 Yuki Saito1,2†
Yuki Saito1,2† Maki Miyajima2
Maki Miyajima2 Sena Yamamoto2Tsukasa Sato2Norihiro Miura2Mineko Fujimiya1
Sena Yamamoto2Tsukasa Sato2Norihiro Miura2Mineko Fujimiya1 Takako S. Chikenji1,2*†
Takako S. Chikenji1,2*†- 1Department of Anatomy, Sapporo Medical University School of Medicine, Sapporo, Japan
- 2Department of Health Sciences, School of medicine, Hokkaido University, Sapporo, Japan
Neuropsychiatric manifestations targeting the central, peripheral, and autonomic nervous system are common in systemic lupus erythematosus (SLE); collectively, these symptoms are termed neuropsychiatric SLE (NPSLE). Among a wide variety of neuropsychiatric symptoms, depression is observed in about 24-39% of SLE patients. Several cytokines and chemokines have been identified as biomarkers or therapeutic targets of NPSLE; in particular, the levels of type 1 interferons, TNFs, and IL-6 are elevated in SLE patient’s cerebrospinal fluid (CSF), and these factors contribute to the pathology of depression. Here, we show that senescent neural cells accumulate in the hippocampal cornu ammonis 3 (CA3) region in MRL/lpr SLE model mice with depressive behavior. Furthermore, oral administration of fisetin, a senolytic drug, reduced the number of senescent neural cells and reduced depressive behavior in the MRL/lpr mice. In addition, transcription of several senescence and senescence-associated secretory phenotype (SASP) factors in the hippocampal region also decreased after fisetin treatment in the MRL/lpr mice. These results indicate that the accumulation of senescent neural cells in the hippocampus plays a role in NPSLE pathogenesis, and therapies targeting senescent cells may represent a candidate approach to treat NPSLE.
Introduction
Systemic lupus erythematosus (SLE) is a currently incurable autoimmune disease characterized by hyperactive immune cells, serum autoantibodies, and multiple organ damage involving the kidney, skin, vasculature, and brain (1). Neuropsychiatric manifestations targeting the central, peripheral, and autonomic nervous system are common in SLE; collectively, these symptoms are called neuropsychiatric SLE (NPSLE). Up to 75% of patients experience central nervous system (CNS) involvement, and 60% of SLE patients experience autonomic symptoms (2–4). Among a wide variety of neuropsychiatric symptoms, depression is observed in about 24-39% of SLE patients (5). Various immune effectors contribute to SLE pathogenesis, including autoantibodies, cytokines, and cell-mediated inflammation (2, 4, 6, 7); however, the detailed mechanism underlying NPSLE remains largely unknown (2, 4, 6, 7).
Cellular senescence is a state of irreversible cell cycle arrest in which an adaptive response is induced by multiple stressors (8, 9). Although senescence serves as a defense mechanism that limits tumorigenesis to maintain tissue homeostasis, accumulation of senescent cells causes age-related disease and chronic inflammation in lung, kidney, heart, and muscle, through the secretion of pro-inflammatory molecules including cytokines, chemokines, and proteases; collectively, these factors are referred to as the senescence-associated secretory phenotype (SASP) (8–13). Prolonged exposure to the SASP leads to pathological changes that contribute to tissue and organ decline (8). Senescent cells contribute to the neurodegeneration and pathogenesis of the brain observed in Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (14–18). For example, in Alzheimer’s disease model mice, astrocytes, microglia, and oligodendrocyte progenitor cells have features of senescence, and elimination of senescent cells via genetic or pharmacological treatment attenuates neuroinflammation and cognitive deficits (17, 18). Chronic neuroinflammation is one of the hallmarks of Parkinson’s disease. The expression levels of pro-inflammatory factors and proteases, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), and interferon-gamma (IFN-γ) and metalloproteinase-3 (MMP-3), which are canonical SASP factors, are elevated in the brains of patients with Parkinson’s disease (19). Furthermore, the number of senescent astrocytes and dopaminergic neurons is elevated, and these senescent cells have the potential to contribute to pathology (14, 15). Although the mechanism by which cellular senescence is linked to neurodegeneration is not fully understood, the accumulation of senescent cells may trigger a chronic inflammatory response that contributes to synapse damage and cognitive decline (20). NPSLE causes a disruption of the blood–brain barrier, which is directly caused by cytokines and complement proteins (21). Pro-inflammatory cytokines and chemokines related to neuroinflammation in NPSLE were identified in the cerebrospinal fluid (CSF) of SLE patients for use as biomarkers or therapeutic targets; in particular, the levels of type 1 interferons, TNFs, and IL-6 are elevated and contribute to the pathology of depression (4, 6, 8, 22). Overexpression of these pro-inflammatory factors in the CSF of NPSLE patients is hypothesized to cause cellular senescence in CNS; however, to the best of our knowledge, cellular senescence in the CNS has not been evaluated in patients with lupus (22–30).
In this study, we sought to determine the relationship between senescence and depression in SLE by investigating cellular senescence in the hippocampus, which is associated with depression (31–34), in MRL/lpr SLE model mice. In addition, we investigated whether senolytics, small molecules that selectively eliminate senescent cells, reduce the observed number of senescent cells and consequently reduce depression symptoms in MRL/lpr SLE model mice.
Materials and Methods
Mice
The Committee of the Animal Experimentation Center of the Sapporo Medical University School of Medicine approved all animal protocols (#17-080 and #21-051). Mice were maintained in an enclosed, specific pathogen–free facility with a 12 h light and dark cycle. Female MRL/lpr mice were used as an SLE mouse model, and haplotype-matched female MRL/MPJ mice were used as phenotypic controls (Sankyo Lab Service, Tokyo, Japan). For pathological analysis, four MRL/MPJ mice and five MRL/lpr mice were used and euthanized at 18 weeks of age. For senolytic treatment, twenty-four MRL/MPJ mice and twenty-four MRL/lpr mice were used and euthanized at 22 weeks of age, and tissue samples were harvested.
Behavioral Analysis
The tail suspension test was performed to assess depression-like behavior (35–39). Mice were suspended by their tails with tape 60 cm above the floor for 6 min, and the time of immobility was measured. Each mouse was tested only once. The time of immobility was defined as the time when the animal stopped struggling for ≥ 1 s, which was measured using a video tracking system (ANY-maze; Muromachi Kikai, Tokyo, Japan).
Cell Culture and Senescence Induction
Neuro-2a cells (Cell No. IFO50081), which are a mouse brain–derived neuroblast cell line, were obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan) and maintained in Eagle’s Minimal Essential Medium with non-essential amino acids and 10% fetal bovine serum. Cells were tested for mycoplasma using the e-Myco Mycoplasma PCR Detection Kit (iNtRON Biotechnology, Seongnam-si, South Korea). Cellular senescence was induced by X-ray irradiation. Neuro-2a cells were exposed to 10 Gy irradiation using an X-Ray Irradiator (MBR-1520-3; HITACHI, Tokyo, Japan), and 3 days later the cells were passaged to avoid confluency. Six days after irradiation, Neuro-2a cells were harvested and subjected to SA-β-Gal staining, PCR analysis, and pharmacological experiments. To detect cellular senescence, we performed senescence-associated β-Galactosidase (SA-β-Gal) staining using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, Danvers, MA, USA). Cells were observed using an inverted microscope (Primovert; ZEISS), and the percentage of SA β-Gal–positive cells was calculated by dividing the number of SA β-Gal–positive cells by the total number of cells observed. The cell size was measured using the ImageJ software (National Institutes of Health). Briefly, the cell body was outlined using the drawing/selection tools, and the area was measured using the analyze tool.
In Vitro Senolytic Treatment
For senolytic treatment, fisetin flavonoid, which is found in many fruits and vegetables and was previously identified as a senolytic compound (40), was used. Fisetin (S2298) was purchased from Selleck (Houston, TX, USA) and dissolved in DMSO before use. Irradiated senescent Neuro-2a cells and non-irradiated Neuro-2a cells were seeded on a 96-well black/clear bottom plate at 40,000 cells and 10,000 cells per well, respectively. After senescence induction for 6 days, 5µM, 10µM, or 20µM fisetin was added and the cells were incubated for 48 h. The concentration of fisetin used was based upon a previous study that reported its senolytic effects (40). Cell number and cellular senescence were determined by DAPI staining and SPiDER-β-Gal staining, respectively. Briefly, cells were washed twice with PBS, fixed in 4% paraformaldehyde at room temperature for 5 min, and washed twice with PBS. Sections were incubated in 20 µM SPiDER-β-gal (Dojindo) in solution in McIlvaine buffer (pH 6.0) for 60 min at 37°C. After washing of tissue sections, nuclei were stained with DAPI. Cells were observed using fluorescence microscopy (Axio Observer7; ZEISS).
In Vivo Senolytic Treatment
Eighteen-week–old MRL/MPJ mice (n = 24) and MRL/lpr mice (n = 24) were randomized for pharmacological treatment analysis. For oral administration, mice were gavaged with 100 mg/kg fisetin (Tokyo Chemical Industry, Tokyo, Japan) (MRL/MPJ: n = 12 and MRL/lpr: n = 12) or vehicle (20% PEG 400) (MRL/MPJ: n = 12 and MRL/lpr: n = 12) for 5 days every week for 4 weeks.
Immunohistochemistry and SPiDER-β-Gal Staining
Brain samples were fixed in 4% paraformaldehyde overnight. The following day, the tissues were transferred to 20% sucrose in phosphate buffer, incubated overnight, frozen in OCT compound in liquid nitrogen, and stored at −80°C until use. Cryosections (8 µm thick) were prepared using a cryostat. The sections were incubated in 0.01 M PBS containing 0.3% Triton-X (PBS-T) and treated with 2% BSA for 60 min at RT. After washing with 0.01 M PBS-T, the sections were incubated with primary antibodies at 4°C overnight, followed by secondary staining. Alexa Fluor 594–conjugated anti-GFAP (1:100; 644708; Biolegend), anti–Iba-1 (1:400; 019-19741; Wako, Osaka, Japan), and anti-NeuN (1:150; 2697501; Proteintech, Rosemont, IL, USA) were used as primary antibodies. Cy3-conjugated IgG (1:400; Jackson ImmunoResearch, West Grove, PA, USA) was used as a secondary antibody, and nuclei were stained with DAPI (1:1000; Dojindo). After washing, tissue sections were mounted with Vectashield (Vector Laboratories). For the SPiDER-β-gal stain, tissue sections were incubated in 20 µM SPiDER-β-gal (Dojindo) in solution in McIlvaine buffer (pH 6.0) for 60 min at 37°C. After washing of tissue sections, nuclei were stained with DAPI, and tissue sections were mounted with VECTASHIELD. Sections were observed by fluorescence microscopy [Axio Observer7 (ZEISS) or BZ-X700 (Keyence)].
RNA Extraction and Quantitative Real-Time PCR
Total RNA was isolated using the Tri Reagent (Molecular Research Center, Cincinnati, OH, USA), and RNA was reverse transcribed into cDNA using the iScript Advanced cDNA Synthesis Kit (1725038; Bio-Rad, Hercules, CA, USA). Quantitative PCR was performed using SsoAdvanced Universal SYBR Green Supermix (172-5270; Bio-Rad) on a QuantStudio3 Real-Time PCR System (Applied Biosystems). Cycling conditions were as follows: 95°C for 20 s, followed by 40 cycles of amplification (95°C for 15 s and 60°C for 1 min). Transcription levels were normalized against the corresponding levels of housekeeping genes listed in Supplementary Table 1. Specific primer sequences used for PCR are listed in Supplementary Table 1. The ΔΔCt method was used to compare data.
Statistical Analysis
Quantitative data are shown as means and standard errors in dot plots generated by ggplot2, a plotting system for R based on The Grammar of Graphics (The R Foundation for Statistical Computing, Vienna, Austria). Normality was assessed using the Shapiro–Wilk test. The pairwise t-test was used for comparison between two groups, and a one-way analysis of variance (ANOVA) was conducted to assess differences among three groups or more. Pairwise comparisons were made only when one-way ANOVA indicated statistical significance. P-values for multiple comparisons were adjusted by the Tukey method. Statistical analyses were performed using EZR, a graphical user interface for R (41). Two-sided P-values less than 0.05 were considered statistically significant.
Results
MRL/lpr Mice Exhibit a Depression-Like Phenotype and Neuroinflammation in the Hippocampus
We used 18-week-old MRL/lpr mice as a SLE model and MRL/MPJ mice as controls. The presence of depression-like behavior was evaluated by tail suspension test. The immobility time observed during the tail suspension test was significantly elevated in MRL/lpr mice relative to the MRL/MPJ mice, which indicated that the MRL/lpr mice exhibited a depression-like phenotype (Figures 1A, B). To determine whether MRL/lpr mice exhibited neuroinflammation, we counted GFAP-positive astrocytes and Iba-1–positive microglia in the hippocampus, which are regions that may be important in regulation of emotion in brains of MRL/lpr mice (42). MRL/lpr mice had more GFAP-positive astrocytes in the cornu ammonis 3 (CA3) region than MRL/MPJ mice (Figures 1C, D; P=0.009). In the dentate gyrus (DG) region, the number of GFAP-positive astrocytes did not significantly differ between the MRL/lpr and MRL/MPJ mice (Figures 1E, F; P=0.108). Senescent cells contribute to neuroinflammation (14–18, 43); therefore, we counted the number of senescent cells, using the marker SPiDER-β-Gal, in GFAP-positive astrocytes. We could not identify SPiDER-β-Gal–positive astrocytes in either MRL/lpr or MRL/MPJ. The number of Iba-1 positive microglia was higher in MRL/lpr mice than in MRL/MPJ mice in the CA3 region (Figures 1G, H; P = 0.033), but not in the DG region (Figures 3C, D; P = 0.429). Small numbers of SPiDER-β-Gal–positive microglia were present in both of MRL/lpr and MRL /MPJ mice; however, microglia were not the major population of SPiDER-β-Gal–positive senescent cells (Figures 1I, J).
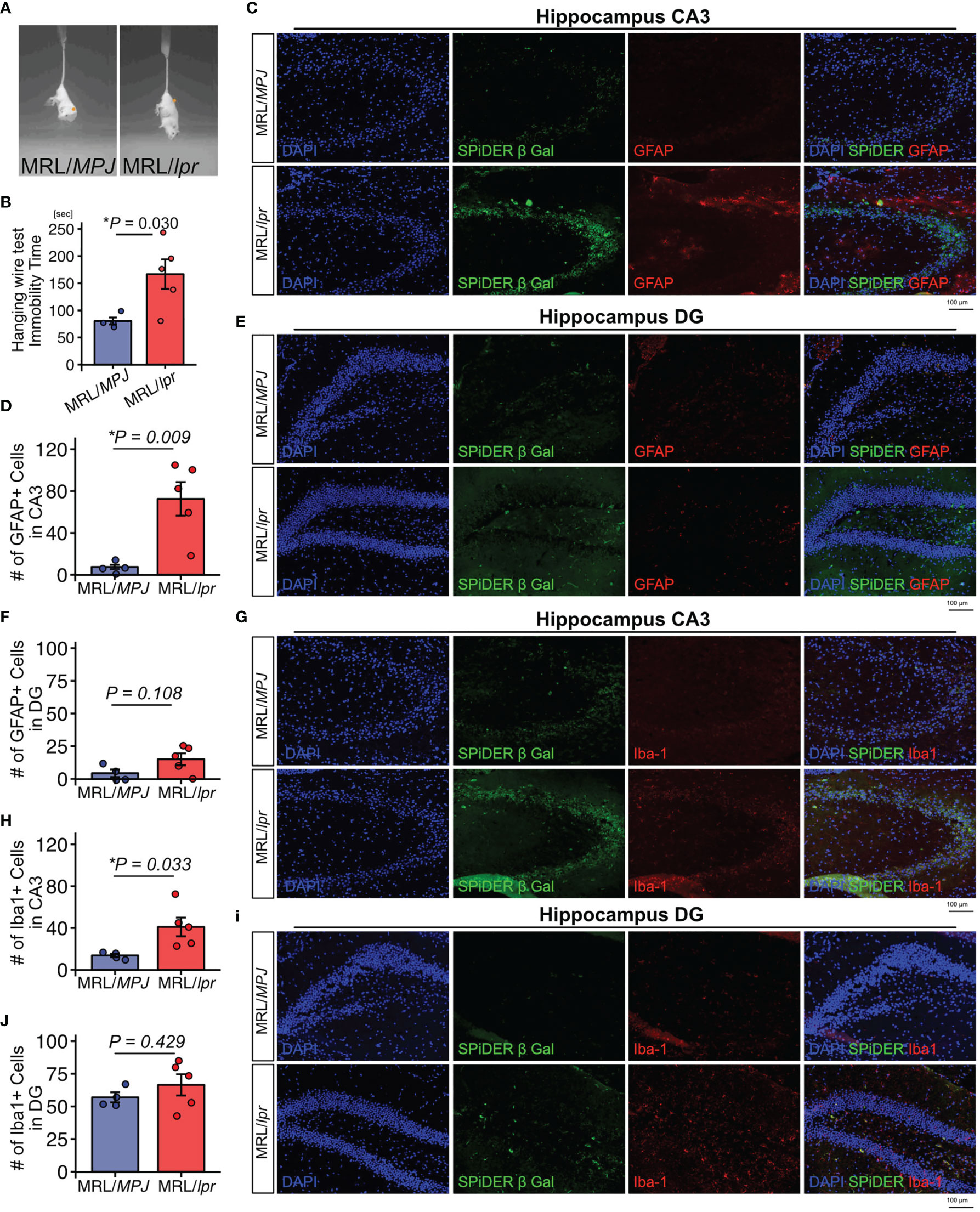
Figure 1 MRL/lpr mice exhibit a depression-like phenotype and have higher proportions of GFAP-positive and Iba-1–positive cells in the hippocampus. (A) Representative images of tail suspension test of MRL/lpr mice (SLE model) and MRL/MPJ mice (controls). (B) Quantitation of immobility time in MRL/MPJ and MRL/lpr mice during tail suspension test. (C) Representative images of GFAP immunostaining of the hippocampus CA3 regions from MRL/MPJ and MRL/lpr mice and (D) the corresponding quantitative data. (E) Representative images of GFAP immunostaining of the hippocampus DG regions from MRL/MPJ and MRL/lpr mice and (F) the corresponding quantitative data. (G) Representative images of Iba-1 immunostaining of the hippocampus CA3 regions from MRL/MPJ and MRL/lpr mice and (H) the corresponding quantitative data. (I) Representative images of Iba-1 immunostaining of the hippocampus DG regions from MRL/MPJ and MRL/lpr mice and (J) the corresponding quantitative data. Quantitative data are shown as means ± SEs in dot plots. P-values were determined by two-tailed Student’s t-test. (*P < 0.05) .
MRL/lpr Mice Have a Higher Proportion of Senescent NeuN+ Cells in CA3 Hippocampus
Next, to determine whether neurons exhibited features of senescence, we performed SPiDER-β-Gal staining and immunofluorescence with a NeuN antibody. SPiDER-β-Gal intensity in NeuN-positive cells was significantly higher in MRL/lpr mice than in MRL/MPJ mice in the hippocampus CA3 region (Figures 2A–C; P = 0.037). By contrast, in the DG region, SPiDER β-Gal expression was not detectable in either MRL/lpr or MRL/MPJ mice (Figure 2B).
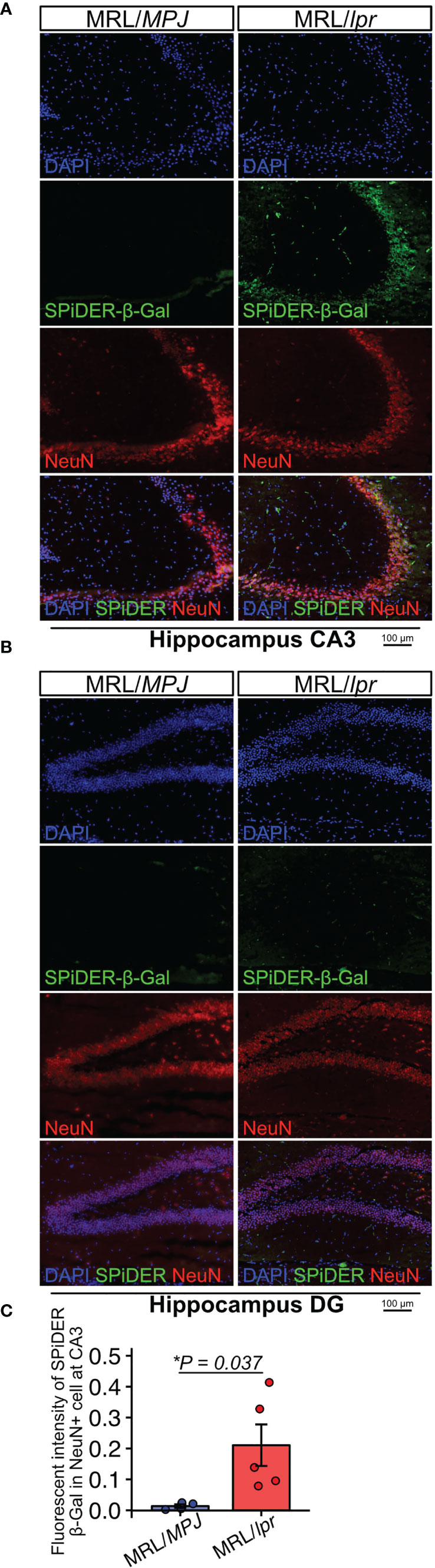
Figure 2 MRL/lpr mice have higher numbers of SPiDER-β-Gal– and NeuN-positive cells in the hippocampus. (A, B) Representative images of NeuN immunostaining and SPiDER-β-Gal staining of the hippocampus CA3 and DG regions in MRL/MPJ and MRL/lpr mice. (C) Quantitation of SPiDER-β-Gal intensity in NeuN-positive cells in MRL/MPJ and MRL/lpr mice. Quantitative data are shown as means ± SEs in dot plots. P-values were determined by two-tailed Student’s t-test. (*P < 0.05).
Neuro-2a Cells Induced to Senesce by Irradiation Exhibit a Neuroinflammatory Phenotype
These histological analyses indicated that neural cells in the CA3 region were a major population of senescent cells in lupus model mice with a depression-like phenotype. We next investigated whether senescent neural cells exhibited the features of cells that induce neuroinflammation in NPSLE. To induce senescence, we exposed Neuro-2a cells to 10 Gy irradiation and passaged them 3 days later to avoid the over-confluency that occurs post-irradiation due to Neuro-2a cell enlargement. Six days after irradiation, we harvested the cells and subjected them to SA-β-Gal staining and quantitative PCR analysis. Irradiated Neuro-2a cells exhibited senescent features including SA-β-Gal expression (Figures 3A, B), elevated cell size (Figure 3C), and upregulation of Cdkn2a (Ink4a and Arf), Cdkn2b, Cdkn1a, and Trp53 (Figure 3D). The irradiated Neuro-2a cells also expressed high levels of genes encoded by SASP factors, including Tnfa, Serpine1, Il6, and Il1b, all of which are also upregulated in NPSLE (Figures 3E, F) (4).
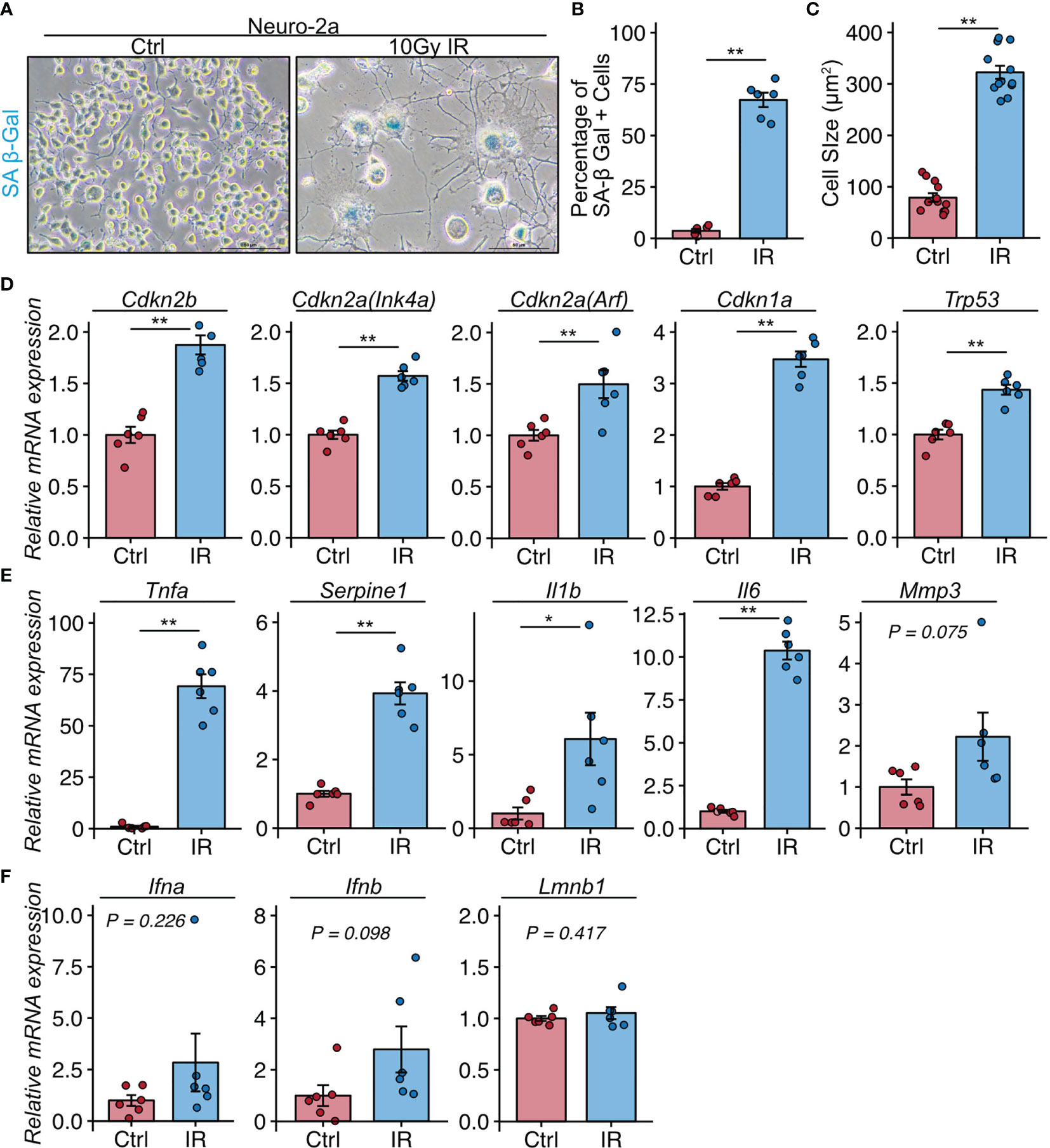
Figure 3 Neuro-2a cells induced to undergo senescence by irradiation exhibit a neuroinflammatory phenotype. (A) Representative images of SA-β-Gal expression after 10 Gy ionizing radiation (IR) in Neuro-2a cells in randomly chosen fields of view (n = 6 per group). (B, C) Quantitation of SA-β-Gal–positive cells and the cell size. (D–F) Relative mRNA expression of senescence and SASP-related genes in Neuro-2a cells with or without 10 Gy IR. Quantitative data are shown as means ± SEs in dot plots. P-values were determined by two-tailed Student’s t-test. (*P < 0.05, **P < 0.01).
Fisetin Treatment Selectively Kills SPiDER-β-Gal–Positive Senescence Neural Cells In Vitro
Next, we investigated whether the senolytic drug fisetin would selectively kill senescent neural cells. Fisetin, a flavonoid found in many fruits and vegetables, was previously identified as a senolytic compound (40). In addition, fisetin exhibits brain uptake potential and penetrates the blood–brain barrier more effectively than other flavonoids, including quercetin, luteolin, and myricetin (44–46). Hence, we used fisetin as a senolytic compound in this study. Unirradiated and irradiated Neuro-2a cells were treated with serial concentrations of fisetin (0–20 µM) for 48 h. The observed number of irradiated Neuro-2a cells significantly decreased in a dose-dependent manner (Figures 4A, B), and the number of control Neuro-2a cells significantly decreased after treatment with 20 µM fisetin (Figures 4A, B). SPiDER-β-Gal expression in irradiated Neuro-2a cells was significantly reduced at doses of 5, 10, and 20 µM (Figure 4C). Doses of 5 and 10 µM fisetin decreased the fraction of SPiDER-β-Gal–positive senescent Neuro-2a cells without affecting non-irradiated proliferating cells.
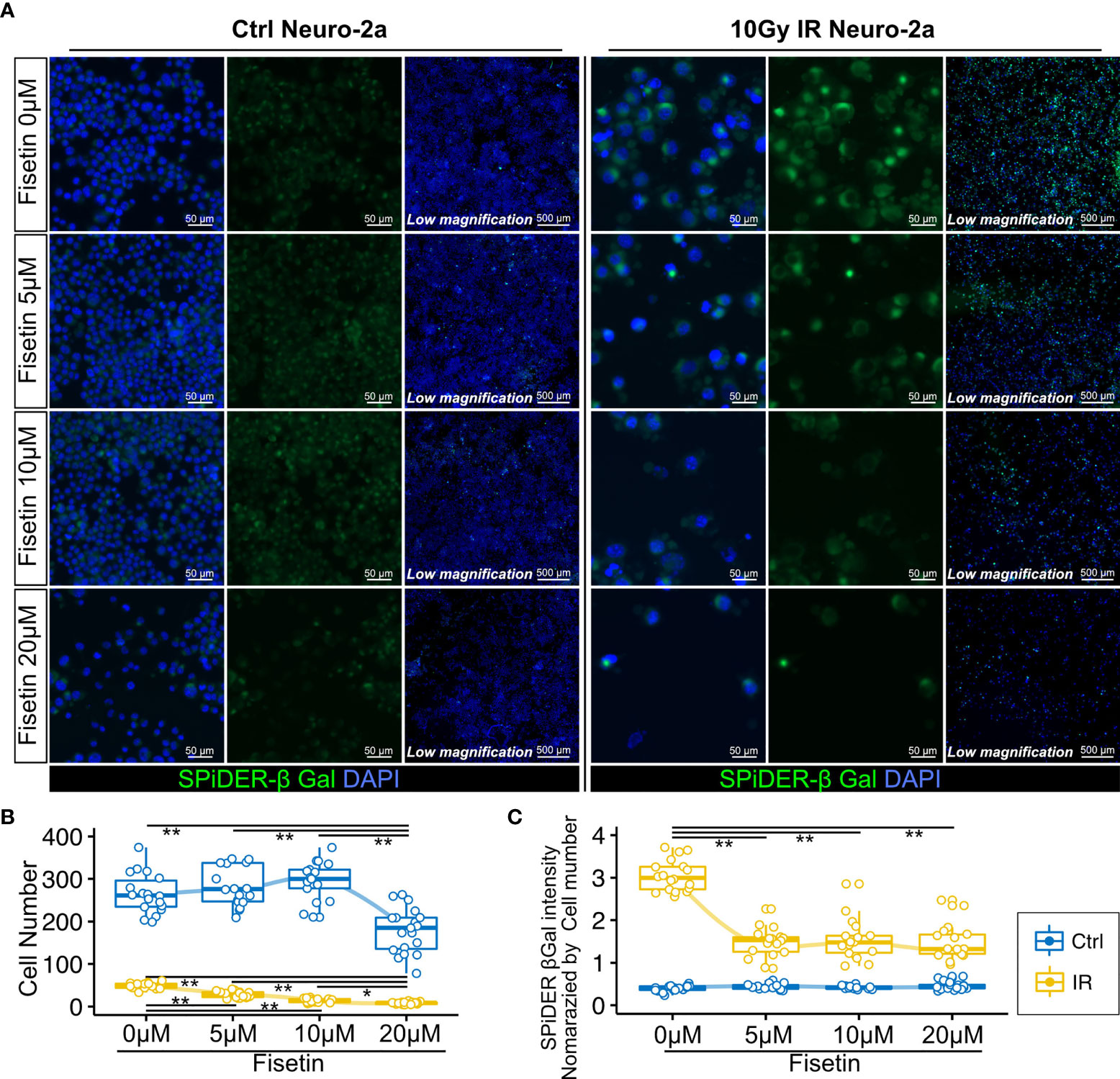
Figure 4 Effect of fisetin treatment on senescent Neuro-2a cells in vitro. (A) Representative images of SPiDER-β-Gal and F-actin staining in Neuro-2a cells with or without 10 Gy IR. (B, C) Quantitation of cell number and SPiDER-β-Gal intensity in Neuro-2a cells treated with the indicated concentrations of fisetin (0–20 µM) for 48 h. Quantitative data are shown as medians with IQRs and 1.5 times the IQR, and are displayed as dot plots and box-and-whisker plots. P-values were determined by one-way ANOVA adjusted by the Tukey method. P-values were determined by two-tailed Student’s t-test. (*P < 0.05 and **P < 0.01).
Fisetin Treatment Reduced the Prevalence of the Depression-Like Phenotype and Number of Senescent Cells In Vivo
To examine the senolytic effect of fisetin in vivo, we orally administered fisetin (100 mg/kg) or 20% PEG400 (as a control) to MRL/lpr (n=12 and 12, respectively) and MRL/MPJ (n=6 and 6, respectively) mice for 5 days every week for 4 weeks (Figure 5A). During this 4-week period, two MRL/lpr mice in the vehicle group died. Fifty percent of MRL/lpr mice die from renal failure by 24 weeks of age (47). After this 4-week period, we found that fisetin treatment reduced the prevalence of depression-like behavior in the MRL/lpr mice (Figure 5B). Fisetin treatment also reduced the observed SPiDER-β-Gal expression level in NeuN+ cells in the CA3 region (Figures 5C, D). PCR analysis showed that fisetin treatment reduced the mRNA transcription levels of Cdkn1a and Cdkn2a(Arf), which are senescence factors, and Ifna and Ifnb, which are known SASP factors, in the hippocampus of the MRL/lpr mice (Figures 5E, F). In addition, vehicle-treated MRL/lpr mice exhibited significantly increased levels of Trp53, Il6, and Mmp3 mRNA transcription relative to the vehicle-treated MRL/MPJ mice, but no significant difference was observed for the fisetin-treated MRL/lpr mice relative to the vehicle-treated MRL/MPJ mice (Figures 5E, F).
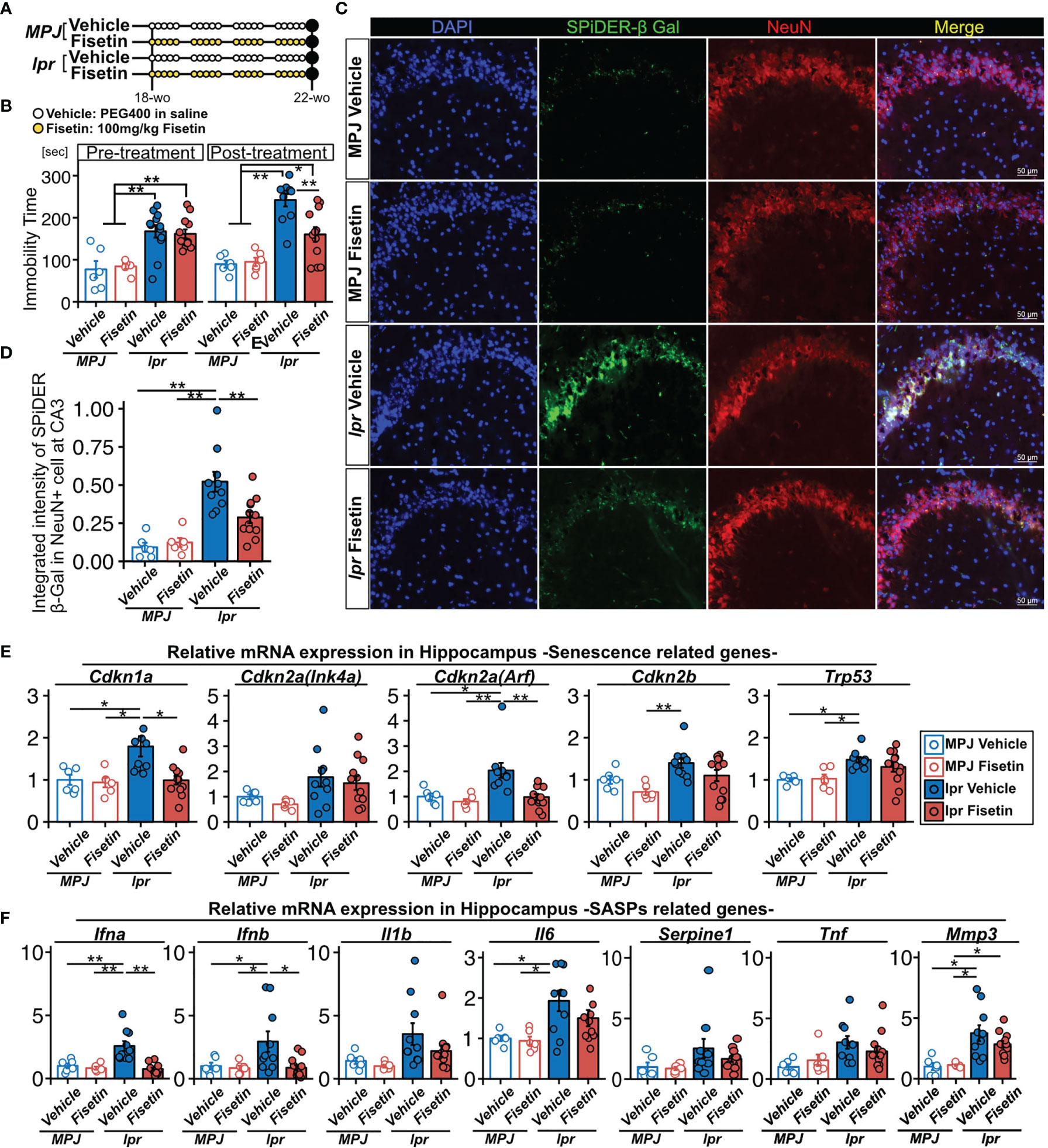
Figure 5 Effect of fisetin treatment on depression-like behavior in MRL/lpr mice. (A) Schematic diagram of the procedure for oral administration of fisetin to MRL/lpr and MRL/MPJ mice. (B) Quantification of the immobility time from the tail suspension test for MRL/MPJ and MRL/lpr mice with or without fisetin treatment. (C) Representative images of NeuN SPiDER-β-Gal immunostaining of the hippocampus CA3 regions and (D) the corresponding quantification of SPiDER-β-Gal intensity in NeuN-positive cells in MRL/MPJ and MRL/lpr mice with or without fisetin treatment. Relative mRNA transcription levels of (E) senescence- and (F) SASP-related genes in hippocampus isolated from MRL/MPJ and MRL/lpr mice with or without fisetin treatment. Quantitative data are shown as means ± SEs in dot plots. P-values were determined by two-tailed Student’s t-test. (*P < 0.05 and **P < 0.01).
Discussion
Senescent cells limit their own proliferation but remain metabolically active, secreting a variety of factors including: inflammatory cytokines such as IL-6, IL-8, and TNF-⍺; chemokines; growth factors such as TGF-β; matrix metalloproteinases (MMPs); and micro-RNAs. Collectively, these secreted factors are referred to as the SASP (48). The SASP is considered a hallmark of cellular senescence when combined with other senescence markers such as the cytoplasmic marker SA-β-gal and the nuclear biomarkers p16INK4a, p21WAF1/Cip1, Ki67, and γH2AX (48–50). In this study, we show that MRL/lpr lupus-prone mice accumulate senescent NeuN-positive cells in the hippocampus. In addition, neural cells induced to undergo senescence increased mRNA expression of genes encoding SASP-related factors such as Tnfa, Serpine1, Il6, and Il1b, all of which are elevated in NPSLE (4). Because neurons are post-mitotic, non-cycling cells (those permanently in the G0 phase of the cell cycle), neuronal senescence, like that observed in other post-mitotic cells, relies on mechanisms other than proliferation arrest. Although it is not a fully specific marker, SA-β-Gal is considered to be a useful marker of cellular senescence, and the number of SA-β-Gal–positive neurons increases in aging mice and rats (51, 52). Furthermore, long-term culture–induced senescent neuronal cells exhibit elevated transcription levels of SASP genes (53). Several cytokines and chemokines were identified as biomarkers or therapeutic targets of NPSLE; in particular, type-1 interferons, TNFs, IL-6, and PAI-1, which are major components of the SASP, are present at elevated levels in the CSF of SLE patients (4, 6, 54, 55). Our results showed that the hippocampus isolated from MRL/lpr mice and irradiated senescent Neuro-2a cells exhibit upregulation of the transcription levels of SASP factors, supporting the idea that senescent neural cells contribute to the elevation of cytokines and chemokines in CSF.
In this study, the SA-β-Gal–positive senescent neural cells were observed in the CA3 region of the hippocampus, which is associated with depression (31–34). SLE and MRL/lpr mouse brain have an elevated population of damaged neural cells that express Fluoro Jade B (FJB) and also exhibit upregulation of ubiquitin in the CA3 region (56, 57). FJB dye is an anionic fluorescein derivative used for visualization of neuronal degeneration in brain tissue sections (58, 59), and ubiquitin binds to damaged or misfolded proteins (60). Most protein damage is not reversible, and degradation by the ubiquitin–proteasome system (UPS) eliminates damaged proteins (50, 61). Activation of the UPS is a key characteristic of the senescent state (50, 62).
We also demonstrated that fisetin exerts a potent senolytic effect in neural cells in vivo and in vitro. Several senolytic compounds have been reported, e.g., flavonoids, quercetin, curcumin, and luteolin (12, 13, 40). We used fisetin to target CNS senescence because it has higher brain uptake potential and more effective blood–brain barrier penetration than other flavonoids such as quercetin, luteolin, and myricetin (44–46). In vivo, fisetin treatment reduced the observed depression-like behavior in the mice and the number of senescent cells in the CA3 region of the hippocampus. In this study, fisetin treatment also reduced the transcription levels of several senescence- and SASP-related genes in the hippocampus. For example, the transcription level of the senescence gene Cdkn1a markedly increased in vehicle-treated MRL/lpr mice, and the level decreased after fisetin treatment. The number of p21-expressing NeuN-positive cells increases in older depressed patients relative to non-depressed older patients (63). In our study, type-I interferons, known SASP factors, are upregulated in MRL/lpr mice. Therapeutic administration of type-I interferons to mice with hepatitis C or other malignancies induces SLE-like psychiatric symptoms, including sickness behavior associated with depression, and inhibition of the type-I interferon receptor reduces anxiety-like behavior and cognitive deficits in lupus-prone mice (4, 54). If neural senescent cells produce type-I interferons, thereby exacerbating the development of NPSLE, senolytics targeting the causative cells may be effective treatments for NPSLE. IL-6 is a known pro-inflammatory SASP factor (8–13) that is upregulated in the hippocampus of MRL/lpr mice. The level of IL-6 observed in CFS is higher in NPSLE patients with an acute confusional state (ACS), which includes anxiety disorders, cognitive dysfunction, mood disorders, and psychosis, relative to those with diffuse NPSLE, states other than ACS, or those with focal NPSLE, which suggests that the IL-6 level observed in CFS may indicate the severity of NPSLE (64, 65). In this study, fisetin administration reduced the high transcription level of IL-6 in the hippocampus of MRL/lpr mice. Fisetin treatment causes a reduction of the transcription level of IL-6 in senescent cells in pulmonary fibrosis and aging-related pathology (40, 66). Although the varied and complex pathogenic pathways complicate the development of NPSLE therapies, our data indicate that fisetin treatment targeted specifically to NPSLE senescent neural cells results in inhibition of SASP factors such as type-I interferon and IL-6, suggesting that fisetin is a candidate NPSLE therapeutic. Fisetin not only has potential as a senolytic in neuronal cells, but also acts as a neuroprotective agent via its antioxidant, antitumor, anti-inflammatory, and anti-apoptosis effects (45, 67, 68). Hence, fisetin could be a candidate drug for CNS disorders by targeting neural cell populations. Our findings indicate that neural cells are a major population of senescent cells in the lupus-prone mouse model, whereas other studies reported that the major populations of senescent cells in Alzheimer’s model mice are astrocytes, microglia, and oligodendrocyte progenitor cells (17, 18). Furthermore, those studies used other senolytic compounds, dasatinib and quercetin (D+Q) and ABT263, to treat Alzheimer’s model mice, and administration of both senolytic compounds alleviated cognitive deficits and decreased the abundance of senescent cells in the brain (17, 18). Further study will be needed to identify the most effective senolytic compound for NPSLE.
In conclusion, our study highlights the accumulation of senescent neural cells in hippocampus of lupus-prone model mice. Oral administration of fisetin, a senolytic drug, reduced the number of senescent neural cells observed, the SASP expression level, and depressive behavior in MRL/lpr mice. These results indicate that the accumulation of senescent neural cells in the hippocampus plays a role in NPSLE pathogenesis, and therapies targeting senescent cells may represent candidate therapeutics for the treatment of NPSLE.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by The Committee of the Animal Experimentation Center of the Sapporo Medical University School of Medicine.
Author Contributions
YS: Conception and design, Collection and/or assembly of data, Manuscript writing, MM: Collection and/or assembly of data, SY: Data collection, TS: Data collection, NM: Data collection, MF: Manuscript writing, TSC: Conception and design, Collection and/or assembly of data, Manuscript writing, Final approval of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by JSPS KAKENHI (Grant Number JP21H03049) and LEOC Co. Ltd. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Naoko Sai, Yumiko Takagi, Kozue Kamiya, Yuko Hayakawa, and Tatsuya Shiraishi for their technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.692321/full#supplementary-material
References
1. Tsokos GC. Systemic Lupus Erythematosus. New Engl J Med (2011) 365:2110–21. doi: 10.1056/nejmra1100359
2. Jeltsch-David H, Muller S. Neuropsychiatric Systemic Lupus Erythematosus: Pathogenesis and Biomarkers. Nat Rev Neurol (2014) 10:579–96. doi: 10.1038/nrneurol.2014.148
3. Haghighat S, Fatemi A, Andalib S. The Autonomic Dysfunction in Patients With Lupus Disease: An Electrophysiological Study. Adv BioMed Res (2016) 5:102. doi: 10.4103/2277-9175.183662
4. Schwartz N, Stock AD, Putterman C. Neuropsychiatric Lupus: New Mechanistic Insights and Future Treatment Directions. Nat Rev Rheumatol (2019) 15:137–52. doi: 10.1038/s41584-018-0156-8
5. Zhang L, Fu T, Yin R, Zhang Q, Shen B. Prevalence of Depression and Anxiety in Systemic Lupus Erythematosus: A Systematic Review and Meta-Analysis. BMC Psychiatry (2017) 17:70. doi: 10.1186/s12888-017-1234-1
6. Rönnblom L, Alm GV, Eloranta M-L. Type I Interferon and Lupus. Curr Opin Rheumatol (2009) 21:471–7. doi: 10.1097/bor.0b013e32832e089e
7. Santer DM, Yoshio T, Minota S, Möller T, Elkon KB. Potent Induction of IFN-α and Chemokines by Autoantibodies in the Cerebrospinal Fluid of Patients With Neuropsychiatric Lupus. J Immunol (2009) 182:1192–201. doi: 10.4049/jimmunol.182.2.1192
8. Deursen JMv. The Role of Senescent Cells in Ageing. Nature (2014) 509:439–46. doi: 10.1038/nature13193
9. Muñoz-Espín D, Serrano M. Cellular Senescence: From Physiology to Pathology. Nat Rev Mol Cell Bio (2014) 15:482–96. doi: 10.1038/nrm3823
10. McHugh D, Gil J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J Cell Biol (2018) 217:65–77. doi: 10.1083/jcb.201708092
11. Sturmlechner I, Durik M, Sieben CJ, Baker DJ, Deursen JMv. Cellular Senescence in Renal Ageing and Disease. Nat Rev Nephrol (2017) 13:77–89. doi: 10.1038/nrneph.2016.183
12. Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. Ebiomedicine (2017) 21:21–8. doi: 10.1016/j.ebiom.2017.04.013
13. Kirkland JL, Tchkonia T. Senolytic Drugs: From Discovery to Translation. J Intern Med (2020) 288:518–36. doi: 10.1111/joim.13141
14. Riessland M, Kolisnyk B, Kim TW, Cheng J, Ni J, Pearson JA, et al. Loss of SATB1 Induces P21-Dependent Cellular Senescence in Post-Mitotic Dopaminergic Neurons. Cell Stem Cell (2019) 25:514–530.e8. doi: 10.1016/j.stem.2019.08.013
15. Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep (2018) 22:930–40. doi: 10.1016/j.celrep.2017.12.092
16. Nicaise AM, Wagstaff LJ, Willis CM, Paisie C, Chandok H, Robson P, et al. Cellular Senescence in Progenitor Cells Contributes to Diminished Remyelination Potential in Progressive Multiple Sclerosis. Proc Natl Acad Sci (2019) 116:201818348. doi: 10.1073/pnas.1818348116
17. Bussian TJ, Aziz A, Meyer CF, Swenson BL, Deursen JMv, Baker DJ. Clearance of Senescent Glial Cells Prevents Tau-Dependent Pathology and Cognitive Decline. Nature (2018) 562:578–82. doi: 10.1038/s41586-018-0543-y
18. Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. Senolytic Therapy Alleviates Aβ-Associated Oligodendrocyte Progenitor Cell Senescence and Cognitive Deficits in an Alzheimer’s Disease Model. Nat Neurosci (2019) 22:719–28. doi: 10.1038/s41593-019-0372-9
19. Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s Disease and Its Potential as Therapeutic Target. Transl Neurodegener (2015) 4:19. doi: 10.1186/s40035-015-0042-0
20. Saez-Atienzar S, Masliah E. Cellular Senescence and Alzheimer Disease: The Egg and the Chicken Scenario. Nat Rev Neurosci (2020) 21:433–44. doi: 10.1038/s41583-020-0325-z
21. Delgado NPD, Vásquez G, Ortiz-Reyes BL. Blood-Brain Barrier Disruption and Neuroinflammation as Pathophysiological Mechanisms of the Diffuse Manifestations of Neuropsychiatric Systemic Lupus Erythematosus. Autoimmun Rev (2019) 18:426–32. doi: 10.1016/j.autrev.2018.12.004
22. Gao L, Slack M, McDavid A, Anolik J, Looney RJ. Cell Senescence in Lupus. Curr Rheumatol Rep (2019) 21:1. doi: 10.1007/s11926-019-0800-6
23. Li X, Liu L, Meng D, Wang D, Zhang J, Shi D, et al. Enhanced Apoptosis and Senescence of Bone-Marrow-Derived Mesenchymal Stem Cells in Patients With Systemic Lupus Erythematosus. Stem Cells Dev (2012) 21:2387–94. doi: 10.1089/scd.2011.0447
24. Gu Z, Jiang J, Tan W, Xia Y, Cao H, Meng Y, et al. P53/P21 Pathway Involved in Mediating Cellular Senescence of Bone Marrow-Derived Mesenchymal Stem Cells From Systemic Lupus Erythematosus Patients. Clin Dev Immunol (2013) 2013:1–13. doi: 10.1155/2013/134243
25. Gu Z, Cao X, Jiang J, Li L, Da Z, Liu H, et al. Upregulation of P16ink4a Promotes Cellular Senescence of Bone Marrow-Derived Mesenchymal Stem Cells From Systemic Lupus Erythematosus Patients. Cell Signal (2012) 24:2307–14. doi: 10.1016/j.cellsig.2012.07.012
26. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, et al. Rapamycin Reverses the Senescent Phenotype and Improves Immunoregulation of Mesenchymal Stem Cells From MRL/lpr Mice and Systemic Lupus Erythematosus Patients Through Inhibition of the mTOR Signaling Pathway. Aging (2016) 8:1102–14. doi: 10.18632/aging.100925
27. Gu Z, Tan W, Feng G, Meng Y, Shen B, Liu H, et al. Wnt/β-Catenin Signaling Mediates the Senescence of Bone Marrow-Mesenchymal Stem Cells From Systemic Lupus Erythematosus Patients Through the P53/P21 Pathway. Mol Cell Biochem (2014) 387:27–37. doi: 10.1007/s11010-013-1866-5
28. Gao L, Bird AK, Meednu N, Dauenhauer K, Liesveld J, Anolik J, et al. Bone Marrow–Derived Mesenchymal Stem Cells From Patients With Systemic Lupus Erythematosus Have a Senescence-Associated Secretory Phenotype Mediated by a Mitochondrial Antiviral Signaling Protein–Interferon-β Feedback Loop. Arthritis Rheumatol (2017) 69:1623–35. doi: 10.1002/art.40142
29. Luo C, Zhou S, Zhou Z, Liu Y, Yang L, Liu J, et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J Am Soc Nephrol (2018) 29:1238–56. doi: 10.1681/asn.2017050574
30. Yang C, Xue J, An N, Huang X, Wu Z, Ye L, et al. Accelerated Glomerular Cell Senescence in Experimental Lupus Nephritis. Med Sci Monitor (2018) 24:6882–91. doi: 10.12659/msm.909353
31. Hashikawa N, Utaka Y, Ogawa T, Tanoue R, Morita Y, Yamamoto S, et al. HSP105 Prevents Depression-Like Behavior by Increasing Hippocampal Brain-Derived Neurotrophic Factor Levels in Mice. Sci Adv (2017) 3:e1603014. doi: 10.1126/sciadv.1603014
32. Berton O, Nestler EJ. New Approaches to Antidepressant Drug Discovery: Beyond Monoamines. Nat Rev Neurosci (2006) 7:137–51. doi: 10.1038/nrn1846
33. Campbell S, Macqueen G. The Role of the Hippocampus in the Pathophysiology of Major Depression. J Psychiatry Neurosci Jpn (2004) 29:417–26.
34. MacQueen G, Frodl T. The Hippocampus in Major Depression: Evidence for the Convergence of the Bench and Bedside in Psychiatric Research? Mol Psychiatr (2011) 16:252–64. doi: 10.1038/mp.2010.80
35. Ueno H, Takahashi Y, Suemitsu S, Murakami S, Kitamura N, Wani K, et al. Effects of Repetitive Gentle Handling of Male C57BL/6NCrl Mice on Comparative Behavioural Test Results. Sci Rep-uk (2020) 10:3509. doi: 10.1038/s41598-020-60530-4
36. Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, Gould TD. The Tail Suspension Test. J Vis Exp (2011) 59:e3769. doi: 10.3791/3769
37. Zhang J, Rong P, Zhang L, He H, Zhou T, Fan Y, et al. IL4-Driven Microglia Modulate Stress Resilience Through BDNF-Dependent Neurogenesis. Sci Adv (2021) 7:eabb9888. doi: 10.1126/sciadv.abb9888
38. Rizzo SJS, Neal SJ, Hughes ZA, Beyna M, Rosenzweig-Lipson S, Moss SJ, et al. Evidence for Sustained Elevation of IL-6 in the CNS as a Key Contributor of Depressive-Like Phenotypes. Transl Psychiat (2012) 2:e199–9. doi: 10.1038/tp.2012.120
39. Steru L, Chermat R, Thierry B, Simon P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology (1985) 85:367–70. doi: 10.1007/bf00428203
40. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin Is a Senotherapeutic That Extends Health and Lifespan. Ebiomedicine (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
41. Kanda Y. Investigation of the Freely Available Easy-to-Use Software “EZR” for Medical Statistics. Bone marrow Transplant (2013) 48:452–8. doi: 10.1038/bmt.2012.244
42. Gao H-X, Campbell SR, Cui M-H, Zong P, Hwang J-H, Gulinello M, et al. Depression Is an Early Disease Manifestation in Lupus-Prone MRL/lpr Mice. J Neuroimmunol (2009) 207:45–56. doi: 10.1016/j.jneuroim.2008.11.009
43. Martínez-Cué C, Rueda N. Cellular Senescence in Neurodegenerative Diseases. Front Cell Neurosci (2020) 14:16. doi: 10.3389/fncel.2020.00016
44. Krasieva TB, Ehren J, O’Sullivan T, Tromberg BJ, Maher P. Cell and Brain Tissue Imaging of the Flavonoid Fisetin Using Label-Free Two-Photon Microscopy. Neurochem Int (2015) 89:243–8. doi: 10.1016/j.neuint.2015.08.003
45. Zhang S, Xue R, Geng Y, Wang H, Li W. Fisetin Prevents HT22 Cells From High Glucose-Induced Neurotoxicity via PI3K/Akt/CREB Signaling Pathway. Front Neurosci Switz (2020) 14:241. doi: 10.3389/fnins.2020.00241
46. He W, Abe K, Akaishi T. Oral Administration of Fisetin Promotes the Induction of Hippocampal Long-Term Potentiation In Vivo. J Pharmacol Sci (2018) 136:42–5. doi: 10.1016/j.jphs.2017.12.008
47. Theofilopoulos AN, Dixon FJ. Murine Models of Systemic Lupus Erythematosus11This Is Publication No. 3665IMM From the Department of Immunology, Research Institute of Scripps Clinic, 10666 North Torrey Pines Road, La Jolla, California 92037. Our Work Cited Herein was Supported, in Part, by National Institutes of Health Grants AI-07007, AM-31023, AM-33826, National Cancer Institute Grants CA-27489 and AG-01743 and the Cecil H. And Ida M. Green Endowment Fund. Adv Immunol (1985) 37:269–390. doi: 10.1016/s0065-2776(08)60342-9
48. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PloS Biol (2008) 6:e301. doi: 10.1371/journal.pbio.0060301
49. Childs BG, Durik M, Baker DJ, Deursen JMv. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat Med (2015) 21:1424–35. doi: 10.1038/nm.4000
50. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular Senescence: Defining a Path Forward. Cell (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
51. Ishikawa S, Ishikawa F. Proteostasis Failure and Cellular Senescence in Long-Term Cultured Postmitotic Rat Neurons. Aging Cell (2020) 19:e13071. doi: 10.1111/acel.13071
52. Geng Y-Q, Guan J-T, Xu X-H, Fu Y-C. Senescence-Associated Beta-Galactosidase Activity Expression in Aging Hippocampal Neurons. Biochem Bioph Res Co (2010) 396:866–9. doi: 10.1016/j.bbrc.2010.05.011
53. Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, et al. Postmitotic Neurons Develop a P21-Dependent Senescence-Like Phenotype Driven by a DNA Damage Response. Aging Cell (2012) 11:996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
54. Karageorgas TP, Tseronis DD, Mavragani CP. Activation of Type I Interferon Pathway in Systemic Lupus Erythematosus: Association With Distinct Clinical Phenotypes. J BioMed Biotechnol (2011) 2011:273907. doi: 10.1155/2011/273907
55. Kwieciński J, Kłak M, Trysberg E, Blennow K, Tarkowski A, Jin T. Relationship Between Elevated Cerebrospinal Fluid Levels of Plasminogen Activator Inhibitor 1 and Neuronal Destruction in Patients With Neuropsychiatric Systemic Lupus Erythematosus. Arthritis Rheumatism (2009) 60:2094–101. doi: 10.1002/art.24603
56. Ballok DA, Woulfe J, Sur M, Cyr M, Sakic B. Hippocampal Damage in Mouse and Human Forms of Systemic Autoimmune Disease. Hippocampus (2004) 14:649–61. doi: 10.1002/hipo.10205
57. Ballok DA, Millward JM, Sakic B. Neurodegeneration in Autoimmune MRL-Lpr Mice as Revealed by Fluoro Jade B Staining. Brain Res (2003) 964:200–10. doi: 10.1016/s0006-8993(02)03980-x
58. Schmued LC, Hopkins KJ. Fluoro-Jade B: A High Affinity Fluorescent Marker for the Localization of Neuronal Degeneration. Brain Res (2000) 874:123–30. doi: 10.1016/s0006-8993(00)02513-0
59. Hopkins KJ, Wang G-J, Schmued LC. Temporal Progression of Kainic Acid Induced Neuronal and Myelin Degeneration in the Rat Forebrain. Brain Res (2000) 864:69–80. doi: 10.1016/s0006-8993(00)02137-5
60. Schmidt MF, Gan ZY, Komander D, Dewson G. Ubiquitin Signalling in Neurodegeneration: Mechanisms and Therapeutic Opportunities. Cell Death Differ (2021) 28:570–90. doi: 10.1038/s41418-020-00706-7
61. Deschênes-Simard X, Gaumont-Leclerc M-F, Bourdeau V, Lessard F, Moiseeva O, Forest V, et al. Tumor Suppressor Activity of the ERK/MAPK Pathway by Promoting Selective Protein Degradation. Gene Dev (2013) 27:900–15. doi: 10.1101/gad.203984.112
62. Ogrodnik M, Salmonowicz H, Gladyshev VN. Integrating Cellular Senescence With the Concept of Damage Accumulation in Aging: Relevance for Clearance of Senescent Cells. Aging Cell (2019) 18:e12841. doi: 10.1111/acel.12841
63. Epp JR, Beasley CL, Galea LA. Increased Hippocampal Neurogenesis and P21 Expression in Depression: Dependent on Antidepressants, Sex, Age, and Antipsychotic Exposure. Neuropsychopharmacol (2013) 38:2297–306. doi: 10.1038/npp.2013.132
64. Katsumata Y, Harigai M, Kawaguchi Y, Fukasawa C, Soejima M, Takagi K, et al. Diagnostic Reliability of Cerebral Spinal Fluid Tests for Acute Confusional State (Delirium) in Patients With Systemic Lupus Erythematosus: Interleukin 6 (IL-6), IL-8, Interferon-Alpha, IgG Index, and Q-Albumin. J Rheumatol (2007) 34:2010–7.
65. Fragoso-Loyo H, Richaud-Patin Y, Orozco-Narváez A, Dávila-Maldonado L, Atisha-Fregoso Y, Llorente L, et al. Interleukin-6 and Chemokines in the Neuropsychiatric Manifestations of Systemic Lupus Erythematosus. Arthritis Rheumatism (2007) 56:1242–50. doi: 10.1002/art.22451
66. Zhang L, Tong X, Huang J, Wu M, Zhang S, Wang D, et al. Fisetin Alleviated Bleomycin-Induced Pulmonary Fibrosis Partly by Rescuing Alveolar Epithelial Cells From Senescence. Front Pharmacol (2020) 11:553690. doi: 10.3389/fphar.2020.553690
67. Zhang L, Wang H, Zhou Y, Zhu Y, Fei M. Fisetin Alleviates Oxidative Stress After Traumatic Brain Injury via the Nrf2-ARE Pathway. Neurochem Int (2018) 118:304–13. doi: 10.1016/j.neuint.2018.05.011
Keywords: systemic lupus erythematosus, senescence, depression, inflammation, SASP (senescence-associated secretory phenotype)
Citation: Saito Y, Miyajima M, Yamamoto S, Sato T, Miura N, Fujimiya M and Chikenji TS (2021) Accumulation of Senescent Neural Cells in Murine Lupus With Depression-Like Behavior. Front. Immunol. 12:692321. doi: 10.3389/fimmu.2021.692321
Received: 08 April 2021; Accepted: 19 October 2021;
Published: 03 November 2021.
Edited by:
Andras Perl, Upstate Medical University, United StatesReviewed by:
Melissa Anne Cunningham, Medical University of South Carolina, United StatesRille Pullerits, Sahlgrenska University Hospital, Sweden
Copyright © 2021 Saito, Miyajima, Yamamoto, Sato, Miura, Fujimiya and Chikenji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takako S. Chikenji, Y2hpa2VuamlAcG9wLm1lZC5ob2t1ZGFpLmFjLmpw
†ORCID: Yuki Saito, orcid.org/0000-0002-7949-1628 Takako S. Chikenji, orcid.org/0000-0003-2832-3656