 Cristina Casals
Cristina Casals María A. Campanero-Rhodes
María A. Campanero-Rhodes Belén García-Fojeda
Belén García-Fojeda Dolores Solís
Dolores Solís- 1Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Instituto de Salud Carlos III, Madrid, Spain
- 2Departamento de Bioquímica y Biología Molecular, Universidad Complutense de Madrid, Madrid, Spain
- 3Instituto de Química Física Rocasolano, CSIC, Madrid, Spain
Different families of endogenous lectins use complementary defense strategies against pathogens. They may recognize non-self glycans typically found on pathogens and/or host glycans. The collectin and galectin families are prominent examples of these two lectin categories. Collectins are C-type lectins that contain a carbohydrate recognition domain and a collagen-like domain. Members of this group include surfactant protein A (SP-A) and D (SP-D), secreted by the alveolar epithelium to the alveolar fluid. Lung collectins bind to several microorganisms, which results in pathogen aggregation and/or killing, and enhances phagocytosis of pathogens by alveolar macrophages. Moreover, SP-A and SP-D influence macrophage responses, contributing to resolution of inflammation, and SP-A is essential for tissue-repair functions of macrophages. Galectins also function by interacting directly with pathogens or by modulating the immune system in response to the infection. Direct binding may result in enhanced or impaired infection of target cells, or can have microbicidal effects. Immunomodulatory effects of galectins include recruitment of immune cells to the site of infection, promotion of neutrophil function, and stimulation of the bactericidal activity of infected macrophages. Moreover, intracellular galectins can serve as danger receptors, promoting autophagy of the invading pathogen. This review will focus on the role of collectins and galectins in pathogen clearance and immune response activation in infectious diseases of the respiratory system.
Introduction
Host defense in the lung is exceptionally, if not uniquely, challenging. The alveolar boundary is clearly the most vulnerable body interface. There are at least three important differences among the alveolar boundary and the upper respiratory tract, gut, and skin interfaces. First, the surface area to be defended is greater in the alveolar boundary (90 m2) than in the gut (10 m2) or skin (2 m2) (1). Second, compared with the skin, gut, and upper respiratory tract, the bacterial biomass in the alveoli of healthy lungs is low (2). Third, there are physical barriers or harsh chemical environments in the skin (cornified epithelial layers) and gut (regular secretion of bile, which acts as an antiseptic detergent) but not in the delicate alveolar space. In addition, there is higher risk of pathogen dissemination at the alveolar boundary than at any other environmental boundary, since only two cell layers (the alveolar epithelium and the capillary endothelium) separate the invader from the bloodstream in order to facilitate gas exchange.
The innate immune system in the alveolar space is made up of a cellular arm [mainly alveolar macrophages (aMΦ) (1), but also epithelial (AEC), dendritic, and T cells (1, 3, 4)] and a humoral arm composed of antimicrobial proteins and peptides present in the alveolar fluid such as surfactant protein A (SP-A) and D (SP-D), lactoferrin, lysozyme, fibronectin, immunoglobulins, complement components, defensins, and cathelicidins, among others (5, 6). In this review we focus on the role of lung soluble collagenous C-type lectins (SP-A and SP-D) and galectins. SP-A and SP-D are principally secreted to the alveolar fluid by type II AECs and to the airway lumen by Club cells and submucosal cells (7). They are also detected in the trachea (8) and nasal mucosa (9), where they provide immune protection. Galectins are expressed in the lung by innate immune cells and epithelial cells. Galectins are present in the cytoplasm and nucleus, as well as extracellular space, although galectins lack a typical secretion signal peptide. They are secreted by direct translocation across the plasma membrane or through release in extracellular vesicles (10). Thus, they can function both inside and outside cells. The review describes biochemical and structural aspects of lung collectins and their role in antimicrobial immunity and alveolar immune homeostasis, and the involvement of galectins in the response to respiratory infectious diseases, including expression, binding to pathogens, modulatory effects on immune cells, and intracellular functions.
Collectins
Biochemical and Structural Aspects
Collectins or collagenous C-type lectins are a family of proteins that contain a Ca2+-dependent carbohydrate recognition domain (CRD) contiguous to a collagen-like triple helical domain. In humans, members of this group include SP-A and SP-D, secreted by the alveolar epithelium, nonciliated bronchiolar cells and other mucosal surfaces exposed to the external environment (7, 8, 11), mannan-binding lectin (MBL) secreted by hepatocytes to serum, and the recently discovered CL-L1, CL-K1, and CL-P1, present in the serum and several tissues (12, 13) (Figure 1A). Collectins are well-conserved oligomeric proteins, assembled in trimers or multiples of three subunits due to their collagen domains. The primary structure of each subunit consists of an N-terminal segment containing cysteine residues involved in oligomerization followed by a collagen-like region, an alpha helical coiled neck region, and a globular CRD with a calcium ion at the lectin site (Figure 1A). Lung collectins are modified after translation (cleavage of the signal peptide, proline hydroxylation, and N-linked glycosylation) (7, 15) and intracellularly assembled into oligomeric structures that, in the case of SP-A, resemble a flower bouquet of six trimers, while the assembly of SP-D resembles a cruciform of four trimers (Figure 1A). Supratrimeric oligomerization of lung collectins appears to be needed for many of their functions (16, 17) since it facilitates multivalent binding and increases the functional affinity of the globular domain for their ligands.
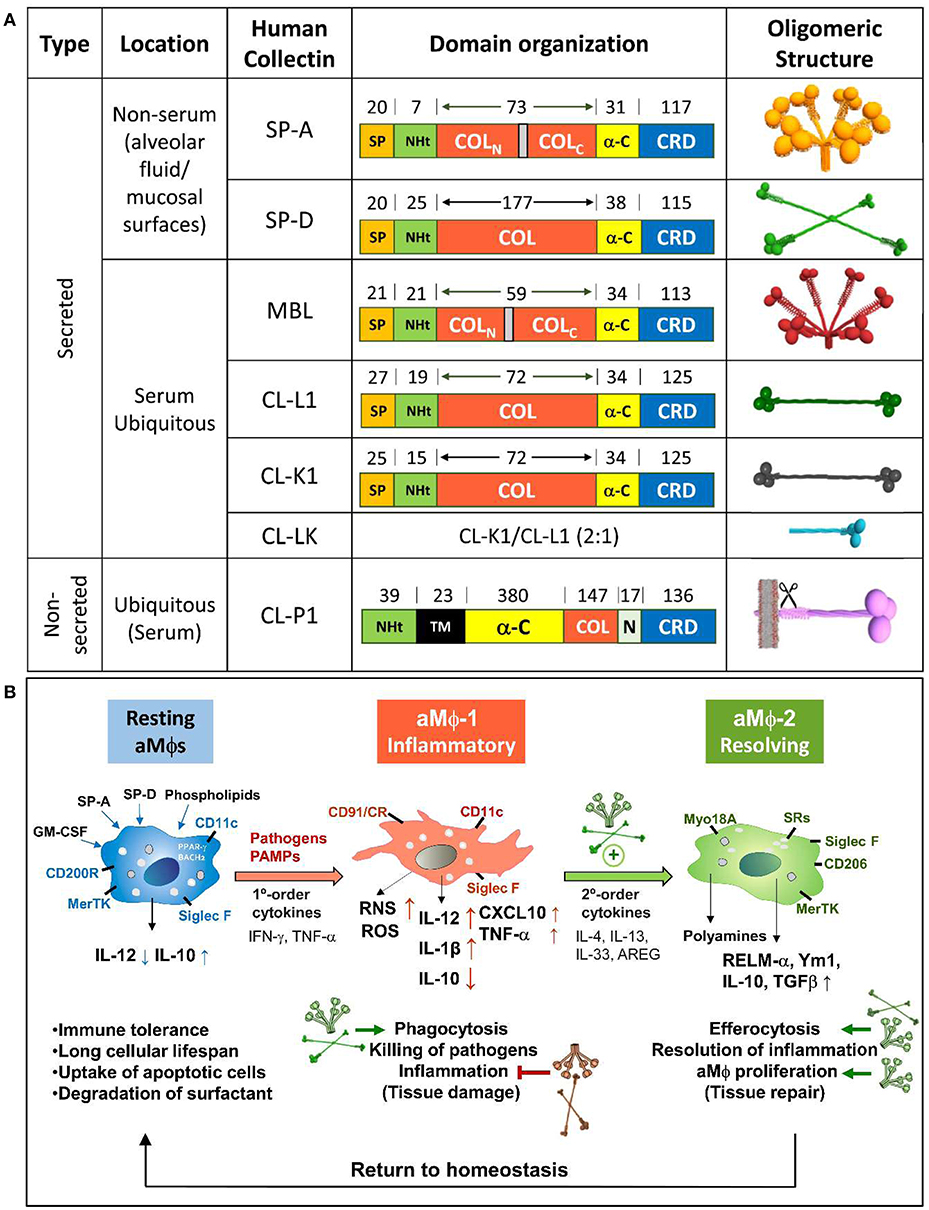
Figure 1. (A) Structural analysis of human collectins. Domain organization of human collectin polypeptide chains and the number of amino acids covering each domain are shown. Interruptions in the collagen domain of SP-A and MBL are indicated. Three-dimensional models of collectin oligomers are also shown. Trimers of collectins are each built up by the association of three polypeptide chains, the collagen regions of which intertwine to form a collagen triple helix. Whereas all other collectins are soluble, CL-P1 is a transmembrane protein orientated with its N-terminal toward the cytosol. CL-P1 may be regarded as both a collectin and a scavenger receptor. The scissors symbol means the shedding of a soluble form of CL-P1 by a hitherto unknown mechanism, which results in the presence of soluble CL-P1 in the circulation. The molecules are not drawn to scale. SP, signal peptide; NHt, N-terminal domain; COL, collagen-like domain; α-C, α-helical coiled-coil domain; CRD, carbohydrate recognition domain; TM, transmembrane domain. (B) Role of lung collectins on sequential type 1 and type 2 immune responses following respiratory infection. Respiratory pathogens are detected by AECs and aMΦs, initiating an innate immune response to clear localized infections. The type 1 response is essential in controlling infection but also induces tissue damage. Stimulated tissue-resident lymphoid cells and AECs release appropriate second-order cytokines that initiate a two-tiered response. The type 2 response, amplified by lung collectins (14), modulates aMΦs toward an anti-inflammatory resolving phenotype involved in lung repair. The role of lung collectins in these homeostatic changes is shown by small green or red arrows, which mean SP-A/D-mediated activation or inhibition, respectively.
The CRDs of lung collectins bind to mannose and mannose-rich microbial glycoconjugates, such as yeast mannans and mycobacterial lipoarabinomannan. Besides, lung collectins recognize a wide variety of carbohydrates present in the surface of several microorganisms, including glucose, fucose, N-acetylglucosamine, and N-acetylmannosamine. Their globular domains recognize not only carbohydrates but also a broader repertoire of ligands, including proteins, nucleic acids, and lipids (7, 18, 19). Despite their similar CRDs, SP-A and SP-D show significant differences in ligand preferences, with SP-A ligands generally being more amphipathic (19, 20), and SP-D ligands richer in carbohydrates (20). These different preferences likely serve to extend the range of innate immune surveillance in the lung.
The collagen-like domains of collectins not only function as scaffolding that amplifies the ligand binding activities of globular domains but also are responsible for collectin binding to receptors in immune cells. Such receptors are involved in phagocytosis and clearance of microorganisms (7, 8, 12, 13) and biological/abiotic particles from the pulmonary environment (21–23), and in efferocytosis of dead cells (apoptotic/necrotic) (24, 25). Collagen-dependent functions of collectins are shared with other secreted defense collagens (C1q, ficolins, and adiponectin) (26). This group of proteins has a dual capacity to promote pathogen elimination and control inflammation (27–30). Moreover, they seem to activate molecular and cellular mechanisms that force a return to homeostasis (14).
Antimicrobial Immunity
SP-A and/or SP-D recognize a wide range of respiratory pathogens, including influenza A virus, respiratory syncytial virus, Mycobacterium tuberculosis, Aspergillus fumigatus, Pseudomonas aeruginosa, Haemophilus influenzae [see (8, 31) for reviews], and the parasitic helminth Nippostrongylus brasiliensis (32). SP-A- or SP-D-deficient mice show decreased microbe clearance and increased tissue markers of inflammation (14, 30–33), suggesting lung collectins' protective role in lung immune defense.
Lung collectins enhance the clearance of pathogens by four different mechanisms: (i) By aggregating pathogens to which they bind, which hinders their entry into epithelial cells and facilitates their removal, either by mucociliary clearance or by phagocytosis by aMΦs and recruited neutrophils (7, 8, 30, 31, 34). (ii) By binding to neutrophil extracellular traps (NET)-DNA and to bacteria simultaneously, thereby promoting bacterial trapping by the NETs (35). (iii) By enhancing phagocytosis of IgG-opsonized particles (36) and complement-coated particles (36, 37). (iv) By up-regulating expression of cell-surface receptors involved in microbial recognition, such as mannose receptor (38) and scavenger receptor SR-AI/II (39).
Data supporting direct antimicrobial activity of SP-A and SP-D are sparse (8, 31). Most respiratory pathogenic bacteria and fungi are resistant to SP-A and SP-D (8, 34, 40, 41). However, it is possible that cooperative interactions of lung collectins with other lung antimicrobial peptides enhance the microbicidal defense of the lungs. In this regard, we recently discovered synergic action between SP-A and SP-BN, a secreted anionic antimicrobial peptide derived from SP-B proprotein. Interaction between SP-A and SP-BN confers new antimicrobial properties, including the ability to bind, kill, and enhance phagocytosis of pathogenic K. pneumoniae K2 that is otherwise resistant to either protein alone (34). Moreover, therapeutic treatment with SP-A and SP-BN protects against K. pneumoniae K2 infection in vivo due to SP-A/SP-BN capability to both kill bacteria and modulate host inflammatory response (34). Yet a promising field to explore is the interaction of lung collectins with other lung antimicrobial peptides and antibiotics and the potential relevance of these interactions in innate host defense in the lung.
Alveolar Immune Homeostasis
The niche in which alveolar macrophages exist, rich in surfactant lipids, SP-A, and SP-D (42), has a considerable influence on many aspects of aMΦ phenotype (1, 43). Alveolar MΦs function as sentinels of a healthy state, promoting immune tolerance to innocuous antigens. During an infection, aMΦs recognize alarm signals such as IFN-γ and PAMPs, initiating proinflammatory responses and pathogen clearance (MΦ-1 phenotype) (Figure 1B), and collectins promote phagocytosis of pathogens by binding to the CD91/calreticulin receptor on aMΦs (44). However, host defense requires a balance between decreasing microbial burden and restricting tissue damage caused directly by pathogens or indirectly by the immune response (45). In this vein, lung collectins influence aMΦ responses to limit inflammation. First, they block the binding of TLR ligands to their receptors by direct interaction with TLR4, TLR2, the TLR co-receptor MD2, and CD14 (17, 30, 46) or by binding to TLR4/CD14 ligands (47, 48), acting as LPS scavengers in vivo (49). Second, they modify aMΦ response to TLR ligands by modulating signaling cascades. For example, SP-A and SP-D bind to SIRPα through their globular heads to initiate an SHP-1-dependent signaling pathway that blocks proinflammatory mediator production (44). In addition, SP-A increases the expression of negative regulators of TLR-signaling, such as IRAK-M (50) and β-arrestin 2 (51), and inhibits activation of NFκB, ERK, p38, and Akt in aMΦs (52, 53). Third, they reduce the production of reactive oxygen intermediates (54, 55) and for SP-D, this effect is mediated through its binding to the inhibitory receptor LAIR-1 (56). Fourth, SP-A limits inflammation by binding to IFN-γ, suppressing IFN-γ interaction with its receptor IFN-γR1 (57).
Besides limiting inflammation, lung collectins activate different mechanisms that contribute to disease resolution. After proinflammatory type 1 responses against invading pathogens, repair-associated type 2 responses must be initiated (58, 59). The tissue repair response is classically associated with the production of IL-4/IL-13 cytokines and the induction of alternatively activated macrophages (MΦ-2 phenotype) (Figure 1B). We recently found that defense collagens (SP-A and C1q) enhance IL-4/13-dependent alternative activation, proliferation, and tissue-repair functions of macrophages through binding to the myosin 18A receptor by their collagen domains (14). Loss of function studies using SP-A- and C1q-deficient mice demonstrated that SP-A and C1q are necessary to promote tissue repair during infection with the parasite N. brasiliensis and the Gram positive bacterium Listeria monocytogenes, respectively (14). SP-D also seems to be an important modulator of protective IL-4/13-induced aMΦ responses against N. brasiliensis (32). Interestingly, IL-4Rα signaling requires concomitant recognition of apoptotic cells to induce the tissue repair program in macrophages (60), suggesting that tissue repair is restricted to the damaged site. SP-A, SP-D, and C1q assist the recognition and clearance of apoptotic neutrophils by macrophages (24, 25, 61, 62), a mechanism that differs from classical phagocytosis and that leads to the production of anti-inflammatory cytokines (IL-10 and TGFβ) (63), contributing to host tolerance during lung infection.
In conclusion, pulmonary collectins provide immune protection against respiratory pathogens, promoting pathogen clearance, limiting inflammation, and activating molecular and cellular mechanisms that help to restore homeostasis. Much of what we know about the protective role of SP-A and SP-D has arisen from studies using SP-A– or SP-D–deficient mice in murine models of respiratory infections (14, 31–33) and other respiratory diseases (30, 31, 64).
Galectins
Galectins are a family of lectins sharing a CRD with β-sandwich fold and β-galactoside-binding ability (65, 66). Nevertheless, the glycan-binding preferences of different galectins may differ significantly, leading to functional divergences. To date, 16 mammalian galectins have been described, of which galectins 5, 6 (both found in rodents), 11, and 15 (found in ruminants) are not present in humans (Figure S1). Based on their structural organization (Figure S1), galectins are classified as proto type, composed of one or two identical CRDs forming non-covalent homodimers (e.g., Gal-1), chimera type, composed of one CRD linked to a non-lectin N-terminal region (Gal-3), and tandem-repeat type, containing two different CRDs covalently connected by a linker peptide (e.g., Gal-8 and -9). Galectins are widely expressed in epithelial and immune cells, and participate in different biological phenomena, including inflammation and immunity (67, 68).
The Expression of Galectins is Altered in Respiratory Infections
An archetypal example is the accumulation of Gal-3 in the alveolar space of Streptococcus pneumoniae-infected mice (69). Gal-3 release also increases in the lungs of mice lethally infected with Francisella novicida (70). In patients infected with M. tuberculosis, the plasma levels of Gal-9 are significantly increased (71). However, Gal-9 expression in macrophages generated in the presence of M. tuberculosis lipoarabinomannan is down-regulated, favoring bacterial intracellular growth (72).
The expression of galectins is also affected by respiratory viruses. As an example, Gal-1 is up-regulated in the lungs of influenza virus-infected mice, in correlation with the viral load (73). Interestingly, Gal-1 is differentially expressed in bronchoepithelial cells infected with 2009 A or seasonal H1N1 influenza virus, revealing strain-specific responses (74). Moreover, patients carrying genetic variants associated with higher Gal-1 expression are less susceptible to infection by avian influenza A (75).
Gal-3 levels in serum and lungs are augmented in infections by the fungus Cryptococcus neoformans (76), while plasma levels of Gal-9 are higher in severe infections by the parasite Plasmodium falciparum (77), and mRNA levels of Gal-9 in the lungs of P. berghei-infected mice are also increased (78).
Thus, the expression of galectins is altered in bacterial, viral, fungal, and parasitic respiratory infections, conceivably correlating with galectin-mediated defense mechanisms.
Galectins Bind to Different Respiratory Pathogens
Gal-3 binds mycolic acids (Figure 2A), the major constituents of mycobacterial cell wall, and could participate in their interaction with host cells (79). Gal-3 also binds lipopolysaccharides from different bacteria, including K. pneumoniae (80) and P. aeruginosa (81). Moreover, Gal-3 and Gal-8 bind to K. pneumoniae O1 cells and decrease bacterial viability, and the same occurs for Gal-8 and strain 2019 of non-typeable H. influenzae (NTHi) (82). Recently, binding of Gal-8 to other six different NTHi clinical isolates was detected (83), suggesting that this could be a general trait.
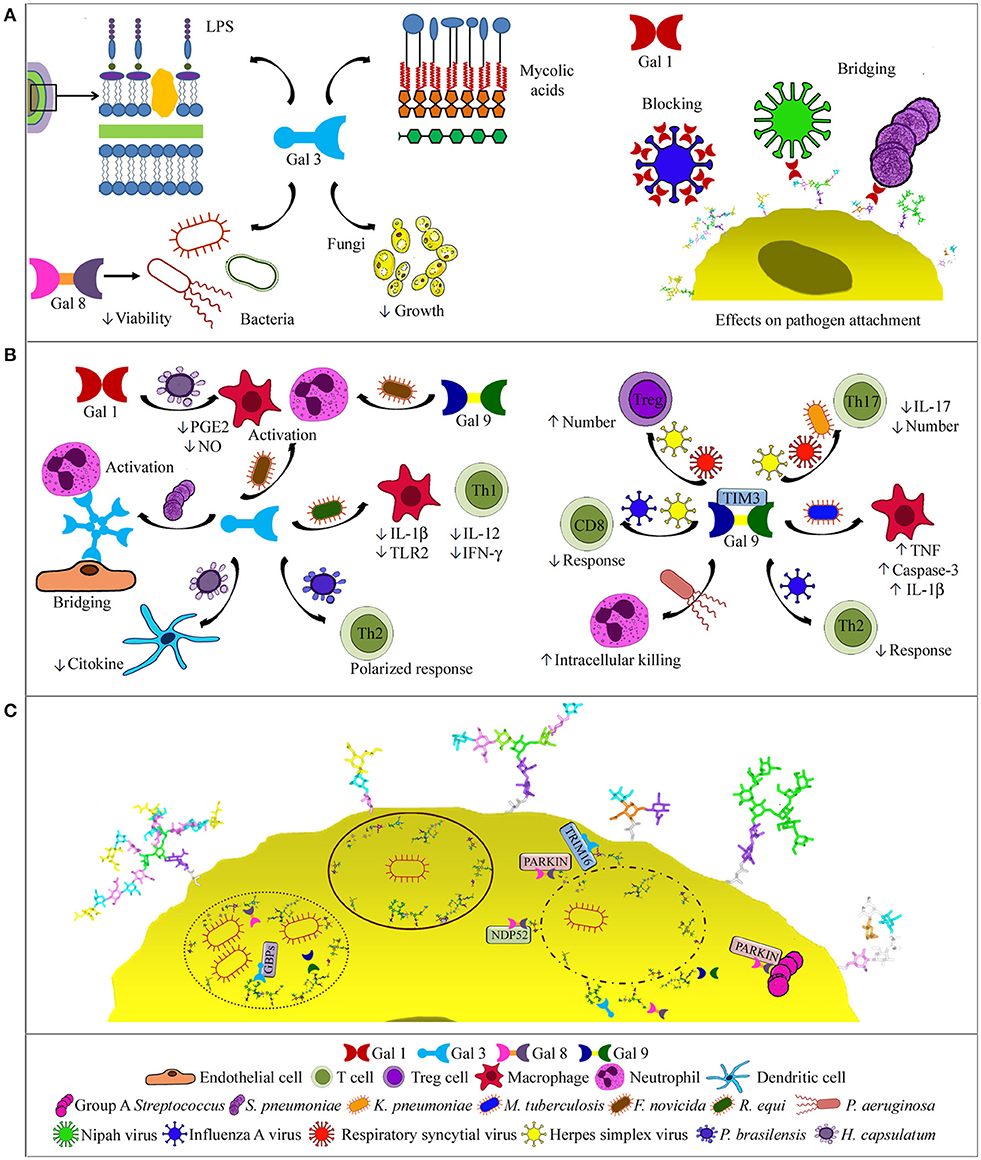
Figure 2. Galectin activities in respiratory infections. (A) Binding to pathogens. Gal-3, the only chimera-type galectin described to date, binds bacterial mycolic acids, lipopolysaccharides, and cells, and also C. neoformans cells with antifungal effects. Gal-8 binds NTHi, decreasing bacterial viability (left side). Gal-1 binding to influenza virus blocks infection, while binding to NiV and S. pneumoniae bridges pathogen and host glycans (right side). (B) Effects on immune cells. Oligomerized Gal-3 can bridge neutrophils to endothelial cells. Depending on the pathogen, Gal-3 drives a Th2-polarized response, decreases macrophage and Th1 cell responses, or activates macrophages and/or neutrophils, similarly to Gal-9. In histoplasmosis, Gal-3 decreases cytokine production by dendritic cells, while Gal-1 modulates PGE2 and NO levels (left). Via TIM-3 binding, Gal-9 may promote bacterial killing by neutrophils or macrophages, decrease humoral and CD8+ cell responses or Th17 cells and IL-17 levels, and increase Treg cells (right). (C) Intracellular functions. Gal-3, -8, and -9 bind to host glycans in the luminal side of lysed phagosomes or permeable replicative vacuoles, and contribute to the autophagic response by recruiting NDP52, parkin, GBPs, or TRIM-16. Gal-8 also recruits parkin to group A Streptococcus.
At the initial phase of infection by Nipah virus (NiV), Gal-1 bridges glycans of the envelope glycoprotein F (NiV-F) with those of host cells, thereby enhancing virus attachment. However, Gal-1 secreted in response to infection reduces NiV-F-mediated syncytia formation and production of progeny virus (84–86). This is a remarkable example of opposing effects of the same galectin on infection by a given pathogen. Moreover, Gal-1 binds to envelope glycoproteins of influenza virus, impairing infection. Cells treated with Gal-1 generate lower viral yields, and treatment of infected mice reduces viral load and lung inflammation (73). Conversely, Gal-1 could account for the increased susceptibility of influenza patients to subsequent infection with pneumococcus. Influenza infection results in desialylation of epithelial cell glycans and exposure of galactosyl moieties that serve as galectin ligands. As Gal-1 also binds to S. pneumoniae, it crosslinks the bacteria to the airway epithelial surface, enhancing pneumococcal adhesion (87).
As final example, Gal-3 binds to C. neoformans cells and delays fungal growth. Moreover, it exerts a lytic effect on fungal extracellular vesicles (76). Thus, Gal-3 has direct anti-C. neoformans effects.
Galectins Modulate the Immune Response to Infection
Accumulation of Gal-3 in the alveoli of pnemococcus-infected mice correlates with neutrophil extravasation (69). Consistently, less neutrophils are recruited in Gal-3−/− mice, which develop more severe pneumonia, and treatment with Gal-3 reduces the severity of infection (88, 89). Gal-3 bridges neutrophils to endothelial cells and activate neutrophils (Figure 2B), augmenting pneumococcus phagocytosis and delaying apoptosis. These effects are disabled by Staphylococcus aureus via degradation of Gal-3 with a bacterial protease (90). In contrast, Gal-3 deficiency confers resistance to Rhodococcus equi (91). Thus, Gal-3−/− mice exhibit higher bacteria lethal doses and production of IL-12 and IFN-γ. Moreover, Gal-3−/− macrophages show decreased bacterial replication and survival, and enhanced production of IL-1β and TLR2. Gal-3−/− mice lethally infected with F. novicida, however, show significantly reduced inflammatory response and leukocyte infiltration in the lungs, in parallel to improved lung architecture and survival (70), and the same is observed for Gal-9−/− mice (92). Thus, Gal-3 and Gal-9 function as proinflammatory alarmins in F. novicida infection.
Gal-9 also modulates the immune response through binding to TIM-3 receptor on neutrophils, macrophages and lymphocytes. For example, P. aeruginosa opsonization with Gal-9 enhances neutrophil-mediated killing via TIM-3 interactions inducing intracellular Ca2+ mobilization, neutrophil degranulation, and NADPH oxidase activity (93). Binding of Gal-9 expressed by M. tuberculosis-infected macrophages to TIM-3 also leads to restriction of intracellular bacterial growth through secretion of IL-1β, upregulation of TNF, and activation of caspase-3 (94–96). In contrast, Gal-9–TIM-3 binding decreases the levels of IL-17 in serum of mice infected with K. pneumoniae, resulting in reduced bacterial clearance (97).
Gal-9 binding to TIM-3 receptor on T lymphocytes decreases the immune response against viral infections. Influenza A virus-infected Gal-9−/− mice generate stronger humoral and CD8+ T-cell responses and cleared virus more rapidly than Gal-9+/+ mice. Accordingly, selective blocking of the Gal-9–TIM-3 interaction in Gal-9+/+ mice boosts the immune response (98). On the other hand, Gal-9 administered to mice infected with respiratory syncytial virus decreases the severity of lung pathology by increasing Tregs number and reducing the number of Th17 cells, IL-17 levels, and CD8+ T cell apoptosis (99). Similarly, Gal-9 injection into herpes simplex virus-infected mice increases Tregs number and decreases the levels of pro-inflammatory cytokines, improving the symptoms of inflammation (100), while intraperitoneal infusion of lactose, which prevents Gal-9 binding to TIM-3, reduces Treg function and augments CD8+ T cell responses (101).
In respiratory fungal infections, Gal-1 and Gal-3 play differential roles. Gal-1 modulates prostaglandin E2 and nitric oxide levels in H. capsulatum infection, contributing to phagocyte responses and thus exerting a protective effect (102). In contrast, Gal-3−/− mice clear H. capsulatum infection more efficiently than Gal-3+/+ mice, likely due to a negative regulatory role of Gal-3 on cytokine production by dendritic cells (103). However, Gal-3−/− mice are more susceptible to P. brasiliensis infection and present a Th2-polarized immune response, clearly showing that Gal-3 effects depend on the particular pathogen (104).
Intracellular Activities of Galectins
After internalization into host cells, many bacteria lyse the phagosome and escape to the cytosol for establishing a replicative niche (Figure 2C). Galectins 3, 8, and 9 bind to damaged vacuoles that expose host glycans in the luminal side of the phagosome membrane (105, 106). Moreover, Gal-8 recruits the autophagy NDP52 receptor, activating phagosome degradation (106). Gal-8 also binds parkin, which targets damaged vesicles and bacteria for ubiquitination. Interestingly, Gal-3 diminishes the recruitment of Gal-8 and parkin to group A Streptococcus, which does not replicate in endothelial cells and organs of Gal-3−/− mice (107). Gal-8 also targets for degradation damaged endosomes in picornavirus and adenovirus infections (108, 109).
Other bacteria replicate within the phagosomes, as e.g., Coxiella burnetii. Yet, galectins 3, 8, and 9 accumulate in the luminal side of the vacuole membrane, revealing membrane permeability (110). Gal-3 and Gal-8 are also detected in replicative vacuoles of Legionella pneumophila (111) and mediate delivery of guanylate binding proteins, a family of antimicrobial GTPases induced by IFN-γ (112). Moreover, Gal-3 binds TRIM-16, further contributing to organizing the autophagic response. The Gal-3-TRIM-16 system operates in macrophages infected with M. tuberculosis strains causing phagosome damage, and is required for bacteria translocation to lysosomes. Accordingly, Gal-3 protects mice in acute and chronic M. tuberculosis infection (113, 114).
Summarizing, galectins play diverse roles in respiratory infections with sometimes disparate effects, which may benefit the host or the pathogen, depending on the specific galectin, pathogen, and host context.
Concluding Remarks
This review touched briefly on the important role of collectins and galectins in pathogen clearance and immune response activation. Lung collectins are critical in mediating a variety of immune and physiological responses during health and disease. Galectins also mediate effective antimicrobial and immunoregulatory activities but, if activated inappropriately, can act as potent inducers of immunopathology. It remains to be determined whether collectins and galectins can interact with each other and whether such collaborations harness a beneficial immune response to pathogens. A more complete understanding of the host factors that control microbial colonization will lead to improved therapies for respiratory infections.
Author Contributions
CC, MC-R, BG-F, and DS contributed equally to the writing of this review article. CC and DS are co-corresponding authors.
Funding
This study was supported by the Spanish Ministerio de Economía y Competitividad through Grants SAF2015-65307-R (to CC) and BFU2015-70052-R (to DS) and Instituto de Salud Carlos III (CIBERES-CB06/06/0002 to CC and CB06/06/1102 to DS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.01998/full#supplementary-material
References
1. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol. (2015) 16:36–44. doi: 10.1038/ni.3052
2. Marsland BJ, Gollwitzer ES. Host-microorganism interactions in lung diseases. Nat Rev Immunol. (2014) 14:827–35. doi: 10.1038/nri3769
3. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. (2015) 16:27–35. doi: 10.1038/ni.3045
4. Chiu C., Openshaw PJ. Antiviral B cell and T cell immunity in the lungs. Nat Immunol. (2015) 16:18–26. doi: 10.1038/ni.3056
5. Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. (2000) 173:39–51. doi: 10.1034/j.1600-065X.2000.917306.x
6. Iwasaki A, Foxman EF, Molony RD. Early local immune defences in the respiratory tract. Nat Rev Immunol. (2017) 17:7–20. doi: 10.1038/nri.2016.117
7. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. (2005) 5:58–68. doi: 10.1038/nri1528
8. Ordonez SR, Veldhuizen EJA, van Eijk M, Haagsman HP. Role of soluble innate effector molecules in pulmonary defense against fungal pathogens. Front Microbiol. (2017) 8:2098. doi: 10.3389/fmicb.2017.02098
9. Kim JK, Kim SS, Rha KW, Kim CH, Cho JH, Lee CH, et al. Expression and localization of surfactant proteins in human nasal epithelium. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L879–84. doi: 10.1152/ajplung.00156.2006
10. Popa SJ, Stewart SE, Moreau K. Unconventional secretion of annexins and galectins. Semin Cell Dev Biol. (2018). doi: 10.1016/j.semcdb.2018.02.022. [Epub ahead of print].
11. Ujma S, Horsnell WG, Katz AA, Clark HW, Schäfer G. Non-pulmonary immune functions of surfactant proteins A and D. J Innate Immun. (2017) 9:3–11. doi: 10.1159/000451026
12. Hansen SW, Ohtani K, Roy N, Wakamiya N. The collectins CL-L1, CL-K1 and CL-P1, and their roles in complement and innate immunity. Immunobiology (2016) 221:1058–67. doi: 10.1016/j.imbio.2016.05.012
13. Howard M, Farrar CA, Sacks SH. Structural and functional diversity of collectins and ficolins and their relationship to disease. Semin Immunopathol. (2018) 40:75–85. doi: 10.1007/s00281-017-0642-0
14. Minutti CM, Jackson-Jones LH, García-Fojeda B, Knipper JA, Sutherland TE, Logan N, et al. Local amplifiers of IL-4Rα-mediated macrophage activation promote repair in lung and liver. Science (2017) 356:1076–80. doi: 10.1126/science.aaj2067
15. García-Verdugo I, Sánchez-Barbero F, Bosch FU, Steinhilber W, Casals C. Effect of hydroxylation, oligomerization, and N187-linked glycosylation on structural and biochemical properties of recombinant human SP-A1. Biochemistry (2003) 42:9532–42. doi: 10.1021/bi0347196
16. Sanchez-Barbero F, Rivas G, Steinhilber W, Casals C. Structural and functional differences among human surfactant proteins SP-A1, SP-A2, and coexpressed SP-A1/SP-A2. Role of supratrimeric oligomerization. Biochem J. (2007) 406:479–89. doi: 10.1042/BJ20070275
17. Yamada C, Sano H, Shimizu T, Mitsuzawa H, Nishitani C, Himi T, et al. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J Biol Chem. (2006) 281:21771–80. doi: 10.1074/jbc.M513041200
18. Palaniyar N, Nadesalingam J, Clark H, Shih MJ, Dodds AW, Reid KB. Nucleic acid is a novel ligand for innate, immune pattern recognition collectins surfactant proteins A and D and mannose-binding lectin. J Biol Chem. (2004) 279:32728–36. doi: 10.1074/jbc.M403763200
19. Casals C. Role of surfactant protein A (SP-A)/lipid interactions for SP-A functions in the lung. Pediatr Pathol Mol Med. (2001) 20:249–68. doi: 10.1080/15513810109168821
20. Rynkiewicz MJ, Wu H, Cafarella TR, Nikolaidis NM, Head JF, Seaton BA, et al. Differential ligand binding specificities of the pulmonary collectins are determined by the conformational freedom of a surface loop. Biochemistry (2017) 56:4095–105. doi: 10.1021/acs.biochem.6b01313
21. Erpenbeck VJ, Malherbe DC, Sommer S, Schmiedl A, Steinhilber W, Ghio AJ, et al. Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L692–8. doi: 10.1152/ajplung.00362.2004
22. Ruge CA, Kirch J, Ca-adas O, Schneider M, Perez-Gil J, Schaefer UF, et al. Uptake of nanoparticles by alveolar macrophages is triggered by surfactant protein A. Nanomedicine (2011) 7:690–3. doi: 10.1016/j.nano.2011.07.009
23. Lopez-Sanchez A, Sáenz A, Casals C. Surfactant protein A (SP-A)-tacrolimus complexes have a greater anti-inflammatory effect than either SP-A or tacrolimus alone on human macrophages-like U937 cells. Eur J Pharm Biopharm. (2011) 77:384–91. doi: 10.1016/j.ejpb.2010.12.013
24. Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol. (2002) 169:3978–86. doi: 10.4049/jimmunol.169.7.3978
25. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. (2016) 38:409–23. doi: 10.1007/s00281-016-0555-3
26. Tenner AJ. Membrane receptors for soluble defense collagens. Curr Opin Immunol. (1999) 11:34–41. doi: 10.1016/S0952-7915(99)80007-7
27. Orgeig S, Hiemstra PS, Veldhuizen EJ, Casals C, Clark HW, Haczku A, et al. Recent advances in alveolar biology: evolution and function of alveolar proteins. Respir Physiol Neurobiol. (2010) 173:S43–54. doi: 10.1016/j.resp.2010.04.023
28. Bohlson SS, O'Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, C1q, and C1q-related molecules regulate macrophage polarization. Front Immunol. (2014) 5:402–30. doi: 10.3389/fimmu.2014.00402
29. Nathan N, Taytard J, Duquesnoy P, Thouvenin G, Corvol H, Amselem S, et al. Surfactant protein A: a key player in lung homeostasis. Int J Biochem Cell Biol. (2016) 81:151–5. doi: 10.1016/j.biocel.2016.11.003
30. Sorensen GL. Surfactant protein D in respiratory and non-respiratory diseases. Front Med. (2018) 5:18. doi: 10.3389/fmed.2018.00018
31. Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front Immunol. (2012) 3:131. doi: 10.3389/fimmu.2012.00131
32. Thawer S, Auret J, Schnoeller C, Chetty A, Smith K, Darby M, et al. Surfactant protein-D is essential for immunity to helminth infection. PLoS Pathog. (2016) 12:e1005461. doi: 10.1371/journal.ppat.1005461
33. LeVine AM, Whitsett JA. Pulmonary collectins and innate host defense of the lung. Microbes Infect. (2001) 3:161–6. doi: 10.1016/S1286-4579(00)01363-0
34. Coya JM, Akinbi HT, Sáenz A, Yang L, Weaver TE, Casals C. Natural anti-infective pulmonary proteins: in vivo cooperative action of surfactant protein SP-A and the lung antimicrobial peptide SP-BN. J Immunol. (2015) 195:1628–36. doi: 10.4049/jimmunol.1500778
35. Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol. (2011) 187:1856–65. doi: 10.4049/jimmunol.1004201
36. Tenner AJ, Robinson SL, Borchelt J, Wright JR. Human pulmonary surfactant protein (SP-A), a protein structurally homologous to C1q, can enhance FcR- and CR1-mediated phagocytosis. J Biol Chem. (1989) 64:13923–8.
37. Gil M, McCormack FX, Levine AM. Surfactant protein A modulates cell surface expression of CR3 on alveolar macrophages and enhances CR3-mediated phagocytosis. J Biol Chem. (2009) 284:7495–504. doi: 10.1074/jbc.M808643200
38. Beharka AA, Crowther JE, McCormack FX, Denning GM, Lees J, Tibesar E, et al. Pulmonary surfactant protein A activates a phosphatidylinositol 3-kinase/calcium signal transduction pathway in human macrophages: participation in the up-regulation of mannose receptor activity. J Immunol. (2005) 175:2227–36. doi: 10.4049/jimmunol.175.4.2227
39. Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota S, et al. Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem. (2004) 279:21421–30. doi: 10.1074/jbc.M312490200
40. Zhang S, Chen Y, Potvin E, Sanschagrin F, Levesque RC, McCormack FX, et al. Comparative signature-tagged mutagenesis identifies Pseudomonas factors conferring resistance to the pulmonary collectin SP-A. PLoS Pathog. (2005) 1:259–68. doi: 10.1371/journal.ppat.0010031
41. Kuang Z, Hao Y, Hwang S, Zhang S, Kim E, Akinbi H, et al. The Pseudomonas aeruginosa flagellum confers resistance to pulmonary surfactant protein-A by impacting the production of exoproteases through quorum-sensing. Mol Microbiol. (2011) 79:1220–35. doi: 10.1111/j.1365–2958.2010.07516.x
42. Casals C, Cañadas O. Role of lipid ordered/disordered phase coexistence in pulmonary surfactant function. Biochim Biophys Acta (2012) 1818:2550–62. doi: 10.1016/j.bbamem.2012.05.024
43. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
44. Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell (2003) 115:13–23. doi: 10.1016/S0092-8674(03)00758-X
45. Meunier I, Kaufmann E, Downey J, Divangahi M. Unravelling the networks dictating host resistance versus tolerance during pulmonary infections. Cell Tissue Res. (2017) 367:525–36. doi: 10.1007/s00441-017-2572-5
46. Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem. (2000) 275:22442–51. doi: 10.1074/jbc.M001107200
47. Kuan S, Rust K, Crouch E. Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J Clin Invest. (1992) 90:97–106. doi: 10.1172/JCI115861
48. García-Verdugo I, Sánchez-Barbero F, Soldau K, Tobias PS, Casals C. Interaction of surfactant protein A (SP-A) with bacterial rouge lipopolysaccharide (Re-LPS) and effects of SP-A on the binding of Re-LPS to CD14 and LPS binding protein. Biochem J. (2005) 391:115–24. doi: 10.1042/BJ20050529
49. van Rozendaal BA, van de Lest CH, van Eijk M, van Golde LM, Voorhout WF, van Helden HP, et al. Aerosolized endotoxin is immediately bound by pulmonary surfactant protein D in vivo. Biochim Biophys Acta (1999) 1454:261–9. doi: 10.1016/S0925-4439(99)00042-3
50. Nguyen HA, Rajaram MV, Meyer DA, Schlesinger LS. Pulmonary surfactant protein A and surfactant lipids upregulate IRAK-M, a negative regulator of TLR-mediated inflammation in human macrophages. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L608–16. doi: 10.1152/ajplung.00067.2012
51. Sender V, Lang L, Stamme C. Surfactant protein-A modulates LPS-induced TLR4 localization and signaling via β-arrestin 2. PLoS ONE (2013) 8:e59896. doi: 10.1371/journal.pone.0059896
52. Moulakakis C, Steinhäuser C, Biedziak D, Freundt K, Reiling N, Stamme C. Surfactant protein A enhances constitutive immune functions of clathrin heavy chain and clathrin adaptor protein 2. Am J Respir Cell Mol Biol. (2016) 55:92–104. doi: 10.1165/rcmb.2015-0219OC
53. Henning LN, Azad AK, Parsa KV, Crowther JE, Tridandapani S, Schlesinger LS. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J Immunol. (2008) 180:7847–58. doi: 10.4049/jimmunol.180.12.7847
54. Crowther JE, Kutala VK, Kuppusamy P, Ferguson JS, Beharka AA, Zweier JL, et al. Pulmonary surfactant protein A inhibits macrophage reactive oxygen intermediate production in response to stimuli by reducing NADPH oxidase activity. J Immunol. (2004) 172:6866–74. doi: 10.4049/jimmunol.172.11.6866
55. Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappaB and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol. (2001) 166:7514–9. doi: 10.4049/jimmunol.166.12.7514
56. Olde Nordkamp MJ, van Eijk M, Urbanus RT, Bont L, Haagsman HP, Meyaard L. Leukocyte-associated Ig-like receptor-1 is a novel inhibitory receptor for surfactant protein D. J Leukoc Biol. (2014) 96:105–11. doi: 10.1189/jlb.3AB0213-092RR
57. Minutti CM, García-Fojeda B, Saenz A, De Las Casas-Engel M, Guillamat-Prats R, De Lorenzo A, et al. Surfactant protein A prevents IFN-gamma/IFN-gammaR interaction and attenuates classical activation of human alveolar macrophages. J Immunol. (2016) 197:590–8. doi: 10.4049/jimmunol.1501032
58. Allen JE, Sutherland TE. Host protective roles of type 2 immunity: parasite killing and tissue repair, flip sides of the same coin. Semin Immunol. (2014) 26:329–40. doi: 10.1016/j.smim.2014.06.003
59. Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42:145–58. doi: 10.1016/j.immuni.2014.12.020
60. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science (2017) 356:1072–6. doi: 10.1126/science.aai8132
61. Jäkel A, Clark H, Reid KB, Sim RB. The human lung surfactant proteins A (SP-A) and D (SP-D) interact with apoptotic target cells by different binding mechanisms. Immunobiology (2010) 215:551–8. doi: 10.1016/j.imbio.2009.09.005
62. Jäkel A, Clark H, Reid KB, Sim RB. Surface-bound myeloperoxidase is a ligand for recognition of late apoptotic neutrophils by human lung surfactant proteins A and D. Protein Cell (2010) 1:563–72. doi: 10.1007/s13238-010-0076-0
63. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev. (2016) 273:357–70. doi: 10.1111/imr.12453
64. Ledford JG, Addison KJ, Foster MW, Que LG. Eosinophil-associated lung diseases. A cry for surfactant proteins A and D help? Am J Respir Cell Mol Biol. (2014) 51:604–14. doi: 10.1165/rcmb.2014-0095TR
65. Johannes L, Jacob R, Leffler H. Galectins at a glance. J Cell Sci. (2018) 131:jcs208884. doi: 10.1242/jcs.208884
66. Dings RPM, Miller MC, Griffin RJ, Mayo KH. Galectins as molecular targets for therapeutic intervention. Int J Mol Sci. (2018) 19:E905. doi: 10.3390/ijms19030905
67. Liu FT, Rabinovich GA. Galectins: regulators of acute and chronic inflammation. Ann N Y Acad Sci. (2010) 1183:158–82. doi: 10.1111/j.1749-6632.2009.05131.x
68. Cerliani JP, Blidner AG, Toscano MA, Croci DO, Rabinovich GA. Translating the sugar code into immune and vascular signaling programs. Trends Biochem Sci. (2017) 42:255–73. doi: 10.1016/j.tibs.2016.11.003
69. Sato S, Ouellet N, Pelletier I, Simard M, Rancourt A, Bergeron MG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. (2002) 168:1813–22. doi: 10.4049/jimmunol.168.4.1813
70. Mishra BB, Li Q, Steichen AL, Binstock BJ, Metzger DW, Teale JM, et al. Galectin-3 functions as an alarmin: pathogenic role for sepsis development in murine respiratory tularemia. PLoS ONE (2013) 8:e59616. doi: 10.1371/journal.pone.0059616
71. Shiratori B, Zhao J, Okumura M, Chagan-Yasutan H, Yanai H, Mizuno K, et al. Immunological roles of elevated plasma levels of matricellular proteins in Japanese patients with pulmonary tuberculosis. Int J Mol Sci. (2016) 18:E19. doi: 10.3390/ijms18010019
72. Chavez-Galan L, Ramon-Luing L, Carranza C, Garcia I, Sada-Ovalle I. Lipoarabinomannan decreases galectin-9 expression and tumor necrosis factor pathway in macrophages favoring Mycobacterium tuberculosis intracellular growth. Front Immunol. (2017) 8:1659. doi: 10.3389/fimmu.2017.01659
73. Yang ML, Chen YH, Wang SW, Huang YJ, Leu CH, Yeh NC, et al. Galectin-1 binds to influenza virus and ameliorates influenza virus pathogenesis. J Virol. (2011) 85:10010–20. doi: 10.1128/JVI.00301-11
74. Fang S, Zhang K, Wang T, Wang X, Lu X, Peng B, et al. Primary study on the lesions and specific proteins in BEAS-2B cells induced with the 2009 A (H1N1) influenza virus. Appl Microbiol Biotechnol. (2014) 98:9691–701. doi: 10.1007/s00253-014-5852-y
75. Chen Y, Zhou J, Cheng Z, Yang S, Chu H, Fan Y, et al. Functional variants regulating LGALS1 (Galectin 1) expression affect human susceptibility to influenza A(H7N9). Sci Rep. (2015) 5:8517. doi: 10.1038/srep08517
76. Almeida F, Wolf JM, da Silva TA, DeLeon-Rodriguez CM, Rezende CP, Pessoni AM, et al. Galectin-3 impacts Cryptococcus neoformans infection through direct antifungal effects. Nat Commun. (2017) 8:1968. doi: 10.1038/s41467-017-02126-7
77. Dembele BP, Chagan-Yasutan H, Niki T, Ashino Y, Tangpukdee N, Shinichi E, et al. Plasma levels of Galectin-9 reflect disease severity in malaria infection. Malar J. (2016) 15:403. doi: 10.1186/s12936-016-1471-7
78. Liu J, Huang S, Su XZ, Song J, Lu F. Blockage of Galectin-receptor interactions by alpha-lactose exacerbates plasmodium berghei-induced pulmonary immunopathology. Sci Rep. (2016) 6:32024. doi: 10.1038/srep32024
79. Barboni E, Coade S, Fiori A. The binding of mycolic acids to galectin-3: a novel interaction between a host soluble lectin and trafficking mycobacterial lipids? FEBS Lett. (2005) 579:6749–55. doi: 10.1016/j.febslet.2005.11.005
80. Mey A, Leffler H, Hmama Z, Normier G, Revillard JP. The animal lectin galectin-3 interacts with bacterial lipopolysaccharides via two independent sites. J Immunol. (1996) 156:1572–7.
81. Gupta SK, Masinick S, Garrett M, Hazlett LD. Pseudomonas aeruginosa lipopolysaccharide binds galectin-3 and other human corneal epithelial proteins. Infect Immun. (1997) 65:2747–53.
82. Stowell SR, Arthur CM, McBride R, Berger O, Razi N, Heimburg-Molinaro J, et al. Microbial glycan microarrays define key features of host-microbial interactions. Nat Chem Biol. (2014) 10:470–6. doi: 10.1038/nchembio.1525
83. Kalograiaki I, Euba B, Proverbio D, Campanero-Rhodes MA, Aastrup T, Garmendia J, et al. Combined bacteria microarray and quartz crystal microbalance approach for exploring glycosignatures of nontypeable Haemophilus influenzae and recognition by host lectins. Anal Chem. (2016) 88:5950–7. doi: 10.1021/acs.analchem.6b00905
84. Levroney EL, Aguilar HC, Fulcher JA, Kohatsu L, Pace KE, Pang M, et al. Novel innate immune functions for galectin-1: galectin-1 inhibits cell fusion by Nipah virus envelope glycoproteins and augments dendritic cell secretion of proinflammatory cytokines. J Immunol. (2005) 175:413–20. doi: 10.4049/jimmunol.175.1.413
85. Garner OB, Aguilar HC, Fulcher JA, Levroney EL, Harrison R, Wright L, et al. Endothelial galectin-1 binds to specific glycans on Nipah virus fusion protein and inhibits maturation, mobility, and function to block syncytia formation. PLoS Pathog. (2010) 6:e1000993. doi: 10.1371/journal.ppat.1000993
86. Garner OB, Yun T, Pernet O, Aguilar HC, Park A, Bowden TA, et al. Timing of galectin-1 exposure differentially modulates Nipah virus entry and syncytium formation in endothelial cells. J Virol. (2015) 89:2520–9. doi: 10.1128/JVI.02435-14
87. Nita-Lazar M, Banerjee A, Feng C, Amin MN, Frieman MB, Chen WH, et al. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol Immunol. (2015) 65:1–16. doi: 10.1016/j.molimm.2014.12.010
88. Nieminen J, St-Pierre C, Bhaumik P, Poirier F, Sato S. Role of galectin-3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae. J Immunol. (2008) 180:2466–73. doi: 10.4049/jimmunol.180.4.2466
89. Farnworth SL, Henderson NC, Mackinnon AC, Atkinson KM, Wilkinson T, Dhaliwal K, et al. Galectin-3 reduces the severity of pneumococcal pneumonia by augmenting neutrophil function. Am J Pathol. (2008) 172:395–405. doi: 10.2353/ajpath.2008.070870
90. Elmwall J, Kwiecinski J, Na M, Ali AA, Osla V, Shaw LN, et al. Galectin-3 is a target for proteases involved in the virulence of Staphylococcus aureus. Infect Immun. (2017) 85:e00177–17. doi: 10.1128/IAI.00177-17
91. Ferraz LC, Bernardes ES, Oliveira AF, Ruas LP, Fermino ML, Soares SG, et al. Lack of galectin-3 alters the balance of innate immune cytokines and confers resistance to Rhodococcus equi infection. Eur J Immunol. (2008) 38:2762–75. doi: 10.1002/eji.200737986
92. Steichen AL, Simonson TJ, Salmon SL, Metzger DW, Mishra BB, Sharma J. Alarmin function of galectin-9 in murine respiratory tularemia. PLoS ONE (2015) 10:e0123573. doi: 10.1371/journal.pone.0123573
93. Vega-Carrascal I, Bergin DA, McElvaney OJ, McCarthy C, Banville N, Pohl K, et al. Galectin-9 signaling through TIM-3 is involved in neutrophil-mediated Gram-negative bacterial killing: an effect abrogated within the cystic fibrosis lung. J Immunol. (2014) 192:2418–31. doi: 10.4049/jimmunol.1300711
94. Jayaraman P, Sada-Ovalle I, Beladi S, Anderson AC, Dardalhon V, Hotta C, et al. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J Exp Med. (2010) 207:2343–54. doi: 10.1084/jem.20100687
95. Sada-Ovalle I, Chavez-Galan L, Torre-Bouscoulet L, Nava-Gamino L, Barrera L, Jayaraman P, et al. The Tim3-galectin 9 pathway induces antibacterial activity in human macrophages infected with Mycobacterium tuberculosis. J Immunol. (2012) 189:5896–902. doi: 10.4049/jimmunol.1200990
96. Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, et al. IL-1beta promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol. (2013) 190:4196–204. doi: 10.4049/jimmunol.1202688
97. Wang F, Xu J, Liao Y, Wang Y, Liu C, Zhu X, et al. Tim-3 ligand galectin-9 reduces IL-17 level and accelerates Klebsiella pneumoniae infection. Cell Immunol. (2011) 269:22–8. doi: 10.1016/j.cellimm.2011.03.005
98. Sharma S, Sundararajan A, Suryawanshi A, Kumar N, Veiga-Parga T, Kuchroo VK, et al. T cell immunoglobulin and mucin protein-3 (Tim-3)/Galectin-9 interaction regulates influenza A virus-specific humoral and CD8 T-cell responses. Proc Natl Acad Sci USA. (2011) 108:19001–6. doi: 10.1073/pnas.1107087108
99. Lu X, McCoy KS, Xu J, Hu W, Chen H, Jiang K, et al. Galectin-9 ameliorates respiratory syncytial virus-induced pulmonary immunopathology through regulating the balance between Th17 and regulatory T cells. Virus Res. (2015) 195:162–71. doi: 10.1016/j.virusres.2014.10.011
100. Shim JA, Park S, Lee ES, Niki T, Hirashima M, Sohn S. Galectin-9 ameliorates herpes simplex virus-induced inflammation through apoptosis. Immunobiology (2012) 217:657–66. doi: 10.1016/j.imbio.2011.11.002
101. Sehrawat S, Reddy PB, Rajasagi N, Suryawanshi A, Hirashima M, Rouse BT. Galectin-9/TIM-3 interaction regulates virus-specific primary and memory CD8 T cell response. PLoS Pathog. (2010) 6:e1000882. doi: 10.1371/journal.ppat.1000882
102. Rodrigues LC, Secatto A, Sorgi CA, Dejani NN, Medeiros AI, Prado MK, et al. Protective Effect of galectin-1 during Histoplasma capsulatum infection is associated with prostaglandin E2 and nitric oxide modulation. Mediators Inflamm. (2016) 2016:5813794. doi: 10.1155/2016/5813794
103. Wu SY, Yu JS, Liu FT, Miaw SC, Wu-Hsieh BA. Galectin-3 negatively regulates dendritic cell production of IL-23/IL-17-axis cytokines in infection by Histoplasma capsulatum. J Immunol. (2013) 190:3427–37. doi: 10.4049/jimmunol.1202122
104. Ruas LP, Bernardes ES, Fermino ML, de Oliveira LL, Hsu DK, Liu FT, et al. Lack of galectin-3 drives response to Paracoccidioides brasiliensis toward a Th2-biased immunity. PLoS ONE (2009) 4:e4519. doi: 10.1371/journal.pone.0004519
105. Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, et al. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol. (2010) 12:530–44. doi: 10.1111/j.1462-5822.2009.01415.x
106. Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature (2012) 482:414–8. doi: 10.1038/nature10744
107. Cheng YL, Wu YW, Kuo CF, Lu SL, Liu FT, Anderson R, et al. Galectin-3 inhibits galectin-8/parkin-mediated ubiquitination of group A Streptococcus. MBio (2017) 8:e00899-17. doi: 10.1128/mBio.00899-17
108. Staring J, von Castelmur E, Blomen VA, van den Hengel LG, Brockmann M, Baggen J, et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature (2017) 541:412–6. doi: 10.1038/nature21032
109. Montespan C, Marvin SA, Austin S, Burrage AM, Roger B, Rayne F, et al. Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog. (2017) 13:e1006217. doi: 10.1371/journal.ppat.1006217
110. Mansilla Pareja ME, Bongiovanni A, Lafont F, Colombo MI. Alterations of the Coxiella burnetii replicative vacuole membrane integrity and interplay with the autophagy pathway. Front Cell Infect Microbiol. (2017) 7:112. doi: 10.3389/fcimb.2017.00112
111. Truchan HK, Christman HD, White RC, Rutledge NS, Cianciotto NP. Type II secretion substrates of Legionella pneumophila translocate out of the pathogen-occupied vacuole via a semipermeable membrane. MBio (2017) 8:e00870-17. doi: 10.1128/mBio.00870-17
112. Feeley EM, Pilla-Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, et al. Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc Natl Acad Sci USA. (2017) 114:E1698–706. doi: 10.1073/pnas.1615771114
113. Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell (2016) 39:13–27. doi: 10.1016/j.devcel.2016.08.003
Keywords: respiratory pathogens, infection, inflammation, surfactant proteins, alternatively activated macrophages, autophagy, tissue repair, lung homeostasis
Citation: Casals C, Campanero-Rhodes MA, García-Fojeda B and Solís D (2018) The Role of Collectins and Galectins in Lung Innate Immune Defense. Front. Immunol. 9:1998. doi: 10.3389/fimmu.2018.01998
Received: 17 June 2018; Accepted: 14 August 2018;
Published: 04 September 2018.
Edited by:
Jesús Gonzalo-Asensio, Universidad de Zaragoza, SpainReviewed by:
Kenneth Reid, University of Oxford, United KingdomUday Kishore, Brunel University London, United Kingdom
Henk Peter Haagsman, Utrecht University, Netherlands
Copyright © 2018 Casals, Campanero-Rhodes, García-Fojeda and Solís. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Casals, Y2Nhc2Fsc2NAdWNtLmVz
Dolores Solís, ZC5zb2xpc0BpcWZyLmNzaWMuZXM=