-2075.1989.tb03534.x
Espinosa-Cantú, A., Ascencio, D., Barona-Gómez, F., and DeLuna, A. (2015). Gene duplication and the evolution of moonlighting proteins. Front. Genet. 6:227. doi: 10.3389/fgene.2015.00227
Evans, B. J. (2008). Genome evolution and speciation genetics of clawed frogs (Xenopus and Silurana). Front. Biosci. 13:4687–4706. doi: 10.2741/3033
Farrè, D., and Albà, M. M. (2010). Heterogeneous patterns of gene-expression diversification in mammalian gene duplicates. Mol. Biol. Evol. 27, 325–335. doi: 10.1093/molbev/msp242
Feiner, N., Motone, F., Meyer, A., and Kuraku, S. (2019). Asymmetric paralog evolution between the “cryptic” gene Bmp16 and its well-studied sister genes Bmp2 and Bmp4. Sci. Rep. 9:3136. doi: 10.1038/s41598-019-40055-1
Ferrier, D. E. K. (2016). Evolution of Homeobox Gene Clusters in Animals: The Giga-Cluster and Primary vs. Secondary Clustering. Front. Ecol. Evol. 4, 1–13. doi: 10.3389/fevo.2016.00036
Ferrier, D. E. K. (2019). “Space and time in Hox / ParaHox gene cluster evolution,” in Perspectives on Evolutionary and Developmental Biology: Essays for Alessandro Minelli, ed. G. Fusco (Paduva: Padova University Press), 245–258.
Fontana, F., Congiu, L., Mudrak, V. A., Quattro, J. M., Smith, T. I. J., Ware, K., et al. (2008). Evidence of hexaploid karyotype in shortnose sturgeon. Genome 51, 113–119. doi: 10.1139/G07-112
Force, A., Amores, A., and Postlethwait, J. H. (2002). Hox cluster organization in the jawless vertebrate Petromyzon marinus. J. Exp. Zool. 294, 30–46. doi: 10.1002/jez.10091
Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y., and Postlethwait, J. (1999). Preservation of Duplicate Genes by Complementary, Degenerative Mutations. Genetics 151, 1531–1545.
Fried, C., Prohaska, S. J., and Stadler, P. F. (2003). Independent Hox-cluster duplications in lampreys. J. Exp. Zool. 299B, 18–25. doi: 10.1002/jez.b.37
Furlong, R. F., Younger, R., Kasahara, M., Reinhardt, R., Thorndyke, M., and Holland, P. W. H. (2007). A Degenerate ParaHox Gene Cluster in a Degenerate Vertebrate. Mol. Biol. Evol. 24, 2681–2686. doi: 10.1093/molbev/msm194
Gans, C., and Northcutt, R. G. (1983). Neural Crest and the Origin of Vertebrates: A New Head. Science 220, 268–273. doi: 10.1126/science.220.4594.268
Garcia-Fernàndez, J., and Holland, P. W. H. (1994). Archetypal organization of the amphioxus Hox gene cluster. Nature 370, 563–566. doi: 10.1038/370563a0
Gaunt, S. J. (2018). Hox cluster genes and collinearities throughout the tree of animal life. Int. J. Dev. Biol. 62, 673–683. doi: 10.1387/ijdb.180162sg
Geraldo, M. T., Valente, G. T., Braz, A. S. K., and Martins, C. (2013). The discovery of Foxl2 paralogs in chondrichthyan, coelacanth and tetrapod genomes reveals an ancient duplication in vertebrates. Heredity. 111, 57–65. doi: 10.1038/hdy.2013.19
Glasauer, S. M. K., and Neuhauss, S. C. F. (2014). Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics 289, 1045–1060. doi: 10.1007/s00438-014-0889-2
Graham, A., Papalopulu, N., and Krumlauf, R. (1989). The murine and Drosophila homeobox gene complexes have common features of organization and expression. Cell 57, 367–378. doi: 10.1016/0092-8674(89)90912-4
Green, S. A., and Bronner, M. E. (2014). The lamprey: A jawless vertebrate model system for examining origin of the neural crest and other vertebrate traits. Differentiation 87, 44–51. doi: 10.1016/j.diff.2014.02.001
Hao, Y., Lee, H. J., Baraboo, M., Burch, K., Maurer, T., Somarelli, J. A., et al. (2020). Baby Genomics: Tracing the Evolutionary Changes That Gave Rise to Placentation. Genome Biol. Evol. 12, 35–47. doi: 10.1093/gbe/evaa026
Hellsten, U., Khokha, M. K., Grammer, T. C., Harland, R. M., Richardson, P., and Rokhsar, D. S. (2007). Accelerated gene evolution and subfunctionalization in the pseudotetraploid frog Xenopus laevis. BMC Biol. 5:31. doi: 10.1186/1741-7007-5-31
Hernández-Hernández, J. M., García-González, E. G., Brun, C. E., and Rudnicki, M. A. (2017). The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 72, 10–18. doi: 10.1016/j.semcdb.2017.11.010
Hoegg, S., Brinkmann, H., Taylor, J. S., and Meyer, A. (2004). Phylogenetic timing of the fish-specific genome duplication correlates with the diversification of teleost fish. J. Mol. Evol. 59, 190–203. doi: 10.1007/s00239-004-2613-z
Hoegg, S., and Meyer, A. (2005). Hox clusters as models for vertebrate genome evolution. Trends Genet. 21, 421–424. doi: 10.1016/j.tig.2005.06.004
Holland, L. Z., Albalat, R., Azumi, K., Benito-Gutiérrez, È, Blow, M. J., Bronner-Fraser, M., et al. (2008). The amphioxus genome illuminates vertebrate origins and cephalochordate biology. Genome Res. 18, 1100–1111. doi: 10.1101/gr.073676.107
Holland, L. Z., and Holland, N. D. (2001). Evolution of neural crest and placodes: amphioxus as a model for the ancestral vertebrate? J. Anat. 199, 85–98. doi: 10.1046/j.1469-7580.199.parts1-2.8.x
Holland, L. Z., and Ocampo Daza, D. (2018). A new look at an old question: when did the second whole genome duplication occur in vertebrate evolution? Genome Biol. 19:209. doi: 10.1186/s13059-018-1592-0
Holland, P. W. H. (2013). Evolution of homeobox genes. Wiley Interdiscip. Rev. Dev. Biol. 2, 31–45. doi: 10.1002/wdev.78
Hughes, T., Ekman, D., Ardawatia, H., Elofsson, A., and Liberles, D. A. (2007). Evaluating dosage compensation as a cause of duplicate gene retention in Paramecium tetraurelia. Genome Biol. 2007:213. doi: 10.1186/gb-2007-8-5-213
Iimura, T., and Pourquié, O. (2006). Collinear activation of Hoxb genes during gastrulation is linked to mesoderm cell ingression. Nature 442, 568–571. doi: 10.1038/nature04838
Janvier, P. (2007). Living Primitive Fishes and Fishes From Deep Time. Fish Physiol. 26, 1–51. doi: 10.1016/S1546-5098(07)26001-7
Jeffery, W. R., Strickler, A. G., and Yamamoto, Y. (2004). Migratory neural crest-like cells form body pigmentation in a urochordate embryo. Nature 431, 696–699. doi: 10.1038/nature02975
Jeffery, W. R. (2006). Ascidian neural crest-like cells: Phylogenetic distribution, relationship to larval complexity, and pigment cell fate. J. Exp. Zool. Part B Mol. Dev. Evol. 306, 470–480. doi: 10.1002/jez.b.21109
Jimenez-Delgado, S., Pascual-Anaya, J., and Garcia-Fernandez, J. (2009). Implications of duplicated cis-regulatory elements in the evolution of metazoans: the DDI model or how simplicity begets novelty. Briefings Funct. Genomics Proteomics 8, 266–275. doi: 10.1093/bfgp/elp029
Kassar-Duchossoy, L., Gayraud-Morel, B., Gomès, D., Rocancourt, D., Buckingham, M., Shinin, V., et al. (2004). Mrf4 determines skeletal muscle identity in Myf5:Myod double-mutant mice. Nature 431, 466–471. doi: 10.1038/nature02876
Kawasaki, K., Lafont, A. G., and Sire, J. Y. (2011). The evolution of milk casein genes from tooth genes before the origin of mammals. Mol. Biol. Evol. 28, 2053–2061. doi: 10.1093/molbev/msr020
Kleinjan, D. A., Bancewicz, R. M., Gautier, P., Dahm, R., Schonthaler, H. B., Damante, G., et al. (2008). Subfunctionalization of Duplicated Zebrafish pax6 Genes by cis-Regulatory Divergence. PLoS Genet. 4:29. doi: 10.1371/journal.pgen.0040029
Kmita, M., and Duboule, D. (2003). Organizing axes in time and space; 25 years of colinear tinkering. Science 301, 331–333. doi: 10.1126/science.1085753
Kobel, H. R., and Du Pasquier, L. (1986). Genetics of polyploid Xenopus. Trends Genet. 2, 310–315. doi: 10.1016/0168-9525(86)90286-6
Koh, E. G. L., Lam, K., Christoffels, A., Erdmann, M. V., Brenner, S., and Venkatesh, B. (2003). Hox gene clusters in the Indonesian coelacanth, Latimeria menadoensis. Proc. Natl. Acad. Sci. 100, 1084–1088. doi: 10.1073/pnas.0237317100
Krumlauf, R. (1994). Hox genes in vertebrate development. Cell 78, 191–201. doi: 10.1016/0092-8674(94)90290-9
Krumlauf, R. (2018). Hox genes, clusters and collinearity. Int. J. Dev. Biol. 62, 659–663. doi: 10.1387/ijdb.180330rr
Kugler, J. E., Kerner, P., Bouquet, J. M., Jiang, D., and Di Gregorio, A. (2011). Evolutionary changes in the notochord genetic toolkit: A comparative analysis of notochord genes in the ascidian Ciona and the larvacean Oikopleura. BMC Evol. Biol. 11:21. doi: 10.1186/1471-2148-11-21
Kuraku, S., Feiner, N., Keeley, S. D., and Hara, Y. (2016). Incorporating tree-thinking and evolutionary time scale into developmental biology. Dev. Growth Differ. 58, 131–142. doi: 10.1111/dgd.12258
Kuraku, S., Meyer, A., and Kuratani, S. (2009). Timing of genome duplications relative to the origin of the vertebrates: Did cyclostomes diverge before or after? Mol. Biol. Evol. 26, 47–59. doi: 10.1093/molbev/msn222
Ledje, C., Kim, C.-B., and Ruddle, F. H. (2002). Characterization of Hox genes in the Bichir, Polypterus palmas. J. Exp. Zool. 294, 107–111. doi: 10.1002/jez.10152
Lefèvre, C. M., Sharp, J. A., and Nicholas, K. R. (2009). Characterisation of monotreme caseins reveals lineage-specific expansion of an ancestral casein locus in mammals. Reprod. Fertil. Dev. 21, 1015–1027. doi: 10.1071/RD09083
Lemaire, P. (2011). Evolutionary crossroads in developmental biology: The tunicates. Development 138, 2143–2152. doi: 10.1242/dev.048975
Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., Crandall, K. A., et al. (2018). Earth BioGenome Project: Sequencing life for the future of life. Proc. Natl. Acad. Sci. U. S. A. 115, 4325–4333. doi: 10.1073/pnas.1720115115
Lien, S., Koop, B. F., Sandve, S. R., Miller, J. R., Kent, M. P., Nome, T., et al. (2016). The Atlantic salmon genome provides insights into rediploidization. Nature 533, 200–205. doi: 10.1038/nature17164
Lynch, M., and Conery, J. S. (2000). The Evolutionary Fate and Consequences of Duplicate Genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Macqueen, D. J., and Johnston, I. A. (2014). A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc. R. Soc. B Biol. Sci. 281:2014. doi: 10.1098/rspb.2013.2881
Marlétaz, F., Firbas, P. N., Maeso, I., Tena, J. J., Bogdanovic, O., Perry, M., et al. (2018). Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 564, 64–70. doi: 10.1038/s41586-018-0734-6
Martin, K. J., and Holland, P. W. H. (2014). Enigmatic Orthology Relationships between Hox Clusters of the African Butterfly Fish and Other Teleosts Following Ancient Whole-Genome Duplication. Mol. Biol. Evol. 31, 2592–2611. doi: 10.1093/molbev/msu202
Martin, K. J., and Holland, P. W. H. (2017). Diversification of Hox gene clusters in osteoglossomorph fish in comparison to other teleosts and the spotted gar outgroup. J. Exp. Zool. Part B Mol. Dev. Evol. 328, 638–644. doi: 10.1002/jez.b.22726
Mehta, T. K., Ravi, V., Yamasaki, S., Lee, A. P., Lian, M. M., Tay, B.-H., et al. (2013). Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc. Natl. Acad. Sci. 110, 16044–16049. doi: 10.1073/pnas.1315760110
Meyer, A., and Schartl, M. (1999). Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 11, 699–704. doi: 10.1016/S0955-0674(99)00039-3
Meyer, A., Schloissnig, S., Franchini, P., Du, K., Woltering, J. M., Irisarri, I., et al. (2021). Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 590, 284–289. doi: 10.1038/s41586-021-03198-8
Meyer, A., and Van de Peer, Y. (2005). From 2R to 3R: evidence for a fish-specific genome duplication (FSGD). BioEssays 27, 937–945. doi: 10.1002/bies.20293
Minguillón, C., Ferrier, D. E. K., Cebrián, C., and Garcia-Fernàndez, J. (2002). Gene duplications in the prototypical cephalochordate amphioxus. Gene 287, 121–128. doi: 10.1016/S0378-1119(01)00828-9
Moncaut, N., Rigby, P. W. J., and Carvajal, J. J. (2013). Dial M(RF) for myogenesis. FEBS J. 280, 3980–3990. doi: 10.1111/febs.12379
Monteiro, A. S., and Ferrier, D. E. K. (2006). Hox genes are not always Colinear. Int. J. Biol. Sci. 2, 95–103. doi: 10.7150/ijbs.2.95
Mungpakdee, S., Seo, H.-C., Angotzi, A. R., Dong, X., Akalin, A., and Chourrout, D. (2008). Differential Evolution of the 13 Atlantic Salmon Hox Clusters. Mol. Biol. Evol. 25, 1333–1343. doi: 10.1093/molbev/msn097
Nakai, Y., Kubota, S., Goto, Y., Ishibashi, T., Davison, W., and Kohno, S. (1995). Chromosome elimination in three Baltic, south Pacific and north-east Pacific hagfish species. Chromosom. Res. 3, 321–330. doi: 10.1007/BF00713071
Nakai, Y., Kubota, S., and Kohno, S. (1991). Chromatin diminution and chromosome elimination in four japanese hagfish species. Cytogenet. Genome Res. 56, 196–198. doi: 10.1159/000133087
Nakatani, Y., Takeda, H., Kohara, Y., and Morishita, S. (2007). Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 17, 1254–1265. doi: 10.1101/gr.6316407
Navratilova, P., Fredman, D., Hawkins, T. A., Turner, K., Lenhard, B., and Becker, T. S. (2009). Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev. Biol. 327, 526–540. doi: 10.1016/j.ydbio.2008.10.044
Near, T. J., Eytan, R. I, Dornburg, A., Kuhn, K. L., Moore, J. A., Davis, M. P., et al. (2012). Resolution of ray-finned fish phylogeny and timing of diversification. Proc. Natl. Acad. Sci. U. S. A 109, 13698–13703. doi: 10.1073/pnas.1206625109
Noonan, J. P., Grimwood, J., Danke, J., Schmutz, J., Dickson, M., Amemiya, C. T., et al. (2004). Coelacanth genome sequence reveals the evolutionary history of vertebrate genes. Genome Res. 14, 2397–2405. doi: 10.1101/gr.2972804
Pascual-Anaya, J., Sato, I., Sugahara, F., Higuchi, S., Paps, J., Ren, Y., et al. (2018). Hagfish and lamprey Hox genes reveal conservation of temporal colinearity in vertebrates. Nat. Ecol. Evol. 2, 859–866. doi: 10.1038/s41559-018-0526-2
Putnam, N. H., Butts, T., Ferrier, D. E. K., Furlong, R. F., Hellsten, U., Kawashima, T., et al. (2008). The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071. doi: 10.1038/nature06967
Qian, W., Liao, B. Y., Chang, A. Y. F., and Zhang, J. (2010). Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 26, 425–430. doi: 10.1016/j.tig.2010.07.002
Robertson, F. M., Gundappa, M. K., Grammes, F., Hvidsten, T. R., Redmond, A. K., Lien, S., et al. (2017). Lineage-specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Biol. 18:111. doi: 10.1186/s13059-017-1241-z
Romanenko, S. A., Biltueva, L. S., Serdyukova, N. A., Kulemzina, A. I., Beklemisheva, V. R., Gladkikh, O. L., et al. (2015). Segmental paleotetraploidy revealed in sterlet (Acipenser ruthenus) genome by chromosome painting. Mol. Cytogenet. 8:90. doi: 10.1186/s13039-015-0194-8
Sacerdot, C., Louis, A., Bon, C., Berthelot, C., and Roest Crollius, H. (2018). Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 19, 1–15. doi: 10.1186/s13059-018-1559-1
Santini, F., Harmon, L. J., Carnevale, G., and Alfaro, M. E. (2009). Did genome duplication drive the origin of teleosts? A comparative study of diversification in ray-finned fishes. BMC Evol. Biol. 9:194. doi: 10.1186/1471-2148-9-194
Sato, Y., and Nishida, M. (2010). Teleost fish with specific genome duplication as unique models of vertebrate evolution. Environ. Biol. Fishes 88, 169–188. doi: 10.1007/s10641-010-9628-7
Satou, Y., Sato, A., Yasuo, H., Mihirogi, Y., Bishop, J., Fujie, M., et al. (2021). Chromosomal inversion polymorphisms in two sympatric ascidian lineages. Genome Biol. Evol. 2021:068. doi: 10.1093/gbe/evab068
Schnapp, E., Pistocchi, A. S., Karampetsou, E., Foglia, E., Lamia, C. L., Cotelli, F., et al. (2009). Induced early expression of mrf4 but not myog rescues myogenesis in the myod/myf5 double-morphant zebrafish embryo. J. Cell Sci. 122, 481–488. doi: 10.1242/jcs.038356
Schubert, M., Escriva, H., Xavier-Neto, J., and Laudet, V. (2006). Amphioxus and tunicates as evolutionary model systems. Trends Ecol. Evol. 21, 269–277. doi: 10.1016/j.tree.2006.01.009
Schubert, M., Meulemans, D., Bronner-Fraser, M., Holland, L. Z., and Holland, N. D. (2003). Differential mesodermal expression of two amphioxus MyoD family members (AmphiMRF1 and AmphiMRF2). Gene Expr. Patterns 3, 199–202. doi: 10.1016/S1567-133X(02)00099-6
Sekigami, Y., Kobayashi, T., Omi, A., Nishitsuji, K., Ikuta, T., Fujiyama, A., et al. (2017). Hox gene cluster of the ascidian, Halocynthia roretzi, reveals multiple ancient steps of cluster disintegration during ascidian evolution. Zool. Lett. 3:17. doi: 10.1186/s40851-017-0078-3
Sémon, M., and Wolfe, K. H. (2008). Preferential subfunctionalization of slow-evolving genes after allopolyploidization in Xenopus laevis. Proc. Natl. Acad. Sci. U. S. A. 105, 8333–8338. doi: 10.1073/pnas.0708705105
Session, A. M., Uno, Y., Kwon, T., Chapman, J. A., Toyoda, A., Takahashi, S., et al. (2016). Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343. doi: 10.1038/nature19840
Simakov, O., Marlétaz, F., Yue, J., O’Connell, B., Jenkins, J., Brandt, A., et al. (2020). Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830. doi: 10.1038/s41559-020-1156-z
Smith, J. J., Baker, C., Eichler, E. E., and Amemiya, C. T. (2012). Genetic consequences of programmed genome rearrangement. Curr. Biol. 22, 1524–1529. doi: 10.1016/j.cub.2012.06.028
Smith, J. J., and Keinath, M. C. (2015). The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 25, 1081–1090. doi: 10.1101/gr.184135.114
Smith, J. J., Kuraku, S., Holt, C., Sauka-spengler, T., Jiang, N., Campbell, M. S., et al. (2013). Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 45, 415–421. doi: 10.1038/ng.2568
Smith, J. J., Timoshevskaya, N., Ye, C., Holt, C., Keinath, M. C., Parker, H. J., et al. (2018). The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 50, 270–277. doi: 10.1038/s41588-017-0036-1
Somorjai, I., Bertrand, S., Camasses, A., Haguenauer, A., and Escriva, H. (2008). Evidence for stasis and not genetic piracy in developmental expression patterns of Branchiostoma lanceolatum and Branchiostoma floridae, two amphioxus species that have evolved independently over the course of 200 Myr. Dev. Genes Evol. 218, 703–713. doi: 10.1007/s00427-008-0256-6
Steinke, D., Salzburger, W., Braasch, I., and Meyer, A. (2006). Many genes in fish have species-specific asymmetric rates of molecular evolution. BMC Genomics 7, 1–18. doi: 10.1186/1471-2164-7-20
Stolfi, A., Ryan, K., Meinertzhagen, I. A., and Christiaen, L. (2015). Migratory neuronal progenitors arise from the neural plate borders in tunicates. Nature 527, 371–374. doi: 10.1038/nature15758
Tan, X., Zhang, P. J., and Du, S. J. (2014). Evolutionary aspects of a new MyoD gene in amphioxus (Branchiostoma belcheri) and its promoter specificity in skeletal and cardiac muscles. Biol. 69, 1210–1221. doi: 10.2478/s11756-014-0427-z
Tapscott, S. J. (2005). The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695. doi: 10.1242/dev.01539
Taylor, J. S., Van De Peer, Y., and Meyer, A. (2001). Genome duplication, divergent resolution and speciation. Trends Genet. 17, 299–301. doi: 10.1016/S0168-9525(01)02318-6
Timoshevskiy, V. A., Lampman, R. T., Hess, J. E., Porter, L. L., and Smith, J. J. (2017). Deep ancestry of programmed genome rearrangement in lampreys. Dev. Biol. 429, 31–34. doi: 10.1016/j.ydbio.2017.06.032
Tschopp, P., Tarchini, B., Spitz, F., Zakany, J., and Duboule, D. (2009). Uncoupling Time and Space in the Collinear Regulation of Hox Genes. PLoS Genet. 5:e1000398. doi: 10.1371/journal.pgen.1000398
Uno, Y., Nishida, C., Takagi, C., Ueno, N., and Matsuda, Y. (2013). Homoeologous chromosomes of Xenopus laevis are highly conserved after whole-genome duplication. Heredity 111, 430–436. doi: 10.1038/hdy.2013.65
Urano, A., Suzuki, M. M., Zhang, P., Satoh, N., and Satoh, G. (2003). Expression of muscle-related genes and two MyoD genes during amphioxus notochord development. Evol. Dev. 5, 447–458. doi: 10.1046/j.1525-142X.2003.03051.x
Vasil’yev, V. P., Solokov, L. I., and Serebryakova, Y. V. (1980). Karyotypes of the Siberian sturgeon, Acipenser baeri, of the Lena river and some aspects of karyotype evolution in Acipenseriformes. J. Ichthyol. 20, 37–45.
Veyrunes, F., Waters, P. D., Miethke, P., Rens, W., McMillan, D., Alsop, A. E., et al. (2008). Bird-like sex chromosomes of platypus imply recent origin of mammal sex chromosomes. Genome Res. 18, 965–973. doi: 10.1101/gr.7101908
Wang, K., Wang, J., Zhu, C., Yang, L., Ren, Y., Ruan, J., et al. (2021). African lungfish genome sheds light on the vertebrate water-to-land transition. Cell 184, 1362.e–1376.e. doi: 10.1016/j.cell.2021.01.047
Warren, W. C., Hillier, L. D. W., Marshall Graves, J. A., Birney, E., Ponting, C. P., Grützner, F., et al. (2008). Genome analysis of the platypus reveals unique signatures of evolution. Nature 453, 175–183. doi: 10.1038/nature06936
Werner, H. B. (2013). Do we have to reconsider the evolutionary emergence of myelin? Front. Cell. Neurosci. 6, 415–421. doi: 10.3389/fncel.2013.00217
Whittington, C. M., Papenfuss, A. T., Bansal, P., Torres, A. M., Wong, E. S. W., Deakin, J. E., et al. (2008). Defensins and the convergent evolution of platypus and reptile venom genes. Genome Res. 18, 986–994. doi: 10.1101/gr.7149808
Yu, J.-K., Meulemans, D., McKeown, S. J., and Bronner-Fraser, M. (2008). Insights from the amphioxus genome on the origin of vertebrate neural crest. Genome Res. 18, 1127–1132. doi: 10.1101/gr.076208.108
Yuan, J., Zhang, S., Liu, Z., Luan, Z., and Hu, G. (2003). Cloning and phylogenetic analysis of an amphioxus myogenic bHLH gene AmphiMDF. Biochem. Biophys. Res. Commun. 301, 960–967. doi: 10.1016/S0006-291X(03)00081-0
Zhang, J. (2003). Evolution by gene duplication: An update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/S0169-5347(03)00033-8
Zhang, Q.-L., Zhang, G.-L., Yuan, M.-L., Dong, Z.-X., Li, H.-W., Guo, J., et al. (2018). A Phylogenomic Framework and Divergence History of Cephalochordata Amphioxus. Front. Physiol. 9:1833. doi: 10.3389/fphys.2018.01833
Zhou, Y., Shearwin-Whyatt, L., Li, J., Song, Z., Hayakawa, T., Stevens, D., et al. (2021). Platypus and echidna genomes reveal mammalian biology and evolution. Nature 2021, 1–7. doi: 10.1038/s41586-020-03039-0
Keywords: amphioxus, tunicates, lamprey, ohnologues, subfunctionalisation, rediploidization, allopolyploidy, autopolyploidy
Citation: Aase-Remedios ME and Ferrier DEK (2021) Improved Understanding of the Role of Gene and Genome Duplications in Chordate Evolution With New Genome and Transcriptome Sequences. Front. Ecol. Evol. 9:703163. doi: 10.3389/fevo.2021.703163
Received: 30 April 2021; Accepted: 08 June 2021;
Published: 29 June 2021.
Edited by:
Juan Pascual-Anaya, Malaga University, SpainReviewed by:
Ricard Albalat, University of Barcelona, SpainIgnacio Maeso, Andalusian Center for Development Biology (CABD), Spain
Copyright © 2021 Aase-Remedios and Ferrier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David E. K. Ferrier, ZGVrZkBzdC1hbmRyZXdzLmFjLnVr