 Silke Machata1
Silke Machata1 Sravya Sreekantapuram1
Sravya Sreekantapuram1 Kerstin Hünniger2,3Oliver Kurzai2,3Christine Dunker1Katja Schubert1Wibke Krüger1Bianca Schulze-Richter1†
Kerstin Hünniger2,3Oliver Kurzai2,3Christine Dunker1Katja Schubert1Wibke Krüger1Bianca Schulze-Richter1† Cornelia Speth4
Cornelia Speth4 Günter Rambach4
Günter Rambach4 Ilse D. Jacobsen1,5*
Ilse D. Jacobsen1,5*- 1Research Group Microbial Immunology, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Jena, Germany
- 2Research Group Fungal Septomics, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Jena, Germany
- 3Institute for Hygiene and Microbiology, University of Würzburg, Würzburg, Germany
- 4Institute of Hygiene and Medical Microbiology, Medical University of Innsbruck, Innsbruck, Austria
- 5Institute of Microbiology, Friedrich Schiller University Jena, Jena, Germany
Murine infection models are widely used to study systemic candidiasis caused by C. albicans. Whole-blood models can help to elucidate host-pathogens interactions and have been used for several Candida species in human blood. We adapted the human whole-blood model to murine blood. Unlike human blood, murine blood was unable to reduce fungal burden and more substantial filamentation of C. albicans was observed. This coincided with less fungal association with leukocytes, especially neutrophils. The lower neutrophil number in murine blood only partially explains insufficient infection and filamentation control, as spiking with murine neutrophils had only limited effects on fungal killing. Furthermore, increased fungal survival is not mediated by enhanced filamentation, as a filament-deficient mutant was likewise not eliminated. We also observed host-dependent differences for interaction of platelets with C. albicans, showing enhanced platelet aggregation, adhesion and activation in murine blood. For human blood, opsonization was shown to decrease platelet interaction suggesting that complement factors interfere with fungus-to-platelet binding. Our results reveal substantial differences between murine and human whole-blood models infected with C. albicans and thereby demonstrate limitations in the translatability of this ex vivo model between hosts.
Introduction
Dissemination of pathogens from a primary site of colonization or infection can occur via different routes, including lymphatic vessels and the blood stream (1). The hematogenous is by far the most frequent route for systemic infections of various bacterial and fungal pathogens, in the most severe cases leading to blood stream infections (2). Survival in blood can thus be considered a major virulence trait in the development of systemic infections. However, our understanding of how pathogens interact with cellular and humoral host factors in blood is limited, mainly due to technical issues: While it is relatively easy to study the interaction of pathogens with isolated blood cells, or their survival in serum or plasma, such approaches lack the complexity of interactions between different types of immune cells and additional factors, e.g., complement, present in blood. Assessing host-pathogen interactions in vivo in patients is challenging due to ethical and logistic limitations. In mice, as the most commonly used laboratory animal for in vivo experiments, the blood volume that can be withdrawn repeatedly is very limited and thereby hampers in depth analysis of interactions within blood. Furthermore, if bacteremia or fungemia occurs transiently or intermittingly, pathogens might not be detectable in every blood sample during hematogenous dissemination (3). Therefore, we previously established an ex vivo human whole-blood infection model that allowed us to define which immune cells interact with the human fungal pathogen Candida albicans (4), to identify cross-talk between different components of the host response (5), and to detect substantial differences between related fungal pathogens (6).
Candida infections (candidiasis) caused by C. albicans commonly arise from endogenous strains that colonize mucosal surfaces as a commensal in healthy individuals. Risk factors for candidiasis are microbiota imbalance, impaired mucosal barrier function, and immunosuppression (7, 8). In the majority of cases the fungus causes relatively benign mucosal infections such as oral and vaginal candidiasis (9). Life-threatening infections arise from dissemination via the blood stream resulting in deep-seated or systemic candidiasis (8). Disseminated candidiasis is associated with high mortality rates that can exceed 50% despite antifungal therapy (10, 11). Dissemination from the gut into internal organs can be triggered in mice by a combination of intestinal barrier disruption and immunosuppression (12), but the most commonly used model to study systemic candidiasis is intravenous infection of mice (13). This model is considered a gold standard tool for detailed investigations of fungal virulence and of host immune responses, and largely resembles catheter-associated disseminated candidiasis in men (13, 14). However, the initial interaction of C. albicans with murine blood and its impact on the development of systemic candidiasis is not well understood. Following intravenous infection, C. albicans rapidly disappears from circulating peripheral blood (15, 16), yet it is unknown whether this is due to killing of circulating fungal cells, adhesion to endothelium, rapid invasion of internal organs, or a combination of these factors.
In the human whole-blood model, neutrophils predominantly associate with and phagocytose C. albicans (4). Fungal killing in the human whole-blood model is to 98% accountable to neutrophils (4) and mediated by phagocytosis, degranulation and formation of extracellular traps. However, murine blood differs significantly from human blood in the abundance of neutrophils: These cells are the most abundant leukocytes in human blood (55–70%) while murine blood is dominated by lymphocytes (70–80%) with neutrophils accounting only for 8% to 24% of all leukocytes (17). To elucidate whether these differences affect interaction of C. albicans with whole blood, we adapted our protocol established for human blood for application to mice. Our results show significant differences between humans and mice in the ex vivo model, including lower association of the fungus with leukocytes, higher association with thrombocytes, and substantially less killing of C. albicans in murine blood.
Materials and Methods
Ethics Statement
Human peripheral blood was collected from healthy volunteers with written informed consent. This study was conducted in accordance with the Declaration of Helsinki and all protocols were approved by the Ethics Committee of the University Hospital Jena (permit number: 273-12/09). All animals used for the project were held in accordance with the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes and the experiments performed in accordance with European and German regulations. The sacrifice of mice for blood withdrawal was performed under §4 “removal of organs” of the German Animal Welfare Act and was approved by the local animal welfare officer (no specific permit number issued). The systemic candidiasis model was approved by the Thuringian authority and ethics committee (Thüringer Landesamt für Verbraucherschutz, permit number 03-007/13 and HKI-19-003).
Strains and Culture Conditions
C. albicans GFP-expressing strains M137 (18) (pACT1-GFP) and C. albicans Δefg1Δcph1 + pADH1-GFP were used. To construct C. albicans Δefg1Δcph1 + pADH1-GFP a CaGFP-caSAT1 construct with homology regions for the integration into the C. albicans ADH1 locus was excised with AscI/SacI from the plasmid pSK-ADH1prom-CaGFP-SAT1 (4) and then transformed into the EFG1/CPH1 double mutant HLC52 (19). Transformation was performed with the established lithium acetate protocol (20). Transformants were grown for two days on YPD with 200 mg/ml nourseothricine and verified by PCR and microscopy. For experiments C. albicans over-night cultures grown in YPD medium (2% peptone, 1% yeast extract, 2% glucose) at 30°C and 180 rpm were diluted 1:50 in YPD medium and sub-cultured for another 3 to 4 h into the mid-log phase under the same conditions. Yeast cells were washed three times with PBS, counted and diluted in PBS to 5 × 107/ml or 5 × 106/ml, as indicated.
GFP-expressing S. aureus [6850/pALC1743 (21, 22)] and Escherichia coli ATCC 25922 (23) were cultivated overnight at 37°C, 180 rpm in LB medium. The overnight culture was inoculated 1:100 into fresh LB medium and incubated at 37°C, 180 rpm until O.D600 0.6 to 0.7 was reached. The cultures were then washed three times with PBS and bacterial cell numbers were calculated based on OD600–CFU correlations. Cultures were diluted to desired concentrations with PBS before inoculation of whole blood.
Whole-Blood Model
Human peripheral blood from healthy donors was collected in hirudin-monovettes (Sarstedt, Germany, 2.7 ml volume). Hirudin was used as an anti-coagulant as it was previously shown to have no effect on complement activation (24). Due to their size, direct use of the hirudin-monovettes was not feasible for the collection of murine blood. Therefore, three hirudin-monovettes were rinsed with total volume of 500 µl sterile saline (0.9% NaCl) to solubilize the hirudin. Needles and syringes rinsed with hirudin-NaCl were then used to collected blood from female BALB/c mice (8–12 weeks old, Charles River, Germany) or from C57BL/6J mice (8–12 weeks old, Service Unit Experimental Biomedicine, Friedrich-Schiller-University Jena, Germany) by heart puncture immediately after sacrifice by intraperitoneal application of an overdose of ketamine (500 mg/kg) and xylazin (25 mg/kg). The blood was then immediately transferred to a falcon tube containing 500 µl hirudin-NaCl, in which blood from ten animals was pooled for each experiment. Pilot experiments analyzing fungal CFU in whole blood of female and male C57BL/6J mice revealed no sex-specific differences; thus, both male and female mice were used for the experiments.
The human whole-blood infection assay was performed as described previously (4), and murine blood was infected in the same way. Briefly, 1/50 volume fungal or bacterial suspensions prepared as described above were added to murine or human blood, resulting in an infectious dose of 1 × 106/ml for most experiments or 1 × 105/ml for lower infection dose experiments. The incubation of the whole blood was carried out at 37°C under slow constant motion on a rolling device (Phoenix instrument RS_TR05) for time points from 10 min to 480 min. Survival of the pathogens was determined by plating on agar plates (YPD for C. albicans, LB for E. coli and S. aureus) using serial dilutions in PBS and counting colony forming units. To determine the effect of ketamine/xylazin on phagocytosis and association of C. albicans with immune cells, 40 µg/ml ketamine and 2 µg/ml xylazin were added to human blood directly before the experiment. 40 µg/ml ketamine is equivalent to fivefold the maximal serum concentration in mice after intraperitoneal application of 100 mg/kg ketamine (25).
Blood Analysis and Flow Cytometry
Generally hematology analysis of murine samples was performed using a BC-5300Vet (Mindray) configured for murine blood. For murine samples, blood smears were prepared for each time point and stained by May-Gruenwald-Giemsa staining (Roth). Filamentation and interaction with platelets were observed by light microscopy. Further analyses were performed by differential staining and subsequent measurement with a flow cytometer. For murine blood, differential staining was performed with CD45-PerCP (Cat. no. 557235, clone 30-F11, 12 µg/ml, BD Biosciences), CD19-APC-Cy7 (Cat. no. 115530, clone 1D3; 12 µg/ml, Biolegend) for B-cells, CD3e-V500 (Cat. no. 560771, clone 500A2, 12 µg/ml, BD Biosciences) for T-cells, Ly6G-PE (Cat. no. 127608, clone 1A8, 12 µg/ml final conc; Biolegend) for neutrophils, NK1.1-BV421 (for C57BL/6J mice: Cat. no. 108731, clone PK136, 12 µg/ml, Biolegend) or CD335-BV421 (for BALB/C mice: Cat. no. 562850, Clone 29A1.4, 12 µg/ml, BD Biosciences) for NK cells, CD11b-APC (Cat. no. 130-091-241, clone M1/70.15.11.5, 30 µg/ml, Miltenyi) for monocytes and neutrophils, CD41 (Cat. no. 133906, clone MWReg30, 12 µg/ml, Biolegend) for platelets, and CD69-PE vio770 (Cat. no. 130-103-944, clone H1.2F3, 30 µg/ml, Miltenyi) and CD62-PE-Vio779 (Cat. no. 130-105-537, clone REA 344, 30 µg/ml, Miltenyi) as platelet activation markers. In parallel, staining with the appropriate isotype controls (APC Rat IgG2b, κ Isotype Control, clone RTK5430, Cat. no. 400611, Biolegend; V500: Syrian hamster IgG2 κ Isotype Control, clone B81-3, Cat. no. 560785, BD Bioscience; PE: rat IgG2a κ Isotype Control, clone RTK2758, Cat. no. 400507, Biolegend; PerCP rat IgG2b κ Isotype Control, clone A95-1, Cat. no. 552991, BD Bioscience; APC-Cy7 rat IgG2a κ Isotype Control, clone RTK2758, Cat. no. 400524, Biolegend; V450 rat IgG2a κ Isotype Control, clone R35-95, Cat. no. 560377, BD Bioscience; V450 mouse IgG2a κ Isotype Control, clone G155-178, Cat. no. 560550, BD Bioscience; PE Vio770 hamster IgG1 Isotype Control, clone G235-2356, Cat. no. 553956, BD Bioscience; PE rat IgG1 κ Isotype Control, clone RTK2071, Cat. no. 400407, Biolegend; APC human IgG1 Isotype Control, clone QA16A12, Cat. no. 403505, Biolegend) was performed as a binding specificity control. After staining the erythrocytes were removed using the BD FACS Lysing solution and fixed samples were analyzed on a FACSVerse (BD Biosciences) flow cytometer after two washing steps in PBS supplemented with 3% FCS.
Human whole-blood was analyzed with a BD FACSCanto II flow cytometer. Platelets were specifically identified by CD42b+ staining (mouse anti-human CD42b-APC antibody, clone HIP-1, Cat. no. 303912, BioLegend). Co-staining with mouse anti-human CD66b-V450 antibody (clone G10F5, Cat. no. 561649, BD Bioscience) identified platelets associated with neutrophils. Activation of human platelets was investigated by changes in surface CD62P expression (mouse anti-human CD62P-PE antibody, clone AK-4, Cat. no. 555524, BD Bioscience). In parallel, staining with the appropriate isotype controls (APC mouse IgG1, κ Isotype Ctrl, clone MOPC-21, Cat. no. 400122, BioLegend; V450 mouse IgM, κ Isotype Control, clone G155-228, Cat. no. 560861, BD Bioscience and PE Mouse IgG1, κ Isotype Control, clone MOPC-21, Cat. no. 555749, BD Bioscience) was performed as a binding specificity control. Stained blood samples were treated with BD FACS Lysing solution followed by washing and harvesting cells in BD CellWASH solution. FlowJo10 was used for analysis of all samples.
Neutrophil Isolation From Murine Bone Marrow
Neutrophils were isolated from bone marrow of femur and tibia of three to four female 8- to 12-week-old BALB/c mice as previously described (26). Briefly, bone marrow was flushed with RPMI 1640 supplemented with Penicillin/Streptomycin (Sigma) and homogenized using a 40 µm cell strainer. Lysis of erythrocytes was carried out for 1 min on ice using a lysis buffer containing 8 mg/ml NH4Cl, 1 mg/ml K2CO3 and 0.01% EDTA. Neutrophils were purified using a Percoll gradient of 52%, 69%, and 78% in PBS and cells were collected from the 69% to 78% interface. After washing with HBSS neutrophils were resuspended in HBSS, analyzed on a BC-5300Vet (Mindray) and stored on ice until usage for a maximum of 1 h. The purity of isolated neutrophils was roughly 70%.
Analysis of Platelets From Mice With Systemic Candidiasis
Six- to eight-week-old female specific-pathogen-free BALB/c mice (16 to 18 g) purchased from Charles River (Germany) were housed in groups of five in individually ventilated cages with free access to food and water. Mice were infected with 2.5 × 104 C. albicans CFU/g body weight in 100 µl DPBS via the lateral tail vein at day 0. Groups of mice were sacrificed at the indicated time points post infection by intraperitoneal application of an overdose of ketamin (500 mg/kg) and xylazin (25 mg/kg). 100 µl of blood were collected under terminal anesthesia by retro-orbital bleeding, immediately transferred into a tube containing 10 µl EDTA solution (1.6 mg/ml), and gently mixed. 20 µl of the sample were analyzed on a BC-5300Vet (Mindray) to determine platelet counts and mean platelet volume. Platelet-rich plasma (RPP) was prepared from 40 µl of whole-blood by centrifugation at 135 g for 15 min at room temperature. To detect platelet activation, platelets were stained for 30 min with fluorescence-labeled antibodies (BioLegend) directed against CD41 (clone HIP8, Cat. no. 303710, 0.1 µg/ml) as platelet marker and CD63 (clone H5C6, Cat. no. 353006, 8 µg/ml) as activation marker, followed by fixation with 1% formaldehyde. Surface expression of CD62P, fibrinogen binding and C3c binding were determined as described for CD63 using fluorescence-labeled antibodies (CD62P: BioLegend, clone AK4, Cat. no. 304906, 1.5 µg/ml; fibrinogen: BioRad, polyclonal, Cat. no. 4440-8004F, 100 µg/ml; C3c: Dako, polyclonal, Cat. no. F0201, 400 µg/ml). Non-activated unstained cells and non-activated and activated stained cells were used to calibrate the flow cytometer using single stains and combined stains. Plasma was prepared by centrifugation of 40 µl whole-blood at 1500 g for 15 min at room temperature. Plasma concentrations of soluble CD62P were determined using the Quantikine® ELISA Mouse sP-Selectin/CD62P kit (R&D Systems, USA) performed according to manufacturer instructions.
Isolation of Platelets and Confrontation Assay
Venous blood of healthy human volunteers was collected in sodium citrate monovettes (Sarstedt) and directly centrifuged at 80 × g for 20 min (without break). Supernatants were collected and treated with 0.25 mg/ml acetylsalicylic acid (Sigma Aldrich) for 30 min followed by addition of 1 mM Prostaglandin E1 (Sigma Aldrich), both to prevent pre-activation of the containing platelets. Centrifugation at 400 × g for 8 min pelleted platelets that were afterwards suspended in HEPES-Tyrodes buffer (10 mM HEPES, 137 mM NaCl, 2.8 mM KCl, 1 mM MgCl26H2O, 12 mM NaHCO3, 0.4 mM Na2HPO42H2O, 5.5 mM Glucose, 0.35% BSA, without Ca2+). Purified platelets were treated again with 1 mM Prostaglandin E1, centrifuged at 400 × g for 10 min and suspended either in RPMI 1640 medium containing 20% autologous active or heat-inactivated plasma. Autologous human plasma was collected after centrifugation of Hirudin-anticoagulated blood from the same donor at 16,000 × g for 10 min. To inactivate complement proteins, autologous human plasma was incubated for 1 h at 56°C. Murine platelets were prepared from freshly collected blood of healthy mice as concentrates by thrombocytapheresis with Amicus cell separator (Baxter, Vienna, Austria) by the Department of Immunology and Blood Transfusion (Innsbruck Medical University, Innsbruck, Austria). The platelet concentration was determined by a hemocytometer and adjusted to a concentration of 1.2 to 1.4 × 109/ml. Murine serum was collected by centrifugation of blood samples from (i) SPF mice without any treatment, (ii) mice systemically infected with a sublethal dose of 1 × 104 C. albicans CFU/g body weight on days 8 to 21 after infection (see 2.6), (iii) C3-deficient mice (B6.129S4-C3tm1Crr/J). Pooled serum of 5 to 12 mice per group was used for experiments with isolated platelets.
Confrontation of isolated platelets with C. albicans was performed at a ratio of platelets to fungal cells of 10:1 for the indicated time points (37°C, constant rolling at 5 rpm). Mock-infected platelets with the two different media served as controls.
Quantification of Cytokines
Plasma samples were generated from whole-blood aliquots that were incubated on ice for 45 min to 1 h and subsequently centrifuged at 4°C for 10 min at 10,000 × g. Supernatants were stored at −80°C until further use. Concentration of IFN-γ, IL-1β, TNF-α, IL-6, and KC (CXCL1) in murine samples was determined by ELISA (Invitrogen, Thermo Fisher Scientific) performed according to manufacturer’s instructions.
Results
C. albicans Survives in Murine Whole Blood
The fate of C. albicans upon exposure to murine blood was analyzed in whole blood collected from two of the most commonly used mouse strains for infection experiments, BALB/c and C57BL/6J. As described by Hünniger et al. for the human whole-blood model (4), whole blood was inoculated with 1 × 106/ml of yeast-grown C. albicans and fungal survival was determined by counting colony forming units (CFU) at various time points over a time course of 3 h. In contrast to human blood in which a 50% decrease of the initial CFUs within the first hour of contact was observed (4), the fungal burden did not decrease during incubation in neither BALB/c nor C57BL/6J mouse blood but remained stable throughout the experiment (Figures 1A, B). Of note, ketamine and xylazin were used to euthanize mice prior to blood collection. As ketamine has been shown to affect antimicrobial functions of macrophages and neutrophils in a dose-dependent manner (27–30), we added ketamine and xylazin to human blood and quantified fungal killing. At the used dose (40 µg/ml ketamine and 2 µg/ml xylazin) ketamine/xylazin treatment did not affect fungal killing (Figure S1).
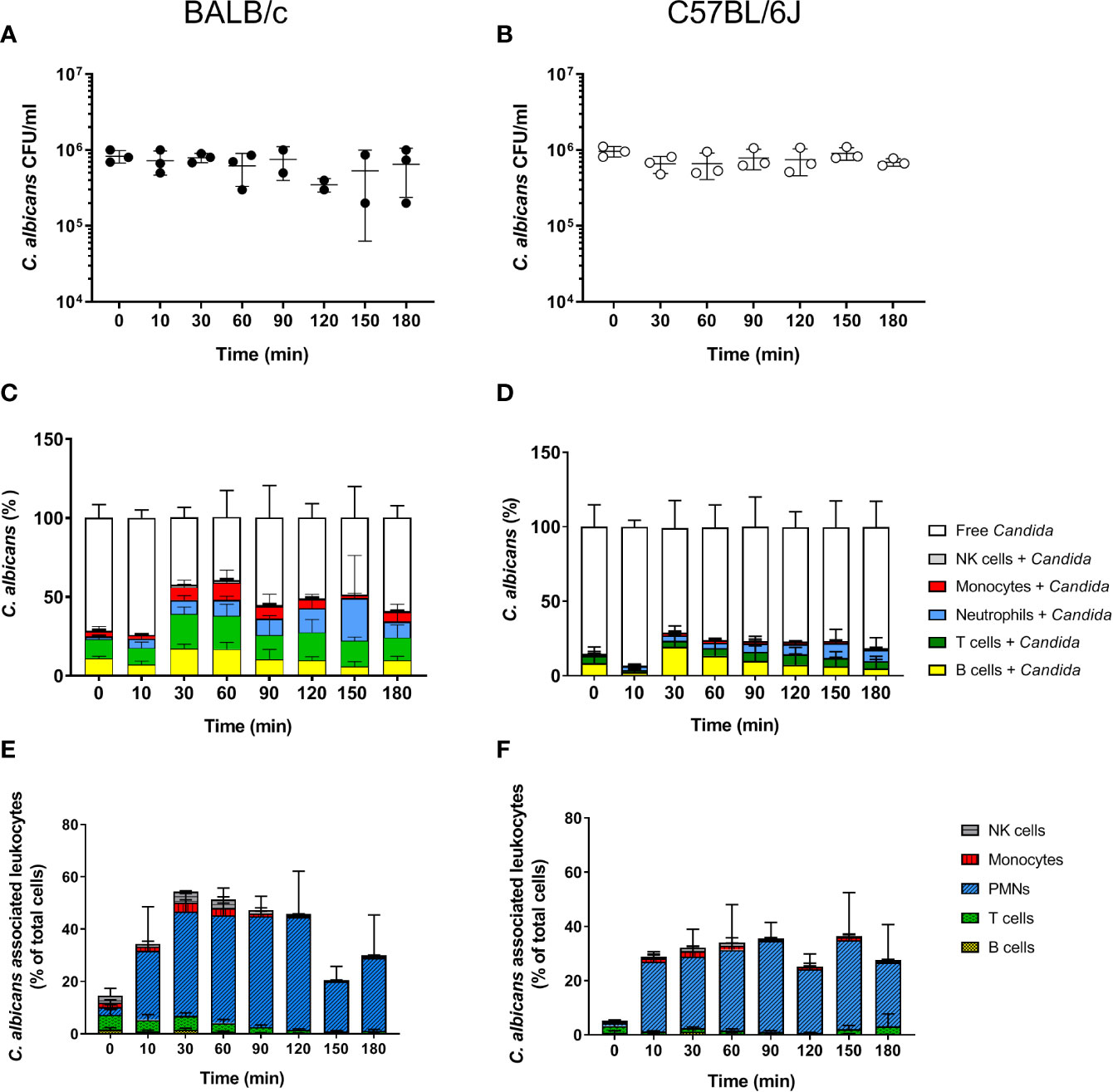
Figure 1 Survival and association of C. albicans with leukocytes in murine whole blood. Colony forming units (CFU) were determined from infected blood of BALB/c (A) and C57BL/6J (B) mice. Each dot represents the data from an independent experiment; lines represent mean ± standard deviation (n = 3, except BALB/c 90 min and 120 min: n = 2). (C, D) The percentage of GFP-expressing Candida binding to immune cells of blood was calculated relative to total C. albicans cells in infected blood (set to 100%) during a time course of 3 h. All values correspond to the means of three independent experiments with pooled whole blood from 10 mice. Left: BALB/c; right: C57BL/6. (E, F) The percentage of various immune cells associated with Candida was determined relative to their overall cell population in the blood. All values correspond to the means of three independent experiments with pooled whole blood from 10 mice. Left: BALB/c; right: C57BL/6.
Less Immune Cells Interact With C. albicans in Murine Compared to Human Blood
In order to determine whether the higher fungal survival in murine compared to human blood was associated with differences in the interaction with immune cells, we analyzed the number of immune cells and association of C. albicans with leukocytes. Quantification of total leukocyte numbers using a hematology analyzer showed a moderate, non-significant decline over time in both mouse strains (Figure S2A). While the numbers of CD45+ leukocytes in relation to all blood cells showed no obvious changes throughout the infection (Figure S2B), the numbers of neutrophils decreased by almost 50% within the first 10 min after addition of C. albicans (Figure S2C). This was not the case for mock-infected control samples (Figure S2D). In contrast, the number of other types of leukocytes remained stable over time (Figure S3). Note that the relative abundance of neutrophils was significantly lower in the blood of B57BL/6J (4.56 ± 1.3%) than of BALB/c mice (8.63 ± 1.58%; p = 0.02).
Using a strain constitutively expressing green fluorescent protein the association of C. albicans with leukocytes was determined by flow cytometry. Within 30 min, more than 50% of C. albicans cells were associated with immune cells in BALB/c whole blood (Figure 1C). The proportion of fungal cells associated with immune cells remained relatively stable until the end of the experiments (Figure 1C). A similar trend was observed in blood of C57BL/6J mice; however, the rate of association was lower and did not exceed 40%. Surprisingly, a large proportion of C. albicans cells were found to be associated with B- and T-cells in both mouse lines (Figures 1C, D). This contrasts results from the human whole-blood model in which monocytes and neutrophils were the dominant types of immune cells physically interacting with C. albicans (4). Since the ratio of the different immune cell populations in blood differs significantly between humans and mice, we speculated that the association to lymphocytes could be the consequence of the higher abundance of these types of immune cells in murine blood rather than the result of specific interactions. We thus calculated the percentage of host cells interacting with C. albicans within the different immune cell populations (Figures 1E, F). Although only a small fraction of C. albicans cells were associated with neutrophils, up to 50% and 40% of all neutrophils associated to fungal cells in blood from BALB/c and C57BL/6J mice, respectively. In contrast, only a minor fraction of the B- and T-cell populations interacted with the fungus, suggesting that the observed association is indeed a stochastic physical event rather than the result of specific interactions.
The Reduced Fungal Killing in Murine Blood Is Only Partially Due to Lower Neutrophil Numbers
Neutrophils are the most active immune cells interacting with C. albicans in both human and murine blood, and have been shown to be critical for killing of the fungus in the human whole-blood model (4). However, the absolute number of neutrophils is approximately 10-fold lower in murine (0.4 × 106/ml) than human blood (4 × 106/ml; according to reference values and determined by a hematology analyzer). Using the same infection dose (1 × 106/ml) in both models thus resulted in a pathogen to neutrophil ratio of 2.5 in murine but 0.25 in human blood. We hypothesized that the higher microbe to neutrophil ratio in the murine whole-blood model overwhelmed the capacity of the neutrophils to interact with and control C. albicans. Therefore, we analyzed fungal survival in whole blood of BALB/c mice using a 10-fold lower infection dose (1 × 105/ml), which is comparable to the ratio used previously in human whole blood (4). With the reduced infection dose there was a moderate reduction of the fungal burden by 30% within 3 h of infection (Figure 2A). A similar effect was observed when murine blood was supplemented with neutrophils isolated from bone marrow to reach cell numbers comparable to human blood (4.4 ± 0.51 × 109/L of cells, determined by a hematology analyzer) (Figure 2B). Thus, both the addition of external neutrophils and the lower infection dose led to increased fungal killing, but survival of C. albicans after 180 min was still substantially higher (>40%) than in a human whole-blood model (10% (4);). Furthermore, fungal burden decreased only slowly in murine blood, whereas 50% of fungal cells were killed within the first 60 min in human blood (4). To exclude effects mediated by ketamine/xylazin, ketamine and xylazin were added to human blood; this did not affect association rates with neutrophils and monocytes (Figure S4).
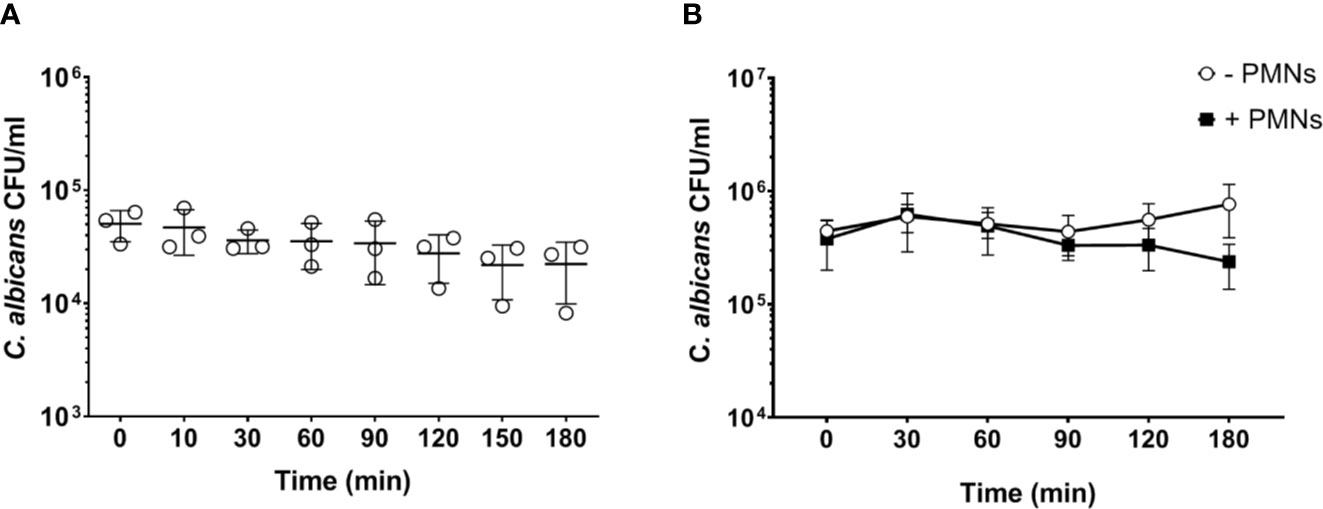
Figure 2 Increased PMN - to - Candida ratio leads to a non-significant decrease of the fungal burden after 180 min. (A) Colony forming units were determined on YPD plates from infected blood samples at various time points for 3 h. All values correspond to the means of three independent experiments with pooled whole blood from 10 BALB/c mice infected ex vivo with 1 × 105/ml of C. albicans::gfp. Each dot represents the mean of an independent experiment ± standard deviation (n = 3). (B) Colony forming units were counted in murine whole blood as described for (A) with the following modifications: the infection dose was 1 × 106/ml and murine whole blood was supplemented with 4.5 × 106/ml neutrophils that were previously isolated from bone marrow of BALB/c mice. Although differences between control and neutrophil supplemented blood were observed at later time points, these were not statistically significant (p>0.05, Wilcoxon matched-pairs signed rank test).
Filamentation of C. albicans in Whole Blood Is Not Effectively Inhibited by Murine Neutrophils but Fungal Survival Does Not Require Filamentation
The lower number of neutrophils in murine compared to human blood did not fully explain the lower killing of C. albicans in murine blood. However, functional differences between human and murine neutrophils have been described: A previous study from Ermert et al. (31) described a reduced efficiency of murine neutrophils to kill Candida species due to the fact that mice lack β-defensins and produce lower amounts of myeloperoxidase. They also showed that internalized C. albicans can escape from murine but not from human neutrophils by outgrowing neutrophils through filamentation and subsequent rupture of the neutrophil membrane (31). We therefore analyzed C. albicans morphology in whole blood by microscopy. Following infection with yeast cells, after 60 min the majority of C. albicans (~70%) in murine blood were hyphae and after 180 min nearly all (>90%) fungal cells grew as filaments (Figure 3A). In contrast, hyphal morphology was observed only for 10% and 20% of all C. albicans cells in human blood after 60 and 180 min, respectively (Figure 3B).
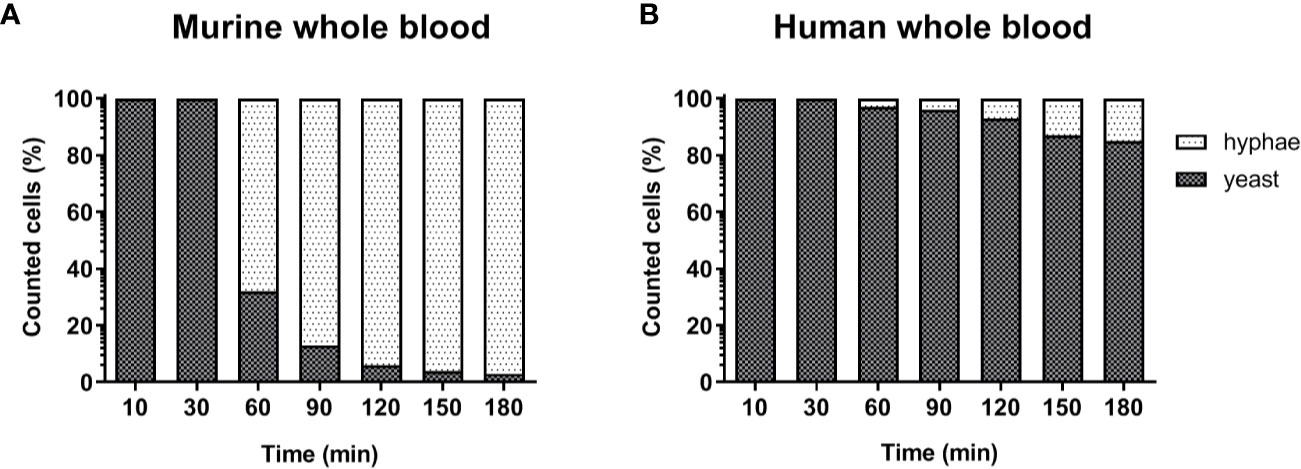
Figure 3 Morphology of C. albicans in murine and human blood during the course of infection. Blood smears were prepared at the given time points and the state of fungal filamentation was determined by counting yeast or hyphae form of Candida (n = 100 per time point) on an Axioplan 2 microscope (Zeiss) after staining of cells with Mai-Gruenwald- Giemsa.
C. albicans morphology has been shown to affect recognition by neutrophils (5, 32) and monocytes (33). Furthermore, while human neutrophils prevent C. albicans filamentation and escape following phagocytosis (31), human monocytes and macrophages do not, and intracellular filamentation leads to immune cell lysis and fungal escape (34). Given the higher rate of C. albicans filamentation observed in murine blood, and the role of morphology for immune escape, we used a yeast-locked C. albicans Δefg1/Δcph1 mutant expressing GFP (19) in the murine whole-blood infection model to determine if the higher filamentation rate could explain higher survival of C. albicans in murine blood. Surprisingly, no decrease in CFU counts was observed with this strain and fungal numbers even increased 2.5-fold from 60 to 180 min (Figure 4A). The increasing fungal burden correlated with a decrease in CD45+ cells (Figure 4B), and especially neutrophils (Figure 4C), at later time points (150 min and 180 min). The percentage of C. albicans Δefg1/Δcph1 cells not associated with any of type of immune cell (~80%, Figure 4D) was significantly higher compared to the wild type strain (~50%, Figure S2A). This is in contrast to experiments with human blood, in which similar association of wild type and mutant with immune cells was observed (4, 5). However, more than 25% of all neutrophils were associated with this mutant after 2 h (Figure 4E), and the release of cytokines upon infection with the mutant was comparable to or higher than C. albicans wild type infection (Figure S4). Thus, the large number of free Δefg1/Δcph1 cells cannot be explained by an inability of immune cells to recognize mutant yeast cells.
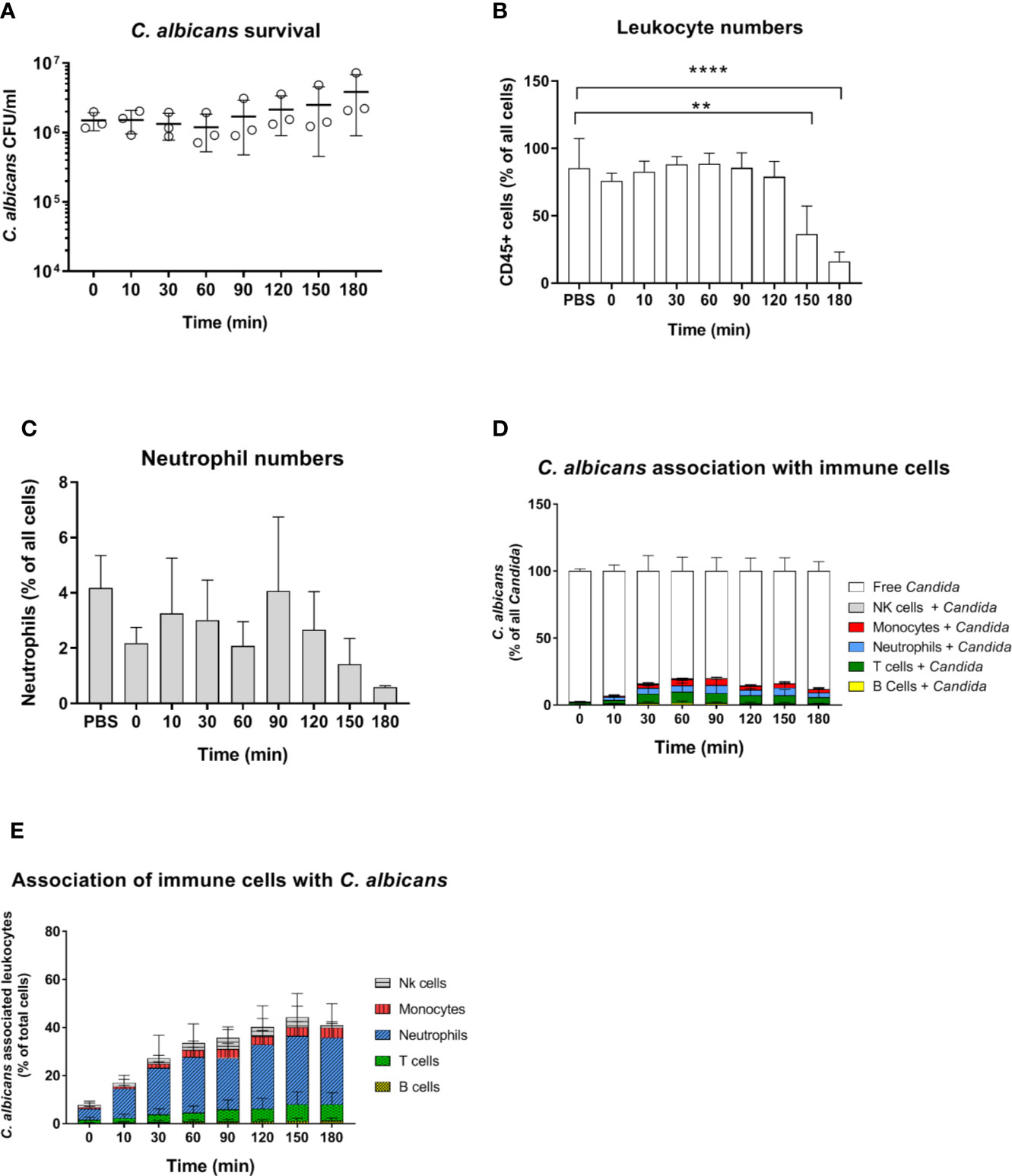
Figure 4 Survival and immune cell association of the yeast-locked C. albicans strain Δefg1Δcph1 + pADH1-GFP in murine whole-blood of BALB/c mice. (A) Whole blood was pooled from 10 mice and infected ex vivo with 1 × 106/ml of the GFP-expressing C. albicans mutant. CFU were determined from infected blood at indicated time points. Each dot represents the mean of an independent experiment ± standard deviation (n = 3). (B, C) Immune cell stability during the course of infection, (B) relative number CD45+ cells and (C) neutrophils as % of all cells. Asterisks indicate significant differences (**p<0.005, ****p<0.0005; 1-Way ANOVA and Holm-Sidak’s multiple comparison test to compare each infected time point to 0 min). (D) The percentage of GFP-expressing Candida binding to immune cells was calculated relative to total C. albicans cells in infected blood (set to 100%) during a time course of 3 h. All values correspond to the means of three independent experiments with pooled whole blood from 10 mice. (E) The percentage of various immune cells associated with Candida was determined relative to their overall cell population in the blood. All values correspond to the means of three independent experiments with pooled whole blood from 10 mice.
Rapid Platelet Aggregation and Activation in Murine Blood Upon Exposure to C. albicans
In addition to fungal filamentation, microscopic analysis revealed aggregates of platelets around C. albicans cells (Figure 5A). This interaction was confirmed and quantified by flow cytometry (Figure 5B): Within 10 min almost 60% of C. albicans cells were associated with platelets. The association rate slowly decreased over time to 40% after 3 h. At early time points, over 35% of the fungal-associated platelets expressed the activation marker CD69 on their surface (Figure 5C). We also observed increased expression of CD62 on the surface of C. albicans bound platelets, however in only 20% of all platelets (Supplementary Figure S6). To exclude that platelet activation was an artefact of the ex vivo situation, blood samples of intravenously (i. v.) infected mice were analyzed. In vivo, platelet counts increased significantly over a course of three days post infection (Figure 6A). Transient platelet activation was observed at 6 h post infection characterized by a significant increase of complement C3c, indicating opsonization, the platelet aggregation factor fibrinogen, and the activation markers CD63 and CD62P on the surface of platelets (Figures 6B–E). While at later time points expression of these markers was comparable to control samples, increased concentrations of soluble CD62 were detectable in the plasma of infected mice from 1 to 3 days post infection by ELISA (Figure 6F). Taken together, these data demonstrate interaction and activation of platelets following infection with C. albicans in murine blood ex vivo and in vivo.
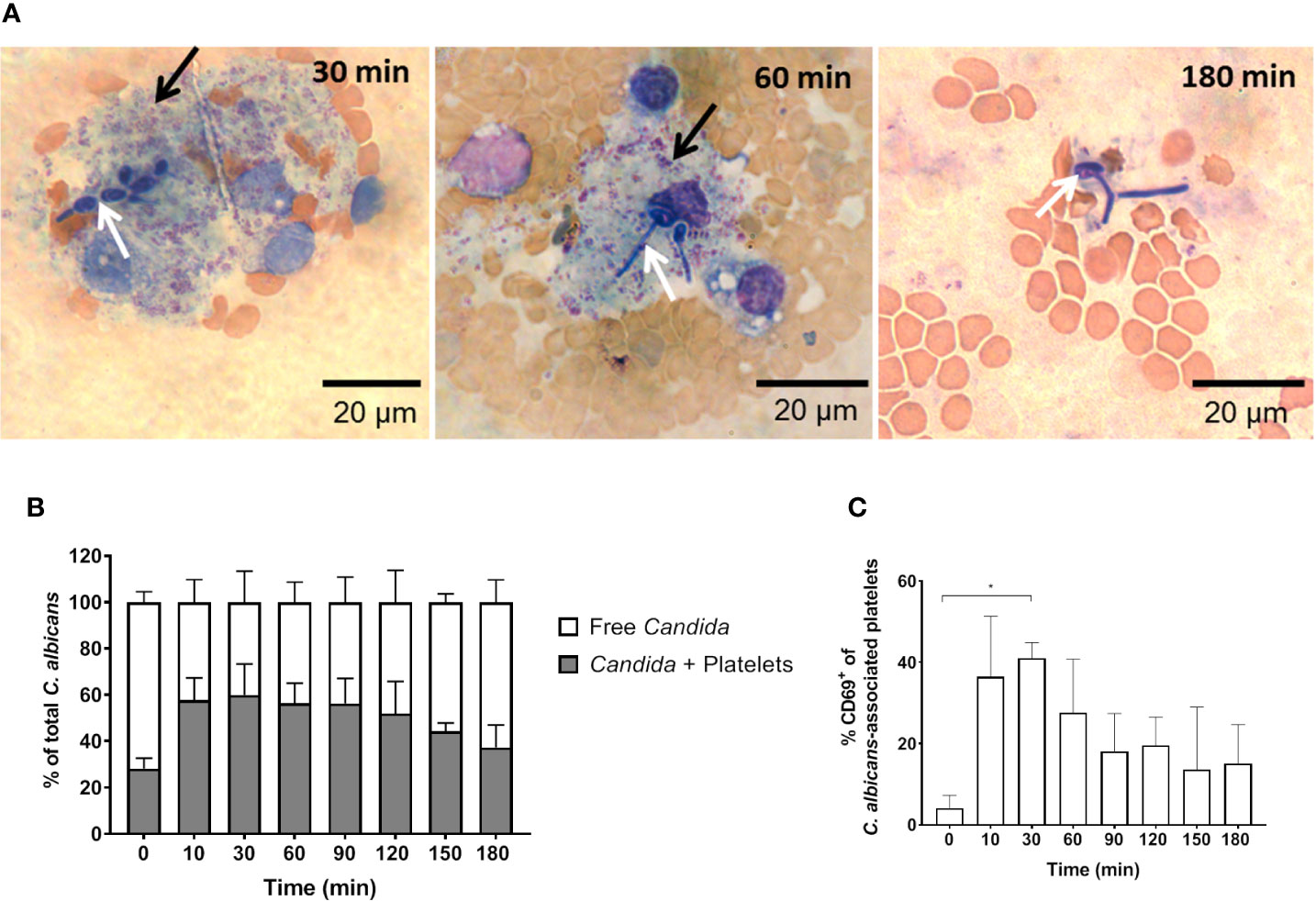
Figure 5 C. albicans-induced platelet activation and aggregation (A) Fungal interactions with platelets were visualized in blood smears of ex vivo infected murine whole blood after staining with Mai-Gruenwald- Giemsa. Aggregates of thrombocytes (black arrow) surround Candida (white arrow) mostly during the first hour of infection. Flow cytometry analysis shows (B) strong binding of C. albicans to platelets almost immediately after infection and (C) induced expression of CD69 on the surface of platelets. Asterisks indicate significant differences (p<0.05; 1-Way ANOVA and Holm-Sidak’s multiple comparison test to compare each infected time point to 0 min).
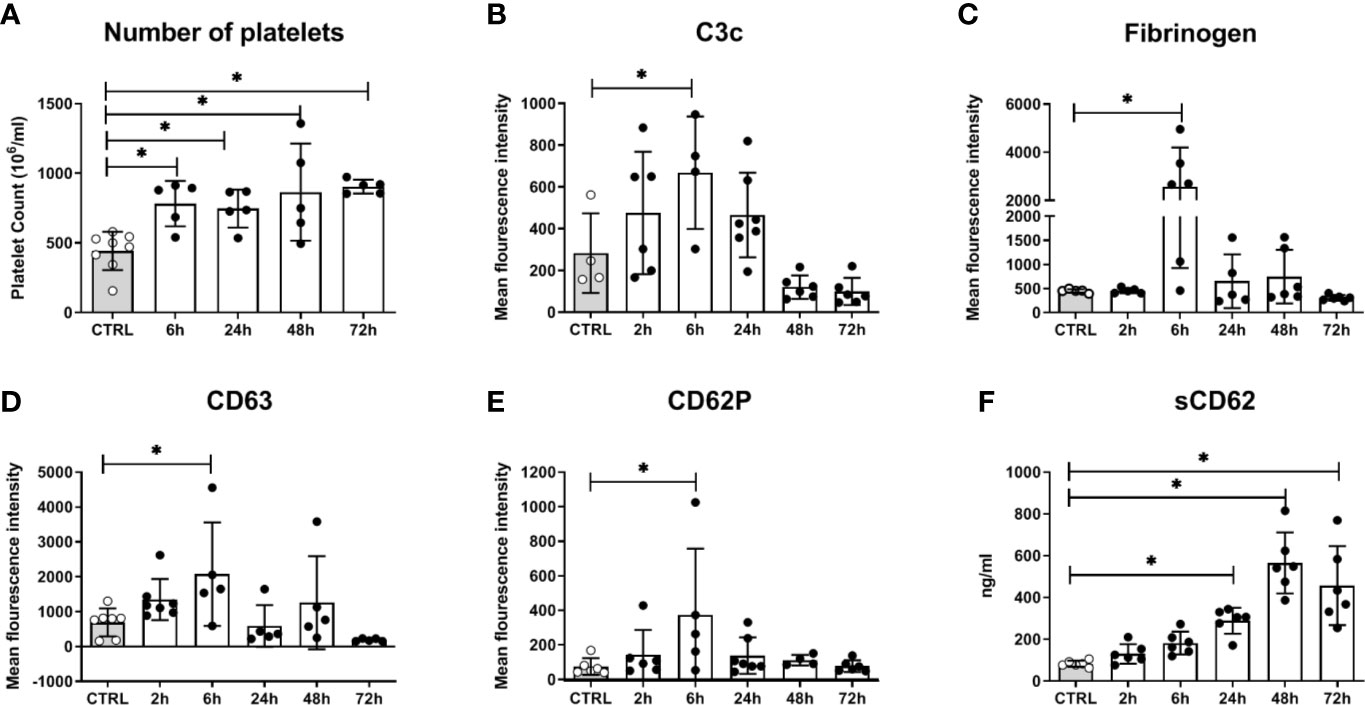
Figure 6 Platelet activation in mice systemically infected with C. albicans. (A) The number of circulating platelets in peripheral murine blood was determined with an automated hematology analyzer. Deposition of complement factor C3 (B), fibrinogen (C), and surface expression of CD63 (D) and CD62P (E) was quantified by flow cytometry. (F) Soluble CD62 was measured in plasma by ELISA. CTRL: Samples from non-infected control animals. Time on X axis: Time after infection. Four to seven samples were analyzed per time point, data is presented as individual data points with bars indicating the mean ± standard deviation. Asterisks indicate significant differences (p<0.05; 1-Way ANOVA and Holm-Sidak’s multiple comparison test to compare each infected group to the control).
Opsonization of C. albicans in Human Blood Leads to Low Platelet Association
Our next aim was to determine if interaction with thrombocytes also occurs in human blood. Consistent with previous findings (4), we observed decreasing numbers of C. albicans cells not associated with host cells in human blood over time (Figure 7A). Only a small proportion (less than 10%) of fungal cells directly associated with platelets (Figure 7B). This was in sharp contrast to the results we obtained in murine blood. To investigate the potential influence of human blood components on fungal-platelet interaction, confrontation of isolated human platelets with C. albicans cells was performed in the presence of non-treated (active) or heat-inactivated autologous plasma and showed a time-dependent increase in platelet-C. albicans interaction when plasma proteins were inactivated (Figure 7C). In contrast, the presence of non-treated plasma containing active complement proteins resulted in a low platelet association. Consequently, we tested if opsonization of fungal cells interferes with platelet binding. Indeed, pre-opsonized C. albicans showed a significantly reduced interaction with isolated platelets compared to non-opsonized C. albicans (Figure 7D) indicating that opsonization of extracellular C. albicans during human whole-blood infection prevents the binding of platelets. In a comparable experiment using isolated murine thrombocytes and murine serum, active but not inactive serum significantly increased the binding of thrombocytes to C. albicans (Figure 8A).
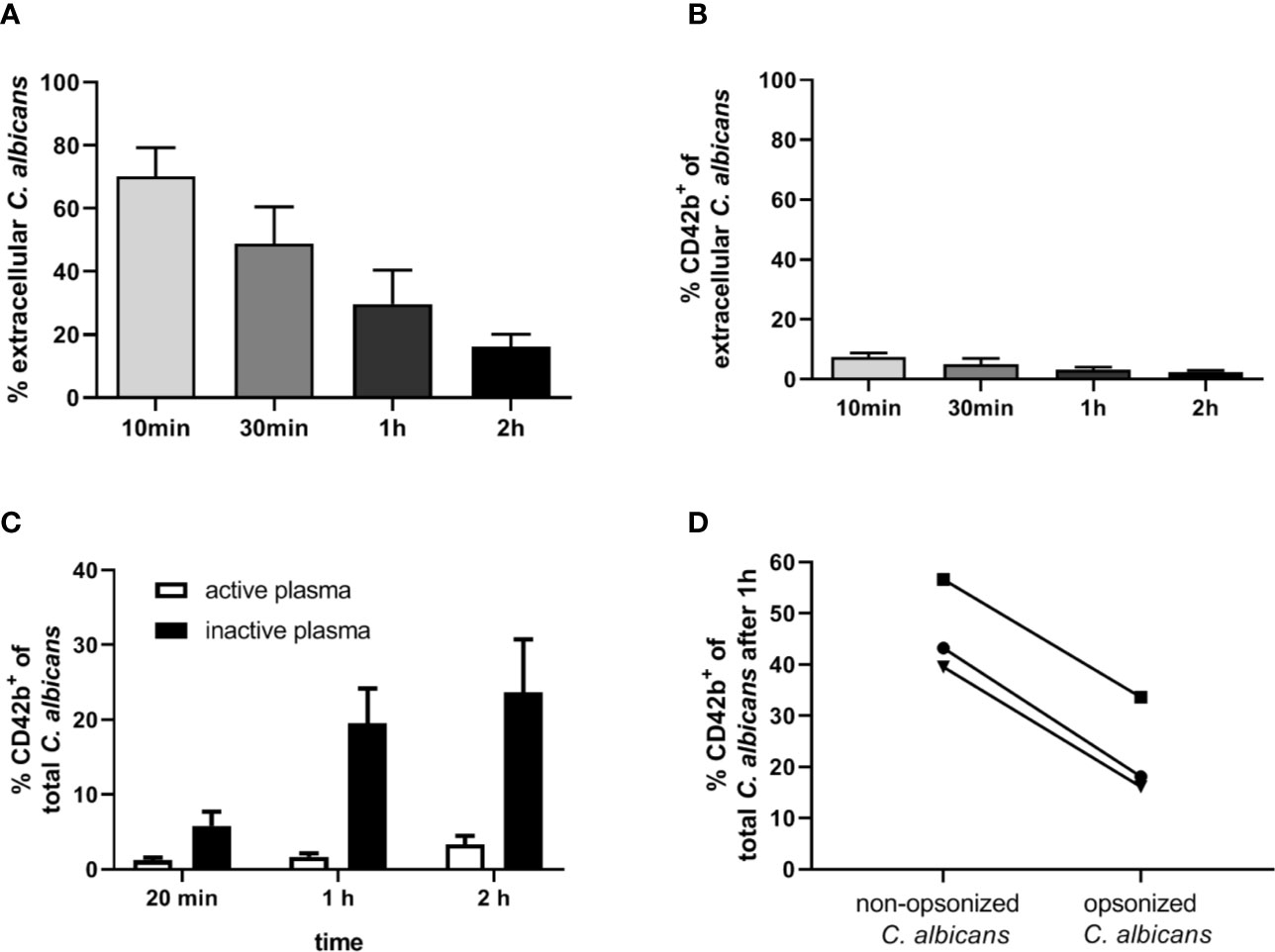
Figure 7 Interaction of human platelets with C. albicans during whole-blood infection is influenced by complement opsonization. (A) The percentage of extracellular C. albicans during human whole-blood infection was investigated via flow cytometry and calculated relative to total fungal cells in blood (set to 100%) of five independent experiments with blood from different donors. A steady reduction of extracellular fungi over time was observed (1-Way ANOVA and Test for linear trend, p<0.0001). (B) Less than 10% of extracellular C. albicans cells showed a CD42b+ staining at each time point, reflecting a low platelet binding to C. albicans in human whole-blood (n = 5). (C) Isolated human platelets were confronted with C. albicans either in the presence of non-treated (active plasma) or heat-inactivated autologous plasma (inactive plasma). Co-incubation in medium containing active plasma prevented the time-dependent increase in platelet-C. albicans interaction as observed for heat-inactivated plasma. Significantly less CD42b-positive fungal cells were observed with active compared to inactive plasma at each time point (unpaired, two-tailed t-test, p<0.0001, n = 7). (D) C. albicans cells were either pre-incubated in non-treated (C3b-opsonized C. albicans) or heat-inactivated autologous plasma (non-opsonized C. albicans, no surface C3b) before confrontation with isolated human platelets in medium with inactive plasma for 1 h. Opsonization of C. albicans resulted in a markedly lower association with platelets compared non-opsonized C. albicans (p = 0.035, unpaired, two-tailed t-test).
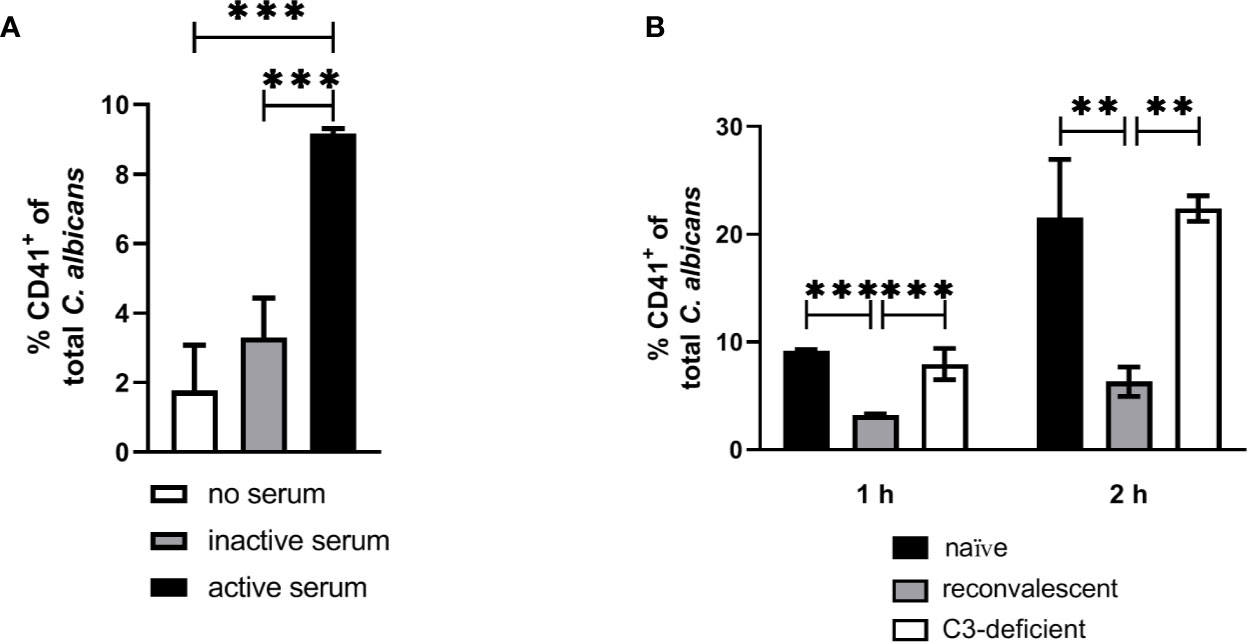
Figure 8 Interaction of isolated murine platelets with C. albicans. (A) Binding of isolated platelets to C. albicans cells in the presence of heat-inactivated serum (inactive serum) or non-treated serum (active serum). The percentage of CD41-positive C. albicans cells was determined after 1 h by flow cytometry. Active serum led to a significantly higher interaction with platelets compared inactive serum and a control without serum (asterisks indicate significant differences (p<0.05) determined by 1-Way ANOVA and Holm-Sidak’s multiple comparison test for pairwise comparisons of all groups). (B) Binding of isolated platelets to C. albicans cells in the presence of active serum from naïve mice, serum of mice collected after infection with C. albicans (reconvalescent) and serum from naïve C3-deficient mice analyzed after 1 h and 2 h. Reconvalescent serum led to significantly lower binding (significance determined by 1-Way ANOVA and Holm-Sidak’s multiple comparison test for pairwise comparisons of all groups within one time point and indicated by asterisks).
The observation that inactivation of human plasma increases interaction of thrombocytes with C. albicans suggests that complement factors interfere with binding of human platelets to the fungus. While it remains unclear how complement becomes activated in active human plasma upon contact with C. albicans, the classical, antibody-mediated complement activation pathway might be involved. Specific-pathogen free mice, as used in this study, are usually not colonized by C. albicans (35) and show comparatively low colonization with fungi (36), resulting in very low detectable anti-Candida antibodies in SPF mice (37). Thus, due to the absence of antibodies, whole murine blood and serum might lack the level of opsonization mediated by normal human plasma and, in consequence, lower opsonization could result in higher interaction with platelets. To test this hypothesis, we compared the binding of isolated thrombocytes to C albicans in the presence of active serum from normal SPF mice (naïve mice) and mice that survived a sublethal systemic C. albicans infection (reconvalescent mice). The binding of thrombocytes to fungal cells was significantly higher with serum from naïve mice; furthermore, naïve serum and serum from C3-deficient mice induced similar binding of thrombocytes to C. albicans (Figure 8B), supporting the hypothesis that the lack of prior contact to the fungus in SPF mice contributes to the observed differences between human and murine whole blood regarding interaction with thrombocytes.
Whole Murine Blood Is Not Able to Clear Bacterial Infections Ex Vivo
In order to determine whether the inability of murine blood to control infections ex vivo is restricted to C. albicans, we performed infections with S. aureus and E. coli as models for Gram-positive and Gram-negative bacteria, respectively. Similar to our observations with the filament-deficient C. albicans strain, CFU counts for both bacterial pathogens remained stable for the first 90 min followed by a steady increase (Figure 9A). This increase in pathogen load was even more pronounced than that observed for C. albicans Δefg1 Δcph1 (Figure 4A), and was also seen with a 10-fold lower infection dose, albeit at a lesser extent (Figure S7). Also similar to infection with the yeast-locked C. albicans mutant was the reduction of leukocyte numbers (Figure 9B), and for S. aureus especially neutrophil counts (Figure 9C), at late time points. The association of bacteria to immune cells varied quite drastically between both bacterial strains: While almost 60% of neutrophils were rapidly bound to S. aureus within 10 min and almost all neutrophils were attached to these Gram-positive bacteria 1.5 h post infection (Figure 9D), bacterial host cell association occurred much later for E. coli and a maximum of 50% of neutrophils were attached to bacteria at 2 h post infection (Figure 9E).
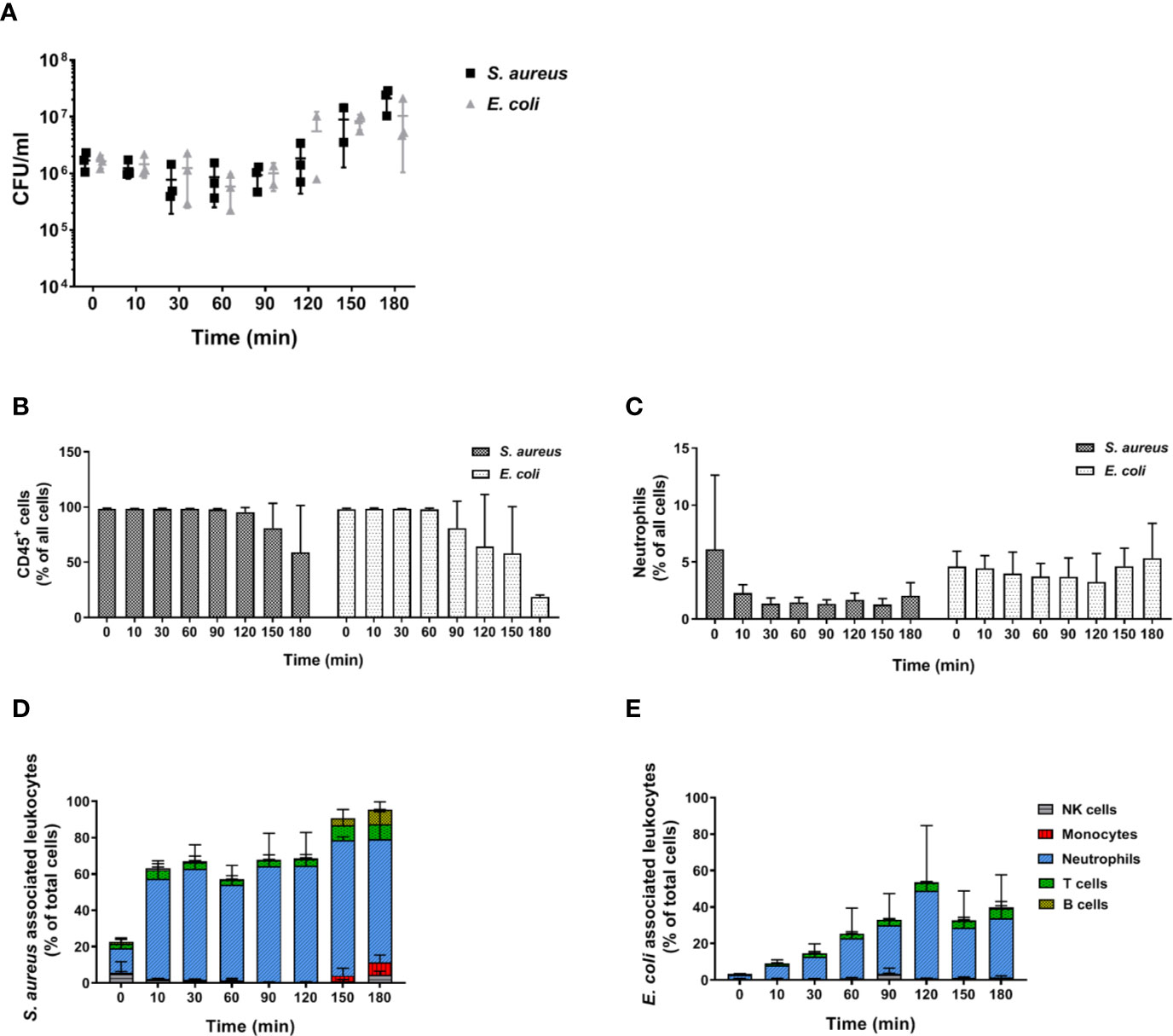
Figure 9 Bacterial survival and immune cell association in murine whole blood. (A) Colony forming units of S. aureus and E. coli were determined from infected murine blood. All values correspond to the means of three independent experiments with pooled whole-blood from 10 BALB/c mice infected ex vivo with 1 × 106/ml of GFP-expressing bacteria. Each dot represents the mean of an independent experiment +/- standard deviation (n = 3). The relative numbers of CD45+ cells (B) and of (C) neutrophils (Ly6G+/CD11b+) during the course of infection were measured by flow cytometry. (D) The percentage of various immune cells associated with GFP-expressing S. aureus or (E) with GFP-expressing E. coli was determined relative to their overall cell population in the blood. All values correspond to the means of three independent experiments with pooled whole blood from 10 mice.
Discussion
Intravenous infection of mice is the most commonly used model to study candidiasis (13). It is known that C. albicans disappears quickly from circulating blood within a few minutes to hours after infection (15, 16, 38), but the mechanisms underlying this phenomenon and the fungal interaction with blood components are not fully understood. We have previously employed a human whole-blood model to determine the interaction of C. albicans with leukocytes and the fate of the fungus. In this study, the model was adapted to mice to elucidate host-fungal interactions in whole blood over a period of 3 h.
One of the most striking differences observed between the murine and human whole-blood experiments was the lack of C. albicans killing in murine blood, both at the infectious dose used in our previous study on human blood (1 × 106/ml) and at a 10-fold lower dose (1 x 105/ml). A commonly used infectious dose in the intravenous mouse model is 2.5 × 104 CFU/g body weight, equivalent to 5 × 105 CFU for a 20 g mouse (13, 16). The approximate blood volume of a 20 g mouse is 1.6 ml (39), resulting in approximately 3 × 105 CFU injected per ml of blood. Thus, the doses used in this study are relevant for the in vivo mouse model. Consequently, the inability of murine blood to effectively eliminate the fungus cannot be explained by an inadequately high infectious dose. Furthermore, as comparable results were obtained with blood from BALB/c and C57BL/6 mice, low fungal killing is unlikely to be a consequence of the specific genetic background.
Differences were also observed in the interaction with immune cells: A higher proportion of fungal cells was not associated to any leukocytes in murine (40–50%) compared to human blood (10–20% (4),). Especially the proportion of C. albicans cells associated with neutrophils was substantially lower in mouse blood (<30% compared to >60% (4),). Neutrophils are known to be the major effector cells mediating C. albicans killing in human whole-blood infection (4), and they are essential for host defense against disseminated candidiasis both in humans (40, 41) and mice (42, 43). It is unlikely that the low number of C. albicans interacting with neutrophils is due to insufficient recognition, as the high proportion of neutrophils associated with C. albicans indicated that these leukocytes responded to the fungus. The lower absolute numbers, however, lead to a pathogen-to-neutrophil ratio that might have been too high to allow efficient infection control. Contrary to this hypothesis, reducing the infectious dose did not substantially increase fungal killing. It is possible that the abundance of other immune cells blocked physical contact between neutrophils and Candida. Consistent with this, many fungal cells were associated with lymphocytes during high dose infection, even though the proportion of lymphocytes interacting with Candida was low, indicating limited directed activity. We therefore increased the absolute number of murine neutrophils to that in human blood, thereby also altering the neutrophil-lymphocyte ratio. As this had only a minor impact on fungal killing, functional differences between murine and human immune cells are likely to contribute to fungal survival in murine blood.
While human and murine neutrophils share many characteristics, they differ in some functional aspects. For example, murine neutrophils do not produce defensins (44), express less myeloperoxidase and lysozyme (45), and differ in the expression and production of pattern recognition receptors, cytokines, and chemokines [summarized in (46)]. Ermert et al. showed that this translates into functional differences: Isolated murine neutrophils were less efficient in killing C. albicans in vitro than their human counterparts (31). Furthermore, while neutrophils were shown to be responsible for preventing hyphae development in human blood (18), isolated murine neutrophils fail to inhibit filamentation of phagocytosed yeast, resulting in fungal escape (31). Consistent with this, we observed substantial filamentation of C. albicans in murine, but not human blood. Thus, the higher filamentation rate in murine blood might be the consequence of an inability of murine neutrophils to control filamentation not only in vitro but also in the more complex ex vivo model. Killing of neutrophils by C. albicans filamentation after phagocytosis would furthermore explain the decline of neutrophil numbers over time in infected murine blood. However, extracellular filamentation has been shown to also occur in human blood (4), and we can therefore not exclude that the increased filamentation observed in murine blood is due to the lower number of fungal cells interacting with immune cells.
Although long filaments are more difficult to phagocytose due to their size (47), it is unlikely that filamentation alone is responsible for the lack of fungal killing, as survival and even proliferation in murine whole blood was observed for a filament-deficient C. albicans mutant. This was surprising because mutants lacking either of the genes (EFG1 and CPH1) deleted in the mutant used here were previously reported to display reduced survival in human blood (18), and the double mutant is avirulent in a systemic mouse infection model (48). The association rate of this mutant with leukocytes was however lower compared to wild type fungi (<25% compared to up to 50%). This is indicative of lower phagocytosis rates and likely leads to less phagocytosis-dependent fungal killing. The lower association rates could be due to the generally less efficient recognition of yeast versus hyphal cells by neutrophils (32, 49). The specific gene deletions furthermore affect the exposure of cell wall components serving as pathogen-associated molecular patterns (50–52). As the percentage of neutrophils interacting with the non-filamentous mutant was however only slightly less than for the wild type, it appears unlikely that altered recognition alone is responsible for proliferation of the filament-deficient mutant. This is supported by the cytokine measurements that demonstrate similar or higher responses of murine whole blood to the mutant compared to the wildtype, and is consistent with the higher cytokine release by monocytes and PBMCs in response to yeast cells observed by others (33, 53).
While our results overall suggest a very limited capacity of murine neutrophils to control C. albicans in murine blood, it should be noted that we observed a reduction of fungal burden at late time points in murine whole blood with added neutrophils. Although not statistically significant, this is in agreement with other studies demonstrating some efficacy of isolated murine neutrophils against this fungus (31, 54). In this context it should be noted that neutrophils are primed during recruitment to a site of inflammation, leading to increased expression of pattern recognition receptors and enhanced effector functions (55, 56). Thus, the limited impact of neutrophils in murine whole blood cannot be transferred to neutrophil efficacy in tissue in vivo. However, our results suggest that the rapid decline of C. albicans CFU numbers in the peripheral blood of intravenously infected mice is not due to fungal killing within circulating blood. Possibly, interaction between C. albicans and murine blood leukocytes in vivo occurs only after fungal attachment to endothelial cells (57, 58).
Host-dependent differences in the interaction of C. albicans with leukocytes were also observed for platelets: In the murine whole-blood model, aggregation of platelets around and adhesion to fungal cells was observed which was associated with platelet activation. Transient activation of platelets was also observed in vivo in mice, consistent with previous observations by others (38). In contrast, only few platelets associated with C. albicans in human blood, supporting recent findings by Eberl et al. (59). The absence of the formation of obvious platelet aggregates around fungal elements in human blood is consistent with other studies (60, 61), and might be mediated by fungal chitin (62). Importantly, others showed that only those human platelets that bound to C. albicans became activated (59). The low number of bound platelets likely results in a very small increase of activation in the overall platelet population. This could explain why several studies described that this fungus does not activate human platelets (60, 63, 64), whereas we detected a significant increase of released platelet effector molecules. Interestingly, we found that the physical interaction between C. albicans and human platelets was enhanced by plasma inactivation, suggesting that complement factors interfere with binding of human platelets to the fungus. In this context our observation that prior exposure of mice to C. albicans significantly affected the impact of their serum on the rate of interaction between the fungus and thrombocytes might be of relevance not only for this, but also for other studies and might have implications beyond C. albicans infection: SPF mice, which are commonly used to study infections, lack contact to common opportunistic pathogens and thus adaptive immune response to these microbes. As demonstrated here, this could explain some of the differences observed between mice and men, and might also contribute to the limited efficacy of murine whole blood to eliminate E. coli and S. aureus shown in this study. Even though S. aureus has been isolated from laboratory SPF mice colonies, over 70% of tested animals were found to be free of this opportunistic pathogen (65). It can also be assumed that SPF mouse colonies are free from pathogenic E. coli and thus naïve to the strain used in this study. Further research is clearly needed to investigate to which extent the lack of prior exposure affects host responses of mice in infection models.
In summary, we present here a thorough analysis of host-pathogen interaction in a murine whole-blood model, the first point of fungal interaction with the host immune system in experimental candidiasis, and provide time-resolved data on fungal survival and association to immune cells. Our results indicate substantial differences to the host-fungal interactions shown in a human whole-blood model (summarized in Table 1), thereby limiting the translatability of data obtained in a murine whole-blood model to human infections—even though the murine systemic infection model recapitulates several important aspects of systemic infection in humans. This could be at least in part due to the lack of C. albicans colonization of SPF mice.
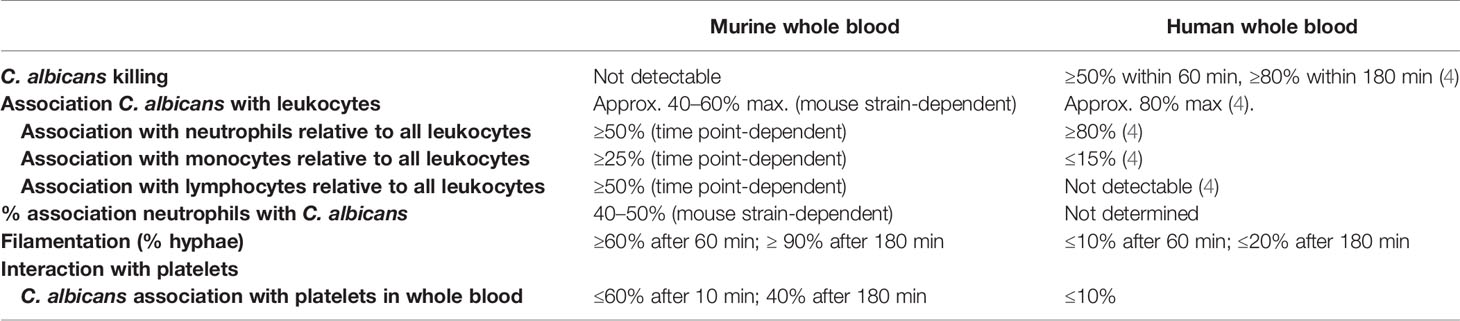
Table 1 Summary of the key differences between the murine and human whole-blood model for C. albicans.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the University Hospital Jena, permit number: 273-12/09. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Thüringer Landesamt für Verbraucherschutz, permit number 03-007/13 and HKI-19-003.
Author Contributions
SM, SS, IJ, BS-R, OK, KH, and CS conceived the study. SM, SS, CD, KS, WK, KH, CS, and GR performed the experiments. SM, KH, CS, and IJ analyzed the data. SM, CD, and IJ drafted the manuscript. SM, SS, KH, OK, CD, KS, WK, BS-R, CS, GR, and IJ revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Part of this project was funded by the funding line Strategic Networking in the Leibniz Association within the framework of the Leibniz Science Campus “InfectoOptics” (Project BLOODi to IJ) in Jena. CS was supported by the FWF Austrian Science Fund (Project Nr. P26117-B20). Work in the lab of OK and IJ was supported by the German Research Foundation (DFG; TRR 124 FungiNet, “Pathogenic fungi and their human host: Networks of Interaction,” DFG project number 210879364, Project C3 to OK and C5 to IJ). Funders had no role in study design, analyses and interpretation of data, in the writing of the report, and in the decision to submit the article for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Joanna M. Niemiec for help with neutrophil isolation, and Phillipp Kaemmer for technical assistance. SS is a member of the Jena School for Microbial Communication (JSMC).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.565869/full#supplementary-material
References
1. Watson KG, Holden DW. Dynamics of growth and dissemination of Salmonella in vivo. Cell Microbiol (2010) 12:1389–97. doi: 10.1111/j.1462-5822.2010.01511.x
2. Martinez RM, Wolk DM. Bloodstream Infections. Microbiol Spectr (2016) 4. doi: 10.1128/microbiolspec.DMIH2-0031-2016
3. Reimer LG, Wilson ML, Weinstein MP. Update on detection of bacteremia and fungemia. Clin Microbiol Rev (1997) 10:444–65. doi: 10.1128/CMR.10.3.444
4. Hünniger K, Lehnert T, Bieber K, Martin R, Figge MT, Kurzai O. A virtual infection model quantifies innate effector mechanisms and Candida albicans immune escape in human blood. PLoS Comput Biol (2014) 10:e1003479. doi: 10.1371/journal.pcbi.1003479
5. Hünniger K, Bieber K, Martin R, Lehnert T, Figge MT, Löffler J, et al. A second stimulus required for enhanced antifungal activity of human neutrophils in blood is provided by anaphylatoxin C5a. J Immunol (Baltimore Md: 1950) (2015) 194:1199–210. doi: 10.4049/jimmunol.1401845
6. Duggan S, Essig F, Hünniger K, Mokhtari Z, Bauer L, Lehnert T, et al. Neutrophil activation by Candida glabrata but not Candida albicans promotes fungal uptake by monocytes. Cell Microbiol (2015) 17:1259–76. doi: 10.1111/cmi.12443
7. Viscoli C. Bloodstream Infections: The peak of the iceberg. Virulence (2016) 7:248–51. doi: 10.1080/21505594.2016.1152440
8. Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, Kullberg BJ. Invasive candidiasis. Nat Rev Dis Primers (2018) 4:18026. doi: 10.1038/nrdp.2018.26
9. Vazquez JA, Sobel JD. Mucosal candidiasis. Infect Dis Clin North Am (2002) 16:793–820, v. doi: 10.1016/S0891-5520(02)00042-9
10. Perlroth J, Choi B, Spellberg B. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med Mycol (2007) 45:321–46. doi: 10.1080/13693780701218689
11. Pfaller M, Neofytos D, Diekema D, Azie N, Meier-Kriesche HU, Quan SP, et al. Epidemiology and outcomes of candidemia in 3648 patients: data from the Prospective Antifungal Therapy (PATH Alliance(R)) registry, 2004-2008. Diagn Microbiol Infect Dis (2012) 74:323–31. doi: 10.1016/j.diagmicrobio.2012.10.003
12. Koh AY, Köhler JR, Coggshall KT, Van Rooijen N, Pier GB. Mucosal damage and neutropenia are required for Candida albicans dissemination. PloS Pathog (2008) 4:e35. doi: 10.1371/journal.ppat.0040035
13. MacCallum DM. Hosting infection: experimental models to assay Candida virulence. Int J Microbiol (2012) 2012:363764. doi: 10.1155/2012/363764
14. Lionakis MS. New insights into innate immune control of systemic candidiasis. Med Mycol (2014) 52:555–64. doi: 10.1093/mmy/myu029
15. Lionakis MS, Lim JK, Lee CCR, Murphy PM. Organ-Specific Innate Immune Responses in a Mouse Model of Invasive Candidiasis. J Innate Immun (2011) 3:180–99. doi: 10.1159/000321157
16. MacCallum DM, Odds FC. Temporal events in the intravenous challenge model for experimental Candida albicans infections in female mice. Mycoses (2005) 48:151–61. doi: 10.1111/j.1439-0507.2005.01121.x
17. Doeing DC, Borowicz JL, Crockett ET. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin Pathol (2003) 3:3. doi: 10.1186/1472-6890-3-3
18. Fradin C, De Groot P, MacCallum D, Schaller M, Klis F, Odds FC, et al. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol Microbiol (2005) 56:397–415. doi: 10.1111/j.1365-2958.2005.04557.x
19. Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. albicans mutants are avirulent. Cell (1997) 90:939–49. doi: 10.1016/S0092-8674(00)80358-X
20. Walther A, Wendland J. An improved transformation protocol for the human fungal pathogen Candida albicans. Curr Genet (2003) 42:339–43. doi: 10.1007/s00294-002-0349-0
21. Balwit JM, van Langevelde P, Vann JM, Proctor RA. Gentamicin-Resistant Menadione and Hemin Auxotrophic Staphylococcus aureus Persist within Cultured Endothelial Cells. J Infect Dis (1994) 170:1033–7. doi: 10.1093/infdis/170.4.1033
22. Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, et al. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun (2000) 68:5385–92. doi: 10.1128/IAI.68.9.5385-5392.2000
23. Sreekantapuram S, Lehnert T, Prauße MTE, Berndt A, Berens C, Figge MT, et al. Dynamic Interplay of Host and Pathogens in an Avian Whole-Blood Model. Front Immunol (2020) 11:500. doi: 10.3389/fimmu.2020.00500
24. Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood (2002) 100:1869–77.
25. Ganguly S, Panetta JC, Roberts JK, Schuetz EG. Ketamine Pharmacokinetics and Pharmacodynamics Are Altered by P-Glycoprotein and Breast Cancer Resistance Protein Efflux Transporters in Mice. Drug Metab Dispos (2018) 46:1014–22. doi: 10.1124/dmd.117.078360
26. Ermert D, Urban CF, Laube B, Goosmann C, Zychlinsky A, Brinkmann V. Mouse neutrophil extracellular traps in microbial infections. J Innate Immun (2009) 1:181–93. doi: 10.1159/000205281
27. Davidson JA, Boom SJ, Pearsall FJ, Zhang P, Ramsay G. Comparison of the effects of four i.v. anaesthetic agents on polymorphonuclear leucocyte function. Br J Anaesth (1995) 74:315–8. doi: 10.1093/bja/74.3.315
28. Heller A, Heller S, Blecken S, Urbaschek R, Koch T. Effects of intravenous anesthetics on bacterial elimination in human blood in vitro. Acta Anaesthesiol Scand (1998) 42:518–26. doi: 10.1111/j.1399-6576.1998.tb05160.x
29. Nishina K, Akamatsu H, Mikawa K, Shiga M, Maekawa N, Obara H, et al. The inhibitory effects of thiopental, midazolam, and ketamine on human neutrophil functions. Anesth Analg (1998) 86:159–65. doi: 10.1097/00000539-199801000-00032
30. Chang Y, Chen TL, Sheu JR, Chen RM. Suppressive effects of ketamine on macrophage functions. Toxicol Appl Pharmacol (2005) 204:27–35. doi: 10.1016/j.taap.2004.08.011
31. Ermert D, Niemiec MJ, Rohm M, Glenthoj A, Borregaard N, Urban CF. Candida albicans escapes from mouse neutrophils. J Leukoc Biol (2013) 94:223–36. doi: 10.1189/jlb.0213063
32. Wozniok I, Hornbach A, Schmitt C, Frosch M, Einsele H, Hube B, et al. Induction of ERK-kinase signalling triggers morphotype-specific killing of Candida albicans filaments by human neutrophils. Cell Microbiol (2008) 10:807–20. doi: 10.1111/j.1462-5822.2007.01086.x
33. Mukaremera L, Lee KK, Mora-Montes HM, Gow NAR. Candida albicans Yeast, Pseudohyphal, and Hyphal Morphogenesis Differentially Affects Immune Recognition. Front Immunol (2017) 8:629. doi: 10.3389/fimmu.2017.00629
34. Jiménez-López C, Lorenz MC. Fungal immune evasion in a model host-pathogen interaction: Candida albicans versus macrophages. PLoS Pathog (2013) 9:e1003741. doi: 10.1371/journal.ppat.1003741
35. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science (New York NY) (2012) 336:1314–7. doi: 10.1126/science.1221789
36. Yeung F, Chen YH, Lin JD, Leung JM, McCauley C, Devlin JC, et al. Altered Immunity of Laboratory Mice in the Natural Environment Is Associated with Fungal Colonization. Cell Host Microbe (2020) 27(5):809–22. doi: 10.1016/j.chom.2020.02.015
37. Huertas B, Prieto D, Pitarch A, Gil C, Pla J, Díez-Orejas R. Serum Antibody Profile during Colonization of the Mouse Gut by Candida albicans: Relevance for Protection during Systemic Infection. J Proteome Res (2017) 16:335–45. doi: 10.1021/acs.jproteome.6b00383
38. Robert R, Nail S, Marot-Leblond A, Cottin J, Miegeville M, Quenouillere S, et al. Adherence of platelets to Candida species in vivo. Infect Immun (2000) 68:570–6. doi: 10.1128/IAI.68.2.570-576.2000
39. Mitruka BM, Rawnsley HM. Clinical biochemical and hematological reference values in normal experimental animals. New York: Masson Publishing USA Inc. (1977).
40. Horn DL, Neofytos D, Anaissie EJ, Fishman JA, Steinbach WJ, Olyaei AJ, et al. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis (2009) 48:1695–703. doi: 10.1086/599039
41. Gazendam RP, van de Geer A, Roos D, van den Berg TK, Kuijpers TW. How neutrophils kill fungi. Immunol Rev (2016) 273:299–311. doi: 10.1111/imr.12454
42. Romani L, Mencacci A, Cenci E, Puccetti P, Bistoni F. Neutrophils and the adaptive immune response to Candida albicans. Res Immunol (1996) 147:512–8. doi: 10.1016/S0923-2494(97)85216-9
43. Han Y, Cutler JE. Assessment of a mouse model of neutropenia and the effect of an anti-candidiasis monoclonal antibody in these animals. J Infect Dis (1997) 175:1169–75. doi: 10.1086/516455
44. Eisenhauer PB, Lehrer RI. Mouse neutrophils lack defensins. Infect Immun (1992) 60:3446–7. doi: 10.1128/IAI.60.8.3446-3447.1992
45. Rausch PG, Moore TG. Granule enzymes of polymorphonuclear neutrophils: A phylogenetic comparison. Blood (1975) 46:913–9. doi: 10.1182/blood.V46.6.913.bloodjournal466913
46. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol (2014) 5:508. doi: 10.3389/fimmu.2014.00508
47. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15:1017–25. doi: 10.1038/ni.2987
48. Lo HJ, Köhler JR, DiDomenico B, Loebenberg D. A. Cacciapuoti, and G.R. Fink, Nonfilamentous C. albicans mutants are avirulent. Cell (1997) 90:939–49. doi: 10.1016/S0092-8674(00)80358-X
49. Rudkin FM, Bain JM, Walls C, Lewis LE, Gow NA, Erwig LP. Altered dynamics of Candida albicans phagocytosis by macrophages and PMNs when both phagocyte subsets are present. mBio (2013) 4:e00810–13. doi: 10.1128/mBio.00810-13
50. Zavrel M, Majer O, Kuchler K, Rupp S. Transcription factor Efg1 shows a haploinsufficiency phenotype in modulating the cell wall architecture and immunogenicity of Candida albicans. Eukaryot Cell (2012) 11:129–40. doi: 10.1128/EC.05206-11
51. Cottier F, Sherrington S, Cockerill S, Del Olmo Toledo V, Kissane S, Tournu H, et al. Remasking of Candida albicans β-Glucan in Response to Environmental pH Is Regulated by Quorum Sensing. mBio (2019) 10. doi: 10.1128/mBio.02347-19
52. Wartenberg A, Linde J, Martin R, Schreiner M, Horn F, Jacobsen ID, et al. Microevolution of Candida albicans in macrophages restores filamentation in a nonfilamentous mutant. PLoS Genet (2014) 10:e1004824. doi: 10.1371/journal.pgen.1004824
53. Torosantucci A, Chiani P, Cassone A. Differential chemokine response of human monocytes to yeast and hyphal forms of Candida albicans and its relation to the beta-1,6 glucan of the fungal cell wall. J Leukoc Biol (2000) 68:923–32.
54. Behnsen J, Narang P, Hasenberg M, Gunzer F, Bilitewski U, Klippel N, et al. Environmental dimensionality controls the interaction of phagocytes with the pathogenic fungi Aspergillus fumigatus and Candida albicans. PLoS Pathog (2007) 3:e13. doi: 10.1371/journal.ppat.0030013
55. Yao Y, Matsushima H, Ohtola JA, Geng S, Lu R, Takashima A. Neutrophil priming occurs in a sequential manner and can be visualized in living animals by monitoring IL-1β promoter activation. J Immunol (Baltimore Md: 1950) (2015) 194:1211–24. doi: 10.4049/jimmunol.1402018
56. Miralda I, Uriarte SM, McLeish KR. Multiple Phenotypic Changes Define Neutrophil Priming. Front Cell Infect Microbiol (2017) 7:217. doi: 10.3389/fcimb.2017.00217
57. Grubb SE, Murdoch C, Sudbery PE, Saville SP, Lopez-Ribot JL, Thornhill MH. Candida albicans-endothelial cell interactions: a key step in the pathogenesis of systemic candidiasis. Infect Immun (2008) 76:4370–7. doi: 10.1128/IAI.00332-08
58. Filler SG, Pfunder AS, Spellberg BJ, Spellberg JP, Edwards JE Jr. Candida albicans stimulates cytokine production and leukocyte adhesion molecule expression by endothelial cells. Infect Immun (1996) 64:2609–17. doi: 10.1128/IAI.64.7.2609-2617.1996
59. Eberl C, Speth C, Jacobsen ID, Hermann M, Hagleitner M, Deshmukh H, et al. Candida: Platelet Interaction and Platelet Activity in vitro. J Innate Immun (2019) 11:52–62. doi: 10.1159/000491030
60. Schultz CM, Goel A, Dunn A, Knauss H, Huss C, Launder D, et al. Stepping Up to the Plate(let) against Candida albicans. Infect Immun (2020) 88:00784-19. doi: 10.1128/IAI.00784-19
61. Willcox MD, Webb BC, Thakur A, Harty DW. Interactions between Candida species and platelets. J Med Microbiol (1998) 47:103–10. doi: 10.1099/00222615-47-2-103
62. Leroy J, Bortolus C, Lecointe K, Parny M, Charlet R, Sendid B, et al. Fungal Chitin Reduces Platelet Activation Mediated via TLR8 Stimulation. Front Cell Infect Microbiol (2019) 9:383. doi: 10.3389/fcimb.2019.00383
63. Bertling A, Niemann S, Uekötter A, Fegeler W, Lass-Flörl C, von Eiff C, et al. Candida albicans and its metabolite gliotoxin inhibit platelet function via interaction with thiols. Thromb Haemost (2010) 104:270–8. doi: 10.1160/TH09-11-0769
64. Woth G, Tőkés-Füzesi M, Magyarlaki T, Kovács GL, Vermes I, Mühl D. Activated platelet-derived microparticle numbers are elevated in patients with severe fungal (Candida albicans) sepsis. Ann Clin Biochem (2012) 49:554–60. doi: 10.1258/acb.2012.011215
65. Schulz D, Grumann D, Trübe P, Pritchett-Corning K, Johnson S, Reppschläger K, et al. Laboratory Mice Are Frequently Colonized with Staphylococcus aureus and Mount a Systemic Immune Response-Note of Caution for In vivo Infection Experiments. Front Cell Infect Microbiol (2017) 7:152. doi: 10.3389/fcimb.2017.00152
Keywords: whole blood ex vivo model, host-pathogen interaction, Candida albicans, neutrophils, mice
Citation: Machata S, Sreekantapuram S, Hünniger K, Kurzai O, Dunker C, Schubert K, Krüger W, Schulze-Richter B, Speth C, Rambach G and Jacobsen ID (2021) Significant Differences in Host-Pathogen Interactions Between Murine and Human Whole Blood. Front. Immunol. 11:565869. doi: 10.3389/fimmu.2020.565869
Received: 26 May 2020; Accepted: 30 November 2020;
Published: 15 January 2021.
Edited by:
Markus M. Heimesaat, Charité-Universitätsmedizin Berlin, GermanyReviewed by:
Caroline N. Jones, The University of Texas at Dallas, United StatesSunil Shaw, Brown University, United States
Copyright © 2021 Machata, Sreekantapuram, Hünniger, Kurzai, Dunker, Schubert, Krüger, Schulze-Richter, Speth, Rambach and Jacobsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilse D. Jacobsen, aWxzZS5qYWNvYnNlbkBsZWlibml6LWhraS5kZQ==
†Present address: Bianca Schulze-Richter, Institute of Immunology, Molecular Pathogenesis, Center for Biotechnology and Biomedicine (BBZ), College of Veterinary Medicine, University of Leipzig, Leipzig, Germany