 Gathoni Kamuyu1,2†
Gathoni Kamuyu1,2† James Tuju1,3†
James Tuju1,3† Rinter Kimathi1
Rinter Kimathi1 Kennedy Mwai1
Kennedy Mwai1 James Mburu1
James Mburu1 Nelson Kibinge1
Nelson Kibinge1 Marisa Chong Kwan4
Marisa Chong Kwan4 Sam Hawkings4
Sam Hawkings4 Reuben Yaa1
Reuben Yaa1 Emily Chepsat1James M. Njunge1
Emily Chepsat1James M. Njunge1 Timothy Chege1Fatuma Guleid5Micha Rosenkranz2Christopher K. Kariuki6,7Roland Frank2
Timothy Chege1Fatuma Guleid5Micha Rosenkranz2Christopher K. Kariuki6,7Roland Frank2 Samson M. Kinyanjui1,3,8Linda M. Murungi1Philip Bejon1
Samson M. Kinyanjui1,3,8Linda M. Murungi1Philip Bejon1 Anna Färnert9,10
Anna Färnert9,10 Kevin K. A. Tetteh11
Kevin K. A. Tetteh11 James G. Beeson12,13,14
James G. Beeson12,13,14 David J. Conway15Kevin Marsh1,8,16Julian C. Rayner17
David J. Conway15Kevin Marsh1,8,16Julian C. Rayner17 Faith H. A. Osier1,2,3,8*
Faith H. A. Osier1,2,3,8*- 1KEMRI-Wellcome Trust Research Programme, Centre for Geographic Medicine Research—Coast, Kilifi, Kenya
- 2Centre for Infectious Diseases, Parasitology, Heidelberg University Hospital, Heidelberg, Germany
- 3Department of Biochemistry, Pwani University, Kilifi, Kenya
- 4Arrayjet, Innovative Microarray Solutions, Edinburgh, United Kingdom
- 5Department of Pathology, University of Cape Town, Cape Town, South Africa
- 6Department of Tropical and Infectious Diseases, Institute of Primate Research, Nairobi, Kenya
- 7Cellular and Molecular Immunology, Vrije Universiteit Brussels, Brussels, Belgium
- 8Centre for Tropical Medicine and Global Health, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom
- 9Division of Infectious Diseases, Department of Medicine Solna, Karolinska Institutet, Stockholm, Sweden
- 10Department of Infectious Diseases, Karolinska University Hospital, Stockholm, Sweden
- 11Immunology and Infection Department, London School of Hygiene and Tropical Medicine, London, United Kingdom
- 12Burnet Institute, Melbourne, VIC, Australia
- 13Central Clinical School, Monash University, Melbourne, VIC, Australia
- 14Department of Medicine, University of Melbourne, Melbourne, VIC, Australia
- 15Pathogen Molecular Biology Department, London School of Hygiene and Tropical Medicine, London, United Kingdom
- 16African Academy of Sciences, Nairobi, Kenya
- 17Wellcome Sanger Institute, Hinxton, Cambridge, United Kingdom
Passive transfer studies in humans clearly demonstrated the protective role of IgG antibodies against malaria. Identifying the precise parasite antigens that mediate immunity is essential for vaccine design, but has proved difficult. Completion of the Plasmodium falciparum genome revealed thousands of potential vaccine candidates, but a significant bottleneck remains in their validation and prioritization for further evaluation in clinical trials. Focusing initially on the Plasmodium falciparum merozoite proteome, we used peer-reviewed publications, multiple proteomic and bioinformatic approaches, to select and prioritize potential immune targets. We expressed 109 P. falciparum recombinant proteins, the majority of which were obtained using a mammalian expression system that has been shown to produce biologically functional extracellular proteins, and used them to create KILchip v1.0: a novel protein microarray to facilitate high-throughput multiplexed antibody detection from individual samples.
The microarray assay was highly specific; antibodies against P. falciparum proteins were detected exclusively in sera from malaria-exposed but not malaria-naïve individuals. The intensity of antibody reactivity varied as expected from strong to weak across well-studied antigens such as AMA1 and RH5 (Kruskal–Wallis H test for trend: p < 0.0001). The inter-assay and intra-assay variability was minimal, with reproducible results obtained in re-assays using the same chip over a duration of 3 months. Antibodies quantified using the multiplexed format in KILchip v1.0 were highly correlated with those measured in the gold-standard monoplex ELISA [median (range) Spearman's R of 0.84 (0.65–0.95)]. KILchip v1.0 is a robust, scalable and adaptable protein microarray that has broad applicability to studies of naturally acquired immunity against malaria by providing a standardized tool for the detection of antibody correlates of protection. It will facilitate rapid high-throughput validation and prioritization of potential Plasmodium falciparum merozoite-stage antigens paving the way for urgently needed clinical trials for the next generation of malaria vaccines.
Introduction
Protein microarrays are increasingly used in the “omic” era of research in multiple formats that share the basic requirement to investigate interactions of tens to thousands of proteins simultaneously (1). They have had important translational applications in biomarker discovery to guide patient diagnosis, treatment and prognosis, as well as in drug discovery and vaccine antigen identification (2). Protein microarrays have facilitated a rapid, systematic and high-throughput approach to probing an entire pathogens' proteome or fraction thereof for immunoreactivity, in an approach that forms part of a reverse vaccinology workflow. These have aided in the discovery of potential diagnostic markers for Mycobacterium tuberculosis and SARS-coronavirus as well as potential vaccine candidates in over 30 human pathogens including Plasmodium falciparum (2, 3).
P. falciparum malaria causes ~450,000 deaths per year (4), and is of major public health importance to sub-Saharan Africa (5). Recent gains in reducing the burden appear to have stalled despite ongoing control efforts (4, 6). Efforts to design a highly effective vaccine that would protect against this disease have been hampered by the complexity of the organism and its' multi-stage life cycle: its genome encodes >5,300 proteins that are expressed variably in different tissues as the infection develops in the host (7). Coupled to this is an impressive array of strategies for generating protein polymorphisms or protein variants and redundant erythrocyte invasion pathways, which facilitate immune evasion (8–10). Consequently, although efforts to develop a highly effective malaria vaccine have been on-going for over a century, this goal has yet to be achieved. The current leading vaccine candidate against P. falciparum malaria has limited efficacy and induces only short-lived protective immunity (11, 12).
Multiple P. falciparum and/or P. vivax protein arrays have been designed over the past decade to help identify and prioritize potential malaria vaccine antigen candidates. The majority of these arrays have been manufactured using either the E. coli-based or the wheat germ cell free in-vitro transcription/translation expression system, with the largest to date including ~30% of the entire P. falciparum proteome (13–17). Protein selection was based on stage-specific transcription or protein expression, sub-cellular localization, secondary protein structures or documented immunogenicity in human and animal models. However, the in-vitro transcription/translation systems are relatively poor at generating functional surface proteins, which frequently require disulphide bonding and/or post-translational modification to attain their correct three-dimensional structure. Nevertheless, subsequent studies have down-selected proteins from this initial panel (18–31), indicating that essentially >75% of the parasite genome has yet to be evaluated in the context of immunity. A few additional proteins have been tested independently in smaller scale studies accounting for only a marginal increase in the proportion of the parasite proteome evaluated to date (32–34). These studies have rationally selected merozoite proteins that were established or plausible targets of antibodies, and evaluated antibody associations with protection in longitudinal studies using standard ELISA-based approaches (32, 33). They highlighted the importance of evaluating a broad repertoire of antigens and combinations of antibody responses in studies of acquired immunity. However, there still remains a need for a common platform with standardized protein expression and high-throughput antibody detection methods that can be applied widely across different clinical studies (35). This would accelerate identification of protective antibody targets and facilitate the comparisons between studies and populations.
To contribute to vaccine candidate discovery, as well as the validation and prioritization of existing candidates for clinical trials, we designed a novel protein microarray. We focused on the merozoite stage that is a target of immunity that can prevent or reduce the clinical symptoms of malaria. As per the case with other infectious diseases (36, 37), we hypothesized that proteins on or associated with the surface of the invasive P. falciparum merozoite would be accessible targets for protective antibodies (33). We mined the literature to identify multiple potential surface-associated merozoite proteins (32–34, 38–43) and added new proteins that were identified as immunogenic in adults from malaria-endemic countries and had proteomic and/or bioinformatic features suggestive of merozoite surface-localization, secretion and/or involvement in erythrocyte invasion (44). We expressed and purified these proteins and printed them on a custom microarray, which we refer to as KILchip v1.0 for its origin at the KEMRI-Wellcome Trust Research Programme in Kilifi, Kenya where the majority of the work was carried out. We demonstrate that KILchip v1.0 is highly specific, has minimal inter- and intra-assay variation, and is strongly correlated with equivalent data acquired using the gold-standard monoplex ELISA.
Materials and Methods
Protein Selection
We aimed to design a microarray that would include proteins already considered as vaccine candidates, as well as novel proteins that had not been studied in the context of protective immunity. This would serve multiple purposes: (i) validation of existing vaccine candidates in new sample sets, (ii) identification of novel potential candidates and (iii) facilitation of head-to-head comparisons of all selected candidates in the same experiment. To this end, we selected a panel of antigens previously published in Zenonos et al. (38), Richards et al. (32), Crosnier et al. (39), Tetteh et al. (34), Raj et al. (43), Polley et al. (40, 45), Kimbi et al. (46), Metzger et al. (47), Taylor et al. (41), and Burghaus and Holder (48), for inclusion in the KILchip v1.0. These proteins are known or predicted to be anchored or associated with the surface of merozoites, secreted from its apical organelles or involved in erythrocyte invasion and have been shown to correlate with protection from clinical malaria (32, 33). We also included 28 novel proteins selected through a protein discovery pipeline that employed proteomic approaches for the detection of proteins that were either immunogenic or located on the surface of merozoites (44). Down-selection criteria for the novel proteins included either (i) the presence of a predicted N-terminal signal peptide and/or transmembrane domain(s), (ii) upregulated transcription at the late stages of the asexual life cycle, (iii) a predicted role in merozoite invasion (49) and (iv) novel with regards to a potential role in protective immunity.
Recombinant P. falciparum Protein Expression
Plasmids containing codon-optimized genes of interest were either obtained from the plasmid repository Addgene (https://www.addgene.org) or newly synthesized by GeneartAG as has been previously published (38, 39). Briefly, predicted signal peptides and transmembrane domains were excluded and the serine or threonine amino acid residues in all potential N-linked glycosylation sites (NXS/T) were substituted with alanine. Codon-optimized genes of interest were sub-cloned into a derivative of the pTT3 expression vector (also obtained from Addgene) that contained an N-terminal signal peptide derived from the mouse kappa light chain to drive secretion of antigen and a rat Cd4 domains 3 and 4 tag followed by a hexa-histidine tag for protein purification (39). Proteins were subsequently expressed using the Expi293 expression system (Invitrogen) according to manufacturer's instructions. Briefly, Expi293F cells were cultured to a density of 2.0 × 106 cells/ml and transfected with expression vectors using the Expifectamine 293 transfection reagent (Invitrogen). Cells were then incubated at 37°C with 8% CO2 in an orbital shaker at 125 rpm. Culture supernatants were harvested 6 days post-transfection and proteins purified using Ni-NTA purification columns (Invitrogen). The majority of proteins included in KILchip v1.0 were expressed using the above mammalian expression system.
A minority of proteins were expressed in E. coli using pGEX-2T and pMAL-c2X vectors to produce fusion proteins with the carriers glutathione-S-transferase (GST) and maltose-binding protein (MBP), respectively. These were transformed into BL21 (DE3) E. coli cells and expressed as previously described (34, 40–42). The gene encoding Pf SEA1 protein was amplified from P. falciparum 3D7 cDNA using previously described primers (43). The PCR products were cloned into pEXP5-NT/TOPO expression vector and transformed into BL21 (DE3) pLysS E.coli cells. Cell expansion, induction of protein expression and subsequent purification was performed as previously published (34, 40–42). Purified recombinant proteins were dialysed into phosphate buffered saline and quantified using NanoDrop (Thermo scientific) before printing onto nitrocellulose slides. Further details are provided in the Supplementary information.
LC-MS/MS Analysis, Protein Validation, and Circular Dichroism Spectroscopy
Five to fifteen μg of purified recombinant proteins were denatured in 50 mM Tris-HCL pH 8.0 (Sigma) containing 8 M urea (Sigma). Proteins were reduced with 40 mM dithiothreitol (Sigma), alkylated with 80 mM iodoacetamide (Sigma) and precipitated using cold acetone. Pelleted proteins were resuspended in 15 μl of 6 M urea in 50 mM Tris-HCL pH 8.0 and digested with trypsin/Lys-C mix (Promega) according to manufacturer's instructions using the two step in-solution digestion. Peptides were desalted using P10 c18 pipette ZipTips (Millipore), dried using the Speedvac concentrator (Thermo Scientific) and resuspended in 15 μl of 99% H20, 1% acetonitrile, and 0.1% formic acid. Peptides (5 μl) were loaded using a Dionex Ultimate 3,000 nano-flow ultra-high-pressure liquid chromatography system (Thermo Scientific) on to a 75 μm × 2 cm C18 trap column (Thermo Scientific). Chromatographic separation of peptides was carried out on a reverse-phase 50 cm-long column (Thermo Scientific) maintained at 40°C over a 60-min elution gradient (2–40% of mobile phase B; 80% acetonitrile with 0.1% formic acid) at a flow rate of 0.3 μl/min. Peptides were measured using LC instrumentation consisting of a Dionex Ultimate 3,000 nano-flow ultra-high-pressure liquid chromatography system (Thermo Scientific) coupled via a nano-electrospray ion source (Thermo Scientific) to a Q Exactive Orbitrap mass spectrometer (Thermo Scientific). The ms1 settings were: Resolution, 70,000; Automatic gain control (AGC) target, 3e6; maximum injection time, 100 ms; scan range, 380–1600 m/z; while the ms2 settings were: Resolution, 17,500; AGC target, 5e4; maximum injection time, 100 ms; isolation window, 1.6 m/z. The top 10 most intense ions were selected for ms2 and fragmented with higher-energy collision fragmentation using normalized collision energy of 28 and these ions were subsequently excluded for the next 20 s. Mass spectrometry raw files were searched on Proteome Discoverer software version 1.3.0.339 (Thermo Scientific) using the Mascot server (Matrix Science) using a concatenated database of human and 3D7 Plasmodium falciparum protein FASTA sequences. Cysteine carbamidomethylation was set as a fixed modification and deamidation of asparagine or glutamine and methionine oxidation as variable modifications. The false discovery rate (FDR) was set to 0.01 for both proteins and peptides and a maximum of two missed cleavages were allowed in the database search. A minimum of two unique peptides for a protein was considered a positive identification.
Two proteins were selected for circular dichroism spectroscopy analysis (50). Briefly, phosphate buffer (50 mM NaH2PO4 pH 8.0, 0.1 M NaCl) and samples in phosphate buffer were degassed in a vacuum at room temperature and the CD spectra recorded on a J-715 spectropolarimeter (Jasco). Six accumulations were taken per protein with continuous scans taken using a 1 mm (0.1 cm) quartz cuvette, a scan rate of 50 nm/min, a band width of 1.0 nm and a resolution of 0.5 nm. The raw CD data (ellipticity θ in mDeg) were normalized for protein concentration and the number of residues yielding the mean residue ellipticity [θ] in mDeg·cm2·dmol−1·res−1.
Data Availability
The mass spectrometry raw files generated and analyzed in the current study have been deposited to the ProteomeXchange Consortium51 (PXD011746), via the MassIVE partner repository (MSV000083144), under the following title: KILchip v1.0 A novel Plasmodium falciparum merozoite protein microarray to facilitate malaria vaccine candidate prioritization. The FTP for the dataset is available here: ftp://massive.ucsd.edu/MSV000083144.
KILchip v1.0 Protein Microarray Assay
Overview
We designed our protein microarray to test 21 unique serum samples per slide with a customized barcode for slide identification. Four slides fitted into a 4 × 24 hybridization cassette (Arrayit Corporation ARYC), thus accommodating 84 samples per cassette and 336 samples per hybridization workstation (ARYC), each of which contains 4 hybridization cassettes (Figure 1).
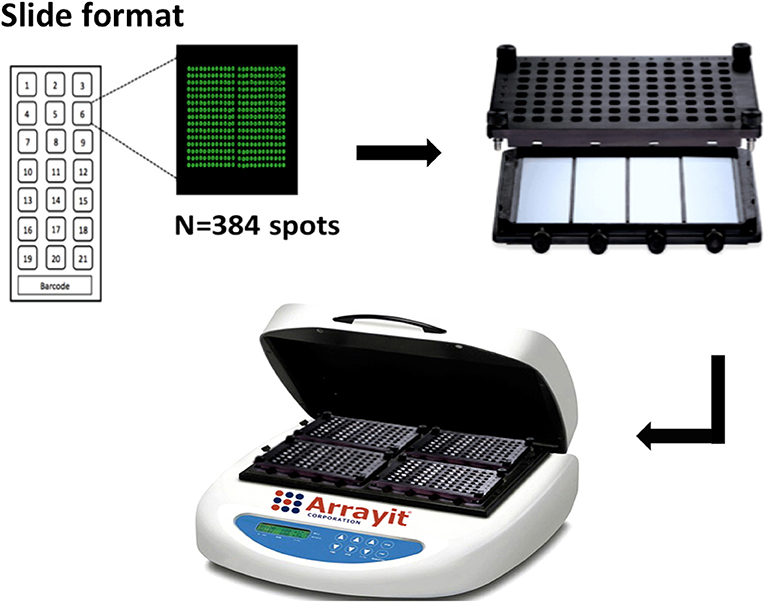
Figure 1. Configuration of KILchip v1.0. Individual slides contain 21 identical mini-arrays comprised of protein spots and a barcode for identification. Each mini-array contains 384 individual spots that include proteins and controls printed in triplicate. Four slides are assembled onto a hybridization cassette, four of which are simultaneously processed in a microarray hybridization work-station (image from Arrayit Corporation, used with permission).
Microarray Protein Map
Each slide contained 21 identical protein mini-arrays. Each mini-array had 384 features (printed spots) at a volume of 400 pl per spot. Recombinant P. falciparum proteins (n = 111) and controls (n = 17) were printed on each mini-array in triplicate, at the same concentration, using the same printer (Ultra Marathon micro-arrayer, ArrayJet) and printing protocol. The first control spots were Alexafluor647 human IgG (Jackson ImmunoResearch) that served as landmarks demarcating the four edges of each mini-array (4 spots to match the edges of each mini-array). Purified human IgG (Jackson ImmunoResearch, 1 spot), served as the second control that confirmed the activity of the secondary antibody utilized in the assay. Protein printing buffer (30% glycerol/PBS, 9 spots) was used as the third set of controls that allowed for the monitoring of background reactivity and for the detection of any potential protein carryover during printing. The last sets of controls were the CD4, MBP, and GST proteins (1 spot each) to control for any potential antibody reactivity against the tags, each of which is present in one or more of the recombinant P. falciparum antigens. Thus, each mini-array contained a total of 384 features: 333 derived from 111 P. falciparum recombinant proteins (109 proteins, 2 of which were included twice) and 51 from 17 controls, all printed in triplicate.
Microarray Printing
We optimized the concentrations for individual proteins, serum samples and the secondary antibody by checkerboard titration. Proteins were printed on nitrocellulose slides (ONCYTE SuperNOVA, GraceBio) at a concentration of 250 μg/ml using the Ultra Marathon micro-arrayer (ArrayJet) with the Inkjet printing technology and the command center 1.5.0.1 (ArrayJet). Printing was carried out at 50% relative humidity and at 18°C. As a drying step, slides were incubated overnight at 18°C in the arrayer after printing, before storage in slide boxes with dessicant at 4°C until use. A salt scan was carried out at a high photomultiplier (PMT) at the 532 nm wavelength (green channel) to verify post-printing quality and at the 635 nm wavelength (red channel) to allow visualization of the landmark spots.
Antibody Detection
Printed slides were carefully assembled onto the hybridization cassette and sealed using silicone gaskets (ARYC) to form leak-proof individual wells. We modified a published protocol for the detection of antibodies (51). Briefly, wells were washed thrice with 0.1% Tween 20/HEPES buffered saline (1.4 M NaCl, 50 mM KCl, 20 mM CaCl2, 10 mM MgCl2, 100 mM HEPES; HBS) followed by HBS to remove any unbound proteins. Non-specific binding to the slide surface was prevented by blocking with 200 μl of 2% BSA/0.1% Tween 20/HBS for 2 h at room temperature while rotating on a microarray hybridization station (ARYC) at 350 rpm. Wells were subsequently washed thrice and incubated overnight at 4°C with 150 μl of serum diluted 1:400 and rotating at 350 rpm on the hybridization station. Thereafter, wells were washed as described above and incubated with 150 μl of donkey anti-human IgG-Fcγ fragment specific Alexafluor647 for 3 h at room temperature followed by three washes. Slides were carefully disassembled from the hybridization cassettes, rinsed thrice in distilled water and dried by centrifugation at 300 g for 5 min using a combiSlide adapter (Eppendorf) and stored in slide boxes in the dark. Slides were scanned using a Genepix 4,000 B scanner coupled to the GenePix Pro & Microarray Acquisition and Analysis Software (Molecular Devices).
ELISA
A standard protocol for an indirect ELISA was performed as has been previously described (52–54). Briefly, a pre-determined concentration for each recombinant protein was either heat-treated at 80°C for 10 min or left untreated and coated overnight at 4°C on 96-microwell ELISA plates (Dynex 4HBX Immunolon). Wells were washed four times in PBST (phosphate-buffered saline/0.05 Tween 20) and blocked at room temperature with 1% skimmed milk (Marvel)/PBST for 5 h. Individual wells were washed and incubated with 100 μl of either test sera or a panel of monoclonal antibodies overnight at 4°C, before being washed four times in PBST and incubated for 3 h at room temperature with 100 μl of the respective horseradish peroxidase-conjugated secondary antibody diluted in 1%Marvel/PBST. Wells were washed four times in PBST and incubated at room temperature with 100 μl of substrate development buffer consisting of H2O2 and o-phenylenediamine dihydrochloride (SigmaFAST). The reaction was stopped with 30μl of 2M H2SO4 per well and the absorbance read at 492 nm.
Serum Samples
Ethics Statement
This study was carried out in accordance with the recommendations of “the Declaration of Helsinki, and the Kenyan National Scientific and Ethical Review Unit (SERU)” with written informed consent from all subjects. The protocol was approved by SERU, reference number KEMRI/SERU/CGMR-C/001/3139.
Kilifi Adults
Samples from life-long adult residents of Junju in Kilifi, Kenya collected in 2008 were used for assay optimization (n = 66). These sera were assayed for antibody responses to well-studied recombinant merozoite proteins using monoplex ELISA assays, and the data compared with that generated from the same proteins printed on KILchip v1.0. Five serum samples from adults residing in Sweden who reported no travel to malaria-endemic regions and designated malaria naïve sera (MNS) were used as negative controls. Purified malaria immunoglobulins (MIG) were obtained from a pool of healthy Malawian adults and were used to generate a standard curve as previously described (54). An additional serum pool from Kenyan adult residents of Kilifi, Kenya was designated malaria immune sera (MIS) and served as a second positive control.
Statistical Analysis
Spot intensities were acquired using the GenePix scanner (Molecular Devices). Background, pre-scan and purification-tag intensities were subtracted (55) before analysis of within sample variability using the Coefficient of Variation (CV)
where σ is the standard deviation and μ the mean fluorescent intensity (MFI).
We used a two-step variance-stabilizing normalization to minimize the systematic variation commonly observed with this type of data. The first step was to handle the differences that could have occurred with different batches of data (machines or day) using the ComBat (SVA package in R). In the second step, variance-stabilizing normalization was used to minimize the systematic variation commonly observed with this type of data (55–58).
Results
Recombinant Proteins
One hundred and ten P. falciparum merozoite proteins were selected from the literature based on their surface localization, known or predicted roles in erythrocyte invasion and associations with protective immunity (32–34, 38–42). Twenty-eight additional novel proteins were identified using a combination of immuno-proteomics and bioinformatics. Of these, 111 P. falciparum proteins, 21 of which were novel targets, were successfully printed onto KILchip v1.0 (Figure 2 and Supplementary Table 1). These included 82 full-length ectodomains or the largest predicted extracellular loop of multi-membrane proteins and 29 protein fragments obtained from different regions of eight unique proteins. Thirteen protein fragments corresponded to polymorphic variants of MSP1 (n = 7), MSP2 (n = 3), MSP3 (n = 1) and SURFIN4.2 (n = 2) (Figure 2 and Supplementary Table 1). All the remaining proteins were based on the P. falciparum 3D7 sequence and in total, 87 unique P. falciparum merozoite proteins are printed on KILchip v1.0. Two antigens (MSP2-CH150/9 and PF3D7_0424400) were printed twice to serve as additional internal controls.
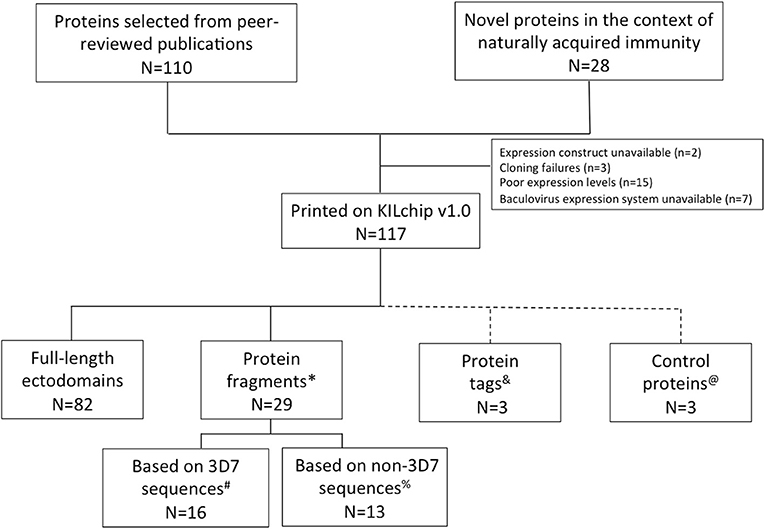
Figure 2. P. falciparum merozoite protein panel included in KILchip v1.0. Proteins were either selected from the literature or from a combination of proteomics and bioinformatics analysis. Details of the parasite proteins are provided in Supplementary Table 1. *Protein fragments refers to specific amino acid regions selected from within a full-length protein ectodomain. #The protein fragments based on the 3D7 allele include MSP1-19 (Block 17), MSP1 Block 2 full, MSP1 Block 2 repeat, MSP3, MSPDBL1 N-terminus, MSPDBL1 C-terminus, MSPDBL2 N-terminus, MSPDBL2 C-terminus, 2 extracellular loops of PF3D7_0629500, Pf SEA1, SURFIN 4.2 3D7A, SURFIN 4.2 3D7B and the C-terminus of SURFIN 4.2. %Thirteen polymorphic variants of the P. falciparum merozoite proteins MSP1, MSP2, MSP3, and SURFIN 4.2 were included in KILchip v1.0. These included MSP1 Block 2 from the K1, MAD20, PaloAlto, Wellcome and RO33 alleles, the CH150/9 and DD2 alleles of MSP2, the K1 allele of MSP3 and the K1A and K1B alleles of SURFIN 4.2. &Protein tags refer to specific amino acids or polypeptides fused to target proteins to facilitate their subsequent affinity purification. These include the CD4 hexa-histidine, MBP and GST tags. @Technical controls for the assay included Alexafluor647 human IgG, purified human IgG and protein printing buffer.
Protein Quality
The quality of the majority of the recombinantly expressed proteins/protein fragments included in KILchip v1.0 have been validated elsewhere, including the demonstration of specific protein-protein interactions (34, 38–41, 43, 45–48). A subset of the well-studied proteins namely: AMA1, EBA175, MSP1, MSP4, and RH5 were evaluated using monoclonal antibodies targeting conformational and disulfide-constrained epitopes in these proteins. Recombinant proteins were tested against humAbAMA1 (59), mAb R217 (60), mAb R218 (60), mAb 2.44 (61), mAb 5.2 (62), mAb 2AC7 (63, 64), and mAb QA1 (64). As shown in Figure 3A, each monoclonal antibody was highly reactive with its respective antigen and showed no reactivity when the target antigen was heat-denatured, confirming the presence of conformational and disulfide-constrained epitopes in the panel of recombinant proteins. As expected, low or negligible reactivity was observed between monoclonal antibodies and off-target recombinant proteins. The presence of heat-labile epitopes was further confirmed by testing native and heat-denatured recombinant proteins for their reactivity with MIS. As shown in Figure 3B, a decrease in immunoreactivity was observed when the proteins were heat-denatured. However, for MSP4, only a minimal drop in immunoreactivity was observed, suggesting the presence of linear epitopes. Collectively, these results suggest that proteins included in KILchip v1.0 were folded correctly and contained heat-labile, conformational epitopes. In addition, circular dichroism analysis of MSP3 and SPATR indicated that these proteins appear to be folded (Supplementary Figure 1).
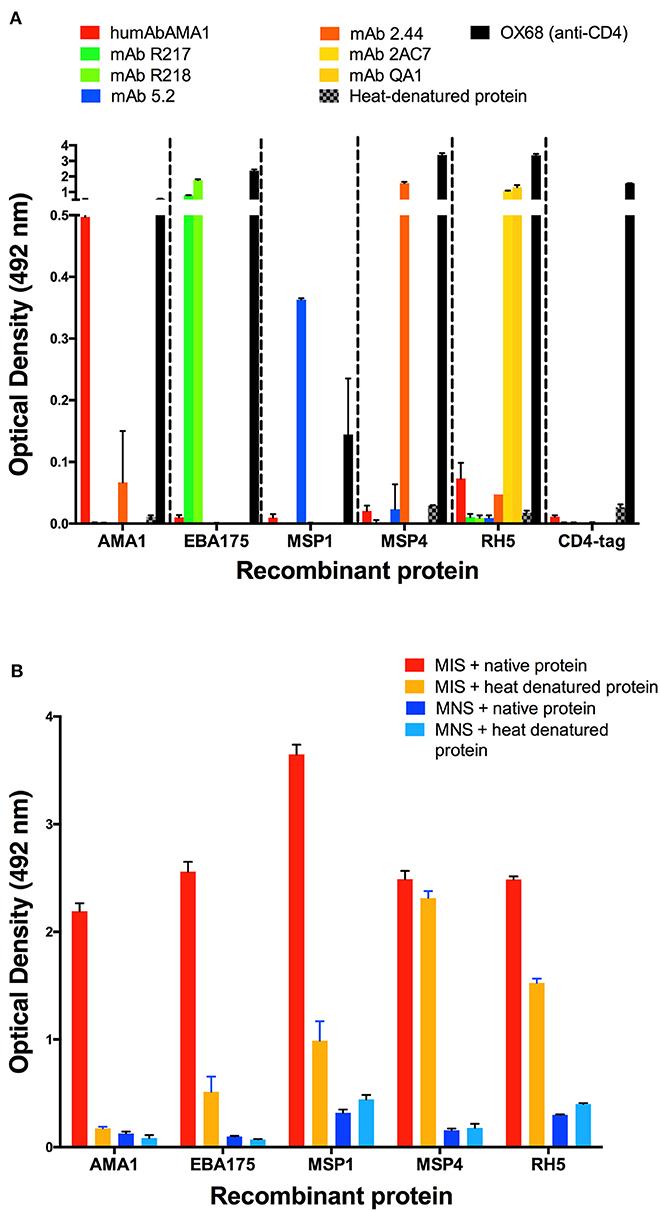
Figure 3. Detection of heat-labile and conformational epitopes in a subset of recombinant proteins. Native and heat-denatured recombinant proteins were tested for reactivity against malaria immune sera (MIS), malaria naïve sera (MNS) and 5 monoclonal antibodies targeting conformational-dependent epitopes. (A) The monoclonal antibodies targeting conformational and disulfide-constrained epitopes on AMA1 (humAbAMA1), the F2 domain of EBA175 region II (mAb R217), the F1 domain of EBA175 region II (mAb R218), MSP1 (mAb 5.2), MSP4 (mAb 2.44), RH5 (mAb QA1 and mAb 2AC7) and rat CD4 domain (OX68) were used to measure reactivity against recombinant proteins by ELISA. All recombinant proteins demonstrated high reactivity with their respective monoclonal antibodies only. Low/negligible reactivity was observed after heat-denaturation of the recombinant proteins. (B) High antibody reactivity was detected with native recombinant protein. Decreased reactivity was observed with the heat-denatured proteins. Low reactivity was detected with malaria naïve sera (MNS) in both native and heat-denatured proteins.
The novel proteins reported here were expressed using Expi293F cells under the same conditions, lending support to their quality with regards to post-translational modifications and disulfide bond formation. The purity of the novel proteins was assessed using reducing SDS gels and mass-spectrometry analysis. Nineteen (90%) of the novel proteins (n = 21) included in KILchip v1.0 were readily detectable on Coomassie stained SDS gels and migrated at a size compatible with their predicted molecular weights (Supplementary Figure 2). Mass-spectrometry analysis confirmed the identity of the majority of recombinant proteins (Supplementary Table 2).
KILchip v1.0
Specificity
To confirm the specificity of antibody detection, individual mini-arrays were probed with the following positive and negative controls: (i) malaria immune sera (MIS), (ii) malaria naïve sera (MNS) and (iii) sample buffer consisting of 2% BSA/0.1% Tween 20/HBS. As expected, strong fluorescence was detected on each mini-array for the landmarks (blue) and commercial human IgG (green), whilst none was detected against the printing buffer (yellow) (Figure 4A, all panels). When probed with MIS, high levels of antibody reactivity against multiple P. falciparum proteins were clearly visible (Figure 4A, panel 1). In contrast, when probed with MNS, negligible antibody reactivity was observed. Similarly, when probed with sample buffer, nil or minimal reactivity was observed against P. falciparum proteins (Figure 4A, panel 2 and 3 respectively).
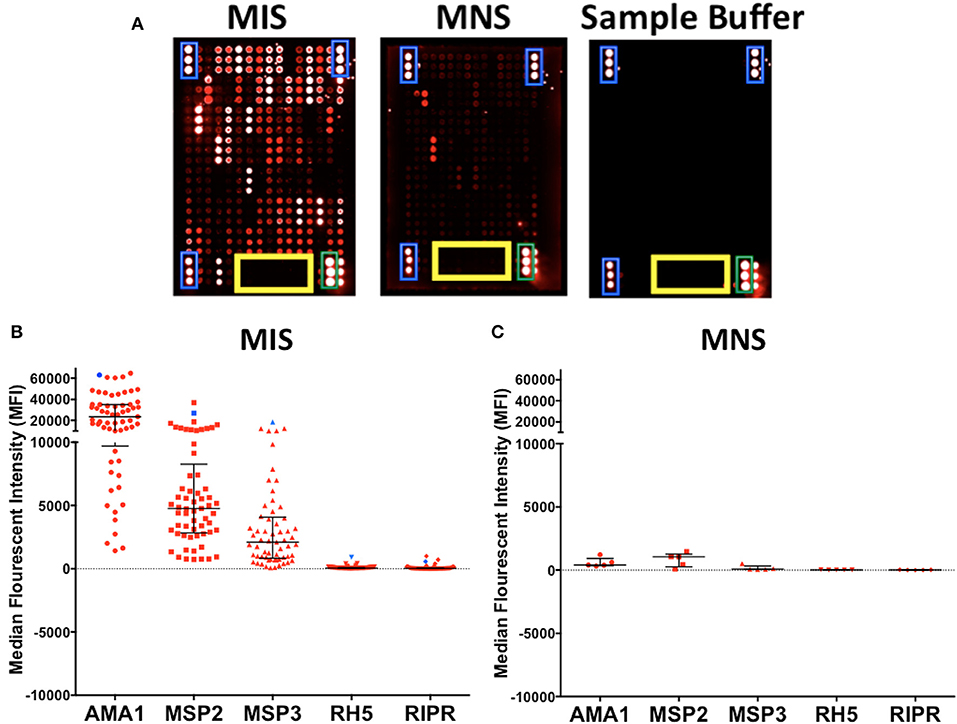
Figure 4. Specificity of KILchip v1.0. Positive and negative controls were used to confirm specificity. (A) High antibody reactivity was detected using malaria immune sera (MIS) and low or negligible reactivity was observed with malaria naïve sera (MNS). No reactivity was observed with the sample buffer with the exception of the technical controls. Blue, landmarks; green, commercial human IgG and yellow, printing buffer. (B) Antibody reactivity to selected well-characterized merozoite antigens AMA1, MSP2 (3D7), MSP3 (3D7), Rh5, and RIPR varied as expected in MIS (blue) and serum samples from adult residents of Kilifi. Kenya (red). High reactivity to AMA1, MSP2 (3D7), and MSP3 (3D7) and low reactivity to Rh5 and RIPR was observed in the serum samples. (C) Antibody reactivity to well-characterized antigens AMA1, MSP2, MSP3, Rh5, and RIPR were all low/negligible when tested in serum samples from Swedish residents.
We further explored the specificity by comparing antibody reactivity to selected, previously characterized proteins in individual samples from Kenyan adults (n = 66) and malaria naïve Swedes (n = 5). The intensity of antibody reactivity varied between these selected proteins in the order AMA1 > MSP2 > MSP3 > Pf RH5 > RIPR (Figure 4B and Supplementary Figure 3). The median (minimum-maximum) fluorescence intensity (MFI) responses to AMA1, MSP2, and MSP3 were 23629 (1426-64840), 4762 (733-37025), and 2105 (72-4069), respectively in the Kenyan adults. MFI responses to RH5 and RIPR were low at 44 (4-914) and 28 (-2-994), respectively (Figure 4B). In contrast, reactivity against all P. falciparum proteins was low or negligible when probed with the non-malaria exposed serum from Swedish adults (Figure 4C and Supplementary Figure 3). AMA1, MSP2, and MSP3 are well-characterized immunodominant merozoite antigens while RH5 and RIPR appear to not be primary targets of naturally acquired antibody responses in some studies (32, 33, 65, 66). A direct comparison of antibody responses to multiple antigens can be evaluated using KILchip v1.0 as each protein was printed at the same concentration (250 μg/ml), and would be tested simultaneously with the same sera, providing information on the relative immunogenicity of this panel of merozoite antigens (Supplementary Figure 4).
Intra- and Inter-assay Variability
Intra-assay variability was tested using 66 serum samples obtained from Kenyan adults resident in Kilifi. Antibody measurements between protein spot replicates tested in the same assay and on the same day were strongly positively correlated, Spearman's R > 0.9770, p < 0.0001 (Figure 5). Inter-assay variability was tested using the positive and negative control sera. Antibody reactivity was measured repeatedly on five separate microarray slides printed over five consecutive days using identical printing conditions. As shown in Figure 6, the average signal intensities against individual proteins were highly reproducible.
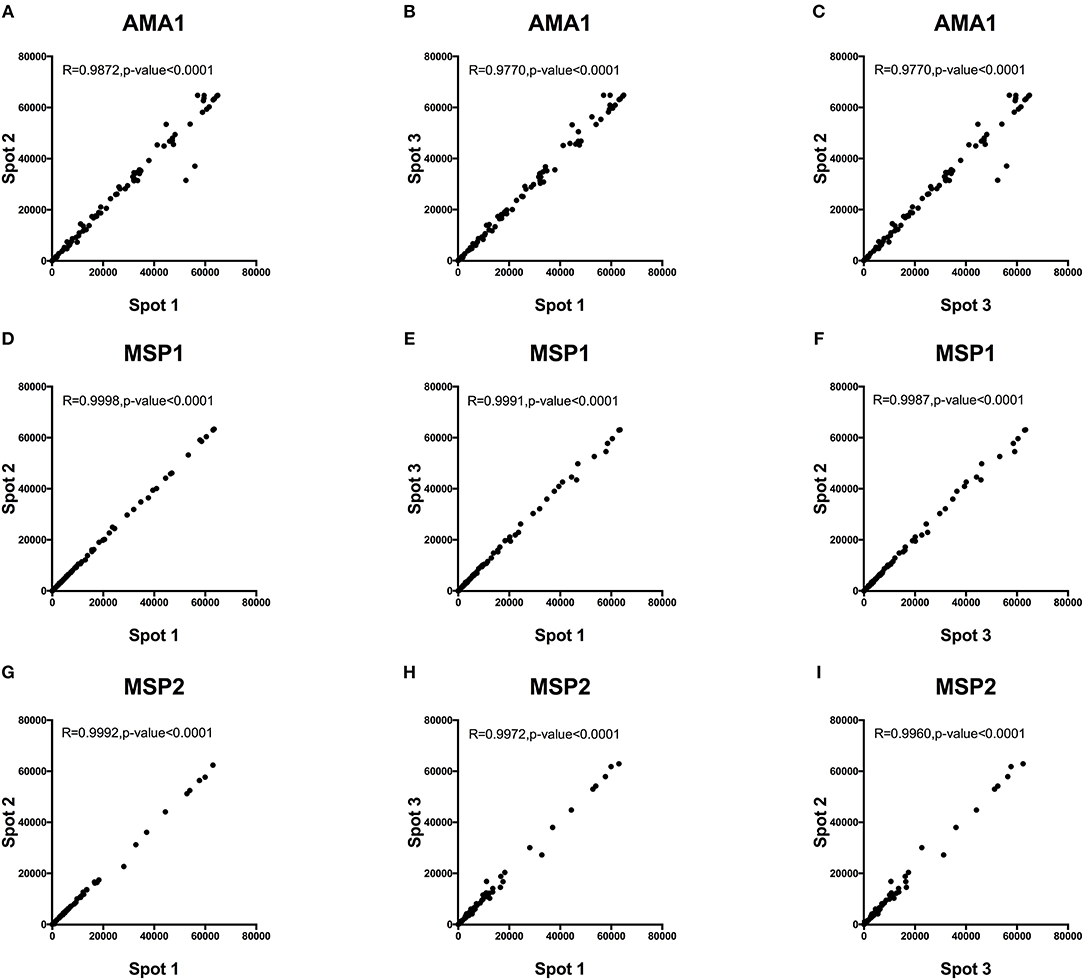
Figure 5. Intra-assay variability of antibody detection by KILchip v1.0. A comparison of antibody responses to triplicate readings of AMA1, MSP1, and MSP2 was measured in 66 serum samples from adults living in the malaria endemic region of Kilifi, Kenya. A strong positive correlation of >0.98 was observed in all three-way scatter plots tested for the three antigens. Correlation between triplicate readings for recombinant AMA1 (A–C), MSP1 (D–F) and MSP2 (G–I).
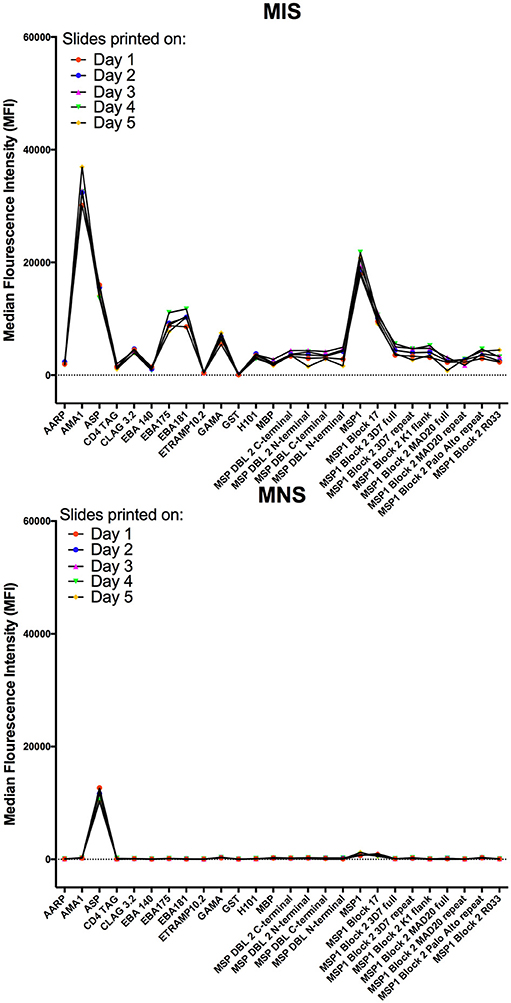
Figure 6. Inter-assay variability of antibody detection by KILchip v1.0. Positive and negative controls were used to measure antibody responses to 5 protein arrays printed on 5 consecutive days using identical printing conditions and the same batch of recombinant proteins. Data presented are the mean MFI values of triplicate readings obtained with MIS (top graph) and MNS (bottom graph) to a subset of the proteins (n = 26) printed on each mini-array. Individual proteins are represented as dots and lines have been included to aid visualization.
Protein Stability on KILchip v1.0
To determine the durability of the microarray slides, we measured responses to the same batch of slides over a 3 months period using the reference reagent, MIG. These slides were printed on the same day, with identical printing conditions and the same batch of recombinant proteins. A tripling dilution of MIG was used to measure responses to each protein and to generate a standard curve. We performed a pairwise comparison of the slopes for each of the sigmoidal 5-parameter curves generated for each antigen over the 3-months period (n = 4 sigmoidal curves; 6 pairwise comparisons per antigen) (67, 68). We observed no significant differences in the slopes for all 6 pairwise comparisons for 79/111 (71%) proteins (T-test; p > 0.05). Of the 32 proteins whose slopes differed significantly from each other, eight proteins had a single curve that differed while 24 proteins had 2 or more significantly different slopes. Results from 12/111 (10%) of the proteins printed on the array are shown in Figure 7 and demonstrate that the curves to the majority of proteins were comparable indicating the detection of responses up to 3 months post printing without significant variation.
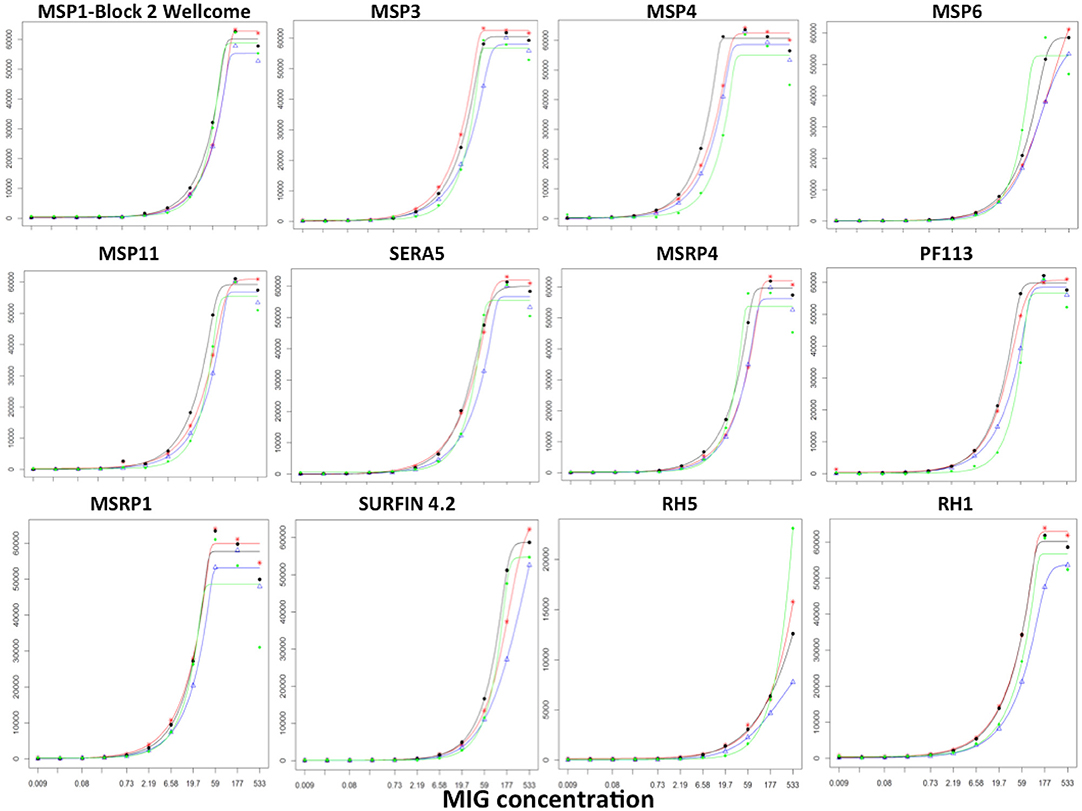
Figure 7. Stability of KILchip v1.0. A reference reagent, MIG, consisting of purified immunoglobulins (98% Total IgG) was used to generate a standard curve by testing a tripling dilution against all antigens printed on the protein array. A batch of slides printed at the same time was tested 24, 40, 54, and 86 days post printing. The standard curve showed good concordance without significant variation. Red, blue, black, and green lines were obtained from measurements obtained on day 24, day 40, day 54, and day 86 post printing of the slides.
Strong Correlations Between KILchip v1.0 and the Monoplex ELISA
We compared measurements obtained using 12/111 proteins (representing ~10% of the P. falciparum proteins included in the array) printed on KILchip v1.0 vs. the identical proteins using our standardized ELISA in 66 samples from Kenyan adults. As shown in Figures 8A–L, a strong positive Spearman's correlation coefficient R of between 0.65 and 0.95 was observed between antibodies measured by KILchip v1.0 and by the standard ELISA, P < 0.0001. Sixty-seven percent of the proteins tested had a correlation coefficient R above 0.8 (Figures 8A–H). A wider dynamic range of antibody measurement was evident in the protein array assay as observed for AMA1 (Figure 8B), where a subset of samples whose MFI values ranged from 40,000 to 60,000 all had an optical density value of 4.0 (red box), the upper limit detectable by ELISA.
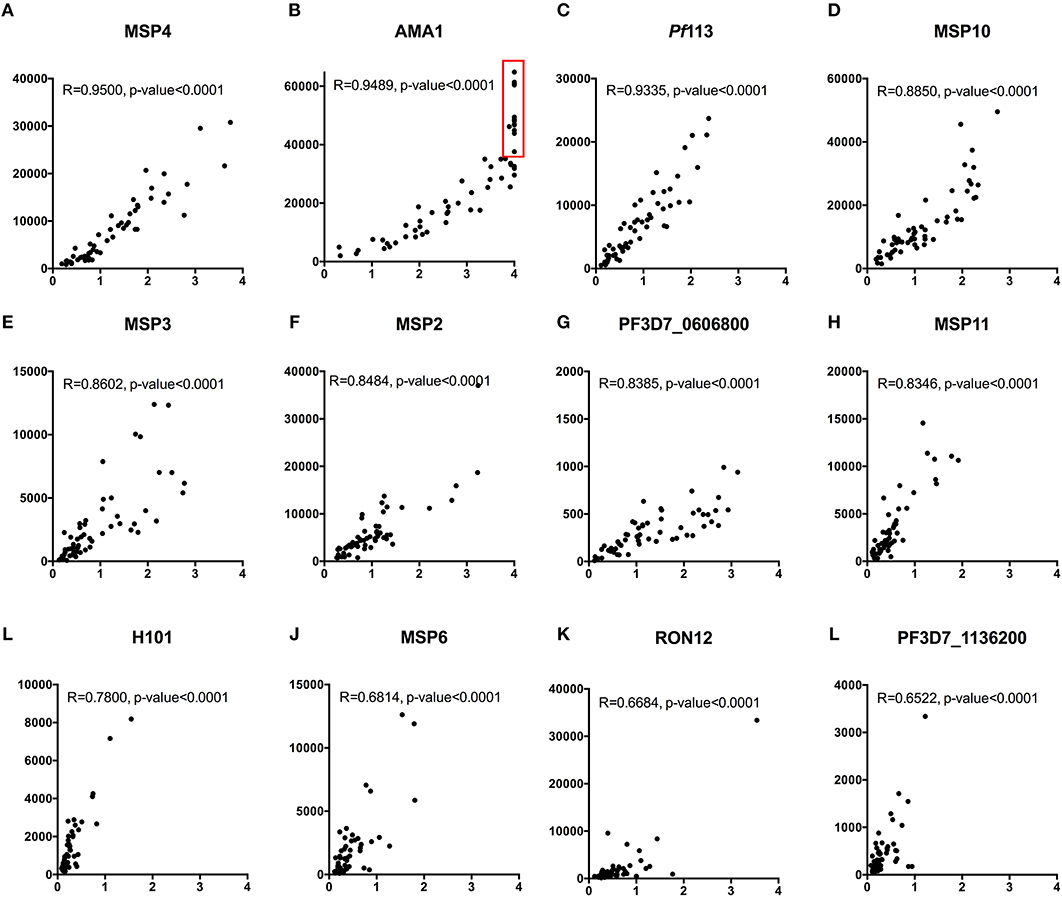
Figure 8. Concordance between KILchip v1.0 and the gold standard ELISA. A comparison of antibody responses to 12 antigens measured simultaneously in KILchip v1.0 and individually by ELISA. Sixty-six serum samples from adults living in the malaria endemic region of Kilifi, Kenya were used to facilitate comparisons. A strong positive correlation of >0.8 was observed in 8 of the antigens (A–H) while four antigens (I–L) showed a correlation coefficient between 0.65 and 0.78. Red box - serum samples in which antibody responses reached the upper limit of detection when measured by ELISA, yet distinguishable by microarray with MFI values ranging from 40,000 to 60,000.
Discussion
KILchip v1.0 is a new protein microarray designed to simultaneously quantify antibodies against multiple P. falciparum merozoite proteins and is currently configured to include >100 proteins, the majority of which are full-length ectodomains from secreted and surface exposed proteins. This facilitates the standardized measurement of antibodies against multiple merozoite proteins in cohort studies or in controlled human malaria infections, both of which are currently used to identify and prioritize potential vaccine candidates. It can be utilized to evaluate antibody dynamics and to monitor antibody decay or longevity. The chip is flexible and can be adapted to include fewer or more proteins, allelic variants of selected proteins, full-length proteins or functionally relevant domains of proteins of interest. Adaptations of the chip could expand from the current species-specific focus on P. falciparum in KILchip v1.0, and be designed to test additional Plasmodium species singly or in parallel. Of particular importance to the research setting in sub-Saharan Africa, the chip is durable, easily stored and transported, and can be shared between partnering laboratories. With modest investments, this tool could transform the pace at which P. falciparum antibody response data is generated across the African continent and the same principles could be applied to other infections, ultimately contributing to improvements in health through the development of diagnostics and vaccines.
Although other protein microarrays have been in existence since the early 2,000 s, none has been designed and developed from Africa specifically to study naturally acquired immunity against P. falciparum malaria merozoites (13, 15, 69, 70). In addition, KILchip v1.0 consists predominantly of merozoite specific proteins, the majority of which were full-length ectodomains, a considerable improvement from evaluating segments of proteins, as is the case for other pre-existing protein microarrays available for P. falciparum. This was enabled by the use of a recently published method for transient protein expression of P. falciparum surface proteins in mammalian cells (39). Many of the proteins included in KILchip v1.0 are known to be immunogenic to varying degrees during natural exposure to malaria parasites, but have only been evaluated in a handful of cohort studies (32, 33). Typically, the exact nature, size and quality of antigens tested in these studies vary considerably, making it difficult to accurately compare results (35). Consequently, although some antigens have been studied for nearly 30 years, it is still not clear which proteins should be prioritized as vaccine candidates in clinical trials. KILchip v1.0 now provides a standardized tool that enables head-to-head comparisons of the immunoreactivity of a large number of proteins in multiple cohorts. One such study is already underway in which samples from at least 15 distinct geographical locations spread across 7 African countries have been tested using KILchip v1.0 (SMART: South-South Malaria Antigen Research Partnership) (71) and highlights the potential strength of KILchip v1.0.
We demonstrate that KILchip v1.0 is a sensitive tool for the detection of P. falciparum specific antibody responses. It is highly reproducible within and between assays and a strong concordance with the gold-standard monoplex ELISA was observed, comparable to the results reported for the different P. falciparum microarrays currently in use (13). Crucially, antibody measurements were stable on KILchip v1.0 up to 3 months post-printing, providing a suitable time frame for the testing of multiple samples. This is probably due to the addition of 60% glycerol to the recombinant proteins at the time of printing which provides a stabilizing effect on proteins preventing degradation (72). Slides are conveniently stored with desiccant at 4°C.
In comparison to our standard ELISA protocol (52, 54), our microarray utilized 0.375 μl of serum to measure responses to all proteins simultaneously, compared to 23 μl required for ELISA assays of the same number of proteins. Similarly, over 100-fold less recombinant protein was required and the laser scanning and data acquisition by the GenePix 4,000 B scanner allowed for much wider dynamic ranges of antibody measurement. Lastly, this is a custom microarray format that could be scaled upwards or downwards and can be adapted to meet specific requirements. In the current version of KILchip v1.0, a single slide has 21 mini-arrays each containing 384 protein spots. This can be re-designed to include more or less proteins, to focus on allelic versions of specific proteins or to facilitate the simultaneous characterization of stage or species-specific antibody responses.
Although we selected 138 proteins/protein fragments for inclusion in KILchip v1.0, we were unable to obtain recombinant protein for 27 targets due to challenges in protein expression. These include targets such as RH4, RAP1-2, RhopH3, GLURP, SERA1, and SERA6 that have also been reported to be difficult to obtain in recombinant form in previous studies (38, 39). Also, the DBL domains of MSPDBL1 and MSPDBL2 have been previously obtained in soluble recombinant form using the baculovirus expression system but this has not yet been established within our laboratories (34). We also observed low antibody responses to some of the merozoite proteins such as RH5 and RIPR, similar to responses measured in adults from Kenya (65) and Mali (66). However, a higher response to these proteins expressed in the wheat germ cell-free expression system has been reported in children in Papua New Guinea (73), suggesting that the protein expression system used may influence the antibody responses measured. Another limitation of the KILchip v1.0 is that the majority of proteins are based on the 3D7 P. falciparum isolate which may underestimate responses to highly polymorphic proteins and limit the evaluation of antigenic diversity on humoral responses and immunity. Efforts are underway to generate an additional protein array that will include allelic variants of specific proteins that warrant further study. Lastly, adsorption of proteins onto nitrocellulose-coated slides may interfere with protein structure and consequently with the detection of conformation-dependent antibody responses (74). However, these and other solid-surface platforms such as in ELISAs are widely utilized for antibody detection for vaccine candidate discovery and prioritization in multiple infectious and non-infectious diseases.
Current and future versions of KILchip v1.0 will be essential to multi-center prospective cohort studies designed to identify correlates of protection, allowing the research community to rapidly compare results head-to-head, and fast track the prioritization of new and old potential vaccine candidates. This would bridge an important gap for the urgently needed evidence base that could guide the development of the next generation of malaria vaccines. Efforts are underway to make this a resource that could be provided at a minimal cost to the malaria research community.
Author Contributions
FO, JR, and KevM conceived the study. GK and JT designed the array. GK, JT, RK, MC, SH, JN, CK, MR, RF, RY, EC, TC, JM, and FG performed the experiments, GK wrote the paper with contributions from JT, KenM, NK, and FO. SK, LM, and PB provided helpful discussions. AF, KT, JB, and DC provided reagents for the array design and testing. All authors read and approved the final manuscript.
Funding
This work was supported by an MRC/DFID African Research Leader Award jointly funded by the UK Medical Research Council (MRC) and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreement (MR/L00450X/1); an EDCTP Senior Fellowship (TMA 2015 SF1001); and a Sofja Kovalevskaja Award from the Alexander von Humboldt Foundation (3.2 - 1184811 - KEN - SKP) awarded to FHAO. JB was supported by a Senior Research Fellowship from the National Health and Medical Research Council of Australia. Additional support was obtained from TIBA (Tackling Infections to Benefit Africa, Project Number is 16/136/33) in collaboration with the IDeAL and DELTAS Africa Initiative [DEL-15-003]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [107769/Z/10/Z] and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was published with permission from the office of the Director of KEMRI. We thank Dr. Gavin Wright and Dr. Cecile Crosnier of the Wellcome Trust Sanger Institute and Prof. Ray Owens of The Oxford Protein Production Facility (OPPP) in Oxford, UK for helpful discussions and training on transient protein expression. We are grateful to Dr. Rolf Fendel of Fraunhofer IME, Aachen and the Institute of Tropical Medicine (ITM), Tübingen, Germany and Dr. Torsten Klockenbring of Fraunhofer IME, Aachen, Germany, who provided the HumAbAMA1 and mAb 2.44 monoclonal antibodies. We thank Prof. Simon Draper of the Jenner Institute, University of Oxford, United Kingdom who provided the mAb 2AC7 and mAb QA1 monoclonal antibodies. Monoclonal antibody R217 and R218 were obtained through BEI resources, and were contributed by EntreMed/NIAID. We thank Prof Stefan Magez of Vrije Universiteit Brussels and Ghent University Global Campus, and Dr. Yann Sterckx of Universiteit Antwerpen, for helpful discussions on the circular dichroism spectroscopy experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02866/full#supplementary-material
Supplementary Table 1. List of P. falciparum merozoite proteins included in KILchip v1.0. The details of the P. falciparum merozoite proteins provided include the expression system used, the region of the protein expressed, the P. falciparum strain the protein was based on and the recombinant expression levels achieved. The expression levels were categorized as low, intermediate and high based on the concentration of purified protein obtained following transfection of a standard concentration of 50.0 × 106 Expi293F cells. The protein concentration following purification and concentration of low, intermediate and high expressors were < 100 μg/ml, 100–250 μg/ml, and >250 μg/ml, respectively. The expression levels are given as guide only given the variability that may be observed with different expression batches. We observed no differences in the background signals when malaria naïve sera were tested against recombinant proteins grouped according to their expression levels (Kruskal-Wallis H Test for trend: p-value = 0.5975) (data not shown).
Supplementary Table 2. Mass-spectrometry confirmation of novel Plasmodium falciparum recombinant proteins. Mass-spectrometry confirmation of the novel Plasmodium falciparum recombinant proteins included in KILchip v1.0. Data for PF3D7_0830500, PF3D7_1025300, PF3D7_1229300, PF3D7_1252300, PF3D7_1237900, PF3D7_1343700, and PF3D7_0629500_SEG2 are not yet available.
Supplementary Figure 1. Circular dichroism spectroscopy on recombinant SPATR and MSP3. Circular Dichroism (CD) spectra graphs for SPATR (A) and MSP3 (B) proteins are plotted from mean residual ellipticity [θ] (mDeg · cm2·dmol−1·res−1) as a function of the wavelength, λ (nM). The ellipticity for the blank (phosphate buffer (50mM NaH2PO4 pH 8.0, 0.1M NaCl)) was subtracted from the ellipticity for the protein samples before calculation of the mean residue ellipticity [θ].
Supplementary Figure 2. Purity of novel Plasmodium falciparum proteins. Coomassie stained SDS gels showing 19 purified recombinant proteins. The numbers in bracket indicate the predicted molecular masses (in KDa) for each protein. Protein bands for PF3D7_1343700 and PF3D7_0629500_SEG2 were not readily visible on a coomassie stained SDS gel. Protein bands for AMA1 and CD4-hexa-histidine tags were included as controls.
Supplementary Figure 3. Antibody responses to previously characterized merozoite proteins. The protein spots corresponding to previously characterized merozoite antigens AMA1, MSP2 (3D7), MSP3 (3D7), Rh5, and RIPR are shown in protein arrays probed with MIS and MNS sera. Red-AMA1, Yellow-MSP2, Blue-MSP3, Orange-RH5, and Green-RIPR.
Supplementary Figure 4. Immunoreactivity of all the recombinant proteins printed on KILchip v1.0 measured using a pool of sera from naturally exposed adults. The immunoreactivity of the 111 novel proteins was tested using malaria immune sera (MIS, red bars) and malaria naïve sera (MNS, blue bars). Bar charts show mean plus standard deviation; n = 2.
References
1. Kingsmore SF. Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nat Rev Drug Discov. (2006) 5:310–20. doi: 10.1038/nrd2006
2. Yu X, Petritis B, LaBaer J. Advancing translational research with next-generation protein microarrays. Proteomics (2016) 16:1238–50. doi: 10.1002/pmic.201500374
3. Davies DH, Duffy P, Bodmer JL, Felgner PL, Doolan DL. Large screen approaches to identify novel malaria vaccine candidates. Vaccine (2015) 33:7496–505. doi: 10.1016/j.vaccine.2015.09.059
5. Nkumama IN, O'Meara WP, Osier FHA. Changes in malaria epidemiology in africa and new challenges for elimination. Trends Parasitol. (2016) 33:128–40. doi: 10.1016/j.pt.2016.11.006
6. Snow RW, Sartorius B, Kyalo D, Maina J, Amratia P, Mundia CW, et al. The prevalence of Plasmodium falciparum in sub-Saharan Africa since 1900. Nature (2017) 550:515–8. doi: 10.1038/nature24059
7. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature (2002) 419:498–511. doi: 10.1038/nature01097
8. Rénia L, Goh YS. Malaria Parasites: the great escape. Front Immunol. (2016) 7:463. doi: 10.3389/fimmu.2016.00463
9. Gomes PS, Bhardwaj J, Rivera-Correa J, Freire-De-Lima CG, Morrot A. Immune escape strategies of malaria parasites. Front Microbiol. (2016) 7:1617. doi: 10.3389/fmicb.2016.01617
10. Persson KE, McCallum FJ, Reiling L, Lister NA, Stubbs J, Cowman AF, et al. Variation in use of erythrocyte invasion pathways by Plasmodium falciparum mediates evasion of human inhibitory antibodies. J Clin Invest. (2008) 118:342–51. doi: 10.1172/JCI32138
11. RTS S. Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet (2015) 386:31–45. doi: 10.1016/S0140-6736(15)60721-8
12. Olotu A, Fegan G, Wambua J, Nyangweso G, Leach A, Lievens M, et al. Seven-year efficacy of RTS,S/AS01 malaria vaccine among young african children. N Engl J Med. (2016) 374:2519–29. doi: 10.1056/NEJMoa1515257
13. Crompton PD, Kayala MA, Traore B, Kayentao K, Ongoiba A, Weiss GE, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci USA. (2010) 107:6958–63. doi: 10.1073/pnas.1001323107
14. Trieu A, Kayala MA, Burk C, Molina DM, Freilich DA, Richie TL, et al. Sterile protective immunity to malaria is associated with a panel of novel P. falciparum antigens. Mol Cell Proteomics (2011) 10:M111 007948. doi: 10.1074/mcp.M111.007948
15. Finney OC, Danziger SA, Molina DM, Vignali M, Takagi A, Ji M, et al. Predicting antidisease immunity using proteome arrays and sera from children naturally exposed to malaria. Mol Cell Proteomics (2014) 13:2646–60. doi: 10.1074/mcp.M113.036632
16. Kanoi BN, Takashima E, Morita M, White MT, Palacpac NM, Ntege EH, et al. Antibody profiles to wheat germ cell-free system synthesized Plasmodium falciparum proteins correlate with protection from symptomatic malaria in Uganda. Vaccine (2017) 35:873–81. doi: 10.1016/j.vaccine.2017.01.001
17. Morita M, Takashima E, Ito D, Miura K, Thongkukiatkul A, Diouf A, et al. Immunoscreening of Plasmodium falciparum proteins expressed in a wheat germ cell-free system reveals a novel malaria vaccine candidate. Sci Rep. (2017) 7:46086. doi: 10.1038/srep46086
18. Tan X, Traore B, Kayentao K, Ongoiba A, Doumbo S, Waisberg M, et al. Hemoglobin S and C heterozygosity enhances neither the magnitude nor breadth of antibody responses to a diverse array of Plasmodium falciparum antigens. J Infect Dis. (2011) 204:1750–61. doi: 10.1093/infdis/jir638
19. Nnedu ON, O'Leary MP, Mutua D, Mutai B, Kalantari-Dehaghi M, Jasinskas A, et al. Humoral immune responses to Plasmodium falciparum among HIV-1-infected Kenyan adults. Proteomics Clin Appl. (2011) 5:613–23. doi: 10.1002/prca.201100021
20. Baum E, Badu K, Molina DM, Liang X, Felgner PL, Yan G. Protein microarray analysis of antibody responses to Plasmodium falciparum in western Kenyan highland sites with differing transmission levels. PLoS ONE (2013) 8:e82246. doi: 10.1371/journal.pone.0082246
21. Felgner PL, Roestenberg M, Liang L, Hung C, Jain A, Pablo J, et al. Pre-erythrocytic antibody profiles induced by controlled human malaria infections in healthy volunteers under chloroquine prophylaxis. Sci Rep. (2013) 3:3549. doi: 10.1038/srep03549
22. Arama C, Skinner J, Doumtabe D, Portugal S, Tran TM, Jain A, et al. Genetic resistance to malaria is associated with greater enhancement of immunoglobulin (Ig)M than IgG responses to a broad array of plasmodium falciparum antigens. Open Forum Infect Dis. (2015) 2:ofv118. doi: 10.1093/ofid/ofv118
23. Campo JJ, Aponte JJ, Skinner J, Nakajima R, Molina DM, Liang L, et al. RTS,S vaccination is associated with serologic evidence of decreased exposure to Plasmodium falciparum liver- and blood-stage parasites. Mol Cell Proteomics (2015) 14:519–31. doi: 10.1074/mcp.M114.044677
24. Helb DA, Tetteh KK, Felgner PL, Skinner J, Hubbard A, Arinaitwe E, et al. Novel serologic biomarkers provide accurate estimates of recent Plasmodium falciparum exposure for individuals and communities. Proc Natl Acad Sci USA. (2015) 112:E4438–47. doi: 10.1073/pnas.1501705112
25. Baum E, Sattabongkot J, Sirichaisinthop J, Kiattibutr K, Davies DH, Jain A, et al. Submicroscopic and asymptomatic Plasmodium falciparum and Plasmodium vivax infections are common in western Thailand - molecular and serological evidence. Malar J. (2015) 14:95. doi: 10.1186/s12936-015-0611-9
26. Torres KJ, Castrillon CE, Moss EL, Saito M, Tenorio R, Molina DM, et al. Genome-level determination of Plasmodium falciparum blood-stage targets of malarial clinical immunity in the Peruvian Amazon. J Infect Dis. (2015) 211:1342–51. doi: 10.1093/infdis/jiu614
27. Dent AE, Nakajima R, Liang L, Baum E, Moormann AM, Sumba PO, et al. Plasmodium falciparum protein microarray antibody profiles correlate with protection from symptomatic malaria in kenya. J Infect Dis. (2015) 212:1429–38. doi: 10.1093/infdis/jiv224
28. Baum E, Sattabongkot J, Sirichaisinthop J, Kiattibutr K, Jain A, Taghavian O, et al. Common asymptomatic and submicroscopic malaria infections in Western Thailand revealed in longitudinal molecular and serological studies: a challenge to malaria elimination. Malar J. (2016) 15:333. doi: 10.1186/s12936-016-1393-4
29. King CL, Davies DH, Felgner P, Baum E, Jain A, Randall A, et al. Biosignatures of Exposure/Transmission and immunity. Am J Trop Med Hyg. (2015) 93(3 Suppl.):16–27. doi: 10.4269/ajtmh.15-0037
30. Portugal S. Treatment of chronic asymptomatic plasmodium falciparum infection does not increase the risk of clinical malaria upon reinfection. Clin Infect Dis. (2017) 64:645–53. doi: 10.1093/cid/ciw849
31. Uplekar S, Rao PN, Ramanathapuram L, Awasthi V, Verma K, Sutton P, et al. Characterizing antibody responses to plasmodium vivax and plasmodium falciparum antigens in india using genome-scale protein microarrays. PLoS Negl Trop Dis. (2017) 11:e0005323. doi: 10.1371/journal.pntd.0005323
32. Richards JS, Arumugam TU, Reiling L, Healer J, Hodder AN, Fowkes FJ, et al. Identification and prioritization of merozoite antigens as targets of protective human immunity to Plasmodium falciparum malaria for vaccine and biomarker development. J Immunol. (2013) 191:795–809. doi: 10.4049/jimmunol.1300778
33. Osier FH, Mackinnon MJ, Crosnier C, Fegan G, Kamuyu G, Wanaguru M, et al. New antigens for a multicomponent blood-stage malaria vaccine. Sci Transl Med. (2014) 6:247ra102. doi: 10.1126/scitranslmed.3008705
34. Tetteh KK, Osier FH, Salanti A, Kamuyu G, Drought L, Failly M, et al. Analysis of antibodies to newly described Plasmodium falciparum merozoite antigens supports MSPDBL2 as a predicted target of naturally acquired immunity. Infect Immun. (2013) 81:3835–42. doi: 10.1128/IAI.00301-13
35. Fowkes FJ, Richards JS, Simpson JA, Beeson JG. The relationship between anti-merozoite antibodies and incidence of Plasmodium falciparum malaria: a systematic review and meta-analysis. PLoS Med. (2010) 7:e1000218. doi: 10.1371/journal.pmed.1000218
36. Pizza M, Scarlato V, Masignani V, Giuliani MM, Aricò B, Comanducci M, et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science (2000) 287:1816–20. doi: 10.1126/science.287.5459.1816
37. Rodríguez-Ortega MJ, Norais N, Bensi G, Liberatori S, Capo S, Mora M, et al. Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat Biotechnol. (2006) 24:191–7. doi: 10.1038/nbt1179
38. Zenonos ZA, Rayner JC, Wright GJ. Towards a comprehensive Plasmodium falciparum merozoite cell surface and secreted recombinant protein library. Malar J. (2014) 13:93. doi: 10.1186/1475-2875-13-93
39. Crosnier C, Wanaguru M, McDade B, Osier FH, Marsh K, Rayner JC, et al. A library of functional recombinant cell-surface and secreted P. falciparum merozoite proteins. Mol Cell Proteomics (2013) 12:3976–86. doi: 10.1074/mcp.O113.028357
40. Polley SD, Tetteh KK, Lloyd JM, Akpogheneta OJ, Greenwood BM, Bojang KA, et al. Plasmodium falciparum merozoite surface protein 3 is a target of allele-specific immunity and alleles are maintained by natural selection. J Infect Dis. (2007) 195:279–87. doi: 10.1086/509806
41. Taylor RR, Smith DB, Robinson VJ, McBride JS, Riley EM. Human antibody response to Plasmodium falciparum merozoite surface protein 2 is serogroup specific and predominantly of the immunoglobulin G3 subclass. Infect Immun. (1995) 63:4382–8.
42. Cavanagh DR, McBride JS. Antigenicity of recombinant proteins derived from Plasmodium falciparum merozoite surface protein 1. Mol Biochem Parasitol. (1997) 85:197–211. doi: 10.1016/S0166-6851(96)02826-5
43. Raj DK, Nixon CP, Nixon CE, Dvorin JD, DiPetrillo CG, Pond-Tor S, et al. Antibodies to PfSEA-1 block parasite egress from RBCs and protect against malaria infection. Science (2014) 344:871–7. doi: 10.1126/science.1254417
44. Kamuyu G. Identifying the Merozoite Targets of Protective Immunity to Plasmodium Falciparum Malaria. PhD thesis The Open University (2017).
45. Polley SD, Tetteh KK, Cavanagh DR, Pearce RJ, Lloyd JM, Bojang KA, et al. Repeat sequences in block 2 of Plasmodium falciparum merozoite surface protein 1 are targets of antibodies associated with protection from malaria. Infect Immun. (2003) 71:1833–42. doi: 10.1128/IAI.71.4.1833-1842.2003
46. Kimbi HK, Tetteh KK, Polley SD, Conway DJ. Cross-sectional study of specific antibodies to a polymorphic Plasmodium falciparum antigen and of parasite antigen genotypes in school children on the slope of Mount Cameroon. Trans R Soc Trop Med Hyg. (2004) 98:284–9. doi: 10.1016/S0035-9203(03)00068-3
47. Metzger WG, Okenu DM, Cavanagh DR, Robinson JV, Bojang KA, Weiss HA, et al. Serum IgG3 to the Plasmodium falciparum merozoite surface protein 2 is strongly associated with a reduced prospective risk of malaria. Parasite Immunol. (2003) 25:307–12. doi: 10.1046/j.1365-3024.2003.00636.x
48. Burghaus PA, Holder AA. Expression of the 19-kilodalton carboxy-terminal fragment of the Plasmodium falciparum merozoite surface protein-1 in Escherichia coli as a correctly folded protein. Mol Biochem Parasitol. (1994) 64:165–9. doi: 10.1016/0166-6851(94)90144-9
49. Hu G, Cabrera A, Kono M, Mok S, Chaal BK, Haase S, et al. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat Biotechnol. (2010) 28:91–8. doi: 10.1038/nbt.1597
50. Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. (2006) 1:2876–90. doi: 10.1038/nprot.2006.202
51. Sun Y, Gallagher-Jones M, Barker C, Wright GJ. A benchmarked protein microarray-based platform for the identification of novel low-affinity extracellular protein interactions. Anal Biochem. (2012) 424:45–53. doi: 10.1016/j.ab.2012.01.034
52. Osier FH, Fegan G, Polley SD, Murungi L, Verra F, Tetteh KK, et al. Breadth and magnitude of antibody responses to multiple Plasmodium falciparum merozoite antigens are associated with protection from clinical malaria. Infect Immun. (2008) 76:2240–8. doi: 10.1128/IAI.01585-07
53. Rono J, Färnert A, Murungi L, Ojal J, Kamuyu G, Guleid F, et al. Multiple clinical episodes of Plasmodium falciparum malaria in a low transmission intensity setting: exposure versus immunity. BMC Med. (2015) 13:114. doi: 10.1186/s12916-015-0354-z
54. Murungi LM, Kamuyu G, Lowe B, Bejon P, Theisen M, Kinyanjui SM, et al. A threshold concentration of anti-merozoite antibodies is required for protection from clinical episodes of malaria. Vaccine (2013) 31:3936–42. doi: 10.1016/j.vaccine.2013.06.042
55. Sundaresh S, Doolan DL, Hirst S, Mu Y, Unal B, Davies DH, et al. Identification of humoral immune responses in protein microarrays using DNA microarray data analysis techniques. Bioinformatics (2006) 22:1760–6. doi: 10.1093/bioinformatics/btl162
56. Sboner A, Karpikov A, Chen G, Smith M, Mattoon D, Freeman-Cook L, et al. Robust-linear-model normalization to reduce technical variability in functional protein microarrays. J Proteome Res. (2009) 8:5451–64. doi: 10.1021/pr900412k
57. Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics (2002) 18(Suppl. 1):S96–104. doi: 10.1093/bioinformatics/18.suppl_1.S96
58. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics (2007) 8:118–27. doi: 10.1093/biostatistics/kxj037
59. Maskus DJ, Królik M, Bethke S, Spiegel H, Kapelski S, Seidel M, et al. Characterization of a novel inhibitory human monoclonal antibody directed against Plasmodium falciparum Apical Membrane Antigen 1. Sci Rep. (2016) 6:39462. doi: 10.1038/srep39462
60. Sim BK, Narum DL, Chattopadhyay R, Ahumada A, Haynes JD, Fuhrmann SR, et al. Delineation of stage specific expression of Plasmodium falciparum EBA−175 by biologically functional region II monoclonal antibodies. PLoS ONE (2011) 6:e18393. doi: 10.1371/journal.pone.0018393
61. Kapelski S, Boes A, Spiegel H, de Almeida M, Klockenbring T, Reimann A, et al. Fast track antibody V-gene rescue, recombinant expression in plants and characterization of a PfMSP4-specific antibody. Malar J. (2015) 14:50. doi: 10.1186/s12936-015-0577-7
62. Chang SP, Gibson HL, Lee-Ng CT, Barr PJ, Hui GS. A carboxyl-terminal fragment of Plasmodium falciparum gp195 expressed by a recombinant baculovirus induces antibodies that completely inhibit parasite growth. J Immunol. (1992) 149:548–55.
63. Douglas AD, Williams AR, Knuepfer E, Illingworth JJ, Furze JM, Crosnier C, et al. Neutralization of Plasmodium falciparum merozoites by antibodies against PfRH5. J Immunol. (2014) 192:245–58. doi: 10.4049/jimmunol.1302045
64. Wright KE, Hjerrild KA, Bartlett J, Douglas AD, Jin J, Brown RE, et al. Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature (2014) 515:427–30. doi: 10.1038/nature13715
65. Douglas AD, Williams AR, Illingworth JJ, Kamuyu G, Biswas S, Goodman AL, et al. The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nat Commun. (2011) 2:601. doi: 10.1038/ncomms1615
66. Tran TM, Ongoiba A, Coursen J, Crosnier C, Diouf A, Huang CY, et al. Naturally acquired antibodies specific for Plasmodium falciparum reticulocyte-binding protein homologue 5 inhibit parasite growth and predict protection from malaria. J Infect Dis. (2014) 209:789–98. doi: 10.1093/infdis/jit553
67. Ritz C, Streibig JC. Bioassay analysis using R. J Stat Softw. (2005) 12:30764. doi: 10.18637/jss.v012.i05
68. Jensen SM, Ritz C, Baty F, Gerhard D. Dose response analysis using R. PLoS ONE (2015) 10:e0146021. doi: 10.1371/journal.pone.0146021
69. Gray JC, Corran PH, Mangia E, Gaunt MW, Li Q, Tetteh KK, et al. Profiling the antibody immune response against blood stage malaria vaccine candidates. Clin Chem. (2007) 53:1244–53. doi: 10.1373/clinchem.2006.081695
70. Fan YT, Wang Y, Ju C, Zhang T, Xu B, Hu W, et al. Systematic analysis of natural antibody responses to P. falciparum merozoite antigens by protein arrays. J Proteomics (2013) 78:148–58. doi: 10.1016/j.jprot.2012.11.020
71. SMART. Availabe online at: https://www.smartpartnership.net/
72. Vagenende V, Yap MG, Trout BL. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry (2009) 48:11084–96. doi: 10.1021/bi900649t
73. Weaver R, Reiling L, Feng G, Drew DR, Mueller I, Siba PM, et al. The association between naturally acquired IgG subclass specific antibodies to the PfRH5 invasion complex and protection from Plasmodium falciparum malaria. Sci Rep. (2016) 6:33094. doi: 10.1038/srep33094
Keywords: Plasmodium falciparum, merozoite, antibodies, vaccine candidates, protein microarray, bioinformatics
Citation: Kamuyu G, Tuju J, Kimathi R, Mwai K, Mburu J, Kibinge N, Chong Kwan M, Hawkings S, Yaa R, Chepsat E, Njunge JM, Chege T, Guleid F, Rosenkranz M, Kariuki CK, Frank R, Kinyanjui SM, Murungi LM, Bejon P, Färnert A, Tetteh KKA, Beeson JG, Conway DJ, Marsh K, Rayner JC and Osier FHA (2018) KILchip v1.0: A Novel Plasmodium falciparum Merozoite Protein Microarray to Facilitate Malaria Vaccine Candidate Prioritization. Front. Immunol. 9:2866. doi: 10.3389/fimmu.2018.02866
Received: 26 June 2018; Accepted: 21 November 2018;
Published: 11 December 2018.
Edited by:
Ashraful Haque, QIMR Berghofer Medical Research Institute, AustraliaReviewed by:
Giampietro Corradin, Université de Lausanne, SwitzerlandTakafumi Tsuboi, Ehime University, Japan
Copyright © 2018 Kamuyu, Tuju, Kimathi, Mwai, Mburu, Kibinge, Chong Kwan, Hawkings, Yaa, Chepsat, Njunge, Chege, Guleid, Rosenkranz, Kariuki, Frank, Kinyanjui, Murungi, Bejon, Färnert, Tetteh, Beeson, Conway, Marsh, Rayner and Osier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Faith H. A. Osier, Rk9zaWVyQGtlbXJpLXdlbGxjb21lLm9yZw==;ZmFpdGgub3NpZXJAbWVkLnVuaS1oZWlkZWxiZXJnLmRl
†These authors have contributed equally to this work