 Sarah Nersesian
Sarah Nersesian Haley Glazebrook
Haley Glazebrook Jay Toulany
Jay Toulany Stephanie R. Grantham
Stephanie R. Grantham Jeanette E. Boudreau
Jeanette E. Boudreau- 1Department of Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
- 2Department of Medicine, Dalhousie University, Halifax, NS, Canada
- 3Department of Pathology, Dalhousie University, Halifax, NS, Canada
Ovarian cancer (OC) is diagnosed in ~22,000 women in the US each year and kills 14,000 of them. Often, patients are not diagnosed until the later stages of disease, when treatment options are limited, highlighting the urgent need for new and improved therapies for precise cancer control. An individual's immune function and interaction with tumor cells can be prognostic of the response to cancer treatment. Current emerging therapies for OC include immunotherapies, which use antibodies or drive T cell-mediated cancer recognition and elimination. In OC, these have been limited by adverse side effects and tumor characteristics including inter- and intra-tumoral heterogeneity, lack of targetable antigens, loss of tumor human leukocyte antigen expression, high levels of immunosuppressive factors, and insufficient immune cell trafficking. Natural killer (NK) cells may be ideal as primary or collateral effectors to these nascent immunotherapies. NK cells exhibit multiple functions that combat immune escape and tumor relapse: they kill targets and elicit inflammation through antigen-independent pathways and detect loss of HLA as a signal for activation. NK cells are efficient mediators of tumor immune surveillance and control, suppressed by the tumor microenvironment and rescued by immune checkpoint blockade. NK cells are regulated by a variety of activating and inhibitory receptors and already known to be central effectors across an array of existing therapies. In this article, we highlight interactions between NK cells and OC and their potential to change the immunosuppressive tumor microenvironment and participate in durable immune control of OC.
Ovarian Cancer
Ovarian cancer (OC) is the leading cause of death from gynecologic malignancies with a 5-year survival of <50% (1). The majority of cases are diagnosed at advanced stages (III and IV), when treatment becomes especially challenging, earning OC its “silent killer” moniker. Beyond stage III, OC disseminates into the peritoneum, and patients present with bloating from a buildup of ascitic fluid in the abdomen (2). Current standard of care includes de-bulking surgery followed by a combination of platinum- and taxane-based chemotherapy. Despite these aggressive treatments, recurrence occurs in 60–70% of patients within 2–5 years; most will eventually succumb to OC (3).
Epithelial OC represents the majority of malignant ovarian tumors and arises from the epithelium of the ovary and fallopian tube. OC tumors are broadly divided into two subtypes: type I and II; these classifications carry both prognostic and predictive value (4). These subtypes are differentially responsive to the available treatments, and whether they may inform the development and application of more precise treatment, including immunotherapy, is the subject of active investigation. Type I OC includes low-grade serous, endometrioid, clear cell, and mucinous carcinomas that together account for ~30% of ovarian tumors (4, 5). This subset is typically genetically stable and slow-growing. By contrast, type II OC is genetically unstable, more aggressive and accounts for the majority of OC mortality (4, 5). This subtype exclusively includes high-grade serous carcinomas, represents 70% of all ovarian tumors, and will be the focus of this review (6–8).
NK Cells
NK cells are lymphocytes that reside in the peripheral blood and are highly efficient anti-tumor effectors (9–11). They comprise 5–10% of circulating lymphocytes and kill target cells without previous sensitization. NK cells differentiate from the common lymphoid progenitor in the bone marrow, are most closely related to T cells, and have similar abilities to expel perforin and granzymes for direct target cell killing (12, 13). In addition, they signal for target cell apoptosis through Fas and TRAIL pathways and secrete pro-inflammatory cytokines including IFN-γ and TNF (12). NK cells can be characterized into an array of categories based on the presence and density of surface markers expressed with some studies reporting up to 30,000 in one individual (14). At a superficial level they can be categorized into two main populations based on CD16 and CD56–expression; with CD56bright/CD16– functioning to primarily produce cytokines in the circulating blood, and CD56dim/CD16+ performing cytotoxicity in the tissues (12).
Rather than direct detection of antigens through a germline-rearranged receptor (the domain of T and B cells), NK cells recognize putative targets based on their expression of stress-induced ligands, upregulated on the cell surface consequent to DNA damage and heat shock, and in response to stimulation by environmental factors, including cytokines and chemokines (15–17). To avoid unwanted auto-aggression, NK cells are also sensitive to inhibition by “self” human leukocyte antigen (HLA) class I molecules; it is the net outcome of incoming activating and inhibitory signals that determines the NK cell response to a putative target (12). A complete summary of the receptors and cytokines produced by NK cells is beyond the scope of this review and can be found elsewhere (18–20). Here, we limit our discussion to those identified as relevant in OC, summarized in Table 1.
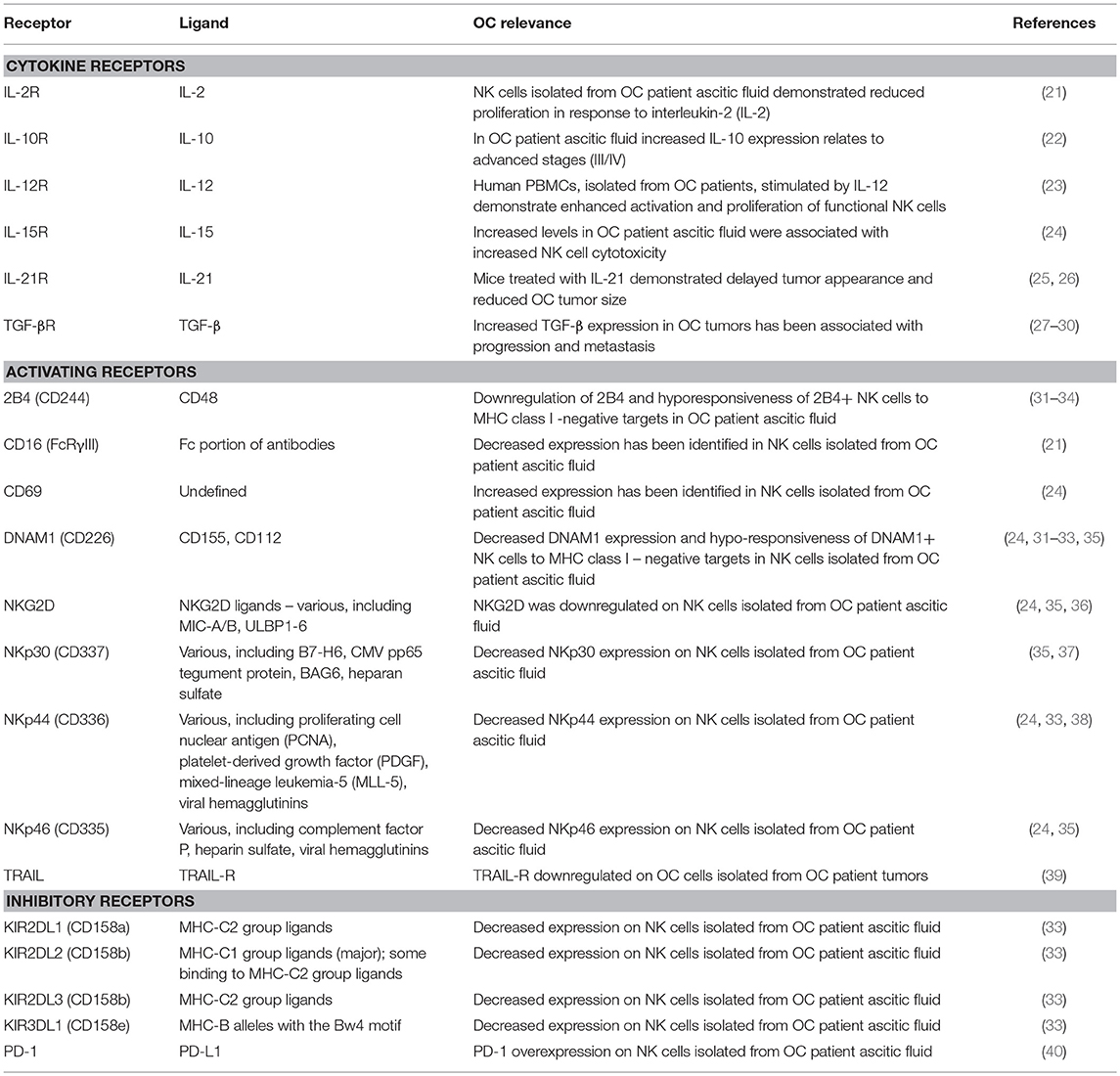
Table 1. NK cell receptors and their relevance to ovarian cancer.
NK cell responsiveness varies within and between individuals, via a process called “education,” “licensing,” or “arming.” The killer immunoglobulin-like receptors (KIR) interact with conserved epitopes on HLA (41). KIR and HLA genes are both highly polymorphic and polygenic and their loci segregate independently (42). Thus, the availability of binding pairs differs between people, with consequences on NK cell reactive potential (42). NK cells expressing KIR that can bind to “self” HLA (licensed) are highly responsive to targets lacking “self” HLA—a process termed “missing self” recognition. NK cells that do not express self-HLA specific receptors are “unlicensed” and require potent stimulation for reactivity. Unlicensed NK cells are especially protective against HLA-expressing tumors because they are refractory to the inhibitory signals sent by HLA (43, 44). Thus, education potentiates a spectrum of NK cell reactivity, establishing, at the repertoire level, an array of functions to target HLA-sufficient and HLA-deficient target cells (19, 20).
NK cells are capable of memory or adaptive functions, whereby previous sensitization leads to expansion and retention of a population of NK cells that respond more rapidly and robustly to secondary challenges with the same virus or hapten (45, 46). These “adaptive” NK cells have undergone epigenetic alterations leading to a distinct phenotype and enhanced function (47–49). While additional research is required to fully characterize this cell population, unbound inhibitory receptors, NKp44, NKp46, NKG2C, and CD57 receptors are consistently overexpressed (47–50). Noteworthy, these same adaptive features can be achieved by cytokine cocktail stimulation in the absence of a specific antigen; cytokine-induced adaptive cells exhibit increased expression of activating receptors including NKp30, NKG2D, NKp44, NKp46, and TRAIL (51). Thus, while a memory response may be generated toward a specific pathogen, it may be the environment or steric changes in the HLA-peptide complex that drive NK cell adaptive functions (45). For cancer immunotherapy, this is noteworthy as it opens the possibility to train highly effective NK cells without strict requirements for antigen restriction.
The Tumor Microenvironment and Immunosuppression in Ovarian Cancer
The tumor microenvironment (TME) includes the tumor, stroma, and local immune cells. The TME is dynamic and can promote or suppress tumor invasion and metastasis. The immune composition in the TME is now known to be an important predictor of response to therapy in a variety of solid tumor types, including OC. Several factors, processes, and cellular subsets contribute to the development of the TME including innate and adaptive immune cells, intercellular signaling, and tumor intrinsic factors such as gene mutational burden (Figure 1) (52).
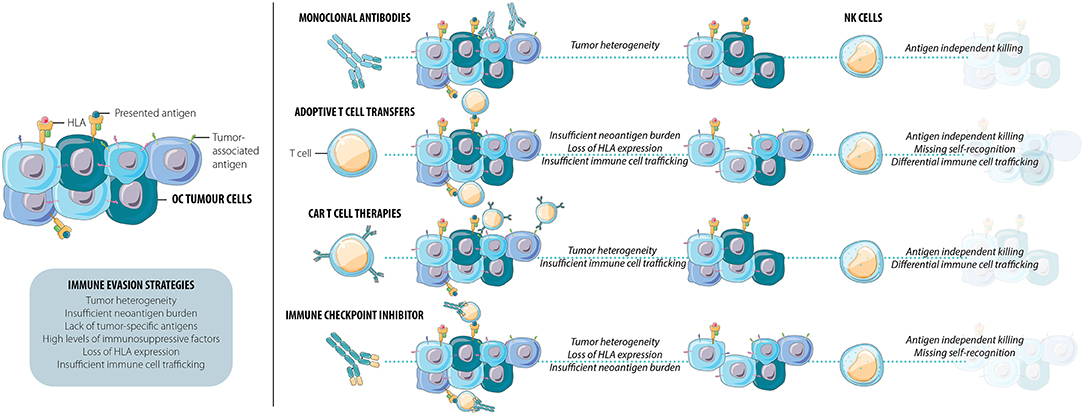
Figure 1. Current immunotherapies frequently result in the persistence of a resistant cell population. This immune evasion of OC tumor cells can be facilitated by tumor heterogeneity, insufficient neoantigen burden, lack of tumor-specific antigens, high levels of immunosuppressive factors, loss of HLA, and/or insufficient immune cell trafficking. NK cells exhibit multiple functions that combat immune escape and tumor relapse: they kill targets and elicit inflammation through both antigen-specific and antigen-independent pathways and detect loss of HLA as a signal for activation. As efficient mediators of tumor immune surveillance and control, NK cells may be able to kill the cells many current immunotherapies leave behind.
Good prognosis for OC and its treatment are correlated with immune cell infiltration in the TME, and the majority of studies focused on infiltrating T cells (53) (Figure 2). These so-called “hot” tumors represent two scenarios: (1) immune inflamed tumors, where T cells can directly contact malignant cells; and (2) immune excluded tumors, where T cells are restricted to the stroma surrounding malignant cells, with limited access for ligation and killing. In contrast, “cold” or “non-inflamed” tumors lack T cell infiltration. Reproducibly, patients whose OC tumors are infiltrated by T cells respond better to therapy and have better prognosis (54, 55): 55% of patients with T cell infiltration reached a 5-year survival of 38% compared to just 4.5% in patients without T cell infiltration (54).
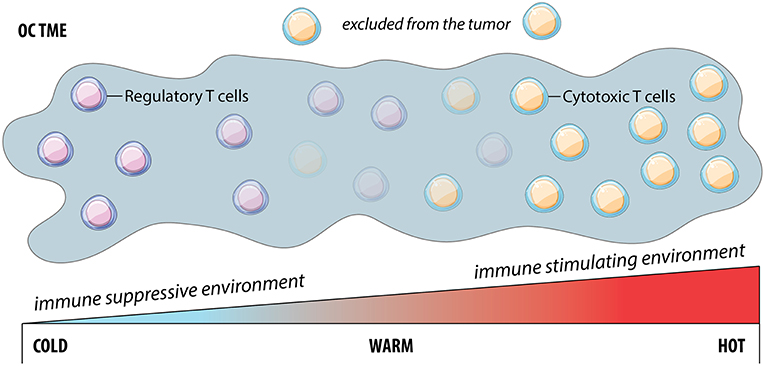
Figure 2. The cold, warm or hot tumor microenvironment is a continuum. Cold tumors are characterized by the lack of cytotoxic T cells and are typically associated with an immune suppressive environment. Conversely, hot tumors are well infiltrated by cytotoxic T cells and are associated with an immune stimulating environment and better prognosis than the prior. However, the characterization of tumors is not dichotomous, but rather exists as a sliding scale of cytotoxic T cell infiltration with “warm” tumors representing a situation where although T cells exist, they are excluded and therefore ineffective at producing an efficient anti-tumor response.
Tumor transcriptome analysis has revealed correlations of OC sub-classifications with T cell infiltration, neoantigen burden and patient prognosis, termed C1, C2, C4, and C5 (8). Among them, the C2 “immunoreactive” subtype is well-infiltrated and associated with the best prognosis (8). Intermediate T cell infiltration and prognoses are associated with the C1 “mesenchymal” and C4 “differentiated” subtypes, and the C5 “proliferative” has the lowest T cell infiltration and conveys the poorest prognosis (8). Despite the mutational burden found in many OC—a feature that can predict priming and infiltration of lymphocytes—they remain immunologically “cold,” and infiltrating T cells are not universally reactive against tumor antigens (56, 57). This highlights an urgent need to better understand how other cellular and acellular players contribute to tumor growth and treatment success.
An additional barrier to immunotherapy and effective anti-cancer reactivity is the immunosuppressive nature of the TME, which advances with OC progression (58). Dendritic cells (DCs) are required for the activation of anti-tumor T cells but also have the ability to release immunosuppressive cytokines. In OC, DCs are dysfunctional and immature due to high levels of VEGF and IL-10 in the TME (59–61). These DCs are not only unable to activate cytotoxic T cells, but also function to induce regulatory T cell (Treg) differentiation, further promoting immune suppression (62). Moreover, patient ascitic fluid contains high levels of the Treg recruiting chemokine, CCL22 (63). Unsurprisingly, Treg accumulate in the ascites isolated from late stage OC patients (64, 65). Treg release IL-6 and IL-10 which induces expression of B7-H4 on macrophages and subsequently leads to cytotoxic T cell cycle arrest (65). Discussed in the following section, these impacts extend beyond suppression of T cell immune responses and likely also interfere with productive, anti-tumor NK cell function (Figure 3).
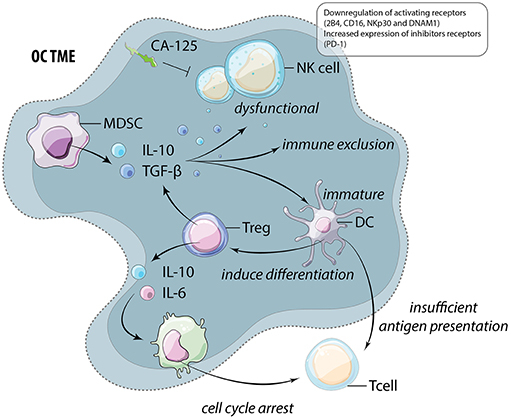
Figure 3. The immunosuppressive TME of OC. Myeloid derived suppressor cells (MDSC) and regulatory T cells (Treg) release high levels of immunosuppressive cytokines including IL-10 and TGF-β. These cytokines function to alter and suppress the function of multiple cells within the TME including natural killer (NK) cells, dendritic cells (DC), and macrophages (M). These cells frequently feedback onto lymphocytes by inducing further Treg differentiation and preventing anti-tumor cytotoxic T cell function through insufficient antigen presentation and the induction of cell cycle arrest.
NK Cells in the OC Tumor Microenvironment
The important contributions of NK cells to cancer control were identified through mouse models deficient in NK cells or key NK cell activating receptors (66, 67). The protective effect of NK cells was further supported by studies in humans that correlated poor NK cell function to cancer susceptibility, progression and metastases in a variety of both solid and hematologic cancers (68, 69). In OC, the prognostic value of infiltrating NK cells has been debated—NK cells and the NK cell-like population, innate-like lymphocytes (ILCs), have been associated with both tumor progression and control (70–72). NK cells co-infiltrate with cytotoxic T cells and are strongly associated with patient survival (71, 73). One study stratified OC patients into three subgroups based on infiltration of T cells and other lymphocytes, primarily NK cells, and reported differential 5-year survival rates: T + NK cells (90%), NK cells only (63%) and neither T nor NK cell infiltration (0%) (70). Even when they are present in OC, NK cells are dysfunctional: they exhibit reduced proliferation, decreased cytolytic function and decreased inflammatory cytokine production compared to the same patient's peripheral blood NK cells (74, 75) (Summarized in Table 1). Hence, a better understanding of how to enumerate, assess and control NK cells will be required to maximize their infiltration, function and reactive potential in the TME.
Alterations in NK cell phenotype and function occur as a result of the products of a growing ovarian tumor, related ascites, and a variety of immunosuppressive cytokines produced by myeloid derived suppressor cells (MDSC) and Treg (76). For example, macrophage migration inhibitory factor (MIF) overexpression has been reported in OC and correlates with tumor progression (77). MIF downregulates transcription of the NK activating receptor NKG2D (37, 77, 78). MIF is also associated with increased expression of the inhibitory checkpoint, B7-H6, which is associated with overall poorer prognosis in OC (34, 79). Similarly, TGF-β overexpression can suppress CD16-triggered NK cell IFN-γ production (80) and, together with IL-10, has been shown to decrease the inflammatory cytokine production and cytotoxicity of various effector cells including NK cells (81–84). These alterations include a downregulation of activating receptors 2B4, CD16, NKp30, DNAM1, and an upregulation of the inhibitory checkpoint receptor PD-1 (21, 31, 32, 37, 40). Finally, despite upregulation of the early activation marker, CD69, NK cells expressing it remain poorly cytolytic (31). In addition to cytokine mediated suppression, CA-125, an antigen overexpressed in 80% of OC, can directly protect tumor cells from NK cell-mediated cytotoxicity by preventing the formation of an immune synapse, regardless of the repertoire of receptors expressed on the NK cells (79, 85).
Taken together, the available research indicates that NK cells interact dynamically with the OC TME and are highly sensitive to the immunoregulation it drives. Infiltration of NK cells into a tumor may only partially predict outcomes of OC. Better understanding the mechanisms through which the TME contributes to immune exclusion and dysregulation will be required for precise tumor control and to maximize NK cell reactivity.
OC Immunotherapy: A Focus on the Roles and Potential of NK Cells
Successful immunotherapy requires restoration of immunity in the immunosuppressive TME and complete targeting of a heterogeneous tumor. Anti-tumor immunity can be driven by ex vivo re-stimulation of lymphocytes, engineering cells for direct targeting of specific tumor-associated antigens or turning off immune suppression (86–90). Antibody-based therapies can redirect immune cells by blocking their function, or for antibody-dependent cell-mediated cytotoxicity (ADCC), a process for which NK cells are major effectors. While the majority of immunotherapeutic approaches have been developed with a goal of supporting or reinvigorating antigen-specific anticancer activity, they can also support the function of NK cells, whose functional features can complement and extend the breadth of OC immunotherapy (Figure 1). In the following sections, we highlight current approaches to cancer immunotherapy, their potential interactions with NK cells and the opportunities to maximize anti-tumor immunity by recruiting NK cells.
Cytokine-Based Immunomodulation
Recognizing that immunosuppression is a major hindrance for lymphocytes to proceed in anti-cancer activity, approaches with cytokines to induce local and/or systemic inflammation have been tested. A strategy to elicit and improve immune cell activation in humans was first attempted using a variety of activating cytokines including IL-2, IL-12, IL-15, IFN-α, and IFN-γ (91).
IL-2 was one of the earliest cytokines tested for improving anti-tumor immunity. Although early clinical trials were limited by toxicity and activation of Treg, they provided an important proof of concept that stimulating T and NK cells can impact tumor progression. Since then, research has focused on strategies to improve IL-2 safety including low-dose IL-2. In patients with platinum-sensitive advanced OC, low-dose IL-2 in combination with 13-cis-retinoic acid improved clinical outcomes and increased lymphocyte and NK cell counts (92). As low-dose IL-2 can activate Treg, current efforts are testing constructs that selectively bind to NK cells to support anti-tumor immunity without driving Treg proliferation (93, 94).
Similar disappointing and toxicity-related issues were reported in many trials of activating cytokines. Research resulting in the development of analogs and oncolytic strategies for local delivery may provide the required specificity to bring cytokines safely into clinical use. IL-15 is similar to IL-2 but more specific in that it binds cytotoxic T cells and non-terminally differentiated NK cells to enhance cell cytotoxicity and proliferation. Further, the toxicity of IL-15 is less than that of IL-2, but the concentrations of IL-15 required to drive efficient anti-tumor function remain toxic. Ongoing efforts involve IL-15 “superagonists,” which deliver the IL-15 signal in complex with the IL-15 receptor alpha subunit or its biologically-relevant fragments, and/or fused in dimers with an IgG1Fc molecule to stabilize the complex. In each instance, these superagonists more closely replicate the biologically-potent delivery of IL-15, exhibit longer in vivo half-lives, and drive lymphocytes (including NK cells) for anti-cancer activity without marked toxicity (95).
ALT-803 is an IL-15 superagonist that potently enhances NK functionality in vitro and in vivo against OC cell lines (96). After ALT-803 treatment, NK cells isolated from OC patient ascitic fluid exhibited greater degranulation (CD107a) and IFN-γ production (24). Several clinical trials are ongoing evaluating the efficacy of ALT-803 and other IL-15-based therapies, alone and in combination with other immunotherapies including three for patients with OC: NCT03054909, NCT03197584, and NCT03127098 (97). It is expected that the addition of IL-15 and its related superagonists will support NK cell proliferation and development. Metrics to understand NK cell recruitment to the OC TME, persistence and NK cell reactivity (i.e., with ex vivo restimulation) will enlighten subsequent clinical trials by indicating how NK cell reactivity can be improved further. Furthermore, studies to understand whether the cytokine milieu varies with defined OC subtypes might help to predict how NK cells will be recruited and effective in patients with OC.
Checkpoint Blockage and Antigen Insufficiency
A high mutational burden creates a challenge for antigen-targeted immunotherapies, but it creates an opportunity for immune-mediated OC recognition. Tumors with high mutational burdens may have increased neoantigen levels, against which antigen-specific T cells may be activated. Many studies have predicted that resistant tumors may lack the neoantigen burden required to mount an effective T cell response (98). One recent investigation profiling tumor and T-cells isolated from the ascites of three OC patients identified that while a high mutational burden was indeed present, only 1.3% of these mutations were recognized by tumor-associated T cells (99). As expected, the presence of neoantigen-reactive T cells (rather than T cells with non-specific reactivity) is predictive of improved prognosis in OC (99, 100). This highlights the importance of the immunogenicity of antigens and an adequate repertoire of tumor-reactive T cells, rather than the overall tumor mutational burden in predicting OC outcomes (101). However, the immune-suppressive TME can still interfere with T cell-mediated tumor recognition.
High mutational burdens have been associated with improved responses to immune checkpoint therapies in both melanoma and non-small cell lung cancers (102, 103). Unexpectedly, despite a significant mutational load being present in pre-treatment surgical samples of OC patients, overall response rates to anti-PD1 treatment in clinical trials have been largely disappointing, ranging from 11–24% (104, 105). These clinical trials were mainly conducted in OC patients that progressed despite conventional treatments—these tumors likely had established immune evasion strategies; earlier intervention with immunotherapy may have achieved better outcomes. Regardless, these clinical trials highlight that a subset of OC patients can respond to anti-PD1 treatment, but the majority have innate or acquired resistance or lack T cells with appropriate anti-tumor reactivity (106).
Various changes in the TME have been identified that may be associated with anti-PD1 resistance including genomic mutations and downregulation of HLA and its associated processing and presentation pathway components (106, 107). These alterations have been reported in both melanoma and lung tumors treated with anti-PD1 (106, 107). Specifically, the acquired genomic mutations provided protection against T-cell mediated killing via loss of IFN-γ or HLA class I components (108, 109). It is not yet known whether anti-PD1 therapy drives similar genetic alterations in OC, but in recurrent OC the expression of HLA genes was negatively correlated with expression of PD-L1, suggesting two mutually exclusive pathways to immune evasion (110). This loss of HLA class I expression, together with insufficient T cell-mediated recognition of tumor antigens, may contribute to the insufficiency of anti-PD1 for complete OC clearance. Strategies to augment NK cell function, particularly those NK cell populations that recognize cells with DNA damage and loss of components of the HLA processing and presentation pathway (i.e., the “licensed” NK cell population), may improve OC treatment and the outcome of anti-PD1 therapy, and complement existing strategies aimed at maximizing T cell-mediated OC control. Likewise, phenotyping of tumor-infiltrating NK cells or the tumors themselves for expression of checkpoints and HLA expression may assist in predicting how NK cells may be functional or inhibited against tumor killing.
Checkpoint inhibitors interfere with inhibitory signaling that prevents anti-tumor reactivity; their application enables lymphocytes to proceed in anticancer cytotoxicity. Although anti-PD1 therapies were designed to rescue T cells from immunosuppression, PD-1 has been found to be expressed on NK cells isolated from OC and other tumor types (40, 111, 112). PD-1 expression is not universal on NK cells however, with studies reporting highly variable PD-1 expression peripheral NK cells in healthy donors, from 0 to 50% (40, 113). In patients with OC however, PD-1 expression on peripheral blood NK cells is increased suggesting that the presence of a tumor could induce its expression (40).
Despite inconsistent expression on NK cells, anti-PD1 therapies have demonstrated the potential to simultaneously support T and NK cell responses in the TME, suggesting that PD-1 blockade could indirectly influence NK cell function, or that PD-1 expression could be dynamic on NK cells in response to the TME. A recent study investigated the therapeutic effect of anti-PD1 therapy on NK cells using several mouse cancer models and concluded that NK cells were crucial to anti-tumor responses (114). Additionally, OC xenograft studies have demonstrated that both NK cell persistence and cytotoxicity can be improved with PD-L1 blockade (115). Hence, NK cells may be contributing to the outcomes of checkpoint inhibition therapy, even if they have not been expressly studied for this purpose. Recognizing this potentially important feature, clinical trials have also begun to investigate the impact of anti-PD-1 treatment on preventing or reversing NK cell exhaustion in the TME (NCT03241927), but this is not yet standard in clinical trials. Given that NK cells may contribute to the anti-tumor immunity driven by checkpoint inhibition therapies, metrics to assess their prevalence and function may help to illuminate the larger picture of anti-OC immunity.
Some studies have identified an immunoregulatory population of NK cells that exhibit the PD-1 ligand PD-L1 in patients with cancer, but its function has not been determined (116). In antigen presenting cells cis expression of PD-L1 and PD-1 permits regulation of PD-1 signaling (117); whether this also occurs in NK cells is unknown. An alternative mechanism, demonstrated in a mouse model, involves PD-L1-mediated editing of dendritic cells, limiting the extent of their interactions with T cells and development of productive anti-tumor T cell response (118). With this in mind, expression of PD-L1 (and its control by anti-PDL1 antibodies) may have significant impacts on direct NK cell function and its interactions with neighboring cells; this warrants further investigation.
In addition to PD-1, other immune checkpoints, including KIR, NKG2A, and TIGIT are being explored as targets for immunotherapy (119, 120). Like PD-1, TIGIT is expressed on both T and NK cells and suppresses anti-tumor effector function in both (120). While its mechanism of action in T cells was recognized, it was only recently identified that they also prevent or reverse NK cell exhaustion (120). TIGIT blockade improved prognosis in murine T and B-deficient, NK-sufficient xenograft models against several human tumor cell lines including colon, breast, melanoma, and fibrosarcoma (120). Anti-TIGIT antibodies are now undergoing clinical trials and just one includes OC (NCT036286770). This trial does not plan measurements of NK cell phenotype or function; future studies should include this as an important outcome measure.
Like TIGIT and PD-1, NKG2A, and the inhibitory subset of KIRs prevent excessive inflammation but may interfere with productive anti-cancer activity. For NK cells, NKG2A and KIR convey an important signal of “self” upon binding with HLA class I. The importance of preventing NK cell inhibition is clear in patients undergoing hematopoietic cell transplantation for acute myelogenous leukemia, where “uninhibitable” populations of NK cells predict for less relapse and greater overall survival (19, 121–123). These observations inspired the creation of antibodies against KIR and NKG2A, which are now being tested against other hematologic and solid malignancies, but not yet in OC.
HLA-E, the ligand for NKG2A, is a non-classical and ubiquitous HLA molecule that has been found at high expression levels in OC tumors (124). As HLA-E overexpression is negatively correlated with survival and exhibited in the majority of tumor types, blocking this NKG2A/HLA-E inhibition could enhance immunotherapy across an array of tumors, including OC (125, 126). In tumors exhibiting high-density HLA-E expression, infiltrating CD8+T and NK cells exhibit high NKG2A and PD-1, suggesting adoption of a phenotype highly sensitive to inhibition in the TME (126). Although OC-infiltrating NK cells' NKG2A expression has not been expressly measured, the abundance of HLA-E on OC tumors would suggest that preventing inhibition of NK cells via NKG2A may enable strong anti-tumor reactivity.
Experimentally, NKG2A surface expression has been eliminated on NK cells by blocking its export from the endoplasmic reticulum using a protein expression blocker construct (126). Primary human NK cells engineered in this way lacked NKG2A surface expression and more efficiently controlled growth of human tumor xenografts in mice (126). Toward a similar goal, a monoclonal antibody, anti-NKG2A (monalizumab) has been delivered for direct delivery to patients. In addition to boosting NK cell cytotoxicity against targets expressing HLA-E, anti-NKG2A has also been demonstrated to augment the function of T cells expressing NKG2A, providing an opportunity to activate both NK and T cells; both were shown to contribute to control of tumor xenografts in mice (127). Inclusion of NKG2A and HLA-E measurements in OC tumors from patients could help to ascertain whether a patient might benefit from anti-NKG2A therapy.
The anti-KIR antibody IPH2101 (lirilumab) aims to interfere with inhibition via the KIR2DL1/2/3 receptors. In vitro, lirilumab functions to enhance killing of tumor cells by NK cells (128). Unfortunately, efficacy for this monoclonal antibody has been poor in clinical trials for patients with hematologic malignancies (129), a finding that corresponds with decreased surface density of KIR molecules and “detuning” or diminishment of missing self-responsiveness (130). IPH2101 has not yet been tested against OC, but the available information suggests that further improvements, such as interrupting inhibitory signaling without altering receptor expression, preventing the loss of cell surface KIR through the trogocytosis prompted by IPH2101, or stratifying patients based on particular KIR haplotypes or KIR allotypes likely to be sensitive to lirilumab, will be necessary to gain efficacy against OC.
By combining checkpoint inhibitors, it may be possible to augment the NK cell anti-tumor response—by relieving two or more inhibitory signals, or by rebalancing immunity toward activation by blocking inhibition while triggering activation. These approaches could lower the threshold for NK cell activation and provide a failsafe to target a tumor that evolves away from dependence on PD-L1 and/or HLA expression. Unlike T cells, where TIGIT and PD-1 are often co-expressed, TIGIT and PD-1 expression was nearly mutually exclusive on NK cells, suggesting that checkpoint blockade for both molecules simultaneously may permit rescue of a larger population of NK cells from inhibition and exhaustion (120). Simultaneous blockade of NKG2A and triggering of ADCC using the anti-EGFR antibody cetuximab enhanced in vivo control of tumor xenografts more efficiently than either antibody alone (127). In a phase II trial (NCT02643550), monalizumab was given to patients with squamous cell carcinoma of the head and neck after chemotherapy and alongside standard-of-care treatment with cetuximab (127). This combination was deemed safe, with interim results indicating an improvement from the addition of monalizumab compared with historical control subjects treated with cetuxumab only, but further studies will be required to formally draw this conclusion.
Clinical trials are now testing combinations of anti-PD1 with novel checkpoint inhibitor therapies including anti-KIR2D antibody (lirilumab) (NCT01714739), anti-NKG2A (monalizumab) (NCT02671435, NCT03822351, NCT02557516, NCT03794544, NCT03833440), and anti-TIGIT (MTIG7192A) (NCT02794571, NCT03119428, NCT03563716, NCT03628677) (127). Theoretically, further strategies to appraise the tumor's expression of PD-L1 (which is the current standard of care for checkpoint therapies in other cancers), HLA and TIGIT ligands may inform the rational combination of checkpoint inhibitors to maximize NK cell function.
Adoptive and Adaptive NK Cell Therapies
Recognizing the potential for NK cells to participate in immune-mediated cancer control, a thrust in NK cell-based cancer therapies is adoptive transfer of NK cells (131, 132). Since NK cells do not require HLA matching to a specific patient, it is feasible and safe to transfer cells across allogeneic barriers. This opens the possibility of transferring NK cell lines (i.e., NK-92) or ex vivo-expanded NK cells from third-party donors (25). Efforts are underway to create cell lines—including those based on NK-92 cells—which may enable direct targeting of OC based on defined criteria (133, 134). In addition, clinical protocols are in place for virtually unlimited expansion of primary NK cells for adoptive transfer. Together, these efforts open the possibility of off-the-shelf NK immunotherapy. A complete summary of all clinical trials employing NK and NK-related cells for treatment of OC is shown in Table 2.
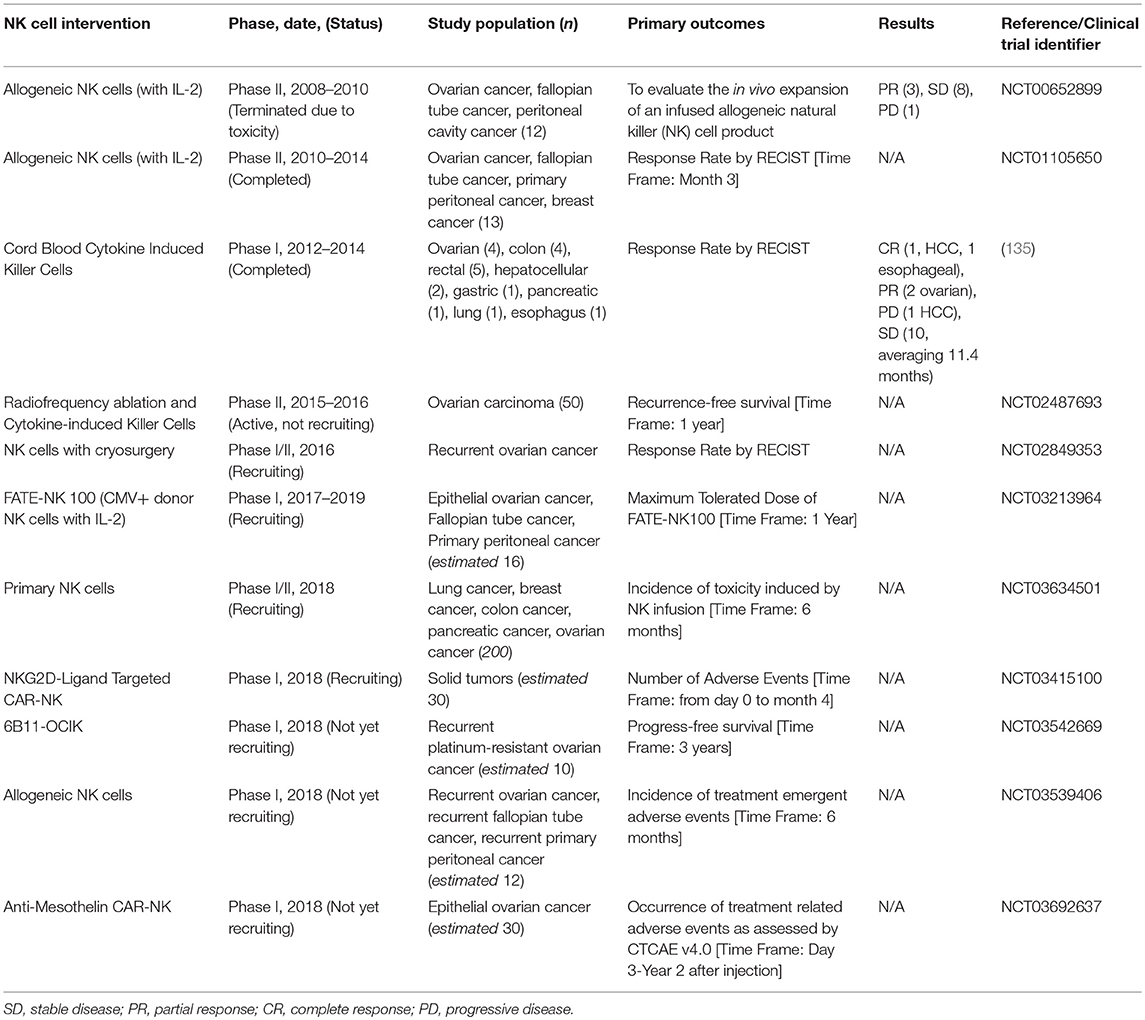
Table 2. Current NK cell-based adoptive cell immunotherapies under clinical trial for the treatment of ovarian cancer.
Early attempts at inducing NK cells for anti-cancer function included priming of lymphokine activated killer (LAK) and cytokine induced killer (CIK) cells. LAK and CIK cells originate from naïve lymphocytes which are “activated” or “induced” by IL-2 alone (LAK) or following IFN-γ stimulation ex vivo (CIK) (136, 137). In clinical trials, LAK cells exhibited limited clinical response and high rates of peritoneal fibrosis (138–140). The addition of IFN-γ stimulation to LAK cells to create CIK cells substantially improved both proliferation and cytotoxicity. CIK cells are characterized by the expression of a CD3+CD56+ cell phenotype and functional properties of NK cells (141). CIK cells were used in a recent phase III study of adoptive transfer following primary debulking and carboplatin/paclitaxel chemotherapy of OC (141, 142). Results from this clinical trial were positive with progression free survival improving from 22.2 months in the control group to 37.7 months among CIK-treated patients (142).
A completed early phase II study used allogeneic, haploidentical donor NK cells in combination with high-intensity chemotherapy to treat patients with recurrent OC (143). NK cell effects were difficult to differentiate from those of the chemotherapy and limited efficacy was attributed by the authors to a significantly increased number of Treg. Overcoming immunosuppression, including that driven by Treg, will indeed be important to ensuring the efficacy of NK cell-based immunotherapy. One strategy is to combine haploidentical donor NK cells with cytokines, with the goal of reversing the suppressive immune TME, enabling NK cell anticancer reactivity to proceed. Supporting this, the authors of the aforementioned phase II study have recently reported the ability to stimulate NK cells to overcome the soluble immunosuppressive environment from OC patient ascites in vitro with a combination of stimulatory IL-12, IL-15, and IL-18 (144). Indeed, this cytokine cocktail has been known to induce an “adaptive” population of highly-functional NK cells (145).
“Adaptive” NK cell population, first identified following cytomegalovirus infection but now known to be long-lived effector cells with potent cytotoxic ability (47). A recent study converted NK cells from a patient with OC to a cytotoxic CD56superbrightCD16+ subset that upon autologous transfer efficiently controlled growth of an autologous OC xenograft in a mouse (146). These adaptive NK cells are refractory to inhibition by Treg, implying a mechanism for their enhanced function (147). In a current Phase I clinical trial, FATE-NK100 cells, which are primary NK cells isolated from haploidentical cytomegalovirus-seropositive donors, are being transferred to patients with OC (NCT03213964).
Identifying the ideal source of NK cells for adoptive immunotherapy is a field of active study. Compared with mature and in vitro differentiated NK cells, an alternate approach is to transfer NK cells derived from umbilical cord blood (UCB) stem cells (NCT03539406). UCB contains a high proportion of immunologically-naïve NK cells that can be easily recovered and exhibit functions similar to peripheral blood NK cells. They produce similar amounts of IFN-γ and TNF (148–150). Research has also highlighted some potential weaknesses of UCB-derived NK cells: the relative immaturity of this NK cell population is associated with lower cytotoxicity, lower expression of perforin, granzyme B and KIR, and higher expression of NKG2A (150, 151). The processes for NK cell differentiation, the relative immaturity of NK cells from UCB and how they can be further differentiated or potentiated using cytokine cocktails or stimulation remain to be studied.
While selection of where to source NK cells from continues to elicit debate, once transferred into a patient, persistence, expansion, and homing/trafficking of the NK cells has proven an additional challenge. Unsurprisingly these factors have been identified as key to anti-tumor efficacy (143, 152, 153). Several factors including modifying cryopreservation and cytokine stimulation techniques can have profound impacts on homing, persistence and expansion of NK cells in in vivo (154). Indeed, a variety of strategies are being tested to improve NK cell potency in vivo. Interestingly, a recent study describes the use of a NK-cell-recruiting protein conjugated antibody that is cleaved into CXCL16 upon interaction with the tumor surface. This created a chemokine gradient resulting in increased NK cell tumor infiltration in a mouse model of pancreatic cancer (155). Similar strategies could be employed for other solid tumors including OC. Likewise, inclusion of extensive immune phenotyping, including that which would identify the type(s) and relative differentiation of NK cells invading tumors in patients, with stratifications based on outcomes and tumor subtypes, might better inform precise application of adoptive NK cell therapies.
Antigen Targeting With Antibodies and CAR-NK Cells
T cells engineered to express a chimeric antigen receptor (CAR)-T cells targeting CD19, a tumor antigen present on B-cell malignancies, provided the groundwork, and rationale for development of T cells engineered to target a specific antigen. Likewise, antibodies against CD20 and the HER2 receptor on lymphoma and breast cancer, respectively, provided the first proof-of-principle that tumors can be tagged for recognition and elimination by the immune system. Noteworthy, many of these antibody therapies rely on NK cells to mediate ADCC, and their efficacy has been inversely correlated to the extent of inhibitory KIR expression on NK cells (43, 156, 157). The list of targetable antigens in OC has been growing, and currently includes CA-125, FOLR1, EPCAM, MUC-1, and NY-ESO-1 (158, 159). Unfortunately, immunotherapies targeting these specific antigens have been largely ineffective. Currently, two CAR-T cell therapies have been approved by the FDA (160); neither is applicable to OC. For patients with OC, CAR-T cells against CA-125 are being developed and have shown promise against human xenograft models and plans to evaluate their safety in in-human phase I clinical trials have been reported (161, 162).
There are limitations to the universal use of CAR-T cells, which may be directly addressed by NK cells: CAR-T cells take weeks to produce, are difficult to generate as autologous, and expensive, making them impractical for patients requiring quick treatment for aggressive tumors or standard of care therapy (163). Moreover, allogeneic CAR-T cells pose the risk of graft-vs.-host disease (GvHD), even when HLA-matched, due to minor mismatches (164).
Since NK cells can be delivered from allogeneic sources, they are readily available and relatively more cost-effective. Importantly, NK cells are not associated with GvHD, and therefore CAR-NK cells may be a safer alternative to CAR-T cells for engineered cell therapy. Preliminary data supports the safety and efficacy of CAR-NK cell therapy, which may be attributed to the relatively short lifespan of NK cells, which lowers the risk of long-term autoimmunity and toxicity (165). Moreover, the pre-existing tolerance conveyed by germline-encoded NK cell inhibitory receptors (i.e., KIR, NKG2A) may restrict their reactivity to the tumor and damaged cells, making them less likely to convey off-target and toxic effects than CAR-T cells. Two CAR-NK cells currently under development include CARs against CD24 and mesothelin.
CD24, a cancer stem cell marker, is rarely expressed in non-hematologic healthy tissue, and associated with poor clinical outcome in OC patients (166, 167). Recently published work tested a genetically engineered CAR-NK92 against CD24, and demonstrated its to ability kill OC cells in vitro, in addition to producing high levels of IFN-γ upon co-culture with CD24 expressing OC cell lines (168). Researchers indicate that future in vivo experiments will be conducted to further evaluate efficacy.
One of the most developed CAR-NK cells is engineered against mesothelin, the receptor for CA-125. CA-125 is overexpressed in ~80% of patients and elicits an effective T cell response in vitro (169). A recent study evaluating various CAR constructs against mesothelin demonstrated their cytotoxic potential in vitro and further proved to be less toxic than their CAR-T cell counterparts in vivo while retaining similar anti-tumor effects (170). Based on early success in vivo, there are plans to evaluate mesothelin targeting CAR-NK cells in a phase I clinical trial (NCT03692637).
Despite its high expression on OC tumors, antibodies against CA-125 have not proven efficacious in OC patients (171), suggesting that exclusively targeting CA-125 is insufficient to target the heterogeneity associated with primary OC. “Antigen escape” in which the target antigen is lost through downregulation, acquired gene mutation or outgrowth of tumor subpopulations, occurs as a result of the selective pressure applied by antigen-targeting therapies like CAR-T cells and antibodies. This likely underlies the incomplete tumor control by existing antigen-specific immunotherapies and is facilitated by the tumor heterogeneity (172, 173). Complete control of OC will likely require simultaneous targeting of more than one feature.
In addition to CA-125, several therapeutics targeting NK cells to specific OC-associated antigens to activate ADCC are emerging. These include monoclonal and bi-specific antibodies, antibody-drug conjugates alone, and in combination with CAR-engineered NK cells (165, 174). One notable study found that the bispecific monoclonal antibody anti-EpCAM x anti-CD3 simultaneously activated T and NK cells with a strong enough interaction to overcome the immunosuppressive TME found in malignant ascites (175). Antibody-drug conjugates have also been developed for OC patients (176). Of these, Mirvetuximab Soravtasine (IMGN83), an anti-folate receptor alpha antibody conjugated to a cytotoxic maytansinoid, is the most developed and has demonstrated efficacy as a single agent in vivo (177). Unfortunately, this efficacy did not translate into clinical efficacy: a phase III study in platinum resistant OC patients concluded that progression free survival was not superior to standard chemotherapy (178, 179). Limited efficacy in antibody-based therapies, including IMGN83, may be due to one or several strategies including acquired resistance and/or an insufficient immune response.
NK cells from cancer patients often exhibit downregulation of the Fc receptor CD16 required for ADCC, resulting in reduced efficacy of antibody-based approaches (180, 181). To overcome this challenge and augment NK cell-mediated ADCC, researchers are inhibiting the metalloproteinases that cleave CD16 (182), or specifically engineering NK cells or cell lines for permanent expression of CD16 to facilitate ADCC (183).
The combination of antigen-agnostic reactivity (i.e., through germline-encoded activating receptors) together with the abilty to recruit NK cells for responsiveness against specific tumor antigens (i.e., through CAR or antibodies) may provide the required heterogeneous immune response required to combat the highly heterogeneous cancer that is OC. Cancer phenotyping to understand this heterogeneity—with extensions in predicting the ideal configuration of NK immunity—would assist in developing more precise approaches to OC immunotherapy.
Future Recommendations
Immunotherapies that focus and predict the specific ligands and ligand combinations for NK cells are likely to enhance OC clearance and control. NK cells can provide a multifaceted approach to meet the challenge of heterogeneity in OC tumors. That NK cells can serve both antigen-specific (i.e., ADCC, CAR) and antigen-agnostic roles (i.e., DNA damage, cell stress) (15, 184), in detecting and eliminating tumors is a strength of NK-based immunotherapies, especially against the highly-heterogeneous OC.
Enhancing or replacing NK cell function in OC is both a feasible and logical strategy that complements several existing immunotherapies. To maximize NK function and activation within the TME, further research is warranted to first identify the relevance of NK cells for the outcome of OC patients. Toward the inclusion of NK cells as key players in immunotherapy we have synthesized the following recommendations for those researching how the OC TME interacts with cancer therapies. These recommendations should be considered when developing fundamental, translational and clinical studies to contribute to the growing body of knowledge surrounding NK cell relevance in OC.
1. NK cells as outcome measures
It is crucial that NK cells be included as outcome measures in clinical trials evaluating immunotherapies. For example, anti-PD-1 therapies characterize T cell populations and neglect the evaluation of other lymphocytes. Likewise, NK cells are important mediators of ADCC; therefore, NK cell function should be considered in antibody-based approaches.
2. Further characterization of NK cells in OC
Identifying phenotypic classification and prognostic value of NK in OC may also aid in the stratification of patients for therapy. For example, identifying KIR immunogenomic status may allow identification of immunotherapeutic responders from non-responders based on immunogenetics and NK cell licensing. Inspiration for this can be drawn from investigations in other cancers that have shown important contributions of NK cell education and inhibition in contributing to therapeutic outcomes (19).
3. Measure leukocyte reactivity and compositions in the TME
Efficient tumor control by immune mechanisms involves the collaboration of innate and adaptive lymphocytes. Thus, strategies to better understand immune function in the tumor will identify shortcomings in the current approaches and keys to next-generation immunotherapies.
Conclusions
Limited treatment options, no effective screening strategies, high recurrence, and poor overall survival emphasize the need for improving therapeutic strategies to combat OC. It is clear that current treatments are unable to control OC progression. As with several other hard-to-treat tumors, researchers and oncologists are turning to immunotherapy to treat OC. Immune cell infiltration carries both predictive and prognostic value in OC; however, the complex relationship between the immune system and tumor remains a topic of active study. This investigation has revealed an extremely immunosuppressive TME in OC that results in the dysregulation of immune effector cells leading to immune evasion and tumor progression.
While immunotherapies have encountered several challenges, strategies are being developed to improve their efficacy. Cytokine treatments now focus on enhancing specificity and safety. The development of cytokine superagonists and oncolytic virus delivery strategies can theoretically provide the required specificity to bring these into clinical use. Similarly, adoptive cell transfer therapies are being enhanced to establish feasibility and efficacy. Most promising are combination immunotherapies designed to target and activate multiple immune pathways.
These advances demonstrate promise to improve immunotherapies and minimize associated toxicities; however, without strategies to efficiently identify OC responders, immunotherapies will continue to yield disappointing results. The ability to properly stratify patients relies on understanding of a patient's underlying immunity and the pre-existing immune TME. It also requires an in depth understanding into how these factors interact with and respond to chemo-, radio- and immuno-therapies. By focusing and targeting immunotherapies to individual pathways, including those which enhance functionality of just one cell type or one immune cell pathway, the therapeutic impact may be limited. The broad functions of NK cells make them amenable for immunotherapy because they can mediate tumor killing using a variety of mechanisms, including ADCC and germline-encoded receptors, with minimal toxicity. Alone, or in combination with existing strategies, NK cells hold great promise for treatment of OC.
Author Contributions
JB and SN conceptualized the review. SN, HG, JT, SG, and JB wrote and edited the review. SN, HG, and SG generated the tables.
Funding
This work was supported by a Terry Fox Research Institute New Investigator Grant to JB, the Banting Research Foundation and the Dalhousie Medical Research Foundation Cameron Cancer Chair.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Designs that cell (www.designsthatcell.ca) created the figures for publication.
References
1. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. (2018) 68:284–96. doi: 10.3322/caac.21456
2. Bast RC Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. (2009) 9:415–28. doi: 10.1038/nrc2644
3. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. (2003) 3:502–16. doi: 10.1038/nrc1123
4. Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int. (2014) 2014:934261. doi: 10.1155/2014/934261
5. Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. (2013) 123:517–25. doi: 10.1172/JCI65833
6. Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. (2012) 460:237–49. doi: 10.1007/s00428-012-1203-5
7. McCluggage WG. Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology. (2011) 43:420–32. doi: 10.1097/PAT.0b013e328348a6e7
8. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. (2011) 474:609–15. doi: 10.1038/nature10166
9. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. (1975) 16:230–9. doi: 10.1002/ijc.2910160205
10. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol. (2007) 7:329–39. doi: 10.1038/nri2073
11. Ljunggren HG, Karre K. In search of the ‘missing self': MHC molecules and NK cell recognition. Immunol Today. (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
12. Kannan GS, Aquino-Lopez A, Lee DA. Natural killer cells in malignant hematology: a primer for the non-immunologist. Blood Rev. (2017) 31:1–10. doi: 10.1016/j.blre.2016.08.007
13. Martinez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. (2015) 21:5047–56. doi: 10.1158/1078-0432.CCR-15-0685
14. Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med. (2013) 5:208ra145. doi: 10.1126/scitranslmed.3006702
15. Gasser S, Raulet D. The DNA damage response, immunity and cancer. Semin Cancer Biol. (2006) 16:344–7. doi: 10.1016/j.semcancer.2006.07.004
16. Fine JH, Chen P, Mesci A, Allan DS, Gasser S, Raulet DH, et al. Chemotherapy-induced genotoxic stress promotes sensitivity to natural killer cell cytotoxicity by enabling missing-self recognition. Cancer Res. (2010) 70:7102–13. doi: 10.1158/0008-5472.CAN-10-1316
17. Le Bert N, Lam AR, Ho SS, Shen YJ, Liu MM, Gasser S. STING-dependent cytosolic DNA sensor pathways regulate NKG2D ligand expression. Oncoimmunology. (2014) 3:e29259. doi: 10.4161/onci.29259
18. Caligiuri MA. Human natural killer cells. Blood. (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
19. Boudreau JE, Hsu KC. Natural killer cell education in human health and disease. Curr Opin Immunol. (2018) 50:102–11. doi: 10.1016/j.coi.2017.11.003
20. Boudreau JE, Hsu KC. Natural killer cell education and the response to infection and cancer therapy: stay tuned. Trends Immunol. (2018) 39:222–39. doi: 10.1016/j.it.2017.12.001
21. Lai P, Rabinowich H, Crowley-Nowick PA, Bell MC, Mantovani G, Whiteside TL. Alterations in expression and function of signal-transducing proteins in tumor-associated T and natural killer cells in patients with ovarian carcinoma. Clin Cancer Res. (1996) 2:161–73. doi: 10.1042/bst025218s
22. Lane D, Matte I, Garde-Granger P, Bessette P, Piche A. Ascites IL-10 promotes ovarian cancer cell migration. Cancer Microenviron. (2018) 11:115–24. doi: 10.1007/s12307-018-0215-3
23. Dowell AC, Oldham KA, Bhatt RI, Lee SP, Searle PF. Long-term proliferation of functional human NK cells, with conversion of CD56(dim) NK cells to a CD56 (bright) phenotype, induced by carcinoma cells co-expressing 4-1BBL and IL-12. Cancer Immunol Immunother. (2012) 61:615–28. doi: 10.1007/s00262-011-1122-3
24. Hoogstad-van Evert JS, Maas RJ, van der Meer J, Cany J, van der Steen S, Jansen JH, et al. Peritoneal NK cells are responsive to IL-15 and percentages are correlated with outcome in advanced ovarian cancer patients. Oncotarget. (2018) 9:34810–20. doi: 10.18632/oncotarget.26199
25. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE. (2012) 7:e30264. doi: 10.1371/journal.pone.0030264
26. Hu W, Wang J, Dou J, He X, Zhao F, Jiang C, et al. Augmenting therapy of ovarian cancer efficacy by secreting IL-21 human umbilical cord blood stem cells in nude mice. Cell Transplant. (2011) 20:669–80. doi: 10.3727/096368910X536509
27. Do TV, Kubba LA, Du H, Sturgis CD, Woodruff TK. Transforming growth factor-beta1, transforming growth factor-beta2, and transforming growth factor-beta3 enhance ovarian cancer metastatic potential by inducing a Smad3-dependent epithelial-to-mesenchymal transition. Mol Cancer Res. (2008) 6:695–705. doi: 10.1158/1541-7786.MCR-07-0294
28. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. doi: 10.4049/jimmunol.172.12.7335
29. Ortaldo JR, Mason AT, O'Shea JJ, Smyth MJ, Falk LA, Kennedy IC, et al. Mechanistic studies of transforming growth factor-beta inhibition of IL-2-dependent activation of CD3- large granular lymphocyte functions. Regulation of IL-2R beta (p75) signal transduction. J Immunol. (1991) 146:3791–8.
30. Malygin AM, Meri S, Timonen T. Regulation of natural killer cell activity by transforming growth factor-beta and prostaglandin E2. Scand J Immunol. (1993) 37:71–6. doi: 10.1111/j.1365-3083.1993.tb01667.x
31. Bellora F, Castriconi R, Dondero A, Pessino A, Nencioni A, Liggieri G, et al. TLR activation of tumor-associated macrophages from ovarian cancer patients triggers cytolytic activity of NK cells. Eur J Immunol. (2014) 44:1814–22. doi: 10.1002/eji.201344130
32. Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, et al. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunol. (2009) 183:4921–30. doi: 10.4049/jimmunol.0901226
33. Belisle JA, Gubbels JA, Raphael CA, Migneault M, Rancourt C, Connor JP, et al. Peritoneal natural killer cells from epithelial ovarian cancer patients show an altered phenotype and bind to the tumour marker MUC16 (CA125). Immunology. (2007) 122:418–29. doi: 10.1111/j.1365-2567.2007.02660.x
34. Tangye SG, Cherwinski H, Lanier LL, Phillips JH. 2B4-mediated activation of human natural killer cells. Mol Immunol. (2000) 37:493–501. doi: 10.1016/S0161-5890(00)00076-6
35. Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human tumour immune evasion via TGF-beta blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS ONE. (2011) 6:e22842. doi: 10.1371/journal.pone.0022842
36. Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Honig A, Hausler S, et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol. (2008) 180:7338–48. doi: 10.4049/jimmunol.180.11.7338
37. Pesce S, Tabellini G, Cantoni C, Patrizi O, Coltrini D, Rampinelli F, et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology. (2015) 4:e1001224. doi: 10.1080/2162402X.2014.1001224
38. Gaggero S, Bruschi M, Petretto A, Parodi M, Zotto GD, Lavarello C, et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. Oncoimmunology. (2018) 7:e1470730. doi: 10.1080/2162402X.2018.1470730
39. Horak P, Pils D, Haller G, Pribill I, Roessler M, Tomek S, et al. Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Mol Cancer Res. (2005) 3:335–43. doi: 10.1158/1541-7786.MCR-04-0136
40. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–46.e3. doi: 10.1016/j.jaci.2016.04.025
41. Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. (2005) 436:709–13. doi: 10.1038/nature03847
42. Kim S, Sunwoo JB, Yang L, Choi T, Song YJ, French AR, et al. HLA alleles determine differences in human natural killer cell responsiveness and potency. Proc Natl Acad Sci USA. (2008) 105:3053–8. doi: 10.1073/pnas.0712229105
43. Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest. (2012) 122:3260–70. doi: 10.1172/JCI62749
44. Boudreau JE, Giglio F, Gooley TA, Stevenson PA, Le Luduec JB, Shaffer BC, et al. KIR3DL1/ HL A-B subtypes govern acute myelogenous leukemia relapse after hematopoietic cell transplantation. J Clin Oncol. (2017) 35:2268–78. doi: 10.1200/JCO.2016.70.7059
45. Min-Oo G, Kamimura Y, Hendricks DW, Nabekura T, Lanier LL. Natural killer cells: walking three paths down memory lane. Trends Immunol. (2013) 34:251–8. doi: 10.1016/j.it.2013.02.005
46. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. (2009) 457:557–61. doi: 10.1038/nature07665
47. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. (2015) 42:443–56. doi: 10.1016/j.immuni.2015.02.008
48. Cichocki F, Miller JS, Anderson SK, Bryceson YT. Epigenetic regulation of NK cell differentiation and effector functions. Front Immunol. (2013) 4:55. doi: 10.3389/fimmu.2013.00055
49. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity. (2015) 42:431–42. doi: 10.1016/j.immuni.2015.02.013
50. Luetke-Eversloh M, Hammer Q, Durek P, Nordstrom K, Gasparoni G, Pink M, et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog. (2014) 10:e1004441. doi: 10.1371/journal.ppat.1004441
51. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
52. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
53. Galon J, Pages F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med. (2012) 10:205. doi: 10.1186/1479-5876-10-205
54. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. (2003) 348:203–13. doi: 10.1056/NEJMoa020177
55. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. (2005) 102:18538–43. doi: 10.1073/pnas.0509182102
56. Cai DL, Jin LP. Immune Cell Population in Ovarian Tumor Microenvironment. J Cancer. (2017) 8:2915–23. doi: 10.7150/jca.20314
57. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
58. Fialova A, Partlova S, Sojka L, Hromadkova H, Brtnicky T, Fucikova J, et al. Dynamics of T-cell infiltration during the course of ovarian cancer: the gradual shift from a Th17 effector cell response to a predominant infiltration by regulatory T-cells. Int J Cancer. (2013) 132:1070–9. doi: 10.1002/ijc.27759
59. Chae CS, Teran-Cabanillas E, Cubillos-Ruiz JR. Dendritic cell rehab: new strategies to unleash therapeutic immunity in ovarian cancer. Cancer Immunol Immunother. (2017) 66:969–77. doi: 10.1007/s00262-017-1958-2
60. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. (1996) 2:1096–103. doi: 10.1038/nm1096-1096
61. De Smedt T, Van Mechelen M, De Becker G, Urbain J, Leo O, Moser M. Effect of interleukin-10 on dendritic cell maturation and function. Eur J Immunol. (1997) 27:1229–35. doi: 10.1002/eji.1830270526
62. Min WP, Zhou D, Ichim TE, Strejan GH, Xia X, Yang J, et al. Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J Immunol. (2003) 170:1304–12. doi: 10.4049/jimmunol.170.3.1304
63. Idorn M, Olsen M, Halldorsdottir HR, Skadborg SK, Pedersen M, Hogdall C, et al. Improved migration of tumor ascites lymphocytes to ovarian cancer microenvironment by CXCR2 transduction. Oncoimmunology. (2018) 7:e1412029. doi: 10.1080/2162402X.2017.1412029
64. Landskron J, Helland O, Torgersen KM, Aandahl EM, Gjertsen BT, Bjorge L, et al. Activated regulatory and memory T-cells accumulate in malignant ascites from ovarian carcinoma patients. Cancer Immunol Immunother. (2015) 64:337–47. doi: 10.1007/s00262-014-1636-6
65. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. (2001) 61:4766–72.
66. Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. (2001) 13:459–63. doi: 10.1093/intimm/13.4.459
67. Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol. (2009) 182:2221–30. doi: 10.4049/jimmunol.0801878
69. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. (2000) 356:1795–9. doi: 10.1016/S0140-6736(00)03231-1
70. Bosmuller HC, Wagner P, Peper JK, Schuster H, Pham DL, Greif K, et al. Combined immunoscore of CD103 and CD3 identifies long-term survivors in high-grade serous ovarian cancer. Int J Gynecol Cancer. (2016) 26:671–9. doi: 10.1097/IGC.0000000000000672
71. Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. (2014) 20:434–44. doi: 10.1158/1078-0432.CCR-13-1877
72. Crome SQ, Nguyen LT, Lopez-Verges S, Yang SY, Martin B, Yam JY, et al. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat Med. (2017) 23:368–75. doi: 10.1038/nm.4278
73. Dong HP, Elstrand MB, Holth A, Silins I, Berner A, Trope CG, et al. NK- and B-cell infiltration correlates with worse outcome in metastatic ovarian carcinoma. Am J Clin Pathol. (2006) 125:451–8. doi: 10.1309/15B66DQMFYYM78CJ
74. Miescher S, Whiteside TL, Carrel S, von Fliedner V. Functional properties of tumor-infiltrating and blood lymphocytes in patients with solid tumors: effects of tumor cells and their supernatants on proliferative responses of lymphocytes. J Immunol. (1986) 136:1899–907.
75. Moy PM, Holmes EC, Golub SH. Depression of natural killer cytotoxic activity in lymphocytes infiltrating human pulmonary tumors. Cancer Res. (1985) 45:57–60.
76. Stojanovic A, Cerwenka A. Natural killer cells and solid tumors. J Innate Immun. (2011) 3:355–64. doi: 10.1159/000325465
77. Zhou Y, Xu Y, Chen L, Xu B, Wu C, Jiang J. B7-H6 expression correlates with cancer progression and patient's survival in human ovarian cancer. Int J Clin Exp Pathol. (2015) 8:9428–33.
78. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. (2002) 419:734–8. doi: 10.1038/nature01112
79. Patankar MS, Jing Y, Morrison JC, Belisle JA, Lattanzio FA, Deng Y, et al. Potent suppression of natural killer cell response mediated by the ovarian tumor marker CA125. Gynecol Oncol. (2005) 99:704–13. doi: 10.1016/j.ygyno.2005.07.030
80. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
81. Yigit R, Massuger LF, Figdor CG, Torensma R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol Oncol. (2010) 117:366–72. doi: 10.1016/j.ygyno.2010.01.019
82. Badger AM, Oh SK, Moolten FR. Differential effects of an immunosuppressive fraction from ascites fluid of patients with ovarian cancer on spontaneous and antibody-dependent cytotoxicity. Cancer Res. (1981) 41:1133–9.
83. Berek JS, Bast RC Jr, Lichtenstein A, Hacker NF, Spina CA, Lagasse LD, et al. Lymphocyte cytotoxicity in the peritoneal cavity and blood of patients with ovarian cancer. Obstet Gynecol. (1984) 64:708–14.
84. Heo DS, Whiteside TL, Kanbour A, Herberman RB. Lymphocytes infiltrating human ovarian tumors. I. Role of Leu-19 (NKH1)-positive recombinant IL-2-activated cultures of lymphocytes infiltrating human ovarian tumors. J Immunol. (1988) 140:4042–9.
85. Gubbels JA, Felder M, Horibata S, Belisle JA, Kapur A, Holden H, et al. MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol Cancer. (2010) 9:11. doi: 10.1186/1476-4598-9-11
86. Song DG, Ye Q, Santoro S, Fang C, Best A, Powell DJ Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Ther. (2013) 24:295–305. doi: 10.1089/hum.2012.143
87. Kandalaft LE, Powell DJ Jr, Coukos G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J Transl Med. (2012) 10:157. doi: 10.1186/1479-5876-10-157
88. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. (2015) 33:4015–22. doi: 10.1200/JCO.2015.62.3397
89. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA. (2003) 100:4712–7. doi: 10.1073/pnas.0830997100
90. Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci USA. (2008) 105:3005–10. doi: 10.1073/pnas.0712237105
91. Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers. (2011) 3:3856–93. doi: 10.3390/cancers3043856
92. Recchia F, Di Orio F, Candeloro G, Guerriero G, Piazze J, Rea S. Maintenance immunotherapy in recurrent ovarian cancer: long term follow-up of a phase II study. Gynecol Oncol. (2010) 116:202–7. doi: 10.1016/j.ygyno.2009.09.042
93. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. (2014) 20:2215–25. doi: 10.1158/1078-0432.CCR-13-3205
94. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine'. Nature. (2012) 484:529–33. doi: 10.1038/nature10975
95. Guo Y, Luan L, Patil NK, Wang J, Bohannon JK, Rabacal W, et al. IL-15 Enables septic shock by maintaining NK cell integrity and function. J Immunol. (2017) 198:1320–33. doi: 10.4049/jimmunol.1601486
96. Felices M, Chu S, Kodal B, Bendzick L, Ryan C, Lenvik AJ, et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol Oncol. (2017) 145:453–61. doi: 10.1016/j.ygyno.2017.02.028
97. Robinson TO, Schluns KS. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol Lett. (2017) 190:159–68. doi: 10.1016/j.imlet.2017.08.010
98. Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, et al. Low mutation burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines. PLoS ONE. (2016) 11:e0155189. doi: 10.1371/journal.pone.0155189
99. Wick DA, Webb JR, Nielsen JS, Martin SD, Kroeger DR, Milne K, et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin Cancer Res. (2014) 20:1125–34. doi: 10.1158/1078-0432.CCR-13-2147
100. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. (2014) 24:743–50. doi: 10.1101/gr.165985.113
101. Ilyas S, Yang JC. Landscape of tumor antigens in T cell immunotherapy. J Immunol. (2015) 195:5117–22. doi: 10.4049/jimmunol.1501657
102. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. (2015) 350:207–11. doi: 10.1126/science.aad0095
103. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
104. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. (2013) 499:214–8. doi: 10.1038/nature12213
105. Emens LA, Kok M, Ojalvo LS. Targeting the programmed cell death-1 pathway in breast and ovarian cancer. Curr Opin Obstet Gynecol. (2016) 28:142–7. doi: 10.1097/GCO.0000000000000257
106. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118:9–16. doi: 10.1038/bjc.2017.434
107. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. (2017) 7:264–76. doi: 10.1158/2159-8290.CD-16-0828
108. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
109. Smahel M. PD-1/PD-L1 blockade therapy for tumors with downregulated MHC class I expression. Int J Mol Sci. (2017) 18:E1331. doi: 10.3390/ijms18061331
110. Li Y, Sun R. Tumor immunotherapy: new aspects of natural killer cells. Chin J Cancer Res. (2018) 30:173–96. doi: 10.21147/j.issn.1000-9604.2018.02.02
111. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
112. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. (2017) 36:6143–53. doi: 10.1038/onc.2017.209
113. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. (2014) 2:410–22. doi: 10.1158/2326-6066.CIR-13-0171
114. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128:4654–68. doi: 10.1172/JCI99317
115. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. (2018) 7:e1509819. doi: 10.1080/2162402X.2018.1509819
116. Fuertes MB, Iraolagoitia XLR, Ziblat A, Nunez SY, Torres NI, Secchiari F, et al. Tumor-experienced human NK cells express PD-L1 and display immunoregulatory functions. J. Immunol. (2017) 198(Suppl. 1):56.2.
117. Zhao YL, Harrison DL, Song YR, Ji J, Huang J, Hui EF. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Reports. (2018) 24:379–90.e6. doi: 10.1016/j.celrep.2018.06.054
118. Iraolagoitia XL, Spallanzani RG, Torres NI, Araya RE, Ziblat A, Domaica CI, et al. NK cells restrain spontaneous antitumor CD8+ T cell priming through PD-1/PD-L1 interactions with dendritic cells. J Immunol. (2016) 197:953–61. doi: 10.4049/jimmunol.1502291
119. Kwon HJ, Kim N, Kim HS. Molecular checkpoints controlling natural killer cell activation and their modulation for cancer immunotherapy. Exp Mol Med. (2017) 49:e311. doi: 10.1038/emm.2017.42
120. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
121. Cooley S, Miller JS. “Self”-reflection by KIR. Blood. (2009) 114:2–3. doi: 10.1182/blood-2009-04-214312
122. Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, et al. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood. (2005) 105:4878–84. doi: 10.1182/blood-2004-12-4825
123. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. (2007) 110:433–40. doi: 10.1182/blood-2006-07-038687
124. Gooden M, Lampen M, Jordanova ES, Leffers N, Trimbos JB, van der Burg SH, et al. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8(+) T lymphocytes. Proc Natl Acad Sci USA. (2011) 108:10656–61. doi: 10.1073/pnas.1100354108
125. Cichocki F, Miller JS. Setting traps for NKG2A gives NK cell immunotherapy a fighting chance. J Clin Invest. (2019) 130:1839–41. doi: 10.1172/JCI128480
126. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. (2019) 130:2094–106. doi: 10.1172/JCI123955
127. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both t and NK cells. Cell. (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
128. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. (2014) 123:678–86. doi: 10.1182/blood-2013-08-519199
129. Benson DM Jr, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res. (2015) 21:4055–61. doi: 10.1158/1078-0432.CCR-15-0304
130. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. (2016) 22:5211–22. doi: 10.1158/1078-0432.CCR-16-1108
131. Hu LJ, Zhao XY, Yu XX, Lv M, Han TT, Han W, et al. Quantity and quality reconstitution of NKG2A(+) natural killer cells are associated with graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2019) 25:1–11. doi: 10.1016/j.bbmt.2018.08.008
132. Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol. (2017) 8:465. doi: 10.3389/fimmu.2017.00465
133. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. (1994) 8:652–8.
134. Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, et al. NK-92: an 'off-the-shelf therapeutic' for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. (2016) 65:485–92. doi: 10.1007/s00262-015-1761-x
135. Zhang Z, Wang L, Luo Z, Zhao X, Huang J, Li H, et al. Efficacy and safety of cord blood-derived cytokine-induced killer cells in treatment of patients with malignancies. Cytotherapy. (2015) 17:1130–8. doi: 10.1016/j.jcyt.2015.04.002
136. Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. (1982) 155:1823–41. doi: 10.1084/jem.155.6.1823
137. Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. (1991) 174:139–49. doi: 10.1084/jem.174.1.139
138. Steis RG, Urba WJ, VanderMolen LA, Bookman MA, Smith JW II, Clark JW, et al. Intraperitoneal lymphokine-activated killer-cell and interleukin-2 therapy for malignancies limited to the peritoneal cavity. J Clin Oncol. (1990) 8:1618–29. doi: 10.1200/JCO.1990.8.10.1618
139. Stewart JA, Belinson JL, Moore AL, Dorighi JA, Grant BW, Haugh LD, et al. Phase I trial of intraperitoneal recombinant interleukin-2/lymphokine-activated killer cells in patients with ovarian cancer. Cancer Res. (1990) 50:6302–10.
140. Kim HM, Kang JS, Lim J, Park SK, Lee K, Yoon YD, et al. Inhibition of human ovarian tumor growth by cytokine-induced killer cells. Arch Pharm Res. (2007) 30:1464–70. doi: 10.1007/BF02977372
141. Herzyk DJ, Haggerty HG. Cancer immunotherapy: factors important for the evaluation of safety in nonclinical studies. AAPS J. (2018) 20:28. doi: 10.1208/s12248-017-0184-3
142. Liu J, Li H, Cao S, Zhang X, Yu J, Qi J, et al. Maintenance therapy with autologous cytokine-induced killer cells in patients with advanced epithelial ovarian cancer after first-line treatment. J Immunother. (2014) 37:115–22. doi: 10.1097/CJI.0000000000000021
143. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. (2011) 13:98–107. doi: 10.3109/14653249.2010.515582
144. Uppendahl LD, Felices M, Bendzick L, Ryan C, Kodal B, Hinderlie P, et al. Cytokine-induced memory-like natural killer cells have enhanced function, proliferation, and in vivo expansion against ovarian cancer cells. Gynecol Oncol. (2019) 153:149–57. doi: 10.1016/j.ygyno.2019.01.006
145. Wagner JA, Berrien-Elliott MM, Rosario M, Leong JW, Jewell BA, Schappe T, et al. Cytokine-induced memory-like differentiation enhances unlicensed natural killer cell antileukemia and FcγRIIIa-triggered responses. Biol Blood Marrow Transplant. (2017) 23:398–404. doi: 10.1016/j.bbmt.2016.11.018
146. Poznanski SM, Nham T, Chew MV, Lee AJ, Hammill JA, Fan IY, et al. Expanded CD56(superbright)CD16(+) NK cells from ovarian cancer patients are cytotoxic against autologous tumor in a patient-derived xenograft murine model. Cancer Immunol Res. (2018) 6:1174–85. doi: 10.1158/2326-6066.CIR-18-0144
147. Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol Res. (2018) 6:766–75. doi: 10.1158/2326-6066.CIR-17-0498
148. Nguyen S, Kuentz M, Vernant JP, Dhedin N, Bories D, Debre P, et al. Involvement of mature donor T cells in the NK cell reconstitution after haploidentical hematopoietic stem-cell transplantation. Leukemia. (2008) 22:344–52. doi: 10.1038/sj.leu.2405041
149. Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol. (2012) 73:248–57. doi: 10.1016/j.humimm.2011.12.015
150. Wang Y, Xu H, Zheng X, Wei H, Sun R, Tian Z. High expression of NKG2A/CD94 and low expression of granzyme B are associated with reduced cord blood NK cell activity. Cell Mol Immunol. (2007) 4:377–82.
151. Cooley S, McCullar V, Wangen R, Bergemann TL, Spellman S, Weisdorf DJ, et al. KIR reconstitution is altered by T cells in the graft and correlates with clinical outcomes after unrelated donor transplantation. Blood. (2005) 106:4370–6. doi: 10.1182/blood-2005-04-1644
152. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
153. Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother. (2010) 59:1739–44. doi: 10.1007/s00262-010-0896-z
154. Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol Blood Marrow Transplant. (2014) 20:1252–7. doi: 10.1016/j.bbmt.2014.05.004
155. Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res. (2019) 7:219–29. doi: 10.1158/2326-6066.CIR-18-0317
156. Forlenza CJ, Boudreau JE, Zheng J, Le Luduec JB, Chamberlain E, Heller G, et al. KIR3DL1 allelic polymorphism and HLA-B epitopes modulate response to anti-GD2 monoclonal antibody in patients with neuroblastoma. J Clin Oncol. (2016) 34:2443–51. doi: 10.1200/JCO.2015.64.9558
157. Terszowski G, Klein C, Stern M. KIR/HLA interactions negatively affect rituximab- but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J Immunol. (2014) 192:5618–24. doi: 10.1158/1538-7445.AM2014-650
158. Kloudova K, Hromadkova H, Partlova S, Brtnicky T, Rob L, Bartunkova J, et al. Expression of tumor antigens on primary ovarian cancer cells compared to established ovarian cancer cell lines. Oncotarget. (2016) 7:46120–6. doi: 10.18632/oncotarget.10028
159. Szender JB, Papanicolau-Sengos A, Eng KH, Miliotto AJ, Lugade AA, Gnjatic S, et al. NY-ESO-1 expression predicts an aggressive phenotype of ovarian cancer. Gynecol Oncol. (2017) 145:420–5. doi: 10.1016/j.ygyno.2017.03.509
160. American Association for Cancer Research. FDA approves second CAR T-cell therapy. Cancer Discov. (2018) 8:5–6. doi: 10.1158/2159-8290.CD-NB2017-155
161. Chekmasova AA, Rao TD, Nikhamin Y, Park KJ, Levine DA, Spriggs DR, et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res. (2010) 16:3594–606. doi: 10.1158/1078-0432.CCR-10-0192
162. Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. (2015) 13:102. doi: 10.1186/s12967-015-0460-x
163. Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T cells: more than ease of access? Cells. (2018) 7:E155. doi: 10.3390/cells7100155
164. Bykova NA, Malko DB, Efimov GA. In silico analysis of the minor histocompatibility antigen landscape based on the 1000 Genomes Project. Front Immunol. (2018) 9:1819. doi: 10.3389/fimmu.2018.01819
165. Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. (2018) 9:283. doi: 10.3389/fimmu.2018.00283
166. Muinao T, Deka Boruah HP, Pal M. Diagnostic and prognostic biomarkers in ovarian cancer and the potential roles of cancer stem cells - an updated review. Exp Cell Res. (2018) 362:1–10. doi: 10.1016/j.yexcr.2017.10.018
167. Fang X, Zheng P, Tang J, Liu Y. CD24: from A to Z. Cell Mol Immunol. (2010) 7:100–3. doi: 10.1038/cmi.2009.119
168. Klapdor R, Wang S, Morgan M, Dork T, Hacker U, Hillemanns P, et al. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci. (2019) 20:E660. doi: 10.3390/ijms20030660
169. Schuster H, Peper JK, Bosmuller HC, Rohle K, Backert L, Bilich T, et al. The immunopeptidomic landscape of ovarian carcinomas. Proc Natl Acad Sci USA. (2017) 114:E9942–51. doi: 10.1073/pnas.1707658114
170. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92.e5. doi: 10.1016/j.stem.2018.06.002
171. Paijens ST, Leffers N, Daemen T, Helfrich W, Boezen HM, Cohlen BJ, et al. Antigen-specific active immunotherapy for ovarian cancer. Cochrane Database Syst Rev. (2018) 9:CD007287. doi: 10.1002/14651858.CD007287.pub4
172. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. (2018) 9:282. doi: 10.1038/s41419-018-0278-6
173. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
174. Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. (2014) 123:3016–26. doi: 10.1182/blood-2013-10-533398
175. Fossati M, Buzzonetti A, Monego G, Catzola V, Scambia G, Fattorossi A, et al. Immunological changes in the ascites of cancer patients after intraperitoneal administration of the bispecific antibody catumaxomab (anti-EpCAMxanti-CD3). Gynecol Oncol. (2015) 138:343–51. doi: 10.1016/j.ygyno.2015.06.003
176. Stewart D, Cristea M. Antibody-drug conjugates for ovarian cancer: current clinical development. Curr Opin Obstet Gynecol. (2019) 31:18–23. doi: 10.1097/GCO.0000000000000515
177. Ab O, Whiteman KR, Bartle LM, Sun X, Singh R, Tavares D, et al. IMGN853, a folate receptor-alpha (FRalpha)-targeting antibody-drug conjugate, exhibits potent targeted antitumor activity against FRalpha-expressing tumors. Mol Cancer Ther. (2015) 14:1605–13. doi: 10.1158/1535-7163.MCT-14-1095
178. Moore KN, Vergote I, Oaknin A, Colombo N, Banerjee S, Oza A, et al. FORWARD I: a Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncol. (2018) 14:1669–78. doi: 10.2217/fon-2017-0646
179. ImmunoGen Announces Top-Line Results from Phase 3 FORWARD I Study of Mirvetuximab Soravtansine in Ovarian Cancer [Press Release]. Business Wire (2019).
180. Watanabe M, Kono K, Kawaguchi Y, Mizukami Y, Mimura K, Maruyama T, et al. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis Esophagus. (2010) 23:675–81. doi: 10.1111/j.1442-2050.2010.01073.x
181. Petricevic B, Laengle J, Singer J, Sachet M, Fazekas J, Steger G, et al. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Transl Med. (2013) 11:307. doi: 10.1186/1479-5876-11-307
182. Mentlik James A, Cohen AD, Campbell KS. Combination immune therapies to enhance anti-tumor responses by NK cells. Front Immunol. (2013) 4:481. doi: 10.3389/fimmu.2013.00481
183. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM II, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
Keywords: natural killer cell, immunotherapy, ovarian cancer immunology, oncoimmunology, tumor microenvironment, high grade serous ovarian cancer
Citation: Nersesian S, Glazebrook H, Toulany J, Grantham SR and Boudreau JE (2019) Naturally Killing the Silent Killer: NK Cell-Based Immunotherapy for Ovarian Cancer. Front. Immunol. 10:1782. doi: 10.3389/fimmu.2019.01782
Received: 01 May 2019; Accepted: 15 July 2019;
Published: 09 August 2019.
Edited by:
Alberto Anel, University of Zaragoza, SpainReviewed by:
Lana E. Kandalaft, Université de Lausanne, SwitzerlandJulian Pardo, Fundacion Agencia Aragonesa Para la Investigacion y el Desarrollo, Spain
Robert J. Canter, University of California, Davis, United States
Copyright © 2019 Nersesian, Glazebrook, Toulany, Grantham and Boudreau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeanette E. Boudreau, amVhbmV0dGUuYm91ZHJlYXVAZGFsLmNh