 Annesa Das1
Annesa Das1 Himanshu Kumar
Himanshu Kumar Prafullakumar Tailor
Prafullakumar Tailor- 1Laboratory of Innate Immunity, National Institute of Immunology, New Delhi, India
- 2Department of Biological Sciences, Laboratory of Immunology and Infectious Disease Biology, Indian Institute of Science Education and Research (IISER) Bhopal, Bhopal, India
- 3Special Centre for Systems Medicine (SCSM), Jawaharlal Nehru University, New Delhi, India
Plasmacytoid dendritic cells (pDCs) are the key producers of type I interferons (IFNs), thus playing a central role in initiating antiviral immune response. Besides robust type I IFN production, pDCs also act as antigen presenting cells post immunogenic stimulation. Transcription factor Irf8 is indispensable for the development of both pDC and cDC1 subset. However, the mechanism underlying the differential regulation by IRF8 in cDC1- and pDC-specific genomic architecture of developmental pathways still remains to be fully elucidated. Previous studies indicated that the Irf8R294C mutation specifically abrogates development of cDC1 without affecting that of pDC. In the present study using RNA-seq based approach, we have found that though the point mutation Irf8R294C did not affect pDC development, it led to defective type I IFN production, thus resulting in inefficient antiviral response. This observation unraveled the distinctive roles of IRF8 in these two subpopulations—regulating the development of cDC1 whereas modulating the functionality of pDCs without affecting development. We have reported here that Irf8R294C mutation also caused defect in production of ISGs as well as defective upregulation of costimulatory molecules in pDCs in response to NDV infection (or CpG stimulation). Through in vivo studies, we demonstrated that abrogation of type I IFN production was concomitant with reduced upregulation of costimulatory molecules in pDCs and increased NDV burden in IRF8R294C mice in comparison with wild type, indicating inefficient viral clearance. Further, we have also shown that Irf8R294C mutation abolished the activation of type I IFN promoter by IRF8, justifying the low level of type I IFN production. Taken together, our study signifies that the single point mutation in Irf8, Irf8R294C severely compromised type I IFN-mediated immune response by murine pDCs, thereby causing impairment in antiviral immunity.
Introduction
Dendritic cells (DCs) are professional antigen-presenting cells that can be subdivided into several classes based on expression of surface markers, transcription factors, and functional specificity (1, 2). DCs are classified broadly into classical dendritic cells (cDCs) and plasmacytoid DCs (pDCs). cDCs are further divided into cDC1 (comprising of splenic CD8α+ DCs and tissue resident CD103+ DCs) and cDC2 (comprising of splenic CD4+ DCs and tissue resident CD11b+ DCs) (3, 4). cDC1 plays an important role in antigen cross-presentation, cytokine interleukin 12 (IL12) production, tumor rejection, protection against Toxoplasma gondii (5–9), whereas cDC2 has a predominant role in antigen presentation by MHC II, tolerance, cytokine IL6 and IL23 production (1, 10, 11). pDCs are the key players in mounting antiviral responses as they are the major producers of type I interferons (IFNs) upon detecting PAMPs (pathogen-associated molecular patterns) by Toll-like receptor 7 (TLR7) and TLR9. Type I IFN response mediated by pDCs also depends on the virus and the route of infection (12, 13). Type I IFN production subsequently leads to induction of ISGs (interferon stimulated genes), which in turn initiate a series of downstream signaling cascades essential for mounting efficient antiviral immune response (14).
In course of DC development and diversification from a common DC progenitor, the presence of subset specific transcription factors and cytokines in the developmental milieu triggers the lineage-specific developmental program (15–17). In vitro bone marrow–derived dendritic cell cultures by employing cytokines such as FLT3L and GM-CSF have aided the studies related to DC development; GMDC (GM-CSF-induced bone marrow–derived DC) cultures give rise to the heterogeneous cell types with similarities to CD11b+ cDCs and macrophages, whereas FLDC (FLT3 ligand–induced bone marrow–derived DC) cultures would preferentially develop into pDCs and cDCs (18–23). Study employing Csf2r null (Csf2rb−/−Csf2rb2−/−) mice demonstrated significance of GM-CSF signaling in survival and homeostasis of non-lymphoid tissue-resident CD103+ and CD11b+ DCs (24). Transcription factors Irf4, Relb, Notch, Klf4, and different members of NF-κB family play a lead role in cDC2 development (11, 16, 25, 26); whereas Irf8, Batf3, Id2, Zbtb46, Nfil3, Bcl6 are important for cDC1 development (15, 25, 27–29). pDC development requires the transcription factors Irf8, Spib, Ikaros, Runx2, E2.2, etc. (13, 16, 30–33). Earlier studies revealed that transcription factor Irf8 is essential for development of both cDC1 and pDC (30); however, transcriptional program that leads to generation of these separate DC subpopulations, which vary in morphology and functionality, is still obscure. A previous study reported that Irf8R294C, a point mutation in Irf8, abolishes cDC1 subset development without affecting pDC developmental program (34). During DC lineage diversification, among all the cells expressing IRF8, the balance of Id2 and E2.2 decides direction of lineage development. Expression of higher amount of E2.2 over Id2 together with Irf8 shifts developmental regime towards pDC (1, 15, 16, 35, 36). It is intriguing that IRF8R294C mice retain the intact pDC population since previous studies indicated that Irf8 plays a central role in controlling fate of pDC development and function (30, 34). Recent studies have found that pDCs also develop from lymphoid progenitor (37, 38). But, how Irf8 plays a role in regulating the divergence of subset specific identity of cDC1 and pDC requires further elucidation. An enhancer element was identified at +32 Kb of Irf8 which displayed binding of BATF3-IRF8 complex leading to autoactivation of Irf8 in cDC1 population, suggesting a critical role of IRF8-BATF3 interaction in cDC1 commitment as against the pDC development (39). Recent study describing Irf8VENUS reporter mouse strain having expression of Irf8 from three intact copies of IRF8 present in the BAC reporter transgenes demonstrated that its over expression could bypass the requirement of Batf3 for cDC1 development (40). IRF8R294C mice develop spontaneously myeloid leukemia, exhibit splenomegaly, are defective in IL12p40 production, and are more prone to M. bovis infection (34, 41, 42). Some of these defects were attributed to cDC1 deficiency; however, further understanding of the mechanism of action and the effect of the mutation Irf8R294C especially in pDC subset in mounting immune responses will provide valuable insights in pDC biology. In depth analysis of DC subset development resulting from Itgax-cre mediated deletion of Irf8 expression revealed that it did not affect pDC development or maintenance of fully developed pDCs but caused a major shift in their function (43). In the present study, to better understand the impact of Irf8R294C mutation in the pDC subset, using RNA-seq approach we compared the splenic pDC transcriptome of IRF8R294C with IRF8WT (wild type) mice for the first time. We report that Ifnb1 gene (encoding IFN-β) is downregulated in pDCs from IRF8R294C mice. This observation is of great significance since pDC is characterized as the major type I IFN-producing cells, and dysregulation of pDCs has been associated with several autoimmune diseases such as psoriasis, systemic lupus erythematosus (SLE), rheumatoid arthritis, etc. (44–48). Further, the role of pDCs is also recognized in viral infection such as hepatitis B as pDC impairment is observed in chronic HBV patients (49).
It was reported that IRF8R291Q mutation in human, orthologous to murine IRF8R294C mutation, results in several immunological consequences (50), suggesting the importance of this point mutation regarding functioning of immune system. So, understanding the pDC development and function in context of Irf8R294C mutation can be of utmost importance to diagnostics and in developing the course of therapeutics. Here, using NDV (Newcastle disease virus) infection and CpG stimulation model, we have demonstrated that Irf8R294C mutation leads to impairment in type I IFN production. In addition to this, we have determined that the impairment in type I IFN production aggravates the defect in production of ISGs and upregulation of costimulatory molecules in pDCs from IRF8R294C mice. Further, using IRF8R294C mice we have shown that under in vivo experimental condition also, IRF8R294C mice are defective in type I IFN production, upregulation of costimulatory molecules in pDC, and exhibit increased NDV burden. Collectively, this study contributes to better understanding of the novel role of Irf8R294C mutation in pDC functionality especially in interferon production and antiviral response. These findings unravel differential role of Irf8 due to point mutation Irf8R294C in pDC function, casting importance in therapeutic implications.
Materials and Methods
Mice and Cell Culture
All animal experiments were conformed to the guidelines of the animal ethics committee at National Institute of Immunology (NII) (IAEC approval# 536/19) and biosafety committee guidelines (IBSC approval# 413/20). IRF8−/− and IRF8R294C mice were procured from Jackson Laboratory. IRF8R294C mice were crossed with C57BL/6 wild-type female for at least three generations to eliminate the vertical transmission of Murine Leukemia Virus (MuLV). Progenies were crossed to generate the mice carrying homozygous alleles for the mutation, and all the mice were used at the age of 8–12 weeks. Bone marrow mononuclear cells were isolated from the femur and tibia and cultured in the presence of FLT3L (100 ng/ml, PeproTech) as described previously (51). After day 8, loosely adherent cells were harvested and used for different treatments, stimulations, and experiments. For stimulation with CpG (1826 ODN, Merck), as employed earlier (23, 27, 43), cells were stimulated with 1 μg/ml CpG for the mentioned time period and were analyzed for transcript or proceeded to flow cytometry. For the 7-AAD experiment, cells were harvested after mentioned time point and stained with different fluorochrome conjugated antibodies and finally with 7-AAD before analyzing through flow cytometry. NDV (LaSota strain) was a kind gift from Dr. Himanshu Kumar (IISER Bhopal). NDV infections were set up in serum-free media for 90 min at MOI 1 followed by replenishment with fresh culture media for the mentioned time period after washing with PBS. DCs were stained for surface markers with the following immunophenotyping antibodies: anti-B220 FITC (RA3-6B2), anti-CD11c APC (N418), anti-CD24 PE (M1/69), anti-CD80 V450 (16-10A1), anti-CD8α PE (53-6.7), anti-CD4 APC Cy7 (GK1.5), anti-Sirpα BV421 (P84) (all from BD biosciences); anti-CD40 FITC (HM40-3) (from eBioscience); anti-CD86 PE (GL-1) (from BioLegend), and biotin-labeled anti-SiglecH (551.3D3) (Miltenyi Biotech). Events were captured in FACS CantoII using FACS Diva software, and data were analyzed using Flowjo software (version 9.3.2).
HEK293T and U2OS cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 50 μg/ml streptomycin. Retroviral vectors expressing murine IRF8WT (wild type) and IRF8R294C were generated, and HEK293T cells were seeded in six-well dish for retroviral transduction with viral supernatants and 4 μg/ml polybrene by spinoculation as described previously (52). Transduced cells were then used for further experiments.
RNA Sequencing
Spleens were harvested from 8- to 12-week-old mice, and splenocytes were isolated. Briefly, dendritic cell-enriched, low-density cell fraction from spleen digested by Liberase (Roche laboratory) was isolated using 30% BSA in PBS as described previously (25). The isolated cells were stained using anti-CD11c, anti-B220, anti-CD8α, anti-CD4 (all from BD biosciences), and biotin-labeled anti-SiglecH (Miltenyi Biotech) antibodies; and CD11c+B220+SiglecH+ pDCs were sorted using FACS Aria III cell sorter. Sorted cells were lysed in RNAiso Plus (Takara), and RNA was isolated using RNeasy mini kit (Qiagen). After the QC procedures, mRNA was enriched using oligo(dT) beads. Then, the mRNA was fragmented randomly by adding fragmentation buffer, and cDNA was synthesized using mRNA template and random hexamer primers, after which a custom second-strand synthesis buffer (Illumina), dNTPs, RNaseH, and DNA polymerase I were added to initiate the second-strand synthesis. Followed by terminal repair, A ligation, and sequencing adaptor ligation, the double-stranded cDNA library was completed through size selection and PCR enrichment. Finally, RNA sequencing was carried out in Illumina platform (150 bp paired end), and generated paired end reads were used for downstream analysis. Raw data QC was carried out to check data quality. The quality trimmed reads were aligned using HISAT2 aligner (v.2.1.0) and mapped to GRCm39 as reference genome. HTSeq-count (v.0.12.4) was utilized to generate read count, and DESeq2 (v.1.30.1) R-package was employed for differential gene expression analysis.
RNA Extraction and qRT-PCR
RNA was isolated using Nucleospin RNA isolation kit (Machery-Nagel), and cDNA was prepared using high-capacity reverse transcription kit (Applied Biosystems) according to the manufacturer’s protocol. cDNA was diluted appropriately, and qRT-PCR was performed using SYBR Green PCR master mix and 7500 Fast RT PCR machine (Applied Biosystems). Relative mRNA expression was calculated using delta Ct method, and to normalize the variability in expression level, Gapdh was used as internal control. Primers used for qRT-PCR are listed in Supplementary Table 1.
Dual Luciferase Reporter Assay
U2OS cells seeded in 24-well plates were co-transfected with 100 ng of reporter plasmid pGL4.20-Luc (Blank), pGL4.20-Ifnβ-Luc containing promoter region of murine Ifnb promoter alone (control), pGL4.20-Ifnα6-Luc containing promoter region of murine Ifna6 promoter alone (control) or along with pcDNA-IRF8WT or IRF8R294C (300ng) expressing plasmids. Transfection was carried out using Lipofectamine 2000 according to the manufacturer’s protocol and as transfection control Renilla luciferase plasmid pRL-TK (Promega, 50 ng) was used. Cells were harvested 24 h post-transfection, and protein activity of firefly and Renilla luciferase was quantified using dual-luciferase reagents (Promega) and Sirius Luminometer V3.2. Renilla luciferase was used to normalize firefly luciferase activity.
In Vivo NDV Infection Experiment
NDV stock was prepared in embryonated chicken eggs and titrated by hemagglutination assay as described previously (53) in Dr. Himanshu Kumar’s laboratory in IISER Bhopal. Then 1,200 hemagglutination assay (HA) unit of NDV was used for the infection where HA unit denotes the HA titer of the virus calculated as reciprocal of highest dilution at which complete agglutination of RBC was observed in hemagglutination assay. Eight- to 12-week-old mice were injected with PBS (control) or 1,200 HA NDV intravenously and kept for 8 h. Then blood was collected and spleens were harvested. DC-enriched low-density cell fraction from spleen was isolated as mentioned above. Blood was centrifuged at 2,000 g for 15 min, and serum was collected and stored for further analysis.
ELISA
FLDC culture supernatants after 24 h treatment or serum from in vivo experiment were subjected to IFN-α1 and IFN-β ELISA using Abcam IFN-α1 (ab252352) and Abcam IFN-β (ab252363) ELISA kits respectively according to the manufacturer’s protocol.
Statistical Analysis
Statistical analysis was performed using unpaired Student’s t test, and significance is represented as *p < 0.05, **p < 0.01, ***p < 0.001, and ns means non-significant.
Results
Irf8R294C Mutation Alters the Transcriptome of pDC Subset
Numerous studies established that Irf8 is a key transcription factor for cDC1 and pDC development (1, 25, 30, 54). The characterization of splenic DCs and FLDC cultures from Irf8−/− mice exhibited complete absence of both cDC1 and pDC populations; however, Irf8R294C mutation selectively abrogated cDC1 development, without affecting that of pDC (Supplementary Figures 1A, B) in coherence with previous studies (30, 34, 54). This intriguing phenomenon of selective loss of cDC1 population in IRF8R294C but retention of pDC population led us to investigate whether Irf8 plays a differential role in cDC1 and pDC development and function. We found that transcript level of Irf8 is elevated in pDCs from IRF8R294C mice in comparison with IRF8WT (wild type) mice (Supplementary Figure 1C). The increased level of Irf8 transcript in IRF8R294C pDCs led us to further explore the role of IRF8 in pDC biology.
To investigate this, we sorted pDCs (CD11c+ B220+ SiglecH+) from IRF8WT and IRF8R294C mice spleen, carried out transcriptome analysis, and studied differentially expressed genes (Supplementary Figure 2A). Principal component analysis (PCA) of RNA-seq data showed differential segregation of clusters of IRF8WT and IRF8R294C pDCs (Figure 1A). Additional analysis demonstrated a total of 3,494 genes were differentially expressed between these two groups after adjusting the p value cutoff <0.05. Further setting the next cutoff parameter of Log2FC value implemented at <=−1 or >=1, the number of differentially regulated genes were reduced to 2,542. Out of 2,542 genes, 898 genes were found to be upregulated in IRF8WT and the rest of the genes (1,644) were upregulated in IRF8R294C. To visualize gene expression data sets, the two-dimensional (2D) scatter MA plot (Bland Altman plot, “M” denotes log ratio and “A” denotes mean average) was constructed (Figure 1B). Global gene expression values, which were obtained from analysis of IRF8WT and IRF8R294C transcriptomic data, were represented in volcano plot (Figure 1C).
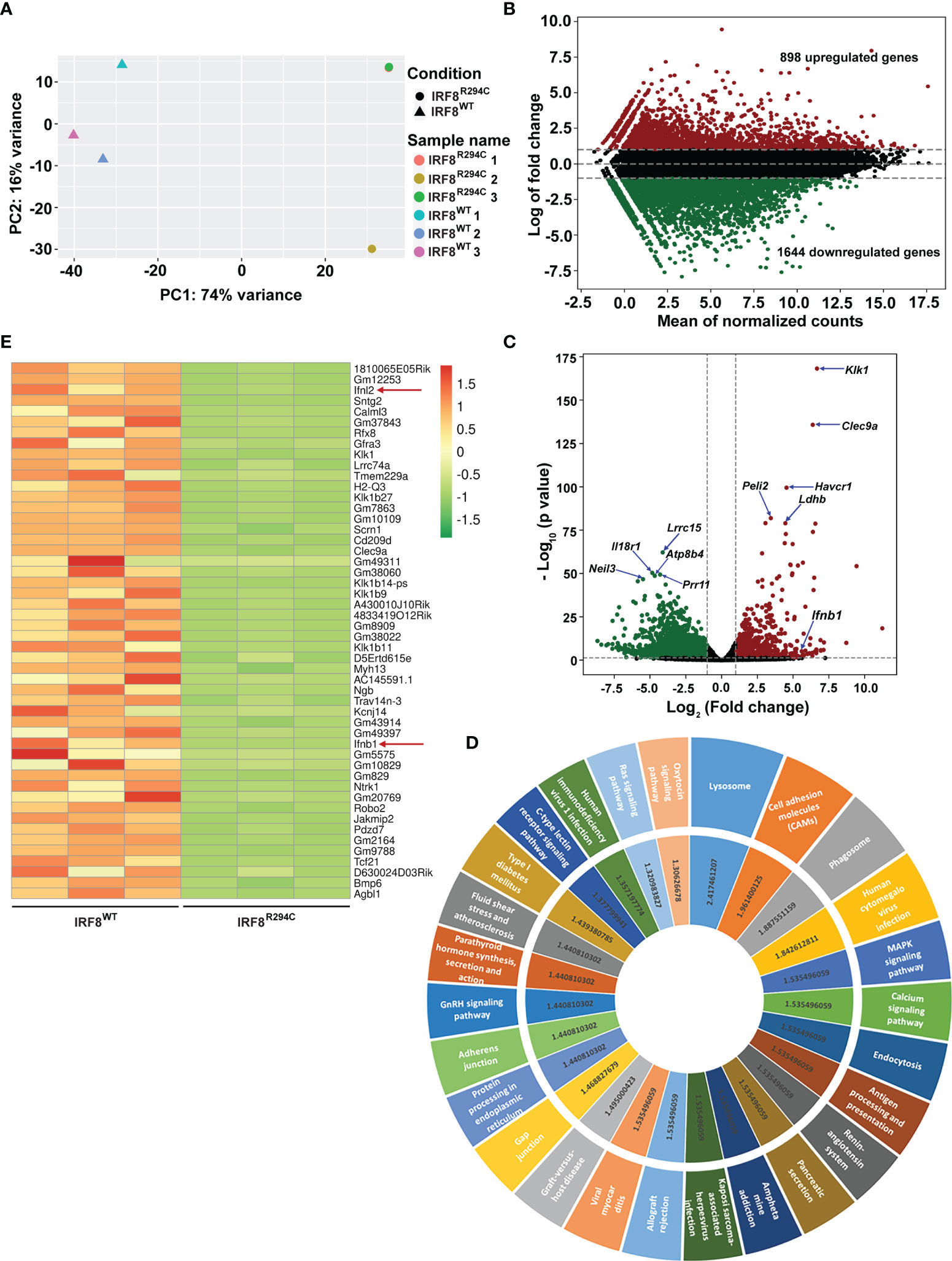
Figure 1 Irf8R294C mutation leads to alteration in pDC transcriptome. (A) Plot showing principal component analysis of six samples; distance between the samples depicts the difference in gene expression among the two sets (IRF8R294C samples 1 and 3 are overlapping, hence appearing as a single dot). (B) MA plot presenting differential gene expression, indicating upregulated (in red) and downregulated (in green) genes in IRF8WT. (C) Volcano plot displaying the differential gene expression between the two groups of samples with upregulated and downregulated genes in IRF8WT illustrated in red and green, respectively. (D) KEGG pathway analysis of upregulated genes in IRF8WT; the outer circle represents top upregulated pathways, and the inner circle indicates corresponding –log10FDR value of respective KEGG pathway. (E) Heatmap depicting relative expression of top 50 genes across six samples with red and green color denoting upregulated and downregulated genes in IRF8WT, respectively.
To further understand the ontological features of differentially regulated genes, we implemented KEGG pathway analysis on divergently upregulated (Figure 1D) and downregulated (Supplementary Figure 2B) genes in IRF8WT pDCs. Genes significantly enriched in KEGG pathway analysis included those involved in different pathways related to immune response and viral infections such as antigen processing and presentation, human cytomegalovirus infection, human immunodeficiency virus 1 infection, etc., and genes regulating essential signaling mechanisms such as MAPK signaling pathway, calcium signaling pathway, Ras signaling pathway, etc. Genes that showed differential expression in IRF8WT and IRF8R294C pDCs were represented in heatmaps (Supplementary Figures 2C, D and Figure 1E). Intriguingly, among various genes, the gene Ifnb1 (also known as Ifnb), which encodes interferon β (IFN-β), was found to be significantly downregulated in pDCs from IRF8R294C than IRF8WT mice (Figures 1C, E). IFN-β, a key cytokine belonging to type I IFN family, plays pivotal role in pDC biology and is well established for its indispensability in mediating antiviral immune response (12, 13). Also, Ifnl2, which encodes interferon lambda 2 (also known as IL28A), was found to be downregulated in IRF8R294C (Figure 1E). IFNL2, distantly related to type I IFN and IL10 family, is important for host defense from antiviral challenge and is involved in antitumor activity (55–58). From these analyses, we can clearly infer that although the point mutation Irf8R294C does not affect development of pDC subset, it results in significant changes at the transcriptome level.
Since Irf8 is a fundamental transcription factor involved in DC development and the fact that pDCs, which are the key producers of type I IFN, are showing decreased level of Ifnb in IRF8R294C mice strongly suggest the probable impact of this specific point mutation in changing the pDC transcriptome and immune response. So, in IRF8R294C pDCs, the decreased expression of Ifnb further prompted us to investigate the effect of Irf8R294C mutation in immune response to gain mechanistic insight of the expression and regulation of downstream signaling molecules.
IRF8R294C pDCs Are Defective in Type I IFN Production
First, to confirm observed reduction of Ifnb expression in RNA-seq data, we determined the transcript level of Ifnb from FLDC cultures derived from IRF8WT and IRF8R294C mice by qRT-PCR and found that indeed Ifnb was downregulated in IRF8R294C compared to its wild-type counterpart (Figure 2A). Next, we wanted to examine the impact of Irf8R294C mutation on type I IFN production upon CpG stimulation (TLR9 agonist) in FLDC cultures as pDCs induce type I IFN transcription upon encountering CpG by TLR9. It was found that induction of both Ifnb and Ifna (type I IFN) transcripts were greatly reduced in IRF8R294C DCs post-stimulation (Figures 2B, C). As pDCs play a major role in initiating antiviral response through type I IFN production, we wanted to determine the type I IFN production upon viral infection. To address this, FLDC cultures from IRF8WT and IRF8R294C mice were infected by a single-stranded RNA virus—Newcastle disease virus (NDV). NDV infection in IRF8WT led to marked augmentation of type I IFN transcript level, whereas in IRF8R294C there was abrogation of type I IFN expression (Figures 2D, E), which was consistent with the result from CpG stimulation.
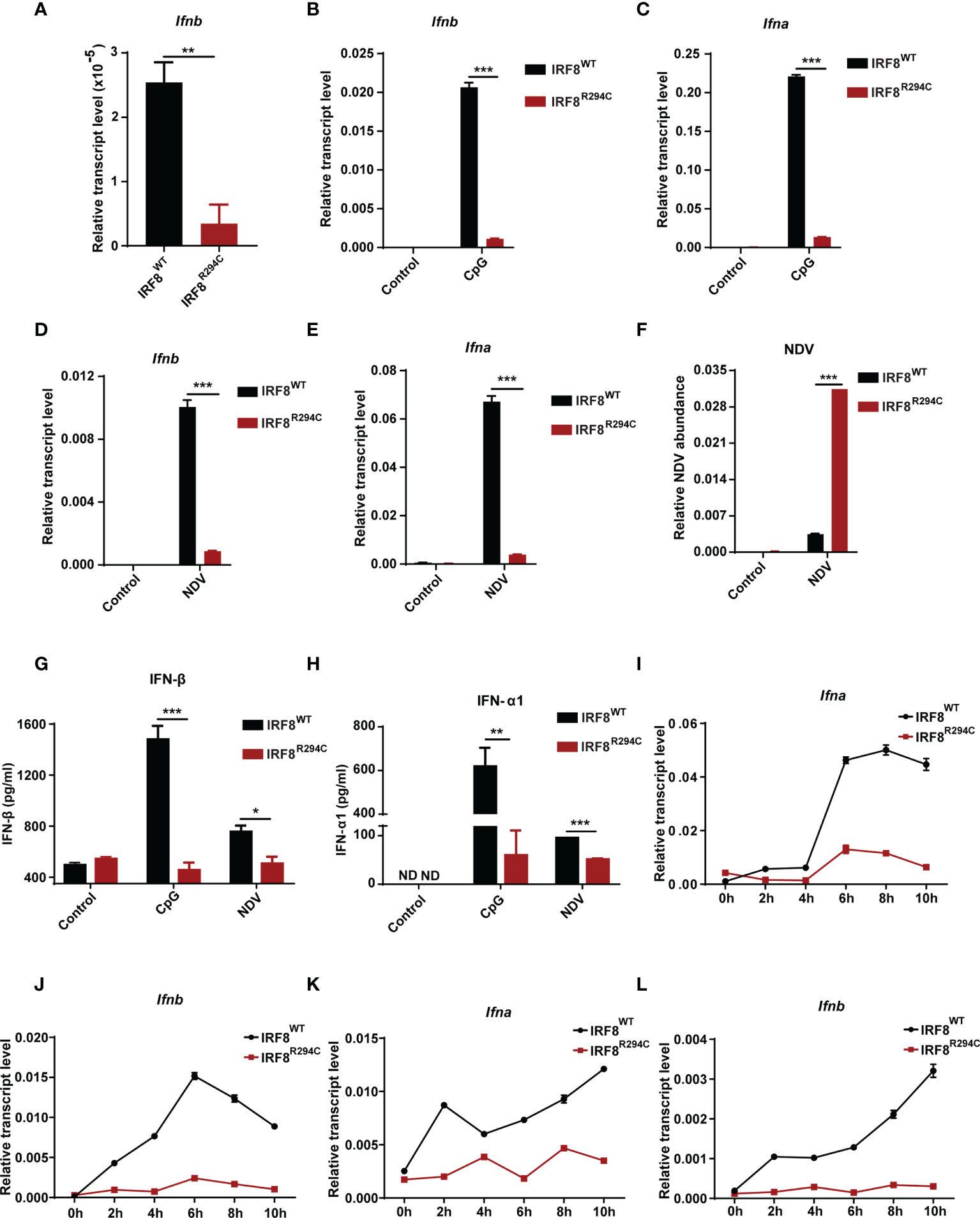
Figure 2 IRF8R294C pDCs show abrogation of type I IFN production in vitro. (A) RNA was isolated from IRF8WT and IRF8R294C FLDCs and analyzed by qRT-PCR to measure Ifnb expression. FLDC cultures from IRF8WT and IRF8R294C mice were stimulated with CpG (1 μg/ml) for 8 h, and level of Ifnb (B) and Ifna (C) transcript expression was checked by qRT-PCR. FLDC cultures from IRF8WT and IRF8R294C mice infected with NDV for 8 h were checked for expression of Ifnb (D), Ifna (E) transcripts, and NDV abundance (F) by qRT-PCR. FLDC culture supernatants from IRF8WT and IRF8R294C mice stimulated with CpG or infected with NDV for 24 h were examined for expression of IFN-β (G), IFN-α1 (H) cytokine by ELISA. FLDC cultures from IRF8WT and IRF8R294C mice stimulated with CpG (1 μg/ml) for indicated time points were analyzed for Ifna (I) and Ifnb (J) transcripts by qRT-PCR. FLDC cultures from IRF8WT and IRF8R294C mice infected with NDV for indicated time points were analyzed for Ifna (K) and Ifnb (L) transcripts by qRT-PCR. Data (A–F) are representative of three independent experiments with error bar representing + standard error of mean (SEM). Data (G, H) are mean of three independent experiments with error bar representing + standard error of mean (SEM). Data (I–L) are representative of three independent experiments with error bar representing ± standard error of mean (SEM). ND, not detected; *p < 0.05, **p < 0.01, ***p < 0.001; p value obtained from Student’s t test.
Further, defective induction of type I IFN transcripts in IRF8R294C FLDCs was concomitant with increased NDV burden in comparison with IRF8WT (Figure 2F). This observation was further confirmed in the protein level by ELISA as we observed IFN-β and IFN-α1 were greatly declined in FLDC culture supernatants from IRF8R294C mice compared to IRF8WT upon CpG stimulation as well as NDV infection (Figures 2G, H). So, these experiments infer that the pDCs from IRF8R294C mice are defective in type I IFN production. Further, we wanted to ascertain the effect of this mutation on the time kinetics of type I IFN production upon immune challenge. FLDCs from IRF8R294C mice stimulated with CpG were found to be defective in type I IFN production starting from early time points to late time points (Figures 2I, J). Next, we examined time kinetics of type I IFN production in FLDCs infected with NDV and found that in this scenario also, type I IFN production was greatly hindered in IRF8R294C in comparison with IRF8WT (Figures 2K, L). In summary, these data conclude that IRF8R294C pDCs are defective in type I IFN production from very early time points to late time points.
We speculated that the defect of type I IFN production in IRF8R294C FLDCs may be explained by either increased cell death in IRF8R294C or due to defect in pDC transcriptional program regulated by IRF8 or any other plausible mechanism mediated by the point mutation Irf8R294C. To examine if the decreased production of type I IFN was due to increased cell death in IRF8R294C, we stimulated FLDCs derived from IRF8WT and IRF8R294C mice with CpG for 6 h and 24 h and stained the treated as well as untreated cells with viability dye 7-AAD. Flow cytometric analysis revealed that the live pDC percentages were similar in IRF8R294C with that of IRF8WT mice under untreated as well as CpG stimulated condition for both 6 h and 24 h time points (Figures 3A–D). This experiment clearly refuted the hypothesis that reduced type I IFN production in IRF8R294C was due to increased cell death.
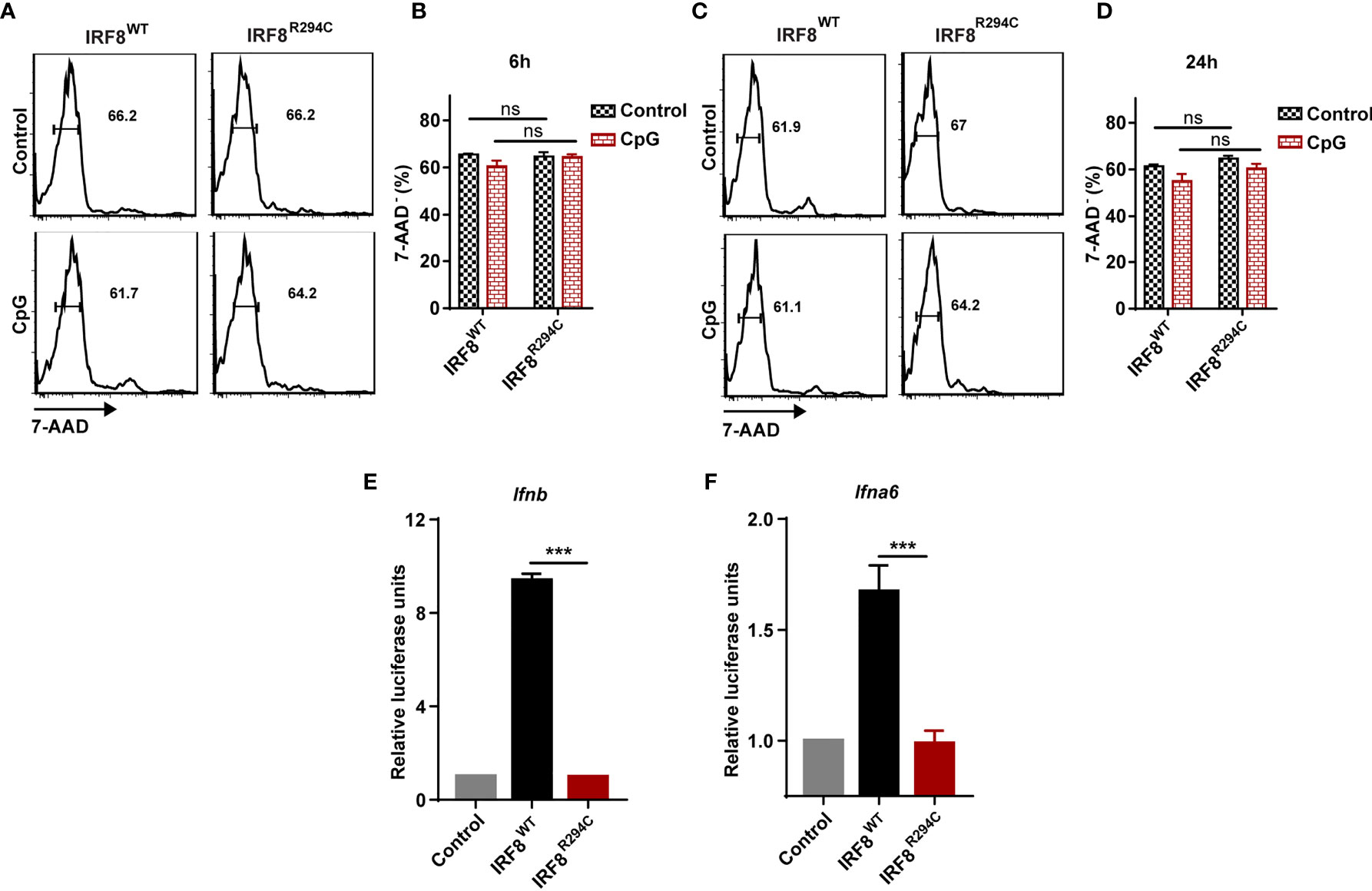
Figure 3 Defect of type I IFN production by IRF8R294C pDCs is not due to increased cell death. FLDC cultures from IRF8WT and IRF8R294C mice were kept unstimulated or stimulated with CpG and stained for different surface markers followed by 7-AAD staining. Cells with negative staining for 7-AAD were considered as live cells. Flow cytometric analysis (A) and quantitation (B) of live CD11c+ B220+ SiglecH+ pDCs from IRF8WT and IRF8R294C FLDC cultures after 6 h time point. Flow cytometric analysis (C) and quantitation (D) of live CD11c+ B220+ SiglecH+ pDCs from IRF8WT and IRF8R294C FLDC cultures after 24 h time point. IRF8WT activates expression of luciferase reporter construct driven by Ifnb (E) and Ifna6 (F) promoters, while IRF8R294C was unable to induce the expression of promoters. Data (A, C) are representative of three independent experiments. Data (B, D, E, F) are mean of three independent experiments with error bar representing + SEM and ***p < 0.001, ns, non-significant. p value obtained from Student’s t test.
Next, we wanted to decipher if this impairment of type I IFN production in IRF8R294C pDCs was an effect of indirect regulation of other pDC-specific factors or due to any mechanism mediated by mutation Irf8R294C itself. RIG-I like receptor (RLR) is a family of cytosolic pathogen recognition receptor (PRR) which recognizes viral RNA and induces type I IFN production (59). Fibroblasts are known to mediate antiviral response through RLR signaling (60), and mechanism for sensing NDV in HEK293T by RLR differs from that of pDCs mediated by TLR. HEK293T cell line was employed previously for studying interferon induction in response to viral infection (61). Hence, to further investigate, we infected HEK293T cells stably expressing IRF8WT, IRF8R294C separately with NDV, studied time kinetics of type I IFN transcript level (Supplementary Figures 3A, B) and found that type I IFN induction was curtailed in IRF8R294C-expressing cells. This experiment indicated that point mutation Irf8R294C directly affects the type I IFN production.
Defect in Type I IFN Production by IRF8R294C pDCs Leads to Defective Induction of ISGs
Next, we wanted to investigate the mechanism and the downstream effect of impaired type I IFN production. We hypothesized that the inability of type I IFN production in IRF8R294C could be due to ineffective type I IFN promoter activation. Through promoter assay we demonstrated that, IRF8WT induced Ifnb and Ifna6 promoters, but induction of Ifnb and Ifna6 promoters was defective in IRF8R294C-expressing cells (Figures 3E, F). This observation suggested that inefficient promoter activation by IRF8R294C translated to abrogated type I IFN production. Upon generation, type I IFN further promotes transcription of other effector molecules of several downstream signaling cascades which carry out different aspects of immune response (14). So, we speculated the defective type I IFN production may result in ineffective downstream signaling subsequently. We observed that, indeed, the production of ISGs was declined significantly in FLDCs from IRF8R294C mice stimulated with CpG compared to IRF8WT (Figure 4A). Similar observations were made in FLDCs from IRF8R294C mice infected with NDV (Figure 4B). IRF7 plays an important role in type I IFN-mediated signaling pathway and is known to be one of the major regulators of the pathway (59). IFIT3 and other ISGs—OAS3, OAS2, OAS1a, OAS1g, OASL2—which are 2’-5’ oligoadenylate synthetases, are involved in mounting antiviral innate immune response by restricting viral replication (62–64). Overall, these results suggest that the reduced expression of ISGs reflects the inability in induction of type I IFNs, and reduced production of these ISGs due to impaired type I IFN production in IRF8R294C may further compromise the response to CpG stimulation and NDV infection.
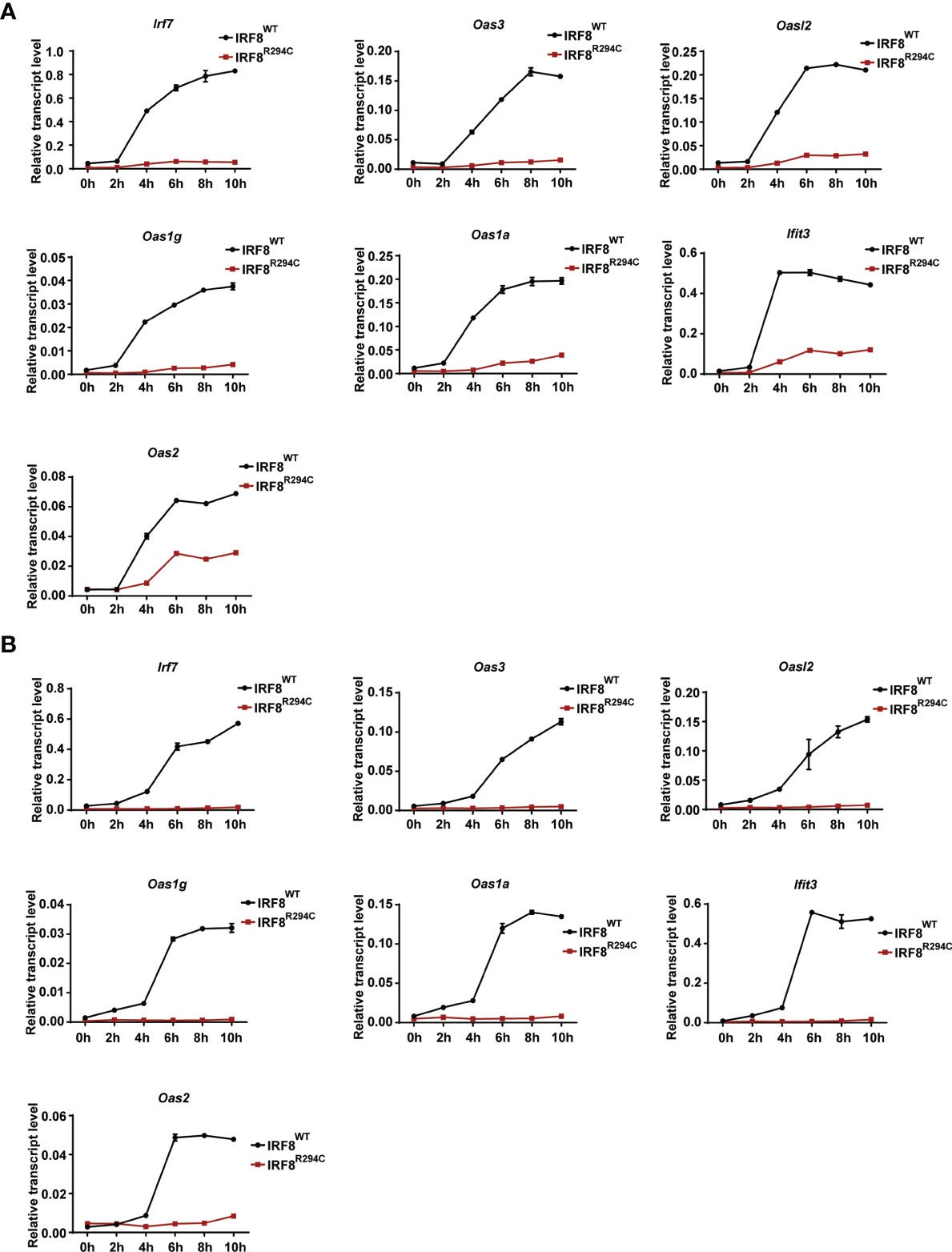
Figure 4 Impaired type I IFN production in IRF8R294C pDCs leads to impairment in induction of ISGs. Transcript analysis of Irf7, Oas3, Oasl2, Oas1g, Oas1a, Ifit3, and Oas2 by qRT-PCR in FLDC cultures from IRF8WT and IRF8R294C mice stimulated with CpG (A) or infected with NDV (B) at indicated time points. Data (A, B) are representative of three independent experiments with error bar representing ± SEM.
pDCs in resting state express the costimulatory molecules in low level, but once pDCs are activated, they upregulate several costimulatory molecules (CD40, CD86, CD80) and initiate a cascade of activities leading to instigating adaptive immune response (13, 65, 66). So, to characterize pDC activation, we next assessed the upregulation of costimulatory molecules in pDCs from IRF8WT and IRF8R294C mice stimulated with CpG or infected with NDV. In comparison to IRF8WT, the IRF8R294C pDCs displayed significant partial but not complete reduction in upregulation of CD40, CD86, and CD80 expression upon CpG stimulation as well as NDV infection (Figures 5A–F). Taken together, these experiments suggest that Irf8R294C point mutation renders pDCs defective in activation as the consequence of impaired type I IFN induction, leading to deficient expression of costimulatory molecules.
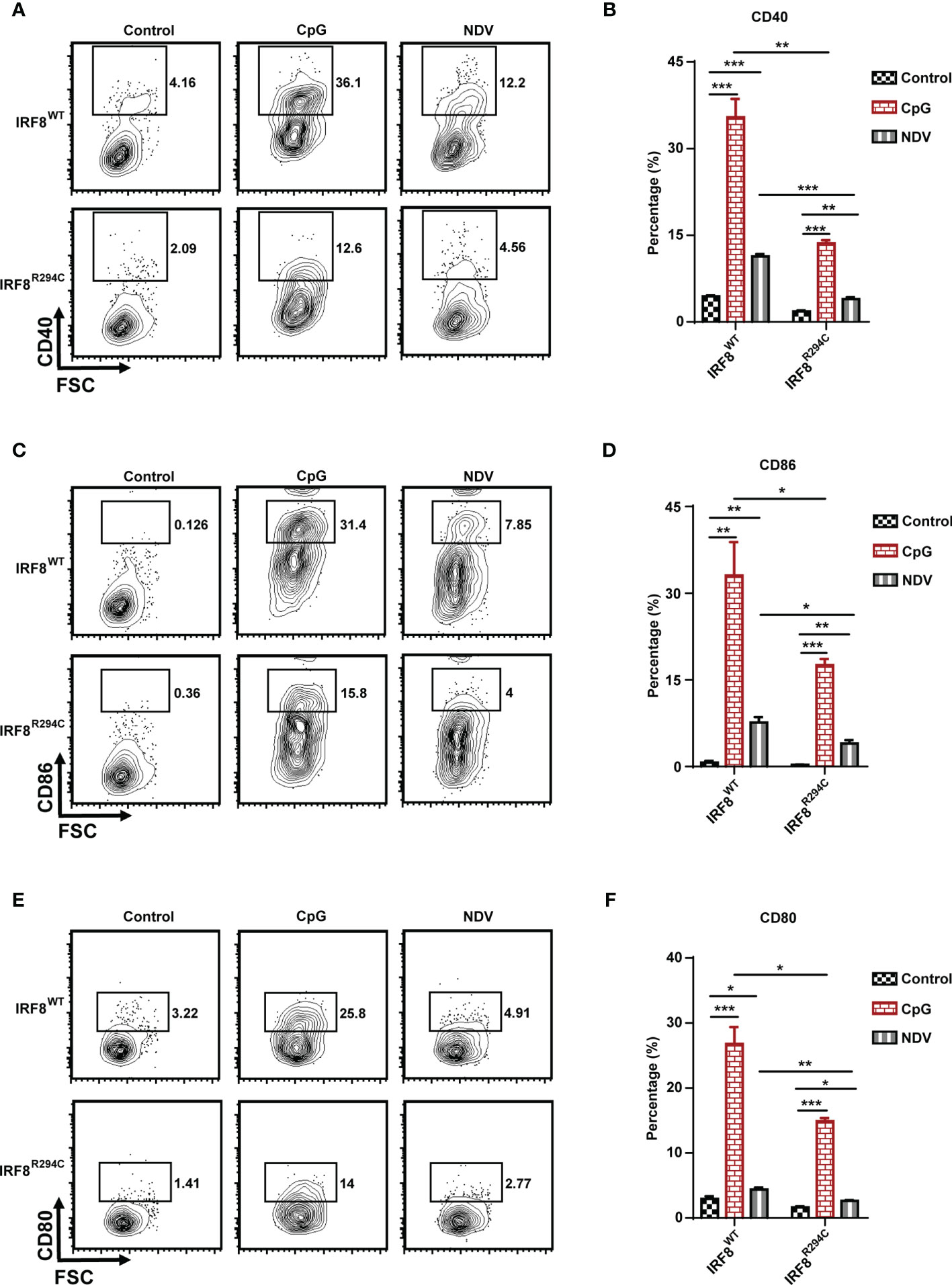
Figure 5 IRF8R294C pDCs are defective in activation upon immune challenges. FLDCs from IRF8WT and IRF8R294C mice were kept unstimulated or stimulated with CpG or infected with NDV, and CD11c+SiglecH+ pDCs were analyzed for expression of costimulatory molecules. Flow cytometric analysis of expression of (A) CD40, (C) CD86, (E) CD80 markers, and quantitation of expression of the respective markers (B, D, F) are shown. Data (A, C, E) are representative of three independent experiments. Data (B, D, F) are mean of three independent experiments with error bar representing + SEM and *p < 0.05, **p < 0.01, ***p < 0.001. p value obtained from Student’s t test.
In Vivo Study Confirms the Defective Type I IFN Production in IRF8R294C Mice
To corroborate our in vitro studies, we infected both IRF8WT and IRF8R294C mice with NDV (Figure 6A) and investigated type I IFN induction, NDV transcript, and status of costimulatory molecules in spleen, as spleen is the major site to elicit successful antiviral immune response against systemic infection (67, 68). There was significant impairment in production of type I IFN transcripts in NDV-infected IRF8R294C mice splenocytes compared to IRF8WT counterpart (Figures 6B, C). Further, the absence of type I IFN transcript augmentation was associated with increased viral burden in IRF8R294C mice splenocytes (Figure 6D). Next, we examined the concentration of IFN-α1 and IFN-β in sera by ELISA. Consistent with increased transcript levels, the upregulation of serum IFN-α1 and IFN-β in IRF8WT mice infected with NDV but not IRF8R294C counterpart bolstered that our observation from in vitro system indeed translated to in vivo infection condition (Figures 6E, F). Next, we examined the activation status of splenic pDCs from infected and uninfected mice. Flow cytometric analysis of splenic pDCs further revealed that IRF8WT mice infected with NDV showed significant upregulation of all the costimulatory molecules—CD40, CD86, CD80 (Figures 7A–F). In IRF8R294C mice, upregulation of CD40, CD86, and CD80 in pDCs was compromised upon NDV infection. Also, IRF8R294C pDCs, in unstimulated condition, showed lower expression of CD80 than wild-type counterpart in contrast to previous study in Irf8fl/flItgax-cre mice (43), suggesting the difference in IRF8R294C pDCs carrying mutant IRF8 as against its deletion in Irf8fl/flItgax-cre counterpart. We also compared the MFI of these costimulatory molecules to study the average expression of these markers on each cell (Supplementary Figures 4A–C). Expression of CD40, CD80, and CD86 displayed clear shift in histogram of respective markers in IRF8WT mice infected with NDV than all the other experimental groups (Figure 7G). Collectively, these experiments indicate that decreased type I IFN production in IRF8R294C mice further leads to immune suppression.
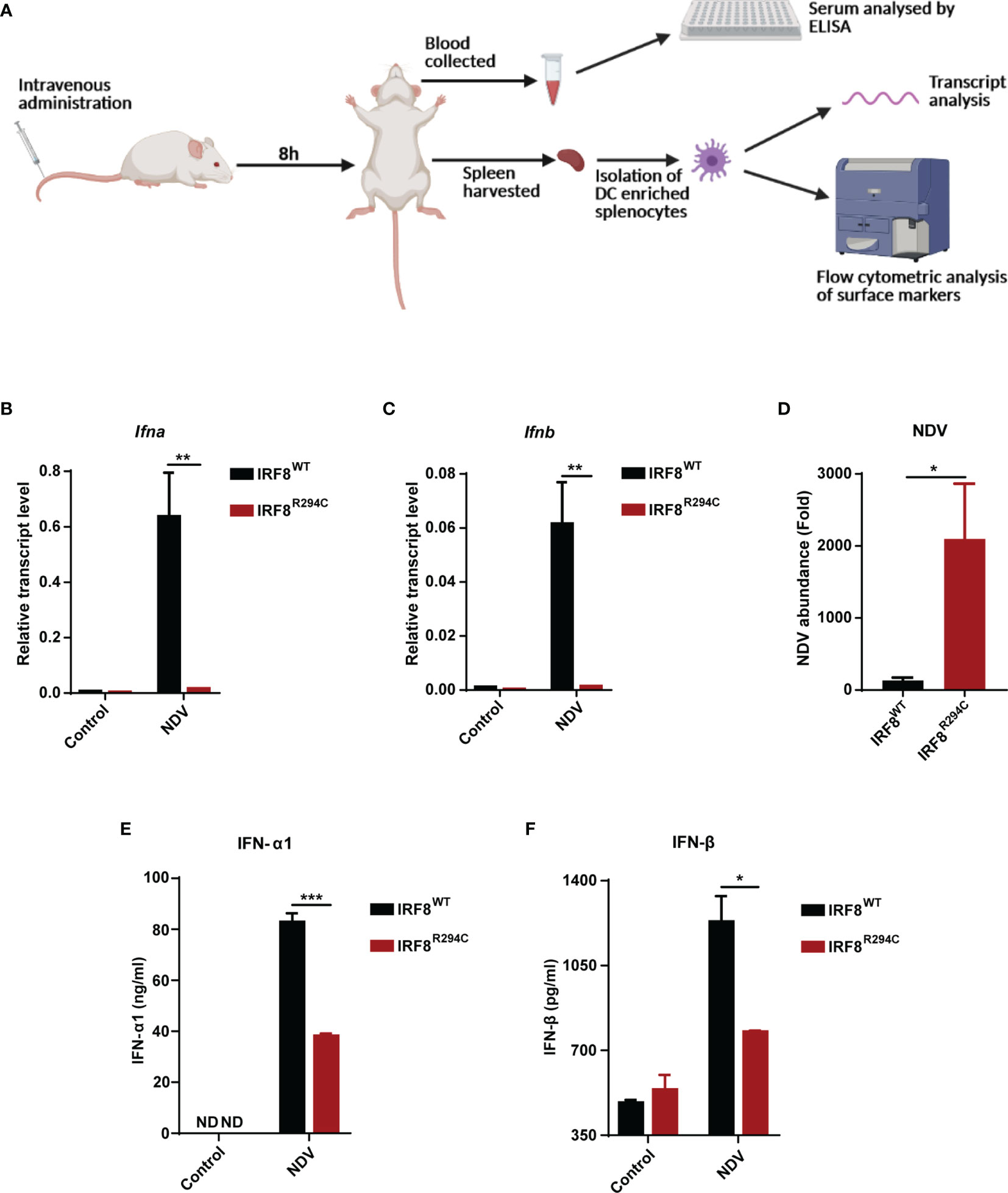
Figure 6 Impairment of type I IFN production in IRF8R294C mice upon NDV infection. (A) Schematic representation of the workflow for in vivo NDV infection. Transcript analysis of Ifna (B), Ifnb (C), and abundance of NDV (D) in splenocytes from uninfected or NDV-infected IRF8WT and IRF8R294C mice by qRT-PCR. IFN-α1 (E) and IFN-β (F) concentration in sera from uninfected or NDV-infected IRF8WT and IRF8R294C mice were quantitated by ELISA. Data (B–D) shown are mean value with error bar representing + SEM (n= 6 mice per group). Data (E, F) shown are mean value with error bar representing + SEM (n= 3 mice per group) and *p < 0.05, **p < 0.01, ***p < 0.001; p value obtained from Student’s t test.
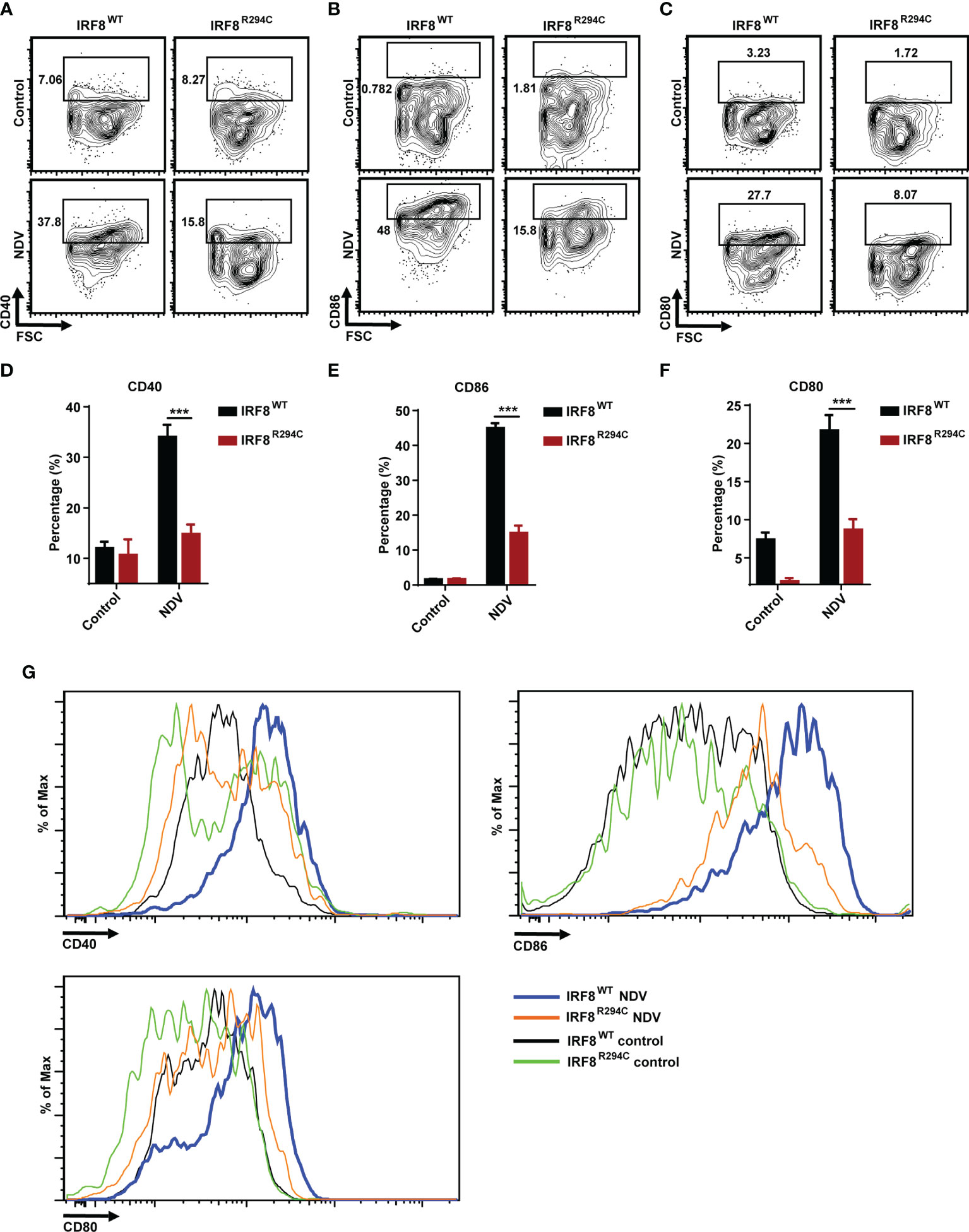
Figure 7 IRF8R294C mice upon NDV infection are inefficient in pDC activation. Mice infected in 6(A) were checked for status of costimulatory molecules among splenic CD11c+SiglecH+ pDCs. Flow cytometric analysis of surface markers CD40 (A), CD86 (B), CD80 (C), and quantitation of expression of the respective markers (D–F) and histogram (G) are shown. Data (D–F) shown are mean value with error bar representing + SEM and ***p < 0.001 (n= 6 mice per group). Data (A–C, G) shown are representative of each group (n= 6 mice per group). p value obtained from Student’s t test.
Conclusively, these results infer that the inability of mutant IRF8R294C to induce type I IFN promoters translates to impaired type I IFN production, which further abrogates production of ISGs and impairs pDC activation, ultimately leading to increased viral burden and futile immune response (Figure 8). Taken together, our study unravels the important role of IRF8 in mediating a successful immune response by controlling pDC function and the differential role played by IRF8 with respect to the cDC1 and pDC subsets. While in cDC1 subset, IRF8 controls the development and differentiation, in pDC subset, IRF8 controls the functionality, and Irf8R294C mutation greatly changes the functions mediated by IRF8.
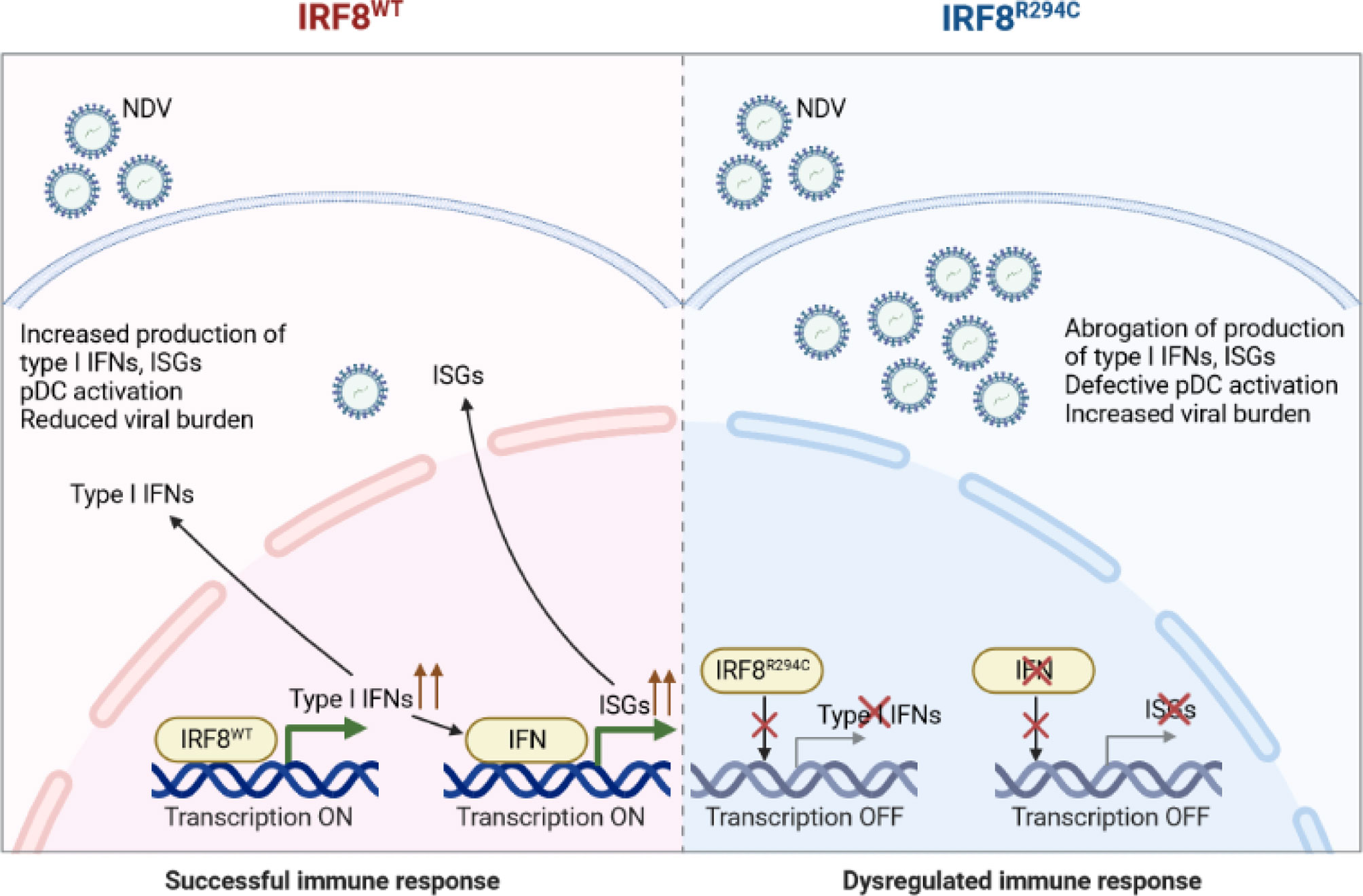
Figure 8 Schematic model of Irf8R294C-mediated impairment in immune response. Upon NDV infection, IRF8WT induces type I IFN promoter and transcription initiates. Type I IFN further results in production of downstream ISGs and pDC activation. Thus, together, type I IFN and ISGs restrict viral replication and mount successful immune response. Whereas, IRF8R294C fails to induce type I IFN promoter, and transcription does not take place. So, the absence of type I IFN leads to impaired pDC activation and abrogation of production of ISGs concomitant with increased NDV burden. Thus, IRF8R294C results in dysregulated immune response.
Discussion
Irf8 is an important member of IRF family that plays a critical role in regulation of DC differentiation (30). Identification of Irf8R294C point mutation emphasized the requirement of subsequent studies regarding the role of IRF8 in DC lineage differentiation program, as IRF8R294C mutation results in obliteration of only cDC1 subset but not pDC population in spite of the previous notion of the importance of IRF8 in pDC development from mice knockout studies (30, 34). To delineate the role of IRF8 in DC development and function, in this study we carried out the RNA-seq analysis to characterize the transcriptome of pDCs derived from IRF8WT and IRF8R294C mice. Our analysis reveals that Ifnb is downregulated in IRF8R294C pDCs than IRF8WT counterpart. pDCs, demonstrating plasma cell–like morphology, secrete massive amounts of type I IFN upon pathogen recognition through TLR7 and TLR9. This extraordinary feature of robust type I IFN production by pDCs further leads to initiation of the downstream signaling to mount host defense against various pathogens (13, 14). pDCs are documented to induce IFN production in response to a wide range of microbial pathogens such as dengue virus, influenza virus, Toxoplasma gondii, etc. (69–71). Recent report suggests that pDC frequency was decreased in PBMCs from individuals infected with SARS-CoV-2 and also, in response to TLR stimuli, pDCs from infected individuals produced decreased amounts of IFN-α (72). These observations indicate the important role of type I IFN response, controlled by pDC, against several immune challenges. Due to this essentiality of type I IFN signaling in pDC biology, decreased basal level of Ifnb in IRF8R294C pDCs is significant to the field and provides some cognizance in the immune-deficient nature of IRF8R294C mice that was previously reported (42).
Our subsequent experiments, which illustrate the drastic hindrance in type I IFN production in IRF8R294C FLDCs upon NDV infection (or CpG mediated TLR9 stimulation), further strengthens the fact that the initial defect in type I IFN production in IRF8R294C is further amplified during the course of viral challenge. The observation of differential expression of Ifnb in pDCs due to a single point mutation Irf8R294C, coupled with the finding of impaired type I IFN production upon immune challenge in IRF8R294C, provides important insight in the role of IRF8 in the regulation of complex transcriptional program of DC development and function. We have demonstrated the inability of IRF8R294C to induce interferon promoters, which might explain defective production of type I IFN in this phenotype. A previous study, which has indicated that IRF8 controls the chromatin access by regulating the stability of transcriptional complexes by recruitment of RNA polymerase II to IFN promoter region, extends support to our finding (52). The differential recruitment of RNA polymerase II by IRF8R294C may have further nullified the type I IFN production by suppressing the feedback response of IFN, which is well established for production of more type I IFNs subsequently (52). How the point mutation IRF8R294C changes the chromatin access and stability of the transcriptional machinery will be an important aspect of future research. IRF8 is well established to control the cDC1 and pDC lineage during developmental regime in precursor cells expressing IRF8 (1, 15). Itgax-cre-mediated deletion of Irf8 identified that Irf8 is important as a terminal selector of the cDC1 lineage but not of the monocyte or pDC lineage and suggested that IRF8 is critical to the functional gene modules in pDCs. The pDC subtype developed in Irf8fl/flItgax-cre mouse model was phenotypically distinct, having higher MHC II, CD11b, Ly6C, and CD11c expression and decreased expression of CD317 (43).
In the current study, for the first time, we report the defect in type I IFN production by IRF8R294C mice upon viral challenge, casting great impact in deciphering the role of IRF8 in controlling developmental and functional paradigm of dendritic cells. It is intriguing that complete absence of IRF8 correlates to loss of pDC and cDC1 subsets, but IRF8R294C mutation specifically blocks the developmental regime in cDC1. Here we have demonstrated that in spite of pDC development, the pDCs in IRF8R294C mice tend to be functionally different than IRF8WT pDCs. Further examinations in molecular level illustrating the mechanistic overview of the differential regulation of IRF8 in cDC1 and pDC will lead to unexplored arena of pDC and DC biology.
Once the IFN receptors are engaged after IFN production, they initiate downstream signaling cascade leading to generation of ISGs (14). Here we addressed the generation of ISGs upon NDV infection (or CpG-mediated TLR9 stimulation) in IRF8WT and IRF8R294C FLDCs and have found that absence of type I IFN production in IRF8R294C further results in reduction of ISGs production, and this reduction in turn leads to immune suppression. However, in IRF8R294C pDCs, studying the status of downstream signaling pathways such as JAK-STAT, which is known to mediate the production of ISGs upon activation by IFN receptor (14, 73), will provide further insight in this field. DCs, upon encountering immune challenges, upregulate the costimulatory molecules, which further helps in mediating adaptive immune response (13, 66). We explored the profile of pDC activation from IRF8WT and IRF8R294C FLDC cultures upon NDV infection (or CpG stimulation) and found that IRF8R294C pDCs expressed a decreased level of costimulatory molecules such as CD40, CD86, and CD80 in comparison with IRF8WT, suggesting the impairment in pDC activation in IRF8R294C mice. Further corroboration of our studies from in vivo model demonstrates the defect in type I IFN production and pDC activation in IRF8R294C mice upon NDV infection. This defect in pDC activation may explain further exaggeration to increased NDV burden in IRF8R294C mice. However, the underlying mechanism leading to the dearth of pDC activation requires additional characterization.
In conclusion, our study demonstrates IRF8 as a critical determinant of pDC functionality as the mutation Irf8R294C in mice does not affect pDC development but severely compromises the type I IFN-mediated crucial functions implemented by pDCs in response to viral pathogen. The consequences of Irf8R294C mutation are defect in three central aspects of innate immune response: (1) type I IFN production, (2) production of ISGs, and (3) pDC activation. These inadequacies in indispensable compartments of innate immune response may culminate to insufficient activation of adaptive immune response, leading to susceptibility of IRF8R294C mice. Finally, IRF8R294C mutant mice thus can be used as a model to elucidate the pDC functionality for designing therapeutic strategies of several autoimmune diseases where pDC dysregulation is associated with worse prognosis. Also, this mice model can help us in understanding the role of type I IFN production during viral infections.
Data Availability Statement
The data presented in the study are deposited in the GEO repository, accession number GSE186051.
Ethics Statement
The animal study was reviewed and approved by the Institute Animal Ethics Committee (IAEC) at National Institute of Immunology.
Author Contributions
AD and PT contributed to study design, data analysis, and data interpretation. AD and KC have performed the experiments. AD, KC, and PT have contributed to manuscript preparation. HK helped in NDV infection studies. All authors contributed to the article and approved the submitted version.
Funding
The authors would like to acknowledge financial support received from NII Core fund. KC was supported by fellowship from ICMR, and work was partially supported by grant #SERB/2016/005634.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
HEK293T cells were brought by PT from Dr. K. Ozato’s laboratory (NIH, MD, USA), and U2OS cells were obtained from Dr. S. Saxena’s laboratory (NII).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.758190/full#supplementary-material
References
1. Merad M, Sathe P, Helft J, Miller J, Mortha A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu Rev Immunol (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
2. Kushwah R, Hu J. Complexity of Dendritic Cell Subsets and Their Function in the Host Immune System. Immunology (2011) 133:409–19. doi: 10.1111/j.1365-2567.2011.03457.x
3. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells Across Tissues and Species. Immunity (2016) 45(3):669–84. doi: 10.1016/j.immuni.2016.08.015
4. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HRB, Schreuder J, Lum J, et al. Identification of Cdc1- and Cdc2-Committed DC Progenitors Reveals Early Lineage Priming at the Common DC Progenitor Stage in the Bone Marrow. Nat Immunol (2015) 16(7):718–28. doi: 10.1038/ni.3200
5. Maldonado-López R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, et al. Cd8α+ and CD8α- Subclasses of Dendritic Cells Direct the Development of Distinct T Helper Cells In Vivo. J Exp Med (1999) 189(3):587–92. doi: 10.1084/jem.189.3.587
6. Belz GT, Shortman K, Bevan MJ, Heath WR. Cd8α+ Dendritic Cells Selectively Present MHC Class I- Restricted Noncytolytic Viral and Intracellular Bacterial Antigens In-Vitro. J Immunol (2005) 175(1):196–200. doi: 10.4049/jimmunol.175.1.196
7. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
8. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of Cdc1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172(5):1022–37. doi: 10.1016/j.cell.2018.01.004
9. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. Cd8α+ Dendritic Cells Are the Critical Source of Interleukin-12 That Controls Acute Infection by Toxoplasma Gondii Tachyzoites. Immunity (2011) 35(2):249–59. doi: 10.1016/j.immuni.2011.08.008
10. Dudziak D, Kampfhorst A, Heidkamp G, Buhcholz V, Trumpfheller C, Yamazaki S, et al. Differential Antigen Processing by Dendritic Cell Subsets In Vivo. Science (2007) 315(5808):107–11. doi: 10.1126/science.1136080
11. Mildner A, Jung S. Development and Function of Dendritic Cell Subsets. Immunity (2014) 40:642–56. doi: 10.1016/j.immuni.2014.04.016
12. Liu YJ. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu Rev Immunol (2005) 23(3):275–306. doi: 10.1146/annurev.immunol.23.021704.115633
13. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid Dendritic Cells: Recent Progress and Open Questions. Annu Rev Immunol (2011) 29:163–83. doi: 10.1146/annurev-immunol-031210-101345
14. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
15. Satpathy AT, Wu X, Albring JC, Murphy KM. Re(De)Fining the Dendritic Cell Lineage. Nat Immunol (2012) 13(12):1145–54. doi: 10.1038/ni.2467
16. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, et al. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
17. Haniffa M, Collin M, Ginhoux F. Ontogeny and Functional Specialization of Dendritic Cells in Human and Mouse. Adv Immunol (2013) 120:1–49. doi: 10.1016/B978-0-12-417028-5.00001-6
18. Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential Development of Murine Dendritic Cells by GM-CSF Versus Flt3 Ligand has Implications for Inflammation and Trafficking. J Immunol (2007) 179(11):7577–84. doi: 10.4049/jimmunol.179.11.7577
19. Na YR, Jung D, Gu GJ, Seok SH. GM-CSF Grown Bone Marrow Derived Cells Are Composed of Phenotypically Different Dendritic Cells and Macrophages. Mol Cells (2016) 39(10):734–41. doi: 10.14348/molcells.2016.0160
20. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c+MHCII+ Macrophages and Dendritic Cells. Immunity (2015) 42(6):1197–211. doi: 10.1016/j.immuni.2015.05.018
21. Guilliams M, Malissen B. A Death Notice for in-Vitro-Generated GM-CSF Dendritic Cells? Immunity (2015) 42(6):988–90. doi: 10.1016/j.immuni.2015.05.020
22. Lutz MB, Inaba K, Schuler G, Romani N. Still Alive and Kicking: In-Vitro-Generated GM-CSF Dendritic Cells! Immunity (2016) 44(1):1–2. doi: 10.1016/j.immuni.2015.12.013
23. Saha I, Jaiswal H, Mishra R, Nel HJ, Schreuder J, Kaushik M, et al. Relb Suppresses Type I Interferon Signaling in Dendritic Cells. Cell Immunol (2020) 349:1–8. doi: 10.1016/j.cellimm.2020.104043
24. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF Controls Nonlymphoid Tissue Dendritic Cell Homeostasis But Is Dispensable for the Differentiation of Inflammatory Dendritic Cells. Immunity (2012) 36(6):1031–46. doi: 10.1016/j.immuni.2012.03.027
25. Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, et al. IFN Regulatory Factor-4 and -8 Govern Dendritic Cell Subset Development and Their Functional Diversity. J Immunol (2005) 174(5):2573–81. doi: 10.4049/jimmunol.174.5.2573
26. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 Expression in Conventional Dendritic Cells Is Required for T Helper 2 Cell Responses. Immunity (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
27. Jaiswal H, Kaushik M, Sougrat R, Gupta M, Dey A, Verma R, et al. Batf3 and Id2 Have a Synergistic Effect on Irf8-Directed Classical CD8α+ Dendritic Cell Development. J Immunol (2013) 191(12):5993–6001. doi: 10.4049/jimmunol.1203541
28. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science (2010) 327(5966):656–61. doi: 10.1126/science.1178331
29. Kashiwada M, Pham NLL, Pewe LL, Harty JT, Rothman PB. NFIL3/E4BP4 is a Key Transcription Factor for CD8α+ Dendritic Cell Development. Blood (2011) 117(23):6193–7. doi: 10.1182/blood-2010-07-295873
30. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, et al. ICSBP Is Essential for the Development of Mouse Type I Interferon-Producing Cells and for the Generation and Activation of CD8α+ Dendritic Cells. J Exp Med (2002) 196(11):1415–25. doi: 10.1084/jem.20021263
31. Schotte R, Nagasawa M, Weijer K, Spits H, Blom B. The ETS Transcription Factor Spi-B Is Required for Human Plasmacytoid Dendritic Cell Development. J Exp Med (2004) 200(11):1503–9. doi: 10.1084/jem.20041231
32. Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, et al. Ikaros Is Required for Plasmacytoid Dendritic Cell Differentiation. Blood (2006) 108:4025–34. doi: 10.1182/blood-2006-03-007757
33. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription Factor E2-2 Is an Essential and Specific Regulator of Plasmacytoid Dendritic Cell Development. Cell (2008) 135(1):37–48. doi: 10.1016/j.cell.2008.09.016
34. Tailor P, Tamura T, Morse HC, Ozato K. The BXH2 Mutation in IRF8 Differentially Impairs Dendritic Cell Subset Development in the Mouse. Blood (2008) 111:1942–5. doi: 10.1182/blood-2007-07-100750
35. Nagasawa M, Schmidlin H, Hazekamp MG, Schotte R, Blom B. Development of Human Plasmacytoid Dendritic Cells Depends on the Combined Action of the Basic Helix-Loop-Helix Factor E2-2 and the Ets Factor Spi-B. Eur J Immunol (2008) 38(9):2389–400. doi: 10.1002/eji.200838470
36. Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous Expression of the Transcription Factor E2-2 Maintains the Cell Fate of Mature Plasmacytoid Dendritic Cells. Immunity (2010) 33(6):905–16. doi: 10.1016/j.immuni.2010.11.023
37. Fernandes Rodrigues P, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct Progenitor Lineages Contribute to the Heterogeneity of Plasmacytoid Dendritic Cells. Nat Immunol (2018) 19:711–22. doi: 10.1038/s41590-018-0136-9
38. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid Dendritic Cells Develop From Ly6D+ Lymphoid Progenitors Distinct From the Myeloid Lineage. Nat Immunol (2019) 20(7):852–64. doi: 10.1038/s41590-019-0420-3
39. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 Maintains Autoactivation of Irf8 for Commitment of a CD8α + Conventional DC Clonogenic Progenitor. Nat Immunol (2015) 16(7):708–17. doi: 10.1038/ni.3197
40. Theisen DJ, Ferris ST, Briseño CG, Kretzer N, Iwata A, Murphy KM, et al. Batf3-Dependent Genes Control Tumor Rejection Induced by Dendritic Cells Independently of Cross-Presentation. Cancer Immunol Res (2019) 7(1):29–39. doi: 10.1158/2326-6066.CIR-18-0138
41. Turcotte K, Gauthier S, Mitsos LM, Shustik C, Copeland NG, Jenkins NA, et al. Genetic Control of Myreloproliferation in BXH-2 Mice. Blood (2004) 103(6):2343–50. doi: 10.1182/blood-2003-06-1852
42. Turcotte K, Gauthier S, Tuite A, Mullick A, Malo D, Gros P. A Mutation in the Icsbp1 Gene Causes Susceptibility to Infection and a Chronic Myeloid Leukemia–Like Syndrome in BXH-2 Mice. J Exp Med (2005) 201(6):881–90. doi: 10.1084/jem.20042170
43. Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity (2016) 45(3):626–40. doi: 10.1016/j.immuni.2016.08.013
44. Li S, Wu J, Zhu S, Liu YJ, Chen J. Disease-Associated Plasmacytoid Dendritic Cells. Front Immunol (2017) 8:1268. doi: 10.3389/fimmu.2017.01268
45. Fei T, Qiumei D, Yong-Jun L. Plasmacytoid Dendritic Cells in Antiviral Immunity and Autoimmunity. Sci China Life Sci (2010) 53(2):172–82. doi: 10.1007/s11427-010-0045-0
46. Kim JM, Park SH, Kim HY, Kwok SK. A Plasmacytoid Dendritic Cells-Type I Interferon Axis Is Critically Implicated in the Pathogenesis of Systemic Lupus Erythematosus. Int J Mol Sci (2015) 16(6):14158–70. doi: 10.3390/ijms160614158
47. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled With Antimicrobial Peptide. Nature (2007) 449(7162):564–9. doi: 10.1038/nature06116
48. Lande R, Giacomini E, Serafini B, Rosicarelli B, Sebastiani GD, Minisola G, et al. Characterization and Recruitment of Plasmacytoid Dendritic Cells in Synovial Fluid and Tissue of Patients With Chronic Inflammatory Arthritis. J Immunol (2004) 173(4):2815–24. doi: 10.4049/jimmunol.173.4.2815
49. Duan XZ, Wang M, Li HW, Zhuang H, Xu D, Wang FS. Decreased Frequency and Function of Circulating Plasmocytoid Dendritic Cells (Pdc) in Hepatitis B Virus Infected Humans. J Clin Immunol (2004) 24(6):637–46. doi: 10.1007/s10875-004-6249-y
50. Bigley V, Maisuria S, Cytlak U, Jardine L, Care MA, Green K, et al. Biallelic Interferon Regulatory Factor 8 Mutation: A Complex Immunodeficiency Syndrome With Dendritic Cell Deficiency, Monocytopenia, and Immune Dysregulation. J Allergy Clin Immunol (2018) 141(6):2234–48. doi: 10.1016/j.jaci.2017.08.044
51. Tsujimura H, Tamura T, Gongora C, Aliberti J, Reis e Sousa C, Sher A, et al. ICSBP/IRF-8 Retrovirus Transduction Rescues Dendritic Cell Development In Vitro. Blood (2003) 101(3):961–9. doi: 10.1182/blood-2002-05-1327
52. Tailor P, Tamura T, Kong HJ, Kubota T, Kubota M, Borghi P, et al. Type I Interferon Induction in Dendritic Cells Requires IRF-8 That Effects the Feedback Phase of Transcription. Immunity (2007) 27(2):228–39. doi: 10.1016/j.immuni.2007.06.009
53. Eisfeld AJ, Neumann G, Kawaoka Y. Influenza a Virus Isolation, Culture and Identification. Nat Protoc (2014) 9(11):2663–81. doi: 10.1038/nprot.2014.180
54. Tsujimura H, Tamura T, Ozato K. Cutting Edge: IFN Consensus Sequence Binding Protein/IFN Regulatory Factor 8 Drives the Development of Type I IFN-Producing Plasmacytoid Dendritic Cells. J Immunol (2003) 170(3):1131–5. doi: 10.4049/jimmunol.170.3.1131
55. Egli A, Santer DM, O’Shea D, Tyrrell DL, Houghton M. The Impact of the Interferon-Lambda Family on the Innate and Adaptive Immune Response to Viral Infections. Emerg Microbes Infect (2014) 3:1–12. doi: 10.1038/emi.2014.51
56. Zhou JH, Wang YN, Chang QY, Ma P, Hu Y, Cao X. Type III Interferons in Viral Infection and Antiviral Immunity. Cell Physiol Biochem (2018) 51(1):173–85. doi: 10.1159/000495172
57. Peterson ST, Kennedy EA, Brigleb PH, Taylor GM, Urbanek K, Bricker TL, et al. Disruption of Type III Interferon (IFN) Genes Ifnl2 and Ifnl3 Recapitulates Loss of the Type III IFN Receptor in the Mucosal Antiviral Response. J Virol (2019) 93:e01073–19. doi: 10.1128/JVI.01073-19
58. Tezuka Y, Endo S, Matsui A, Sato A, Saito K, Semba K, et al. Potential Anti-Tumor Effect of IFN-λ2 (IL-28A) Against Human Lung Cancer Cells. Lung Cancer (2012) 78(3):185–92. doi: 10.1016/j.lungcan.2012.09.005
59. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF Family Transcription Factors in Immunity and Oncogenesis. Annu Rev Immunol (2008) 26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400
60. Kawai T, Akira S. Innate Immune Recognition of Viral Infection. Nat Immunol (2006) 7(2):131–7. doi: 10.1038/ni1303
61. Li P, Wong JJY, Sum C, Sin WX, Ng KQ, Koh MBC, et al. IRF8 and IRF3 Cooperatively Regulate Rapid Interferon-β Induction in Human Blood Monocytes. Blood (2011) 117(10):2847–54. doi: 10.1182/blood-2010-07-294272
62. Samuel CE. Antiviral Actions of Interferons. Clin Microbiol Rev (2001) 14(4):778–809. doi: 10.1128/CMR.14.4.778–809.2001
63. Sadler AJ, Williams BRG. Interferon-Inducible Antiviral Effectors. Nat Rev Immunol (2008) 8(7):559–68. doi: 10.1038/nri2314
64. Vladimer GI, Górna MW, Superti-Furga G. Ifits: Emerging Roles as Key Anti-Viral Proteins. Front Immunol (2014) 5:94. doi: 10.3389/fimmu.2014.00094
65. Karrich JJ, Jachimowski LCM, Uittenbogaart CH, Blom B. The Plasmacytoid Dendritic Cell as the Swiss Army Knife of the Immune System: Molecular Regulation of Their Multifaceted Functions. J Immunol (2014) 193(12):5772–8. doi: 10.4049/jimmunol.1401541
66. Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse Type I IFN-Producing Cells Are Immature Apcs With Plasmacytoid Morphology. Nat Immunol (2001) 2(12):1144–50. doi: 10.1038/ni736
67. Bronte V, Pittet MJ. The Spleen in Local and Systemic Regulation of Immunity. Immunity (2013) 39(5):806–18. doi: 10.1016/j.immuni.2013.10.010
68. Lang KS, Lang PA. Balancing Viral Replication in Spleen and Liver Determines the Outcome of Systemic Virus Infection. Z Gastroenterol (2015) 53(12):1432–5. doi: 10.1055/s-0041-109631
69. Pichyangkul S, Endy TP, Kalayanarooj S, Nisalak A, Yongvanitchit K, Green S, et al. A Blunted Blood Plasmacytoid Dendritic Cell Response to an Acute Systemic Viral Infection Is Associated With Increased Disease Severity. J Immunol (2003) 171(10):5571–8. doi: 10.4049/jimmunol.171.10.5571
70. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid Dendritic Cells Activated by Influenza Virus and CD40L Drive a Potent TH1 Polarization. Nat Immunol (2000) 1(4):305–10. doi: 10.1038/79747
71. Pepper M, Dzierszinski F, Wilson E, Tait E, Fang Q, Yarovinsky F, et al. Plasmacytoid Dendritic Cells Are Activated by Toxoplasma Gondii to Present Antigen and Produce Cytokines. J Immunol (2008) 180(9):6229–36. doi: 10.4049/jimmunol.180.9.6229
72. Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems Biological Assessment of Immunity to Mild Versus Severe COVID-19 Infection in Humans. Science (2020) 369(6508):1210–20. doi: 10.1126/science.abc6261
Keywords: plasmacytoid dendritic cells (pDCs), type I interferons (IFNs), IRF8R294C, antiviral immune response, interferon regulatory factor 8 (IRF8), innate immunity, DC activation, interferon stimulated gene (ISG)
Citation: Das A, Chauhan KS, Kumar H and Tailor P (2021) Mutation in Irf8 Gene (Irf8R294C) Impairs Type I IFN-Mediated Antiviral Immune Response by Murine pDCs. Front. Immunol. 12:758190. doi: 10.3389/fimmu.2021.758190
Received: 13 August 2021; Accepted: 25 October 2021;
Published: 17 November 2021.
Edited by:
Elodie Segura, Institut Curie, FranceReviewed by:
Anne B. Krug, Ludwig Maximilian University of Munich, GermanyKeiko Ozato, National Institutes of Health (NIH), United States
Copyright © 2021 Das, Chauhan, Kumar and Tailor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prafullakumar Tailor, dGFpbG9ycEBuaWkuYWMuaW4=
†Present address: Kuldeep Singh Chauhan, Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO, United States