 Nicole R. Foster1*
Nicole R. Foster1* Kor-jent van Dijk1
Kor-jent van Dijk1 Ed Biffin2
Ed Biffin2 Jennifer M. Young3
Jennifer M. Young3 Vicki A. Thomson1
Vicki A. Thomson1 Bronwyn M. Gillanders1
Bronwyn M. Gillanders1 Alice R. Jones1,4
Alice R. Jones1,4 Michelle Waycott1,2
Michelle Waycott1,2- 1School of Biological Sciences, University of Adelaide, Adelaide, SA, Australia
- 2State Herbarium of South Australia, Botanic Gardens and State Herbarium, Department for Environment and Water, Adelaide, SA, Australia
- 3College of Science and Engineering, Flinders University, Adelaide, SA, Australia
- 4Department for Environment and Water, Adelaide, SA, Australia
Metabarcoding of plant DNA recovered from environmental samples, termed environmental DNA (eDNA), has been used to detect invasive species, track biodiversity changes, and reconstruct past ecosystems. The P6 loop of the trnL intron is the most widely utilised gene region for metabarcoding plants due to the short fragment length and subsequent ease of recovery from degraded DNA, which is characteristic of environmental samples. However, the taxonomic resolution for this gene region is limited, often precluding species level identification. Additionally, targeting gene regions using universal primers can bias results as some taxa will amplify more effectively than others. To increase the ability of DNA metabarcoding to better resolve flowering plant species (angiosperms) within environmental samples, and reduce bias in amplification, we developed a multi-gene targeted capture method that simultaneously targets 20 chloroplast gene regions in a single assay across all flowering plant species. Using this approach, we effectively recovered multiple chloroplast gene regions for three species within artificial DNA mixtures down to 0.001 ng/μL of DNA. We tested the detection level of this approach, successfully recovering target genes for 10 flowering plant species. Finally, we applied this approach to sediment samples containing unknown compositions of eDNA and confidently detected plant species that were later verified with observation data. Targeting multiple chloroplast gene regions in environmental samples, enabled species-level information to be recovered from complex DNA mixtures. Thus, the method developed here, confers an improved level of data on community composition, which can be used to better understand flowering plant assemblages in environmental samples.
Introduction
Environmental DNA (eDNA) is a rapidly growing field of research and has been applied extensively to monitor site-based vegetation change over periods of hundreds to thousands of years using samples from soil cores (Willerslev et al., 2003, 2014; Parducci et al., 2013; Zimmermann et al., 2017; Del Carmen Gomez Cabrera et al., 2019). As plants are sedentary, they are a reliable reflection of their environment at a specific point in time (Yoccoz et al., 2012), therefore, reconstruction of plant communities through time can represent environmental conditions and how they have changed at the sampled location. Such information can be used to predict future changes, inform management (Brown and Blois, 2001; Fordham et al., 2016; Balint et al., 2018) and provide insight to uncover species extinctions and/or introductions, elucidate past climate conditions and assess ecosystem trajectories (Thomsen and Willerslev, 2015; Ruppert et al., 2019). In addition, changes in plant community composition can shed light on historical human impacts and agricultural practices (Giguet-Covex et al., 2014; Pansu et al., 2015).
The ability to take an environmental sample and accurately determine the plant community composition contained within, as remnant fragments of tissue or DNA, is most commonly achieved through the process of DNA metabarcoding. Metabarcoding involves polymerase chain reaction (PCR) amplification of a short, but highly variable, gene region that is flanked by conserved regions. The design of PCR amplification primers target these conserved regions, which allows amplification of the variable region across all plant taxa present in an environmental sample (Murchie et al., 2020). The variable region most often used to recover plant DNA in environmental samples is the P6 loop of the chloroplast trnL (UAA) intron (Taberlet et al., 2006). This gene region was adopted due to being short enough to amplify DNA in environmental samples (10–143 bp), whilst still possessing enough discriminatory information to distinguish many groups of plant taxa. Unfortunately, amplifying a single, short region creates limitations to generating accurate results, as species can only be detected if this region remains present in the genomic DNA extracted, and if the primer binding sites are intact to allow successful amplification. Additionally, primers can preferentially bind to certain taxa, creating an unreliable representation of the vegetation community and biasing results (Pedersen et al., 2015). Further, recovering this short section of the trnL gene region does not ensure resolution to species level (Lamb et al., 2016), especially when compared to other plant barcodes such as rbcL and matK (Hollingsworth et al., 2011; Fahner et al., 2016). However, these barcoding regions are much longer and are difficult to recover from degraded samples (Fahner et al., 2016), in addition to being subject to the same limitations of relying on a single gene region. These shortcomings highlight that a more effective approach is needed to fully utilise plant DNA from environmental samples and obtain detailed information on plant community composition.
Targeted capture (also referred to as hybridisation capture) offers an alternative way to overcome the limitations of single gene metabarcoding and bypass the issues associated with PCR amplification (Lemmon and Lemmon, 2013). Targeted capture uses biotinylated RNA molecules called “baits” that are specifically designed to bind to target DNA regions which are then separated from non-target sequences using a magnet. These baits eliminate the need for universal primers and, compared to methods such as genome skimming or shotgun sequencing, the targeted nature enhances recovery of organisms of interest within a DNA mixture. This approach reduces overall sequencing costs and increases the amount of plant genetic information that can be recovered (Foster et al., 2020). Targeted capture approaches have been shown to recover more taxa from environmental samples compared to PCR-based metabarcoding for a range of different organisms (Dowle et al., 2016; Shokralla et al., 2016; Murchie et al., 2020), and given the potential to recover a large amount of sequence data from multiple gene regions, this approach is also likely to improve taxonomic assignment of sequences from various plant communities.
Here, we employed a targeted capture approach to characterise flowering plant communities in environmental samples, using a universal bait set designed to simultaneously capture 20 chloroplast regions across all flowering plants (angiosperms). By incorporating multiple gene regions, we aimed to accurately reconstruct plant communities in environmental samples to species level identification, removing the reliance on a single, short barcode. The bait set utilised in this study has not previously been applied to environmental samples and it is rare that studies are undertaken targeting multiple gene regions in a single assay from complex DNA mixtures (only; Murchie et al., 2020; Lentz et al., 2021) and never with this many gene regions. We conducted sensitivity and discriminatory power tests on artificial DNA mixtures to assess the threshold of detection and the level of taxonomic resolution offered by the approach before analysing soil samples from a study site where we could verify results using observations from the area.
Materials and Methods
Three trials were conducted to determine the sensitivity, discriminatory power, and demonstrate a proof-of-concept for the multi-gene region targeted capture of chloroplast (plastid) DNA proposed in this study. Each test consisted of a separate experimental setup (see section “Experimental Setup”) but the same library preparation (section “Library Preparation”), multi-gene region bait capture (section “Multi-Gene Region Bait Capture”), read processing and mapping (section “Read Processing and Mapping”), and data analysis (section “Data Analysis”) were conducted for all three trials (Figure 1). Control samples (blanks) were run with each trial sample set to monitor potential contamination and false-positive taxonomic assignments (Ficetola et al., 2016).
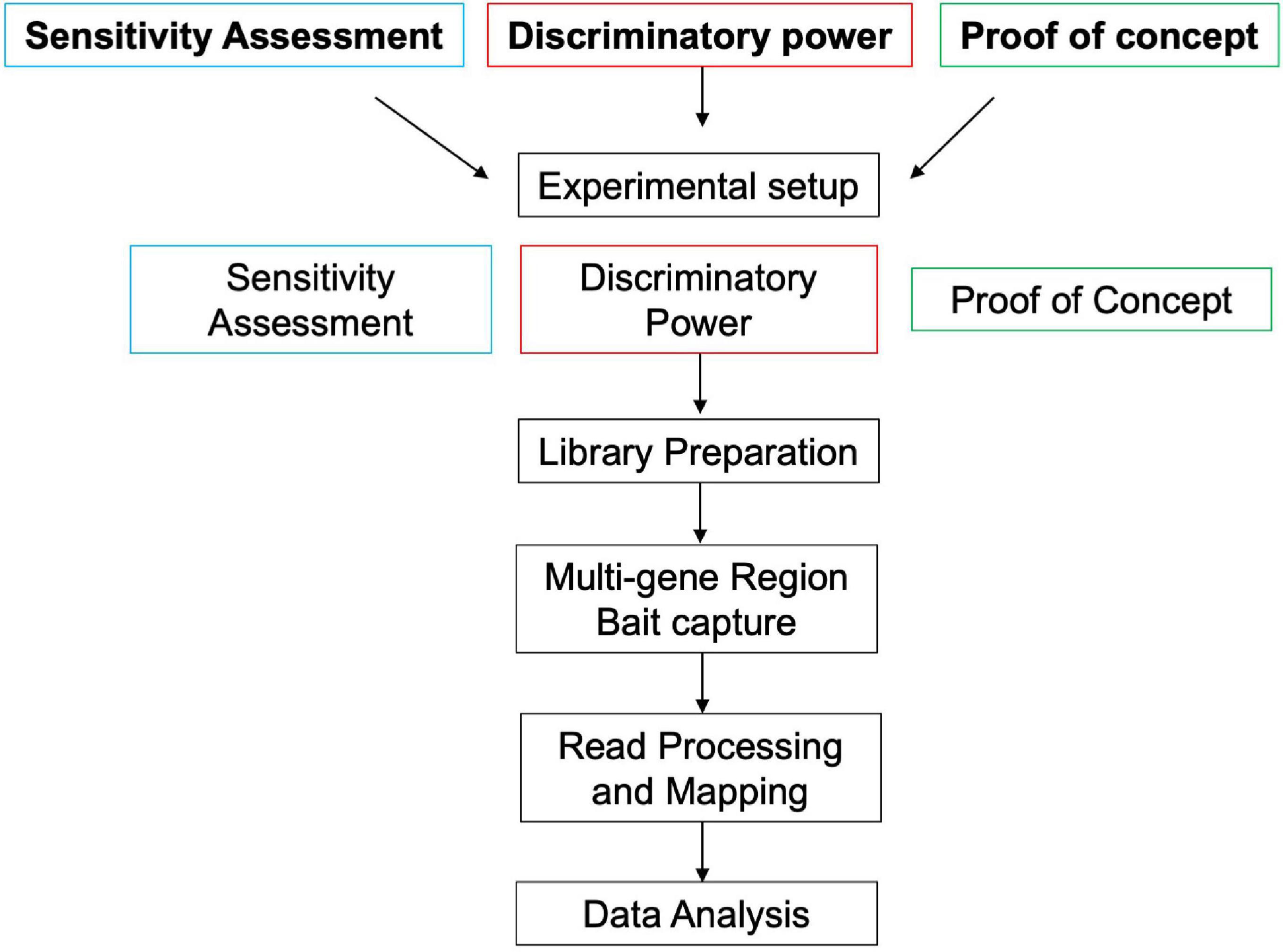
Figure 1. Flow diagram outlining the different tests for this study.
Experimental Setup
Sensitivity Assessment
DNA was extracted from three coastal plant species [Avicennia marina (grey mangrove), Tecticornia flabelliformis (saltmarsh/samphire) and Zostera marina (seagrass)] using the Plant DNeasy Mini Kit (QIAGEN) as per manufacturer’s instructions and quantified using a QuantusTM Fluorometer and QuantiFluor® dsDNA System (Supplementary Table 2). An artificial DNA mixture was prepared by combining 40 μL of each DNA extract into a single stock solution (i.e., species were standardised by volume rather than concentration to mimic an environmental sample) which was quantified as above (5.9 ng/μL). This stock was then diluted to the following concentrations: 1, 0.1, 0.01, 0.001, and 0.0001 ng/μL. Three replicates of each stock concentration (N = 15, volume = 100 μL) were sonicated using a Diagenode Bioruptor® Pico to a size distribution peaking around 400–600 bp (cycle of 15 s on, 90 s off and repeated seven times to obtain the required size distribution). Further sample processing followed the description in section “Data Analysis” (Figure 1).
Discriminatory Power
Total genomic DNA was extracted from 10 coastal plant species (Supplementary Table 2) using the Plant DNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. DNA extracts were quantified individually using a QuantusTM Fluorometer and QuantiFluor® dsDNA System. Five different artificial mixtures (with a total volume of 90 μL) were prepared to include an increasing number of species ranging from 3 to 10, standardised by volume rather than concentration (Table 1). Species were chosen across divergent flowering plant groups including Monocots, Asterids, and Rosids and were iteratively added from 3 to 10 species to document any species/gene dropout. Environmental samples are likely to have a varied number of species present and as such, we acknowledge we may not have reached the limitation of discriminatory ability, however, the 10 species chosen allowed assessment of this method to discern species across divergent groups and multiple taxa. The total DNA concentration of each artificial mixture was quantified as above (5.3, 3.85, 3.06, 5, and 5.3 ng/μL, respectively) and then standardised to 2 ng/μL. Three replicates from each mixture (N = 15, volume = 90 μL) were sonicated using a Diagenode Bioruptor® Pico to a size distribution peaking around 400–600 bp (cycle of 15 s On, 90 s Off, and repeat 7 times to obtain the required size distribution). Further sample processing then proceeded to section “Data Analysis” (Figure 1).

Table 1. Species mixtures and volumes for DNA artificial mixtures to test discriminatory ability of the targeted capture approach.
Proof of Concept
A single sediment core (75 mm wide and 1 m long) was collected as part of a separate study from an intertidal wetland location on Torrens Island, SA, Australia (34° 47.574′ S 138° 31.59′ E). A. marina (grey mangrove) was growing at the site where the core was taken, and saltmarsh habitat and various coastal plants species were observed in the wider area. The core was transported upright and stored at 4°C until sample processing. All equipment and benchtops were cleaned with bleach, ethanol, and water prior to sample processing. Two 250 mg samples (A and B) were collected from the centre of the core at 2.5 cm down the length of the core and DNA was extracted using the DNeasy PowerLyzer PowerSoil Kit (QIAGEN®) with zirconia beads. We chose this extraction kit based on in house trials and previous research (Hermans et al., 2018) and used zirconia beads instead of the standard glass beads to ensure plant cells could be properly lyzed (personal observation). In this instance, sonication was not conducted before library preparation as DNA was assumed to be already fragmented due to the degraded nature of DNA in sediments (Corinaldesi et al., 2008) and therefore, DNA extracts follow the procedure described in section “Data Analysis” (Figure 1).
Library Preparation
An aliquot of the DNA extract was placed into the NEBNext Ultra II Library preparation kit (New England Biolabs®) following manufacturer’s instructions with the following modifications: 1/3 the recommended reaction volume (16.7 μL) and custom-made stubby (incomplete, P5 and P7 indexes missing) Y-adaptors (25 μM) (Glenn et al., 2019) were used at the ligation step. The design of these adapters replaced the uracil excision in the Ultra II protocol as instead, DNA underwent end repair then A-tailing prior to ligating Y-adapters. Each adaptor had a unique eight nucleotide barcode, giving each sample a unique pair of identical internal molecular identifiers (identified as the eight first base calls for each read). Following adapter ligation, libraries were amplified to detectable concentrations using the supplied Q5 Master Mix at the original reaction volume of 50 μL with in-house primers P7 preCap Long and P5 preCap Long [Cycling conditions: (98°C 10 s, 65°C 30 s, 72°C 30 s) × 17 cycles, 72°C 120 s, 4°C hold]. A total of 2 μL of each uniquely indexed library was then visually checked using gel electrophoresis (1 × TE buffer, 1.5% agarose gel for 40 min at 80 V) and pooled according to concentration estimates (determined via visual inspection) into batches of eight samples and then purified using AMPure XP (at 0.8 × volume concentration) to remove remaining primers and other impurities. Pooled libraries then progressed to section “Multi-Gene Region Bait Capture.”
Multi-Gene Region Bait Capture
Bait Design
We used the RefSeq release of plastid sequences1 across ∼160 taxa to design probes targeting a set of 20 plastid gene regions for angiosperms (Supplementary Table 1). These 20 gene regions were chosen to include standard plant barcoding regions (Hollingsworth et al., 2011) and they possessed flanking regions available to amplify the variable regions, in addition, almost all are chloroplast specific. Using Arabidopsis lyrata (Genbank reference NC_034379) as a reference, target regions were extracted from the RefSeq data using Blast (blastn, e-value < 1e–50) and were clustered using CD-HIT (Li and Godzik, 2006) with a 95% identity cut-off, retaining the longest sequence per cluster for probe design. A total of c. 2800 representative sequences, ranging in length from 180 to 900 bp (mean 370 bp) were used to design c. 15,000 120-mer probe sequences with 2X tiling (i.e., each probe overlaps half its length). Further information can be found in Waycott et al. (2021).
Targeted Capture
Targeted capture was performed on each batch of libraries following the myBaits® Targeted NGS Manual Version 4.01 as per the manufacturer’s instructions. The hybridisation temperature/time was 65°C for 48 h. Following hybridisation, the product was amplified using custom P7 and P5 indexed primers designed in-house using cycling conditions: 98°C 120 s, (98°C 20 s, 60°C 30 s, 72°C 45 s) × 17 cycles, 72°C 30 s, 4°C hold. The final product was an Illumina library where each sample had a unique combination of identical internal dual barcodes (incorporated during library preparation) and two indexes (incorporated after hybridisation). Within our laboratory, all dual barcode-Index 1–Index 2 combinations are only used once, thus reducing contamination risk.
Following targeted capture and amplification, the resulting libraries were run on a 2100 Bioanalyzer (Agilent) using the high sensitivity DNA assay and molarity was calculated between 300 and 800 bp. All libraries were then pooled in equimolar concentration and purified using AMPure XP (New England Biolabs) at 0.7 × concentration to remove primer dimer and short sequences. The final library, which included all samples in this study, underwent further size selection using a Pippin Prep (Sage Science) with a 1.5% agarose gel cassette set to select between 300 and 600 bp. The resulting library was quantified using a QuantStudio 6 Flex Real-Time PCR (Thermo Fisher Scientific), diluted to 1.5 nM in 30 μL and sent to the Garvan Institute of Medical Research (Sydney, NSW, Australia) to be sequenced on one lane of an Illumina HiSeq X Ten using 2 × 150 chemistry.
Read Processing and Mapping
Raw sequences were demultiplexed based on indexes using Illumina Bcl2fastq v2.18.0. The output Read 1 and Read 2 fastq.gz files were then demultiplexed based on the Y-adapter internal barcodes using AdapterRemoval v2 (Schubert et al., 2016). PALEOMIX (Schubert et al., 2014) was then used to trim adapters (using AdapterRemoval), discard singletons and sequences less than 25 bp, and trim for ambiguous nucleotides and low-quality base calls. BWA-MEM aligner (Li, 2013) was selected within PALEOMIX as the mapping tool while discarding unmapped reads as this is the most accurate tool for mapping next generation sequencing (NGS) reads of plants (Wu et al., 2019; Schilbert et al., 2020; Yao et al., 2020). A specific reference database was used at this step for the different trials, where “restricted” and “wider” reference databases were used in both the sensitivity and discrimination trials and the “wider” reference database was used for the proof of concept. The wider reference database consisted of 94 coastal temperate plant species (Foster et al., 2021, unpublished data) generated from voucher specimens using the same chloroplast bait set as applied to the test samples (Supplementary Table 1) and combined with references from the National Centre for Biotechnology Information (NCBI Resource Coordinators, 2018) database consisting of all available sequences for the 20 target chloroplast genes of South Australian flora. The restricted database consisted of a subset of this database to only include plant species present in the artificial mixtures to quantify the success of the approach in regard to gene recovery.
Following mapping, Picard Mark Duplicates (Version 4.0.10.1) was used to remove clonality (duplication in read alignment) and the resulting BAM files were then used to generate VCF files using SAMtools mpileup (Li, 2011), specifying ploidy as 1 (as haplotypic organellar DNA) and filtering for base quality <30, mapping quality <30, and depth <50. Variant calls were normalised with BCFtools norm (Li, 2011) and the consensus caller in BCFtools was then used to call final consensus FASTA files, outputting variants with N’s. Output FASTA files were then filtered for length <100 bp.
Data Analysis
For the sensitivity analysis, the number of chloroplast gene regions recovered in each mixture was counted and calculated as a percentage of the total number of regions that were targeted. We then fitted candidate quasibinomial distributed Generalised Linear Models (GLM) with species, concentration and type of mapping reference database as explanatory variables and the percentage of chloroplast gene regions recovered as the response variable. We included different interaction terms between explanatory variables in each candidate GLM and selected the candidate with the greatest model performance (i.e., lowest Akaike Information Criterion). We then conducted an ANOVA on the selected model output to test for significant effects of species, concentration, type of reference and the interactive effect of species and concentration on chloroplast gene region recovery (Supplementary Table 4).
In the discriminatory power analysis, we found that we could still detect all target gene regions for each of the known species up to the 10 species mixture (the maximum number of species in our tested sample mixtures), indicating that there was no decline in gene region recovery as the number of species in the sample increased from 3 to 10. Therefore, we proceeded to focus our subsequent analyses only on the 10 species sample mixture results. We collated scores of presence or absence for each species, gene region and replicate when sequence reads were mapped to the restricted reference database. We then mapped this same 10 species mixture to the wider reference database and combined all replicates to conduct further analyses to observe the level of taxonomic classification achieved for each gene region. We combined replicate FASTA samples (from section “Read Processing and Mapping”) and used CD-HIT-EST (Li and Godzik, 2006; Fu et al., 2012) to cluster sample sequences with the wider reference database at 95% similarity cut-off, removing any samples that did not cluster. We then wrote a custom script in R version 3.5.1 (R Core Team, 2018), to determine the lowest discernible taxonomic rank for clusters containing sample sequences (see Supplementary Methods for details on this). To assess the rate of false-positive assignments we separated these results into (1) species we put into the sample and (2) all species recovered and their taxonomic ranking.
For the proof-of-concept study, we analysed the FASTA files from section “Read Processing and Mapping” as above, using the CD-HIT-EST analysis and custom R script.
Results
Sensitivity Assessment
Using artificial DNA mixtures of decreasing concentration, we identified a minimum detection threshold of 0.001–0.0001 ng/μL total DNA concentration (Figure 2), where below 0.001 ng/μL, the number of target regions recovered across all species was zero. The number of filtered and mapped reads reflects a similar result, as there is a steady decrease in reads with decrease in DNA concentration, and a substantial decline after 0.001 ng/μL (Supplementary Table 3). There was no difference in target gene region recovery between the wider and restricted reference databases (P = 0.85), however, there was an interactive effect between species and concentration (P < 0.05) where the percentage of target regions recovered for Z. marina started decreasing at higher concentrations than the other two species (see Supplementary Table 4).
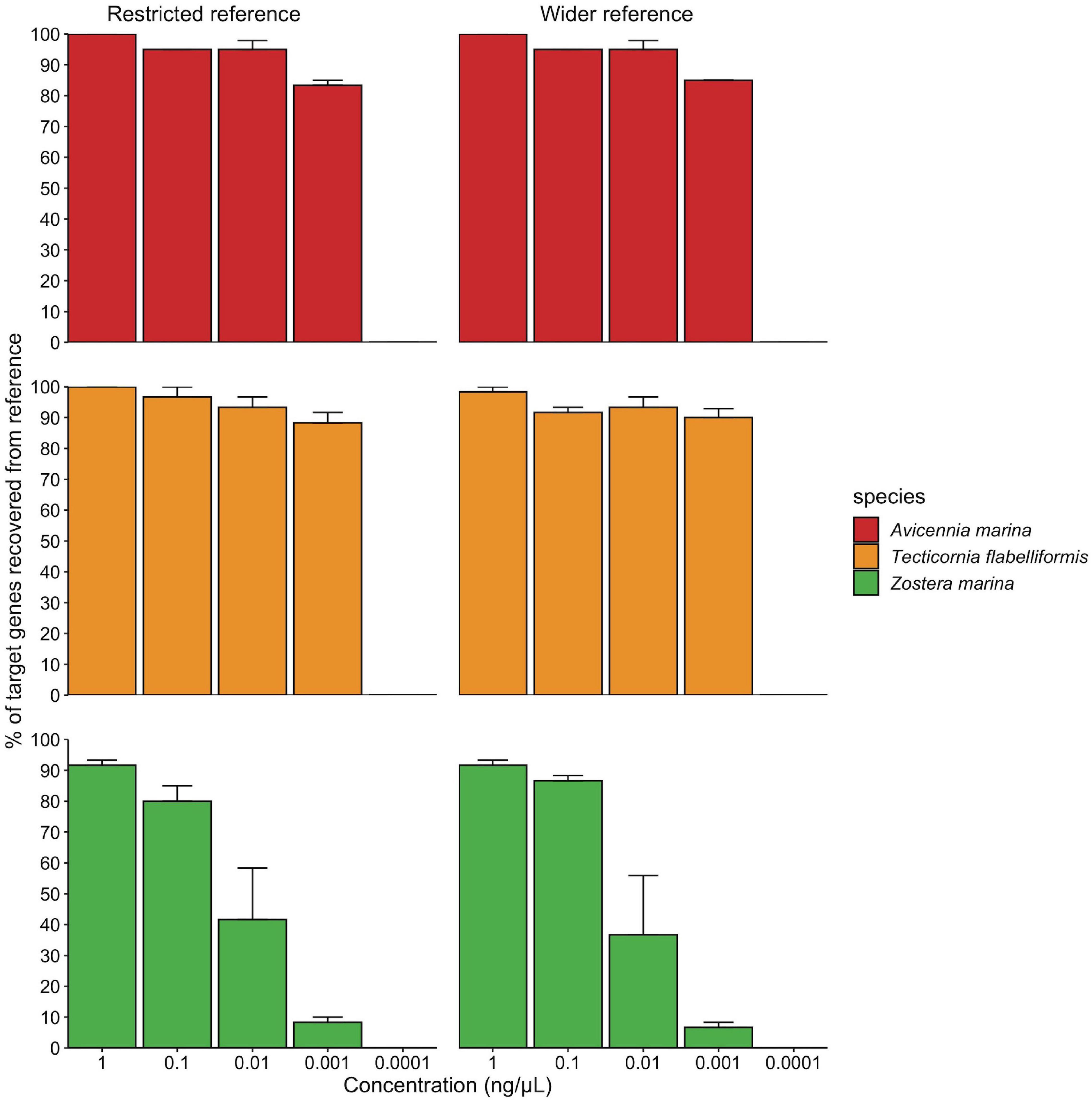
Figure 2. Minimum detection threshold of the targeted capture approach using artificial plant DNA mixtures. All species are in the same mixture but are graphed separately. Concentration reflects the total concentration of the three species mixture, where each species was added in the same volume with a different starting concentration. Restricted and wider reference libraries refer to the different reference databases used in the mapping step.
Discriminatory Power
Mapping the maximum 10 species mixture to the restricted reference database, we found 100% recovery for all 20 target plastid gene regions across all species and replicates with one exception, gene region recovery for Zostera muelleri was 95% and of these, only 53% were recovered in all three replicates (Figure 3A). When the same 10 species artificial DNA mixture was mapped to the wider reference database, and the replicates were pooled, psbE, atpI, and rpl16 were recovered across all 10 species (Figure 3B). The standard barcoding regions, matK and rbcL as well as atpH, psbD, and petA, were recovered across 9 of the 10 species and of these, matK had the greatest discriminatory ability, discerning all 9 detected samples to family level or below. Whilst recovered well, the gene region psbE, had the lowest discriminatory ability, only capable of discerning samples to order level. Six of the 10 species placed in the mixture were recovered at species level resolution (A. marina, Disphyma crassifolium, Frankenia pauciflora, Parapholis incurva, Samolus repens, and Wilsonia humilis) and the other 4 were recovered at genus level (Posidonia australis, Sarcocornia quinqueflora, Tecticornia halocnemoides, and Z. muelleri). In addition to the species that were put in the mixture, additional FASTA files were generated for species that were not placed in the mixture. Taxonomic classification using the clustering algorithm in section “Data Analysis,” showed these all resolved to order, family or genus of our known species. Supplementary Figure 2 shows the results of all taxa recovered from the mixture, where the orders Alismatales, Caryophyllales, Lamiales, and Poales are the plant orders of our known species, as are the families; Aizoaceae, Chenopodiaceae, Poaceae, and Primulaceae. Gene regions resolving to the family Scrophulariaceae, the genus Chenopodium and the species T. flabelliformis, Tecticornia pruinosa, and Tecticornia syncarpa were the only ones recovered but were not placed in the mixture.
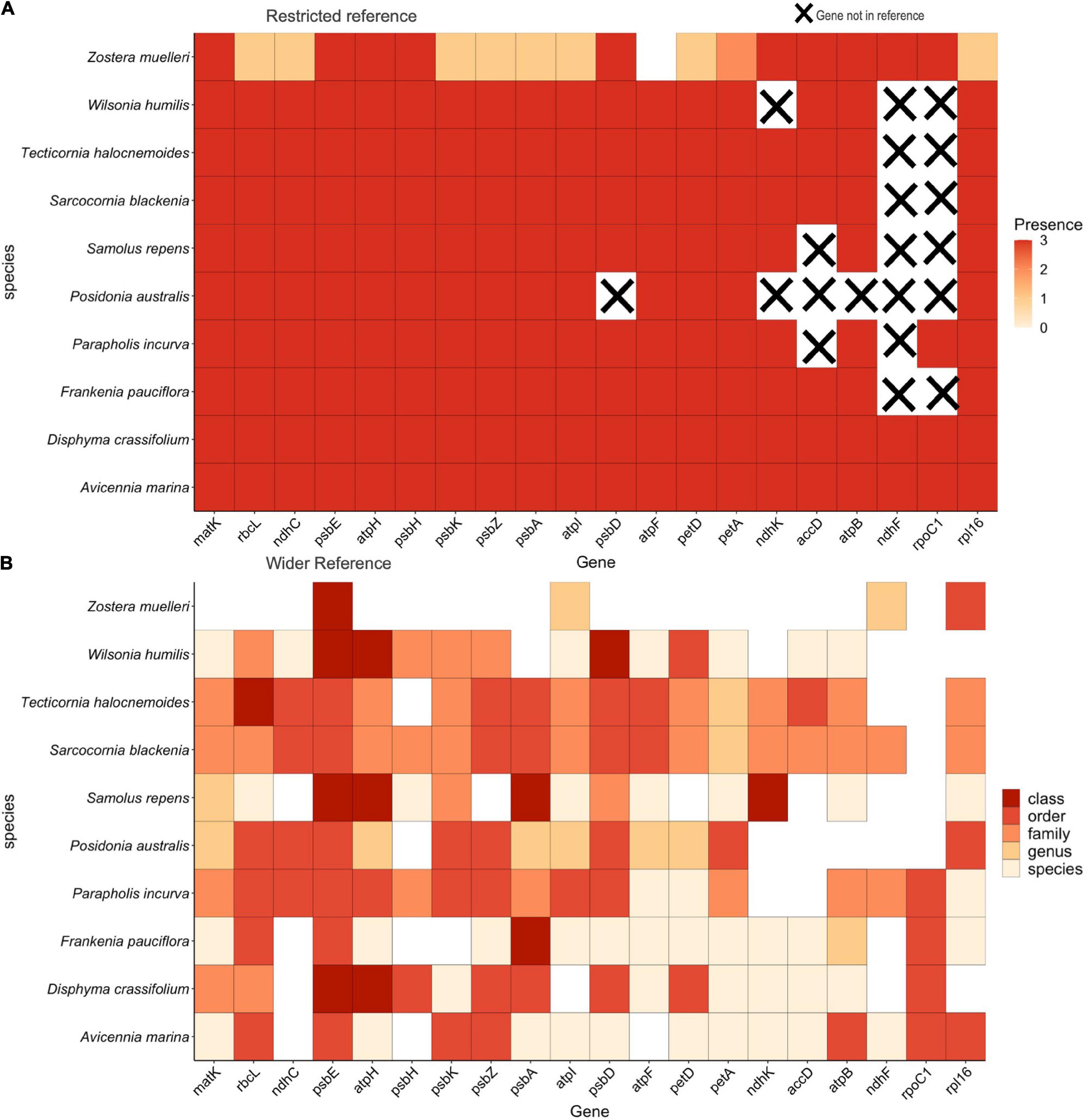
Figure 3. (A) The 10 species DNA mixture mapped to a restricted reference library containing only the species included in the mixture. Presence is defined as whether the target gene region was recovered across replicates. (B) Taxonomic level of classification when the 10 species mixture was mapped to the wider reference library. All replicate samples are combined.
Proof of Concept
Proof of Concept Study
Testing the multi-gene region capture approach developed in previous sections on an environmental sample from a coastal wetland, we were able to recover multiple gene regions and species from sediment samples containing an unknown composition of plant genetic material. Figure 4 shows the results of this trial combining the two replicate samples A and B. Eleven target gene regions recovered A. marina across different A. marina samples in the reference database, and 9 gene regions resolved to the order Lamiales – the order A. marina belongs to. In addition, two gene regions were recovered for the family Scrophulariaceae. Based on these results, we can conclude A. marina presence within the environmental samples tested and some evidence for the presence of species belonging to the family Scrophulariaceae where only two gene regions were recovered for this family. Observational data was able to verify that A. marina was growing at this site but there was no evidence of species belonging to the family Scrophulariaceae, except in the wider area (Myoporum insulare).
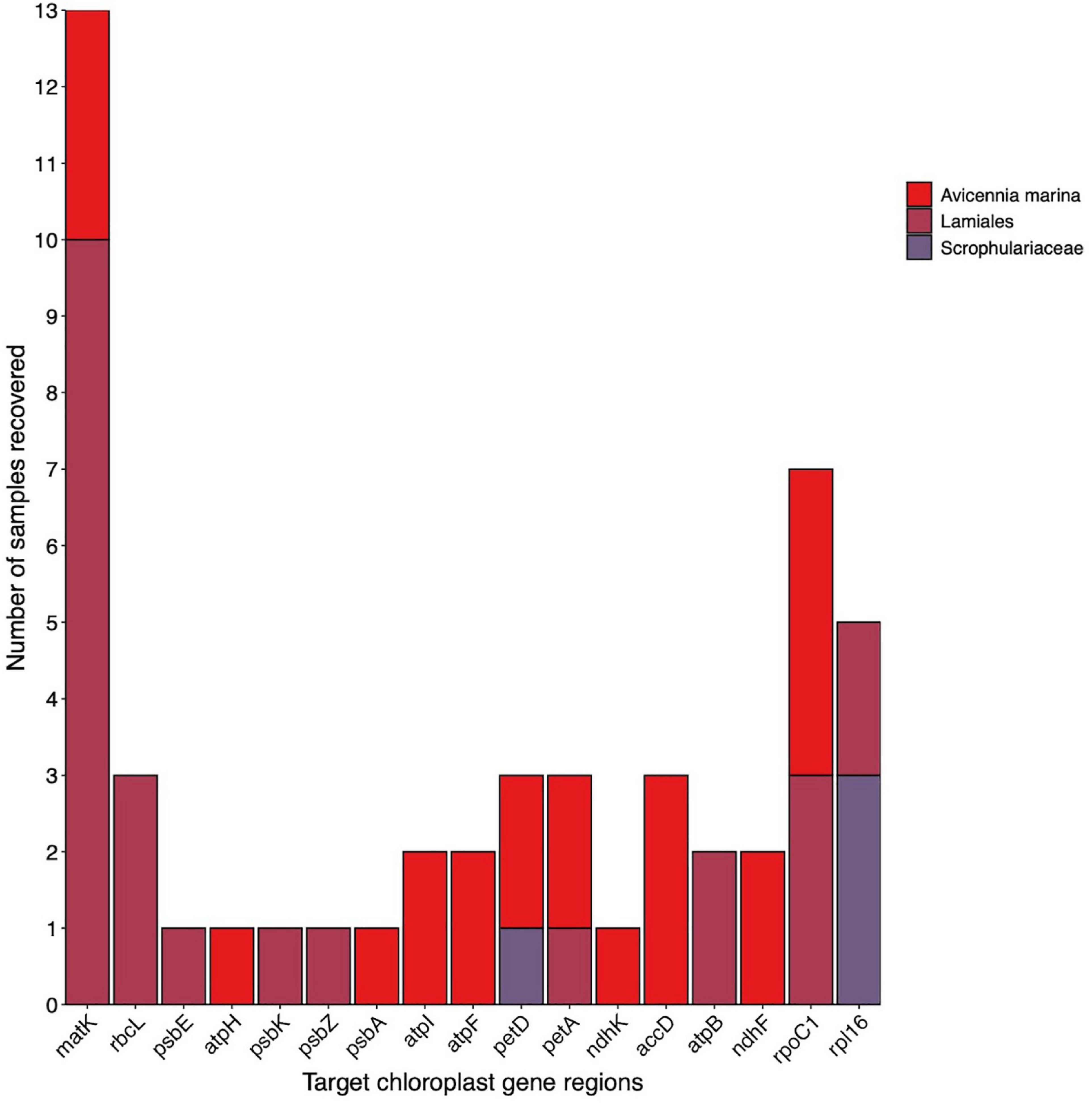
Figure 4. Target chloroplast gene region recovery for plant taxa detected in an unknown sediment sample. Colours indicate level of taxonomic classification. Multiples of the same gene region recovered for A. marina indicate multiple samples in the reference library were recovered.
Controls
Sample processing and bioinformatic analysis of sample blank controls for each stage of this study yielded no matches to either the restricted or wider reference databases and therefore we are confident there has not been any contamination in this study.
Discussion
This study presents a novel approach to detecting and characterising flowering plant DNA in environmental samples. Whilst bait capture has recently been used for eDNA studies on plants (Murchie et al., 2020; Lentz et al., 2021), these studies require knowledge of plant taxa that are present in the area to design the baits. Our study has demonstrated the use of a universal bait set, capable of detecting flowering plants across diverse groups and recover species level identification of plant taxa in sediment samples. A universal bait set increases the likelihood of detecting plants that are not known to be in the area and also enables this method to be applied to environments beyond coastal habitats. We have demonstrated this targeted capture technique and bait set can recover multiple chloroplast gene regions from environmental samples in a single assay. We show that the lowest detection level of this approach is down to 0.001 ng/μL and therefore, this method is sensitive enough to detect low concentrations of DNA that are likely to be found in environmental samples due to the degradation that occurs (Pedersen et al., 2015). Furthermore, we highlight that multi-gene recovery is possible across many divergent taxa (i.e., up to 10 species in the same sample), however, the comprehensiveness of reference databases used for read mapping can influence the number of chloroplast gene regions that are recovered. We also showed that these target chloroplast gene regions have different levels of discrimination for different flowering plant groups. This exemplifies that a multi-gene capture approach as opposed to metabarcoding a single gene region, increases the likelihood of recovering all species present in a sample at either species or genus level. Overall, testing this approach on sediment samples of unknown composition yielded species level assignment of taxa that were verified to be present, and family assignment of plant taxa in the surrounding area.
Detection Threshold When Targeting Multiple Regions and Species in Environmental Samples
Using the multi-gene region targeted approach, our ability to detect all species present in an artificial mixture was significantly impacted by total DNA concentration of the sample. In our sensitivity analysis, the percentage of target gene regions recovered for Z. marina slowly declined below 1 ng/μL to no recovery at 0.0001 ng/μL, whereas the other two species (T. flabelliformis and A. marina) recorded a decline only below 0.001 ng/μL. We attribute this result to the varying DNA input concentrations of species in this trial where Z. marina was present at lower concentration in the starting mixture, contributing to only 1% whereas A. marina and T. flabelliformis contributed 46 and 53%, respectively (Supplementary Table 2). However, despite the lower initial concentration, we were still able to recover >10% of Z. marina target chloroplast regions down to 0.001 ng/μL. The sharp decrease in gene region recovery for all species in the mixture after 0.001 ng/μL implies that total DNA concentration significantly impacts gene region recovery for all species in a mixture, regardless of the amount of DNA contributed for each individual species. As eDNA and ancient samples have characteristically degraded DNA and thus low concentration (Pedersen et al., 2015), a multi-gene region targeted approach seems the best option to increase the chances of capturing the entire plant community present.
The Power of a Multi-Gene Region Approach
We have demonstrated that the multi-gene region capture method developed in this study can successfully recover multiple regions across many species when we have prior knowledge of what is in a sample (Figure 3A). The lower gene region recovery for Z. muelleri in Figure 3, can again be attributed to the low input DNA concentration (Supplementary Table 2). However, the fact that both Zostera species in each of our trials had the lowest number of recovered regions, suggests additional factors may be influencing the amount of DNA recovered for this genus, such as the chloroplast copy number (Sakamoto and Takami, 2018). This highlights the possibility that taxa within environmental samples can have naturally unequal amounts of DNA which can potentially skew the number and type of gene regions recovered for different plant groups. Therefore, targeting a single region may give an inaccurate picture of the plant community composition present in a sample as some species will be missed.
Furthermore, when we then mapped the same 10 species mixture to the wider reference library, we found that, for some gene regions, reads were no longer mapped directly to the expected taxon. It is likely, for these regions, mapping has occurred to closely related species within the wider reference database that otherwise mapped to known taxa in the restricted database or were removed. This highlights the importance of a comprehensive reference database when deciphering plant community presence in environmental samples. Having confidence that the read mapping step has correctly assigned reads to the reference database of species that are present in the sample is important. Furthermore, as we show in Figure 3B, the variability and thus discrimination potential varies between gene regions and between flowering plant groups. Thus, we undertook additional analyses to assign taxonomic classification to sample sequences, so we could ensure sequences mapped uniquely to each species and to increase confidence when assigning species presence to unknown sequences. Overall, we assigned species presence for 6 of the 10 species we placed in the mixture and the rest were assigned to genus level.
The additional plant taxa detected that were not placed in the mixture highlights possible shortcomings of our target bait set (Supplementary Figure 2). The family Scrophulariaceae and the genus Chenopodium were not placed in the mixture but are closely related to the species that were placed in the mixture. This result demonstrates that some of the plastid gene regions used in this study may not contain enough variability to separate all plant groups. Therefore, recovering multiple gene regions for each species increases the likelihood of accurate detection as more genetic data, and subsequent variability between taxa, is captured. As demonstrated here, species that were known to be in the mixture were recovered across far more genes than Scrophulariaceae (three genes) and Chenopodium (one gene). Therefore, examination of the number and type of gene regions recovered can help increase confidence in assigning species presence to environmental samples, which is a possible area of further study and testing. In addition, the detection of T. flabelliformis, T. pruinosa, and T. syncarpa, despite putting T. halocnemoides in the mixture, is likely due to the poor discrimination of these taxa for this genus. The species boundaries for this genus are poorly resolved with chloroplast-based data sets (e.g., Shepherd et al., 2004), and these taxa may be forming hybrids, an observation consistent with other data sets under study (E. Biffin and M. Waycott, personal communication). Unfortunately, due to these factors, we cannot tease apart the exact cause of these false positive detections but, due to the reason listed above, and the fact they are of the same genus, we can still view this as a correct detection within the limits of our bait set.
Proof of Concept
Using the 20 chloroplast gene regions targeted in this study, we were able to accurately determine the presence of A. marina in an unknown environmental sample and detect the family Scrophulariaceae and the order Lamiales. The subsequent verification of the presence of A. marina through observation, demonstrates the power in multiple gene regions to accurately detect flowering plant species within environmental samples. Additionally, detecting the family Scrophulariaceae may mean there are plants from this family present at the site. However, recovering only two gene regions and taxonomically resolving to only family level does not instil confidence in this conclusion. Interestingly, M. insulare, a species from the family Scrophulariaceae is known to inhabit areas close to the study site, and thus is possibly being detected. However, since the evidence is not strong we conclude just the presence of A. marina.
Future Directions
The limitations of PCR-amplifying a single universal barcode for plants means capturing the genetic variation required to disentangle species concepts across all flowering plant groups is difficult. Hence, we have developed a universal bait set that targets multiple regions in a single assay to create redundancy in our detection ability as we do not rely on one region but 20, to provide evidence of species presence. While we acknowledge PCR-amplification can be conducted separately across many genes, this cannot be completed in a single assay, making this a costly and time-consuming process while still being subject to PCR bias and errors. Targeted capture may be more time efficient but is still currently expensive. Fortunately, with technology constantly evolving, costs will start to decrease and already research has been conducted into ways to reduce target capture costs across the whole process (Hale et al., 2020).
Furthermore, current research is exploring the minimum number of gene regions that need to be captured for species level classifications across flowering plant groups (Foster et al., 2021, unpublished data). This can help focus efforts on the most useful gene regions to reduce sequencing genes that are not useful and overall, help to capture more taxa for less sequencing effort. Despite this, chloroplasts may not always be capable of separating some closely related taxa and therefore, future studies could work towards incorporating existing nuclear bait sets, e.g., Angiosperm v.1 kit by Buddenhagen et al. (2016), Angiosperms353 v1 kit by Johnson et al. (2019), or RAPiD genomics Angio408 kit (Gainesville, FL, United States). Consideration will need to be taken when working with nuclear DNA within environmental samples as this is far less abundant that plastid DNA (lower copy number). Therefore, it is advised to conduct separate hybridising reactions for chloroplast and nuclear DNA. This additional data could improve the ability to disentangle more difficult species concepts in unknown mixtures and form more accurate conclusions around flowering plant presence in environmental samples. This can then be used to accurately identify whether species have been lost from a system or detect invasive species as well as track food webs and reconstruct past flowering plant communities.
Conclusion
The proliferation of eDNA studies highlights a growing interest in methods for environmental monitoring (Beng and Corlett, 2020). While current DNA metabarcoding using a single gene region provides a tool for monitoring our natural environments (Balint et al., 2018), the application and interpretation of this data relies on accurate taxonomic identification of sequences recovered from an environmental sample (Pedersen et al., 2015). We have shown that a targeted capture approach can recover multiple species in a mixture and assign taxonomy across a large number of flowering plant groups. Obtaining a suite of genetic data across diverse plant taxa, equips us with the ability to generate reliable conclusions regarding plant communities in environmental samples, which can be used to improve monitoring, management, and conservation outcomes. This study is the first to apply a universal bait set to capture multiple gene regions from environmental samples in a single assay, and has demonstrated the breadth of genetic information that can be recovered, which far outweighs that of metabarcoding a single gene region. Whilst further study is needed to ensure correct interpretation of this data, if applied correctly, it can enable a more reliable and accurate method to determine plant presence in environmental samples.
Data Availability Statement
All raw, demultiplexed and script files are available on Figshare with the DOI: 10.25909/15049151. Sediment samples for the proof-of-concept section are available on Sequence Read Archive server with the BioProject ID: PRJNA749388 with file names 2.5a and 2.5b.
Author Contributions
NF, JY, and MW conceived the ideas. NF, JY, MW, K-JD, and EB designed the methodology. NF collected the data, ran the experiments, and wrote the manuscript. NF and VT analysed the data. MW advised on interpretation of analysis and results. All authors contributed to editing and preparing the manuscript.
Funding
This work was supported by a Herman Slade Foundation grant (HSF 18-2) (University of Adelaide grant 0006005104) awarded to AJ. A Goyder Institute research grant (CA-16-04) (University of Adelaide grant UA170835). A Max Day Environmental Fellowship awarded by the Australian Academy of Science to NF and a Research Training Program Scholarship awarded to NF. Collection of plant and soil samples were conducted under the State Herbarium of South Australia permit G25787-3.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This research was undertaken on the lands of the Kaurna people. We acknowledge and respect their spiritual relationship with their country, their cultural and heritage beliefs, and their rights as the Traditional Owners of the land. We would like to thank Lucy and Emma for their assistance with field work. We would also like to thank the three helpful reviewers who provided their time and expertise to make this a better manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.735744/full#supplementary-material
Footnotes
- ^ https://ftp.ncbi.nlm.nih.gov/refseq/release/plastid/, accessed October 2017.
References
Balint, M., Pfenninger, M., Grossart, H. P., Taberlet, P., Vellend, M., Leibold, M. A., et al. (2018). Environmental DNA time series in ecology. Trends Ecol. Evol. 33, 945–957. doi: 10.1016/j.tree.2018.09.003
Beng, K. C., and Corlett, R. T. (2020). Applications of environmental DNA (eDNA) in ecology and conservation: opportunities, challenges and prospects. Biodivers. Conserv. 29, 2089–2121. doi: 10.1007/s10531-020-01980-0
Brown, S. K., and Blois, J. L. (2016). “Ecological insights from ancient DNA,” In eLS, ed. John Wiley & Sons, Ltd (Chichester: John Wiley & Sons, Ltd). doi: 10.1002/9780470015902.a0026352
Buddenhagen, C., Lemmon, A. R., Lemmon, E. M., Bruhl, J., Cappa, J., Clement, W. L., et al. (2016). Anchored phylogenomics of angiosperms I: assessing the robustness of phylogenetic estimates. BioRxiv [Preprint] doi: 10.1101/086298
Corinaldesi, C., Beolchini, F., and Dell’anno, A. (2008). Damage and degradation rates of extracellular DNA in marine sediments: implications for the preservation of gene sequences. Mol. Ecol. 17, 3939–3951. doi: 10.1111/j.1365-294x.2008.03880.x
Del Carmen Gomez Cabrera, M., Young, J. M., Roff, G., Staples, T., Ortiz, J. C., Pandolfi, J. M., et al. (2019). Broadening the taxonomic scope of coral reef palaeoecological studies using ancient DNA. Mol. Ecol. 28, 2636–2652. doi: 10.1111/mec.15038
Dowle, E. J., Pochon, X., Banks, J. C., Shearer, K., and Wood, S. A. (2016). Targeted gene enrichment and high-throughput sequencing for environmental biomonitoring: a case study using freshwater macroinvertebrates. Mol. Ecol. Resour. 16, 1240–1254. doi: 10.1111/1755-0998.12488
Fahner, N. A., Shokralla, S., Baird, D. J., and Hajibabaei, M. (2016). Large-scale monitoring of plants through environmental DNA metabarcoding of soil: recovery, resolution, and annotation of four DNA markers. PLoS One 11:e0157505. doi: 10.1371/journal.pone.0157505
Ficetola, G. F., Taberlet, P., and Coissac, E. (2016). How to limit false positives in environmental DNA and metabarcoding? Mol. Ecol. Resour. 16, 604–607. doi: 10.1111/1755-0998.12508
Fordham, D. A., Akçakaya, H. R., Alroy, J., Saltré, F., Wigley, T. M. L., and Brook, B. W. (2016). Predicting and mitigating future biodiversity loss using long-term ecological proxies. Nat. Clim. Change 6, 909–916. doi: 10.1038/nclimate3086
Foster, N. R., Gillanders, B. M., Jones, A. R., Young, J. M., and Waycott, M. (2020). A muddy time capsule: using sediment environmental DNA for the long-term monitoring of coastal vegetated ecosystems. Mar. Freshw. Res. 71, 869–876. doi: 10.1071/mf19175
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Giguet-Covex, C., Pansu, J., Arnaud, F., Rey, P. J., Griggo, C., Gielly, L., et al. (2014). Long livestock farming history and human landscape shaping revealed by lake sediment DNA. Nat. Commun. 5:3211.
Glenn, T. C., Nilsen, R. A., Kieran, T. J., Finger, J. W., Pierson, T. W., Bentley, K. E., et al. (2019). Adapterama I: universal stubs and primers for thousands of dual-indexed Illumina libraries (iTru & iNext). BioRxiv [Preprint] doi: 10.1101/049114
Hale, H., Gardner, E. M., Viruel, J., Pokorny, L., and Johnson, M. G. (2020). Strategies for reducing per-sample costs in target capture sequencing for phylogenomics and population genomics in plants. Appl. Plant Sci. 8:e11337.
Hermans, S. M., Buckley, H. L., and Lear, G. (2018). Optimal extraction methods for the simultaneous analysis of DNA from diverse organisms and sample types. Mol. Ecol. Resour. 18, 557–569. doi: 10.1111/1755-0998.12762
Hollingsworth, P. M., Graham, S. W., and Little, D. P. (2011). Choosing and using a plant DNA barcode. PLoS One 6:e19254. doi: 10.1371/journal.pone.0019254
Johnson, M. G., Pokorny, L., Dodsworth, S., Botigue, L. R., Cowan, R. S., Devault, A., et al. (2019). A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Syst. Biol. 68, 594–606. doi: 10.1093/sysbio/syy086
Lamb, E. G., Winsley, T., Piper, C. L., Friedrich, S. A., and Siciliano, S. D. (2016). A high-throughput belowground plant diversity assay using next-generation sequencing of the trnL intron. Plant Soil 404, 361–372. doi: 10.1007/s11104-016-2852-y
Lemmon, E. M., and Lemmon, A. R. (2013). High-throughput genomic data in systematics and phylogenetics. Annu. Rev. Ecol. Evol. Syst. 44, 99–121. doi: 10.1146/annurev-ecolsys-110512-135822
Lentz, D. L., Hamilton, T. L., Dunning, N. P., Tepe, E. J., Scarborough, V. L., Meyers, S. A., et al. (2021). Environmental DNA reveals arboreal cityscapes at the ancient maya center of Tikal. Sci. Rep. 11:12725.
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint] arXiv: 1303.3997,
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659.
Murchie, T. J., Kuch, M., Duggan, A. T., Ledger, M. L., Roche, K., Klunk, J., et al. (2020). Optimizing extraction and targeted capture of ancient environmental DNA for reconstructing past environments using the PalaeoChip Arctic-1.0 bait-set. Quat. Res. 99, 1–24.
NCBI Resource Coordinators (2018). Database resources of the national center for biotechnology information. Nucleic Acids Res. 46, D8–D13.
Pansu, J., Giguet-Covex, C., Ficetola, G. F., Gielly, L., Boyer, F., Zinger, L., et al. (2015). Reconstructing long-term human impacts on plant communities: an ecological approach based on lake sediment DNA. Mol. Ecol. 24, 1485–1498.
Parducci, L., Matetovici, I., Fontana, S. L., Bennett, K. D., Suyama, Y., Haile, J., et al. (2013). Molecular- and pollen-based vegetation analysis in lake sediments from central Scandinavia. Mol. Ecol. 22, 3511–3524.
Pedersen, M. W., Overballe-Petersen, S., Ermini, L., Der Sarkissian, C., Haile, J., Hellstrom, M., et al. (2015). Ancient and modern environmental DNA. Philos. Trans. R. Soc. B 370:20130383.
R Core Team (2018). R: A Language And Environment For Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ruppert, K. M., Kline, R. J., and Rahman, M. S. (2019). Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 17:e00547.
Sakamoto, W., and Takami, T. (2018). Chloroplast DNA dynamics: copy number, quality control and degradation. Plant Cell Physiol. 59, 1120–1127.
Schilbert, H. M., Rempel, A., and Pucker, B. (2020). Comparison of read mapping and variant calling tools for the analysis of plant NGS data. Plants 9:439. doi: 10.3390/plants9040439
Schubert, M., Ermini, L., Der Sarkissian, C., Jonsson, H., Ginolhac, A., Schaefer, R., et al. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using Paleomix. Nat. Protoc. 9, 1056–1082.
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Shepherd, K. A., Waycott, M., and Calladine, A. (2004). Radiation of the Australian Salicornioideae (Chenopodiaceae): based on evidence from nuclear and chloroplast DNA sequences. Am. J. Bot. 91, 1387–1397.
Shokralla, S., Gibson, J. F., King, I., Baird, D. J., Janzen, D. H., Hallwachs, W., et al. (2016). Environmental DNA barcode sequence capture: targeted, PCR-free sequence capture for biodiversity analysis from bulk environmental samples. BioRxiv [Preprint] doi: 10.1101/087437 BioRxiv: 087437,
Taberlet, P., Coissac, E., Pompanon, F., Gielly, L., Miquel, C., Valentini, A., et al. (2006). Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 35, e14–e14.
Thomsen, P. F., and Willerslev, E. (2015). Environmental DNA – An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18.
Waycott, M., Van Dijk, K.-J., and Biffin, E. (2021). A hybrid capture RNA bait set for resolving genetic and evolutionary relationships in angiosperms from deep phylogeny to intraspecific lineage hybridization. bioRxiv [Preprint] bioRxiv: 2021.09.06.456727,
Willerslev, E., Davison, J., Moora, M., Zobel, M., Coissac, E., Edwards, M. E., et al. (2014). Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51.
Willerslev, E., Hansen, A. J., Binladen, J., Brand, T. B., Gilbert, M. T. P., Shapiro, B., et al. (2003). Diverse plant and animal genetic records from holocene and pleistocene sediments. Science 300, 791–795.
Wu, X., Heffelfinger, C., Zhao, H. and Dellaporta, S. L. (2019). Benchmarking variant identification tools for plant diversity discovery. BMC Genomics 20:701.
Yao, Z., You, F. M., N’diaye, A., Knox, R. E., Mccartney, C., Hiebert, C. W., et al. (2020). Evaluation of variant calling tools for large plant genome re-sequencing. BMC Bioinformatics 21:360. doi: 10.1186/s12859-020-03704-1
Yoccoz, N. G., Brathen, K. A., Gielly, L., Haile, J., Edwards, M. E., Goslar, T., et al. (2012). DNA from soil mirrors plant taxonomic and growth form diversity. Mol. Ecol. 21, 3647–3655.
Zimmermann, H. H., Raschke, E., Epp, L. S., Stoof-Leichsenring, K. R., Schwamborn, G., Schirrmeister, L., et al. (2017). Sedimentary ancient DNA and pollen reveal the composition of plant organic matter in late quaternary permafrost sediments of the buor khaya peninsula (North-Eastern Siberia). Biogeosciences 14, 575–596.
Keywords: chloroplast, eDNA, hybridisation capture, metabarcoding, angiosperms
Citation: Foster NR, van Dijk K-j, Biffin E, Young JM, Thomson VA, Gillanders BM, Jones AR and Waycott M (2021) A Multi-Gene Region Targeted Capture Approach to Detect Plant DNA in Environmental Samples: A Case Study From Coastal Environments. Front. Ecol. Evol. 9:735744. doi: 10.3389/fevo.2021.735744
Received: 03 July 2021; Accepted: 27 September 2021;
Published: 25 October 2021.
Edited by:
Lisa Pokorny, National Institute for Agricultural and Food Research and Technology, SpainReviewed by:
Nathan Wales, University of York, United KingdomRoswitha Schmickl, Academy of Sciences of the Czech Republic (ASCR), Czechia
Dario Ojeda Alayon, Norwegian Institute of Bioeconomy Research, Norway
Copyright © 2021 Foster, van Dijk, Biffin, Young, Thomson, Gillanders, Jones and Waycott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole R. Foster, bmljb2xlLmZvc3RlckBhZGVsYWlkZS5lZHUuYXU=