 Nikolaos Skartsis
Nikolaos Skartsis Leonardo M. R. Ferreira
Leonardo M. R. Ferreira Qizhi Tang
Qizhi Tang- 1Division of Nephrology and Hypertension, Department of Medicine, Mayo Clinic, Rochester, MN, United States
- 2Mayo Clinic William J. von Liebig Center for Transplantation and Clinical Regeneration, Mayo Clinic, Rochester, MN, United States
- 3Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, United States
- 4Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, SC, United States
- 5Hollings Cancer Center, Medical University of South Carolina, Charleston, SC, United States
- 6Department of Surgery, University of California, San Francisco, San Francisco, CA, United States
- 7Diabetes Center, University of California, San Francisco, San Francisco, CA, United States
- 8Gladstone University of California San Francisco (UCSF) Institute of Genome Immunology, University of California, San Francisco, San Francisco, CA, United States
TNFa blocking agents were the first-in-class biologic drugs used for the treatment of autoimmune disease. Paradoxically, however, exacerbation of autoimmunity was observed in some patients. TNFa is a pleiotropic cytokine that has both proinflammatory and regulatory effects on CD4+ T cells and can influence the adaptive immune response against autoantigens. Here, we critically appraise the literature and discuss the intricacies of TNFa signaling that may explain the controversial findings of previous studies. The pleiotropism of TNFa is based in part on the existence of two biologically active forms of TNFa, soluble and membrane-bound, with different affinities for two distinct TNF receptors, TNFR1 and TNFR2, leading to activation of diverse downstream molecular pathways involved in cell fate decisions and immune function. Distinct membrane expression patterns of TNF receptors by CD4+ T cell subsets and their preferential binding of distinct forms of TNFα produced by a diverse pool of cellular sources during different stages of an immune response are important determinants of the differential outcomes of TNFa-TNF receptor signaling. Targeted manipulation of TNFa-TNF receptor signaling on select CD4+ T cell subsets may offer specific therapeutic interventions to dampen inflammation while fortifying immune regulation for the treatment of autoimmune diseases.
Introduction
In the late 1970s, tumor necrosis factor alpha (TNFa) was discovered due to its tumoricidal activity, as bacterially contaminated neoplastic tumors would regress in size (1, 2). Over the ensuing years, TNFa was shown to have an important proinflammatory role in a variety of preclinical experimental models (3) and human disease settings (4). Several studies have established an association between genetic and protein processing defects in the NF-κB signaling pathway downstream of TNFa in human autoimmune and inflammatory diseases, such as systemic lupus erythematosus (SLE) (5), Sjögren’s syndrome (6), Crohn’s disease (7), ulcerative colitis (8) and rheumatoid arthritis (RA) (9). In the clinical arena, inhibition of TNFa signaling in otherwise therapy-resistant patients has led to improved treatment outcomes in a variety of immune-mediated diseases, such as RA (10), inflammatory bowel disease (11), adult-onset Still’s disease (12), and psoriasis (13). Surprisingly, however, some patients experienced new-onset autoimmune diseases, such as multiple sclerosis (MS), psoriasis, or lupus-like syndromes following administration of TNFa blocking agents (14–16). This led to an insurgence of studies shedding light on the immunomodulatory role that TNFa exerts on T cells (17, 18). It is now clear that TNFa is a multifaceted cytokine that has both proinflammatory and immunoregulatory roles. The differential outcomes of TNFa signaling depend on the (a) type of receptor TNFa binds to, (b) cell type carrying the specific TNF receptor, (c) cellular source of TNFa production, (d) phase of the immune response, and (e) type of regulatory T cell (Treg) responsible for suppression in each disease setting (Table 1). Given the latest advances in our understanding of the intricacies of TNFa signaling and its central role in auto- and allo-immunity, we are reviewing and critically appraising the literature and discussing potential opportunities to further develop precision medicine approaches for autoimmune diseases and transplant rejection.

Table 1 Factors that influence the impact of TNFa on the outcome of the immune response.
TNFa and TNFR overview
Ligand and receptor interactions
TNFa is a cytokine mainly produced by immune cells, such as macrophages, CD4+ T cells, NK cells, neutrophils, mast cells, and eosinophils. Yet, it is also secreted by non-immune cells, including endothelial cells, fibroblasts, and neurons (19, 20). TNFa is generated in a precursor form as a 233 amino acid type II single-pass transmembrane protein anchored in the cell membrane that is assembled in homotrimers and acts in a paracrine manner (21–23). Known as transmembrane TNFa (tmTNFa, 26kDa), this precursor protein can act both as a ligand and as a receptor during cell-to-cell interactions, thereby mediating both forward and reverse signaling (24, 25). It therefore plays a pivotal role in local inflammation. tmTNFa is processed by TNFa converting enzyme (TACE, also known as ADAM17) to generate soluble TNFa (sTNFa, 17 kDa) homotrimers (26, 27). Following TACE-mediated cleavage, the cytoplasmic domain of tmTNFa is further processed by the aspartyl protease SPPL2b, which then translocates to the nucleus and mediates proinflammatory cytokine production (28–30). The released fragment of sTNFa mediates additional endocrine function of TNFa in remote sites via hematogenous circulation. The individual roles of sTNFa and tmTNFa in mediating the pathogenesis of autoimmunity has been studied in transgenic mice expressing a TACE resistant form of tmTNFa (31). Interestingly, these mice were protected from autoimmune phenomena at a higher rate than TNFa KO animals, highlighting both the pathogenetic role of sTNFa and the partially protective role of tmTNFa (31). TACE inhibition has since emerged as a targeted strategy to preserve favorable inhibitory tmTNFa downstream signaling while efficiently blocking sTNFa deleterious activity (32).
TNFa exerts its effects via binding to either TNFR1 or TNFR2 (Figure 1). TNFR1 is expressed in all nucleated cells in the form of pre-assembled trimers, while TNFR2 is preferentially expressed in immune cells, such as activated CD4+ effector T (Teff) and CD4+ Treg cells (33–35). Importantly, the assembly of TNFR trimers is ligand-independent and is regulated by cysteine-rich domains in the extracellular region of TNFR (36). Mutations in these domains that mediate receptor folding and trafficking lead to systemic inflammation called TNFR-associated periodic syndrome (TRAPS) (37–41). TRAPS-associated mutant TNFR1 is not secreted, therefore does not bind TNF, but, instead, is retained in the endoplasmic reticulum leading to less efficient induction of apoptosis, compared to wild-type TNFR1 (39). Upon binding of a TNFa homotrimer to a TNFR1 or a TNFR2 homotrimer, signaling is initiated after secondary assembly of initial TNFa-TNFR homotrimers (42, 43). Both TNF receptors can bind to both tmTNFa and sTNFa. Yet, tmTNFa has higher affinity to TNFR2 (44), while sTNFa has a more stable association with TNFR1 (45) (Figure 1).
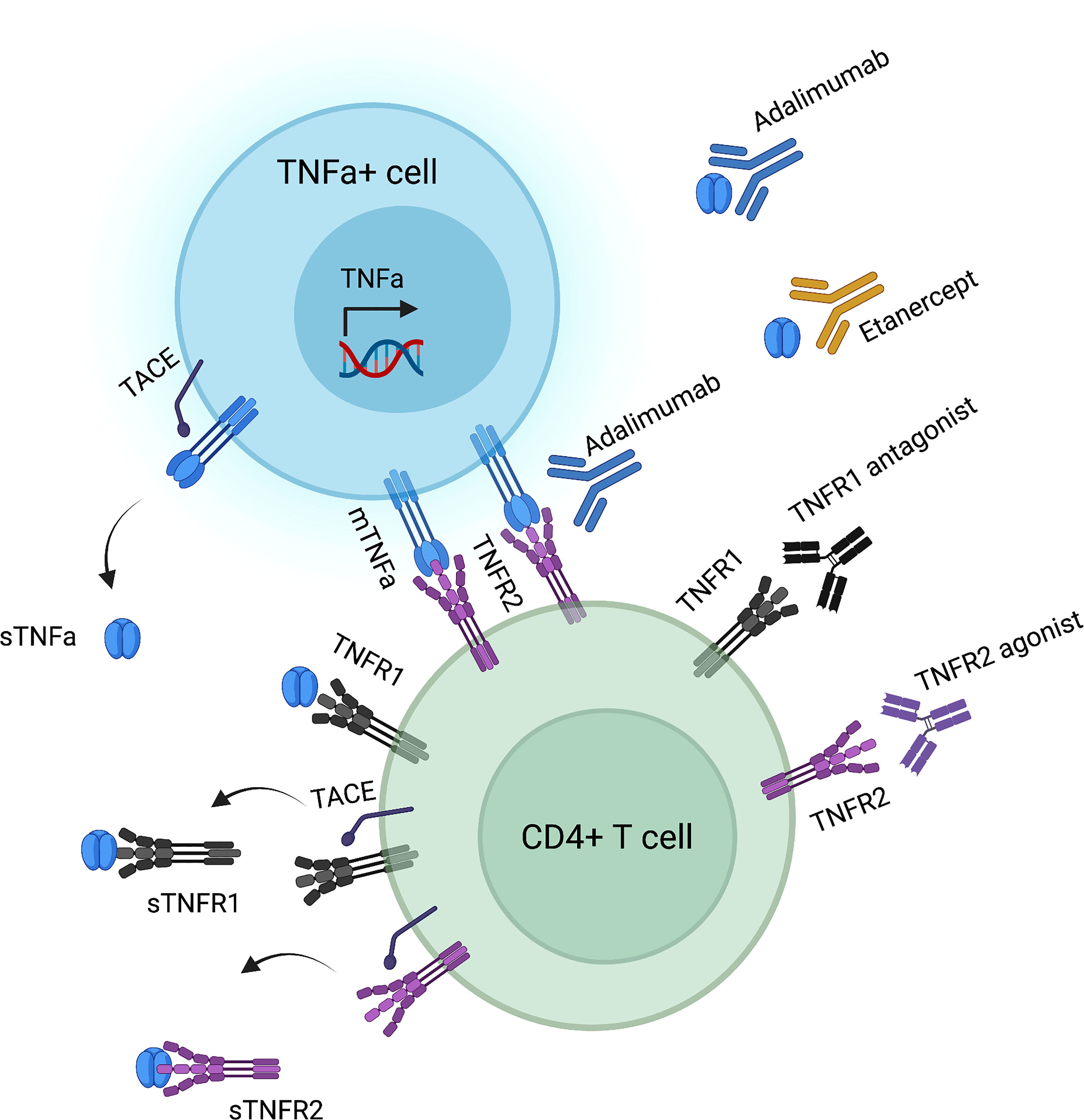
Figure 1 TNFa-TNFR interactions and corresponding biologic agents’ targets. Created by using biorender.com.
Signaling and pathway crosstalk
TNFR1 engagement triggers the transcription of proinflammatory genes via activation of the canonical NF-κB pathway (33, 46, 47). TNFR1 activation induces the recruitment of death domain (DD)-containing adapters, such as TNFR1-associated DD protein (TRADD) and receptor-interacting serine/threonine kinase 1 (RIPK1), with further recruitment of TNFR-associated factor 2 (TRAF2), culminating in the formation of signaling complex I. Associated cellular inhibitor of apoptosis (cIAP) 1/2 proteins ubiquitinate themselves and RIPK1, leading to further recruitment of TAK1/TAB2/TAB3 and linear ubiquitination assembly complex (LUBAC), which in concert polyubiquitinate NEMO and activate the IKK complex. This results in the phosphorylation and proteasomal degradation of IκB with the subsequent nuclear translocation of the active NF-κB complex (p50/p65). Additionally, TAK1 can lead to proinflammatory gene transcription via the phosphorylation of MAP kinases, such as the JNK and p38, which activate the AP-1 complex (48–50). Alternatively, TNFR1 signaling may lead to either apoptosis or necroptosis (51). Destabilization of complex I can lead to formation of complex II as non-ubiquinated RIPK1 and TRADD recruit FADD, C-FLIP and pro-caspase 8, ultimately leading to the activation of effector caspases (52). However, when caspase 8 is inhibited by caspase inhibitors or virally expressed proteins, RIPK1 and RIPK3 associate and either self-phosphorylate or phosphorylate each other, eventually leading to necroptosis (53).
TNFR2, in contrast to TNFR1, does not contain a death domain. Instead, TNFR2 directly recruits TRAF1 or TRAF2 along with cIAP1/2, triggering the recruitment of LUBAC, TAK1/TAB2/TAB3, and NEMO/IKK1/2 complexes, ultimately leading to activation of the canonical NF-kB pathway. tmTNFa binding to TNFR2 results in activation of the non-canonical NF-κB pathway via NIK accumulation, phosphorylation of IKK1 complex and processing of p100, and subsequent formation of p52/RelB heterodimers that translocate to the nucleus to activate transcription of target genes (54, 55). TNFR2 ligation in human Tregs enhances IL-2-induced proliferation mainly via the activation of the non-canonical NF-κB pathway (56). In addition to NF-κB pathway stimulation, TNFR2 crosslinking can also lead to MAPK activation. Indeed, p38 MAPK signaling is key to TNFR2-driven Treg activation and proliferation (57). On the other hand, pro-apoptotic signaling mediated by JNK activation depends on TNFR2 localization, which is regulated by TRAF2 (58) and requires the association of internalized TNFR2 with AIP1 (59).
TNFa binding to TNFR1 or TNFR2 may elicit downstream signaling that is not solely restricted to either type of receptor. Cross-talk between TNFR1 and TNFR2 may occur at multiple levels (60). Ligand passing has been credited with endowing TNFR2 the ability to deliver pro-apoptotic signals (61). According to this mechanism, owing to TNFR2’s more rapid association with TNFa and associated longer half-life of TNFa binding, TNFR2 increases the local TNFa concentration in the vicinity of TNFR1 receptors. These subsequently accept TNFa ligand molecules, inducing apoptosis (61). Under long-term TNFa exposure, TNFR1 and TNFR2 co-expressing cells prevent apoptosis by generating TRAF1 and TRAF2 heterodimers that are more efficient activators of NF-κB pathway signaling (62). Moreover, TNFR2 activation during inflammatory conditions may control TNFR1-induced activation via ASK1 ubiquitination (63).
Role of TNFa-TNFR in T cell development in the thymus
TNFa has been found to be important in many stages of thymic T cell development as it promotes the apoptosis of triple negative CD3-CD4-CD8- (64) and double positive CD4+CD8+ thymocytes (65). Beyond NF-κB’s conventional role in transmitting downstream TCR signals, TNFa can also directly upregulate anti-apoptotic genes, such as cIAP1/2, which is important for the maturation of CD4+ and CD8+ single-positive T cells during the later stages of T cell development (66). Contrary to their conventional counterparts, Treg development relies upon the cooperative activity of several TNF receptor superfamily members (67). Tregs are dedicated to suppressing immune responses, ensuring self-tolerance and immune homeostasis, limiting tissue damage by overactive immune responses (68). Their unique transcriptional program is bestowed on them by the sustained expression of the transcription factor FOXP3 (69). Thymic Treg (tTreg) development is a two-step process (70). First, TCR-CD28 signaling upregulates CD25 (IL2Rα) and CD122 (IL2Rβ) expression along with c-REL-dependent chromatin remodeling at the FOXP3 locus. Second, IL-2 signaling drives FOXP3 expression in a STAT5-dependent manner to endorse full Treg phenotype (70). TNFR2 expression is upregulated in Treg progenitors and serves as a link between TCR signaling strength and augmented IL-2/STAT5 signaling, eventually driving Treg differentiation (67).
Effect on Teffs in the periphery
Teffs not only produce TNFa, but also respond to it. Cell-type specific ablation of TNFa expression showed that myeloid-derived TNFa mediates the pathogenesis of collagen-induced arthritis (CIA), whereas T cell-derived TNFa is protective during the induction phase of arthritis by limiting T cell priming and memory T cell development (71). Interestingly, in a preclinical model of EAE, TNF produced by myeloid cells exacerbated neuroinflammation by driving the recruitment of inflammatory cells in the central immune system (CNS), and TNF produced by T cells further promoted myeloid cell recruitment into the CNS (72). However, in secondary lymphoid organs, TNF derived from myeloid and T cells synergized to dampen encephalitogenic Th1 and Th17 responses by decreasing IL-12 and IL-6 production (72). Another study using CD4+CD45RBhi T cell-induced colitis in lymphopenic mice showed that resident non-T cells are induced by Teffs in situ to produce TNFa, which in turn induced colitis. Of note, TNFa derived from Teffs was neither necessary nor sufficient to induce colitis (73). The exact nature of cell-to-cell interactions or soluble factors that Teffs employ to induce TNFa production by intestinal resident cells remains unclear. To address this, a subsequent study showed that TNFR2-deficient Teffs failed to induce full-fledged colitis in Rag1 KO mice due to their impaired capacity to produce Th1 cytokines, owing to increased p100/p52 ratio and thus defective non-canonical NF-κB signaling (74). Therefore, it seems plausible that TNFa-TNFR2 interactions between Teffs and local resident cells are key to pathogenicity in colitis. The balance between distinct cellular sources of TNFa in each disease setting may influence the outcome of the immune response.
Multiple studies have shown the role of TNFa in driving immune pathology. Local expression of TNFa in neonatal non-obese diabetic (NOD) mouse islets causes an influx of antigen-specific Teffs that precedes the onset of diabetes (75). Nevertheless, TNFa can have a dichotomous role in the pathogenesis of diabetes, depending on the stage of the ongoing autoimmune process. In a transgenic model of virally induced diabetes, early islet-specific TNFa expression augmented diabetes incidence, while late TNFa expression abrogated diabetes (76). It is unclear whether TNFa acts via induction of autoreactive cell apoptosis or promoting the expansion of Tregs at the later stage of the immune response. In that vein, seminal work indicated that Tregs accumulated preferentially in the pancreatic lymph nodes and islets suppressed islet destruction by CD8+ T cells in a TNFa signaling-dependent fashion (77). Another important temporal parameter relating to the diabetogenic potential of TNFa is the duration of exposure; chronic exposure of diabetogenic T cell clones to TNFa results in T cell unresponsiveness (78).
TNFa regulates multiple aspects of Teff fate, including survival, activation, and proinflammatory cytokine production (79–81). TNFa signaling lowers the threshold for TCR-dependent activation by providing TNFR2-mediated costimulation (82). Furthermore, TNFR2 signaling delivers an anti-apoptotic stimulus during the primary T cell response, expanding the resulting memory T cell pool following a second antigen encounter (83). A clinical study showed that infliximab, a TNFa blocking antibody, induces apoptosis in activated T cells isolated from the lamina propria of steroid-refractory Crohn’s disease patients, presumably due to anti-apoptotic NF-κB signaling withdrawal (84). Lastly, TNFa signaling promotes Teff proliferation even in the presence of Treg-mediated suppression (85). However, prolonged TNFa exposure restored Treg suppressive function, suggesting that TNFa promotes an effective immune response early on, yet delivers a delayed immunoregulatory feedback signal to Tregs to restore homeostasis (85).
Effect on Tregs in the periphery
Thymic derived T regulatory cells (tTregs)
The inflammatory microenvironment poses several challenges to Treg stability (86). In the synovial fluid of RA patients, TNFa was found to compromise Treg suppressive capacity via dephosphorylation of a serine residue in the DNA-binding domain of FOXP3 (87). Several studies have focused on TNFa blocking agents. Infliximab has been reported to reverse TNFa-induced suppressive capacity and FOXP3 expression loss in Tregs (88). Adalimumab, but not etanercept, was shown to increase Treg numbers in the synovial fluid of RA patients and improve Tregs’ capacity to suppress IL-17 production (89). Interestingly, adalimumab, in contrast to etanercept, stabilizes tmTNFa on the surface of monocytes; tmTNFa then promotes Treg expansion via TNFR2-mediated IL-2/STAT5 signaling (89). Promoting tolerogenic tmTNFa signaling, while neutralizing sTNFa deleterious actions, may underly the increased efficiency of adalimumab over etanercept and warrants confirmation in larger clinical trials.
Multiple studies have shown that TNFR2 signaling safeguards Treg stability under inflammatory conditions. Adoptively transferred TNFR2-deficient Tregs were unable to confer protection from colitis induced by co-transferred Teffs in Rag1 KO mice, underlining the critical role of TNFR2 in Treg phenotypic and functional stability in inflammatory environments (90). Indeed, TNFR2 expression defines a maximally suppressive Treg subgroup within effector Tregs (eTreg) that accounts for tumor-infiltrating eTreg-mediated immune surveillance evasion by solid tumors (91). Conditional TNFR2 ablation in Tregs led to exacerbated Th17-mediated experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, due to impaired Treg homeostasis (92). TNFa signaling has been shown to be pivotal for maintenance of eTreg phenotype in the periphery (93) and endows Tregs with the ability to suppress IFNg production by Teffs in a TNFR2-dependent manner (94).
In addition to safeguarding Treg lineage under inflammatory conditions, TNFR2 signaling synergizes with IL-2 to boost Treg proliferation via the non-canonical NF-κB signaling pathway (56). In a broader sense, TNFa signaling is used by Tregs to scale to inflammation. In line with this concept, Teff-derived TNFa has been shown to boost Tregs, dependent upon TNFR2 expression on Tregs, in experimental models of diabetes (95) and graft-vs-host disease (GvHD) (96). CD8+ T cells responding to virus infection stimulate a Vb5+ thymic Treg subset that expressed markers of a terminally differentiated effector cell phenotype, which promotes chronic infection in a TNFa-dependent manner (97). While proinflammatory signals enhance Treg proliferation, intense cell cycling increases the risk of Treg lineage destabilization. Indeed, IL-6 and TNFa synergistically drive robust human Treg proliferation in a TNFR2-dependent manner, with genetic deletion of TNFR2 leading to reduced expression of FOXP3 (98). TNFa-TNFR2 form a feedback loop that drives epigenetic changes that stabilize Treg phenotypic identity (99). These properties have been employed to expand Tregs ex vivo by delivering TNFR2 agonistic signals (100). In addition, a selective TNFR2 agonist was used to promote expansion of the endogenous Treg pool, which resulted in significantly reduced GvHD severity and mortality (101).
Induced T regulatory cells (iTregs)
In contrast to tTregs, induced Tregs (iTregs) do not require TNFa for in vivo function (102). While TNFR2 KO tTregs were unable to prevent adoptive T cell transfer-induced colitis in Rag1 KO mice, TNFR2 KO iTregs were fully suppressive (102). Interestingly, pre-treatment of TNFR2 KO tTregs with TGFb restored their suppressive function (102). Even though TGFb had been previously shown to promote iTreg homeostasis in the periphery, this was the first study to show TGFb’s role in restoring TNFR2-deficient tTreg function. In fact, TNFa antagonizes TGF-b-induced iTreg generation (103).
TNFa was shown to exacerbate the development of EAE by impairing differentiation and function of iTregs via TNFR2-mediated activation of Akt, which in turn inhibited TGFb-induced SMAD3 phosphorylation, resulting in decreased Foxp3 expression (103). Interestingly, TNFa does not activate the Akt pathway in tTregs (103), which may explain the dichotomy of the precipitation of autoimmune side effects by anti-TNFa biologics, depending on whether tTregs or iTregs have the predominant regulatory role in the pathogenesis of each disease. In contrast, adoptive transfer of TNFR1 KO iTregs showed improved clinical scores that were associated with sustained elevated TNFR2 expression on their surface (104). Still, TNFR2 KO iTregs were unable to prevent colitis, unlike WT iTregs. The differential results of these studies may be due to differences in experimental methodology, such as the use of different genetic knockout mice, which may alter T cell development, or the use of different protocols to induce Treg differentiation.
Therapeutic opportunities
Current TNFa blockade agents are aimed at preventing ligand binding to TNF receptors without taking into consideration the different forms of the receptor or their cell-specific expression pattern. Therefore, they indiscriminately prevent both the proinflammatory and the immunoregulatory effects of TNFR2 signaling. Novel biologics under development are aimed to selectively inhibit binding of TNFa to TNFR1, whereas selective TNFR2 activation requires both specific binding to the receptor and facilitation of oligomerization of TNFR2 complexes (105). Selective anti-TNFR1 binders under development include humanized mouse monoclonal antibodies (106), nanobodies (107), and small molecule inhibitors (108). Interestingly, XPro1595, a novel class of TNFa inhibitor called signaling-incompetent TNF derivative that inactivates sTNFa through the formation of mixed TNFa heterotrimers, has demonstrated an impressive improvement in EAE models (109). On the other hand, targeted TNFR2 binding has been achieved by generating TNFa muteins by mutagenesis or using phage display (110, 111). Fusion of homotrimeric TNF moieties allows for the generation of a nonvalent molecule capable of TNFR2 clustering (112). Indeed, sTNFa muteins S95C/G148C and TNF07, which display TNFR2 agonist properties due to their stable trimeric structure created by internal covalent cross-linking, expanded Tregs while simultaneously selectively inducing activation-induced cell death (AICD) of autoreactive CD8+ T cells in diabetic patients ex vivo (113).
Conclusion
The outcomes of TNFa signaling in CD4+ T cells depend on multiple parameters, including the phase of the immune response, duration of TNFa exposure, cellular source of TNFa production, and type of TNF receptor expressed on the responding CD4+ T cell subpopulation. The complex role of TNFa in Treg biology holds promise in aiding the development of Treg-based therapies, both in improving ex vivo expansion (98) and in vivo function (114). Incorporating TNFa signaling in synthetic immune receptors, such as chimeric antigen receptors, could help fine tune engineered Treg therapies under development (115–117). Recent advances in our understanding of the biology of TNFa-TNFR signaling provide excellent opportunities to design targeted therapies that inhibit the effector arm of T cell immunity while unleashing the immunoregulatory properties of TNFa signaling for the treatment of autoimmune diseases and transplant rejection.
Author contributions
NS wrote the manuscript and generated the table and the figure. LF and QT reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Human Islet Research Network (HIRN) Emerging Leader in Type 1 Diabetes grant U24DK104162-07 (LMRF), American Cancer Society (ACS) Institutional Research Grant IRG-19-137-20 (LF), South Carolina Clinical and Translational Research (SCTR) Pilot Project Discovery Grant 1TL1TR001451-01 (LF), Diabetes Research Connection (DRC) Grant IPF 22-1224 (LF), and grants from the Beatson Foundation 2021-021 (QT) and the Northern California JDRF Center of Excellence 5-COE-2019-860-S-B (QT).
Acknowledgments
The authors would like to thank Flavio Vincenti for helpful discussions.
Conflict of interest
NS is an inventor in a patent application covering the use of proinflammatory cytokines in ex-vivo Treg expansion. LF is an inventor in patents and patent applications covering genetically engineered effector T cells and regulatory T cells and uses thereof. QT is a co-founder and scientific advisor of Sonoma Biotherapeutics. QT is a scientific advisor of Moderna, eGenesis, Qihan Bio, Minutia and Encellin. QT is an inventor in patents and patent applications on Treg cell therapy and Treg cell engineering.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Matthews N, Watkins JF. Tumour-necrosis factor from the rabbit. i. mode of action, specificity and physicochemical properties. Br J Cancer (1978) 38(2):302–9. doi: 10.1038/bjc.1978.202
2. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U.S.A. (1975) 72(9):3666–70. doi: 10.1073/pnas.72.9.3666
3. Apostolaki M, Armaka M, Victoratos P, Kollias G. Cellular mechanisms of TNF function in models of inflammation and autoimmunity. Curr Dir Autoimmun (2010) 11:1–26. doi: 10.1159/000289195
4. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, et al. The role of tumor necrosis factor alpha (TNF-alpha) in autoimmune disease and current TNF-alpha inhibitors in therapeutics. Int J Mol Sci (2021) 22(5). doi: 10.3390/ijms22052719
5. Wong HK, Kammer GM, Dennis G, Tsokos GC. Abnormal NF-kappa b activity in T lymphocytes from patients with systemic lupus erythematosus is associated with decreased p65-RelA protein expression. J Immunol (1999) 163(3):1682–9. Available at: https://www.jimmunol.org/content/163/3/1682.long.
6. Wang X, Shaalan A, Liefers S, Coudenys J, Elewaut D, Proctor GB, et al. Dysregulation of NF-kB in glandular epithelial cells results in sjogren's-like features. PloS One (2018) 13(8):e0200212. doi: 10.1371/journal.pone.0200212
7. Han YM, Koh J, Kim JW, Lee C, Koh SJ, Kim B, et al. NF-kappa b activation correlates with disease phenotype in crohn's disease. PloS One (2017) 12(7):e0182071. doi: 10.1371/journal.pone.0182071
8. Andresen L, Jorgensen VL, Perner A, Hansen A, Eugen-Olsen J, Rask-Madsen J. Activation of nuclear factor kappaB in colonic mucosa from patients with collagenous and ulcerative colitis. Gut (2005) 54(4):503–9. doi: 10.1136/gut.2003.034165
9. Makarov SS. NF-kappa b in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res (2001) 3(4):200–6. doi: 10.1186/ar300
10. Taylor PC. Pharmacology of TNF blockade in rheumatoid arthritis and other chronic inflammatory diseases. Curr Opin Pharmacol (2010) 10(3):308–15. doi: 10.1016/j.coph.2010.01.005
11. Ngo B, Farrell CP, Barr M, Wolov K, Bailey R, Mullin JM, et al. Tumor necrosis factor blockade for treatment of inflammatory bowel disease: efficacy and safety. Curr Mol Pharmacol (2010) 3(3):145–52. doi: 10.2174/1874467211003030145
12. Dechant C, Schauenberg P, Antoni CE, Kraetsch HG, Kalden JR, Manger B. [Longterm outcome of TNF blockade in adult-onset still's disease]. Dtsch Med Wochenschr (2004) 129(23):1308–12. doi: 10.1055/s-2004-826865
13. Tobin AM, Kirby B. TNF alpha inhibitors in the treatment of psoriasis and psoriatic arthritis. BioDrugs (2005) 19(1):47–57. doi: 10.2165/00063030-200519010-00006
14. Kemanetzoglou E, Andreadou E. CNS demyelination with TNF-alpha blockers. Curr Neurol Neurosci Rep (2017) 17(4):36. doi: 10.1007/s11910-017-0742-1
15. Brown G, Wang E, Leon A, Huynh M, Wehner M, Matro R, et al. Tumor necrosis factor-alpha inhibitor-induced psoriasis: Systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol (2017) 76(2):334–41. doi: 10.1016/j.jaad.2016.08.012
16. Wetter DA, Davis MD. Lupus-like syndrome attributable to anti-tumor necrosis factor alpha therapy in 14 patients during an 8-year period at Mayo clinic. Mayo Clin Proc (2009) 84(11):979–84. doi: 10.1016/S0025-6196(11)60668-X
17. Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine (2018) 101:14–8. doi: 10.1016/j.cyto.2016.08.003
18. Salomon BL. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat Rev Rheumatol (2021) 17(8):487–504. doi: 10.1038/s41584-021-00639-6
19. Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun (2010) 11:27–60. doi: 10.1159/000289196
20. Tsai EY, Yie J, Thanos D, Goldfeld AE. Cell-type-specific regulation of the human tumor necrosis factor alpha gene in b cells and T cells by NFATp and ATF-2/JUN. Mol Cell Biol (1996) 16(10):5232–44. doi: 10.1128/MCB.16.10.5232
21. Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature (1984) 312(5996):724–9. doi: 10.1038/312724a0
22. Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell (1988) 53(1):45–53. doi: 10.1016/0092-8674(88)90486-2
23. Luettig B, Decker T, Lohmann-Matthes ML. Evidence for the existence of two forms of membrane tumor necrosis factor: an integral protein and a molecule attached to its receptor. J Immunol (1989) 143(12):4034–8. Available at: https://www.jimmunol.org/content/jimmunol/143/12/4034.full.pdf.
24. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatol (Oxford) (2010) 49(7):1215–28. doi: 10.1093/rheumatology/keq031
25. Qu Y, Zhao G, Li H. Forward and reverse signaling mediated by transmembrane tumor necrosis factor-alpha and TNF receptor 2: Potential roles in an immunosuppressive tumor microenvironment. Front Immunol (2017) 8:1675. doi: 10.3389/fimmu.2017.01675
26. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature (1997) 385(6618):729–33. doi: 10.1038/385729a0
27. Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature (1997) 385(6618):733–6. doi: 10.1038/385733a0
28. Friedmann E, Hauben E, Maylandt K, Schleeger S, Vreugde S, Lichtenthaler SF, et al. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFalpha in activated dendritic cells to trigger IL-12 production. Nat Cell Biol (2006) 8(8):843–8. doi: 10.1038/ncb1440
29. Fluhrer R, Grammer G, Israel L, Condron MM, Haffner C, Friedmann E, et al. A gamma-secretase-like intramembrane cleavage of TNFalpha by the GxGD aspartyl protease SPPL2b. Nat Cell Biol (2006) 8(8):894–6. doi: 10.1038/ncb1450
30. Domonkos A, Udvardy A, Laszlo L, Nagy T, Duda E. Receptor-like properties of the 26 kDa transmembrane form of TNF. Eur Cytokine Netw (2001) 12(3):411–9. Available at: https://www.jle.com/fr/revues/ecn/e-docs/receptor_like_properties_of_the_26_kda_transmembrane_form_of_tnf_90341/article.phtml.
31. Alexopoulou L, Kranidioti K, Xanthoulea S, Denis M, Kotanidou A, Douni E, et al. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur J Immunol (2006) 36(10):2768–80. doi: 10.1002/eji.200635921
32. Wong E, Cohen T, Romi E, Levin M, Peleg Y, Arad U, et al. Harnessing the natural inhibitory domain to control TNFalpha converting enzyme (TACE) activity in vivo. Sci Rep (2016) 6:35598. doi: 10.1038/srep35598
33. Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev (2014) 25(4):453–72. doi: 10.1016/j.cytogfr.2014.07.016
34. MacEwan DJ. TNF ligands and receptors–a matter of life and death. Br J Pharmacol (2002) 135(4):855–75. doi: 10.1038/sj.bjp.0704549
35. So T, Ishii N. The TNF-TNFR family of Co-signal molecules. Adv Exp Med Biol (2019) 1189:53–84. doi: 10.1007/978-981-32-9717-3_3
36. Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science (2000) 288(5475):2351–4. doi: 10.1126/science.288.5475.2351
37. Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U.S.A. (2010) 107(21):9801–6. doi: 10.1073/pnas.0914118107
38. Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, Oh HH, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet (2001) 69(2):301–14. doi: 10.1086/321976
39. Lobito AA, Kimberley FC, Muppidi JR, Komarow H, Jackson AJ, Hull KM, et al. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in TNFR1-associated periodic fever syndrome (TRAPS). Blood (2006) 108(4):1320–7. doi: 10.1182/blood-2005-11-006783
40. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell (1999) 97(1):133–44. doi: 10.1016/S0092-8674(00)80721-7
41. Todd I, Radford PM, Draper-Morgan KA, McIntosh R, Bainbridge S, Dickinson P, et al. Mutant forms of tumour necrosis factor receptor I that occur in TNF-receptor-associated periodic syndrome retain signalling functions but show abnormal behaviour. Immunology (2004) 113(1):65–79. doi: 10.1111/j.1365-2567.2004.01942.x
42. Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, et al. Solution of the structure of the TNF-TNFR2 complex. Sci Signal (2010) 3(148):ra83. doi: 10.1126/scisignal.2000954
43. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell (2009) 36(5):831–44. doi: 10.1016/j.molcel.2009.10.013
44. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell (1995) 83(5):793–802. doi: 10.1016/0092-8674(95)90192-2
45. Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U.S.A. (1998) 95(2):570–5. doi: 10.1073/pnas.95.2.570
46. Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa b activation. Cell (1995) 81(4):495–504. doi: 10.1016/0092-8674(95)90070-5
47. Atretkhany KN, Gogoleva VS, Drutskaya MS, Nedospasov SA. Distinct modes of TNF signaling through its two receptors in health and disease. J Leukoc Biol (2020) 107(6):893–905. doi: 10.1002/JLB.2MR0120-510R
48. Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol (2014) 26(3):237–45. doi: 10.1016/j.smim.2014.02.009
49. Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell (1996) 87(3):565–76. doi: 10.1016/S0092-8674(00)81375-6
50. Zeke A, Misheva M, Remenyi A, Bogoyevitch MA. JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol Mol Biol Rev (2016) 80(3):793–835. doi: 10.1128/MMBR.00043-14
51. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15(6):362–74. doi: 10.1038/nri3834
52. Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ (2015) 22(4):526–39. doi: 10.1038/cdd.2014.216
53. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
54. Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, et al. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J Biol Chem (2010) 285(10):7394–404. doi: 10.1074/jbc.M109.037341
55. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17(9):545–58. doi: 10.1038/nri.2017.52
56. Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, et al. TNFR2 ligation in human T regulatory cells enhances IL2-induced cell proliferation through the non-canonical NF-kappaB pathway. Sci Rep (2018) 8(1):12079. doi: 10.1038/s41598-018-30621-4
57. He T, Liu S, Chen S, Ye J, Wu X, Bian Z, et al. The p38 MAPK inhibitor SB203580 abrogates tumor necrosis factor-induced proliferative expansion of mouse CD4(+)Foxp3(+) regulatory T cells. Front Immunol (2018) 9:1556. doi: 10.3389/fimmu.2018.01556
58. Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun n-terminal kinase is mediated by TRAF2. EMBO J (1997) 16(5):1080–92. doi: 10.1093/emboj/16.5.1080
59. Ji W, Li Y, Wan T, Wang J, Zhang H, Chen H, et al. Both internalization and AIP1 association are required for tumor necrosis factor receptor 2-mediated JNK signaling. Arterioscler Thromb Vasc Biol (2012) 32(9):2271–9. doi: 10.1161/ATVBAHA.112.253666
60. Naude PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. FEBS J (2011) 278(6):888–98. doi: 10.1111/j.1742-4658.2011.08017.x
61. Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem (1993) 268(25):18542–8. doi: 10.1016/S0021-9258(17)46661-0
62. Wicovsky A, Henkler F, Salzmann S, Scheurich P, Kneitz C, Wajant H. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1-TNFR2 cooperation. Oncogene (2009) 28(15):1769–81. doi: 10.1038/onc.2009.29
63. Zhao Y, Conze DB, Hanover JA, Ashwell JD. Tumor necrosis factor receptor 2 signaling induces selective c-IAP1-dependent ASK1 ubiquitination and terminates mitogen-activated protein kinase signaling. J Biol Chem (2007) 282(11):7777–82. doi: 10.1074/jbc.M609146200
64. Baseta JG, Stutman O. TNF regulates thymocyte production by apoptosis and proliferation of the triple negative (CD3-CD4-CD8-) subset. J Immunol (2000) 165(10):5621–30. doi: 10.4049/jimmunol.165.10.5621
65. Guevara Patino JA, Ivanov VN, Lacy E, Elkon KB, Marino MW, Nikolic-Zugic J. TNF-alpha is the critical mediator of the cyclic AMP-induced apoptosis of CD8+4+ double-positive thymocytes. J Immunol (2000) 164(4):1689–94. doi: 10.4049/jimmunol.164.4.1689
66. Webb LV, Ley SC, Seddon B. TNF activation of NF-kappaB is essential for development of single-positive thymocytes. J Exp Med (2016) 213(8):1399–407. doi: 10.1084/jem.20151604
67. Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol (2014) 15(5):473–81. doi: 10.1038/ni.2849
68. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discovery (2019) 18(10):749–69. doi: 10.1038/s41573-019-0041-4
69. Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol (2008) 9(3):239–44. doi: 10.1038/ni1572
70. Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol (2012) 30:95–114. doi: 10.1146/annurev-immunol-020711-075035
71. Kruglov A, Drutskaya M, Schlienz D, Gorshkova E, Kurz K, Morawietz L, et al. Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann Rheum Dis (2020) 79(11):1453–9. doi: 10.1136/annrheumdis-2019-216068
72. Kruglov AA, Lampropoulou V, Fillatreau S, Nedospasov SA. Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J Immunol (2011) 187(11):5660–70. doi: 10.4049/jimmunol.1100663
73. Corazza N, Eichenberger S, Eugster HP, Mueller C. Nonlymphocyte-derived tumor necrosis factor is required for induction of colitis in recombination activating gene (RAG)2(-/-) mice upon transfer of CD4(+)CD45RB(hi) T cells. J Exp Med (1999) 190(10):1479–92. doi: 10.1084/jem.190.10.1479
74. Chen X, Nie Y, Xiao H, Bian Z, Scarzello AJ, Song NY, et al. TNFR2 expression by CD4 effector T cells is required to induce full-fledged experimental colitis. Sci Rep (2016) 6:32834. doi: 10.1038/srep32834
75. Green EA, Eynon EE, Flavell RA. Local expression of TNFalpha in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity (1998) 9(5):733–43. doi: 10.1016/S1074-7613(00)80670-6
76. Christen U, Wolfe T, Mohrle U, Hughes AC, Rodrigo E, Green EA, et al. A dual role for TNF-alpha in type 1 diabetes: islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J Immunol (2001) 166(12):7023–32. doi: 10.4049/jimmunol.166.12.7023
77. Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4(+)CD25(+) treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity (2002) 16(2):183–91. doi: 10.1016/S1074-7613(02)00279-0
78. Lee LF, Lih CJ, Huang CJ, Cao T, Cohen SN, McDevitt HO. Genomic expression profiling of TNF-alpha-treated BDC2.5 diabetogenic CD4+ T cells. Proc Natl Acad Sci U.S.A. (2008) 105(29):10107–12. doi: 10.1073/pnas.0803336105
79. Scheurich P, Thoma B, Ucer U, Pfizenmaier K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)-alpha: induction of TNF receptors on human T cells and TNF-alpha-mediated enhancement of T cell responses. J Immunol (1987) 138(6):1786–90. Available at: https://www.jimmunol.org/content/138/6/1786.long.
80. Yokota S, Geppert TD, Lipsky PE. Enhancement of antigen- and mitogen-induced human T lymphocyte proliferation by tumor necrosis factor-alpha. J Immunol (1988) 140(2):531–6. Available at: https://www.jimmunol.org/content/140/2/531.long.
81. Banerjee D, Liou HC, Sen R. C-rel-dependent priming of naive T cells by inflammatory cytokines. Immunity (2005) 23(4):445–58. doi: 10.1016/j.immuni.2005.09.012
82. Kim EY, Teh HS. TNF type 2 receptor (p75) lowers the threshold of T cell activation. J Immunol (2001) 167(12):6812–20. doi: 10.4049/jimmunol.167.12.6812
83. Kim EY, Priatel JJ, Teh SJ, Teh HS. TNF receptor type 2 (p75) functions as a costimulator for antigen-driven T cell responses in vivo. J Immunol (2006) 176(2):1026–35. doi: 10.4049/jimmunol.176.2.1026
84. ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in crohn's disease. Gut (2002) 50(2):206–11. doi: 10.1136/gut.50.2.206
85. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol (2007) 179(1):154–61. doi: 10.4049/jimmunol.179.1.154
86. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol (2013) 13(6):461–7. doi: 10.1038/nri3464
87. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19(3):322–8. doi: 10.1038/nm.3085
88. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108(1):253–61. doi: 10.1182/blood-2005-11-4567
89. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med (2016) 213(7):1241–53. doi: 10.1084/jem.20151255
90. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory t. cell phenotype in the inflammatory environment. J Immunol (2013) 190(3):1076–84. doi: 10.4049/jimmunol.1202659
91. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol (2008) 180(10):6467–71. doi: 10.4049/jimmunol.180.10.6467
92. Ronin E, Pouchy C, Khosravi M, Hilaire M, Gregoire S, Casrouge A, et al. Tissue-restricted control of established central nervous system autoimmunity by TNF receptor 2-expressing treg cells. Proc Natl Acad Sci U.S.A. (2021) 118(13). doi: 10.1073/pnas.2014043118
93. Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, et al. The TNF receptor superfamily-NF-kappaB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep (2017) 20(12):2906–20. doi: 10.1016/j.celrep.2017.08.068
94. Nagar M, Jacob-Hirsch J, Vernitsky H, Berkun Y, Ben-Horin S, Amariglio N, et al. TNF activates a NF-kappaB-regulated cellular program in human CD45RA- regulatory T cells that modulates their suppressive function. J Immunol (2010) 184(7):3570–81. doi: 10.4049/jimmunol.0902070
95. Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, et al. Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting tregs. J Clin Invest (2010) 120(12):4558–68. doi: 10.1172/JCI42945
96. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood (2016) 128(12):1651–9. doi: 10.1182/blood-2016-02-700849
97. Myers L, Joedicke JJ, Carmody AB, Messer RJ, Kassiotis G, Dudley JP, et al. IL-2-independent and TNF-alpha-dependent expansion of Vbeta5+ natural regulatory T cells during retrovirus infection. J Immunol (2013) 190(11):5485–95. doi: 10.4049/jimmunol.1202951
98. Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, et al. IL-6 and TNFalpha drive extensive proliferation of human tregs without compromising their lineage stability or function. Front Immunol (2021) 12:783282. doi: 10.3389/fimmu.2021.783282
99. Urbano PCM, Koenen H, Joosten I, He X. An autocrine TNFalpha-tumor necrosis factor receptor 2 loop promotes epigenetic effects inducing human treg stability in vitro. Front Immunol (2018) 9:573. doi: 10.3389/fimmu.2018.00573
100. He X, Landman S, Bauland SC, van den Dolder J, Koenen HJ, Joosten I. A TNFR2-agonist facilitates high purity expansion of human low purity treg cells. PloS One (2016) 11(5):e0156311. doi: 10.1371/journal.pone.0156311
101. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J Exp Med (2016) 213(9):1881–900. doi: 10.1084/jem.20151563
102. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol (2011) 186(12):6779–87. doi: 10.4049/jimmunol.1003868
103. Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-alpha impairs differentiation and function of TGF-beta-induced treg cells in autoimmune diseases through akt and Smad3 signaling pathway. J Mol Cell Biol (2013) 5(2):85–98. doi: 10.1093/jmcb/mjs063
104. Yang S, Xie C, Chen Y, Wang J, Chen X, Lu Z, et al. Differential roles of TNFalpha-TNFR1 and TNFalpha-TNFR2 in the differentiation and function of CD4(+)Foxp3(+) induced treg cells in vitro and in vivo periphery in autoimmune diseases. Cell Death Dis (2019) 10(1):27. doi: 10.1038/s41419-019-1883-8
105. Fischer R, Marsal J, Gutta C, Eisler SA, Peters N, Bethea JR, et al. Novel strategies to mimic transmembrane tumor necrosis factor-dependent activation of tumor necrosis factor receptor 2. Sci Rep (2017) 7(1):6607. doi: 10.1038/s41598-017-06993-4
106. Richter F, Zettlitz KA, Seifert O, Herrmann A, Scheurich P, Pfizenmaier K, et al. Monovalent TNF receptor 1-selective antibody with improved affinity and neutralizing activity. MAbs (2019) 11(1):166–77. doi: 10.1080/19420862.2018.1524664
107. Steeland S, Van Ryckeghem S, Van Imschoot G, De Rycke R, Toussaint W, Vanhoutte L, et al. TNFR1 inhibition with a nanobody protects against EAE development in mice. Sci Rep (2017) 7(1):13646. doi: 10.1038/s41598-017-13984-y
108. Lo CH, Vunnam N, Lewis AK, Chiu TL, Brummel BE, Schaaf TM, et al. An innovative high-throughput screening approach for discovery of small molecules that inhibit TNF receptors. SLAS Discovery (2017) 22(8):950–61. doi: 10.1177/2472555217706478
109. Nomura T, Abe Y, Kamada H, Shibata H, Kayamuro H, Inoue M, et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J Control Release (2011) 149(1):8–14. doi: 10.1016/j.jconrel.2009.12.015
110. Loetscher H, Stueber D, Banner D, Mackay F, Lesslauer W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J Biol Chem (1993) 268(35):26350–7. doi: 10.1016/S0021-9258(19)74322-1
111. Ando D, Inoue M, Kamada H, Taki S, Furuya T, Abe Y, et al. Creation of mouse TNFR2-selective agonistic TNF mutants using a phage display technique. Biochem Biophys Rep (2016) 7:309–15. doi: 10.1016/j.bbrep.2016.06.008
112. Fischer R, Maier O, Siegemund M, Wajant H, Scheurich P, Pfizenmaier K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PloS One (2011) 6(11):e27621. doi: 10.1371/journal.pone.0027621
113. Ban L, Kuhtreiber W, Butterworth J, Okubo Y, Vanamee ES, Faustman DL. Strategic internal covalent cross-linking of TNF produces a stable TNF trimer with improved TNFR2 signaling. Mol Cell Ther (2015) 3:7. doi: 10.1186/s40591-015-0044-4
114. Rodriguez-Barbosa JI, Schneider P, Graca L, Buhler L, Perez-Simon JA, Del Rio ML. The role of TNFR2 and DR3 in the in vivo expansion of tregs in T cell depleting transplantation regimens. Int J Mol Sci (2020) 21(9). doi: 10.3390/ijms21093347
115. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
116. Muller YD, Ferreira LMR, Ronin E, Ho P, Nguyen V, Faleo G, et al. Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front Immunol (2021) 12:686439. doi: 10.3389/fimmu.2021.686439
Keywords: tumor necrosis factor alpha, tumor necrosis factor receptor, T effector cells, T regulatory cells, pleiotropism, activation, costimulation, autoimmunity
Citation: Skartsis N, Ferreira LMR and Tang Q (2022) The dichotomous outcomes of TNFα signaling in CD4+ T cells. Front. Immunol. 13:1042622. doi: 10.3389/fimmu.2022.1042622
Received: 12 September 2022; Accepted: 03 November 2022;
Published: 16 November 2022.
Edited by:
Murugaiyan Gopal, Harvard Medical School, United StatesReviewed by:
Maria Bettini, The University of Utah, United StatesJamil R. Azzi, Harvard Medical School, United States
Benoit L. Salomon, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Skartsis, Ferreira and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolaos Skartsis, c2thcnRzaXMubmlrb2xhb3NAbWF5by5lZHU=