 Jordi Paps
Jordi Paps Maria Eleonora Rossi2
Maria Eleonora Rossi2 Alexander M. C. Bowles
Alexander M. C. Bowles Marta Álvarez-Presas
Marta Álvarez-Presas- 1School of Biological Sciences, University of Bristol, Bristol, United Kingdom
- 2School of Earth Sciences, University of Bristol, Bristol, United Kingdom
- 3School of Geographical Sciences, University of Bristol, Bristol, United Kingdom
The Animal Kingdom is an astonishingly diverse group. Together with plants and fungi is one of the three major lineages of multicellular eukaryotes. Due to anthropocentrism and/or genuine scientific interest, their origin and diversification are pivotal to modern evolutionary biology. In the last few decades, dramatic technological advances in molecular biology and computational power have generated new phylogenetic proposals, as well as new tools to compare genomes or study cell type evolution. These new approaches complement the insights from fields such as comparative morphology, evodevo, or palaeontology, which all together provide an integrative view of animal evolution, including major evolutionary transitions such as the origin of animals or the emergence of animals with bilateral symmetry. In this paper, we review recent developments in animal phylogenetics, comparative genomics, and cell type evolution related to these two transitions, and we compare animals to another major lineage of multicellular eukaryotes, plants.
Assembling trees
Animals (Metazoa) are multicellular eukaryotic organisms loosely characterized by their ability to move, sense, and react to their environment, as well as consuming other organisms. They play fundamental roles in the biosphere and in many ecological processes. Most animals display bilateral symmetry, like worms, molluscs, arthropods, or vertebrates, forming a monophyletic group called Bilateria. While other animals —sponges, ctenophores, placozoans, and cnidarians— display other body plans (Paps, 2018).
Paraphrasing Theodosius Dobzhansky, “nothing makes sense in macroevolution except in the light of phylogeny.” A robust evolutionary tree is central to comparative biology, including the study of genomes or cell types. Early phylogenies were based on morphological traits, and the advent of PCR, expanded the range of characters to one or few genes. In the last 20 years (Delsuc et al., 2005; Philippe et al., 2005; Dunn et al., 2008), massive animal phylogenomic matrices containing hundreds or thousands of genes have dominated the narrative of animal evolution. Often, they have improved the resolution of major lineages like molluscs (Kocot et al., 2011; Smith et al., 2011), annelids (Struck et al., 2011), flatworms (Laumer and Giribet, 2014; Egger et al., 2015; Laumer et al., 2015), and insects (Rota-Stabelli et al., 2011; Misof et al., 2014). In contrast, the radiations of early animals and bilaterians have been more problematic, resulting in major controversies.
Previous articles have thoroughly reviewed the phylogeny of animals and its main problems (Edgecombe et al., 2011; Dunn et al., 2014; Jékely et al., 2015; Telford et al., 2015; Giribet, 2016; Ruiz-Trillo and Paps, 2016; Paps, 2018; Giribet and Edgecombe, 2019). Briefly, most pre-phylogenomic trees supported sponges as the sister group to other animals (Figure 1; hypothesis affectionately called #porosis or sponges-first; Hejnol, 2016). However, phylogenomic datasets have invariably been ambiguous with some supporing sponges-first (Philippe et al., 2009; Pick et al., 2010; Nosenko et al., 2013; Simion et al., 2017) and others recovering ctenophores in that same position (#ctenosis or ctenophores-first; Dunn et al., 2008; Hejnol et al., 2009; Ryan et al., 2013; Moroz et al., 2014). Later datasets, sometimes with expanded gene and taxa sampling, have controversially provided support for one hypothesis or the other based on the analytical methods used (see summary in Table 1 in Li et al., 2021). This debate has major consequences in our understanding of the nature of the last common ancestor (LCA) of all animals and the evolution of essential animal traits such as the nervous or muscle systems.
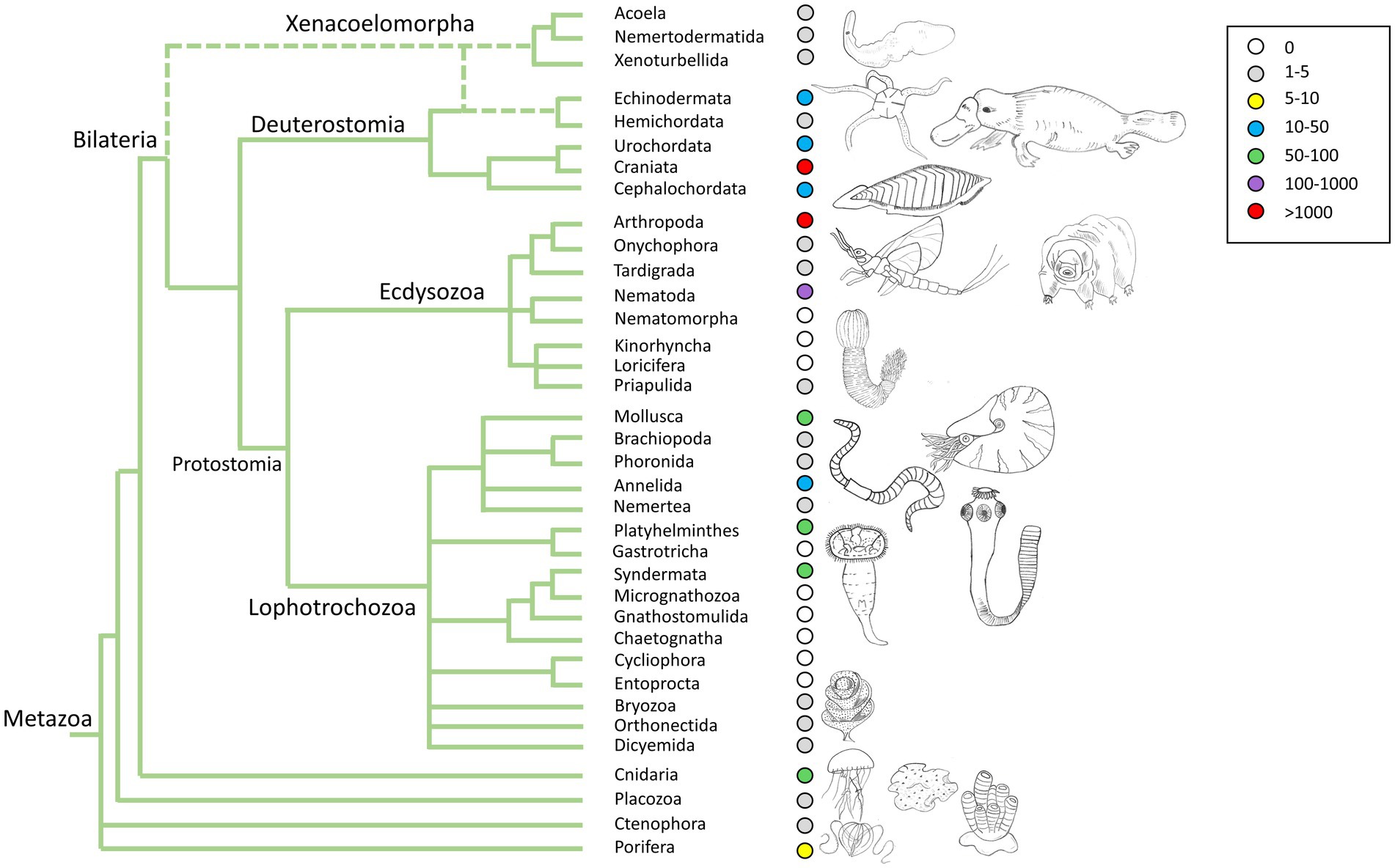
Figure 1. Genome data availability across the animal tree of life.
A similar controversy haunts the origin of bilateral animals (Hejnol and Pang, 2016; Ruiz-Trillo and Paps, 2016; Telford and Copley, 2016; Marlétaz, 2019; Pisani et al., 2022). The Xenoacoelomorpha (Xenoturbellida, Acoela, and Nemertodermatida; XAN from now on) is a lineage of simple animals central to this transition. Often linked to flatworms in morphological trees, early molecular phylogenies placed acoels and nemertodermatids as sister to the other bilaterians (Nephrozoa hypothesis; Ruiz-Trillo et al., 1999, 2002; Paps et al., 2009a), position backed by later phylogenomic analyses and expanded to all XAN (Hejnol et al., 2009; Cannon et al., 2016; Rouse et al., 2016). This would suggest that the first bilaterian was a simple benthic acoelomate animal with direct development and no brain (Baguñà et al., 2001; Baguñà and Riutort, 2004). However, other phylogenomic studies identified XAN as sister to the Ambulacraria (echinoderms and hemichordates; Xenambulacraria hypothesis), which sparked the War of the Worms (Telford and Copley, 2016). These studies recover (1) Xenambulacraria as sister to chordates, together forming the deuterostomes (Brinkmann et al., 2011) or, intriguingly, (2) Xenambulacraria as sister to all the other bilaterians, formed by chordates as sister to protostomes (Marlétaz, 2019; Marlétaz et al., 2019; Philippe et al., 2019). The later would effectively mean the demise of deuterostomes, although these topologies usually show low statistical support. In any case, the Xenambulacraria hypothesis implies that XAN body plans would be the result of body plan simplification rather than the ancestral condition of bilaterians.
These two controversies, origin of animals and bilaterians, share similar challenges. First, they both share branch length issues. Often short branches flank the nodes of interest, probably due to molecular saturation or rapid radiations. In addition, some of the key lineages (e.g., XAN, ctenophores) show fast evolutionary rates, making them susceptible to long branch attraction artifacts (LBA; Felsenstein, 1978). Amongst the different strategies that minimize the impact of LBA (Bergsten, 2005; Paps et al., 2009b), expanding the taxon sampling of fast evolving taxa to shorten the long branches has proven to be effective with different lineages such as nematodes or acoels (Aguinaldo et al., 1997; Ruiz-Trillo et al., 1999). Remarkably, the molecular phylogenies of extant ctenophores resemble a “duster,” with a long branch from their first ancestor to the most recent one, with all the extant taxa radiating in a recent short period of time. This entails a relatively young ctenophoran LCA (Podar et al., 2001), with earlier ctenophore lineages lost to time due to extinction; the recent discoveries of new stem fossil ctenophores seem to support this view (Ou et al., 2015; Zhao et al., 2019). Therefore, expanding the taxon sampling of extant ctenophores may not help to shorten their branch, although new environmental DNA studies indicate that we might be able to find new undescribed lineages (Arroyo et al., 2016; Christianson et al., 2022).
All these branch problems reflect the heterogeneity of molecular data, especially in key lineages like XAN or ctenophores, where the evolutionary models available may fail to adequately describe the data (Giacomelli et al., 2022), possibly resulting in model misspecification and inaccurate trees. Different approaches aim to reduce the impact of compositional rate, including the CAT model (Lartillot and Philippe, 2004; Lartillot Brinkmann et al., 2007; Philippe et al., 2009; Pick et al., 2010; Pisani et al., 2015), recoding (Feuda et al., 2017; Redmond and McLysaght, 2021; Giacomelli et al., 2022), expansion of gene and taxon sampling (Laumer et al., 2019), or —conversely— the significant removal markers that introduce phylogenetic noise (Mulhair et al., 2022; Pisani et al., 2022; McCarthy et al., 2023). In general, these approaches favor sponges-first and Xenambulacraria. However, some of these strategies have been criticized in studies that generally support ctenophores-first (Whelan et al., 2017; Whelan and Halanych, 2017; Hernandez and Ryan, 2021; Li et al., 2021). Moreover, sources of systematics errors have also been identified in the phylogeny of early bilaterians, which require further consideration when tackling these phylogenetic questions (Kapli and Telford, 2020; Natsidis et al., 2021; Kapli et al., 2021a,b).
A second common issue in these controversies stems from the datasets used. Most phylogenomic studies rely on data mostly derived from transcriptomes rather than whole genomes. Transcriptomes are a convenient and affordable way to expand taxon and gene sampling, at the expense of only capturing a subset of all the genes and isoforms present in an organism, the ones highly expressed in the tissue and/or developmental stages (usually adults) sampled to generate the data. Thus, transcriptomes do not reflect the full set of genes in an organism, and some genes will be missing, truncated, or split into two or more transcripts. These issues introduce missing data into matrices and confound the orthology assignment (Paps and Holland, 2018; Bowles et al., 2020; Guijarro-Clarke et al., 2020), an essential step in phylogenetic inference. The use of only complete genomes should overcome these concerns but, unfortunately, not many new genomes for sponges or ctenophores have been released in the recent years, with exceptions (Kenny et al., 2020). Genomes for XAN representatives have started to be available only very recently (Figure 1; Philippe et al., 2019; Martinez et al., 2022; Schiffer et al., 2022).
Generating new data is central to moving away from previous datasets. As a result of the lack of new data, many recent studies reanalyze earlier datasets, some over 10 years old, with new methods rather than compiling new dataset matrices. These analyses provide many interesting insights into methodological issues and datasets behaviors, but overall they do not provide a conclusive answer. For external observers, the succession of publications with opposing outcomes may resemble an endless tennis match with no resolution in sight. The lack of data has also introduced alternative interpretations of the analytical outcomes and the general outlook of ancestors. Sometimes a topology is favored because it is retrieved in most analyses even if suboptimal methods were used, following an implicit “democratic” criterion rather than integrating method optimality into the discrimination of topologies. In others, a topology is rejected because it is recovered by suboptimal approaches affected by systematic errors, or supported by simulations that integrate systematic error; however, in these cases no alternative hypothesis is strongly supported by optimal approaches or empirical data. Even a broken clock is right twice a day, and scientific hypotheses must be rejected using the best methods rather than flawed ones.
More genomes are being quickly produced by initiatives like the Darwin Tree of Life, Earth Biogenome, or the Global Invertebrate Genomics Alliance (Bracken-Grissom et al., 2014; Wellcome Sanger Institute, 2020). This will finally allow the generation of new datasets based on complete genomes, reducing the issues with orthology, hopefully moving the field forward from the current phylotranscriptomics era to a real phylogenomics stage. Complete genomes will also improve the curation of genome databases, reducing issues with contaminant sequences, misannotated genes, genes without functional annotation, etc. Moreover, this new high quality data will offer the chance to exploit genome-level information to reconstruct phylogenies and developing new methods, in addition to classical phylogenomic analyses, fulfilling the promise of rare genomic changes (Rokas and Holland, 2000). These include characters such indels, microRNAs, ancient linkage groups, etc. A recent example of this is the use of gene content to infer the phylogeny of animals, supporting porifera-first and Nephrozoa (Pett et al., 2019; Juravel et al., 2022; but see Schiffer et al., 2022). These recent developments and others to come will certainly move forward our understanding of animal evolution.
Assembling genomes
The characterization of the genetic toolkit in early animals is one of the main research goals of evolutionary biologists (Dunn and Ryan, 2015). Inferring the evolution of gene gains and losses, gene duplications, or horizontal gene transfer at different key points of the tree of life could help to better understand how extant species have evolved. In addition, it might answer some of the questions that have led to debate in recent years and that have been discussed in the previous section, such as whether the sponges or ctenophores came first or what is the phylogenetic position of XAN.
With the advent of next-generation sequencing (NGS) methods, a wealth of genomic data is available for a larger diversity of organisms. However, the taxon sampling is still highly biased in terms of the quality of genomes (referred to as reference genomes) and taxonomic diversity, which hinders the comprehensive comparative study at the kingdom level (Figure 1). As mentioned above, there are several initiatives trying to speed up the sequencing process of all existing animal species’ genomes. However, there are still some underrepresented groups, mainly due to the difficulty in sampling and obtaining good quality data. The comparative genomics methods advance almost at the same speed as the number of available genomes, ranging from whole genome comparisons to analysis of gene expression. The integration of machine and deep learning in the study of genomes is also advancing rapidly, beginning to replace traditional comparison methods, which are much slower and computationally imprecise (Zou et al., 2018). One of the critical points for conducting comparative genomics studies is the definition of orthologs and paralogs. In recent years, new sophisticated but also faster methods have emerged for the detection of orthologs that are facilitating robust genomic comparisons (Paps and Holland, 2018; Emms and Kelly, 2019; Miller et al., 2019; Buchfink et al., 2021; Grau-Bové and Sebé-Pedrós, 2021).
Evolutionary genomics can reconstruct ancestral animal genomes, inferring ancient gene complements or, more recently, even chromosome-level ancestral genomes (Nakatani and McLysaght, 2017; Simakov et al., 2020, 2022; Nakatani et al., 2021). Some preconceptions that have been rejected with the advancement of genomic study are the correlation between the number of genes and complexity (Dunn and Ryan, 2015), although the definition of organismal complexity has been always complex. Still, gene gains and losses play a significant role in evolution, with gene loss being associated with the loss of anatomical structures in evolution, in accordance with the view that evolution can lead to both increases and decreases in complexity (Lankester, 1880). The prevalence of gains and losses of genes and protein domains at the dawn of distinct groups of animals have been demonstrated (Albalat and Cañestro, 2016; Guijarro-Clarke et al., 2020).
A key element of comparative biology is the use of outgroups to polarise evolutionary changes. In recent years, the genomes of the close relatives of animals have become increasingly available. One of the main divisions of the eukaryotic Tree of Life is Opisthokonta, which includes fungi and choanoflagellates, some single-celled taxa such as Ichthyosporea or Filasterea, and of course, metazoans (or animals). Unicellular eukaryotes are key to revealing the origin of animal multicellularity (Paps et al., 2010; Sebé-Pedrós et al., 2017; Paps, 2018). The analysis of animal genomes and their close relatives in a phylogenetic context, facilitated the reconstruction of the minimum gene complement present in the genome of the last common ancestor of all animals, revealing an unprecedented increase in the degree of genomic novelty during the origin of metazoans (Paps and Holland, 2018). This genomic novelty involves biological functions characteristic of animal multicellularity (Brunet and King, 2017), especially gene regulation (e.g., transcription factors, signaling pathways) but also cell adhesion and the cell cycle. However, the functions and roles of these genes in opisthokonts are still pending to be deciphered.
Analyzing a large number of genomes, a reductive evolution pattern was observed on protein-coding genes complement, with a notable loss of genes during the emergence of two main groups of bilateral animals, Ecdysozoa and Deuterostomia (Guijarro-Clarke et al., 2020). At the phylum level, flatworms, nematodes, and tardigrades showed the greatest reduction in genetic complement, along with genetic novelty. A parallel study using different methods and datasets obtained remarkably similar results, describing that the origin of animals was characterized by the duplication of genes (Fernández and Gabaldón, 2020). Using transcriptome data, they incorporated the XAN to the dataset, which was characterized by rampant gene loss. Significant gene loss was also detected in Deuterostomia and Ecdysozoa. Novel genes in all nodes from Metazoa to specific phyla were enriched in functions related to the nervous system, suggesting that this system has been continuously and independently reformed throughout evolution inanimals. Thus, it appears that numerous duplication events occurred at the origin of the animals, followed later by the massive loss of some genes in certain lineages. These big gene losses are contrary to a “supposed” increase in complexity during evolution.
However, the taxon sampling for high quality genomes is still too scarce to be able to draw clear conclusions (Figure 1), and the picture may change with the addition of genomes in key phyla, such as Priapulida, Kinorhyncha, Gnathostomulida, and Chaetognatha, for example. The effect of taxon sampling bias is also a critical issue to address, as it can completely change the results of studies, as well as which external group is used for genomic comparison (Richter et al., 2018) and the detection of gene orthology also present serious challenges (Weisman et al., 2020; Natsidis et al., 2021).
Assembling cells
Understanding the evolution of cell types is key to reconstruct the evolutionary history of animals. We know that one of the major events that shaped animal evolution is the acquisition of multicellularity. In eukaryotes, it has evolved independently at least 25 times (Parfrey and Lahr, 2013), and the last common ancestor of animals was multicellular (Brunet and King, 2017). The diversification of the Animal Kingdom is concomitant to the enormous number of cell types they present. However, how multicellularity evolved and what shaped the diversity of cell types remains an open question.
Multicellularity is the result of a single cell dividing and differentiating into cell types, that differ in functions, morphology, and organization. It requires a process of spatial and temporal differentiation, and the resulting cell identity is regulated by a hierarchy of gene regulation (Arendt, 2008). With the recent development of NGS, cell identity was strictly associated by gene regulatory networks and the expression of specific transcription factors, and it became necessary to extend the definition of cell types, including the cell-type specific regulatory mechanisms (Arendt et al., 2016; Wang et al., 2021). Moreover, it has been suggested that cells undergo evolutionary changes, and they can be considered evolutionary units (Arendt et al., 2016). Historically, similar cell types of different animals have always been compared to one another to highlight similarities, question homology, and finally understand the evolutionary history of animals. This is the case of choanoflagellates compared to the choanocytes of the sponges, and the nervous system in ctenophores and other animals (Mah et al., 2014; Moroz et al., 2014; Jékely et al., 2015; Moroz and Kohn, 2016; Sogabe et al., 2019).
During the last decade, the advent of single cell RNA sequencing (scRNA-seq) made it possible to investigate gene expression at the cellular level. This enabled the mapping of all the different cell types present in an organism or a tissue (i.e., Atlas) using gene regulatory networks (Fiers et al., 2018) and gave the possibility to pose phylogenies of closely related cell types (Kin et al., 2015; Posada, 2020). In addition, it also showed the potential of comparative studies to question the origin of animals and their cell types, when applied to the earliest-branching taxa in the Animal Tree of Life, being sponges, ctenophores, cnidarians, and placozoan (Tanay and Sebé-Pedrós, 2021). The first atlases generated for early metazoans showed a greater variety of cell lineages within species, identified by specific transcription factors. Across species comparison of similar cell types highlighted that most genes involved in co-regulation have evolved independently (e.g., convergent evolution), with only housekeeping function, ribosomal apparatus and flagellar apparatus being conserved. Moreover, genes expressed more broadly across tissues have older phylogenetic origin, while genes expressed in a subset of tissues should be considered more recent; this trend is reflected in the cell lineages as well (Sebé-Pedrós et al., 2018). A similar approach has been used in bilaterians, where muscle cells show conserved markers shared among epithelial cells, like cadherin involved in cell–cell adhesion, but across taxa comparison underline the presence of clade-specific transcripts, as in the case of XAN, which may be linked to specific cell types (Duruz et al., 2021).
Finally, an early hierarchical clustering analysis of cell types has been performed across animals, including the cnidarian Nematostella and bilaterian species model (Wang et al., 2021). Similarities have been found among stromal cells, smooth muscle cells, and endothelial cells which form a cluster with neurons closely related, this entire cluster is related to a second major cluster formed by stem cells, epithelial cells, and striated muscle cells, and highlights the amount of genes shared among different cell types across animals. These recent studies show that scRNA-seq can be a powerful tool to infer the evolutionary history of animals. However, this field is extremely young, and it faces many challenges. First being the restricted taxon sampling available, that does not reflect the diversity in the animal tree of life and in cell categories. The lack of knowledge regarding the gene regulatory networks that does not allow to characterize new cell types, followed by the plethora of sequencing techniques that have been developed in the last 10 years that provide a broad range of sequencing depth and sensibility, which makes it difficult to compare data. Finally, there is no shared consensus across the scientific community regarding the bioinformatic tools needed to correctly handle and analyze the scRNA-seq data under a phylogenomic framework. Nevertheless, scRNA-seq is a useful and powerful technique, that will allow us to understand better how cells function, and possibly disentangle the evolutionary history of animals if applied wisely.
Assembling plants
As highlighted above, insights from the analysis of genes, genomes and cell types have revealed that there are complex mechanisms regulating the development of animals. Plants represent another evolutionary distinct group with complex evolutionary development, with common elements emerging convergently in both plants and animals. Indeed, Szathmáry and Smith (1995) defined the evolution of multicellularity in animals, plant and fungi as a major evolutionary transition. However, the extent to which the mechanisms and innovations governing the origin of animals are unique or ubiquitous across multicellular organisms is only now being understood. As such, evolutionary developmental analyses are contrasted below, with parallels drawn between land plants (embryophytes) and animals.
Land plants are divided into the vascular plants (tracheophytes) and bryophytes, which originated from a single common ancestor that emerged onto land approximately 500 million years ago (Morris et al., 2018). The radiation of land plants changed the biosphere, enabling the establishment of terrestrial animal life (Julca et al., 2021). In comparison to animals, the phylogeny of land plant evolution is reasonably well resolved with bryophytes considered as a monophyletic group that is sister to vascular plants (Leebens-Mack et al., 2019; Harris et al., 2020). The closest relatives of land plants are the streptophyte algae, a paraphyletic group, with Zygnematophyceae identified as sister group to land plants (Wickett et al., 2014; Leebens-Mack et al., 2019).
Common features required for plant life on land and therefore present in the first land plants are rhizoids (root-like structures), stomata (pores) and the alternation of generations (Harrison, 2017). The latter of these involves two distinct phases in the plant life cycle, alternating between sporophyte (non-sexual phase) and gametophyte (sexual phase) forms. Additionally, three-dimensional growth was present in the ancestor of land plants. This is juxtaposed to streptophyte algal relatives which represent a plethora of forms with filamentous Zygnematophyceae, multicellular two-dimensional Coleochaetophyceae, three-dimensional Charophyceae and single-celled Mesostigmatophyceae (Umen, 2014). Therefore, the evolution of multicellularity and terrestrialisation occurred at distinct points in the evolution of plants (Hess et al., 2022), which contrasts with animals, whose obligated multicellular origins coincided with the origin of Metazoa (Bowles et al., 2020). There are additional examples of convergent evolution of multicellularity, with multicellular lineages found outside the streptophytes, in the chlorophytes and rhodophytes (Parfrey and Lahr, 2013; Bowles et al., 2022).
Comparative analysis of transcriptomes and genomes of streptophyte algae and land plants is beginning to reveal the evolutionary novelties associated with life on land. For example, gene content analysis suggests that the transition of plants from water onto land (terrestrialisation) was preceded by major innovations previously thought to be land plant specific (Hori et al., 2014; Nishiyama et al., 2018; Cheng et al., 2019). These include the symbiotic association of plants with beneficial fungi (Delaux et al., 2015), a partial genetic toolkit for directing stress responses (Bowman et al., 2017; de Vries et al., 2018; de Vries and Archibald, 2018), as well as cell wall modifications (Nishiyama et al., 2018; Cheng et al., 2019; Jiao et al., 2020).
Recent genomic studies have shown that the evolution of plants was coordinated by the development of increasingly complex signaling molecules (Bowman et al., 2017) and genetic networks (Catarino et al., 2016). Furthermore, the genomes of early land plants was associated with gene family expansions related to cutin and lignin biosynthesis and phytohormone production (Bowman et al., 2017). Further analysis of diverse plant genomes identified that the origin of land plants was accompanied by an unprecedented level of genomic novelty, with a second smaller burst in the ancestor of streptophytes (Bowles et al., 2020). This is in comparison to a single burst seen in the origin of animals (Paps and Holland, 2018). These patterns are reinforced based on the analysis from the one thousand plant transcriptome project, which identified gene family birth and expansion in some of the largest plant gene families in Streptophyta and land plants (Leebens-Mack et al., 2019).
These molecular innovations led to the evolution of embryogenesis in land plants, one of the defining features of embryophytes. In multicellular organisms, embryogenesis begins simply with a single cell, the zygote. This is true for both multicellular plants and animals, but the subsequent stages of development differ drastically (Radoeva et al., 2019). Comparative analysis of embryonic and post-embryonic transcriptomes in Arabidopsis thaliana revealed a unique transcriptomic profile coordinating embryo development which is distinct from all other tissue types (Hofmann et al., 2019). Importantly for most land plants, after embryogenesis, the mature embryo does not represent the architecture of the adult organism. The stem cells of meristems, undifferentiated plant tissue (e.g., shoots, roots), develop after embryonic development, defining different plant cell and tissue types (Radoeva et al., 2019). Since the origin of land plants, major plant groups have emerged, accompanied by the emergence of distinct plant cell types (e.g., xylem and phloem cells of vascular plants).
One of the main techniques, scRNA-seq, is used to understand animal cell types, as highlighted above, and only very recently in plants (Seyfferth et al., 2021; Otero et al., 2022; Tung et al., 2023). Recent analysis using scRNA-seq of 10,000 A. thaliana root cells identified all major cell and tissue types across multiple developmental stages, including the root cap, epidermis, and endodermis as well as xylem and phloem cells (Ryu et al., 2019). Application of scRNA-seq has also been used to investigate cell differentiation in the shoot apical meristem, the above-ground organ, in maize. Similar to root cells, the shoot apical meristem has distinct transcriptomic signatures, which enabled the identification of cell types including epidermal, tip, meristem, and vascular cells (Satterlee et al., 2021). Both studies aimed to track the developmental trajectory of individual cells identifying key genes involved in cellular development and differentiation (Ryu et al., 2019; Satterlee et al., 2021). Ultimately, these analyses provide insights into the processes governing cell fate specification and the identification of distinct cell types.
Much of the work aiming to understand plant cell types has been completed in model (e.g., A. thaliana) and crop species (e.g., Zea mays; Denyer and Timmermans, 2022). However, there is an important debate about how well cell and tissue development in model and crop species represents processes of more distant evolutionary groups (e.g., bryophytes, lycophytes). To address this question, gene expression atlases were developed for different tissues (e.g., shoot and root meristems, spores, seeds) of 10 species from across the land plant phylogeny. Comparative analysis across species and tissues found highly conserved developmental transcriptomes with many gene groups identified as organ specific across phylogenetic distance (Julca et al., 2021). This is in contrast to the development of some animal cell types where a large number of clade specific genes are responsible for distinct cell types (Duruz et al., 2021). The first land plants evolved new reproductive structures such as spores and embryo sacs through coordinated changes in gene expression and co-option of existing genes. It was identified that cell specific gene groups did not accompany the origin of the corresponding tissue, rather that cell specific gene expression is correlated with the age of the gene group. Based on Gene Ontology annotations, these gene groups also have biological functions relevant with their associated tissues (Julca et al., 2021). These broad patterns of changes in gene expression are familiar to animal cell type evolution (Sogabe et al., 2019; Duruz et al., 2021; Tarashansky et al., 2021).
Conclusion
The incoming wave of genome data and ever-increasing computational power, together with the development of new theoretical and analytical frameworks, will no doubt provide many surprises and new insights into the origins of animals and bilaterians. A comprehensive and representative taxon sampling will always remain central to any comparative study, including phylogenetics, comparative genomics, and cell type analyses. Integrating genome-level phylogenomic trees, comparative genomics, and the study of cell types with comparative morphology, evodevo, and palaeobiology will pose a difficult but interesting challenge. Comparing these findings with the evolution of other eukaryotic groups, plants, and fungi— will unveil the genomic forces driving major evolutionary transitions. Altogether, the near future looks like a great place to be an evolutionary biologist.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
MR is funded by NERC DTP GW4+, AB is funded by the Leverhulme Trust (RPG-2020-199 “iDAPT”), and MÁ-P and JP are supported by the Wellcome Trust (210101/Z/18/Z) and the School of Biological Sciences (University of Bristol).
Acknowledgments
The authors would like to thank Davide Pisani (University of Bristol) and the two reviewers (PM and JM-D) for their feedback on this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguinaldo, A. M., Turbeville, J. M., Linford, L. S., Rivera, M. C., Garey, J. R., Raff, R. A., et al. (1997). Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387, 489–493. doi: 10.1038/387489a0
Albalat, R., and Cañestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17, 379–391. doi: 10.1038/nrg.2016.39
Arendt, D. (2008). The evolution of cell types in animals: emerging principles from molecular studies. Nat. Rev. Genet. 9, 868–882. doi: 10.1038/nrg2416
Arendt, D., Musser, J. M., Baker, C. V. H., Bergman, A., Cepko, C., Erwin, D. H., et al. (2016). The origin and evolution of cell types. Nat. Rev. Genet. 17, 744–757. doi: 10.1038/nrg.2016.127
Arroyo, A. S., López-Escardó, D., de Vargas, C., and Ruiz-Trillo, I. (2016). Hidden diversity of Acoelomorpha revealed through metabarcoding. Biol. Lett. 12:20160674. doi: 10.1098/rsbl.2016.0674
Baguñà, J., and Riutort, M. (2004). The dawn of bilaterian animals: the case of acoelomorph flatworms. BioEssays 26, 1046–1057. doi: 10.1002/bies.20113
Baguñà, J., Ruiz-Trillo, I., Paps, J., Loukota, M., Ribera, C., Baguna, J., et al. (2001). The first bilaterian organisms: simple or complex? New molecular evidence. Int. J. Dev. Biol. 45, 133–134. doi: 10.1387/ijdb.01450133
Bergsten, J. (2005). A review of long-branch attraction. Cladistics 21, 163–193. doi: 10.1111/j.1096-0031.2005.00059.x
Bowles, A. M. C., Bechtold, U., and Paps, J. (2020). The origin of land plants is rooted in two bursts of genomic novelty. Curr. Biol. 30, 530–536.e2. doi: 10.1016/j.cub.2019.11.090
Bowles, A. M. C., Williamson, C. J., Williams, T. A., Lenton, T. M., and Donoghue, P. C. J. (2022). The origin and early evolution of plants. Trends Plant Sci. 28, 312–329. doi: 10.1016/j.tplants.2022.09.009
Bowman, J. L., Kohchi, T., Yamato, K. T., Jenkins, J., Shu, S., Ishizaki, K., et al. (2017). Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cells 171, 287–304. doi: 10.1016/j.cell.2017.09.030
Bracken-Grissom, H., Collins, A. G., Collins, T., Crandall, K., Distel, D., Dunn, C., et al. (2014). The global invertebrate genomics Alliance (GIGA): developing community resources to study diverse invertebrate genomes. J. Hered. 105, 1–18. doi: 10.1093/jhered/est084
Brinkmann, H., Copley, R. R., Moroz, L. L., Nakano, H., Poustka, A. J., Wallberg, A., et al. (2011). Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 470, 255–258. doi: 10.1038/nature09676
Brunet, T., and King, N. (2017). The origin of animal multicellularity and cell differentiation. Dev. Cell 43, 124–140. doi: 10.1016/j.devcel.2017.09.016
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Cannon, J. T., Vellutini, B. C., Smith, J., Ronquist, F., Jondelius, U., and Hejnol, A. (2016). XAN is the sister group to Nephrozoa. Nature 530, 89–93. doi: 10.1038/nature16520
Catarino, B., Hetherington, A. J., Emms, D. M., Kelly, S., and Dolan, L. (2016). The stepwise increase in the number of transcription factor families in the Precambrian predated the diversification of plants on land. Mol. Biol. Evol. 33, 2815–2819. doi: 10.1093/molbev/msw155
Cheng, S., Xian, W., Fu, Y., Marin, B., Keller, J., Wu, T., et al. (2019). Genomes of subaerial Zygnematophyceae provide insights into land plant evolution. Cells 179, 1057–1067. doi: 10.1016/j.cell.2019.10.019
Christianson, L. M., Johnson, S. B., Schultz, D. T., and Haddock, S. H. D. (2022). Hidden diversity of Ctenophora revealed by new mitochondrial COI primers and sequences. Mol. Ecol. Resour. 22:283. doi: 10.1111/1755-0998.13459
de Vries, J., and Archibald, J. M. (2018). Plant evolution: landmarks on the path to terrestrial life. New Phytol. 217, 1428–1434. doi: 10.1111/nph.14975
de Vries, J., Curtis, B. A., Gould, S. B., and Archibald, J. M. (2018). Embryophyte stress signaling evolved in the algal progenitors of land plants. Proc. Natl. Acad. Sci. U. S. A. 115, 3471–3480. doi: 10.1073/pnas.1719230115
Delaux, P. M., Radhakrishnan, G. V., Jayaraman, D., Cheema, J., Malbreil, M., Volkening, J. D., et al. (2015). Algal ancestor of land plants was preadapted for symbiosis. Proc. Natl. Acad. Sci. U. S. A. 112, 13390–13395. doi: 10.1073/pnas.1515426112
Delsuc, F., Brinkmann, H., and Philippe, H. (2005). Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 6, 361–375. doi: 10.1038/nrg1603
Denyer, T., and Timmermans, M. C. P. (2022). Crafting a blueprint for single-cell RNA sequencing. Trends Plant Sci. 27, 92–103. doi: 10.1016/j.tplants.2021.08.016
Dunn, C. W., Giribet, G., Edgecombe, G. D., and Hejnol, A. (2014). Animal phylogeny and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 45, 371–395. doi: 10.1146/annurev-ecolsys-120213-091627
Dunn, C. W., Hejnol, A., Matus, D. Q., Pang, K., Browne, W. E., Smith, S. A., et al. (2008). Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749. doi: 10.1038/nature06614
Dunn, C. W., and Ryan, J. F. (2015). The evolution of animal genomes. Curr. Opin. Genet. Dev. 35, 25–32. doi: 10.1016/j.gde.2015.08.006
Duruz, J., Kaltenrieder, C., Ladurner, P., Bruggmann, R., Martìnez, P., and Sprecher, S. G. (2021). Acoel Single-Cell Transcriptomics: cell type analysis of a deep branching Bilaterian. Mol. Biol. Evol. 38, 1888–1904. doi: 10.1093/molbev/msaa333
Edgecombe, G. D., Giribet, G., Dunn, C. W., Hejnol, A., Kristensen, R. M., Neves, R. C., et al. (2011). Higher-level metazoan relationships: recent progress and remaining questions. Org. Divers. Evol. 11, 151–172. doi: 10.1007/s13127-011-0044-4
Egger, B., Lapraz, F., Tomiczek, B., Müller, S., Dessimoz, C., Girstmair, J., et al. (2015). A transcriptomic-phylogenomic analysis of the evolutionary relationships of flatworms. Curr. Biol. 25, 1347–1353. doi: 10.1016/j.cub.2015.03.034
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Felsenstein, J. (1978). Cases in which parsimony or compatibility methods will be positively misleading. Syst. Biol. 27, 401–410. doi: 10.1093/sysbio/27.4.401
Fernández, R., and Gabaldón, T. (2020). Gene gain and loss across the metazoan tree of life. Nat. Ecol. Evol. 4, 524–533. doi: 10.1038/s41559-019-1069-x
Feuda, R., Dohrmann, M., Pett, W., Philippe, H., Rota-Stabelli, O., Lartillot, N., et al. (2017). Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Curr. Biol. 27, 3864–3870.e4. doi: 10.1016/j.cub.2017.11.008
Fiers, M. W. E. J., Minnoye, L., Aibar, S., Bravo González-Blas, C., Kalender Atak, Z., and Aerts, S. (2018). Mapping gene regulatory networks from single-cell omics data. Brief. Funct. Genomics 17, 246–254. doi: 10.1093/bfgp/elx046
Giacomelli, M., Rossi, M. E., Lozano-Fernandez, J., Feuda, R., and Pisani, D. (2022). Resolving tricky nodes in the tree of life through amino acid recoding. iScience 25:105594. doi: 10.1016/j.isci.2022.105594
Giribet, G. (2016). New animal phylogeny: future challenges for animal phylogeny in the age of phylogenomics. Org. Divers. Evol. 16, 419–426. doi: 10.1007/s13127-015-0236-4
Giribet, G., and Edgecombe, G. D. (2019). “Perspectives in animal phylogeny and evolution: a decade late” in Perspectives on evolutionary and developmental biology essays for Alessandro Minelli. ed. G. Fusco (Padova (Italy): Padova University Press), 167–178.
Grau-Bové, X., and Sebé-Pedrós, A. (2021). Orthology clusters from gene trees with Possvm. Mol. Biol. Evol. 38, 5204–5208. doi: 10.1101/2021.05.03.442399
Guijarro-Clarke, C., Holland, P. W. H., and Paps, J. (2020). Widespread patterns of gene loss in the evolution of the animal kingdom. Nat. Ecol. Evol. 4, 519–523. doi: 10.1038/s41559-020-1129-2
Harris, B. J., Harrison, C. J., Hetherington, A. M., and Williams, T. A. (2020). Phylogenomic evidence for the Monophyly of bryophytes and the reductive evolution of stomata. Curr. Biol. 30, 2001–2012. doi: 10.1016/j.cub.2020.03.048
Harrison, C. J. (2017). Development and genetics in the evolution of land plant body plans. Philos. Trans. R. Soc. B Biol. Sci. 372:e2015.0490. doi: 10.1098/rstb.2015.0490
Hejnol, A. (@Hejnol_Lab) (2016). No “undetected systematic bias” in Chang et al detected so far (for ctenosis). Tweet. Available at: https://twitter.com/Hejnol_Lab/status/697740643209232384 (Accessed May 23, 2023).
Hejnol, A., Obst, M., Stamatakis, A., Ott, M., Rouse, G. W., Edgecombe, G. D., et al. (2009). Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. R. Soc. B Biol. Sci. 276, 4261–4270. doi: 10.1098/rspb.2009.0896
Hejnol, A., and Pang, K. (2016). XAN’s significance for understanding bilaterian evolution. Curr. Opin. Genet. Dev. 39, 48–54. doi: 10.1016/j.gde.2016.05.019
Hernandez, A. M., and Ryan, J. F. (2021). Six-state amino acid recoding is not an effective strategy to offset compositional heterogeneity and saturation in phylogenetic analyses. Syst. Biol. 70, 1–34. doi: 10.1093/sysbio/syab027
Hess, S., Williams, S. K., Busch, A., Irisarri, I., Delwiche, C. F., Vries, S.De, et al. (2022). A phylogenomically informed five-order system for the closest relatives of land plants. Curr. Biol. 32, 4473–4482. doi: 10.1016/j.cub.2022.08.022
Hofmann, F., Schon, M. A., and Nodine, M. D. (2019). The embryonic transcriptome of Arabidopsis thaliana. Plant Reprod. 32, 77–91. doi: 10.1007/s00497-018-00357-2
Hori, K., Maruyama, F., Fujisawa, T., Togashi, T., Yamamoto, N., Seo, M., et al. (2014). Klebsormidium flaccidum genome reveals primary factors for plant terrestrial adaptation. Nat. Commun. 5:3978. doi: 10.1038/ncomms4978
Jékely, G., Paps, J., and Nielsen, C. (2015). The phylogenetic position of ctenophores and the origin(s) of nervous systems. EvoDevo 6:1. doi: 10.1186/2041-9139-6-1
Jiao, C., Sørensen, I., Sun, X., Sun, H., Behar, H., Alseekh, S., et al. (2020). The Penium margaritaceum genome: hallmarks of the origins of land plants. Cells 181, 1097–1111. doi: 10.1016/j.cell.2020.04.019
Julca, I., Flores, M., Proost, S., Lindner, A.-C., Hackenberg, D., Steinbachova, L., et al. (2021). Comparative transcriptomic analysis reveals conserved transcriptional programs underpinning organogenesis and reproduction in land plants. Nat. Plants 7, 1143–1159. doi: 10.1038/s41477-021-00958-2
Juravel, K., Porras, L., Höhna, S., Pisani, D., and Wörheide, G. (2022). Exploring genome gene content and morphological analysis to test recalcitrant nodes in the animal phylogeny. bioRxiv :2021.11.19.469253. doi: 10.1101/2021.11.19.469253
Kapli, P., Flouri, T., and Telford, M. J. (2021a). Systematic errors in phylogenetic trees. Curr. Biol. 31, R59–R64. doi: 10.1016/J.CUB.2020.11.043
Kapli, P., Natsidis, P., Leite, D. J., Fursman, M., Jeffrie, N., Rahman, I. A., et al. (2021b). Lack of support for Deuterostomia prompts reinterpretation of the first Bilateria. Sci. Adv. 7:eabe2741. doi: 10.1126/SCIADV.ABE2741/SUPPL_FILE/ABE2741_SM.PDF
Kapli, P., and Telford, M. J. (2020). Topology-dependent asymmetry in systematic errors affects phylogenetic placement of Ctenophora and XAN. Sci. Adv. 6, 5162–5173. doi: 10.1126/SCIADV.ABC5162/SUPPL_FILE/ABC5162_SM.PDF
Kenny, N. J., Francis, W. R., Rivera-Vicéns, R. E., Juravel, K., de Mendoza, A., Díez-Vives, C., et al. (2020). Tracing animal genomic evolution with the chromosomal-level assembly of the freshwater sponge Ephydatia muelleri. Nat. Commun. 11, 1–11. doi: 10.1038/s41467-020-17397-w
Kin, K., Nnamani, M. C., Lynch, V. J., Michaelides, E., and Wagner, G. P. (2015). Cell-type phylogenetics and the origin of endometrial stromal cells. Cell Rep. 10, 1398–1409. doi: 10.1016/j.celrep.2015.01.062
Kocot, K. M., Cannon, J. T., Todt, C., Citarella, M. R., Kohn, A. B., Meyer, A., et al. (2011). Phylogenomics reveals deep molluscan relationships. Nature 477, 452–456.
Lartillot Brinkmann, H., Philippe, H., Lartillot, N., Brinkmann, H., and Philippe, H. (2007). Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 7:S4. doi: 10.1186/1471-2148-7-S1-S4
Lartillot, N., and Philippe, H. H. (2004). A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109. doi: 10.1093/molbev/msh112
Laumer, C. E., Fernández, R., Lemer, S., Combosch, D., Kocot, K. M., Riesgo, A., et al. (2019). Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. R. Soc. B Biol. Sci. 286:20190831. doi: 10.1098/rspb.2019.0831
Laumer, C. E., and Giribet, G. (2014). Inclusive taxon sampling suggests a single, stepwise origin of ectolecithality in Platyhelminthes. Biol. J. Linn. Soc. 111, 570–588. doi: 10.1111/bij.12236
Laumer, C. E., Hejnol, A., Giribet, G., Appeltans, W., Ahyong, S. T., Anderson, G., et al. (2015). Nuclear genomic signals of the “microturbellarian” roots of platyhelminth evolutionary innovation. elife 4, 2189–2202. doi: 10.7554/eLife.05503
Leebens-Mack, J. H., Barker, M. S., Carpenter, E. J., Deyholos, M. K., Gitzendanner, M. A., Graham, S. W., et al. (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. doi: 10.1038/s41586-019-1693-2
Li, Y., Shen, X. X., Evans, B., Dunn, C. W., and Rokas, A. (2021). Rooting the animal tree of life. Mol. Biol. Evol. 38, 4322–4333. doi: 10.1093/molbev/msab170
Mah, J. L., Christensen-Dalsgaard, K. K., and Leys, S. P. (2014). Choanoflagellate and choanocyte collar-flagellar systems and the assumption of homology. Evol. Dev. 16, 25–37. doi: 10.1111/ede.12060
Marlétaz, F. (2019). Zoology: worming into the origin of Bilaterians. Curr. Biol. 29, R577–R579. doi: 10.1016/j.cub.2019.05.006
Marlétaz, F., Peijnenburg, K. T. C. A., Goto, T., Satoh, N., and Rokhsar, D. S. (2019). A new Spiralian phylogeny places the enigmatic arrow Worms among Gnathiferans. Curr. Biol. 29, 312–318.e3. doi: 10.1016/j.cub.2018.11.042
Martinez, P., Ustyantsev, K., Biryukov, M., Mouton, S., Glasenburg, L., Sprecher, S. G., et al. (2022). Genome assembly of the acoel flatworm Symsagittifera roscoffensis, a model for research on body plan evolution and photosymbiosis. G3: Genes Genomes Genet. 13. doi: 10.1093/G3JOURNAL/JKAC336
McCarthy, C. G. P., Mulhair, P. O., Siu-Ting, K., Creevey, C. J., and O’Connell, M. J. (2023). Improving orthologous signal and model fit in datasets addressing the root of the animal phylogeny. Mol. Biol. Evol. 40, 1–16. doi: 10.1093/molbev/msac276
Miller, J. B., Pickett, B. D., and Ridge, P. G. (2019). JustOrthologs: a fast, accurate and user-friendly ortholog identification algorithm. Bioinformatics 35, 546–552. doi: 10.1093/bioinformatics/bty669
Misof, B., Liu, S., Meusemann, K., Peters, R. S., Donath, A., Mayer, C., et al. (2014). Phylogenomics resolves the timing and pattern of insect evolution. Science 1979, 763–767. doi: 10.1126/SCIENCE.1257570/SUPPL_FILE/1257570S9.XLS
Moroz, L. L., Kocot, K. M., Citarella, M. R., Dosung, S., Norekian, T. P., Povolotskaya, I. S., et al. (2014). The ctenophore genome and the evolutionary origins of neural systems. Nature 510, 109–114. doi: 10.1038/nature13400
Moroz, L. L., and Kohn, A. B. (2016). Independent origins of neurons and synapses: insights from ctenophores. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 371:20150041. doi: 10.1098/rstb.2015.0041
Morris, J. L., Puttick, M. N., Clark, J. W., Edwards, D., Kenrick, P., Pressel, S., et al. (2018). The timescale of early land plant evolution. Proc. Natl. Acad. Sci. 115, 2274–2283. doi: 10.1073/pnas.1719588115
Mulhair, P. O., McCarthy, C. G. P., Siu-Ting, K., Creevey, C. J., and O’Connell, M. J. (2022). Filtering artifactual signal increases support for XAN and Ambulacraria sister relationship in the animal tree of life. Curr. Biol. 32, 5180–5188.e3. doi: 10.1016/j.cub.2022.10.036
Nakatani, Y., and McLysaght, A. (2017). Genomes as documents of evolutionary history: a probabilistic macrosynteny model for the reconstruction of ancestral genomes. Bioinformatics 33, i369–i378. doi: 10.1093/BIOINFORMATICS/BTX259
Nakatani, Y., Shingate, P., Ravi, V., Pillai, N. E., Prasad, A., McLysaght, A., et al. (2021). Reconstruction of proto-vertebrate, proto-cyclostome and proto-gnathostome genomes provides new insights into early vertebrate evolution. Nat. Commun. 12, 1–14. doi: 10.1038/s41467-021-24573-z
Natsidis, P., Kapli, P., Schiffer, P. H., and Telford, M. J. (2021). Systematic errors in orthology inference and their effects on evolutionary analyses. iScience 24:102110. doi: 10.1016/j.isci.2021.102110
Nishiyama, T., Sakayama, H., de Vries, J., Buschmann, H., Saint-Marcoux, D., Ullrich, K. K., et al. (2018). The Chara genome: secondary complexity and implications for plant Terrestrialization. Cells 174, 448–464. doi: 10.1016/j.cell.2018.06.033
Nosenko, T., Schreiber, F., Adamska, M., Adamski, M., Eitel, M., Hammel, J., et al. (2013). Deep metazoan phylogeny: When different genes tell different stories. Mol. Phylogenet. Evol. 67, 223–233. doi: 10.1016/j.ympev.2013.01.010
Otero, S., Gildea, I., Roszak, P., Lu, Y., Vittori, V.Di, Bourdon, M., et al. (2022). A root phloem pole cell atlas reveals common transcriptional states in protophloem-adjacent cells. Nat. Plants 8, 954–970, doi: 10.1038/s41477-022-01178-y
Ou, Q., Xiao, S., Han, J., Sun, G., Zhang, F., Zhang, Z., et al. (2015). A vanished history of skeletonization in Cambrian comb jellies. Sci. Adv. 1, 1–9. doi: 10.1126/sciadv.1500092
Paps, J. (2018). What makes an animal? The molecular quest for the origin of the animal kingdom. Integr. Comp. Biol. 58, 654–665. doi: 10.1093/icb/icy036
Paps, J., Baguñà, J., and Riutort, M. (2009a). Bilaterian phylogeny: a broad sampling of 13 nuclear genes provides a new Lophotrochozoa phylogeny and supports a paraphyletic basal acoelomorpha. Mol. Biol. Evol. 26, 2397–2406. doi: 10.1093/molbev/msp150
Paps, J., Baguñà, J., Riutort, M., and Baguñà, J. (2009b). Lophotrochozoa internal phylogeny: new insights from an up-to-date analysis of nuclear ribosomal genes. Proc. R. Soc. B Biol. Sci. 276, 1245–1254. doi: 10.1098/rspb.2008.1574
Paps, J., and Holland, P. W. H. (2018). Reconstruction of the ancestral metazoan genome reveals an increase in genomic novelty. Nat. Commun. 9:1730. doi: 10.1038/s41467-018-04136-5
Paps, J., Ruiz-Trillo, I., and de Barcelona, U. (2010). “Animals and their unicellular ancestors” in Encyclopedia of Life Sciences (Chichester: John Wiley & Sons Ltd), 1–8.
Parfrey, L. W., and Lahr, D. J. G. (2013). Multicellularity arose several times in the evolution of eukaryotes. BioEssays 35, 339–347. doi: 10.1002/bies.201200143
Pett, W., Adamski, M., Adamska, M., Francis, W. R., Eitel, M., Pisani, D., et al. (2019). The role of homology and orthology in the phylogenomic analysis of metazoan gene content. Mol. Biol. Evol. 36, 643–649. doi: 10.1093/molbev/msz013
Philippe, H. H., Derelle, R., Lopez, P., Pick, K., Borchiellini, C., Boury-Esnault, N., et al. (2009). Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 19, 706–712. doi: 10.1016/j.cub.2009.02.052
Philippe, H. H., Lartillot, N., and Brinkmann, H. (2005). Multigene analyses of Bilaterian animals corroborate the Monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Mol. Biol. Evol. 22, 1246–1253. doi: 10.1093/molbev/msi111
Philippe, H., Poustka, A. J., Chiodin, M., Hoff, K. J., Dessimoz, C., Tomiczek, B., et al. (2019). Mitigating anticipated effects of systematic errors supports sister-group relationship between XAN and Ambulacraria. Curr. Biol. 29, 1818–1826.e6. doi: 10.1016/j.cub.2019.04.009
Pick, K. S., Philippe, H., Schreiber, F., Erpenbeck, D., Jackson, D. J., Wrede, P., et al. (2010). Improved phylogenomic taxon sampling noticeably affects non-bilaterian relationships. Mol. Biol. Evol. 27, 1983–1987. doi: 10.1093/molbev/msq089
Pisani, D., Pett, W., Dohrmann, M., Feuda, R., Rota-Stabelli, O., Philippe, H., et al. (2015). Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl. Acad. Sci. U. S. A. 112, 15402–15407. doi: 10.1073/pnas.1518127112
Pisani, D., Rossi, M. E., Marlétaz, F., and Feuda, R. (2022). Phylogenomics: is less more when using large-scale datasets? Curr. Biol. 32, R1340–R1342. doi: 10.1016/J.CUB.2022.11.019
Podar, M., Haddock, S. H., Sogin, M. L., and Harbison, G. R. (2001). A molecular phylogenetic framework for the phylum Ctenophora using 18S rRNA genes. Mol. Phylogenet. Evol. 21, 218–230. doi: 10.1006/mpev.2001.1036
Posada, D. (2020). CellCoal: coalescent simulation of single-cell sequencing samples. Mol. Biol. Evol. 37, 1535–1542. doi: 10.1093/molbev/msaa025
Radoeva, T., Vaddepalli, P., Zhang, Z., and Weijers, D. (2019). Evolution, initiation, and diversity in early plant embryogenesis. Dev. Cell 50, 533–543. doi: 10.1016/j.devcel.2019.07.011
Redmond, A. K., and McLysaght, A. (2021). Evidence for sponges as sister to all other animals from partitioned phylogenomics with mixture models and recoding. Nat. Commun. 12, 1–14. doi: 10.1038/s41467-021-22074-7
Richter, D. J., Fozouni, P., Eisen, M. B., and King, N. (2018). Gene family innovation, conservation and loss on the animal stem lineage. elife 7:e34226. doi: 10.7554/eLife.34226
Rokas, A., and Holland, P. W. H. (2000). Rare genomic changes as a tool for phylogenetics. Trends Ecol. Evol. 15, 454–459. doi: 10.1016/S0169-5347(00)01967-4
Rota-Stabelli, O., Campbell, L., Brinkmann, H., Edgecombe, G. D., Longhorn, S. J., Peterson, K. J., et al. (2011). A congruent solution to arthropod phylogeny: phylogenomics, microRNAs and morphology support monophyletic Mandibulata. Proc. R. Soc. B Biol. Sci. 278, 298–306. doi: 10.1098/rspb.2010.0590
Rouse, G. W., Wilson, N. G., Carvajal, J. I., and Vrijenhoek, R. C. (2016). New deep-sea species of Xenoturbella and the position of XAN. Nature 530, 94–97. doi: 10.1038/nature16545
Ruiz-Trillo, I., and Paps, J. (2016). Acoelomorpha: earliest branching bilaterians or deuterostomes? Org. Divers. Evol. 16, 391–399. doi: 10.1007/s13127-015-0239-1
Ruiz-Trillo, I., Paps, J., Loukota, M., Ribera, C., Jondelius, U., Baguna, J., et al. (2002). A phylogenetic analysis of myosin heavy chain type II sequences corroborates that Acoela and Nemertodermatida are basal bilaterians. Proc. Natl. Acad. Sci. U. S. A. 99, 11246–11251. doi: 10.1073/pnas.172390199
Ruiz-Trillo, I., Riutort, M., Littlewood, D. T., Herniou, E. A., Baguña, J., and And Baguna, J. (1999). Acoel flatworms: earliest extant bilaterian metazoans, not members of Platyhelminthes. Science 1979, 1919–1923.
Ryan, J. F., Pang, K., Schnitzler, C. E., Nguyen, A.-D., Moreland, R. T., Simmons, D. K., et al. (2013). The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342:1242592. doi: 10.1126/science.1242592
Ryu, K. H., Huang, L., Kang, H. M., and Schiefelbein, J. (2019). Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. 179, 1444–1456. doi: 10.1104/pp.18.01482
Satterlee, J. W., Strable, J., and Scanlon, M. J. (2021). Plant stem-cell organization and differentiation at single-cell resolution. Proc. Natl. Acad. Sci. U. S. A. 117, 33689–33699. doi: 10.1073/PNAS.2018788117
Schiffer, P. H., Natsidis, P., Leite, D. J., Robertson, H. E., Lapraz, F., Marlétaz, F., et al. (2022). The slow evolving genome of the xenacoelomorph worm Xenoturbella bocki. bioRxiv :2022.06.24.497508. doi: 10.1101/2022.06.24.497508
Sebé-Pedrós, A., Chomsky, E., Pang, K., Lara-Astiaso, D., Gaiti, F., Mukamel, Z., et al. (2018). Early metazoan cell type diversity and the evolution of multicellular gene regulation. Nat. Ecol. Evol. 2, 1176–1188. doi: 10.1038/s41559-018-0575-6
Sebé-Pedrós, A., Degnan, B. M., and Ruiz-Trillo, I. (2017). The origin of Metazoa: a unicellular perspective. Nat. Rev. Genet. 18, 498–512. doi: 10.1038/nrg.2017.21
Seyfferth, C., Renema, J., Wendrich, J. R., Eekhout, T., Seurinck, R., Vandamme, N., et al. (2021). Advances and opportunities in single-cell transcriptomics for plant research. Annu. Rev. Plant Biol. 72, 847–866. doi: 10.1146/annurev-arplant-081720-010120
Simakov, O., Bredeson, J., Berkoff, K., Marletaz, F., Mitros, T., Schultz, D. T., et al. (2022). Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 8:5884. doi: 10.1126/SCIADV.ABI5884/SUPPL_FILE/SCIADV.ABI5884_DATA_FILES_S1_TO_S5.ZIP
Simakov, O., Marlétaz, F., Yue, J. X., O’Connell, B., Jenkins, J., Brandt, A., et al. (2020). Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830. doi: 10.1038/s41559-020-1156-z
Simion, P., Philippe, H., Baurain, D., Jager, M., Richter, D. J., Di Franco, A., et al. (2017). A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 27, 958–967. doi: 10.1016/j.cub.2017.02.031
Smith, S. A., Wilson, N. G., Goetz, F. E., Feehery, C., Andrade, S. C. S., Rouse, G. W., et al. (2011). Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature 480, 364–367. doi: 10.1038/nature10526
Sogabe, S., Hatleberg, W. L., Kocot, K. M., Say, T. E., Stoupin, D., Roper, K. E., et al. (2019). Pluripotency and the origin of animal multicellularity. Nature 570, 519–522. doi: 10.1038/s41586-019-1290-4
Struck, T. H., Paul, C., Hill, N., Hartmann, S., Hösel, C., Kube, M., et al. (2011). Phylogenomic analyses unravel annelid evolution. Nature 471, 95–98. doi: 10.1038/nature09864
Szathmáry, E., and Smith, J. M. (1995). The major evolutionary transitions. Nature 374, 227–232. doi: 10.1038/374227a0
Tanay, A., and Sebé-Pedrós, A. (2021). Evolutionary cell type mapping with single-cell genomics. Trends Genet. 37, 919–932. doi: 10.1016/j.tig.2021.04.008
Tarashansky, A. J., Musser, J. M., Khariton, M., Li, P., Arendt, D., Quake, S. R., et al. (2021). Mapping single-cell atlases throughout metazoa unravels cell type evolution. elife 10:e66747. doi: 10.7554/eLife.66747
Telford, M. J., Budd, G. E., and Philippe, H. (2015). Phylogenomic insights into animal evolution. Curr. Biol. 25, 876–887. doi: 10.1016/j.cub.2015.07.060
Telford, M. J., and Copley, R. R. (2016). Zoology: war of the worms. Curr. Biol. 26, R335–R337. doi: 10.1016/j.cub.2016.03.015
Tung, C.-C., Kuo, S.-C., Yang, C.-L., Yu, J.-H., Huang, C.-E., Liou, P.-C., et al. (2023). Single-cell transcriptomics unveils xylem cell development and evolution. Genome Biol. Evol. 24. doi: 10.1186/s13059-022-02845-1
Umen, J. G. (2014). Green algae and the origins of multicellularity in the plant kingdom. Cold Spring Harb. Perspect. Biol. 6:a016170. doi: 10.1101/cshperspect.a016170
Wang, J., Sun, H., Jiang, M., Li, J., Zhang, P., Chen, H., et al. (2021). Tracing cell-type evolution by cross-species comparison of cell atlases. Cell Rep. 34:108803. doi: 10.1016/j.celrep.2021.108803
Weisman, C. M., Murray, A. W., and Eddy, S. R. (2020). Many, but not all, lineage-specific genes can be explained by homology detection failure. PLoS Biol. 18:e3000862. doi: 10.1371/journal.pbio.3000862
Wellcome Sanger Institute (2020). Darwin tree of life. Available at: https://www.darwintreeoflife.org/
Whelan, N. V., and Halanych, K. M. (2017). Who let the CAT out of the bag? Accurately dealing with substitutional heterogeneity in Phylogenomic analyses. Syst. Biol. 66, 232–255. doi: 10.1093/sysbio/syw084
Whelan, N. V., Kocot, K. M., Moroz, T. P., Mukherjee, K., Williams, P., Paulay, G., et al. (2017). Ctenophore relationships and their placement as the sister group to all other animals. Nat. Ecol. Evol. 1, 1737–1746. doi: 10.1038/s41559-017-0331-3
Wickett, N. J., Mirarab, S., Nguyen, N., Warnow, T., Carpenter, E., Matasci, N., et al. (2014). Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. 111, 4859–4868. doi: 10.1073/pnas.1323926111
Zhao, Y., Vinther, J., Parry, L. A., Wei, F., Green, E., Pisani, D., et al. (2019). Cambrian sessile, suspension feeding stem-group ctenophores and evolution of the comb jelly body plan. Curr. Biol. 29, 1112–1125.e2. doi: 10.1016/j.cub.2019.02.036
Keywords: metazoa, comparative genomics, phylogenetics, cell types, plants
Citation: Paps J, Rossi ME, Bowles AMC and Álvarez-Presas M (2023) Assembling animals: trees, genomes, cells, and contrast to plants. Front. Ecol. Evol. 11:1185566. doi: 10.3389/fevo.2023.1185566
Edited by:
Andreas Wanninger, University of Vienna, AustriaReviewed by:
Jose Maria Martin-Duran, Queen Mary University of London, United KingdomPedro Martinez, University of Barcelona, Spain
Copyright © 2023 Paps, Rossi, Bowles and Álvarez-Presas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordi Paps, am9yZGkucGFwc0BicmlzdG9sLmFjLnVr
†Present address: Marta Álvarez-Presas, Institute of Evolutionary Biology (UPF-CSIC), Barcelona, Spain