 Prontip Saelee
Prontip Saelee Alyssa Kearly
Alyssa Kearly Stephen L. Nutt
Stephen L. Nutt Lee Ann Garrett-Sinha
Lee Ann Garrett-Sinha- 1Department of Biochemistry, State University of New York at Buffalo, Buffalo, NY, USA
- 2The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 3Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
Background: The transcription factor Ets1 is highly expressed in B lymphocytes. Loss of Ets1 leads to premature B cell differentiation into antibody-secreting cells (ASCs), secretion of autoantibodies, and development of autoimmune disease. Despite the importance of Ets1 in B cell biology, few Ets1 target genes are known in these cells.
Results: To obtain a more complete picture of the function of Ets1 in regulating B cell differentiation, we performed Ets1 ChIP-seq in primary mouse B cells to identify >10,000-binding sites, many of which were localized near genes that play important roles in B cell activation and differentiation. Although Ets1 bound to many sites in the genome, it was required for regulation of less than 5% of them as evidenced by gene expression changes in B cells lacking Ets1. The cohort of genes whose expression was altered included numerous genes that have been associated with autoimmune disease susceptibility. We focused our attention on four such Ets1 target genes Ptpn22, Stat4, Egr1, and Prdm1 to assess how they might contribute to Ets1 function in limiting ASC formation. We found that dysregulation of these particular targets cannot explain altered ASC differentiation in the absence of Ets1.
Conclusion: We have identified genome-wide binding targets for Ets1 in B cells and determined that a relatively small number of these putative target genes require Ets1 for their normal expression. Interestingly, a cohort of genes associated with autoimmune disease susceptibility is among those that are regulated by Ets1. Identification of the target genes of Ets1 in B cells will help provide a clearer picture of how Ets1 regulates B cell responses and how its loss promotes autoantibody secretion.
Introduction
B cells are crucial contributors to immunity both by secreting specific antibodies and by serving as antigen-presenting cells. B cell immune responses and differentiation to antibody-secreting cells (ASCs) are controlled by the expression and activation of specific transcription factors at different stages and time points in B cell development and functional activation. For instance, the transcription factors Pax5, Ebf1, E2a (Tcf3), and Foxo1 are required for the development of B cell progenitors in the bone marrow and in mature and peripheral B cells (1–7). A different set of transcription factors including Blimp1 (Prdm1), Xbp1, and Irf4 are required for B cells to undergo ASC differentiation. Blimp1 stimulates immunoglobulin secretion and suppresses mature B cell gene expression (8, 9). Xbp1 is required for endoplasmic reticulum remodeling and high-level immunoglobulin production (10, 11). Irf4 is also required for ASC differentiation (12), although the mechanisms by which it does so are still under debate (13).
In addition to the transcription factors summarized above, various members of the Ets gene family also regulate B cell differentiation and function. The roles of six Ets proteins have been studied in detail in B lymphocytes, including Ets1 (14), Fli1 (15, 16), Gabpa (17), and the three related Ets family factors PU.1, SpiB, and SpiC (18, 19). Two additional Ets proteins, Elf3 (Ese1) and Elf4 (MEF) have been reported to affect B cell development or functional responses (20, 21), although their detailed roles in this process have not yet been identified.
Ets1, the prototypical member of the Ets gene family, is highly expressed in B cells and is necessary for maintaining them in a quiescent state, since lack of Ets1 leads to premature differentiation into ASCs (22, 23). This is accompanied by a loss of B cell tolerance to self-antigens (24). Ets1−/− mice also lack marginal zone type B cells, possibly because of depletion due to excessive differentiation to ASCs (23, 25). In keeping with a role for Ets1 in establishing B cell tolerance, Ets1−/− mice develop an autoimmune phenotype (23, 26) and single-nucleotide polymorphisms (SNPs) in the human ETS1 gene have been highly implicated in a variety of autoimmune diseases (27, 28).
The most well-studied function of Ets1 in B cells is in regulating the formation of ASCs. One mechanism by which Ets1 regulates this process is by forming a protein–protein complex with Blimp1 resulting in the inhibition of Blimp1 DNA binding (22, 29). Ets1 may also regulate B cell differentiation through direct binding to target genes. To date, only a few such target genes of Ets1 have been identified in B cells, including Pax5 (22, 29–37), which is crucial for maintaining mature B cell identity. In our study, we identify Ets1-binding sites in mouse B cells using ChIP-sequencing and identify gene expression changes in the absence of Ets1 using RNA-sequencing. Interestingly, many of these target genes are implicated in autoimmune responses, a cohort of which is tested for their ability to restore normal differentiation to Ets1−/− B cells. Restoring the normal expression levels of four of these Ets1 targets (Stat4, Ptpn22, Egr1 and Prdm1) failed to reverse the Ets1−/− B cell phenotype of excessive plasma cell differentiation in response to TLR ligands. Instead, restoring Stat4 and Ptpn22 resulted in increased plasma cell differentiation. Therefore, other targets of Ets1 or the combined actions of multiple targets may be crucial for regulating this B cell differentiation step.
Materials and Methods
Mice
Wild-type (WT) C57BL/6 mice were purchased from Jackson Laboratory. Ets1−/− mice (RRID:MGI: 3833458) and littermate WT controls used in this study were bred in our facility and maintained on a mixed genetic background of C57BL/6 × 129Sv because, on a pure C57BL/6 genetic background, the loss of Ets1 is lethal perinatally. The mutation in the Ets1 locus of these mice has previously been described (23, 38). Mice carrying the Prdm1-green fluorescent protein (GFP) allele that inactivates Blimp1 were obtained from Dr. Stephen Nutt (Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia) (39).
B Cell Purification and ChIP-seq
Wild-type mouse B cells were purified from spleens of 3-month-old C57BL/6 male mice using negative selection with the Easysep mouse B cell isolation kit (Stem Cell Technologies). Purified B cells were rested for 1 h in complete media [RPMI 1640 + 10% fetal bovine serum, 1% pen/strep, 1% Glutamax (Gibco), and 50 μM β-mercaptoethanol] in a tissue culture incubator. After resting, cells were cross-linked by adding formaldehyde to a final concentration of 0.25% for 8 min. Fixed B cells were lysed and chromatin prepared according to the manufacturer’s protocol for the ChIP-IT High Sensitivity Kit (Active Motif). Chromatin was sonicated to yield fragments of an average size ~200–700 bp and immunoprecipitated with a rabbit polyclonal anti-Ets1 antibody (sc-350X, Santa Cruz) that has previously been used in chromatin immunoprecipitation assays (40–42). Two biological replicates were separately prepared and analyzed. The number of uniquely mapped reads for ChIP-seq was between 10 and 27 million for the Ets1 ChIP-seq and between 7 and 10 million for sequencing of the input.
Libraries were generated from the purified chromatin, and ChIP-sequencing was performed on input chromatin and Ets1-precipitated chromatin using an Illumina Hiseq2500 Sequencing System. The ChIP-seq data were found to be of good quality using the normalized strand coefficient and the relative strand correlation parameters as described previously (43). The reads were aligned to mouse mm9 genome assembly using Bowtie (44).
Bioinformatics Analyses of ChIP-seq
We identified Ets1-bound regions using the MACS2 program (45). Peaks identified from each biological replicate were compared to input controls using the irreproducible discovery rate to identify reproducible Ets1-binding sites of which 10,391 were detected. Ets1-bound regions were annotated for enrichment at intergenic regions, promoters, exons, or introns using ChIPseeker (46). Overrepresented motifs in the peaks were analyzed using the findmotifgenome function in Homer (47). Locations of binding sites with respect to potential target genes were visualized in GenomeBrowser (https://genome.ucsc.edu/cgi-bin/hgGateway).
Ets1 consensus binding sites and other transcription factor-binding sites in the Ets1-bound regions were identified using PscanChIP (48). The regions were then aligned centered on the Ets1 consensus motif. ChIP-seq data for acetylated and methylated lysine variants of histone H3 in primary B cells were obtained from the GEO database and are reported in Ref. (49–51). The program ArchTex (52) was then used to map average histone modifications surrounding the Ets1 consensus-binding sites. As a control, ArchTex was also used to align histone modifications surrounding 60,000 random consensus Ets1-binding sites from regions not bound by Ets1 in ChIP-seq assays.
To test if Ets1 might co-regulate some of the same genes as Pax5, Tcf3 (E2A), or Irf4, we obtained ChIP-seq data for these factors in primary mouse splenic B cells, as reported in Ref. (51, 53, 54). We used Bedtools to identify the co-occurrence between peaks and common target sites for these transcription factors.
RNA-seq Analysis
Spleens from female WT (C57BL/6) and Ets1 knockout mice (C57 × 129Sv) were used to prepare single cell suspensions, and quiescent follicular B cells were sorted from each population based on the following markers: B220+ CD23hi CD21low CD80neg IgAneg IgEneg IgG1neg IgG2aneg IgG2bneg IgG3neg using FACSAria II cell sorter. Dead cells were gated out from the sorted population using Live/Dead Fixable Aqua dead cell stain (Molecular Probes). Total RNA was isolated from two biological replicates of sorted B cells and subjected to RNA-sequencing on an Illumina Hiseq2500 Sequencing System. Sequence reads (24–49 million per sample) were aligned to the mm9 genome assembly using the Tophat program, and differences in gene expression between samples were analyzed using Cuffdiff (55). For further analysis, we chose genes that showed a fold change between WT and Ets1−/− B cells of at least twofold [log2(FC) = 1.0 or more] and a q value of 0.05 or less. We excluded genes with a reported Fragments Per Kilobase of transcript per Million mapped reads (FPKM) of less than 1.0 in either genotype. CummeRbund was used to visualize the Cuffdiff analysis, including generating heat maps of gene expression levels.
Genes that showed an alteration in expression as described above were analyzed for their functions by DAVID software (56) to identify the most-relevant gene ontology (GO) terms that were significantly enriched. We used an EASE score equal to 0.05 and a count threshold of 5 to identify enriched pathways.
RNA Isolation and Reverse Transcription-PCR
To validate RNA-seq data, we purified B cells from the spleens of 3-month-old male and female WT and Ets1−/− mice (C57BL/6 × 129Sv genetic background) by first using magnet beads (Stem Cell Technologies mouse B cell purification kit) followed by sorting mature naïve follicular B cell subset B220+ CD23hi CD21low CD11bneg CD80neg IgAneg IgEneg IgG1neg IgG2aneg IgG2bneg IgG3neg. RNA was extracted from sorted B cells (n = 4 of each genotype) using Direct-zol RNA MiniPrep kits (Zymo Research). cDNA was synthesized from equal amounts of RNA using QuantiTect Reverse Transcription kit (Qiagen). Quantitative RT-PCR was performed using iQ SYBR Green Supermix (Bio-Rad). The sequences of primers used for RT-PCR were: Slamf6 (Forward-CAGCTAATGAATGGCGTTCTAGG, Reverse-CTTAGGTTGATAACGAGGGCAG), Egr1 (Forward-AACCGGCCCAGCAAGACACC, Reverse-TGGCAAACTTCCTCCCACAAAT), Ptpn22 (Forward-AGCAAGCCTACAGAACGTG, Reverse-TCCAGAGGTGCGTTACATATTC), Stat4 (Forward-TGGCAACAATTCTGCTTCAAAAC, Reverse-GAGGTCCCTGGATAGGCATGT), Prdm1 (Forward-TGTGGTAATGTCGGGACTTTG, Reverse-TTCCTTTTGGAGGGATTGGAG), Hprt (Forward-CCTCATGGACTGATTATGGACAG, Reverse-TCAGCAAAGAACTTATAGCCCC), Gapdh (Forward-AATGGTGAAGGTCGGTGTG, Reverse-GTGGAGTCATACTGGAACATGTAG), and Actb (β-actin) (Forward-GCAGCTCCTTCGTTGCCGGTC, Reverse-TTTGCACATGCCGGAGCCGTTG). Gene expression was normalized to all three housekeeping genes (Gapdh, Hprt, and Actb) using Bio-Rad CFX Manager Software.
Retroviral Transduction
Plasmids encoding mouse Stat4 (57) or human myc-tagged Ptpn22 (58) were obtained from Dr. John O’Shea (National Institutes of Health, Bethesda, MD, USA) or Dr. Dimitar Efremov (International Centre for Genetic Engineering and Biotechnology, Rome, Italy), respectively. The cDNAs were cut from the original plasmids and sub-cloned into the MIGR1 retroviral plasmid, which contains an internal ribosomal entry site (IRES) followed by GFP to allow easy identification of virally infected cells. The resulting plasmids were confirmed by sequencing. The MIGR1 plasmid encoding mouse Ets1 has been described previously (22). The plasmid used for Egr1 knockdown was generated by cloning a shRNA against Egr1 into the microRNA-30-adapted shRNAmir retroviral vector (MSCV-lmp) from Open Biosystems. Oligonucleotides with sense and antisense strands of an shRNA targeting nucleotides 2,314–2,336 of mouse Egr1 mRNA (NM_007913.5) were cloned into MSCV-lmp and confirmed by sequencing. MSCV-lmp also contains an IRES-GFP module that can be used for detecting transduction. A retroviral plasmid containing a shRNA against Prdm1 in MSCV-lmp, where the GFP gene was substituted with a GFP variant Ametrine (59), was obtained from Dr. Matthew E. Pipkin (The Scripps Research Institute, Jupiter, FL, USA).
Retroviral plasmids were used to transfect the Plat-E packaging cell line along with the plasmid pCL-Eco (which contains additional copies of the viral structural genes) using Fugene-6. The retroviral supernatant was harvested after 48 h of transfection and used to infect WT or Ets1−/− B cells that were purified using anti-B220 microbeads (Miltenyi Biotec) and stimulated with 10 μg/ml of lipopolysaccharide (LPS) for 24 h. The infected B cells were subsequently returned to fresh medium containing LPS. Forty-eight hours later, B cells were stained for flow cytometry with antibodies to B220 and CD138. Just prior to flow cytometry, 7AAD was added to allow exclusion of dead cells from analysis. Flow cytometry data were collected on a BD LSR II flow cytometer and analyzed using FlowJo software.
To test viral gene expression, infected cells were harvested to make lysates. For the Stat4-virus infected cells, we detected expression in the packaging cell line Plat-E as well as in sorted GFP+ virally-infected B cells. For the other constructs, we used unsorted, infected B cells to make lysates. Protein expression was analyzed by Western blotting using the following monoclonal antibodies: anti-Ptpn22 (clone D6D1H; Cell Signaling Technology), anti-Egr1 (clone T.126.1; Thermo Fisher Scientific), anti-Stat4 (clone C46B10; Cell Signaling Technology), anti-Blimp1 (clone 6D3; EMD Millipore), and anti-GAPDH (clone 6C5; EMD Millipore).
Statistical Analysis
Statistical analysis for qPCR and ELISPOT assays were performed using GraphPad Prism software. p-Value was calculated using unpaired Student’s t-test with two-tailed p-value or with the Mann–Whitney U-test.
Results
Identification of Ets1-Binding Sites in the Chromatin of B Cells
To gain insight into how Ets1 mechanistically regulates B cell differentiation, we assessed its genome-wide occupancy by chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) using chromatin derived from mouse mature B cells. Purified splenic B cells showed enhanced phosphorylation of Ets1 based on SDS-PAGE mobility when compared to whole spleen (Figure 1A). This shift in mobility arises from calcium-induced CAM kinase-dependent serine phosphorylation (34, 60) and results in inhibition of Ets1 DNA binding (61). In order to restore Ets1-binding activity, we rested the B cells for 1–2 h, which resulted in normalization of Ets1 phosphorylation status (Figure 1A). After the resting period, purified B cells were fixed and sonicated.
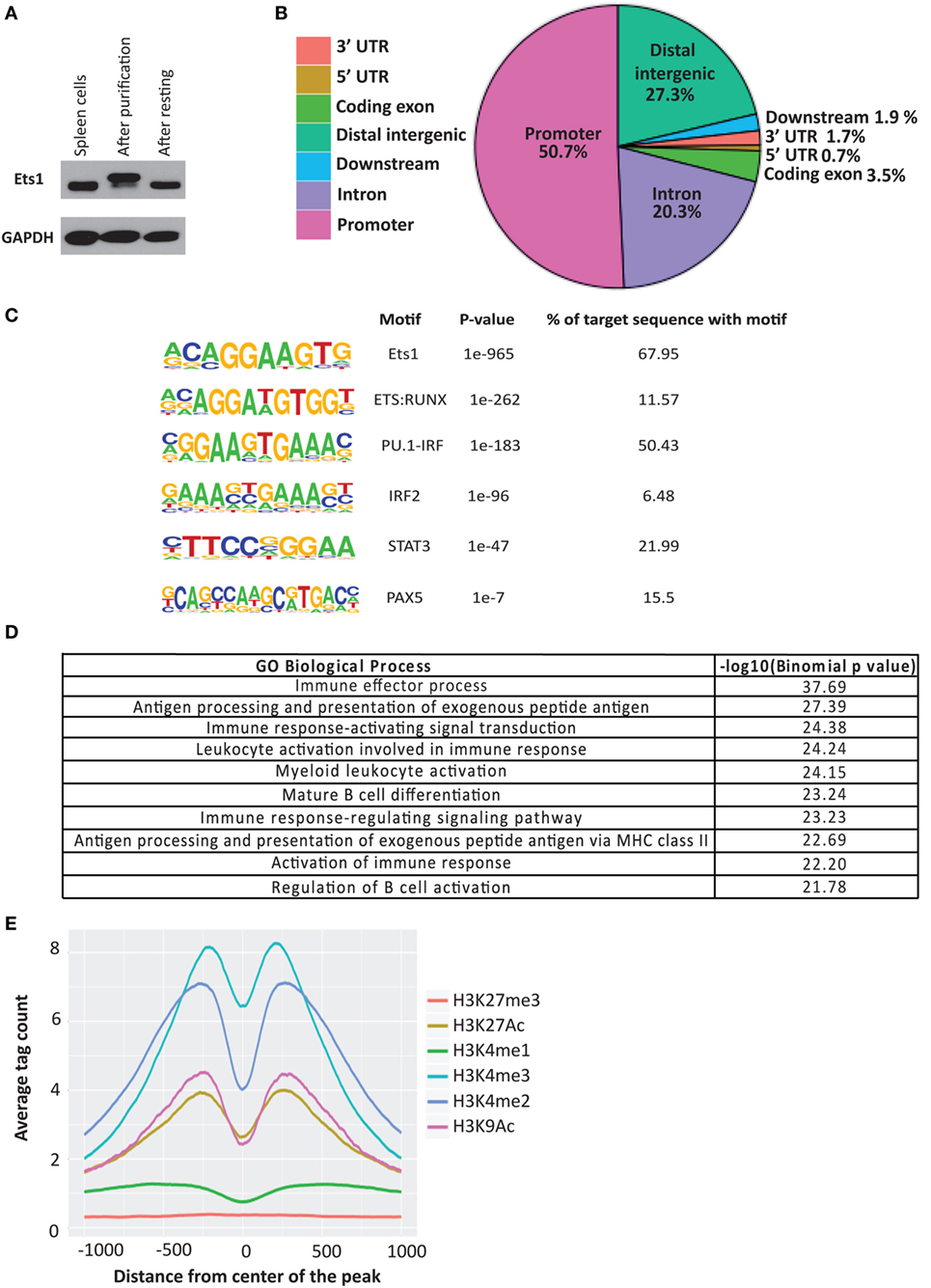
Figure 1. Identification of Ets1-binding sites in mouse B cells. (A) Western blot to show phosphorylation of Ets1 in freshly isolated B cells versus rested B cells. GAPDH serves as loading control. (B) Pie chart of location of Ets1 sites in the genome. (C) Motifs enriched in Ets1-bound regions. Shown are overrepresented transcription factor-binding motifs localized in the Ets1 peaks and the percent of sites with that motif. (D) Gene ontology biological terms associated with Ets1-binding peaks in B cells. (E) Analysis of epigenetic features surrounding Ets1-bound regions by mapping adjacent histone modifications. Data come from the ENCODE Consortium or from the studies described in Ref. (50, 51).
B cell chromatin was immunoprecipitated with an anti-Ets1 antibody that has been used previously in chromatin immunoprecipitation (40–42). We identified 10,391 reproducible peaks of Ets1 binding, which target 8,975 genes in B cells (GEO dataset: GSE83758). Among Ets1-bound regions, approximately half are in the promoters of the genes (within 3 kb of the transcriptional start site). An additional 20% are in the introns of genes and about 27% are located in distal intergenic regions that may represent enhancers or silencers (Figure 1B). Less than 10% of Ets1-binding sites were detected in coding exons, the 5’ or 3’ UTRs of genes or within 3 kb downstream of target genes. We identified the consensus Ets1-binding motif in 68% of target sites (Figure 1C). Additional transcription factor motifs that were enriched in the Ets1-bound regions include combined ETS-RUNX sites, combined PU.1-IRF sites, IRF2 sites, STAT3 sites, and PAX5 sites (Figure 1C).
Pax5 is a key transcription factor that regulates B cell identity and limits differentiation to ASCs (62, 63). We have previously implicated Ets1 in controlling Pax5 levels in B cells (22, 29). Indeed, examination of the ChIP-seq peaks of Ets1 showed several peaks in the Pax5 gene including one in the proximal promoter and several in introns of the gene including two strong peaks in intron 5 where a B cell-specific enhancer of Pax5 has been described (64) (Figure S1 in Supplementary Material). These observations support the idea that Ets1 may directly regulate expression of the Pax5 gene. Several strong Ets1-binding peaks were also detected near the Prdm1 gene, which encodes Blimp1 (Figure S2 in Supplementary Material). Although these regions are not associated with any known regulatory elements, they are enriched in H3K4me1 and H3K27Ac marks indicative that they may represent functional regulatory elements. When examining other genes that have been previously described as Ets1 targets [such as those encoding Cd79a (Igα), H-2Aa and H-2Eb1 (isoforms of MHC II), and Nfkb1 (p50)], we found that each gene contained nearby Ets1-binding sites (Figure S3 in Supplementary Material) and, therefore, is potentially regulated by Ets1 in vivo.
Analysis of Patterns of Ets1 Binding in B Cells
To understand how Ets1 might regulate B cell responses, we analyzed pathways associated with Ets1 binding. Ets1-binding peaks were enriched in genes associated with immune response, antigen processing and presentation, immune signaling pathways, and mature B cell differentiation (Figure 1D). Interestingly, Ets1-binding peaks are located near a large number of genes involved in the BCR signaling cascade (Figure S4 in Supplementary Material). Additional non-immune pathways enriched in Ets1-binding sites included genes involved in processing of non-coding RNA, protein folding, and apoptosis (data not shown).
The ENCODE project has mapped histone modifications and transcription factor-binding sites for various human and mouse cell lines (65, 66). In mature primary mouse B cells, data are available for the H3K27me3 modification (49). In addition, two research groups have mapped histone modifications (H3K4me1, H3K4me2, H3K4me3, H3K9Ac, H3K27Ac, and H3K27me3) in primary mouse B cells (50, 51). We tested whether any of these histone marks showed enrichment near the Ets1-binding peaks as compared to random chromatin. We found that peaks of H3K4me3 (associated with promoters) were strongly enriched on either side of the peak Ets1-binding locus (Figure 1E). Similarly, peaks of H3K4me2, H3K9Ac, and H3K27Ac (associated with both promoters and enhancers) were also strongly enriched flanking the Ets1-binding sites. Weaker enrichment of H3K4me1 (associated with enhancers) was also observed flanking Ets1-binding sites, while there was no enrichment of H3K27me3 (associated with repressed genes) near Ets1-binding peaks. None of the histone marks showed specific enrichment when random chromatin regions were used (not shown). These data indicate that binding of Ets1 is strongly associated with active promoters and enhancers of genes, but is associated weakly if at all with repressed sites.
ChIP-seq datasets are also available for Pax5 in mouse mature splenic B cells (51). We compared the binding sites of Pax5 with those bound by Ets1 to identify genes that might be co-regulated. There were 4,590 overlapping peaks common between Ets1 and Pax5 (Figure 2A), representing ~44% of all Ets1-bound regions and ~45% of all Pax5-bound regions. Among the genes having peaks for both Ets1 and Pax5 are some that encode genes that are crucial for B cell biology including CD79a, ICOS ligand, Foxp1, PLCγ2, and Vav1. Ontology enrichment analysis of the identified common targets of Ets1 and Pax5 indicate that they are associated with immune system activation and effector processes (not shown). This suggests that there is a strong overlap in function of Ets1 and Pax5 in B cells.
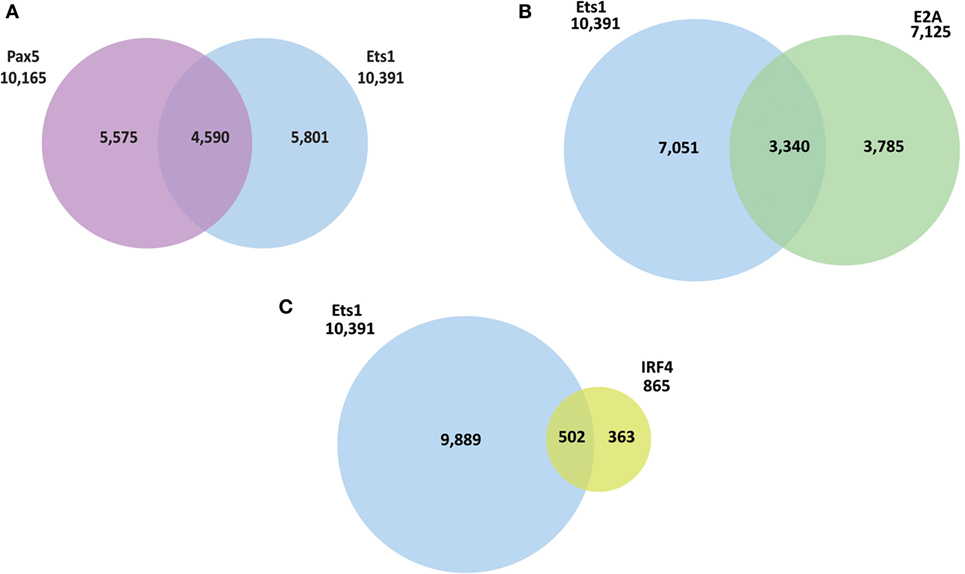
Figure 2. Ets1-binding sites overlap with those of other B cell transcription factors. Venn diagrams to show overlap between Ets1-bound regions in primary mouse B cells with (A) Pax5-bound regions, (B) E2A-bound regions, and (C) Irf4-bound regions. Pax5, E2A, and Irf4 ChIP-seq data come from studies described in Ref. (51, 53, 54).
Binding sites for Tcf3 (E2A) (53) and Irf4 (54) have also been mapped in primary B cells. We found that ~47% of E2A-binding sites overlap with those of Ets1 (Figure 2B). These overlapping binding sites include genes such as Cd19, Blnk, and Rasgrp1. With Irf4, relatively few (865) binding sites were detected in primary naïve B cells (Figure 2C). However, of these 865 sites, ~58% overlap with Ets1-binding sites, including sites in important B cell genes such as Cxcr4, Cxcr5, Lyn, Pax5, Ebf1, and Foxo1. Overall, these studies show that Ets1 is targeted to promoters and/or enhancers of genes important for B cell activation and differentiation and can potentially co-regulate such genes along with other B cell transcription factors like Pax5, E2A, and Irf4.
Identification of Changes in Gene Expression in the Absence of Ets1
The presence of a binding site for a transcription factor does not always correlate with its ability to activate or repress transcription from the gene. To better identify genes in B cells whose expression directly depends on Ets1, we performed RNA-sequencing using RNA isolated from WT and Ets1–/− splenic B cells (GEO Dataset: GSE83797). One potential complicating factor is that Ets1−/− mice have different B cell composition in the spleen, because they lack marginal zone B cells and their follicular B cells (1) have a more activated phenotype, (2) undergo altered isotype-switching, and (3) differentiate at higher rates to ASCs than their WT counterparts (23, 25, 26, 36, 67). Therefore, to compare similar populations of B cells from WT and Ets1−/− mice, we used flow cytometry to isolate follicular B cells (B220+ CD23hi CD21low) that were not activated (CD80neg) and had not undergone class-switching (IgAneg IgEneg IgG1neg IgG2aneg IgG2bneg IgG3neg) (Figure 3A). We avoided sorting B cells based on surface levels of IgM or IgD, because we did not want to activate the cells by cross-linking the antigen receptor. Total RNA was prepared from sorted B cell subsets from each genotype and subjected to deep sequencing.
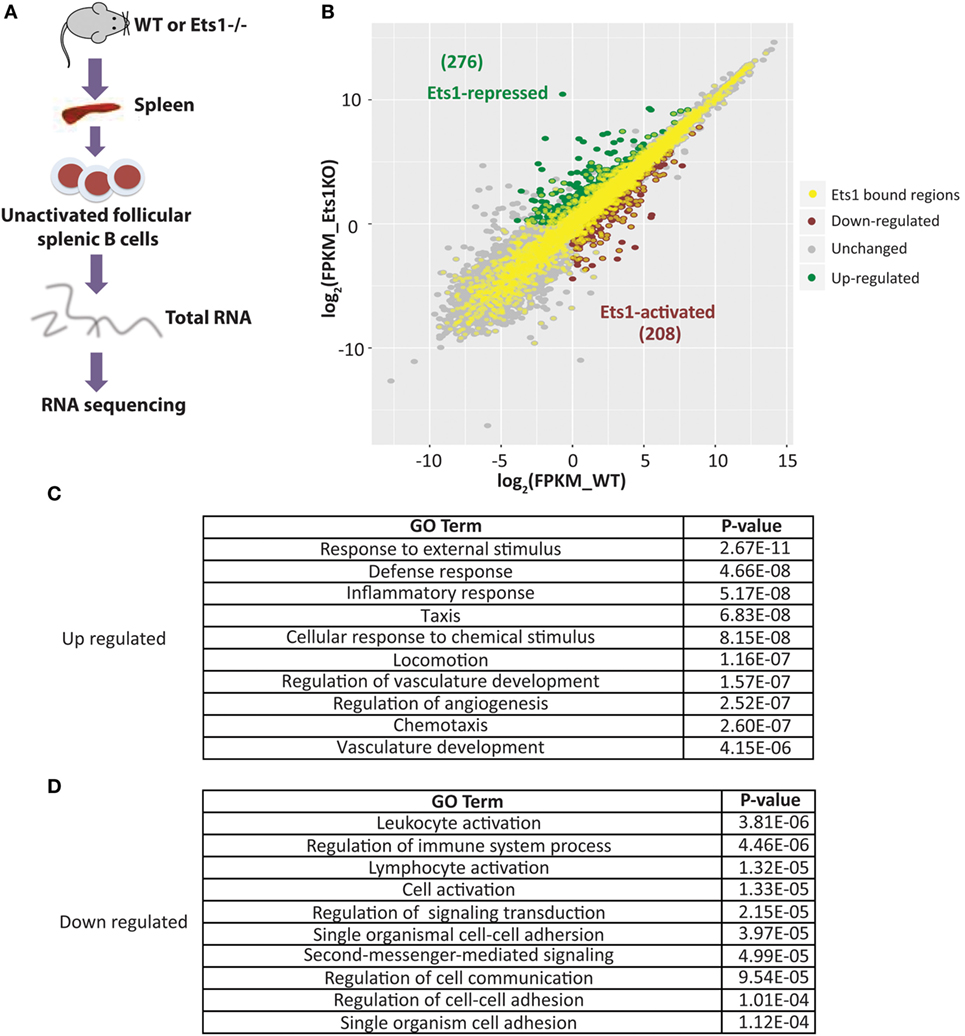
Figure 3. Identification of genes whose expression in B cells requires Ets1. (A) Scheme to isolate naive follicular B cells from the spleens of wild-type (WT) and Ets1−/− mice (n = 2 samples/genotype). (B) Scatterplot analysis of differential gene expression in WT and Ets1−/− B cells. Genes that are upregulated (Ets1-repressed) in Ets1−/− cells are shown as green dots, while genes that are downregulated (Ets1-activated) are shown as red dots. Gray dots are genes whose expression does not change and/or whose expression is less than 1.0 FKPM in both cell types. Dots with yellow centers are the genes that show an associated Ets1-binding site by ChIP-seq. Pathway analysis of genes repressed (C) and activated (D) by Ets1. The top 10 GO terms ranked according to p-value are shown for both Ets1-repressed and Ets1-activated genes.
We identified a list of genes that showed at least a twofold change in expression between WT and Ets1−/− B cells with a q-value of 0.05 or less and with a FPKM value of at least 1.0 in either genotype. Using these criteria, 484 genes showed altered expression in the absence of Ets1 (Figure 3B). Of these, the expression of 208 genes (43%) was downregulated in the absence of Ets1, suggesting that Ets1 activates the expression of those genes. The expression of 276 genes (57%) was upregulated in the absence of Ets1, suggesting that Ets1 might repress these genes (Figure 3B). Overall, the changes in expression of potential Ets1 target genes were mostly moderate (typically in the range of twofold to fourfold differences between WT and Ets1−/−). Unexpectedly, we found that the Pax5 gene was not one of those genes whose expression was altered in the absence of Ets1 (Figure S5 in Supplementary Material). Functional annotation of the molecular pathways that are overexpressed in Ets1−/− B cells (i.e., pathways repressed by Ets1) showed that they were predominantly associated with inflammatory responses and chemotaxis (Figure 3C; Figure S6A in Supplementary Material). In contrast, the molecular pathways that are under-expressed in Ets1−/− B cells (pathways activated by Ets1) were associated with immune cell signal transduction and activation (Figure 3D; Figure S6B in Supplementary Material). Thus, Ets1 is an important regulator of B cell functions by both positively and negatively regulating various aspects of the immune response.
Genes Implicated in Autoimmunity Are among Those Controlled by Ets1
We compared the ChIP-seq dataset that contained binding sites for Ets1 to the RNA-seq dataset that showed which genes changed expression in Ets1−/− B cells to find genes that might be directly regulated by Ets1. There were 263 genes that contained a nearby Ets1-binding peak and showed an alteration in expression in the absence of Ets1 (Figure 4A). This represents ~3% of genes containing Ets1-binding peaks. Among these genes, 137 (~52%) are activated by Ets1 and 126 (~48%) are repressed by Ets1. Examining the list of 263 genes, we identified numerous putative Ets1 target genes where SNPs have been associated with autoimmune disease susceptibility (Table 1). Additional Ets1 target genes include ones important for immune function such as Il5ra, Tlr1, Traf4, and Ltk, but that not yet been implicated as susceptibility loci for autoimmune disease. In further studies, we focused on those genes listed in Table 1, because of their known associations with immune regulation and autoimmune disease susceptibility.
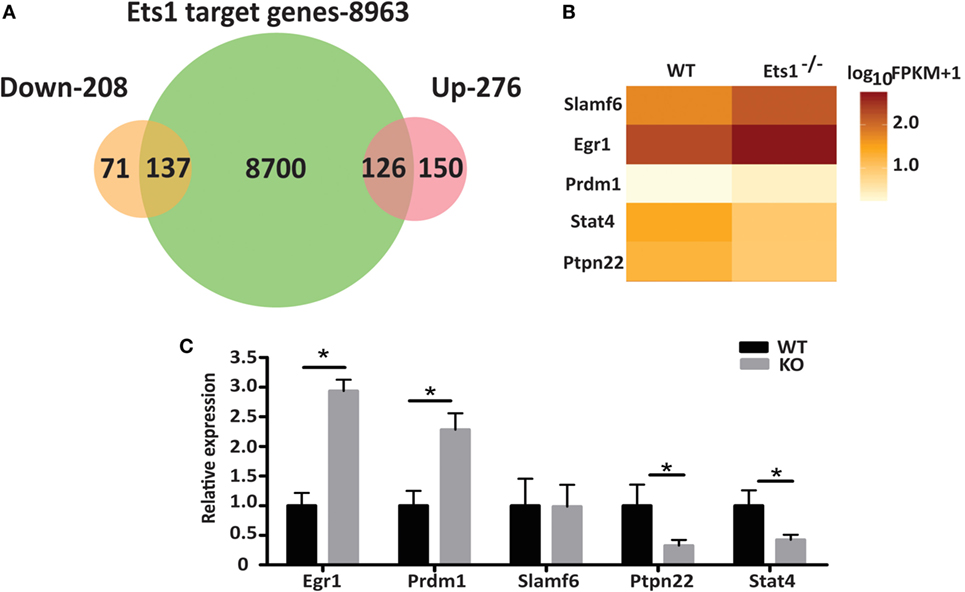
Figure 4. Genes that are potential direct targets of Ets1. (A) Venn diagram of overlap of ChIP-seq dataset with genes upregulated in the absence of Ets1 (276 genes) or downregulated in the absence of Ets1 (208 genes). (B) Heat map of selected Ets1 target genes associated with autoimmunity. (C) qPCR analysis of the expression of the target genes shown in part B (n = 4 for each genotype) using cDNA from sorted (B220+ CD23hi CD21low CD11bneg CD80neg IgAneg IgEneg IgGneg) follicular B cells. Shown is average ±SEM, *p < 0.05.
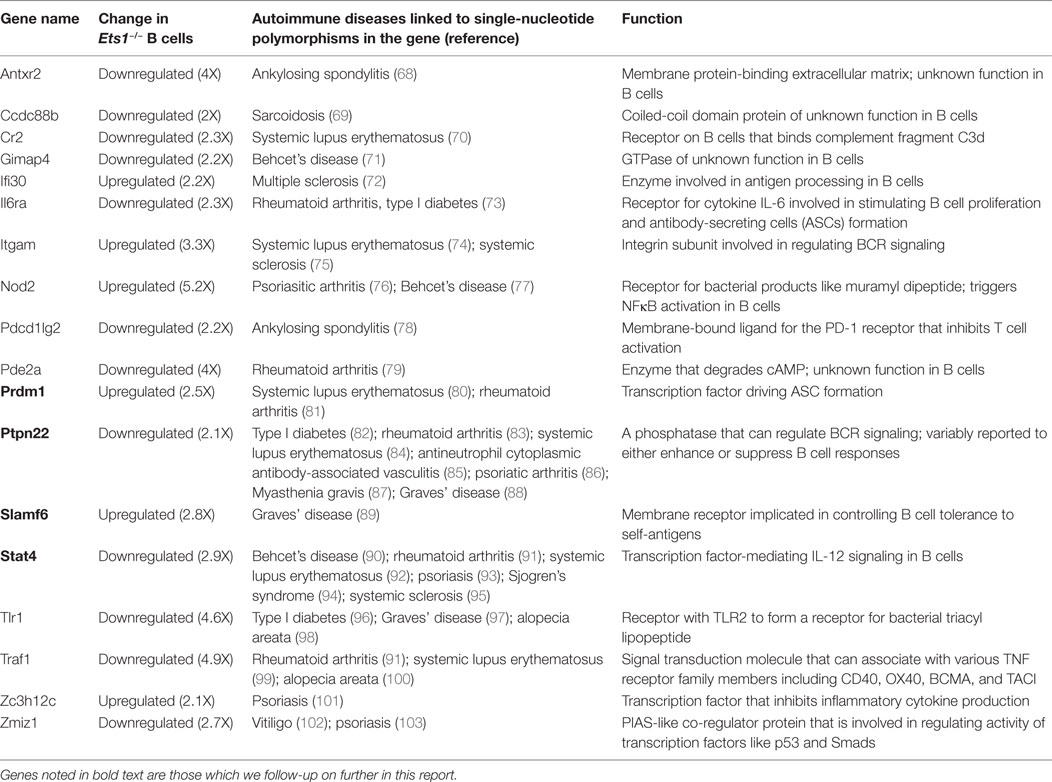
Table 1. List of potential Ets1 target genes associated with autoimmune disease.
We selected a subset of potential Ets1 target genes and designed primers to test their expression in qRT-PCR. The genes selected for further analysis were Egr1, Stat4, Prdm1, Slamf6, and Ptpn22 as shown in Figure 4B and Figure S7 in Supplementary Material. qRT-PCR analysis showed that Egr1, Stat4, Prdm1, and Ptpn22 all displayed a pattern of expression consistent with the RNA-seq results (Figure 4C). On the other hand, expression of Slamf6 did not appear to change in the absence of Ets1 in this analysis.
Stat4 and Ptpn22 showed decreased expression in the absence of Ets1, while Egr1 and Prdm1 are elevated. Therefore, Ets1 may promote the expression of Stat4 or Ptpn22, while suppressing expression of Egr1 or Prdm1. Alternatively, these changes in Stat4, Ptpn22, Egr1, and Prdm1 may instead represent secondary changes in gene expression that reflect the fact that Ets1–/− B cells are primed to become activated and differentiate to ASCs, rather than being important drivers of the Ets1–/− B cell phenotype. If Ptpn22 or Stat4 were target genes that mediated the effects of Ets1 on B cell differentiation, then we would expect that expression of Stat4 and Ptpn22 in B cells might in part mimic the effects of expression of Ets1, such as its ability to suppress ASC generation. To test this, we cloned cDNAs encoding Stat4 and Ptpn22 into a retroviral vector so that we could restore expression of these genes to Ets1−/− B cells and overexpress them in WT B cells. WT or Ets1−/− splenic B cells were stimulated with LPS and infected with control virus (MIGR1), virus-expressing Ets1 (MIGR1-Ets1), or viruses-expressing Stat4 or Ptpn22 (MIGR1-Stat4 or MIGR1-Ptpn22). We monitored cellular differentiation to ASCs (B220low CD138+) using flow cytometry in the GFP+ virally infected cells.
Western blot using extracts of virally infected B cells showed that B cells infected with Ptpn22 virus overexpress Ptpn22 (Figure 5A). Retrovirally expressed Stat4 was also easily detectable in the packaging cell line (Figure 5A), but its overexpression in B cells was less obvious due to the high levels of endogenous Stat4 in this cell type (Figure 5A). Viruses encoding Stat4 or Ptpn22 reproducibly resulted in low percentages of GFP+ virally infected B cells (5–10%) and low intensity of GFP staining, while empty virus or virus encoding Ets1 resulted in high percentages (40–80%) of GFP+ cells and high intensity of GFP staining (Figure 5B). Expression of Ets1 suppresses development of B220loCD138+ plasmablasts compared to B cells infected with empty vector (MIGR1) (Figure 5C). On the other hand, we found that there was a dramatic increase in the B220loCD138+ plasmablast cell numbers in B cells expressing Stat4 or Ptpn22 (Figure 5D), indicating that these genes stimulate the development of ASCs.
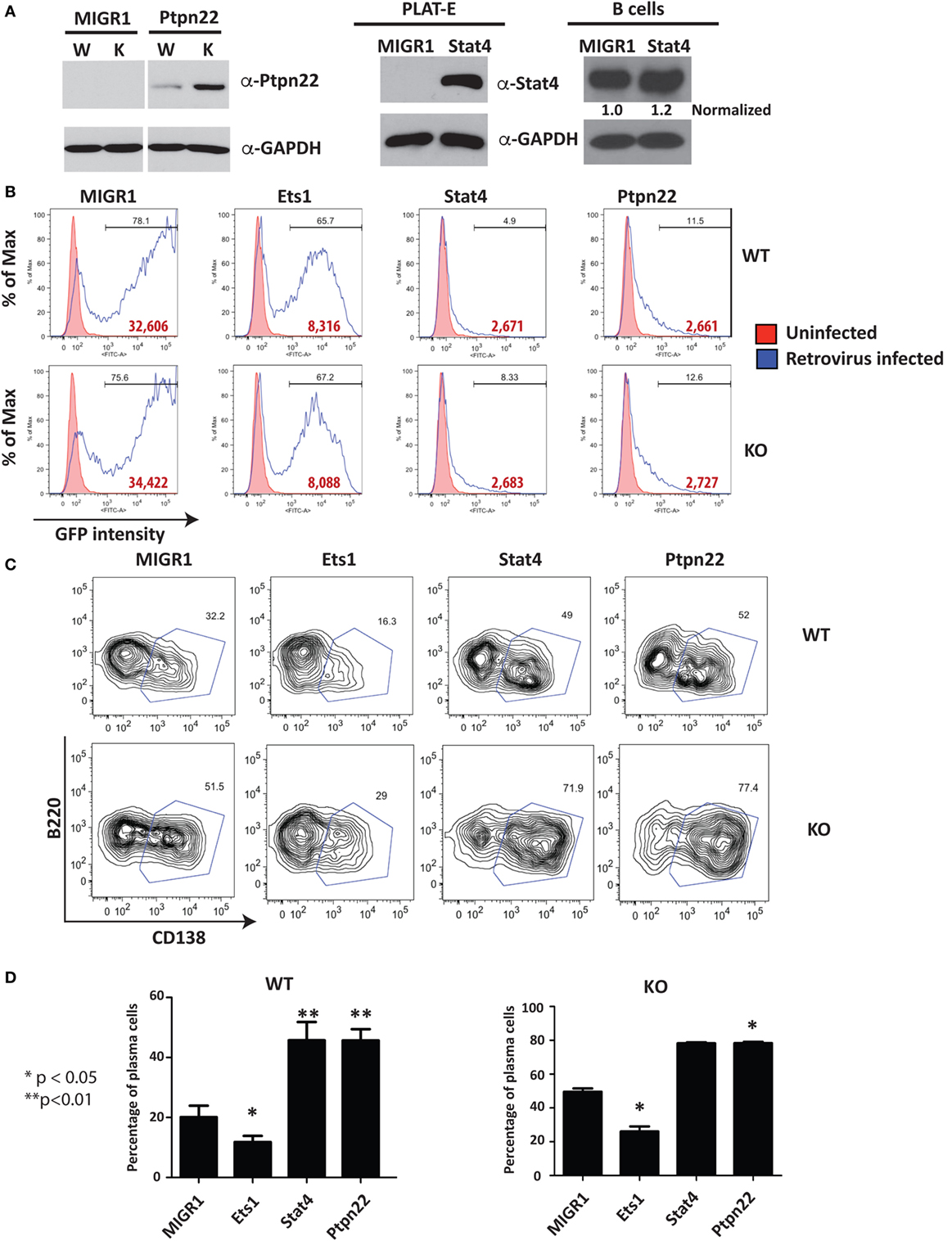
Figure 5. Restoration of selected target genes in Ets1−/− B cells. (A) Western blot analysis of Ptpn22 [with lysates from unsorted virally infected wild-type (W) and knockout (K) B cells] and Stat4 expression [with lysates from the packaging cell line (PLAT-E) or virally infected B cells (B cells); note that the small text under the Stat4 blot represents Stat4 expression levels normalized to GAPDH]. (B) Green fluorescent protein (GFP) profiles of virally infected cells. Red shaded curves are background staining of control non-infected cells, while open black curves are GFP staining in virally infected cells. Bold red type is the mean fluorescent intensity of GFP in the GFP+ gated population. (C) Analysis of GFP+ WT and Ets1−/− B cells to show differentiation to B220-low CD138+ plasmablasts (boxed). (D) Mean ± SEM of the percentage of plasmablasts within the GFP+ population in three independent viral infection experiments.
Unlike Stat4 and Ptpn22, Egr1 and Prdm1 were upregulated in Ets1−/− B cells by about twofold to threefold, suggesting that Ets1 represses expression of these genes. We used a retroviral vector encoding shRNAs to knockdown expression of these genes in B cells (Figure 6A) (59). Expression of shRNA against Egr1 was effective in reducing levels of Egr1 protein in stimulated B cells (Figure 6B), but did not impair formation of B220lowCD138+ plasmablasts (Figures 6C–E). The Prdm1 shRNA was also able to knockdown expression, although it was less efficient in Ets1−/− B cells (Figure 6B and data not shown). The Prdm1 shRNA did not alter ASC differentiation in WT or Ets1−/− B cells (Figures 6C–E), despite the fact that Blimp1 is known to be essential for plasma cell generation. This is likely because sufficient Blimp1 is still expressed to allow ASC differentiation. To further assess the role of Prdm1 in the phenotype of Ets1−/− B cells, we crossed Ets1−/− mice to mice carrying a GFP knock in allele in the Prdm1 locus that disrupts expression of the Prdm1 gene and leads to reduced levels of full-length functional Blimp1 protein (104). Homozygous Prdm1 knockout mice carrying this allele die embryonically due to a combination of developmental defects, but heterozygous mice are viable (104). We generated Ets1−/−Prdm1gfp/+ mice that carry a single copy of Prdm1 and express reduced levels of Blimp1 (Figure 7A). The numbers of ASCs in these mice and control mice was quantitated using ELISPOT, which showed that reduced levels of Prdm1 was not sufficient by itself to restrain excess ASC formation in the absence of Ets1 (Figure 7B). In summary, the changes in expression of the genes we tested (Stat4, Ptpn22, Egr1 and Prdm1) cannot by themselves explain the effects of Ets1 on ASC formation.
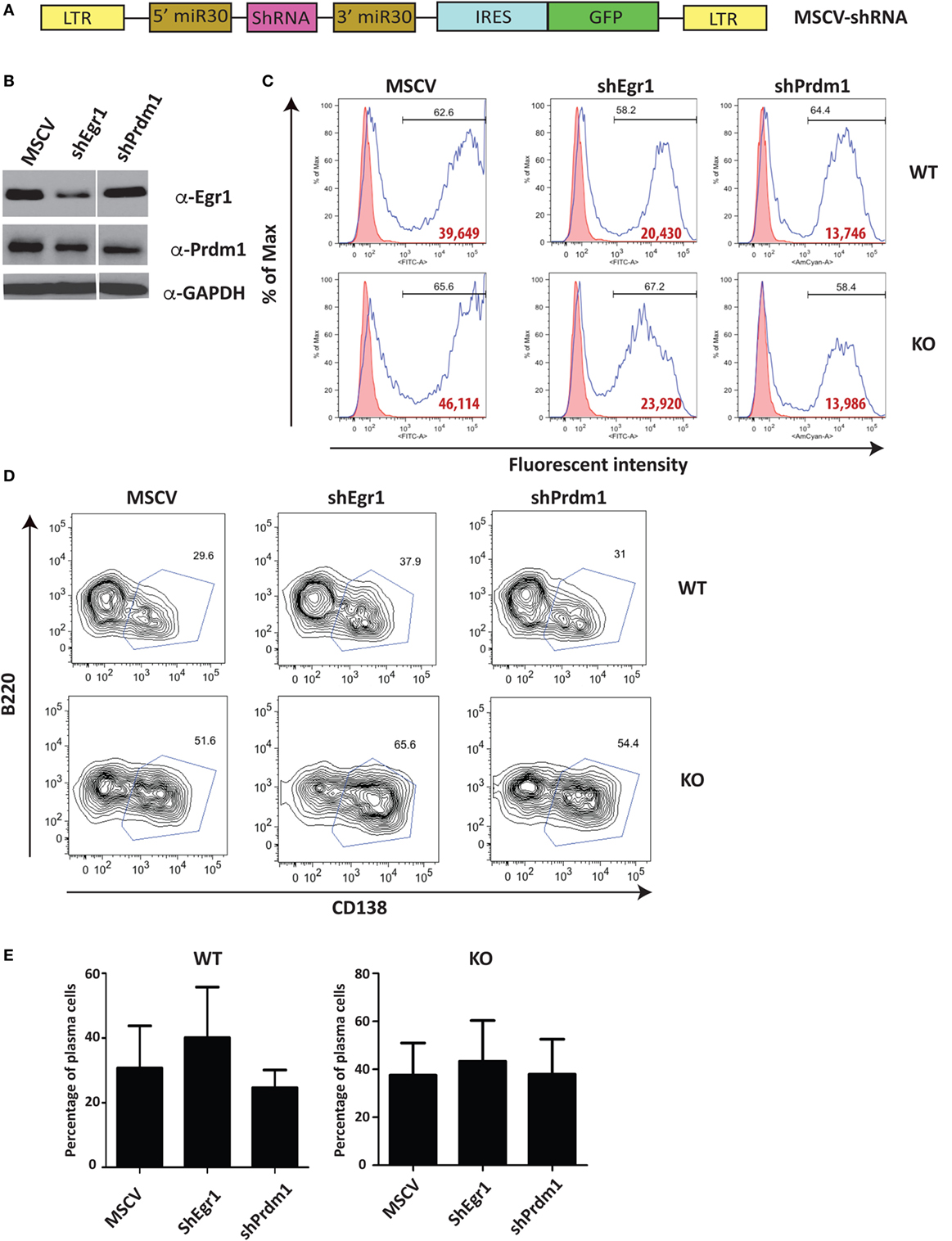
Figure 6. Reduction of selected target genes in Ets1−/− B cells. (A) Diagram of the retroviral construct encoding shRNAs used for knock down of gene expression in B cells. (B) Egr1 and Blimp1 expression analyzed by Western blot in lysates of B cells infected with shRNA viruses. (C) Green fluorescent protein (GFP) (MSCV and Egr1) and Ametrine (Prdm1) profiles of virally infected cells. Bold red type is the mean fluorescent intensity of GFP (or Ametrine) in the GFP+ (or Ametrine+) gated population. (D) B220 versus CD138 staining in the GFP+ (or Ametrine+) cells to show differentiation to B220-low CD138+ plasmablasts (boxed). (E) Mean ± SEM of the percentage of plasmablasts within the GFP+ (or Ametrine+) population in three independent viral infection experiments.
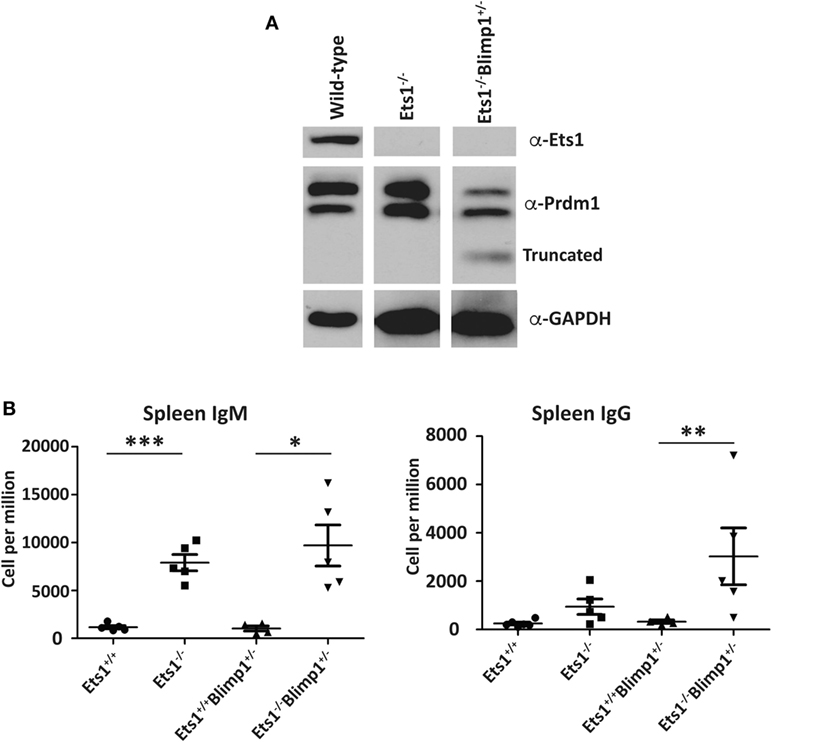
Figure 7. Reduction of selected target genes in Ets1−/− B cells. (A) Analysis of Blimp1 expression in lipopolysaccharide-stimulated Ets1+/+, Ets1−/−, and Ets1−/−Prdm1+/gfp B cells. The location of the full-length wild-type and the truncated proteins is indicated on the panel. (B) ELISPOT analysis of the numbers of IgM- and IgG-secreting cells in the spleens of unchallenged mice of the indicated genotypes (n = 4–5 for each genotype). *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
Over time, evidence has accumulated that Ets1 is a key regulator of B cell differentiation and that reduced levels of Ets1 are associated with the development of autoimmune diseases. We previously demonstrated that Ets1 controls B cell differentiation in part via the ability of Ets1 protein to directly bind to the Blimp1 protein and prevent Blimp1 interaction with DNA (22, 29). However, it remains unclear whether this protein–protein interaction is the main mechanism by which Ets1 inhibits B cell differentiation into ASCs or not. Because Ets1 is a transcription factor, it is important to understand what genes Ets1 regulates in B cells and how they may contribute to the control of B cell differentiation and secretion of autoantibodies. In this study, we show that Ets1 binds to and is required for normal expression of ~260 genes in mature B cells, including a subset of genes highly linked to immune responses and autoimmune disease susceptibility.
A large percentage (~50%) of the Ets1-binding sites, we identified in primary B cells, are localized in proximal promoters of genes in regions enriched for histone marks associated with active promoters (H3K4me3, H3K4me2, H3K9Ac, and H3K27Ac). Such a high percentage of binding sites in the proximal promoter is not always the case in ChIP-seq analyses of transcription factors. For instance, a ChIP-seq study of PU.1 and SpiB binding in the WEHI-279 B cell line found only ~15% of binding sites localized in the proximal promoter (105). Similarly, analysis of Runx1 binding in pro-B cells found that only ~10% of sites were in the proximal promoter of target gene (106). On the other hand, ChIP-seq analysis of Ets1 binding in human hematopoietic and embryonic stem cell lines showed that Ets1 binding was enriched in the promoter regions of genes in these cell types (107), although in the same study Ets1 binding was fairly equally distributed between promoters and enhancers in mouse cell lines. In the human Jurkat T cell line, Ets1 was shown to bind redundantly to proximal promoters of housekeeping genes along with other members of the Ets gene family, but to bind specifically to distal T cell enhancers (108). We found a similar pattern in primary mouse B cells, where Ets1 tends to bind only to the promoter of genes that have housekeeping functions (not shown). However, when Ets1 binds to distal regulatory elements (e.g., enhancer sequences) or to both promoters and distal regulatory sequences, those genes tend to be involved in specific B cell immune response pathways.
We previously showed that Ets1 can bind to the Pax5 gene, transactivate the Pax5 promoter and sustain Pax5 expression in differentiating B cells (22, 29). Here we confirm using ChIP-seq that Ets1 binds to both the Pax5 promoter and to the intron 5 B cell-specific enhancer of Pax5. However, somewhat surprisingly the levels of Pax5 mRNA are not significantly changed in Ets1−/− B cells as compared to WT B cells, as detected by RNA-sequencing. Several explanations could account for this. First, other Ets family members expressed in B cells (such as Fli1, Spi1, Spib, Elk4, Elf1, Elf2, Gabpa, and/or Etv3) could bind to these sites in the absence of Ets1 and compensate to maintain Pax5 expression. Supporting a potentially redundant role for Ets family transcription factors in regulating Pax5 gene expression, the Ets protein PU.1 can also bind to Pax5 enhancer sequences (64, 109). Alternatively, Ets1, specifically, may be required to maintain Pax5 expression, but may only regulate the gene at certain stages of development or in response to certain stimuli. Finally, it is also possible that the Ets-binding sites at these regions might represent fortuitous binding without functional significance. Similar arguments may be made for the many other genes in which we detected Ets1 binding, but whose expression does not change in B cells lacking Ets1. For instance, we found that Ets1-binding sites were detected in many genes involved in the BCR signaling pathway. Yet, the expression of most of these genes was unchanged in the absence of Ets1. Indeed, only ~3% of genes with a nearby Ets1-binding site showed altered expression in mature Ets1 knockout B cells.
Interestingly, among the genes identified that had at least one Ets1-binding site nearby and whose expression changed in Ets1−/− B cells, we found many that have previously been implicated in immune responses and autoimmune disease susceptibility based on genome-wide SNP assays. We chose two genes whose expression is reduced in Ets1−/− B cells (Stat4 and Ptpn22) and restored their expression using retroviral vectors. An unexpected result of our study is that expression of either Stat4 or Ptpn22 has an opposite effect to the expression of Ets1 (i.e., Ets1 expression suppresses LPS-induced plasmablast formation, while Stat4 and Ptpn22 both promote this process). This observation is interesting in light of the fact that Stat4 and Ptpn22 are both strongly associated with susceptibility to multiple different autoimmune diseases (110, 111).
Stat4 is best known as a transcription factor activated downstream of IL-12 signaling where it plays an important role in the production of IFN-γ (112). In B cells, Stat4 and IL-12 signaling have been most closely linked to differentiation of B cells to B effector type 1 (Be1) cells that secrete IFN-γ (113). However, our results suggest that Stat4 has additional B cell-intrinsic effects that promote the formation of ASCs, since retrovirally induced production of Stat4 potentiated LPS-induced formation of B220loCD138+ plasmablasts. This was true even though Stat4 was expressed at low levels in B cells using our retroviral construct. This effect of Stat4 may be independent of its roles in promoting IFN-γ production, although further studies are needed to confirm this. Recent data show that a lupus-associated SNP in the STAT4 gene leads to increased Stat4 expression (114), supporting the idea that higher Stat4 expression may promote autoimmunity. Interestingly, knockout of the Stat4 gene reduces autoantibody production in lupus-prone B6.TC mice (115). These results are consistent with the data in this manuscript that show Stat4 expression is increased in B cells from autoimmune-prone Ets1−/− mice and that Stat4 expression in B cells potentiates development of ASCs.
We found that retroviral expression of Ptpn22 also potentiates formation of plasmablasts in response to LPS. In myeloid cells, Ptpn22 promotes TLR signaling by stimulating Traf3 autoubiquitination (116). It may play a similar role in B cells, resulting in increased LPS-derived signals that promote ASC generation. Altogether, our data indicate that Stat4 and Ptpn22 have opposite roles compared to Ets1 in regulating formation of ASCs in response to LPS stimulation. The low levels of Stat4 and Ptpn22 transcripts in Ets1−/− B cells may, therefore, be due to secondary changes in the cells in an attempt to compensate for excessive B cell activation and differentiation in the absence of Ets1.
We also investigated roles for two genes whose expression is upregulated in Ets1−/− B cells, Egr1, and Prdm1. Recently, B cells lacking Egr1 were shown to undergo reduced differentiation to CD138+ plasmablasts when cultured with LPS (117). Furthermore, Egr1 knockout mice secrete less antigen-specific antibody in response to immunization with a T-dependent antigen (117). Since Egr1 is overexpressed in Ets1−/− B cells, reducing the levels of this gene might be expected to reverse the phenotype of Ets1−/− B cells and reduce ASC numbers. We used shRNA to knockdown Egr1 in B cells undergoing differentiation and found that this did not change ASC generation in either WT or Ets1−/− B cells. Thus, Egr1 appears not to be an essential Ets1 target gene regulating B cell differentiation, but rather is likely upregulated as a secondary consequence of B cell activation.
Ets1-binding sites are found in the Prdm1 locus and Ets1 has previously been suggested to directly repress transcription of the Prdm1 gene in Th1-skewed T cells (118). Since Blimp1, the protein encoded by the Prdm1 gene, is a key transcription factor driving ASC formation, it is reasonable to assume that the approximately twofold upregulation of Prdm1 in Ets1−/− B cells might explain their propensity to differentiate to ASCs. To test this, we used both knockdown of Prdm1 in cultured B cells as well as gene-targeted mice. Knockdown of Prdm1 by shRNA did not impair ASC generation, potentially, because the knockdown did not fully eliminate Blimp1 expression. As an alternative approach to test whether reducing Blimp1 levels could restore normal ASC differentiation to mice lacking Ets1, we crossed Ets1−/− mice with mice carrying a heterozygous GFP knock in mutation in the Blimp1 locus that reduces Blimp1 expression by 50%. In the resulting Ets1−/−Prdm1gfp/+ mice, Blimp1 levels in Ets1−/− B cells are normalized. Yet, these mice still show an excess of splenic ASCs. Therefore, Ets1-dependent repression of Blimp1 expression does not seem to be an essential mechanism for preventing ASC generation.
Conclusion
Our study has provided a genome-wide list of target genes for the key B cell transcription factor Ets1. Given that normal expression of Ets1 is required to prevent autoimmune disease, it is interesting that a number of these Ets1 target genes are genes identified as susceptibility alleles of autoimmune diseases. We expect that our results will be helpful in gaining insight into the molecular mechanisms that contribute to autoimmune disease and the roles of B cells in this process.
Availability of Data and Material
The datasets generated during the current study (RNA-seq and ChIP-Seq of Ets1 in B cells) are available in the GeoDatasets repository accession number GSE83797, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83797.
Ethics Statement
Animal experiments were performed under the approval and guidance of the Institutional Animal Care and Use Committee (IACUC) of Roswell Park Cancer Institute protocol #UB1104M, “Ets Transcription Factors in Hematopoiesis.”
Author Contributions
PS performed most experiments with help from AK. PS, SN, and LG-S wrote the manuscript. All authors read and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the support of the University at Buffalo Genomics Core facility, led by Dr. Michael Buck. We also thank Isha Sethi, Jonathan Bard, Christian Gluck, and Dr. Satrajit Sinha for their helpful discussions and suggestions on bioinformatics analyses.
Funding
This study was supported by NIH grant R01 AI085127.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00383/full#supplementary-material.
Figure S1. Ets1-binding sites in the Pax5 gene. The genomic locus of the mouse Pax5 gene (bottom) with Ets1-binding sites indicated as black bars along the top of the figure and by vertical columns of yellow shading. Note that Ets1 is strongly enriched at the promoter and in Intron 5 where a known B cell-specific enhancer is localized. Also shown are peaks of H3K27 acetylation and H3K4 monomethylation derived from the mouse ENCODE datasets, which mark active enhancers and promoters.
Figure S2. Ets1-binding sites in the Prdm1 gene. The genomic locus of the mouse Prdm1 gene (bottom) with Ets1-binding sites indicated as black bars along the top of the figure and yellow shaded vertical columns. Note that Ets1 is enriched in Intron 3 as well as sequences localized 50 kb downstream of the gene and sequences localized ~180 kb upstream of Prdm1.
Figure S3. Ets1 binding sites in previously described target genes. The genomic loci encoding Nfkb1 (p50), Cd79a (Igα), and H-2Aa and H-2Eb1 (MHC II isoforms), genes that have previously been described as Ets1 targets based on transient transfection assays. Each gene is shown in a format similar to that described for Figures S1 and S2 in Supplementary Material. Note that each gene contains Ets1-binding sites in the promoter and other nearby regions based on ChIP-seq, supporting the idea that these are true functional target genes of Ets1.
Figure S4. Ets1-binding sites are found in a large percentage of genes involved in BCR signaling. BCR signaling cascade analyzed by IPA software. Genes encoding proteins outlined in purple contain one or more Ets1-binding sites nearby.
Figure S5. Expression of Pax5 is not altered in Ets1-deficient B cells. RNA-seq profiles for the Ets1 gene, the Pax5 gene, and a constitutively expressed housekeeping gene Ppia (peptidylprolyl isomerase A). Neither Pax5 nor Ppia shows any change in transcription in Ets1−/− B cells. Overall, Ets1 transcription is also not reduced in Ets1−/− B cells, but the exons that are targeted in the knockout (part of the second exon and the third exon) show reduced/absent transcription.
Figure S6. Heat maps to show expression changes of genes in certain selected pathways identified by gene ontology analysis. Heat maps of the genes identified in the (A) defense response and (B) protein phosphorylation pathways showing clustering of wild-type and knockout B cells. Genes associated with autoimmune disease susceptibility that are further studied in this manuscript are highlighted by red type.
Figure S7. Ets1-binding sites in the Egr1, Stat4, Slamf6, and Ptpn22 genes. The genomic loci of the mouse Egr1, Stat4, Slamf6, and Ptpn22 genes with Ets1-binding regions shown along the top as black bars and as yellow shaded vertical columns. Also shown are peaks of H3K27 acetylation and H3K4 monomethylation derived from the mouse ENCODE datasets, which mark active enhancers and promoters.
Abbreviations
ASCs, antibody-secreting cells; BCR, B cell receptor; ChIP-seq, chromatin immunoprecipitation-sequencing; ENCODE, encyclopedia of DNA elements; FPKM, fragments per kilobase of transcript per million mapped reads; GO, gene ontology; GWAS, genome-wide association study; IRES, internal ribosomal entry site; LPS, lipopolysaccharide; SNPs, single-nucleotide polymorphisms; ShRNA, short hairpin RNA.
References
1. Nutt SL, Urbanek P, Rolink A, Busslinger M. Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev (1997) 11(4):476–91. doi: 10.1101/gad.11.4.476
2. Zandi S, Mansson R, Tsapogas P, Zetterblad J, Bryder D, Sigvardsson M. EBF1 is essential for B-lineage priming and establishment of a transcription factor network in common lymphoid progenitors. J Immunol (2008) 181(5):3364–72. doi:10.4049/jimmunol.181.5.3364
3. Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell (1994) 79(5):875–84. doi:10.1016/0092-8674(94)90076-0
4. Mikkola I, Heavey B, Horcher M, Busslinger M. Reversion of B cell commitment upon loss of Pax5 expression. Science (2002) 297(5578):110–3. doi:10.1126/science.1067518
5. Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature (2007) 449(7161):473–7. doi:10.1038/nature06159
6. Nechanitzky R, Akbas D, Scherer S, Gyory I, Hoyler T, Ramamoorthy S, et al. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat Immunol (2013) 14(8):867–75. doi:10.1038/ni.2641
7. Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, et al. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol (2008) 9(12):1388–98. doi:10.1038/ni.1667
8. Turner CA Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell (1994) 77(2):297–306. doi:10.1016/0092-8674(94)90321-2
9. Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity (2002) 17(1):51–62. doi:10.1016/S1074-7613(02)00335-7
10. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature (2001) 412(6844):300–7. doi:10.1038/35085509
11. Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol (2001) 189(7):3328–38. doi:10.4049/jimmunol.1201042
12. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol (2006) 7(7):773–82. doi:10.1038/ni1357
13. De Silva NS, Simonetti G, Heise N, Klein U. The diverse roles of IRF4 in late germinal center B-cell differentiation. Immunol Rev (2012) 247(1):73–92. doi:10.1111/j.1600-065X.2012.01113.x
14. Garrett-Sinha LA. Review of Ets1 structure, function, and roles in immunity. Cell Mol Life Sci (2013) 70:3375–90. doi:10.1007/s00018-012-1243-7
15. Zhang XK, Moussa O, LaRue A, Bradshaw S, Molano I, Spyropoulos DD, et al. The transcription factor Fli-1 modulates marginal zone and follicular B cell development in mice. J Immunol (2008) 181(3):1644–54. doi:10.4049/jimmunol.181.3.1644
16. Bradshaw S, Zheng WJ, Tsoi LC, Gilkeson G, Zhang XK. A role for Fli-1 in B cell proliferation: implications for SLE pathogenesis. Clin Immunol (2008) 129(1):19–30. doi:10.1016/j.clim.2008.05.010
17. Xue HH, Bollenbacher-Reilley J, Wu Z, Spolski R, Jing X, Zhang YC, et al. The transcription factor GABP is a critical regulator of B lymphocyte development. Immunity (2007) 26(4):421–31. doi:10.1016/j.immuni.2007.03.010
18. Carotta S, Wu L, Nutt SL. Surprising new roles for PU.1 in the adaptive immune response. Immunol Rev (2010) 238(1):63–75. doi:10.1111/j.1600-065X.2010.00955.x
19. DeKoter RP, Geadah M, Khoosal S, Xu LS, Thillainadesan G, Torchia J, et al. Regulation of follicular B cell differentiation by the related E26 transformation-specific transcription factors PU.1, Spi-B, and Spi-C. J Immunol (2010) 185(12):7374–84. doi:10.4049/jimmunol.1001413
20. Lee CM, Gupta S, Parodo J, Wu J, Marshall JC, Hu J. The uncovering of ESE-1 in human neutrophils: implication of its role in neutrophil function and survival. Genes Immun (2015) 16(5):356–61. doi:10.1038/gene.2015.10
21. Lacorazza HD, Miyazaki Y, Di Cristofano A, Deblasio A, Hedvat C, Zhang J, et al. The ETS protein MEF plays a critical role in perforin gene expression and the development of natural killer and NK-T cells. Immunity (2002) 17(4):437–49. doi:10.1016/S1074-7613(02)00422-3
22. John SA, Clements JL, Russell LM, Garrett-Sinha LA. Ets-1 regulates plasma cell differentiation by interfering with the activity of the transcription factor Blimp-1. J Biol Chem (2008) 283(2):951–62. doi:10.1074/jbc.M705262200
23. Wang D, John SA, Clements JL, Percy DH, Barton KP, Garrett-Sinha LA. Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol (2005) 17(9):1179–91. doi:10.1093/intimm/dxh295
24. Russell L, John S, Cullen J, Luo W, Shlomchik MJ, Garrett-Sinha LA. Requirement for transcription factor Ets1 in B cell tolerance to self-antigens. J Immunol (2015) 195(8):3574–83. doi:10.4049/jimmunol.1500776
25. Eyquem S, Chemin K, Fasseu M, Chopin M, Sigaux F, Cumano A, et al. The development of early and mature B cells is impaired in mice deficient for the Ets-1 transcription factor. Eur J Immunol (2004) 34(11):3187–96. doi:10.1002/eji.200425352
26. Mouly E, Chemin K, Nguyen HV, Chopin M, Mesnard L, Leite-de-Moraes M, et al. The Ets-1 transcription factor controls the development and function of natural regulatory T cells. J Exp Med (2010) 207(10):2113–25. doi:10.1084/jem.20092153
27. Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA. Altered sequence of the ETS1 transcription factor may predispose to rheumatoid arthritis susceptibility. Scand J Rheumatol (2013) 42(1):11–4. doi:10.3109/03009742.2012.711367
28. Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet (2010) 6(2):e1000841. doi:10.1371/journal.pgen.1000841
29. John S, Russell L, Chin SS, Luo W, Oshima R, Garrett-Sinha LA. Transcription factor ets1, but not the closely related factor ets2, inhibits antibody-secreting cell differentiation. Mol Cell Biol (2014) 34(3):522–32. doi:10.1128/MCB.00612-13
30. Nikolajczyk BS, Nelsen B, Sen R. Precise alignment of sites required for mu enhancer activation in B cells. Mol Cell Biol (1996) 16(8):4544–54. doi:10.1128/MCB.16.8.4544
31. Nikolajczyk BS, Sanchez JA, Sen R. ETS protein-dependent accessibility changes at the immunoglobulin mu heavy chain enhancer. Immunity (1999) 11(1):11–20. doi:10.1016/S1074-7613(00)80077-1
32. Jabrane-Ferrat N, Peterlin BM. Ets-1 activates the DRA promoter in B cells. Mol Cell Biol (1994) 14(11):7314–21. doi:10.1128/MCB.14.11.7314
33. Maier H, Colbert J, Fitzsimmons D, Clark DR, Hagman J. Activation of the early B-cell-specific mb-1 (Ig-alpha) gene by Pax-5 is dependent on an unmethylated Ets binding site. Mol Cell Biol (2003) 23(6):1946–60. doi:10.1128/MCB.23.6.1946-1960.2003
34. Liu H, Grundstrom T. Calcium regulation of GM-CSF by calmodulin-dependent kinase II phosphorylation of Ets1. Mol Biol Cell (2002) 13(12):4497–507. doi:10.1091/mbc.E02-03-0149
35. Roessler S, Gyory I, Imhof S, Spivakov M, Williams RR, Busslinger M, et al. Distinct promoters mediate the regulation of Ebf1 gene expression by interleukin-7 and Pax5. Mol Cell Biol (2007) 27(2):579–94. doi:10.1128/MCB.01192-06
36. Nguyen HV, Mouly E, Chemin K, Luinaud R, Despres R, Fermand JP, et al. The Ets-1 transcription factor is required for Stat1-mediated T-bet expression and IgG2a class switching in mouse B cells. Blood (2012) 119(18):4174–81. doi:10.1182/blood-2011-09-378182
37. Liu F, Fan H, Ren D, Dong G, Hu E, Ji J, et al. TLR9-induced miR-155 and Ets-1 decrease expression of CD1d on B cells in SLE. Eur J Immunol (2015) 45(7):1934–45. doi:10.1002/eji.201445286
38. Barton K, Muthusamy N, Fischer C, Ting CN, Walunas TL, Lanier LL, et al. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity (1998) 9(4):555–63. doi:10.1016/S1074-7613(00)80638-X
39. Fairfax KA, Corcoran LM, Pridans C, Huntington ND, Kallies A, Nutt SL, et al. Different kinetics of blimp-1 induction in B cell subsets revealed by reporter gene. J Immunol (2007) 178(7):4104–11. doi:10.4049/jimmunol.178.7.4104
40. Hollenhorst PC, Shah AA, Hopkins C, Graves BJ. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev (2007) 21(15):1882–94. doi:10.1101/gad.1561707
41. Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev (2011) 25(20):2147–57. doi:10.1101/gad.17546311
42. Plotnik JP, Budka JA, Ferris MW, Hollenhorst PC. ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res (2014) 42(19):11928–40. doi:10.1093/nar/gku929
43. Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res (2012) 22(9):1813–31. doi:10.1101/gr.136184.111
44. Langmead B. Aligning short sequencing reads with bowtie. Curr Protoc Bioinformatics (2010) Chapter 11:Unit 11.7. doi:10.1002/0471250953.bi1107s32
45. Feng J, Liu T, Zhang Y. Using MACS to identify peaks from ChIP-seq data. Curr Protoc Bioinformatics (2011) Chapter 2:Unit 2.14. doi:10.1002/0471250953.bi0214s34
46. Yu G, Wang LG, He QY. ChIPseeker: an R/bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics (2015) 31(14):2382–3. doi:10.1093/bioinformatics/btv145
47. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell (2010) 38(4):576–89. doi:10.1016/j.molcel.2010.05.004
48. Zambelli F, Pesole G, Pavesi G. PscanChIP: finding over-represented transcription factor-binding site motifs and their correlations in sequences from ChIP-Seq experiments. Nucleic Acids Res (2013) 41(Web Server issue):W535–43. doi:10.1093/nar/gkt448
49. Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol (2012) 13(8):418. doi:10.1186/gb-2012-13-8-418
50. Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science (2014) 345(6199):943–9. doi:10.1126/science.1256271
51. Revilla IDR, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, et al. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J (2012) 31(14):3130–46. doi:10.1038/emboj.2012.155
52. Lai WK, Bard JE, Buck MJ. ArchTEx: accurate extraction and visualization of next-generation sequence data. Bioinformatics (2012) 28(7):1021–3. doi:10.1093/bioinformatics/bts063
53. Wohner M, Tagoh H, Bilic I, Jaritz M, Poliakova DK, Fischer M, et al. Molecular functions of the transcription factors E2A and E2-2 in controlling germinal center B cell and plasma cell development. J Exp Med (2016) 213(7):1201–21. doi:10.1084/jem.20152002
54. Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim HP, Oh J, Tunyaplin C, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity (2009) 31(6):941–52. doi:10.1016/j.immuni.2009.10.008
55. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc (2012) 7(3):562–78. doi:10.1038/nprot.2012.016
56. Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res (2007) 35(Web Server issue):W169–75. doi:10.1093/nar/gkm415
57. Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C, et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci U S A (2002) 99(19):12281–6. doi:10.1073/pnas.182618999
58. Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, et al. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood (2012) 119(26):6278–87. doi:10.1182/blood-2012-01-403162
59. Chen R, Belanger S, Frederick MA, Li B, Johnston RJ, Xiao N, et al. In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity (2014) 41(2):325–38. doi:10.1016/j.immuni.2014.08.002
60. Valentine MA, Czernik AJ, Rachie N, Hidaka H, Fisher CL, Cambier JC, et al. Anti-immunoglobulin M activates nuclear calcium/calmodulin-dependent protein kinase II in human B lymphocytes. J Exp Med (1995) 182(6):1943–9. doi:10.1084/jem.182.6.1943
61. Cowley DO, Graves BJ. Phosphorylation represses Ets-1 DNA binding by reinforcing autoinhibition. Genes Dev (2000) 14(3):366–76.
62. Medvedovic J, Ebert A, Tagoh H, Busslinger M. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol (2011) 111:179–206. doi:10.1016/B978-0-12-385991-4.00005-2
63. Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol (2007) 8(5):463–70. doi:10.1038/ni1454
64. Decker T, Pasca di Magliano M, McManus S, Sun Q, Bonifer C, Tagoh H, et al. Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity (2009) 30(4):508–20. doi:10.1016/j.immuni.2009.01.012
65. Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature (2014) 515(7527):355–64. doi:10.1038/nature13992
66. Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res (2012) 40(Database issue):D912–7. doi:10.1093/nar/gkr1012
67. Bories JC, Willerford DM, Grevin D, Davidson L, Camus A, Martin P, et al. Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature (1995) 377(6550):635–8. doi:10.1038/377635a0
68. Ou Y. Anthrax toxin receptor 2 gene (ANTXR2) rs4333130 is associated with ankylosing spondylitis. Int J Clin Exp Med (2015) 8(5):7679–83.
69. Fischer A, Schmid B, Ellinghaus D, Nothnagel M, Gaede KI, Schurmann M, et al. A novel sarcoidosis risk locus for Europeans on chromosome 11q13.1. Am J Respir Crit Care Med (2012) 186(9):877–85. doi:10.1164/rccm.201204-0708OC
70. Douglas KB, Windels DC, Zhao J, Gadeliya AV, Wu H, Kaufman KM, et al. Complement receptor 2 polymorphisms associated with systemic lupus erythematosus modulate alternative splicing. Genes Immun (2009) 10(5):457–69. doi:10.1038/gene.2009.27
71. Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, et al. Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behcet’s disease. Ann Rheum Dis (2013) 72(9):1510–6. doi:10.1136/annrheumdis-2011-200288
72. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature (2015) 518(7539):337–43. doi:10.1038/nature13835
73. Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet (2013) 9(4):e1003444. doi:10.1371/journal.pgen.1003444
74. Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet (2008) 40(2):152–4. doi:10.1038/ng.71
75. Carmona FD, Simeon CP, Beretta L, Carreira P, Vonk MC, Rios-Fernandez R, et al. Association of a non-synonymous functional variant of the ITGAM gene with systemic sclerosis. Ann Rheum Dis (2011) 70(11):2050–2. doi:10.1136/ard.2010.148874
76. Rahman P, Bartlett S, Siannis F, Pellett FJ, Farewell VT, Peddle L, et al. CARD15: a pleiotropic autoimmune gene that confers susceptibility to psoriatic arthritis. Am J Hum Genet (2003) 73(3):677–81. doi:10.1086/378076
77. Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behcet disease. Proc Natl Acad Sci U S A (2013) 110(20):8134–9. doi:10.1073/pnas.1306352110
78. Huang CH, Wong RH, Wei JC, Tsay MD, Chen WC, Chen HY, et al. Effects of genetic polymorphisms of programmed cell death 1 and its ligands on the development of ankylosing spondylitis. Rheumatology (Oxford) (2011) 50(10):1809–13. doi:10.1093/rheumatology/ker211
79. Okada Y, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet (2012) 44(5):511–6. doi:10.1038/ng.2231
80. Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet (2009) 41(11):1228–33. doi:10.1038/ng.468
81. Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet (2009) 41(12):1313–8. doi:10.1038/ng.479
82. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36(4):337–8. doi:10.1038/ng1323
83. Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet (2004) 75(2):330–7. doi:10.1086/422827
84. Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet (2004) 75(3):504–7. doi:10.1086/423790
85. Carr EJ, Niederer HA, Williams J, Harper L, Watts RA, Lyons PA, et al. Confirmation of the genetic association of CTLA4 and PTPN22 with ANCA-associated vasculitis. BMC Med Genet (2009) 10:121. doi:10.1186/1471-2350-10-121
86. Bowes J, Loehr S, Budu-Aggrey A, Uebe S, Bruce IN, Feletar M, et al. PTPN22 is associated with susceptibility to psoriatic arthritis but not psoriasis: evidence for a further PsA-specific risk locus. Ann Rheum Dis (2015) 74(10):1882–5. doi:10.1136/annrheumdis-2014-207187
87. Vandiedonck C, Capdevielle C, Giraud M, Krumeich S, Jais JP, Eymard B, et al. Association of the PTPN22*R620W polymorphism with autoimmune myasthenia gravis. Ann Neurol (2006) 59(2):404–7. doi:10.1002/ana.20751
88. Heward JM, Brand OJ, Barrett JC, Carr-Smith JD, Franklyn JA, Gough SC. Association of PTPN22 haplotypes with Graves’ disease. J Clin Endocrinol Metab (2007) 92(2):685–90. doi:10.1210/jc.2006-2064
89. Zhao SX, Xue LQ, Liu W, Gu ZH, Pan CM, Yang SY, et al. Robust evidence for five new Graves’ disease risk loci from a staged genome-wide association analysis. Hum Mol Genet (2013) 22(16):3347–62. doi:10.1093/hmg/ddt183
90. Hou S, Yang Z, Du L, Jiang Z, Shu Q, Chen Y, et al. Identification of a susceptibility locus in STAT4 for Behcet’s disease in Han Chinese in a genome-wide association study. Arthritis Rheum (2012) 64(12):4104–13. doi:10.1002/art.37708
91. Zervou MI, Sidiropoulos P, Petraki E, Vazgiourakis V, Krasoudaki E, Raptopoulou A, et al. Association of a TRAF1 and a STAT4 gene polymorphism with increased risk for rheumatoid arthritis in a genetically homogeneous population. Hum Immunol (2008) 69(9):567–71. doi:10.1016/j.humimm.2008.06.006
92. Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med (2007) 357(10):977–86. doi:10.1056/NEJMoa073003
93. Zervou MI, Goulielmos GN, Castro-Giner F, Tosca AD, Krueger-Krasagakis S. STAT4 gene polymorphism is associated with psoriasis in the genetically homogeneous population of Crete, Greece. Hum Immunol (2009) 70(9):738–41. doi:10.1016/j.humimm.2009.05.008
94. Gestermann N, Mekinian A, Comets E, Loiseau P, Puechal X, Hachulla E, et al. STAT4 is a confirmed genetic risk factor for Sjogren’s syndrome and could be involved in type 1 interferon pathway signaling. Genes Immun (2010) 11(5):432–8. doi:10.1038/gene.2010.29
95. Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suarez H, et al. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet (2009) 18(11):2071–7. doi:10.1093/hmg/ddp119
96. Sun C, Zhi D, Shen S, Luo F, Sanjeevi CB. SNPs in the exons of toll-like receptors are associated with susceptibility to type 1 diabetes in Chinese population. Hum Immunol (2014) 75(11):1084–8. doi:10.1016/j.humimm.2014.09.008
97. Xiao W, Liu Z, Lin J, Li J, Wu K, Ma Y, et al. Polymorphisms in TLR1, TLR6 and TLR10 genes and the risk of Graves’ disease. Autoimmunity (2015) 48(1):13–8. doi:10.3109/08916934.2014.939269
98. Seok H, Suh DW, Jo B, Lee HB, Jang HM, Park HK, et al. Association between TLR1 polymorphisms and alopecia areata. Autoimmunity (2014) 47(6):372–7. doi:10.3109/08916934.2014.910769
99. Nishimoto K, Kochi Y, Ikari K, Yamamoto K, Suzuki A, Shimane K, et al. Association study of TRAF1-C5 polymorphisms with susceptibility to rheumatoid arthritis and systemic lupus erythematosus in Japanese. Ann Rheum Dis (2010) 69(2):368–73. doi:10.1136/ard.2008.104315
100. Redler S, Brockschmidt FF, Forstbauer L, Giehl KA, Herold C, Eigelshoven S, et al. The TRAF1/C5 locus confers risk for familial and severe alopecia areata. Br J Dermatol (2010) 162(4):866–9. doi:10.1111/j.1365-2133.2009.09598.x
101. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet (2012) 44(12):1341–8. doi:10.1038/ng.2467
102. Sun Y, Zuo X, Zheng X, Zhou F, Liang B, Liu H, et al. A comprehensive association analysis confirms ZMIZ1 to be a susceptibility gene for vitiligo in Chinese population. J Med Genet (2014) 51(5):345–53. doi:10.1136/jmedgenet-2013-102233
103. Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet (2012) 90(4):636–47. doi:10.1016/j.ajhg.2012.02.020
104. Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med (2004) 200(8):967–77. doi:10.1084/jem.20040973
105. Solomon LA, Li SK, Piskorz J, Xu LS, DeKoter RP. Genome-wide comparison of PU.1 and Spi-B binding sites in a mouse B lymphoma cell line. BMC Genomics (2015) 16:76. doi:10.1186/s12864-015-1303-0
106. Niebuhr B, Kriebitzsch N, Fischer M, Behrens K, Gunther T, Alawi M, et al. Runx1 is essential at two stages of early murine B-cell development. Blood (2013) 122(3):413–23. doi:10.1182/blood-2013-01-480244
107. Cheng Y, Ma Z, Kim BH, Wu W, Cayting P, Boyle AP, et al. Principles of regulatory information conservation between mouse and human. Nature (2014) 515(7527):371–5. doi:10.1038/nature13985
108. Hollenhorst PC, Chandler KJ, Poulsen RL, Johnson WE, Speck NA, Graves BJ. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet (2009) 5(12):e1000778. doi:10.1371/journal.pgen.1000778
109. Carotta S, Willis SN, Hasbold J, Inouye M, Pang SHM, Emslie D, et al. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med (2014) 211(11):2169–81. doi:10.1084/jem.20140425
110. Liang Y, Pan HF, Ye DQ. Therapeutic potential of STAT4 in autoimmunity. Expert Opin Ther Targets (2014) 18(8):945–60. doi:10.1517/14728222.2014.920325
111. Rawlings DJ, Dai X, Buckner JH. The role of PTPN22 risk variant in the development of autoimmunity: finding common ground between mouse and human. J Immunol (2015) 194(7):2977–84. doi:10.4049/jimmunol.1403034
112. Korman BD, Kastner DL, Gregersen PK, Remmers EF. STAT4: genetics, mechanisms, and implications for autoimmunity. Curr Allergy Asthma Rep (2008) 8(5):398–403. doi:10.1007/s11882-008-0077-8
113. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol (2010) 10(4):236–47. doi:10.1038/nri2729
114. Raj P, Rai E, Song R, Khan S, Wakeland BE, Viswanathan K, et al. Regulatory polymorphisms modulate the expression of HLA class II molecules and promote autoimmunity. Elife (2016) 5:e12089. doi:10.7554/eLife.12089
115. Xu Z, Duan B, Croker BP, Morel L. STAT4 deficiency reduces autoantibody production and glomerulonephritis in a mouse model of lupus. Clin Immunol (2006) 120(2):189–98. doi:10.1016/j.clim.2006.03.009
116. Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity (2013) 39(1):111–22. doi:10.1016/j.immuni.2013.06.013
117. Oh YK, Jang E, Paik DJ, Youn J. Early growth response-1 plays a non-redundant role in the differentiation of B cells into plasma cells. Immune Netw (2015) 15(3):161–6. doi:10.4110/in.2015.15.3.161
Keywords: Ets1, target genes, ChIP-seq, RNA-seq, promoter, enhancer, autoimmunity
Citation: Saelee P, Kearly A, Nutt SL and Garrett-Sinha LA (2017) Genome-Wide Identification of Target Genes for the Key B Cell Transcription Factor Ets1. Front. Immunol. 8:383. doi: 10.3389/fimmu.2017.00383
Received: 09 November 2016; Accepted: 17 March 2017;
Published: 07 April 2017
Edited by:
Barbara L. Kee, University of Chicago, USAReviewed by:
Rodney P. DeKoter, University of Western Ontario, CanadaKay L. Medina, Mayo Clinic, USA
Robert Mansson, Karolinska Institutet, Sweden
Copyright: © 2017 Saelee, Kearly, Nutt and Garrett-Sinha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lee Ann Garrett-Sinha, bGVlc2luaGFAYnVmZmFsby5lZHU=