 Brahim Belaid1,2
Brahim Belaid1,2 Lydia Lamara Mahammed1,2
Lydia Lamara Mahammed1,2 Aida Mohand Oussaid2,3
Aida Mohand Oussaid2,3 Melanie Migaud4,5Yasmine Khadri3
Melanie Migaud4,5Yasmine Khadri3 Jean Laurent Casanova4,5,6,7
Jean Laurent Casanova4,5,6,7 Anne Puel4,5,6Nafissa Ben Halla2,3
Anne Puel4,5,6Nafissa Ben Halla2,3 Reda Djidjik1,2*
Reda Djidjik1,2*- 1Department of Medical Immunology, Beni-Messous University Hospital Center, Algiers, Algeria
- 2Faculty of Medicine, Benyoucef Benkhedda University of Algiers 1, Algiers, Algeria
- 3Department of Pediatrics A, Beni-Messous University Hospital Center, Algiers, Algeria
- 4Laboratory of Human Genetics of Infectious Diseases, Necker Hospital for Sick Children, INSERM UMR 1163, Paris, France
- 5Imagine Institute, University of Paris, Paris, France
- 6St Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller University, New York, NY, United States
- 7Howard Hughes Medical Institute, New York, NY, United States
X-linked severe combined immunodeficiency (X-SCID) is caused by mutations of IL2RG, the gene encoding the interleukin common gamma chain (IL-2Rγ or γc) of cytokine receptors for interleukin (IL)-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Hypomorphic mutations of IL2RG may cause combined immunodeficiencies with atypical clinical and immunological presentations. Here, we report a clinical, immunological, and functional characterization of a missense mutation in exon 1 (c.115G>A; p. Asp39Asn) of IL2RG in a 7-year-old boy. The patient suffered from recurrent sinopulmonary infections and refractory eczema. His total lymphocyte counts have remained normal despite skewed T cell subsets, with a pronounced serum IgE elevation. Surface expression of IL-2Rγ was reduced on his lymphocytes. Signal transducer and activator of transcription (STAT) phosphorylation in response to IL-2, IL-4, and IL-7 showed a partially preserved receptor function. T-cell proliferation in response to mitogens and anti-CD3/anti-CD28 monoclonal antibodies was significantly reduced. Further analysis revealed a decreased percentage of CD4+ T cells capable of secreting IFN-γ, but not IL-4 or IL-17. Studies on the functional consequences of IL-2Rγ variants are important to get more insight into the pathogenesis of atypical phenotypes which may lay the ground for novel therapeutic strategies.
Introduction
X-linked severe combined immunodeficiency (X-SCID) is a life-threatening inborn error of immunity, accounts for approximately half of all cases of SCID (1–5). Most infants with SCID die within their first year of life in the absence of immune reconstitution via hematopoietic stem cell transplantation, due to severe and recurrent infections that begin in the first months of life, frequently associated with diarrhea and growth retardation. The infections may be viral, bacterial, and/or fungal, and vaccination with BCG may lead to disseminated infection (6).
X-SCID is caused by hemizygous pathogenic variants of the interleukin 2 receptor gamma (IL2RG) gene, organized in eight exons that encode the common γ chain, (IL-2Rγ or γc, also known as CD132), which is a part of the IL-2 high-affinity receptor and several interleukin receptors, including those for IL-4, IL-7, IL-9, IL-15 and IL-21 (7, 8). The γc protein is expressed on the surface of lymphoid, myeloid, and hematopoietic progenitor cells. The extracellular domain of the chain is encoded by exons 1–5, followed by a transmembrane domain encoded by exon 6, while the two last exons encode the intracellular portion which can cooperate with the Janus kinase family member 3 (JAK3) (9, 10), a signaling kinase that interacts with other JAK and STAT proteins in complex signal transduction through common gamma chain of cytokine receptor subfamily (11). These receptors’ engagement is crucial to lymphocyte activation, proliferation, and function. Impaired signaling downstream of IL-7 (12) and IL-21 (13–17) explains, at least in part, the absence of T cells and impaired B-cell function, respectively, of X-SCID patients. Altogether, the absence of normal IL-4, IL-7, IL-9, IL-15, and IL-21 signaling leads to the almost complete absence of T and NK cells, with nonfunctional B cells observed in the typical T-B+NK- X-SCID patients carrying amorphic mutations of IL2RG (4, 6). On the other hand, atypical cases of X-SCID have been described in male patients carrying hypomorphic mutations of IL2RG, resulting in either a residual expression of γc protein with a weaker affinity for IL-2, or a weaker interaction of γc with the downstream kinase JAK3 leading to an impaired signaling (18–21).
The pathogenic genetic variations causing X-SCID are found throughout the IL2RG sequence, with missense mutations being the most common ones, followed by nonsense mutations and insertions/deletions (7). Mutations resulting in a complete absence of IL-2Rγ expression are likely to result in the classical X-SCID phenotype. On the other hand, missense mutations in the IL2RG locus have been mainly associated with less severe phenotypes referred to as “leaky or atypical X-SCID” (22–24). Furthermore, some atypical X-SCID cases with hypomorphic mutations may display, later in life, chronic lung diseases, warts, and recurrent respiratory and gastrointestinal tract infections, as well as other atypical clinical manifestations (25).
We report the case of a male child with an atypical clinical presentation carrying to a missense mutation of IL2RG gene, with normal T, B, and NK cell counts, high serum IgE levels, persistent eczema, and recurrent sinopulmonary infections. We performed a deep immunophenotyping and functional tests to better characterize the impact of this IL2RG variation. This report adds to the ever-growing knowledge on atypical X-SCID disorders and contributes to a better understanding of the clinical variability associated with the IL2RG gene defect.
Results
Case Presentation
The patient, a 7-year-old Algerian male born to unrelated healthy parents, was admitted to the department of pediatrics at Beni Messous University Hospital due to recurrent sinopulmonary infections and treatment-resistant eczema. He was born at term by cesarean section with a birth weight of 3400g and received all the routine vaccinations with no noticeable complications. Besides moderate growth retardation, his mental and psychomotor development was normal. Recurrent upper and lower respiratory tract infections began at four years of age, requiring antibiotic therapy and hospitalization. In addition, the patient had several episodes of ear, nose, and throat (ENT) infections with a marked increase of C-reactive protein (CRP) concentration. He suffered from severe eczema since the age of 4 years, refractory to topical corticosteroids; one year later, he also developed a cutaneous leishmaniasis treated successfully with pentavalent antimonial salts (meglumine antimoniate) by parenteral administration. Investigation of his family history (Figure 1A) revealed that two of his maternal uncles displayed similar clinical manifestations, with one of them who has been treated for a celiac disease and who died of infectious complications at 22 years of age, while the other one, who was treated for bronchiectasis and severe refractory eczema, died at the age 28 years.
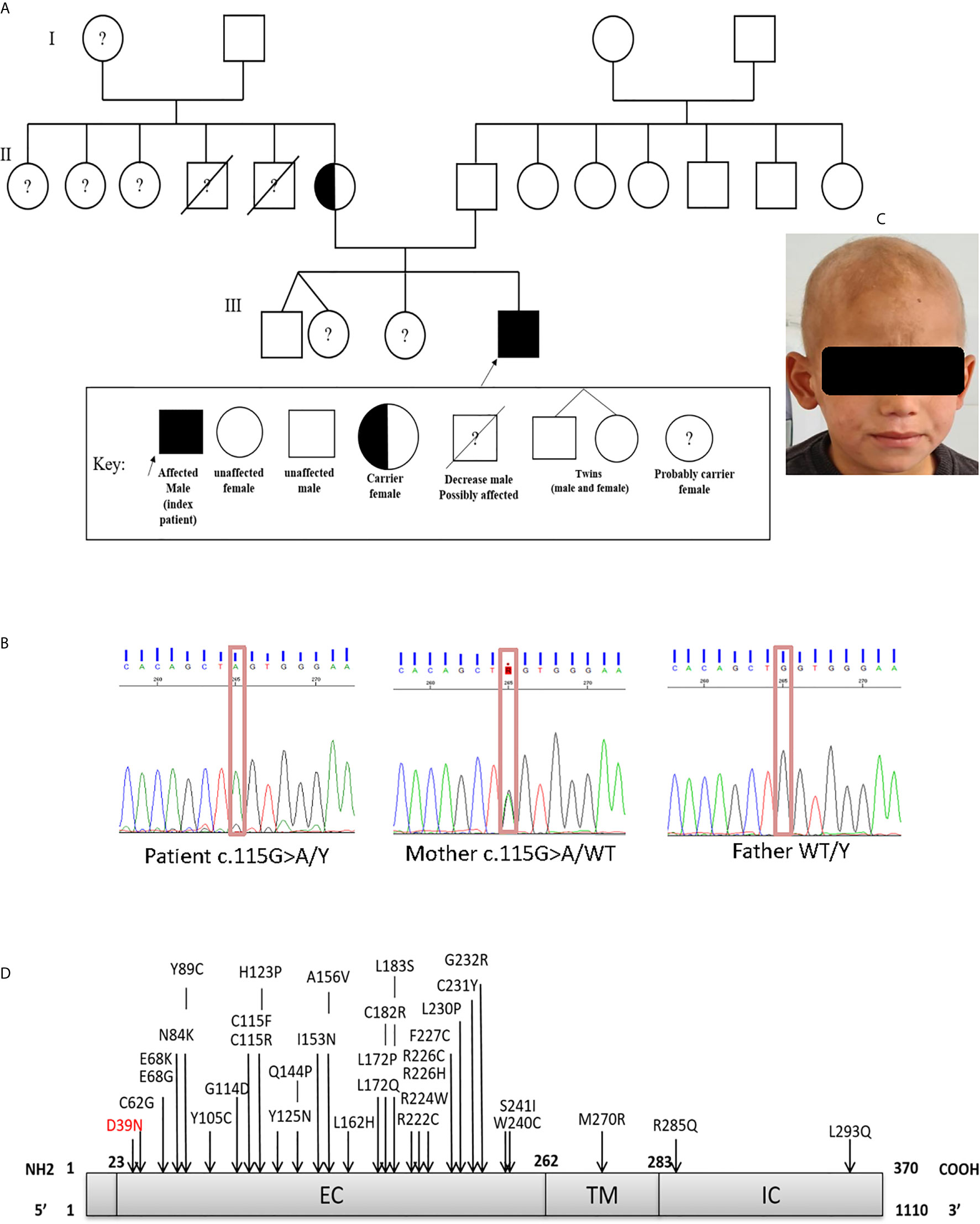
Figure 1 (A) Family pedigree of the patient. (B) Sanger sequencing confirmed the presence of the mutation in exon 1 (c.115G>A; p.Asp39Asn). The heterozygous (mother) and WT states. (C) Scarring alopecia, hypotrichosis with ringworm of the scalp. (D) Schematic diagram of the IL-2RG protein, with its extracellular (EC), transmembrane (TM), intracellular (IC) domains. In red the mutation carried by the patient and in black the positions affected by the mutations already described in the literature.
The patient who was underweight exhibited during the first physical examination, at the age of 6 years, a ringworm of the scalp accompanied by hair loss (Figure 1C), and an eczema present on his ears, elbow folds, and bursae. His first blood test (Table 1) showed a high white blood cell (WBC) count 13800/mm3 (neutrophils 57%, lymphocytes 21%, monocytes 6.5%, eosinophils 15.5%), and normal liver and kidney function tests. Measurement of serum immunoglobulin concentrations showed a pan-hypergammaglobulinemia: IgG 1755 mg/dL (normal or elevated IgG subclasses), IgA 219 mg/dL, IgM 145 mg/dL, and IgE 31120 IU/mL. A flow cytometry analysis of the different lymphocyte subsets (Supplementary Figure S1) revealed normal numbers and frequencies of CD3+ T cells 84% (absolute count 2434/mm3), CD3+CD8+ T cells 61.5% (1782/mm3), CD19+ B cells 9.5% (275/mm3), and CD56+NK cells 6.5% (188/mm3). However, the patient had low numbers of CD3+CD4+ T cells 15.5% (449/mm3), with inverted CD4+/CD8+ ratio (0.25).
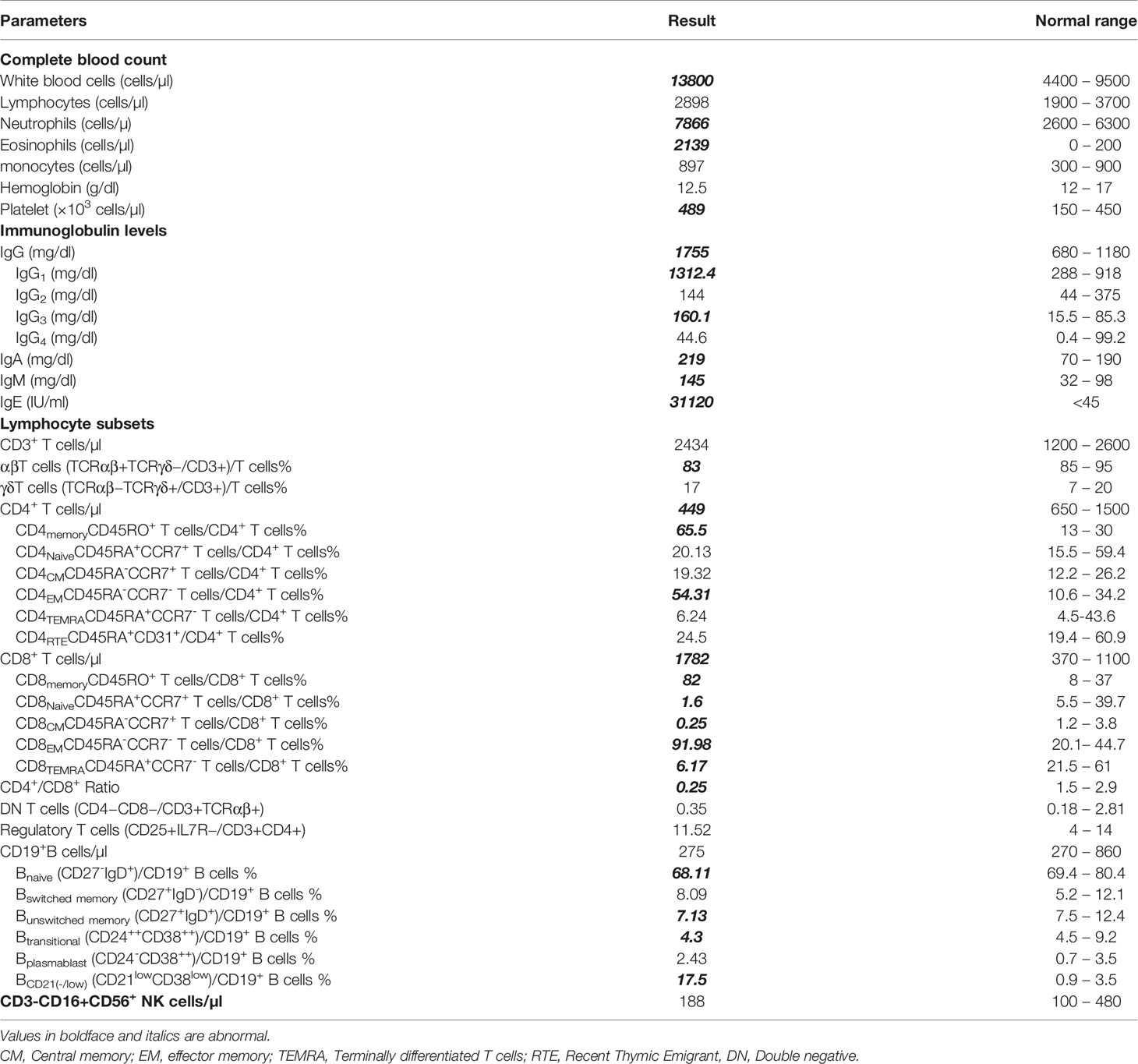
Table 1 Immunologic characteristics of the patient with hypomorphic/atypical X-SCID.
Further characterization of the lymphocyte subpopulations (Table 1) showed a substantial shift towards a memory phenotype (Supplementary Figure S2) with an increased percentage of CD45RO+ CD4+ and CD45RO+ CD8+T cells, low percentages of naïve T cells (CCR7+CD45RA+), and high percentages of CD4+ and CD8+ effector memory T cells (TEM) (CCR7-CD45RA-) (Supplementary Figure S3). By contrast, the frequency of CD4+ T cells displaying the phenotypical characteristics of recent thymic emigrants (RTE) (CD4+CD45RA+CD31+) was in the normal range (Supplementary Figure S4). similarly, the evaluation of peripheral B lymphocytes demonstrated a normal distribution of B cell subsets (Supplementary Figure S5). In fact, despite an expansion of CD19+CD21–/low B cell subset, the Percentages of naïve B cells (CD19+IgD+CD27-), switched and unswitched memory B cells (CD19-CD27+IgD- and CD19+CD27+IgD+, respectively), transitional B cells (CD19+CD24++CD38++), as well as plasmablasts (CD19+CD24-CD38++) did not significantly differ from values observed in healthy age-matched individuals.
Given the presence of refractory eczema associated with high serum IgE concentrations, hypereosinophilia, and recurrent bacterial infections, we initially suspected a hyper-IgE syndrome. Therefore, we used a scoring system which the NIH has developed to help in the diagnosis of these patients based on both the immunologic and clinical features of the syndrome. Indeed, a score of ˃40 points makes the diagnosis of AD-HIES highly probable and unlikely with a score of < 20 points (26). When the scoring criteria were applied to our patient, the overall clinical and immunological presentation was between STAT3-HIES and DOCK8-CID with 39 points in the NIH-HIES score. Therefore, IL-6-induced expression of intracellular phospho-STAT3 (pSTAT3) was determined in CD4+T-cells by whole blood flow cytometric analysis, which included assessment of surface IL-6RA and gp130 expression. No difference for pSTAT3 expression was detected between patient and healthy control (Figure 2B).
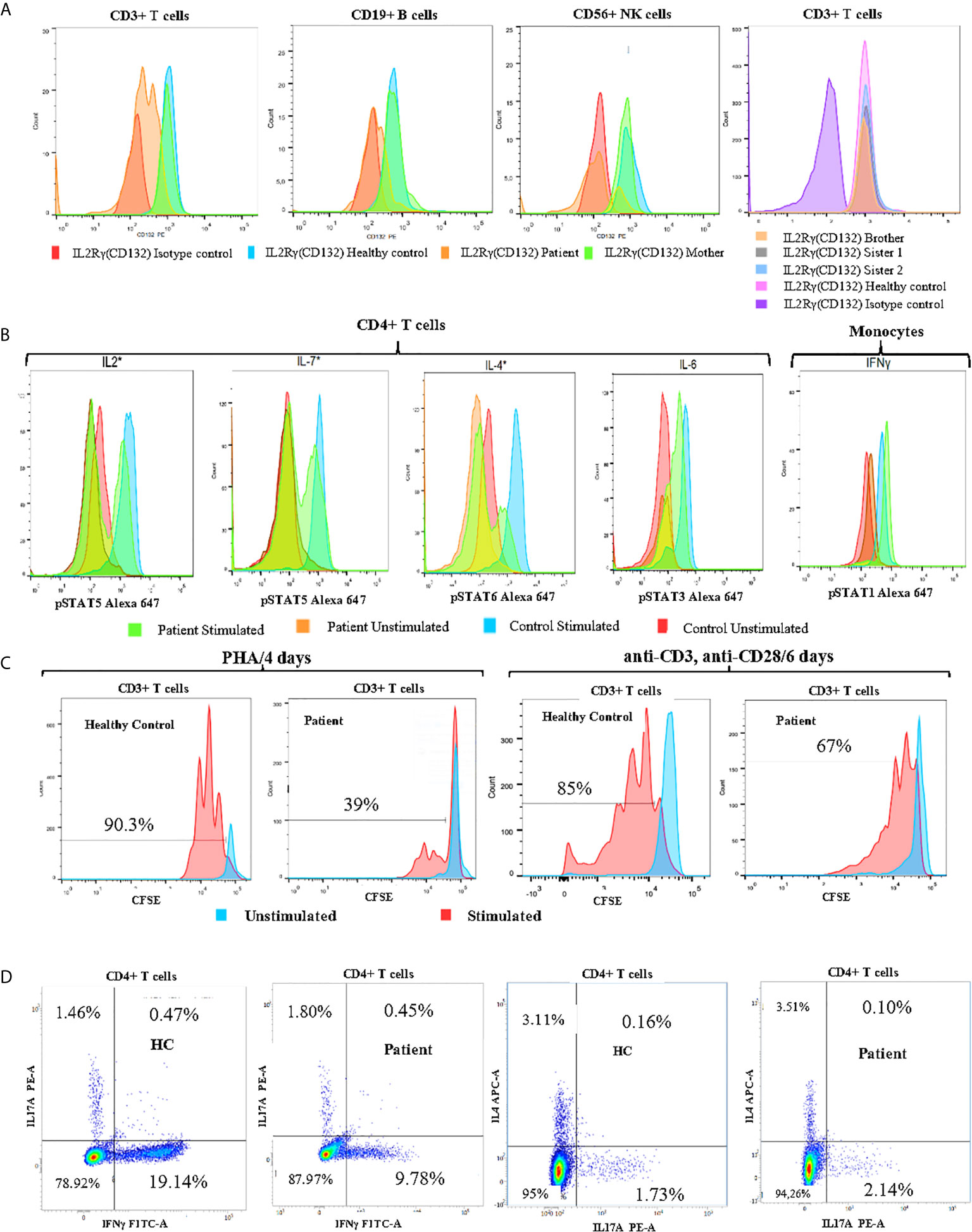
Figure 2 Immune functional and proliferation assays in subsets of lymphocytes. (A) Flow cytometry analysis of CD132 (γc) expression on T, B and NK cells of the patient, his mother, and healthy control, and screening of siblings for CD132 expression on T cells. (B) Histogram of CD4+ T cell and monocyte response with STAT phosphorylation to several γc dependent (IL-2, IL-4, IL-7) and independent (IL-6, IFN-γ) cytokines in whole blood of patient. (C) Analysis of proliferation of healthy control (HC) and patient CFSE labeled cells with the indicated stimuli: 4 days of culture with Phytohemaglutinin (PHA) and 6 days with anti-CD3/anti-CD28 monoclonal antibodies. (D) The percentages of IL-17A, IFNγ and IL-4-producing CD4+ T cells from patient and age-matched control using PMA/Ionomycin stimulation in the presence of monensin for 5 hours. *Gamma chain dependent cytokines; the others are gamma chain independent.
In order to identify the molecular defect behind the pathology, whole-exome sequencing (WES) identified the presence of a hemizygous missense mutation (c.115G>A) in exon 1 of the IL2RG gene. The presence of the missense mutation was further confirmed by Sanger sequencing (Figure 1B). This substitution resulted in an amino acid change from Aspartic Acid to Asparagine at position 39 (p.Asp39Asn) of the mature protein. Bioinformatic analysis predicted that the p.Asp39Asn mutation was predicted to be probably pathogenic and affected a highly conserved amino-acid residue (SIFT-deleterious; PolyPhen2-possibly damaging), (Supplementary Figure S6). The high CADD score of 24.2 (sensitivity: 0.41; specificity: 0.98) also suggested a probably damaging effect of the variant. No additional mutations were found in any other known disease-causing genes (Supplementary Figure S7).
We also found that the patient’s mother was a heterozygous carrier of this variant. An immunostaining with anti-CD132 and flow cytometry analysis showed a significant reduction of γc surface expressions on T, B and NK cells of the patient when compared to his parents, siblings, or healthy control (Figure 2A).
In order to study the impact of Asp39Asn amino acid substitution on immune cell signaling, we measured STAT phosphorylation in CD4+ T cells and monocytes. The mutation was associated with defective phosphorylation of STAT5, STAT6, and STAT5 upon the stimulation of CD4+ T cell with IL-2, IL-4, and IL7, respectively. However, γc independent signaling in monocytes (IFN-γ induced pSTAT1) was not affected (Figure 2B). Evaluation of CD3+ T cell proliferation through carboxyfluorescein succinimidyl ester (CFSE) staining revealed a significant defect of mitogen phytohemagglutinin (PHA)-induced cell proliferation compared to healthy control (39% vs. 90.3%). Similarly, the activation of CD3+ T cells of the patient with anti-CD3 and anti-CD28-coated beads led to a reduced proliferation compared to healthy control (67% vs. 85%) (Figure 2C). Taken together, these data suggest that the patient has a “leaky” form of SCID.
To better characterize the immunologic defects associated with the patient’s mutation, the percentages of CD4+IFNγ+ (Th1), CD4+IL4+ (Th2), and CD4+IL-17A+ (Th17) cells were assessed in PBMCs by flow cytometry. The patient exhibited a significant reduction in the proportion of IFNγ producing CD4+ T cells (Th1) and normal CD4+IL4+(Th2) and CD4+IL17A+(Th17) frequencies when compared to an age-matched control subject (Figure 2D). CD4+CD25highCD127low/-FoxP3+ Regulatory T-cells represent 11,52% of CD4+ T-cells. Based on these findings, we have recommended allogeneic hematopoietic stem cell transplantation as a definitive treatment. Since, the patient regularly performed immunoglobulin replacement therapy and received prophylactic antimicrobial (co-trimoxazole, sulfamethoxazole, and itraconazole). He is checked for signs of malignant disease on a regular basis, but so far, no lymphoma or other tumors have been noted.
Discussion
This report describes the case of a seven-year-old Algerian patient carrying a hemizygous missense (c.115G>A) variant of IL2RG gene, and results in replacement of aspartic acid by asparagine at position 39 (p.Asp39Asn) in the extracellular domain of the protein (Figure 1D). As far as we are aware, only a single other case carrying a same mutation has been reported in the literature (27, 28).
Our patient displayed unusual clinical and immunological features with persistent eczema and ringworm of the scalp, associated with recurrent sinopulmonary infections, high IgE levels, and peripheral blood eosinophilia, which initially suggested a hyper-IgE syndrome. A reported case (27, 28) with the same amino acid substitution and a more severe clinical phenotype (Table 2) was mainly characterized by repetitive infections and protracted diarrhea starting around nine months of age without a history of eczema. Given the different clinical and immunological manifestations, it seems reasonable to hypothesize that other factors, such as epigenetic, environmental, and ethnic contributions, could affect the disease’s evolution (29). These findings emphasize the issues that might be involved in the relationship between the environment and the genome in multifactorial disorders, in which numerous environmental factors are included (30). As reported by Dworkin et al, attempting to understand the molecular interaction of genetic background effects using model organisms, found that were partly associated with differentially expressed genes where quantitative transcription level differences correlated with variation for the phenotype (31). Furthermore, Lachance et al. studied genetic background effects on an X-linked gene in Drosophila melanogaster leading to wing defects. The natural variant was placed into multiple backgrounds, then they assessed penetrance and expressivity of wing defects. They found significant complex interactions that were affected by the genetic background (32).
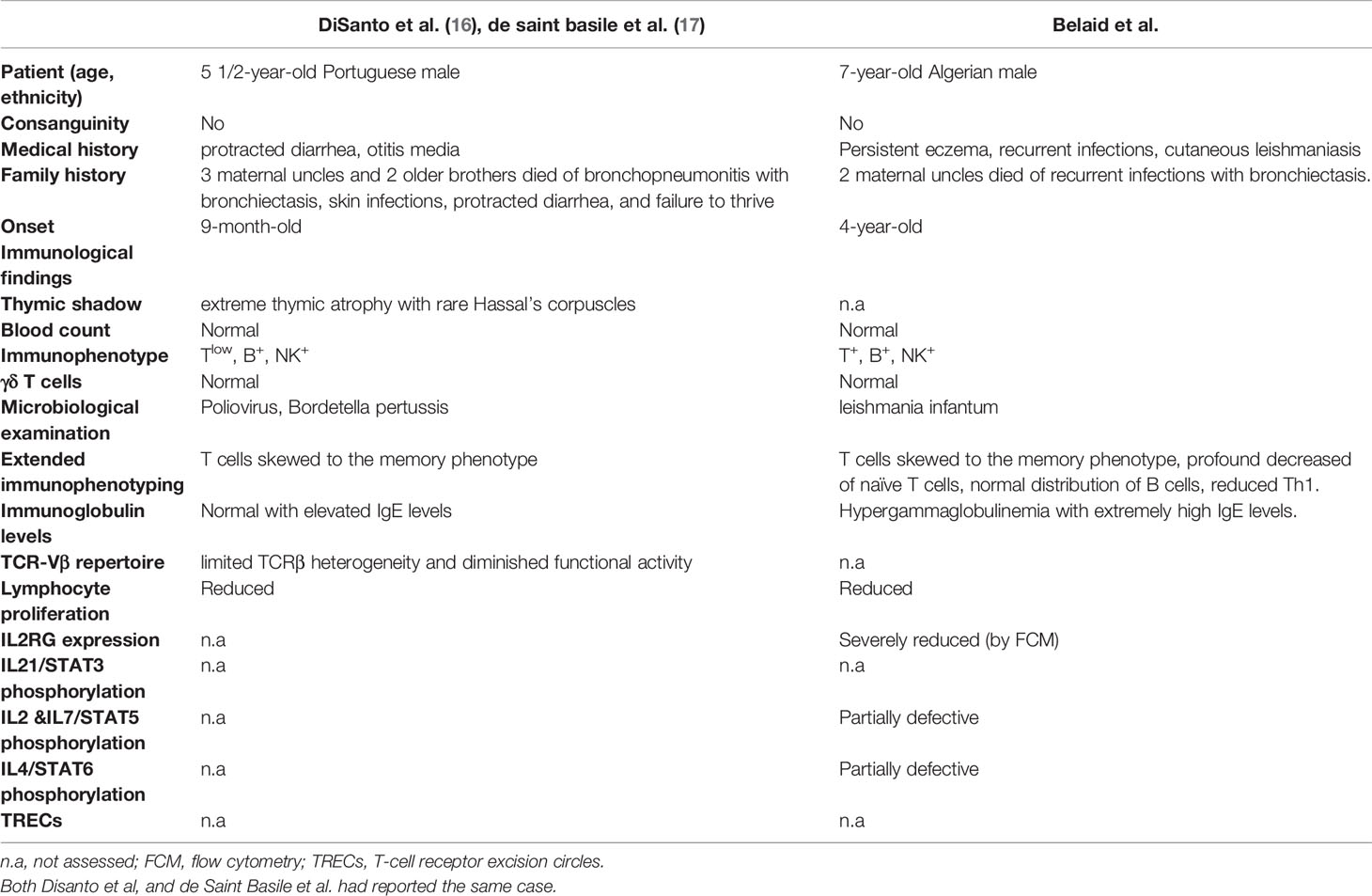
Table 2 Characteristics of reported patient carrying the IL2RG115G>A missense mutation.
Typically, patients with genetic mutations of the IL2RG gene are classically characterized by the absence or severe reduction of T and NK cell numbers, as well as the presence of non-functional B cells (33), these mutations are mainly localized within the exons 3-5 (34, 35). Missense mutations are the most common pathogenic changes observed, followed by nonsense variants and insertions/deletions (35). On the other hand, atypical clinical presentations of X-SCID have been also described in patients carrying missense mutations of IL2RG resulting in the expression of lower amounts of γc protein with conserved binding affinity for IL-2, or in its reduced interaction with JAK3, and thus impairing T cell activation (9, 36). However, such mutation requires assessing a cell line from the affected patient to evaluate γc cell surface expression and/or IL2RG mRNA transcripts as a direct proof if the mutation is deleterious. In the present case, an accurate diagnosis of atypical X-SCID was initially compromised because of the unusual clinical presentation, with almost normal development and growth, a low number of infections during the first 3 years of life. Moreover, the most first-line immunological investigations showed normal percentages and numbers of total T, NK, and B cells, but CD4+ T cell counts decreased while CD8+ T cell counts were expanded, combined with a polyclonal hypergammaglobulinemia; hence, these findings were initially inconsistent with X-SCID. Atypical cases with normal numbers of T and NK cells are very unusual and were previously reported in only few cases with IL2RG mutations (37–41), and some IL2RG mutations can lead to functional lymphocyte abnormalities rather than cell development defects, as some patients with normal lymphocyte differentiation and normal thymus biopsies were reported (37). Such phenotype can be a direct consequence of a residual γc expression providing sufficient signal for normal T and NK cell development. This is in accordance with the preserved signaling by the IL-7R and IL-15R, as it has been shown before by Smyth et al. that signal transduction by the IL-7R is crucial for T-cell development but is dispensable for NK cell development, whereas adequate signaling via the IL-15R is essential for NK cell but not for T-cell development (42, 43). This mechanism corroborates with our findings, suggesting that the amount of γc required for the correct signaling of various signaling pathways is different.
In addition to the aforementioned findings, the T lymphocyte differentiation and maturation skewed toward memory phenotype (CD45RO+), combined with increased counts of total CD8+ T cells as well as expansion of CD45-CCR7- effector memory for both CD4+ and CD8+ T cells. This condition probably is in consistence with persistent viral infection (44), although no virological confirmation was possible. On other hand, thymic stromal lymphopoietin (TSLP) is another cytokine that is not a member of the γc family but has overlapping functions with IL-7 (45). Indeed, whereas the IL-7 receptor contains IL-7Rα and γc, the TSLP receptor consists of IL-7Rα and TSLPR, which is closely related to γc (46, 47). IL2RG−/− mice treated with recombinant TSLP, which cannot respond to IL-7 or other γc family cytokines, lead to a partial increase in CD8+ T cell numbers (48). Moreover, TSLP promotes the survival of CD8+ T cells in both normal and lymphogenic conditions (49).
In addition, our patient displayed moderately reduced capacity of T cells to proliferate in response to PHA or anti-CD3/anti-CD28 stimulation, reflecting disturbed IL-2 signaling. Indeed, IL-2 is key growth factor required for T cell expansion and promotes the proliferation and survival of activated T cells (50). Thus, it influences effector T cell differentiation and promotes fate decisions in activated T cells (51).
Furthermore, our patient had normal percentages of switched memory B cells, transitional B cells, and plasmablasts, but negligible reduction of naïve B cells, and unswitched memory B cells. There may exist a threshold level of γc expression necessary for normal B cell function, consequently, the collaborative IL-4 and IL-21 signaling might be sufficient for humoral responses (52–55). In contrast, we found signs of misguided enhanced B cell activity reflected by a large CD21lowCD38low B cell population, in addition to a polyclonal hypergammaglobulinemia. CD21lowCD38low B cells are a distinct B cell population that is mainly associated with manifestations of chronic immune activation, lymphoproliferation, and autoimmunity (56–58). Interestingly, in our case there was no evidence of conditions considered to be autoimmune or lymphoproliferation. This is in consistence with normal numbers of regulatory T cells (T reg) in indexed patient, although IL-2 is also critical for the development of Tregs in the thymus and for their maintenance and function in the periphery (59). In addition, high IgG1 and IgG3 levels were also observed which can recognize protein and viral antigens (60, 61). In fact, viral infections in general lead to IgG antibodies of the IgG1 and IgG3 subclasses, with IgG3 antibodies appearing first in the course of the infection (60, 61).
In the case reported by DiSanto et al., with the same mutation as the present patient, a reduced capacity to splice a correct-sized transcript, leading to the production of a nonfunctional transcript containing an insertion of 27 bp, and a reduced amount of a normal sized transcript containing a single amino acid substitution has been shown (28). Thus, they demonstrated that splicing of exons 1 and 2 normally generates the codon GAT, but with the base change, the resultant codon becomes AAT, and the point mutation (D39N) appeared not to impair IL-2 binding or its subsequent endocytosis (28). Therefore, a residual expression of IL2RG transcripts with normal length may account for the limited expression of γc detectable at the cell surface. Unfortunately, the size of the transcripts and γc protein were not assessed in the current work. However, our data showed that γc expression and STAT5/STAT6 phosphorylation were reduced but not completely abolished. These findings are consistent with a moderate phenotype suggesting that this mutation (p.Asp39Asn) is hypomorphic.
Interestingly, our patient developed atopic dermatitis-like skin lesions and alopecia associated to IgE hyperproduction (31,120 IU/ml) and hypereosinophilia (2139 cells/µl), such conditions were surprisingly rare in reviewed patients carrying IL2RG hypomorphic mutations. Indeed, only four patients, suffering from eczema or other skin rashes, were reported in the literature (15, 62–64). Milner et al. demonstrated that reduced T-cell activation caused by a weak signaling has been associated with a skew towards the development of T helper type 2 cells (Th2 cells) (62), this may suggest its profile as a default differentiation pathway partly underlying the features of skin lesions in these patients. This is a typical finding in patients with Omenn syndrome in whom expanded T-cell clones were consistently found to be predominantly of TH2 type (65), and to secrete IL-4/IL-13, IL-5 and IL-9, which promote immunoglobulin class-switching to IgE, activates eosinophils, and activates mast cells, respectively (66–69). These results are consistent with our patient, who showed normal percentage of IL-4 producing T cells and low proportions of IFN-γ producing T cells compared to age-matched control subject (Figure 2D), suggesting that Th2-type cytokines are central in the pathogenesis of this hyper-IgE production. Moreover, previous studies have also demonstrated that B cells from X-SCID patients with decreased expression of γc can respond to IL-4 via a type II IL-4R complex composed of IL-4Rα/IL-13R chains (70–72).
In conclusion, X-SCID is an inborn error of immunity that manifests as different clinical phenotypes, from milder to severe disease. Most of the attention focused on the possible relationship between mutations of the IL2RG gene and clinical/immunological features. Further investigations are required to achieve a better classification of the disease. We believe that unusual clinical and laboratory observations may be very useful to unravel complex diseases and help find novel gene-function relationship laying the ground for novel targeted therapeutic approaches.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité d’éthique du CHU Beni Messous. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
BB drafted the manuscript and contributed in collecting and analyzing the data. LM designed and performed experiments, analyzed the data, and reviewed the manuscript critically. AM, YK, and NB, cared for the patient and provided clinical data. AP, MM, and JC reviewed the manuscript critically and suggested changes to the final version and did the genetic testing. RD reviewed the manuscript and conceptualized the paper. All authors contributed to the article and approved the submitted version.
Funding
The French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), ANR-FNS LTh-MSMD-CMCD (ANR-18-CE93-0008-01), The Rockefeller University and the National Institutes of Health (# R01AI127564).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Belgacem Mihi for his critical reading of the manuscript, and we would like to thank the patient and family for providing the samples and their approval to share his clinical course to the healthcare and scientific community.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.696350/full#supplementary-material
Abbreviations
FOXP3, forkhead box P3; HIES, Hyper-IgE syndrome; IFN, Interferon; IL, Interleukin; IL2RG, Interleukin 2 receptor gamma; JAK3, Janus kinase 3; γC, common gamma chain; NK, Natural Killer; PBMC, Peripheral Blood Mononuclear cells; PHA, phytohemagglutinin; PMA, Phorbol 12-myristate 13-acetate; STAT, Signal transducer and activator of transcription.; SCID, Severe combined immunodeficiency; Th, Helper T lymphocyte.
References
1. Stephan JL, Vlekova V, Deist F, Blanche S, Donadieu J, Saint-Basile G, et al. Severe Combined Immunodeficiency: A Retrospective Single-Center Study of Clinical Presentation and Outcome in 117 Patients. J Pediatr (1993) 123:564–72. doi: 10.1016/S0022-3476(05)80951-5
2. Haddad E, Landais P, Friedrich W, Gerritsen B, Cavazzana-Calvo M, Morgan G, et al. Long-Term Immune Reconstitution and Outcome After HLA-Nonidentical T- Cell-Depleted Bone Marrow Transplantation for Severe Combined Immunodeficiency: A European Retrospective Study of 116 Patients. Blood (1998) 91:3646–53. doi: 10.1182/blood.V91.10.3646
3. Bertrand Y, Landais P, Friedrich W, Gerritsen B, Morgan G, Fasth A, et al. Influence of Severe Combined Immunodeficiency Phenotype on the Outcome of HLA Non-Identical, T-Cell- Depleted bone Marrow Transplantation. J Pediatr (1999) 134:740–8. doi: 10.1016/s0022-3476(99)70291-x
4. Antoine C, Müller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-Term Survival and Transplantation of Haemopoietic Stem Cells for Immunodeficiencies: Report of the European Experience 1968-99. Lancet (2003) 361:553–60. doi: 10.1016/S0140-6736(03)12513-5
5. AL Dossari D, Kamal D. Clinical Practice: Clinical Practice. J Gastroenterol Hepatol (2015) 30:13–26. doi: 10.1111/jgh.13088
6. Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 Receptor γ Chain Mutation Results in X-Linked Severe Combined Immunodeficiency in Humans. Cell (1993) 73:147–57. doi: 10.1016/0092-8674(93)90167-O
7. Puck JM, Deschenes SM, Porter JC, Dutra AS, Brown CJ, Willard HF, et al. The Interleukin-2 Receptor γ Chain Maps to Xq13.1 and Is Mutated in X-linked Severe Combined Immunodeficiency, SCIDX1. Hum Mol Genet (1993) 2:1099–104. doi: 10.1093/hmg/2.8.1099
8. Yrina R, Spolski R, WJL. New Insights Into the Regulation of T Cells by γc Family Cytokines. Bone (2008) 23:1–7. doi: 10.1038/nri2580.New
9. Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, et al. Interaction of IL-2Rβ and γc Chains With Jak1 and Jak3: Implications for XSCID and XCID. Science (80-) (1994) 266:1042–5. doi: 10.1126/science.7973658
10. Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, et al. Functional Activation of Jak1 and Jak3 by Selective Association With IL-2 Receptor Subunits. Science (80-) (1994) 266:1045–7. doi: 10.1126/science.7973659
11. Puck JM, Pepper AE, Henthorn PS, Candotti F, Isakov J, Whitwam T, et al. Mutation Analysis of IL2RG in Human X-Linked Severe Combined Immunodeficiency. Blood (1997) 89:1968–77. doi: 10.1182/blood.V89.6.1968
12. Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R Expression in T-B+NK+ Severe Combined Immunodeficiency. Nat Genet (1998) 20:394–7. doi: 10.1038/3877
13. Kotlarz D, Ziętara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, et al. Loss-of-Function Mutations in the IL-21 Receptor Gene Cause a Primary Immunodeficiency Syndrome. J Exp Med (2013) 210:433–43. doi: 10.1084/jem.20111229
14. Erman B, Bilic I, Hirschmugl T, Salzer E, Çagdas D, Esenboga S, et al. Combined Immunodeficiency With CD4 Lymphopenia and Sclerosing Cholangitis Caused by a Novel Loss-of-Function Mutation Affecting IL21R. Haematologica (2015) 100:e216–9. doi: 10.3324/haematol.2014.120980
15. Stepensky P, Keller B, Abuzaitoun O, Shaag A, Yaacov B, Unger S, et al. Extending the Clinical and Immunological Phenotype of Human interleukin-21 Receptor Deficiency. Haematologica (2015) 100:63–7. doi: 10.3324/haematol.2014.112508
16. Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A Recessive Form of hyper-IgE Syndrome by Disruption of ZNF341-Dependent STAT3 Transcription and Activity. Sci Immunol (2018) 3:1–19. doi: 10.1126/sciimmunol.aat4956
17. Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, et al. Il-21 Signalling Via STAT3 Primes Human Näive B Cells to Respond to IL-2 to Enhance Their Differentiation Into Plasmablasts. Blood (2013) 122:3940–50. doi: 10.1182/blood-2013-06-506865
18. Fischer A. Severe Combined Immunodeficiencies (SCID). Clin Exp Immunol (2000) 122:143–9. doi: 10.1046/j.1365-2249.2000.01359.x
19. Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining Combined Immunodeficiency. J Allergy Clin Immunol (2012) 130:177–83. doi: 10.1016/j.jaci.2012.04.029
20. Okuno Y, Hoshino A, Muramatsu H, Kawashima N, Wang X, Yoshida K, et al. Late-Onset Combined Immunodeficiency With a Novel IL2rg Mutation and Probable Revertant Somatic Mosaicism. J Clin Immunol (2015) 35:610–4. doi: 10.1007/s10875-015-0202-0
21. Stevens M, Frobisher C, Hawkins M, Jenney M, Lancashire E, Reulen R, et al. The British Childhood Cancer Survivor Study: Objectives, Methods, Population Structure, Response Rates and Initial Descriptive Information. Pediatr Blood Cancer (2008) 50:1018–25. doi: 10.1002/pbc
22. Fuchs S, Rensing-Ehl A, Erlacher M, Vraetz T, Hartjes L, Janda A, et al. Patients With T+/low Nk+ IL-2 Receptor γ Chain Deficiency Have Differentially-Impaired Cytokine Signaling Resulting in Severe Combined Immunodeficiency. Eur J Immunol (2014) 44:3129–40. doi: 10.1002/eji.201444689
23. Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and Immunological Manifestations of Patients With Atypical Severe Combined Immunodeficiency. Clin Immunol (2011) 141:73–82. doi: 10.1016/j.clim.2011.05.007
24. Disanto JP, Rieux-Laucat F, Dautry-Varsat A, Fischer A, De Saint Basile G. Defective Human Interleukin 2 Receptor γ Chain in An Atypical X Chromosome-Linked Severe Combined Immunodeficiency With Peripheral T Cells. Proc Natl Acad Sci USA (1994) 91:9466–70. doi: 10.1073/pnas.91.20.9466
25. Allenspach E, Rawlings DJ, Scharenberg AM. X-Linked Severe Combined Immunodeficiency (1993). Seattle: University of Washington. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20301584 (Accessed April 15, 2021).
26. Yong PFK, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. An Update on the Hyper- IgE syndromes. Arthritis Res Ther (2012) 14:1–10. doi: 10.1186/ar4069
27. De Saint-Basile G, Le Deist F, Caniglia M, Lebranchu Y, Griscelli C, Fischer A. Genetic Study of a New X-Linked Recessive Immunodeficiency Syndrome. J Clin Invest (1992) 89:861–6. doi: 10.1172/JCI115665
28. Disanto JP, Rieux-laucat F, Dautry-varsatt A, Fischer A. Basile GDE Saint. Chromosome-Linked Peripheral Cells. Sci York (1994) 91:9466–70. doi: 10.1073/pnas.91.20.9466
29. Kuijpers TW, van Leeuwen EMM, Barendregt BH, Klarenbeek P, aan de Kerk DJ, Baars PA, et al. Van Der Burg M. A Reversion of An IL2RG Mutation in Combined Immunodeficiency Providing Competitive Advantage to the Majority of CD8+ T Cells. Haematologica (2013) 98:1030–8. doi: 10.3324/haematol.2012.077511
30. Weatherall DJ. Phenotype-Genotype Relationships in Monogenic Disease: Lessons From the Thalassaemias. Nat Rev Genet (2001) 2:245–55. doi: 10.1038/35066048
31. Dworkin I, Kennerly E, Tack D, Hutchinson J, Brown J, Mahaffey J, et al. Genomic Consequences of Background Effects on Scalloped Mutant Expressivity in the Wing of Drosophila Melamogaster. Genetics (2009) 181:1065–76. doi: 10.1534/genetics.108.096453
32. Lachance J, Jung L, True JR. Genetic Background and GxE Interactions Modulate the Penetrance of a Naturally Occurring Wing Mutation in Drosophila Melanogaster. G3 Genes Genomes Genet (2013) 3:1893–901. doi: 10.1534/g3.113.007831
33. Blanco E, Izotova N, Booth C, Thrasher AJ. Immune Reconstitution After Gene Therapy Approaches in Patients With X-Linked Severe Combined Immunodeficiency Disease. Front Immunol (2020) 11:608653. doi: 10.3389/fimmu.2020.608653
34. Puck JM. IL2RGbase : A Database of γc-Chain Defects Causing Human X-SCID. Trends Immunol Today (1996) 17:507–11. doi: 10.1016/0167-5699(96)30062-5
35. Villa A, Moshous D, de Villartay JP, Notarangelo LD, Candotti F. Severe Combined Immunodeficiencies. In Stiehm’s Immune Deficiencies Elsevier Inc. (2014) pp. 87–141. doi: 10.1016/B978-0-12-405546-9.00004-2
36. Schmalstieg FC, Leonard WJ, Noguchi M, Berg M, Rudloff HE, Denney RM, et al. Missense Mutation in Exon 7 of the Common γ Chain Gene Causes a Moderate Form of X-linked Combined Immunodeficiency. J Clin Invest (1995) 95:1169–73. doi: 10.1172/JCI117765
37. Sharfe N, Shahar M, Roifman CM. An Interleukin-2 Receptor γ Chain Mutation With Normal Thymus Morphology. J Clin Invest (1997) 100:3036–43. doi: 10.1172/JCI119858
38. Illig D, Navratil M, Kelečić J, Conca R, Hojsak I, Jadrešin O, et al. Alternative Splicing Rescues Loss of Common Gamma Chain Function and Results in IL-21R-like Deficiency. J Clin Immunol (2019) 39:207–15. doi: 10.1007/s10875-019-00606-7
39. Stepensky P, Keller B, Shamriz O, von Spee-Mayer C, Friedmann D, Shadur B, et al. T+ NK+ Il-2 Receptor γ Chain Mutation: A Challenging Diagnosis of Atypical Severe Combined Immunodeficiency. J Clin Immunol (2018) 38:527–36. doi: 10.1007/s10875-018-0514-y
40. Tuovinen EA, Grönholm J, Öhman T, Pöysti S, Toivonen R, Kreutzman A, et al. Novel Hemizygous IL2RG P. (Pro58Ser) Mutation Impairs IL-2 Receptor Complex Expression on Lymphocytes Causing X-Linked Combined Immunodeficiency. J Clin Immunol (2020) 40:503–14. doi: 10.1007/s10875-020-00745-2
41. Somech R, Roifman CM. Mutation Analysis Should be Performed to Rule Out γc Deficiency in Children With Functional Severe Combined Immune Deficiency Despite Apparently Normal Immunologic Tests. J Pediatr (2005) 147:555–7. doi: 10.1016/j.jpeds.2005.05.010
42. Smyth CM, Ginn SL, Deakin CT, Logan GJ, Alexander IE. Limiting γc Expression Differentially Affects Signaling Via the Interleukin (IL)-7 and IL-15 Receptors. Blood (2007) 110:91–8. doi: 10.1182/blood-2006-11-055442
43. Waickman AT, Park JY, Park JH. The Common γ-Chain Cytokine Receptor: Tricks-and-treats for T Cells. Cell Mol Life Sci (2016) 73:253–69. doi: 10.1007/s00018-015-2062-4
44. Klenerman P, Oxenius A. T Cell Responses to Cytomegalovirus. Nat Rev Immunol (2016) 16:367–77. doi: 10.1038/nri.2016.38
45. Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, Malefyt RDW, et al. Tslp: An Epithelial Cell Cytokine That Regulates T Cell Differentiation by Conditioning Dendritic Cell Maturation. Annu Rev Immunol (2007) 25:193–219. doi: 10.1146/annurev.immunol.25.022106.141718
46. Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, et al. Cloning of a Receptor Subunit Required for Signaling by Thymic Stromal Lymphopoietin. Nat Immunol (2000) 1:59–64. doi: 10.1038/76923
47. Park LS, Martin U, Garka K, Gliniak B, Di Santo JP, Muller W, et al. Cloning of the Murine Thymic Stromal Lymphopoietin (TSLP) Receptor: Formation of a Functional Heteromeric Complex Requires Interleukin 7 Receptor. J Exp Med (2000) 192:659–69. doi: 10.1084/jem.192.5.659
48. Al-Shami A, Spolski R, Kelly J, Fry T, Schwartzberg PL, Pandey A, et al. A Role for Thymic Stromal Lymphopoietin in CD4+ T Cell Development. J Exp Med (2004) 200:159–68. doi: 10.1084/jem.20031975
49. Rochman Y, Leonard WJ. The Role of Thymic Stromal Lymphopoietin in CD8 + T Cell Homeostasis. J Immunol (2008) 181:7699–705. doi: 10.4049/jimmunol.181.11.7699
50. Rochman Y, Spolski R, Leonard WJ. New Insights Into the Regulation of T Cells by γc Family Cytokines. Nat Rev Immunol (2009) 9:480–90. doi: 10.1038/nri2580
51. Ursini MV, Gaetaniello L, Ambrosio R, Matrecano E, Apicella AJ, Salerno MC, et al. Atypical X-Linked SCID Phenotype Associated With Growth Hormone Hyporesponsiveness. Clin Exp Immunol (2002) 129:502–9. doi: 10.1046/j.1365-2249.2002.01823.x
52. Weinstein JS, Herman EI, Lainez B, Licona-Limón P, Esplugues E, Flavell R, et al. T FH Cells Progressively Differentiate to Regulate the Germinal Center Response. Nat Immunol (2016) 17:1197–205. doi: 10.1038/ni.3554
53. McGuire HM, Vogelzang A, Warren J, Loetsch C, Natividad KD, Chan TD, et al. IL-21 and IL-4 Collaborate To Shape T-Dependent Antibody Responses. J Immunol (2015) 195:5123–35. doi: 10.4049/jimmunol.1501463
54. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. Il-21 Acts Directly on B Cells to Regulate Bcl-6 Expression and Germinal Center Responses. J Exp Med (2010) 207:353–63. doi: 10.1084/jem.20091738
55. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. Il-21 Regulates Germinal Center B Cell Differentiation and Proliferation Through a B Cell-Intrinsic Mechanism. J Exp Med (2010) 207:365–78. doi: 10.1084/jem.20091777
56. Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement Receptor 2/CD21- Human Naive B Cells Contain Mostly Autoreactive Unresponsive Clones. Blood (2010) 115:5026–36. doi: 10.1182/blood-2009-09-243071
57. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass Trial: Defining Subgroups in Common Variable Immunodeficiency. Blood (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
58. Mouillot G, Carmagnat M, Gérard L, Garnier JL, Fieschi C, Vince N, et al. B-Cell and T-Cell Phenotypes in CVID Patients Correlate With the Clinical Phenotype of the Disease. J Clin Immunol (2010) 30:746–55. doi: 10.1007/s10875-010-9424-3
59. Nelson BH. Il-2, Regulatory T Cells, and Tolerance. J Immunol (2004) 172:3983–8. doi: 10.4049/jimmunol.172.7.3983
60. Upton J. Immunodeficiencies With Hypergammaglobulinemia: A Review. LymphoSign J (2015) 2:57–73. doi: 10.14785/lpsn-2014-0019
61. Vidarsson G, Dekkers G, Rispens T. Igg Subclasses and Allotypes: From Structure to Effector Functions. Front Immunol (2014) 5:520. doi: 10.3389/fimmu.2014.00520
62. Milner JD, Fazilleau N, McHeyzer-Williams M, Paul W. Cutting Edge: Lack of High Affinity Competition for Peptide in Polyclonal CD4+ Responses Unmasks IL-4 Production. J Immunol (2010) 184:6569–73. doi: 10.4049/jimmunol.1000674
63. Brooks EG, Schmalstieg FC, Wirt DP, Rosenblatt HM, Adkins LT, Lookingbill DP, et al. A Novel X-Linked Combined Immunodeficiency Disease. J Clin Invest (1990) 86:1623–31. doi: 10.1172/JCI114884
64. Wada T, Yasui M, Toma T, Nakayama Y, Nishida M, Shimizu M, et al. Detection of T Lymphocytes With a Second-Site Mutation in Skin Lesions of Atypical X-linked Severe Combined Immunodeficiency Mimicking Omenn Syndrome. Blood (2008) 112:1872–5. doi: 10.1182/blood-2008-04-149708
65. Schandené L, Ferster A, Mascart-Lemone F, Crusiaux A, Gérard C, Marchant A, et al. T Helper Type 2-Like Cells and Therapeutic Effects of Interferon-γ in Combined Immunodeficiency With Hypereosinophilia (Omenn’s Syndrome). Eur J Immunol (1993) 23:56–60. doi: 10.1002/eji.1830230110
66. Larché M, Akdis CA, Valenta R. Immunological Mechanisms of Allergen-Specific Immunotherapy. Nat Rev Immunol (2006) 6:761–71. doi: 10.1038/nri1934
67. Akdis CA, Akdis M. Mechanisms of Allergen-Specific Immunotherapy. J Allergy Clin Immunol (2011) 127:18–27. doi: 10.1016/j.jaci.2010.11.030
68. Geha RS, Jabara HH, Brodeur SR. The Regulation of Immunoglobulin E Class-Switch Recombination. Nat Rev Immunol (2003) 3:721–32. doi: 10.1038/nri1181
69. Gould HJ, Sutton BJ. Ige in Allergy and Asthma Today. Nat Rev Immunol (2008) 8:205–17. doi: 10.1038/nri2273
70. Matthews DJ, Clark PA, Herbert J, Morgan G, Armitage RJ, Kinnon C, et al. Function of the Interleukin-2 (IL-2) Receptor γ-Chain in Biologic Responses of X-Linked Severe Combined Immunodeficient B Cells to IL-2, Il-4, IL-13, and IL-15. Blood (1995) 85:38–42. doi: 10.1182/blood.v85.1.38.bloodjournal85138
71. Matthews DJ, Hibbert L, Friedrich K, Minty A, Callard RE. X-Scid B Cell Responses to Interleukin-4 and Interleukin-13 Are Mediated by a Receptor Complex That Includes the Interleukin-4 Receptor α Chain (p140) But Not the γc Chain. Eur J Immunol (1997) 27:116–21. doi: 10.1002/eji.1830270118
Keywords: Interleukin-2 receptor gamma, combined immunodeficiency, hypomorphic mutations, hyper-IgE, inborn error of immunity
Citation: Belaid B, Lamara Mahammed L, Mohand Oussaid A, Migaud M, Khadri Y, Casanova JL, Puel A, Ben Halla N and Djidjik R (2021) Case Report: Interleukin-2 Receptor Common Gamma Chain Defect Presented as a Hyper-IgE Syndrome. Front. Immunol. 12:696350. doi: 10.3389/fimmu.2021.696350
Received: 16 April 2021; Accepted: 07 June 2021;
Published: 24 June 2021.
Edited by:
Mohamed-Ridha Barbouche, Pasteur Institute of Tunis, TunisiaReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilLeopoldo Santos-Argumedo, Instituto Politécnico Nacional de México (CINVESTAV), Mexico
Copyright © 2021 Belaid, Lamara Mahammed, Mohand Oussaid, Migaud, Khadri, Casanova, Puel, Ben Halla and Djidjik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reda Djidjik, b3VydGlsYW5lQHlhaG9vLmZy