 Brahim Belaid1
Brahim Belaid1 Lydia Lamara Mahammed1
Lydia Lamara Mahammed1 Ouardia Drali2
Ouardia Drali2 Aida Mohand Oussaid3
Aida Mohand Oussaid3 Nabila Souad Touri4Souhila Melzi5Abdelhak Dehimi6Lylia Meriem Berkani1Fatma Merah7
Nabila Souad Touri4Souhila Melzi5Abdelhak Dehimi6Lylia Meriem Berkani1Fatma Merah7 Zineb Larab7Ines Allam1Ouarda Khemici8Sonya Yasmine Kirane9
Zineb Larab7Ines Allam1Ouarda Khemici8Sonya Yasmine Kirane9 Mounia Boutaba10Reda Belbouab11Hadjira Bekkakcha10Assia Guedouar10Abdelhakim Chelali12
Mounia Boutaba10Reda Belbouab11Hadjira Bekkakcha10Assia Guedouar10Abdelhakim Chelali12 Brahim Baamara12Djamila Noui13Hadda Baaziz13Radia Rezak14Sidi Mohamed Azzouz15Malika Aichaoui16Assia Moktefi16Redha Mohamed Benhatchi16Meriem Oussalah17Naila Benaissa18
Brahim Baamara12Djamila Noui13Hadda Baaziz13Radia Rezak14Sidi Mohamed Azzouz15Malika Aichaoui16Assia Moktefi16Redha Mohamed Benhatchi16Meriem Oussalah17Naila Benaissa18 Amel Laredj18
Amel Laredj18 Assia Bouchetara18Abdelkader Adria19Brahim Habireche20Noureddine Tounsi20Fella Dahmoun21
Assia Bouchetara18Abdelkader Adria19Brahim Habireche20Noureddine Tounsi20Fella Dahmoun21 Rabah Touati21Hamza Boucenna3Fadila Bouferoua3Lynda Sekfali3Nadjet Bouhafs5Rawda Aboura5Sakina Kherra10Yacine Inouri22Saadeddine Dib23Nawel Medouri24
Rabah Touati21Hamza Boucenna3Fadila Bouferoua3Lynda Sekfali3Nadjet Bouhafs5Rawda Aboura5Sakina Kherra10Yacine Inouri22Saadeddine Dib23Nawel Medouri24 Noureddine Khelfaoui24Aicha Redjedal24Amara Zelaci25Samah Yahiaoui26Sihem Medjadj27Tahar Khelifi Touhami28Ahmed Kadi29
Noureddine Khelfaoui24Aicha Redjedal24Amara Zelaci25Samah Yahiaoui26Sihem Medjadj27Tahar Khelifi Touhami28Ahmed Kadi29 Fouzia Amireche30Imane Frada31Shahrazed Houasnia32Karima Benarab33
Fouzia Amireche30Imane Frada31Shahrazed Houasnia32Karima Benarab33 Chahynez Boubidi10Yacine Ferhani11Hayet Benalioua11Samia Sokhal11Nadia Benamar34
Chahynez Boubidi10Yacine Ferhani11Hayet Benalioua11Samia Sokhal11Nadia Benamar34 Samira Aggoune35Karima Hadji36Asma Bellouti37Hakim Rahmoune6Nada Boutrid6kamelia Okka6Assia Ammour38Houssem Saadoune39Malika Amroun22Hayet Belhadj22Amina Ghanem40Hanane Abbaz40Sana Boudrioua41Besma Zebiche42Assia Ayad42Zahra Hamadache42Nassima Ouaras43Nassima Achour43Nadira Bouchair44Houda Boudiaf45Dahila Bekkat-Berkani46Hachemi Maouche35Zahir Bouzrar5Lynda Aissat47Ouardia Ibsaine48Belkacem Bioud6Leila Kedji4Djazia Dahlouk22Manoubia Bensmina49
Samira Aggoune35Karima Hadji36Asma Bellouti37Hakim Rahmoune6Nada Boutrid6kamelia Okka6Assia Ammour38Houssem Saadoune39Malika Amroun22Hayet Belhadj22Amina Ghanem40Hanane Abbaz40Sana Boudrioua41Besma Zebiche42Assia Ayad42Zahra Hamadache42Nassima Ouaras43Nassima Achour43Nadira Bouchair44Houda Boudiaf45Dahila Bekkat-Berkani46Hachemi Maouche35Zahir Bouzrar5Lynda Aissat47Ouardia Ibsaine48Belkacem Bioud6Leila Kedji4Djazia Dahlouk22Manoubia Bensmina49 Abdelkarim Radoui17Mimouna Bessahraoui15Nadia Bensaadi33Azzeddine Mekki2Zoulikha Zeroual10
Abdelkarim Radoui17Mimouna Bessahraoui15Nadia Bensaadi33Azzeddine Mekki2Zoulikha Zeroual10 Koon-Wing Chan50
Koon-Wing Chan50 Daniel Leung50Amar Tebaibia51Soraya Ayoub52Dalila Mekideche53Merzak Gharnaout29
Daniel Leung50Amar Tebaibia51Soraya Ayoub52Dalila Mekideche53Merzak Gharnaout29 Jean Laurent Casanova54,55,56,57
Jean Laurent Casanova54,55,56,57 Anne Puel54,55,56
Anne Puel54,55,56 Yu Lung Lau50Nacira Cherif9Samir Ladj58Leila Smati46
Yu Lung Lau50Nacira Cherif9Samir Ladj58Leila Smati46 Rachida Boukari11Nafissa Benhalla3
Rachida Boukari11Nafissa Benhalla3 Reda Djidjik1*
Reda Djidjik1*- 1Department of Medical Immunology, Beni Messous University Hospital Center, University of Algiers 1, Algiers, Algeria
- 2Department of Pediatrics B, Hussein Dey University Hospital Center, University of Algiers 1, Algiers, Algeria
- 3Department of Pediatrics A, Beni Messous University Hospital Center, University of Algiers 1, Algiers, Algeria
- 4Department of Pediatrics, Blida University Hospital Center, University of Blida, Blida, Algeria
- 5Department of Pediatrics, Bab El Oued University Hospital Center, University of Algiers 1, Algiers, Algeria
- 6Department of Pediatrics, Setif University Hospital Center, University of Setif 1, Setif, Algeria
- 7Department of Medical Immunology, Beni Messous University Hospital Center, Algiers, Algeria
- 8Department of Pediatrics B, Beni Messous University Hospital Center, Algiers, Algeria
- 9Department of Pediatrics B, Beni Messous University Hospital Center, University of Algiers 1, Algiers, Algeria
- 10Department of Pediatrics A, Hussein Dey University Hospital Center, University of Algiers 1, Algiers, Algeria
- 11Department of Pediatrics, Mustapha Pacha University Hospital Center, University of Algiers 1, Algiers, Algeria
- 12Department of Pediatrics, Djelfa Public Hospital Institution, Djelfa, Algeria
- 13Department of Pediatrics, Batna University Hospital center, University of Batna, Batna, Algeria
- 14Department of Pediatric Gastroenterology and Nutrition, Canastel Children’s Hospital, Oran, Algeria
- 15Department of Pediatric Gastroenterology and Nutrition, Canastel Children’s Hospital, University of Oran, Oran, Algeria
- 16Department of Pediatric Pneumo-Allergology, Canastel Children’s Hospital, Oran, Algeria
- 17Department of Pediatric Pneumo-Allergology, Canastel Children’s Hospital, University of Oran, Oran, Algeria
- 18Department of Children’s Infectious Diseases, Canastel Children’s Hospital, University of Oran, Oran, Algeria
- 19Department of Pediatric Hematology, Canastel Children’s Hospital, Oran, Algeria
- 20Department of Pediatrics, El Bayadh Public Hospital Institution, EL Bayadh, Algeria
- 21Department of Pediatrics, Bejaia University Hospital Center, University of Bejaia, Bejaia, Algeria
- 22Department of Pediatrics, Central Hospital of the Army, University of Algiers 1, Algiers, Algeria
- 23Department of Pediatrics, Mother & Child Hospital of Tlemcen, University of Tlemcen, Tlemcen, Algeria
- 24Department of Pediatrics, Saida Public Hospital Institution, Saida, Algeria
- 25Department of Pediatrics, El Oued Public Hospital Institution, El Oued, Algeria
- 26Department of Pediatrics, Barika Public Hospital Institution, Batna, Algeria
- 27Department of Pediatrics, Ghardaia Public Hospital Institution, Ghardaia, Algeria
- 28Private Practitioner, Constantine, Algeria
- 29Department of Pneumology A, Beni Messous University Hospital Center, University of Algiers 1, Algiers, Algeria
- 30Department of Pediatrics, Mother & Child Hospital of EL Mansourah, University of Constantine 3, Constantine, Algeria
- 31Department of Pediatrics, Biskra Public Hospital Institution, Biskra, Algeria
- 32Department of Pediatrics, El Harrouche Public Hospital Institution, Skikda, Algeria
- 33Department of Pediatrics, Tizi Ouzou University Hospital Center, University of Tizi Ouzou, Tizi Ouzou, Algeria
- 34Department of Pediatrics, Tighennif Public Hospital Institution, Mascara, Algeria
- 35Department of Pediatrics, El-Harrach Public Hospital Institution, University of Algiers 1, Algiers, Algeria
- 36Department of Pediatrics, Ain Oulmene Public Hospital Institution, Setif, Algeria
- 37Department of Pediatrics, Ain Azel Public Hospital Institution, Setif, Algeria
- 38Department of Pediatrics, Mother & Child Hospital of Touggourt, Touggourt, Algeria
- 39Department of Pneumology, Mila Public Hospital Institution, Mila, Algeria
- 40Department of Pediatrics, Khenchela Public Hospital Institution, Khenchela, Algeria
- 41Department of Pediatrics, El Khroub Public Hospital Institution, Constantine, Algeria
- 42Department of Pediatrics, Kolea Public Hospital Institution, Tipaza, Algeria
- 43Department of Infectious Diseases, EL Kettar Specialized Hospital, University of Algiers 1, Algiers, Algeria
- 44Department of Pediatrics, Annaba University Hospital Center, University of Annaba, Annaba, Algeria
- 45Department of Pediatric Oncology, Mustapha pacha University Hospital Center, University of Algiers 1, Algiers, Algeria
- 46Department of Pediatrics, Bologhine Public Hospital Institution, University of Algiers 1, Algiers, Algeria
- 47Department of Pediatrics, Mother & Child Hospital of Tipaza, University of Blida, Algiers, Algeria
- 48Department of Pediatrics, Ain Taya Public Hospital Institution, University of Algiers 1, Algiers, Algeria
- 49Department of Pediatrics B, Douera University Hospital Center, University of Blida, Algiers, Algeria
- 50Department of Pediatrics and Adolescent Medicine, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 51Department of Internal Medicine, El Biar Public Hospital Institution, University of Algiers 1, Algiers, Algeria
- 52Department of Internal Medicine, Beni Messous University Hospital Center, University of Algiers 1, Algiers, Algeria
- 53Department of Pneumology B, Beni Messous University Hospital Center, University of Algiers 1, Algiers, Algeria
- 54Laboratory of Human Genetics of Infectious Diseases, Necker Hospital for Sick Children, INSERM UMR 1163, Paris, France
- 55Imagine Institute, University of Paris, Paris, France
- 56St Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller University, New York, NY, United States
- 57Howard Hughes Medical Institute, New York, NY, United States
- 58Department of Pediatrics, El Biar Public Hospital Institution, University of Algiers 1, Algiers, Algeria
Background: Inborn errors of immunity (IEI) predispose patients to various infectious and non-infectious complications. Thanks to the development and expanding use of flow cytometry and increased awareness, the diagnostic rate of IEI has markedly increased in Algeria the last decade.
Aim: This study aimed to describe a large cohort of Algerian patients with probable IEI and to determine their clinical characteristics and outcomes.
Methods: We collected and analyzed retrospectively the demographic data, clinical manifestations, immunologic, genetic data, and outcome of Algerian IEI patients - diagnosed in the department of medical immunology of Beni Messous university hospital center, Algiers, from 2008 to 2021.
Results: Eight hundred and seven patients with IEI (482 males and 325 females) were enrolled, 9.7% of whom were adults. Consanguinity was reported in 50.3% of the cases and a positive family history in 32.34%. The medium age at disease onset was 8 months and at diagnosis was 36 months. The median delay in diagnosis was 16 months. Combined immunodeficiencies were the most frequent (33.8%), followed by antibody deficiencies (24.5%) and well-defined syndromes with immunodeficiency (24%). Among 287 patients tested for genetic disorders, 129 patients carried pathogenic mutations; 102 having biallelic variants mostly in a homozygous state (autosomal recessive disorders). The highest mortality rate was observed in patients with combined immunodeficiency (70.1%), especially in patients with severe combined immunodeficiency (SCID), Omenn syndrome, or Major Histocompatibility Complex (MHC) class II deficiency.
Conclusion: The spectrum of IEI in Algeria is similar to that seen in most countries of the Middle East and North Africa (MENA) region, notably regarding the frequency of autosomal recessive and/or combined immunodeficiencies.
Introduction
Inborn errors of immunity (IEIs), formerly known as primary immunodeficiency disorders (PIDs), form a heterogeneous group of inherited disorders that impair the development and/or function of leukocytes or other cell types involved in immunity, and often predispose patients to recurrent, persistent, or severe infections. Some patients may also display autoimmunity, autoinflammation, allergy, or malignancy (1, 2). During the last decade, advances in understanding human genetics and immunity have led to a better recognition of several immune disorders and their underlying genetic defects.
According to the 2021 IUIS classification (3), over 450 IEI have been identified as underlying diverse clinical phenotypes. These IEI are divided into 10 main categories: (I) immunodeficiencies affecting cellular and humoral immunity, (II) combined immunodeficiencies with associated or syndromic features, (III) predominantly antibody deficiencies, (IV) diseases of immune dysregulation, (V) congenital defects of phagocyte number or function, (VI) defects in intrinsic and innate immunity, (VII) autoinflammatory diseases, (VIII) complement deficiencies, (IX) bone morrow failure, (X) phenocopies of IEI.
Inheritance of IEI can be X-linked, autosomal recessive (AR), or autosomal dominant (AD); the first symptoms can manifest at birth, early childhood, or later in life. Acquired forms of IEI resulting from somatic mutations or anti-cytokines autoantibodies are increasingly identified (4).
The prevalence and incidence of IEI vary depending on the type of disorder, age, sex, ethnicity, and geographic location. At least 1 in 10,000 people are affected by IEI worldwide (1, 2, 5, 6), This number is probably underestimated due in part to the high and precocious mortality of patients before diagnosis, and to the lack of awareness and of dedicated diagnostic tools, leading to a low rate of diagnoses, particularly in developing countries. Moreover, many conditions may result from hitherto unknown IEI, as attested by the increasing speed at which new IEI are being discovered, including IEI underlying common infectious diseases such as tuberculosis or COVID-19 (4, 7–9). However, recent advances in molecular genetic technologies, particularly next-generation sequencing, have greatly improved the identification of a growing number of specific genetic defects of the immune system as well as the number of patients (4). Indeed, recent studies showed a higher prevalence worldwide, suggesting to re-assess the previous estimates of the IEI frequency in the general population (10, 11).
In order to better estimate the prevalence of these IEI in the Middle East and North Africa (MENA) region, a global survey was recently carried out and established the MENA IEI registry (12), similarly to European (13), and North American (14) and South American (15), and Asian (16, 17) series or registries. This survey reported a high frequency of IEI predominantly due to antibody deficiencies in MENA populations (12). In sub-Saharan Africa, data are extremely rare and even unavailable for most countries (18).
Interestingly, populations in the MENA region share a particularly high rate of consanguinity which might exceed 50% in some regions (19, 20). According to the MENA report, combined immunodeficiencies were the most common IEI in North Africa (Tunisia, Algeria) and in some Middle East countries (Saudi Arabia and Kuwait), while primary antibody deficiencies were the dominant disorders in most other MENA countries (12).
Algeria has an estimated population of more than 45 million of inhabitants in 2021, with a high birth rate estimated at 21.5 births per 1000 people (21). Like all MENA countries, Algeria depicts a high level of consanguinity and subsequently of autosomal recessive disorders, similar to those reported in highly consanguineous populations (20, 22).
In the current study, we retrospectively analyzed a cohort of IEI patients to enhance our understanding of IEI and to highlight the fundamental role of immunological and molecular testing to effectively identify major forms of IEI. We also aimed to describe the clinical characteristics and outcomes of patients with IEI in Algeria.
Methods
A single-center retrospective study was carried out at the department of medical immunology of Beni Messous University Hospital Center of Algiers, Algeria, from January 2008 to September 2021. As next generation sequencing is not available routinely in Algeria, most of our patients were diagnosed according to the diagnostic criteria set by the European Society of Immunodeficiency Disorders (ESID) (23) and classified according to the IUIS criteria (1). Immunodeficiencies secondary to other conditions (e.g., human immunodeficiency virus (HIV) infection) were excluded.
Patient Source and Enrollment
The Algerian healthcare system is a wide network of hospitals, clinics, and dispensaries. It is mainly public, accessible and free of charge to all Algerian citizens; with a recent sharp increase in the private sector.
According to their age, patients were initially assessed by pediatricians or other adult specialists, from either outpatient clinics or inpatient wards.
Any patient with clinical signs suggestive of IEI was addressed to the department of medical immunology at Beni Messous University Hospital Center, with a request form and clinical information sheet that may suggest an IEI. Request forms were examined and entered into a dedicated database.
Data Collection and Evaluation
The request forms contained the following information:
i. referral centre’s contact details
ii. patient’s demographics such as full name, date of birth, gender, place of birth, age at onset of suggestive symptoms, parental consanguinity, familial history of IEI, history of suspicious sibling death, patient’s history of severe, atypical, recurrent or persistent infections, patient’s immunizations
iii. clinical data: particular examination findings and any specific clinical phenotype (autoimmunity, lymphoproliferation, allergy, or syndromic features)
iv. previous work up results
v. management and treatments prior to referral
vi. potential suspected diagnosis and investigations requested
Regarding confidentiality and patient data privacy, the collected data were listed in an electronic database with a highly restricted access.
A further written consent was obtained for the patients or their parents (for children < 18 years) before running genetic studies.
Laboratory Assessment
Laboratory tests are a crucial to confirm the initial clinical suspicion and guide further biological investigation of IEI.
A stepwise approach for immunological diagnostic is warranted in our department and we used the following flow chart (Supplementary Figure S1):
Initial Screening Laboratory Tests
Basic screening methods are used with standard techniques such as complete blood counts; peripheral blood smear, total serum immunoglobulins (Ig) levels (i.e. IgM, IgG, IgA, IgD and IgE), IgG subclass levels, measurement of antibody responses to vaccines (mainly in predominantly antibody deficiencies), complement assays, optic microscopy examination of scalp hair (notably in pediatric patients with bamboo hair in Netherton’s Syndrome) and immunophenotype analysis of peripheral blood cells using a flow cytometry: T-cell subsets (CD3+, CD4+, CD8+), B-cells (CD19+) and natural killer (NK) cells (CD56/CD16+).
In-depth Immunophenotyping
An extended phenotyping of peripheral B- and T- cell compartments was performed when indicated, including naïve, effector, and memory T cell subsets (CCR7+/-, CD45RA+/-, CD45RO+/-), recent thymic emigrants (RTE) CD4+ T cells (CD45RA+, CD31+), double negative T cells (TCRαβ+, CD4-, CD8-), regulatory T cells (CD4+, CD127-, CD25+, FOXP3+), class-switched (CD27+, IgD-) and non-switched memory B cell subsets (CD27+, IgD+), transitional B cells (CD24++, CD38++) and plasmablasts (CD24-, CD38++).
Protein Specific Assays
Flow cytometry was also used to get an accurate diagnosis of immunodeficiencies associated with defects in the expression of intracellular or cell surface proteins (Supplementary Table S1). These tests were carried-out according to the results of previous tests or based on the index of suspicion: Human Leukocytes Antigen cells- DR (HLA-DR) expression on B cells and monocytes in case of CD4+ T cell lymphopenia (to rule out MHC class II deficiency); WAS protein expression in T cells in case of micro-thrombocytopenia and eczema (Wiskott Aldrich Syndrome, WAS), common γ chain expression on B cells in case of T-B+NK- SCID in boys; BTK protein in monocytes in case of agammaglobulinemia associated with severe decrease of B cells in males; CD79a, CD79b, CD179α, and Mu chain in bone morrow precursor B cells in case of AR agammaglobulinemia; CD40 on B cells in case of hypogammaglobulinemia with normal or elevated IgM levels, neutropenia, and opportunistic infections (hyper-IgM syndrome, HIMS); interferon-γ receptor 1 (IFN-γR1) on monocytes in case of susceptibility to infections with environmental or low virulent mycobacterial (to rule out Mendelian Susceptibility to Mycobacterial Disease, MSMD); ZAP-70 in T cells and HLA-ABC on lymphocytes in case of severe CD8+ T cell lymphopenia (CID); CD15 and CD18 on granulocytes in case of hyperleukocytosis and skin infections (leukocyte adhesion deficiencies, LAD); SAP and XIAP proteins in lymphocytes in male (in case of Epstein Barr virus infections to rule out X-linked lymphoproliferative (XLP) syndrome XLP1/2); and perforin in NK cells when suspecting a familial hemophagocytic lymphohistiocytosis (F-HLH).
Advanced Immunological and Functional Assays:
More specific tests were performed (Supplementary Table S1), such as in vitro lymphocyte proliferation to assess T cell responsiveness to mitogens and TCR-dependent signaling; protein phosphorylation analysis to unmask defects in the intracellular signaling cascade such as pSTAT1 in inborn defects of the IL-12/IFN-γ axis (for suspected MSMD) or STAT1 gain-of-function (GOF), and pSTAT-3 in hyper-IgE syndrome; induction of specific activation markers such as CD40L after in vitro stimulation in male patients suspected of HIMS, and IL-12RB1 on activated CD4+ T cells in patients suspected with MSMD; dihydrorhodamine (DHR) 123 oxidation test to assess nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity (to assess chronic granulomatous disease, CGD); NK cell degranulation and cytotoxicity assays using K562 target cells to explore F-HLH.
Genetic Analysis
In our series, genomic deoxyribonucleic acid (DNA) was extracted from whole blood according to the previously described protocol (24).
In patients with clinical and laboratory findings indicative of a specific IEI, Sanger sequencing was performed on specific genes with high frequency of mutations and low exon numbers (BTK, WAS, RAG1/2, ITGB1, CYBB, RFXANK). Patients presenting clinical and biological findings suggestive of DiGeorge syndrome or Jacobsen syndrome were subjected to a high-resolution cytogenetic study using G-banding and fluorescence in situ hybridization (FISH) for 22q11.2 deletion or 11q23 deletion, respectively.
For patients with unusual clinical presentation, if Sanger sequencing was unable to detect any candidate rare variants, next generation sequencing (NGS) through targeted gene sequencing panels or whole exome/genome sequencing was then performed.
Statistical Analysis
Statistical analysis was done using a commercially available software package (SPSS Statistics 17.0.0; SPSS, Chicago, Ill). Descriptive statistics were reported for all variables in the cohort. All quantitative variables were reported with mean values, and median for continuous variables. Kaplan-Meier curve plots was used to distinguish survival curves; and log-rank tests were used to compare these survival curves. A p-value of 0.05 or less (p ≤ 0.05) was considered as statistically significant.
Results
Frequency and Distribution of IEIs
From January 2008 to December 2021, more than 5,500 patients were screened for IEI in the department of medical immunology of Beni Messous University Hospital Center of Algiers. Among them, 807 were identified with confirmed IEI and included in the dedicated database. Up to 50 different IEI classified were identified, belonging to nine major categories (Figure 1) and, surprisingly, no patient was identified with an inherited bone marrow failure. The frequency and characteristic phenotypes of patients in each IEI category are represented in Table 1. The predominant category was the combined immunodeficiency (CID) (273 patients, 33.8%), followed by predominantly antibody deficiencies (PAD) (198 patients, 24.5%), combined immunodeficiencies with associated or syndromic features (SyCID) (194 patients, 24%), congenital defects of phagocytosis (57 patients, 7.1%), diseases of immune dysregulation (DID) (50 patients, 6.2%), innate immunity disorders (16 patients, 2%), complement deficiencies (11 patients,1.4%), and, lastly, autoinflammatory diseases and phenocopies of IEI in similar proportions (4 patients, 0.5%). In particular, within the CID category, SCID was the most commonly found in 10.8% (87 patients), with a predominance of T-B-NK+ SCID phenotype 6.8% (55 patients), followed by MHC-II deficiency (55 patients; 6.8%), hypomorphic SCID (47; 5.9%), and Omenn syndrome/SCID-GVHD (24; 2.9%). Some patients (4.7%) did not match with any specific phenotype of the CID according to the IUIS classification and were categorized as unclassified CID (uCID).
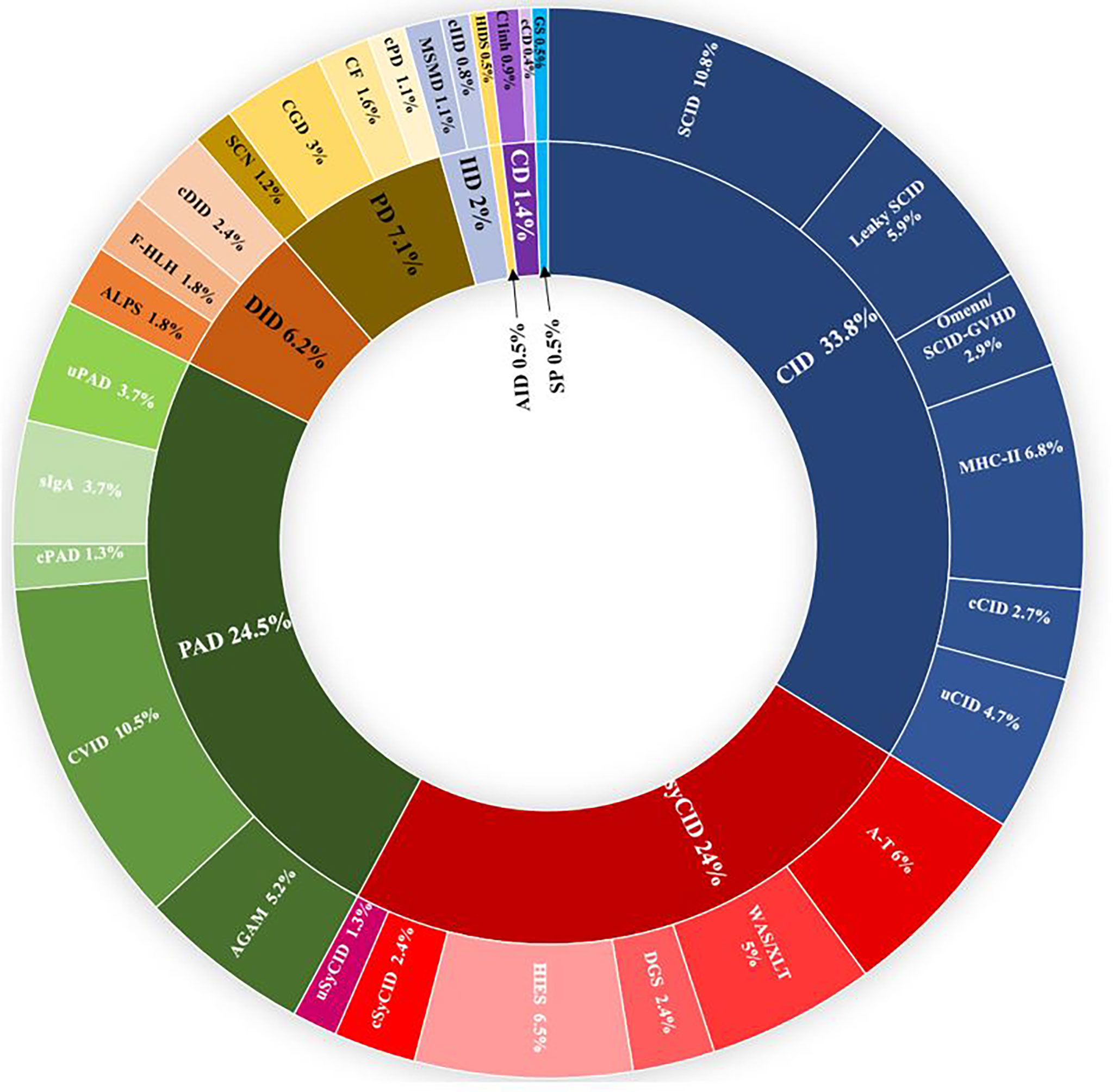
Figure 1 Distribution of the frequencies of IEIs according to IUIS categories in 807 Algerian patients in the study. IEI groups are shown according to IUIS classification, 2021. Total number of patients and percentage of all registered patients are shown for each group. AGAM, agammaglobulinemia; ALPS, autoimmune lymphoproliferative syndrome; AID, autoinflammatory diseases; A-T, ataxia- telangiectasia; c, classified; CD, complement deficiencies; CF, cystic fibrosis; CGD, chronic granulomatous disease; CID, combined immunodeficiencies; CVID, common variable immunodeficiency; DGS, DiGeorge syndrome, DID, Dysregulation of immune diseases; f-HLH, familial Hemophagocytic lymphohistiocytosis; GS, good syndrome; HIES, hyper-IgE syndrome; HIDS, hyper-IgD syndrome; IID, innate immune deficiencies; MSMD, mendelian susceptibility to mycobacterial disease; PAD, predominantly antibody deficiencies; PD, phagocytic deficiencies; SCN, severe congenital neutropenia; sIgA; selective IgA deficiency; SP, Somatic phenocopies; SynCID, syndromic combined immunodeficiencies; u, unclassified; WAS, Wiskott Aldrich syndrome; XLT, X linked thrombocytopenia.
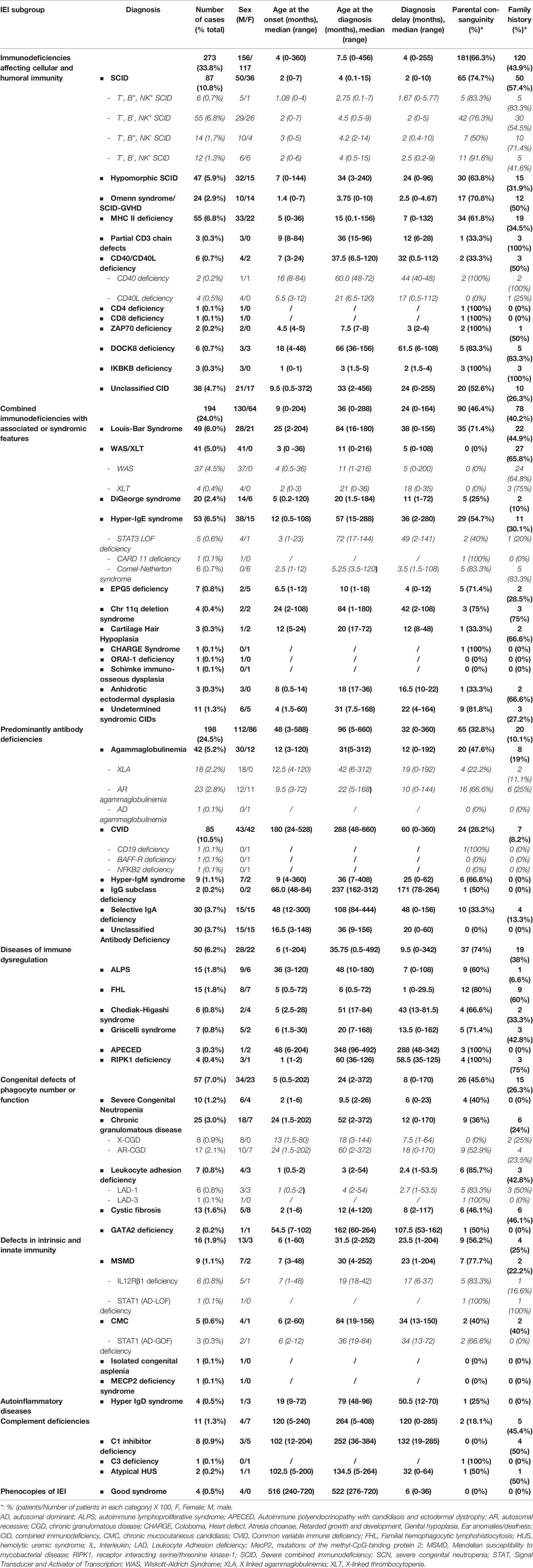
Table 1 General characteristics of patients with different phenotypes of inborn error of immunity.
Among patients with antibody deficiencies, common variable immunodeficiency (CVID) was the most common diagnosis in 10.5% (85 patients), followed by congenital agammaglobulinemia in 5.2% (42 patients). Interestingly, AR agammaglobulinemia was more common than X-linked recessive forms (2.8%, 23 patients versus 2.2%, 18 patients, respectively).
Syndromic combined immunodeficiencies were mainly represented by hyper-IgE syndrome (HIES, 53 cases,6.5%), followed by ataxia-telangiectasia (49 patients; 6.0%) and Wiskott-Aldrich syndrome (37 boys; 4.5%).
Finally, CGD, autoimmune lymphoproliferative syndrome (ALPS), f-HLH, MSMD, hereditary angioedema type I (due to C1-inhibitor deficiency), were the main other IEI.
Patient Characteristics
There were 482 male patients (59.7%) and 325 female patients (40.3%), and all of them were Algerian. The male-to-female ratio (M/F) was close to 1.5:1. The majority of IEI categories were observed in children. However, adult patients only belonged to certain IEI groups such as predominantly antibody deficiencies, diseases of immune dysregulation, phenocopies and complement deficiencies.
Our study indicated that age at onset, age at diagnosis, and diagnosis delay varied significantly for different types of IEI. In all patients, the median age at diagnosis was 36 months (range, 0 days to 60 years). Of these patients, 729 (90.3%) were children (mean age at diagnosis, 46.2 months), and 78 (9.7%) were adults (mean age at diagnosis, 32.9 years). The majority of the children (525 of 729, 72%) were under 5 years old (Figure 2). Overall, the onset of symptoms occurred at the median age of 8 months (range, 0 month to 52 years). In children, the age at onset of symptoms was less than 6 months for 316 of them (43.3%), from 6 months to 3 years for 279 (38.2%) and more than 3 years for 134 (18.3%). The symptoms (Figure 3A) were notably observed earlier in CID, phagocytic defects, diseases of immune dysregulation, and defects in intrinsic and innate immunity with the median onset age of 4, 5, 6 and 6 months, respectively, compared to other categories of IEI. Age at diagnosis was shorter in CID with the median diagnosis time of 7.5 months. Moreover, patients with PAD displayed symptoms remarkably later with the median age at the onset of 48 months among children, whereas in adults, this delay was observed in patients with complement deficiencies and phenocopies with the median age at the onset of 10 years and 43 years respectively (Table 1).
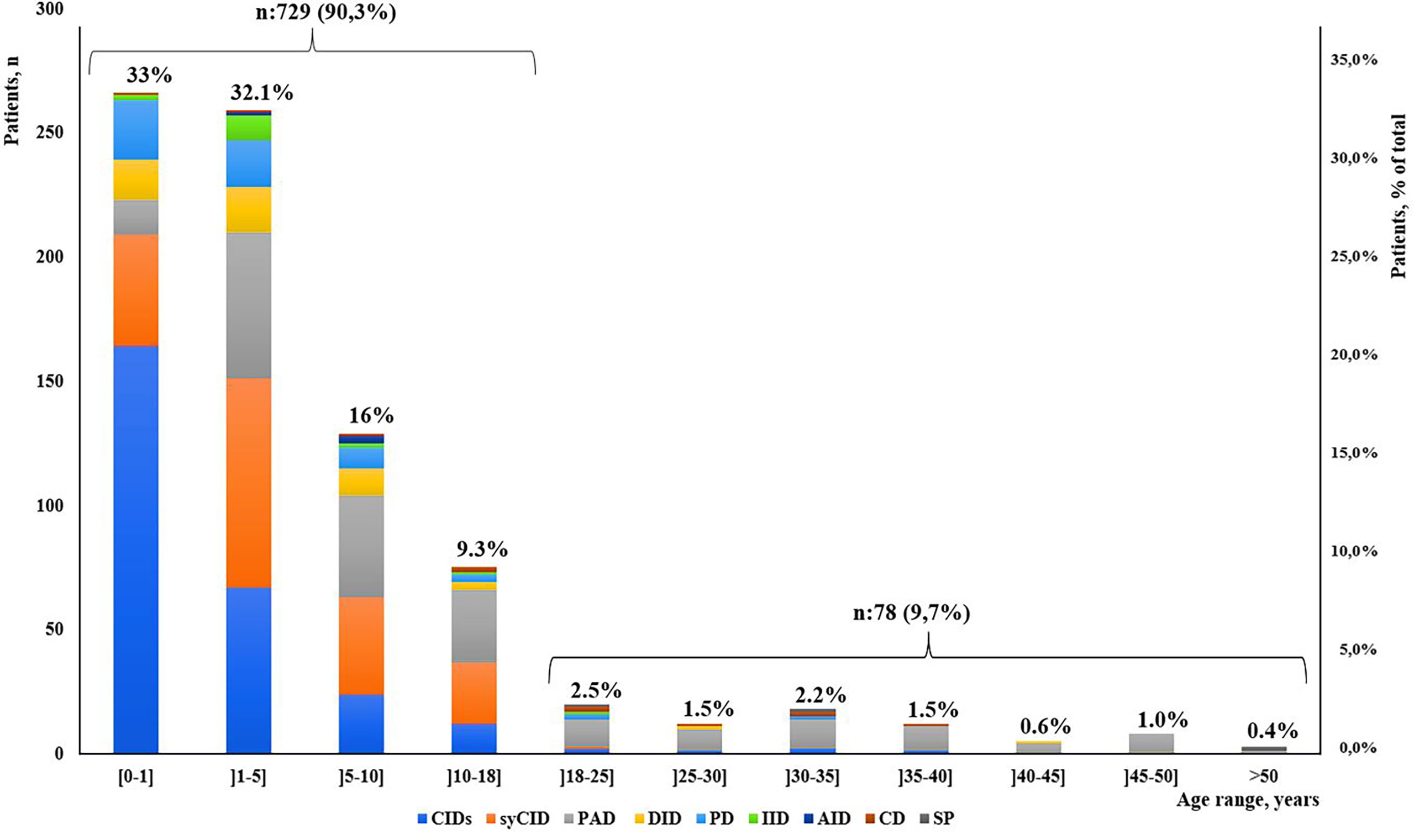
Figure 2 Distribution of patients according to IEI diagnosis age groups and main IEI categories (N = 807).
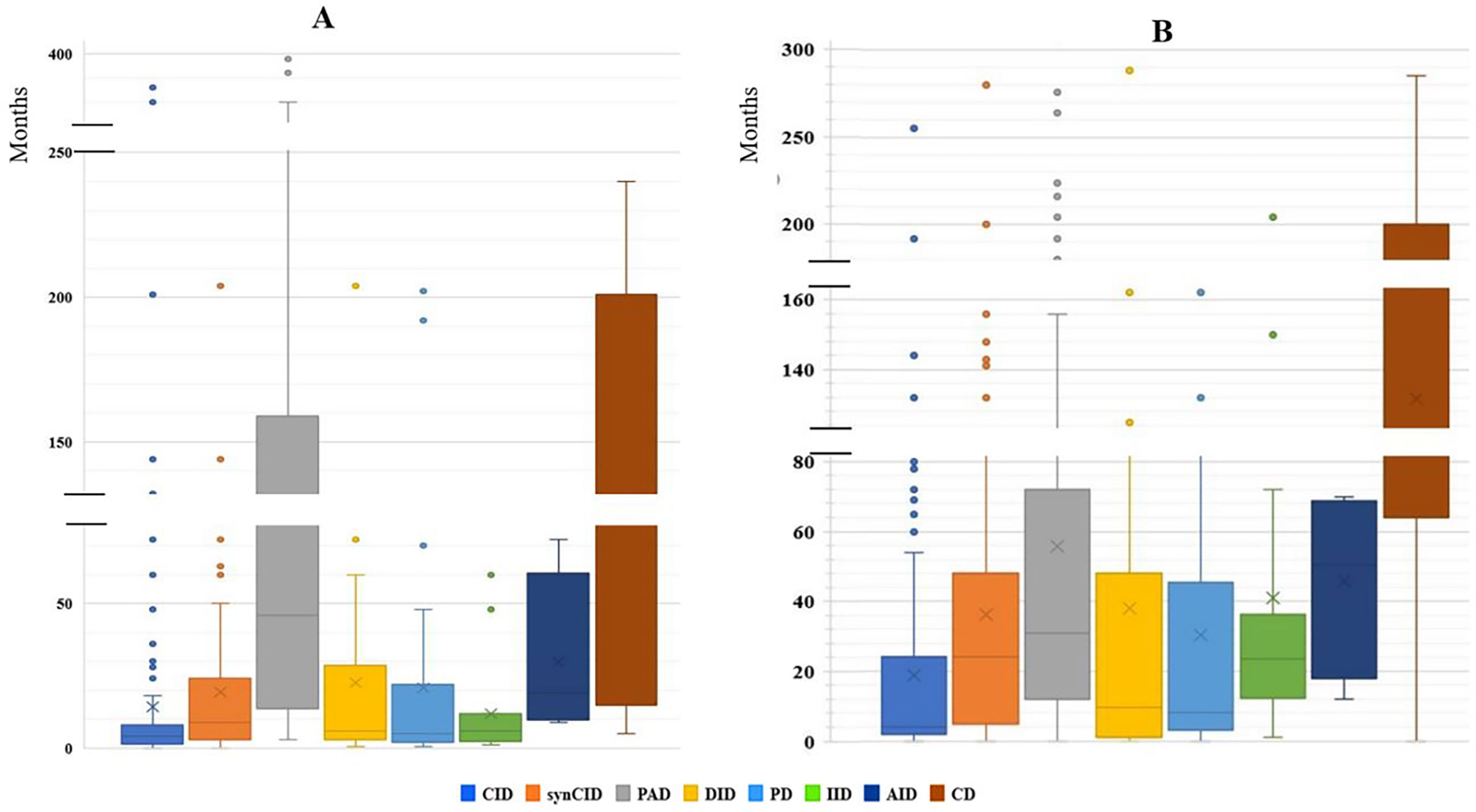
Figure 3 The median age of disease onset and diagnostic delay in the main IEI categories. (A) Median age of disease onset. (B) Median of diagnostic delay.
The diagnostic delay is defined as the time interval between the onset of symptoms and confirmed diagnosis of a disease. Significant diagnostic delay has been noted in Algeria (Figure 3B), with an overall median diagnostic delay of 16 months but over a broad age-range (from less than 1 month to 30 years), and it was more important in adult patients (9 years; 0-30 years) than children (12 months; 0-200 months). There were no antenatal diagnoses among patients registered in the database. The delay in diagnosis was shorter in CID (median: 4 months; range: 0-456 months) when compared to other groups of IEI. Among the most common CID, the shortest mean diagnostic delay was observed in SCID (median: 2 months; range: 0–10 months), followed by the Omenn syndrome/SCID-GVHD (median: 2.5 months, range: 0–4.67 months), MHC class II deficiency (median: 7 months; range: 0-132 months), and hypomorphic SCID (median: 24 months; range: 0-96 months).
Four hundred ten patients (50.3%) were born to consanguineous marriages, in most cases from first cousin parents. The rate of consanguinity varied substantially among the groups of IEI. The highest reported rates of consanguinity have been found in patients with diseases of immune dysregulation at 74%, followed by CIDs at 66.3%, while in the other categories the rates were as follow: defects in intrinsic and innate immunity (56.2%), syndromic CID (46.4%), phagocytic defects (45.6%), and predominantly antibodies deficiencies (32.8%). The lowest rates of consanguinity were recorded for phenocopies, complement deficiencies and autoinflammatory diseases at 0%, 18.1% and 25%, respectively. A positive family history of unexplained death or family member known to have an immunodeficiency disorder was noticed in 261 patients (32.34%), with the highest frequencies in patients with complement deficiencies, CID, and syndromic CID in 5 (45.4%), 120 (43.9%), and 78 (40.2%), respectively.
Epidemiology and Geographical Distribution of IEI Patients
The minimum overall IEI prevalence in the Algerian population was estimated at 1.8/100,000 inhabitants, with drastic variations among the provinces (from 0.38 to 4.2 per 100,000). From 2008 to 2021, an average of 57 new patients were made per year with 29.5% of the cohort being diagnosed from 2008 to 2016, and 70.5% from 2017 to September 2021. From 2019, more than 120 new cases are diagnosed every year (Figure 4; Supplementary Figure S2), the highest number was recorded in 2019. On the basis of the patient’s home address, the geographical distribution of patients was heterogeneous with the large number of cases was found in the following provinces (Figure 5): Algiers, Djelfa, Tizi ouzou, and Batna.
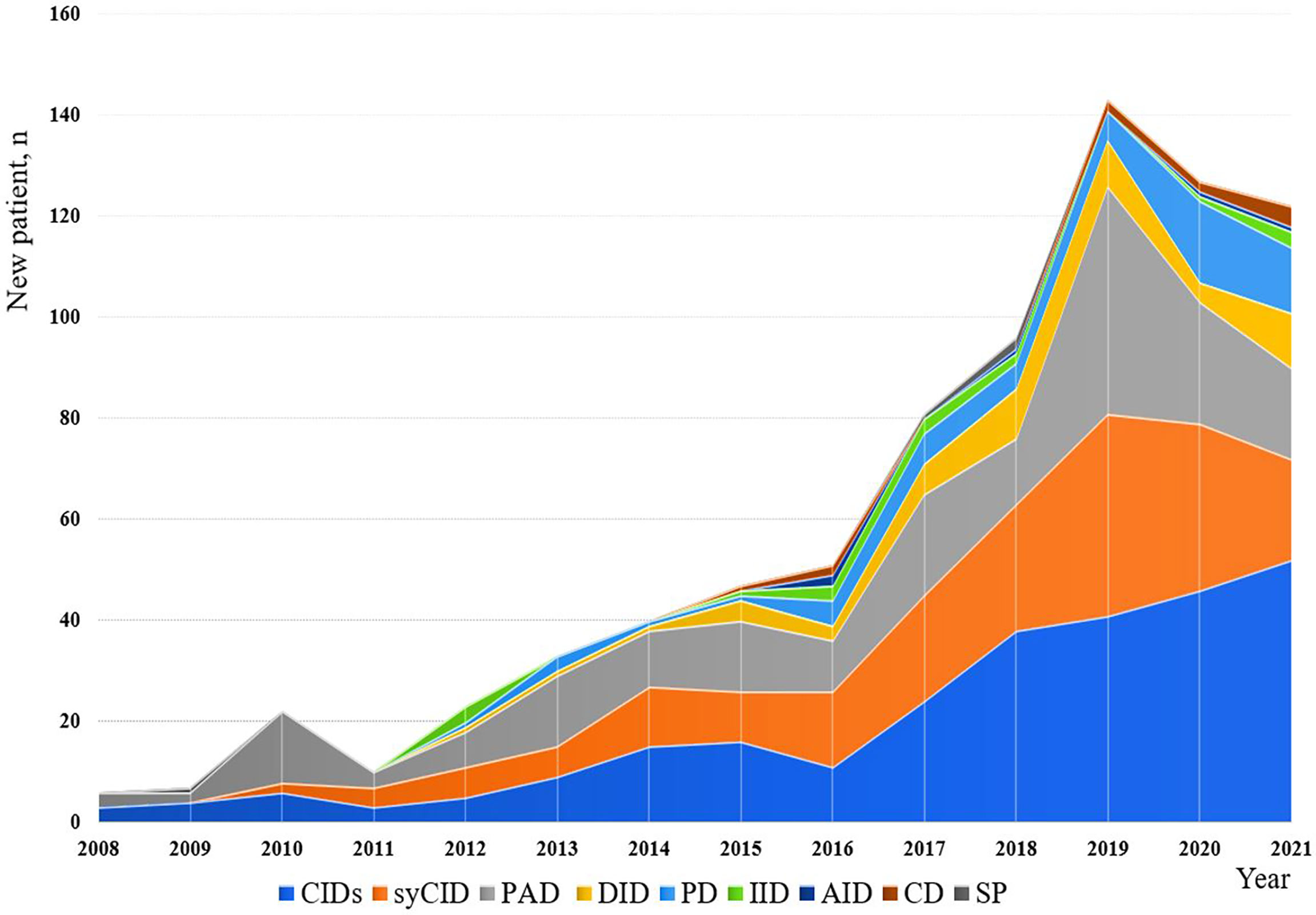
Figure 4 Annual numbers of newly diagnosed IEI cases from 2008 to September 2021. The registration of patients into the database has been stopped in September for the year 2021.
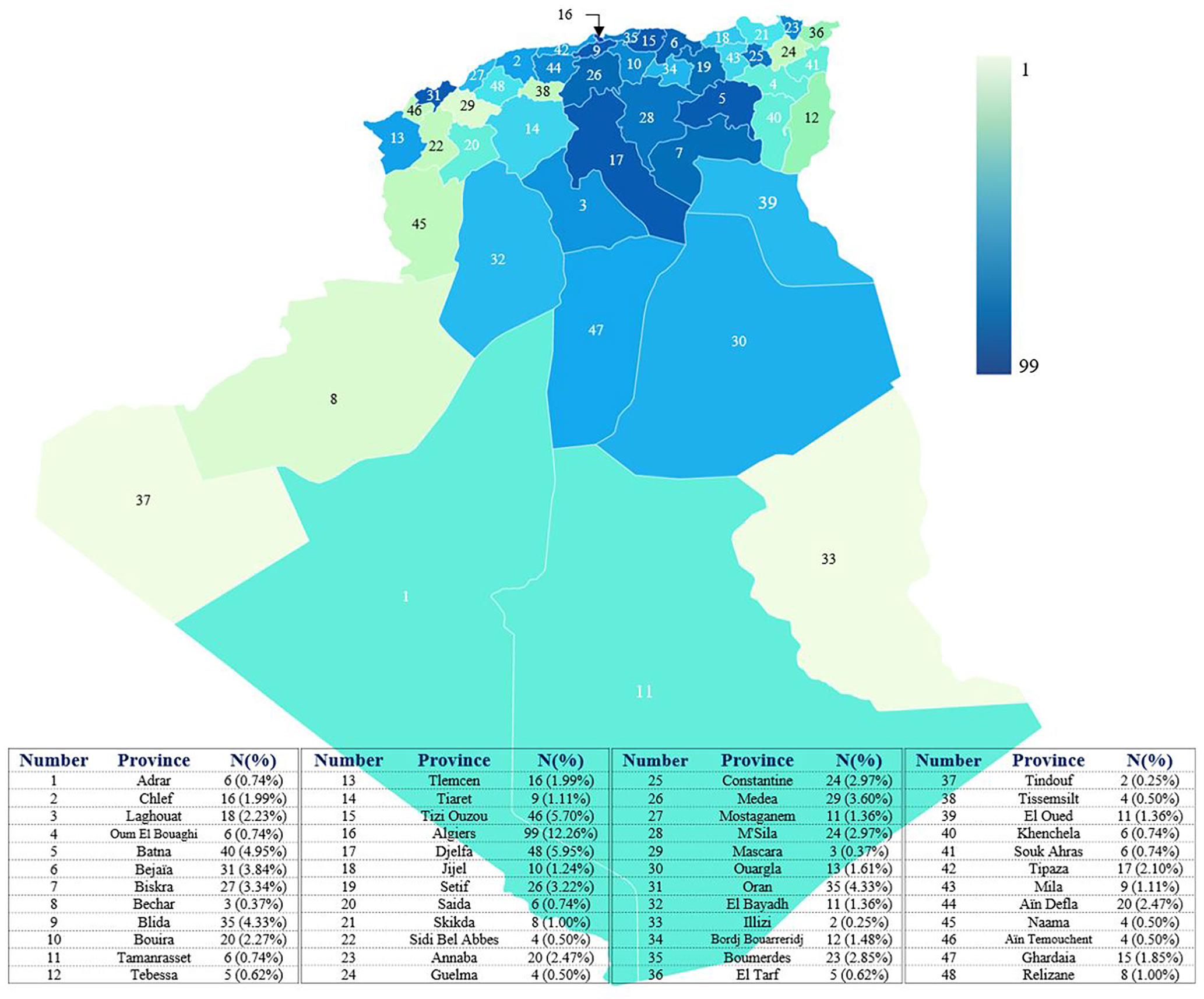
Figure 5 distribution of patients in the different provinces of Algeria. The base map was created with the permission of data wrapper. The intensity of the blue color is proportional to the number of patients. The registered number includes living and deceased patients. Each number on the map corresponds to a province.
Clinical Features
At time of diagnosis, patients with IEI disorders presented with a broad clinical spectrum (Table 2). Recurrent infections were the most common indication for investigation, and respiratory tract infections were the most frequent. The least common infections were osteoarticular infections, septicemia, and meningitis/encephalitis. CID was the most category affected by recurrent infections, followed by PAD and syCID. Lower respiratory tract infections predominated in CIDs, PADs, syCIDs, and phagocytic disorders, with 73.2%, 71.7%, 57.2%, and 43.9%, respectively, whereas, upper respiratory tract infections found more frequently in PADs (54%), gastrointestinal infections in CIDs (45%), and skin infections (usually deep or superficial abscesses) in phagocytic disorders (43.9%). With respect to Mycobacterium bovis disease, as a serious complication of Bacillus Calmette-Guérin (BCG) vaccination, ranging from local disease (known as BCGitis) to disseminated disease (BCGosis), occurred in twelve cases with CID, nine cases with inborn defects of the IL-12/IFN-γ axis, three cases with syCID, and one case with phagocytic disorder. Failure to thrive and growth retardation were observed in 82 patients (10.1%). Bronchiectasis was found to be a common complication of PADs (21.7%).
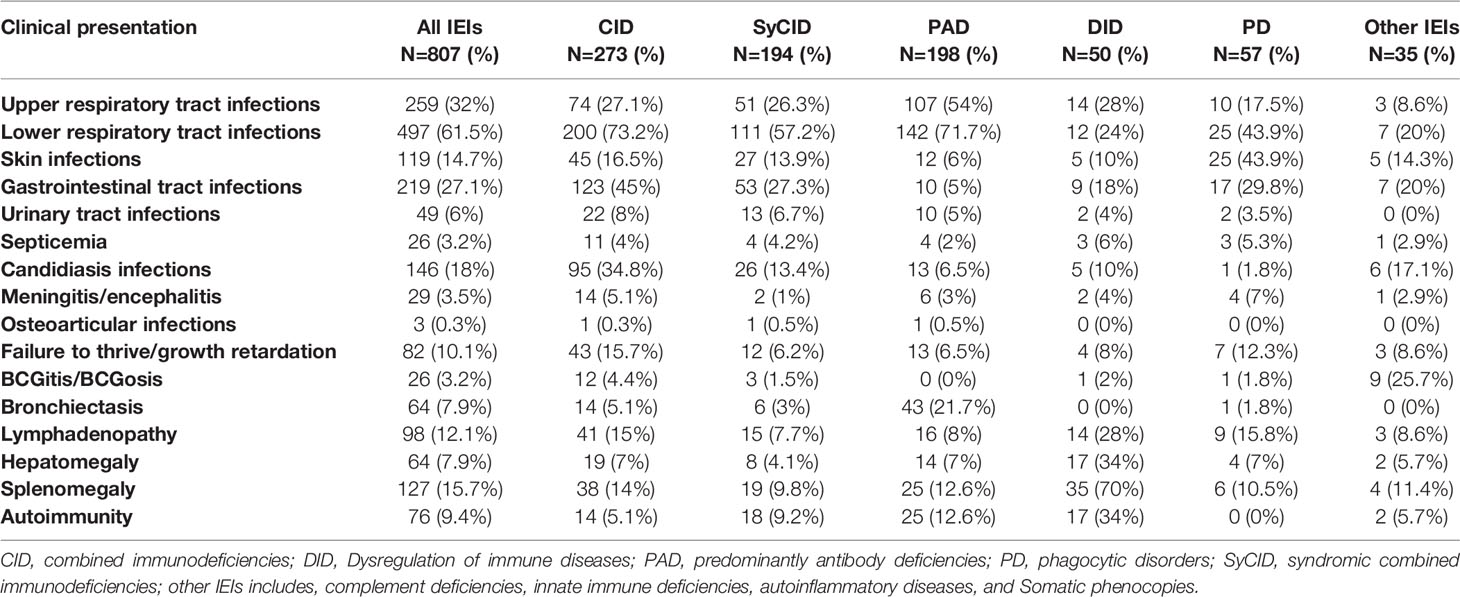
Table 2 Clinical presentations and complications of IEIs observed in Algerian patients.
Autoimmune disorders, including autoimmune hemolytic anemia, thrombocytopenia, neutropenia, organ-specific autoimmune diseases, coeliac disease, Sjogren’s syndrome, were globally less common and were observed in 76 of total (9.4%), while they were relatively more common in immune dysregulation disorders (34%). Patients presented with splenomegaly, often accompanied by hepatomegaly and/or multifocal peripheral and/or internal lymphadenopathy, and these manifestations were found to be more frequent in patients with immune dysregulation disorders than those in other categories.
Mutational Analysis
Genetic testing was performed in 287 patients (35.5%), among them 129 patients were confirmed carrying pathogenic germline variants (Table 3), of which 22 (18.5%) were located in RFXANK, 14 (12.6%) in RAG1, 13 (10.9%) in RAG2, 9 (7.5%) in BTK, 5 (4.2%) in DOCK8, 5 (4.2%) in STAT3, 5 (4.2%) in RFXAP, 5(4.2%) in CFTR, 4 (3.3%) in RIPK1, 4 (3.3%) in ITGB2, 3 (2.5%) in STAT1 (GOF), 3 (2.5%) in RAB27A, 3 (2.5%) in ADA, 3 (2.5%) in IKBKB, 2 (1.7%) in DCLRE1C, 2 (1.7%) in 22q11.2del, 2 (1.7%) in IL12RB1, 2 (1.7%) in CD40, 2 (1.7%) in JAK3 and in other genes (IL2RG, CD40L, CD3Z, 11q23del, CD79A, CARD11, CD19, ORAI1, IGHM, TCF3, NFKB2, TNFRSF6, UNC13D, FERMT3, MVK, CYBB, GATA2, IRF3) 1 (0.8%) for each of these genes. However, in some cases more than one pathogenic mutation in different genes have been identified. The first case was a rare double homozygosity of two different mutations in two brothers, a missense biallelic variation in the CD3G gene and a biallelic nucleotide deletion mutation in the CD3E gene. The two mutations were considered as previously unreported and were expected to cause the disorder. The second case concerned a 16-month-old boy who also carried two variations, a heterozygous nonsense nucleotide substitution mutation in the NOD2 gene, and a homozygous nucleotide deletion mutation in the RFXANK gene, responsible of Blau syndrome and MHC class II deficiency, respectively. Both mutations have been previously reported and recognized to be pathogenic. Most of the remaining patients are still under analysis.
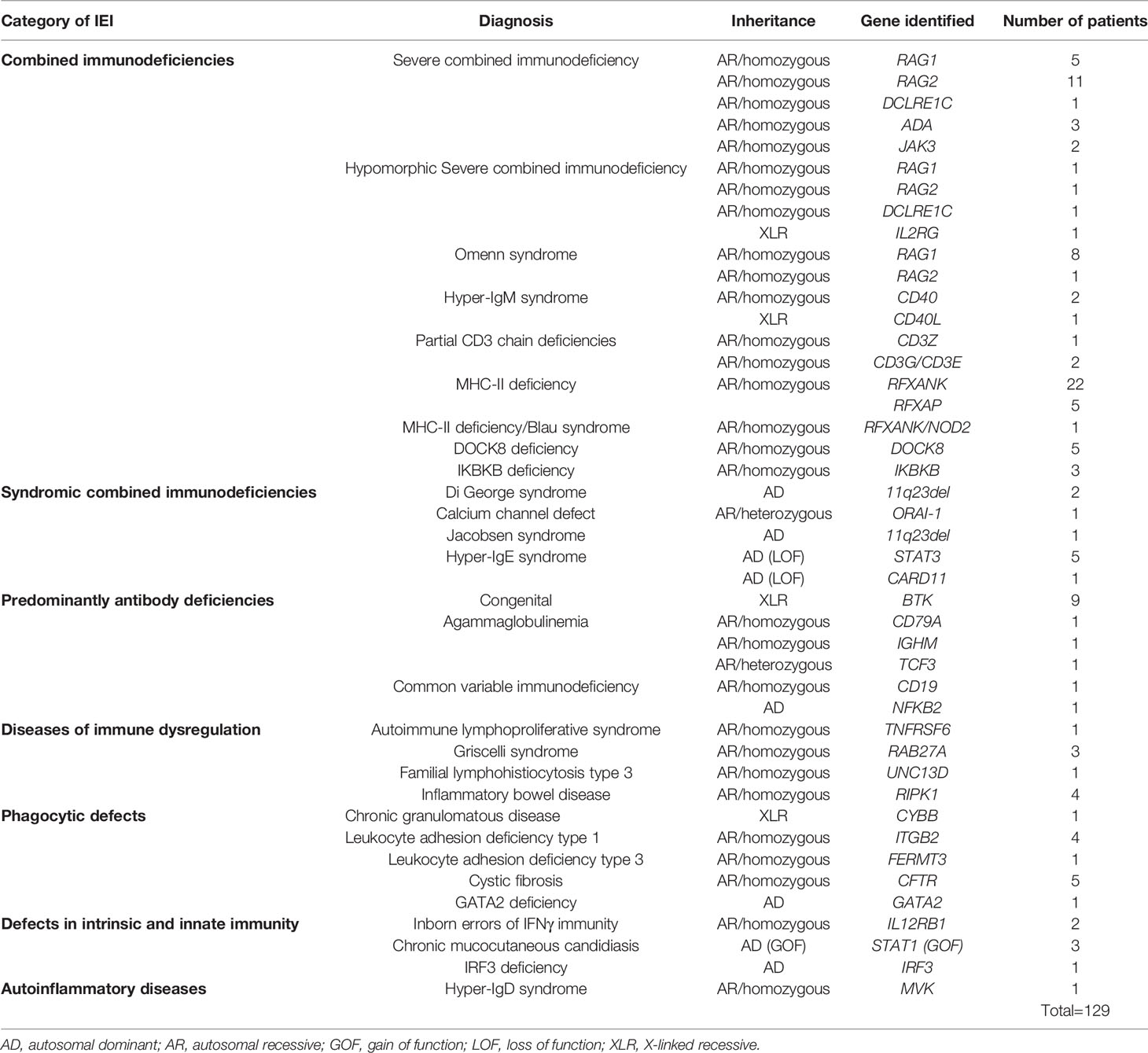
Table 3 distribution of patients according to genetically-confirmed patients.
Mortality
To assess mortality, we excluded 102 patients for whom we were unable to determine their status, then we analyzed the cohort of 705 patients whose status was known, including 474 alive patients and 231 cases confirmed to be dead. Among deceased patients, 219 (94.8%) were children and 12 (5.2%) were adults (Figure 6A). The overall mortality rate was estimated at 32.7%. It ranged from 0.4% to 50.2% in different age ranges; the highest rate was found in children in their first 2 years of life. The majority of infant deaths occurred in patients with SCID and Omenn syndrome (92 of 129; 71.3%). In the two next age ranges (2–5 years and 6-10 years), mortality was higher in patients with CID and syndromic CID. Overall, 75% (172/231) of all IEI-related deaths occurred in patients during their first 5 years of life, whereas, in adult, the mortality rate was significantly lower, with approximately equal proportions in the following seven IEI categories: predominantly antibody deficiencies, somatic phenocopies, CID, syndromic CID, phagocytic deficiencies, complement deficiencies, and innate immune deficiencies.
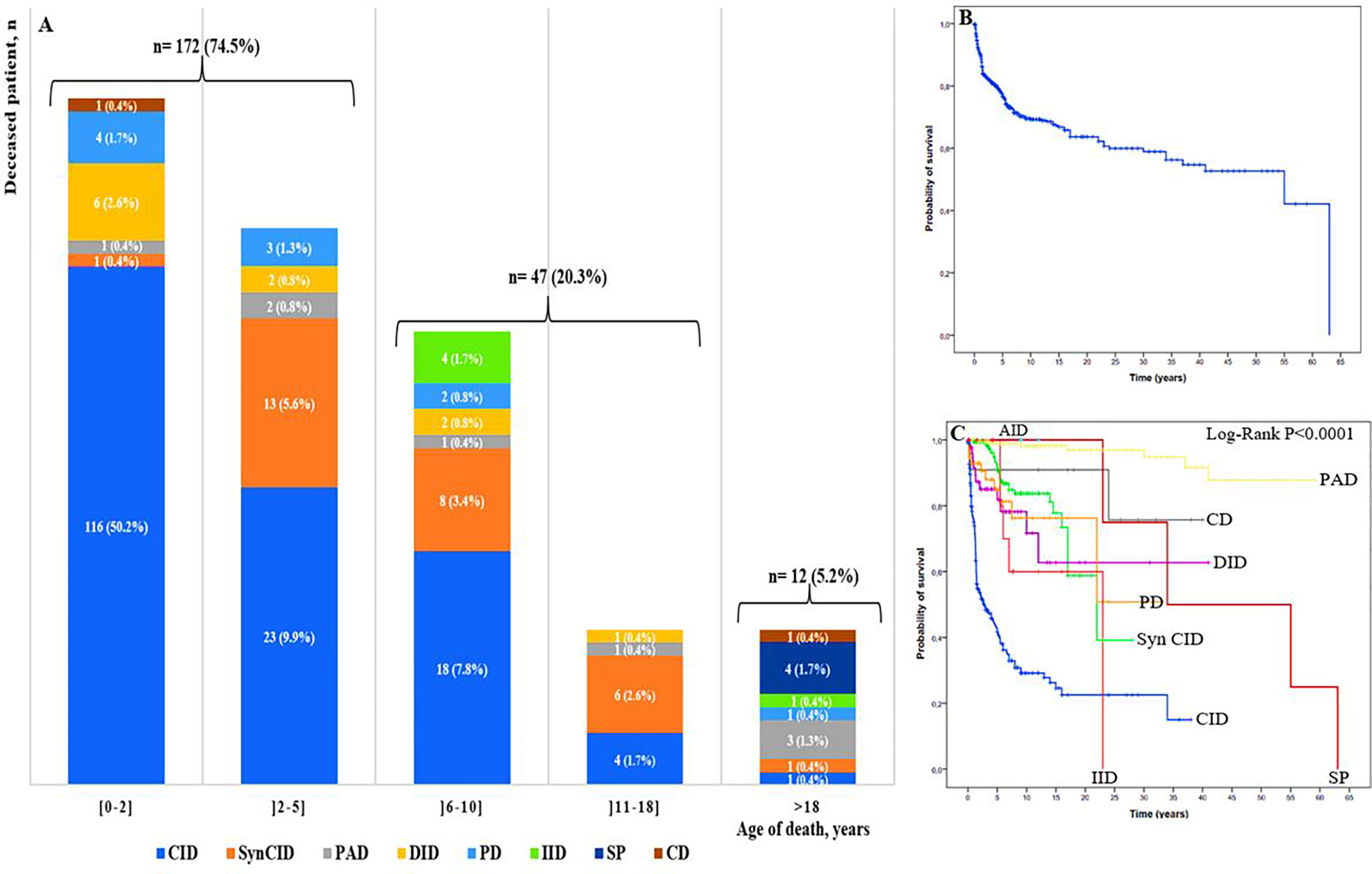
Figure 6 (A) Mortality rate by PID group and age of the deceased patients (n: 231). Colors represent different IEI categories. Numbers in the plots show the number of patients and the ratio of the deceased patients by IEI group to the total number of the deceased patients of the age-group; (B) Overall survival (Kaplan-Meier curve) of patients with IEI showing the probability of survival following diagnosis. (C) Kaplan-Meier curve showing overall survival in the 807 studied patients within the 9 categories of IEI. CID, combined immunodeficiencies; SyCID, syndromic combined immunodeficiencies; PAD, predominantly antibody deficiencies; DID, Dysregulation of immune diseases; PD, phagocytic disorders; CD, complement deficiencies; IID, innate immune deficiencies; AID, autoinflammatory diseases; SP, Somatic phenocopies.
The probability of survival was analyzed using the Kaplan-Meier curve, and the overall probability of survival 5 years after diagnosis was approximately 77% (Figure 6B), while, the lowest survival rate was 42% in CID, when comparing the survival rate in different IEI categories after the first 5 years of diagnosis, for disease of immune dysregulation was 82%, for phagocytic disorders was 84%, for syndromic CIDs and complement deficiencies was 90%, and for the predominantly antibody disorders group was 98%. The difference was found to be statistically significant between at least two categories (P<0.0001) (Figure 6C; Supplementary Table S2).
Discussion
Most IEIs result from inherited defects in development and/or function of immune system; resulting in infections that develop and recur more frequently, autoimmune disorders and an increased risk of malignancies (25). However, acquired forms have also been reported (5, 26).
This current report represents a single center study which provides epidemiologic, clinical, and genetic data on Algerian patients with IEI. At the time of analysis, a total of 807 IEI patients were registered, representing all provinces of the country, thus making this cohort a valid evaluation of the IEI in Algeria. Although our department represents the largest center for immunological investigation of IEIs in Algeria, the number reported in the current study does not represent all cases, therefore we are only able to report the minimal prevalence of these conditions. It is widely accepted that IEIs are underdiagnosed worldwide, particularly in developing countries. The Algeria’s population census (up to 2021) has reported a population 45,002,737 million (21), and the overall IEI prevalence of 1.8 per 100,000 is quite low which shows that our findings certainly underestimated the disease burden in Algeria. Although our department receives the majority of patients referred from all provinces for investigation, the prevalence of these disorders is expected to be much higher in our country. This is due, on one hand, to the fact that some patients with mild forms of IEI are usually managed as outpatients without having recourse to investigation for a probable IEI but, on the other hand, severe forms of IEI such SCID and its variants usually die from severe infections before being formally diagnosed as immunodeficient patients. Also, the existence of a few other centers that participate to the investigation of IEI, all located in Algiers. Consequently, a better recruitment and diagnostic strategy could improve the coverage of diagnosed patients for a better understanding of IEI epidemiology in Algeria.
According to different IEI categories, the distribution of different types of IEI in our cohort is consistent with other reports from MENA countries. Combined immunodeficiencies (≈34%) were the most common entity of IEI, similar to the series from Tunisia, Saudi Arabia, Kuwait and Egypt (27–30), and to the MENA countries’ registry (12). Such high frequency of these disorders in our cohort could be due to ethnic/geographical particularities, and mostly to the local increased prevalence of consanguinity, and these hypotheses are strengthened by close frequencies in neighboring countries like Tunisia (27). Predominantly antibody deficiency and well-defined syndromes with immunodeficiency involved 24.5% and 24% of patients respectively, very close to IEI reports of other North African countries including Morocco, Tunisia, Egypt; except in the slight relative predominance of syndromic combined immunodeficiencies in these countries (27, 30, 31). Congenital defects of phagocytosis and immune dysregulation diseases were the fourth and fifth most common groups of IEI involving 7.1% and 6.2% of patients, respectively.
Regarding phagocytic defects, this frequency is lower than those reported from Asian countries and regions, including Qatar, Oman, China, Korea, Japan, and Asia-pacific region (16, 32–36) as well as those from Europe and Latin America but higher than those from Australia (15, 17, 37), Immune dysregulation diseases frequency is in the same range of previous studies in Saudi Arabia, Russia and Asia pacific region (16, 28, 38), while other studies showed a lower rate (15, 27, 31–33, 39–42). Severe phenotypes of IEI are more represented in our study: SCID, atypical SCID, Omenn syndrome and MHC class II deficiency were more frequent, comprising nearly ≈80% of the total combined immunodeficiencies. This might be explained by a genetic predisposition/background in the Algerian population.
The overall median diagnostic delay in all IEIs (time between symptom onset and diagnosis) was 16 months, close to that of neighboring countries: Morocco (24 months) (31), Tunisia (18 months) (27). However, patients with CID were more promptly diagnosed (mean delay =7.5 months). This is probably due to the early-onset and severe symptoms these patients do present in the first year of life, with rapid referral and a subsequent earlier age at diagnosis.
Similar to various reports (27, 29, 31, 38, 43–47), age at the onset of symptoms and at diagnosis as well as diagnosis delay were very variable according to the IEI groups. Early diagnosis and management of IEI can lead to lower morbidity and mortality rates and better quality of life in IEI patients (48).
In accordance with previous studies (49), the vast majority of IEI patients in our cohort were children (90.3%) and about 65% of patients were diagnosed before the age of 5. In some studies, adult patients (aged 18 or above) represent almost half of all IEI (32, 47). Hence, adults with IEI are the most underdiagnosed group in Algeria. This is supported by the low proportion of predominantly antibody deficiencies in our cohort compared to other countries (13, 50, 51). Of note, most of patients referred to our department came from the pediatric departments, suggesting that education sessions and programs may be required to raise awareness of IEI especially CVID in general practitioners and adult specialists.
An increased frequency of unions between relatives is associated with an increased birth prevalence of children with severe autosomal recessive single-gene disorders (52). Parental consanguinity rate in our cohort was 50.3%; higher rates have also been reported in other MENA countries as in Kuwait (29), Oman (33), Saudi Arabia (28), Egypt (30), Qatar (36), Iran (41), Tunisia (27) and Algeria (53) at a rate of 78, 76, 75, 75.2, 61.1, 60.1, 58.2%, and 52.6%, respectively. But it is significantly lower than that described in other countries such as Russia (38), Germany (47), US (14), UK (46), France (43), South Africa (44), estimated at 1.6, 8, 10, 15, and 1.2%, respectively. There was also a notable difference in consanguinity rates between the nine groups among which the highest level was found in immune dysregulation diseases with 74%, while its frequency was lower in complement deficiencies with 18.1%. Although X-linked conditions such as XLA and X-linked SCID are less common in our cohort, the male ratio in different category of IEI does not differ greatly from previous studies, with males predominating amongst children and adult patients which is in line with worldwide trends (12, 14, 15, 17, 32, 34, 37–39, 41, 44, 46, 47).
The increased diagnosis over the last decade reflects a major and active effort to improve the field of clinical immunology by organizing a working group studying IEIs in our country, the organization of scientific events and campaigns about IEI for a better awareness and understanding of IEI presentations and the existence of a specialized laboratory in the investigation of IEIs at Beni Messous university hospital center in Algiers.
Recurrent and chronic infections, rather than atypical infections, are the main clinical feature of IEI in this cohort. Several cohort studies, including large IEI patients, indicated that the majority of patients had a history of repeated and/or persistent infections before a definitive diagnosis was made (54–56). In consistent with the reported studies, the present cohort revealed that more than 90% of IEI patients presented symptoms of recurrent infections before diagnosis. The most common infections are respiratory tract infections (lower 61.5% and upper 32%), followed by gastrointestinal tract infections (27.1%). It is not surprising that most of infectious agents invade the human body through the respiratory and digestive tract. Indeed, the mucosal surfaces of the body are particularly vulnerable to infection especially in patients with altered immunity such as people with IEI, cancer or HIV/AIDS, because mucosal surfaces especially bronchus-associated lymphoid tissue (BALT) or gastric-associated lymphoid tissue (GALT) are constantly exposed to pathogenic or nonpathogenic micro-organisms (57). This suggests that in such clinical manifestations, immunological assessment is required mainly when they occur early in life. Moreover, gastrointestinal diseases related to IEI are not only caused by infection, but also by inflammatory or autoimmune process, or malignancy. Sometimes, recurrent and/or persistent digestive symptom could be the unique presentation of IEI, thus it will be necessary to pay more attention of the possibility of IEI in patient with chronic diarrhea, growth retardation and failure to thrive especially the ones who do not respond to conventional therapy (58, 59). Most of localized and disseminated BCG infections have occurred in patients with combined immunodeficiency and with inborn defects of the IFNγ/IL12 axis which found to be in agreement with previously reported findings (60–62). As BCG vaccine is given to infants at birth in Algeria, serious adverse reactions after BCG vaccination may serve as a useful clinical signature to consider investigation of IEI in such children (63). BCG vaccination should be avoided if any family history of IEI or early deaths from infections or complication of tuberculosis. In addition to unusual increased susceptibility to infections, various noninfectious disorders can also occur in IEI patients such as autoimmune disorders and malignancies among others (64, 65). In this cohort, autoimmune diseases and lymphoproliferative disorders appeared to be more frequent in diseases of immune dysregulation. Studies of apoptosis and tolerance of T cells especially in patients with monogenic defects allowed us to understand self-tolerance, occurrence of lymphoproliferative syndrome, and autoimmune phenomena (66).
There were no facilities that offer regular genetic testing in Algeria, until recently, where our department was equipped with a platform of molecular genetics and cytogenetics, but these are not yet fully operational. Besides some research projects carried out locally, most molecular diagnostics were performed abroad, through international collaborations. Therefore, only 16% of all patients included in this study had a genetic diagnosis. This percentage is most similar to the Tunisian report (27), but lower compared to other reports (29, 36, 38, 41, 43). This could be due to the fact that the diagnosis is easily made based on clinical and laboratory findings, and identification of the genetic defect will not have any effect on the further treatment. On other hand, genetic testing was not carried out in some IEI such as common variable immunodeficiencies, selective IgA deficiency, and some other unclassified IEIs. This is partly due to the fact that genetic defects often go undetected, even using next generation sequencing methods (67). Unsurprisingly, the majority of the genetic disorders described have autosomal recessive inheritance and occur in the homozygous state for most of the respective mutations. This could be explained by the founder effect, known to underlie several IEI genes in highly consanguineous populations such as ours (68).
In this study, we reported mortality rate of 32.7%, with severe infections as the most leading causes of death. Overall, the mortality of our cohort is comparable to the findings of neighboring countries such as Morocco (31) and Tunisia (27), but it is much higher than to what was reported in European countries (38, 43, 45–47, 69–72) and Asian countries and regions (73–75) (Figure 7). Furthermore, Combined immunodeficiency is the category with the highest mortality rate (70.1%) which is very close to what was found in Kuwait 69% (29). This is due to the high incidence rate of SCID associated with a high frequency and severity of infections. Hence, these patients ultimately die generally during their first year of life, due to the lack of allogeneic stem cell transplantation or gene therapy in our country. The variability in mortality rates, reported in different reports, depends on the type of IEI most common in the patient’s country or region, and also on resource availability for early diagnosis (newborn screening), as well as the supply of proper treatment such as hematopoietic stem cell transplantation (HSCT) which is the mainstay of curative treatment in CID patients. There is little information on survival of patients with IEI. The management of these patients is challenging in developing countries, and this has resulted in high mortality rate. This study showed an overall 5-year survival rate of 77%, ranging from the highest in antibody deficiencies, at 98% and lowest in combined immunodeficiencies, at 42%. Similar findings have been shown in Oman (33) and Iran (41). Such findings are useful when describing resources for IEI patients, awareness raising, and its management.
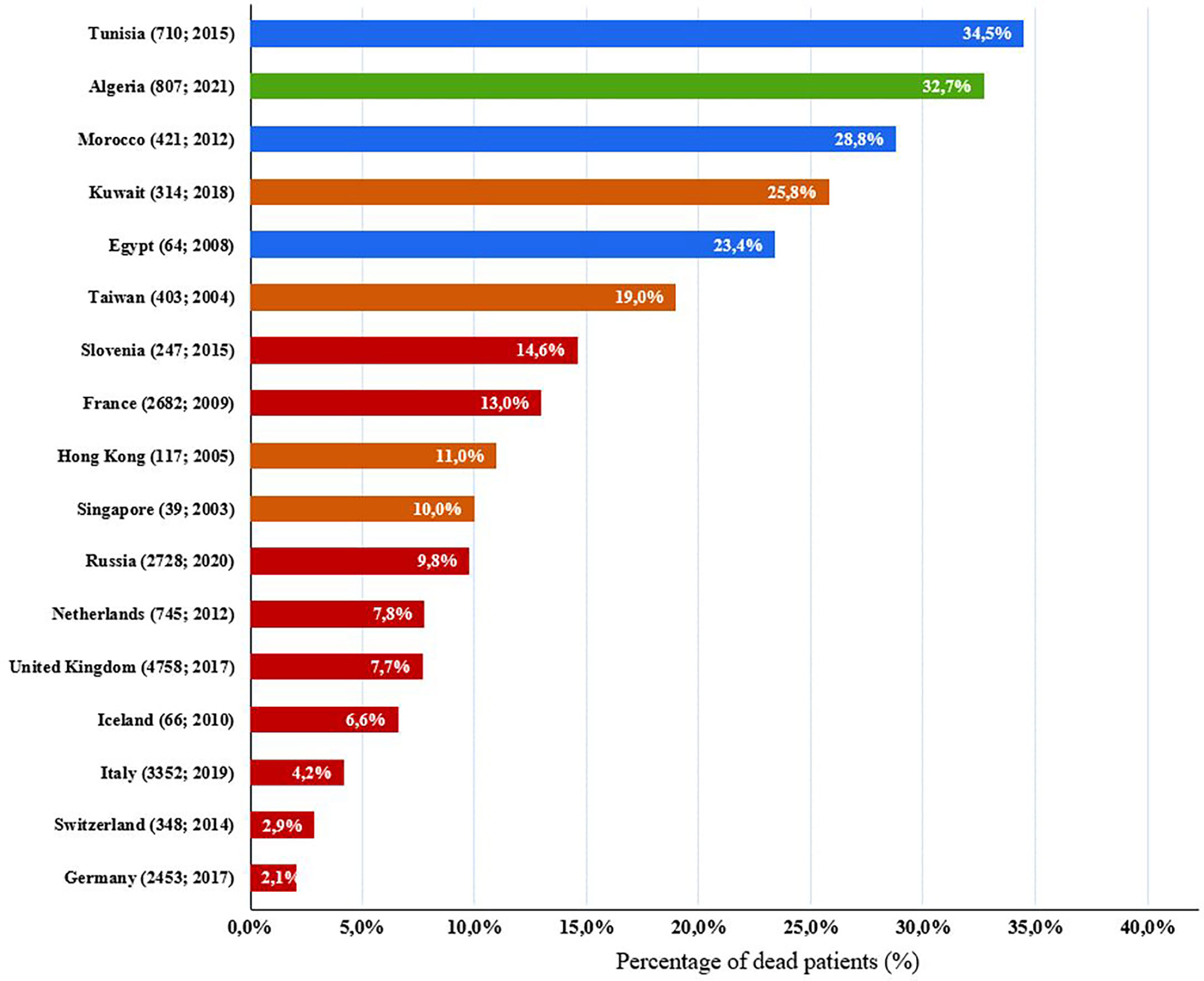
Figure 7 Comparison of IEI death percentages in our cohort with previously published IEI studies of other countries. Algeria: green; European countries: red; Asian countries and regions: brown, north African countries: blue. Parentheses next to each country name correspond to number of patients followed by year of analysis (number of patients; year of analysis).
Conclusion
Our study represents a sample of children and adult Algerian patients carrying a wide spectrum of IEI.
This monocentric cohort showed that IEI mostly appear at an early age, and recurrent/severe infections were the most common clinical manifestations in Algeria, and late diagnosis (or even misdiagnosis) can be disastrous and lead to higher morbidity and mortality.
The study confirmed, in a highly consanguineous population, the increased frequency of the autosomal recessive IEI, along with peculiar clinical features and genetic characteristics.
Increasing awareness and education of medical practitioners, expanding diagnostic tests and establishing a national hematopoietic stem cell transplantation (HSCT), are urgently needed to reduce the heavy burden of IEI in Algeria.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by The ethics committee of the Beni Messous university hospital center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contribution
BBe, RD conceptualized the study. BBe, LL-M, FM, LB, ZL, IA performed data analysis under supervision by RD. LB, K-WC, DL, AP, JC and YL performed genetic study. BBe drafted the manuscript. Other authors referred patients and provided clinical care and clinical data. All authors critically reviewed the manuscript and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patients who participated in the study and made this research study possible. We also would like to thank Prof. Raif Geha and Dr. Janet Chou for providing genetic analyses, and prof Mohamed Ridha Barbouche for his critical reading of the manuscript. Thanks to the Laboratory of Human Genetics of Infectious Diseases which is supported by the Howard Hughes Medical Institute, the Rockefeller University, the St. Giles Foundation, the National Institutes of Health (NIH) (P01AI06109, R01AI095983, R01AI127564, UL1TR001866), the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), ANR-GENMSMD (ANR-16-CE17-0005-01), ANR FNS LTh-MSMD-CMCD (ANR-18-CE93-0008-01), the French Foundation for Medical Research (FRM) (EQU201903007798), the European Union’s Horizon 2020 research and innovation programme under grant agreement No 824110 (EASI-genomics), the Square Foundation, Grandir - Fonds de solidarité pour l’enfance, the SCOR Corporate Foundation for Science, Institut National de la Santé et de la Recherche Médicale (INSERM), and the University Paris Cité.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.900091/full#supplementary-material
Abbreviations
ALPS, autoimmune lymphoproliferative syndrome; AD, Autosomal dominant; AR, Autosomal recessive; CD, cluster of differentiation; CHARGE, coloboma, choanal atresia, heart defects, genital abnormalities, retarded growth, and ear malformation; Chr, Chromosome; CID, combined immunodeficiency; CGD, chronic granulomatous disease; CVID, common variable immunodeficiency; BAFF-R, B cell-activating factor receptor; DHR, Dihydrorhodamin; DOCK8, Dedicator of cytokinesis 8; GOF, Gain of function; HIGMS, Hyper-IgM syndrome; HLA, Human leukocyte Antigen; HLH, hemophagocytic lymphohistiocytosis; IKBKB, Inhibitor Of Nuclear Factor Kappa B Kinase Subunit Beta; IUIS, International union of immunological societies; LAD, leukocyte Adhesion deficiency; LOF, loss of function; MENA, Middle East and North Africa.; MHC, major histocompatibility complex; MSMD, mendelian susceptibility to mycobacterial disease; NFKB, nuclear factor kappa-light-chain-enhancer of activated B cells; PID, primary immune deficiency; SCID, Severe combined immunodeficiency; XLA, X-linked agammaglobulinemia; XLT, X linked thrombocytopenia; WAS, Wiskott-Aldrich Syndrome; ZAP70, Zeta associated protein 70.
References
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
2. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40:66–81. doi: 10.1007/s10875-020-00758-x
3. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41:666–79. doi: 10.1007/s10875-021-00980-1
4. Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human Inborn Errors of Immunity: An Expanding Universe. Sci Immunol (2020) 5:eabb1662. doi: 10.1126/sciimmunol.abb1662
5. Notarangelo LD. Primary Immunodeficiencies. J Allergy Clin Immunol (2010) 125:S182–94. doi: 10.1016/j.jaci.2009.07.053
6. van Zelm MC, Condino-Neto A, Barbouche MR. Editorial: Primary Immunodeficiencies Worldwide. Front Immunol (2020) 10:3148. doi: 10.3389/fimmu.2019.03148
7. Casanova JL, Abel L. Mechanisms of Viral Inflammation and Disease in Humans. Science (2021) 374:1080–6. doi: 10.1126/SCIENCE.ABJ7965
8. Kerner G, Laval G, Patin E, Boisson-Dupuis S, Abel L, Casanova JL, et al. Human Ancient DNA Analyses Reveal the High Burden of Tuberculosis in Europeans Over the Last 2,000 Years. Am J Hum Genet (2021) 108:517–24. doi: 10.1016/j.ajhg.2021.02.009
9. Zhang Q, Bastard P. COVID Human Genetic Effort, Cobat A, Casanova J-L. Human Genetic and Immunological Determinants of Critical COVID-19 Pneumonia. Nature. (2022) 603 (7902):587–98. doi: 10.1038/s41586-022-04447-0
10. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, et al. Primary Immunodeficiency Diseases Worldwide: More Common Than Generally Thought. J Clin Immunol (2013) 33:1–7. doi: 10.1007/s10875-012-9751-7
11. Boyle JM, Buckley RH. Population Prevalence of Diagnosed Primary Immunodeficiency Diseases in the United States. J Clin Immunol (2007) 27:497–502. doi: 10.1007/s10875-007-9103-1
12. Aghamohammadi A, Rezaei N, Yazdani R, Delavari S, Kutukculer N, Topyildiz E, et al. Consensus Middle East and North Africa Registry on Inborn Errors of Immunity. J Clin Immunol (2021) 41:1339–51. doi: 10.1007/s10875-021-01053-z
13. Gathmann B, Grimbacher B, Beauté J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European Internet-Based Patient and Research Database for Primary Immunodeficiencies: Results 2006-2008. Clin Exp Immunol (2009) 157:3–11. doi: 10.1111/j.1365-2249.2009.03954.x
14. Lopes JP, Cunningham-Rundles C. The Importance of Primary Immune Deficiency Registries: The United States Immunodeficiency Network Registry. Immunol Allergy Clin North Am (2020) 40:385–402. doi: 10.1016/j.iac.2020.03.002
15. Leiva LE, Zelazco M, Oleastro M, Carneiro-Sampaio M, Condino-Neto A, Costa-Carvalho BT, et al. Primary Immunodeficiency Diseases in Latin America: The Second Report of the LAGID Registry. J Clin Immunol (2007) 27:101–8. doi: 10.1007/s10875-006-9052-0
16. Leung D, Chua GT, Mondragon AV, Zhong Y, Nguyen-Ngoc-Quynh L, Imai K, et al. Current Perspectives and Unmet Needs of Primary Immunodeficiency Care in Asia Pacific. Front Immunol (2020) 11:1605. doi: 10.3389/fimmu.2020.01605
17. Kirkpatrick P, Riminton S. Primary Immunodeficiency Diseases in Australia and New Zealand. J Clin Immunol (2007) 27:517–24. doi: 10.1007/s10875-007-9105-z
18. Barbouche MR, Eley B. Considerations for Primary Immune Deficiency Disorders in Africa and the Middle East. Stiehm’s Immune Defic (2014) 41:957–64. doi: 10.1016/B978-0-12-405546-9.00054-6
19. Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and Reproductive Health Among Arabs. Reprod Health (2009) 6:1–9. doi: 10.1186/1742-4755-6-17
20. Anwar WA, Khyatti M, Hemminki K. Consanguinity and Genetic Diseases in North Africa and Immigrants to Europe. Eur J Public Health (2014) 24:57–63. doi: 10.1093/eurpub/cku104
21. Birth Rate by Country 2021. Available at: https://worldpopulationreview.com/country-rankings/birth-rate-by-country (Accessed January 27, 2022).
22. Barbouche M-R, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary Immunodeficiencies in Highly Consanguineous North African Populations. Ann N Y Acad Sci (2011) 1238:42–52. doi: 10.1111/j.1749-6632.2011.06260.x
23. ESID - European Society for Immunodeficiencies. Available at: https://esid.org/Education/Diagnostic-Criteria-PID (Accessed March 2, 2021).
24. Miller SA, Dykes DD, Polesky HF. A Simple Salting Out Procedure for Extracting DNA From Human Nucleated Cells. Nucleic Acids Res (1988) 16:1215. doi: 10.1093/nar/16.3.1215
25. Casanova JL, Abel L. Lethal Infectious Diseases as Inborn Errors of Immunity: Toward a Synthesis of the Germ and Genetic Theories. Annu Rev Pathol Mech Dis (2021) 16:23–50. doi: 10.1146/annurev-pathol-031920-101429
26. Chi CY, Lin CH, Ho MW, Ding JY, Huang WC, Shih HP, et al. Clinical Manifestations, Course, and Outcome of Patients With Neutralizing Anti-Interferon-γ Autoantibodies and Disseminated Nontuberculous Mycobacterial Infections. Med (United States) (2016) 95:e392. doi: 10.1097/MD.0000000000003927
27. Mellouli F, Ben MI, Ben KM, Besbes H, Ouederni M, Mekki N, et al. Report of the Tunisian Registry of Primary Immunodeficiencies: 25-Years of Experience (1988–2012). J Clin Immunol (2015) 35:745–53. doi: 10.1007/s10875-015-0206-9
28. Al-Saud B, Al-Mousa H, Al Gazlan S, Al-Ghonaium A, Arnaout R, Al-Seraihy A, et al. Primary Immunodeficiency Diseases in Saudi Arabia: A Tertiary Care Hospital Experience Over a Period of Three Years (2010–2013). J Clin Immunol (2015) 35:651–60. doi: 10.1007/s10875-015-0197-6
29. Al-Herz W, Al-Ahmad M, Al-Khabaz A, Husain A, Sadek A, Othman Y. The Kuwait National Primary Immunodeficiency Registry 2004–2018. Front Immunol (2019) 10:1754. doi: 10.3389/fimmu.2019.01754
30. Galal N, Meshaal S, Elhawary R, ElAziz DA, Alkady R, Lotfy S, et al. Patterns of Primary Immunodeficiency Disorders Among a Highly Consanguineous Population: Cairo University Pediatric Hospital’s 5-Year Experience. J Clin Immunol (2016) 36:649–55. doi: 10.1007/s10875-016-0314-1
31. Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, et al. First Report on the Moroccan Registry of Primary Immunodeficiencies: 15 Years of Experience (1998-2012). J Clin Immunol (2014) 34:459–68. doi: 10.1007/s10875-014-0005-8
32. Ishimura M, Takada H, Doi T, Imai K, Sasahara Y, Kanegane H, et al. Nationwide Survey of Patients With Primary Immunodeficiency Diseases in Japan. J Clin Immunol (2011) 31:968–76. doi: 10.1007/s10875-011-9594-7
33. Al-Tamemi S, Naseem SUR, Al-Siyabi N, El-Nour I, Al-Rawas A, Dennison D. Primary Immunodeficiency Diseases in Oman: 10-Year Experience in a Tertiary Care Hospital. J Clin Immunol (2016) 36:785–92. doi: 10.1007/s10875-016-0337-7
34. Rhim JW, Kim KH, Kim DS, Kim BS, Kim JS, Kim CH, et al. Prevalence of Primary Immunodeficiency in Korea. J Korean Med Sci (2012) 27:788–93. doi: 10.3346/jkms.2012.27.7.788
35. Wang LL, Jin YY, Hao YQ, Wang JJ, Yao CM, Wang X, et al. Distribution and Clinical Features of Primary Immunodeficiency Diseases in Chinese Children (2004-2009). J Clin Immunol (2011) 31:297–308. doi: 10.1007/s10875-010-9493-3
36. Ehlayel MS, Bener A, Laban MA. Primary Immunodeficiency Diseases in Children: 15 Year Experience in a Tertiary Care Medical Center in Qatar. J Clin Immunol (2013) 33:317–24. doi: 10.1007/s10875-012-9812-y
37. ESID - European Society for Immunodeficiencies. Available at: https://esid.org/Working-Parties/Registry-Working-Party/ESID-Registry (Accessed December 25, 2021).
38. Mukhina AA, Kuzmenko NB, Rodina YA, Kondratenko IV, Bologov AA, Latysheva TV, et al. Primary Immunodeficiencies in Russia: Data From the National Registry. Front Immunol (2020) 11:1491. doi: 10.3389/fimmu.2020.01491
39. Kilic SS, Ozel M, Hafizoglu D, Karaca NE, Aksu G, Kutukculer N. The Prevalances and Patient Characteristics of Primary Immunodeficiency Diseases in Turkeytwo Centers Study. J Clin Immunol (2013) 33:74–83. doi: 10.1007/s10875-012-9763-3
40. Reda SM, Afifi HM, Amine MM. Primary Immunodeficiency Diseases in Egyptian Children: A Single-Center Study. J Clin Immunol (2009) 29:343–51. doi: 10.1007/s10875-008-9260-x
41. Abolhassani H, Kiaee F, Tavakol M, Chavoshzadeh Z, Mahdaviani SA. Fourth Update on the Iranian National Registry of Primary Immunodeficiencies: Integration of Molecular Diagnosis J Clin Immunol (2018) 38:816–32. doi: 10.1007/s10875-018-0556-1.
42. Wu J, Zhong W, Yin Y, Zhang H. Primary Immunodeficiency Disease: A Retrospective Study of 112 Chinese Children in a Single Tertiary Care Center. BMC Pediatr (2019) 19:1–7. doi: 10.1186/s12887-019-1729-7
43. The C, Pid F. The French National Registry of Primary Immunodeficiency Diseases. Clin Immunol (2010) 135:264–72. doi: 10.1016/j.clim.2010.02.021
44. Naidoo R, Ungerer L, Cooper M, Pienaar S, Eley BS. Primary Immunodeficiencies: A 27-Year Review at a Tertiary Paediatric Hospital in Cape Town, South Africa. J Clin Immunol (2011) 31:99–105. doi: 10.1007/s10875-010-9465-7
45. Lougaris V, Pession A, Baronio M, Soresina A, Rondelli R, Gazzurelli L, et al. The Italian Registry for Primary Immunodeficiencies (Italian Primary Immunodeficiency Network; IPINet): Twenty Years of Experience (1999–2019). J Clin Immunol (2020) 40:1026–37. doi: 10.1007/s10875-020-00844-0
46. Shillitoe B, Bangs C, Guzman D, Gennery AR, Longhurst HJ, Slatter M, et al. The United Kingdom Primary Immune Deficiency (UKPID) Registry 2012 to 2017. Clin Exp Immunol (2018) 192:284–91. doi: 10.1111/cei.13125
47. El-Helou SM, Biegner AK, Bode S, Ehl SR, Heeg M, Maccari ME, et al. The German National Registry of Primary Immunodeficiencies (2012–2017). Front Immunol (2019) 10:1272. doi: 10.3389/fimmu.2019.01272
48. Zebracki K, Palermo TM, Hostoffer R, Duff K, Drotar D. Health-Related Quality of Life of Children With Primary Immunodeficiency Disease: A Comparison Study. Ann Allergy Asthma Immunol (2004) 93:557–61. doi: 10.1016/S1081-1206(10)61263-X
49. Abolhassani H, Azizi G, Sharifi L, Yazdani R, Mohsenzadegan M, Delavari S, et al. Global Systematic Review of Primary Immunodeficiency Registries. Expert Rev Clin Immunol (2020) 16:717–32. doi: 10.1080/1744666X.2020.1801422
50. Grimbacher B. The European Society for Immunodeficiencies (ESID) Registry 2014. Clin Exp Immunol (2014) 178:18–20. doi: 10.1111/cei.12496
51. Kobrynski L, Powell RW, Bowen S. Prevalence and Morbidity of Primary Immunodeficiency Diseases, United States 2001–2007. J Clin Immunol (2014) 34:954–61. doi: 10.1007/s10875-014-0102-8
52. Darr A, Small N, Ahmad WIU, Atkin K, Corry P, Modell B. Addressing Key Issues in the Consanguinity-Related Risk of Autosomal Recessive Disorders in Consanguineous Communities: Lessons From a Qualitative Study of British Pakistanis. J Community Genet (2016) 7:65–79. doi: 10.1007/s12687-015-0252-2
53. Yagoubi A. Algerian Registry for Inborn Errors of Immunity in Children: Report of 887 Children (1985 - 2021). Res Square (2021) 1–18. doi: 10.21203/rs.3.rs-1287012/v1
54. Modell V, Quinn J, Orange J, Notarangelo LD, Modell F. Primary Immunodeficiencies Worldwide: An Updated Overview From the Jeffrey Modell Centers Global Network. Immunol Res (2016) 64:736–53. doi: 10.1007/s12026-016-8784-z
55. Reisi M, Azizi G, Kiaee F, Masiha F, Shirzadi R, Momen T, et al. Evaluation of Pulmonary Complications in Patients With Primary Immunodeficiency Disorders. Eur Ann Allergy Clin Immunol (2017) 49:122–8.
56. Barlogis V, Mahlaoui N, Auquier P, Fouyssac F, Pellier I, Vercasson C, et al. Burden of Poor Health Conditions and Quality of Life in 656 Children With Primary Immunodeficiency. J Pediatr (2018) 194:211–7.e5. doi: 10.1016/j.jpeds.2017.10.029
57. Randall TD, Mebius RE. The Development and Function of Mucosal Lymphoid Tissues: A Balancing Act With. Mucosal Immunol (2014) 7:455–66. doi: 10.1038/mi.2014.11
58. Palareti G, Legnani C, Cosmi B, Antonucci E, Erba N, Poli D, et al. Comparison Between Different D-Dimer Cutoff Values to Assess the Individual Risk of Recurrent Venous Thromboembolism: Analysis of Results Obtained in the DULCIS Study. Int J Lab Hematol (2016) 38:42–9. doi: 10.1111/ijlh.12426
59. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-Term Follow-Up of IPEX Syndrome Patients After Different Therapeutic Strategies: An International Multicenter Retrospective Study. J Allergy Clin Immunol (2018) 141:1036–49.e5. doi: 10.1016/j.jaci.2017.10.041
60. Al-Hammadi S, Alsuwaidi AR, Alshamsi ET, Ghatasheh GA, Souid AK. Disseminated Bacillus Calmette-Guérin (BCG) Infections in Infants With Immunodeficiency. BMC Res Notes (2017) 10:1–5. doi: 10.1186/s13104-017-2499-7
61. Norouzi S, Aghamohammadi A, Mamishi S, Rosenzweig SD, Rezaei N. Bacillus Calmette-Guérin (BCG) Complications Associated With Primary Immunodeficiency Diseases. J Infect (2012) 64:543–54. doi: 10.1016/j.jinf.2012.03.012
62. Marciano BE, Huang CY, Joshi G, Rezaei N, Carvalho BC, Allwood Z, et al. BCG Vaccination in Patients With Severe Combined Immunodeficiency: Complications, Risks, and Vaccination Policies. J Allergy Clin Immunol (2014) 133:1134–41. doi: 10.1016/j.jaci.2014.02.028
63. Bonilla FA. Update: Vaccines in Primary Immunodeficiency. J Allergy Clin Immunol (2018) 141:474–81. doi: 10.1016/j.jaci.2017.12.980
64. Bonilla FA, Geha RS. 12. Primary Immunodeficiency Diseases. J Allergy Clin Immunol (2003) 111:571–81. doi: 10.1067/mai.2003.86
65. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and Primary Immunodeficiency: Two Sides of the Same Coin? Nat Rev Rheumatol (2018) 14:7–18. doi: 10.1038/nrrheum.2017.198
66. Yamashita M, Inoue K, Okano T, Morio T. Inborn Errors of Immunity—Recent Advances in Research on the Pathogenesis. Inflamm Regener (2021) 41. doi: 10.1186/s41232-021-00159-6
67. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, Azucena González-Navarro E, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
68. Barbouche MR, Mekki N, Ben-Ali M, Ben-Mustapha I. Lessons From Genetic Studies of Primary Immunodeficiencies in a Highly Consanguineous Population. Front Immunol (2017) 8:737. doi: 10.3389/fimmu.2017.00737
69. Marschall K, Hoernes M, Bitzenhofer-Grüber M, Jandus P, Duppenthaler A, Wuillemin WA, et al. The Swiss National Registry for Primary Immunodeficiencies: Report on the First 6 Years’ Activity From 2008 to 2014. Clin Exp Immunol (2015) 182:45–50. doi: 10.1111/cei.12661
70. Ludviksson BR, Sigurdardottir ST, Johannsson JH, Haraldsson A, Hardarson TO. Epidemiology of Primary Immunodeficiency in Iceland. J Clin Immunol (2015) 35:75–9. doi: 10.1007/s10875-014-0107-3
71. Jonkman-Berk BM, van den Berg JM, ten Berge IJM, Bredius RGM, Driessen GJ, Dalm VASH, et al. Primary Immunodeficiencies in the Netherlands: National Patient Data Demonstrate the Increased Risk of Malignancy. Clin Immunol (2015) 156:154–62. doi: 10.1016/j.clim.2014.10.003
72. Blazina Š, Markelj G, Debeljak M, Jeverica AK, Toplak N, Bratanič N, et al. Slovenski Nacionalni Register Bolnikov s Primarno Imunsko Pomanjkljivostjo. Zdr Vestn (2015) 84:797–808. doi: 10.6016/zdravvestn.1321
73. Lim DLC, Thong BY, Ho SY, Shek LPC, Lou J, Leong KP, et al. Primary Immunodeficiency Diseases in Singapore - The Last 11 Years. Singapore Med J (2003) 44:579–86.
74. Lam DST, Lee TL, Chan KW, Ho HK, Lau YL. Primary Immunodeficiency in Hong Kong and the Use of Genetic Analysis for Diagnosis. Hong Kong Med J (2005) 11:90–6.
Keywords: primary immunodeficiency, inborn errors of immunity, Algeria, epidemiology, molecular diagnosis, clinical features, diagnosis
Citation: Belaid B, Lamara Mahammed L, Drali O, Oussaid AM, Touri NS, Melzi S, Dehimi A, Berkani LM, Merah F, Larab Z, Allam I, Khemici O, Kirane SY, Boutaba M, Belbouab R, Bekkakcha H, Guedouar A, Chelali A, Baamara B, Noui D, Baaziz H, Rezak R, Azzouz SM, Aichaoui M, Moktefi A, Benhatchi RM, Oussalah M, Benaissa N, Laredj A, Bouchetara A, Adria A, Habireche B, Tounsi N, Dahmoun F, Touati R, Boucenna H, Bouferoua F, Sekfali L, Bouhafs N, Aboura R, Kherra S, Inouri Y, Dib S, Medouri N, Khelfaoui N, Redjedal A, Zelaci A, Yahiaoui S, Medjadj S, Touhami TK, Kadi A, Amireche F, Frada I, Houasnia S, Benarab K, Boubidi C, Ferhani Y, Benalioua H, Sokhal S, Benamar N, Aggoune S, Hadji K, Bellouti A, Rahmoune H, Boutrid N, Okka k, Ammour A, Saadoune H, Amroun M, Belhadj H, Ghanem A, Abbaz H, Boudrioua S, Zebiche B, Ayad A, Hamadache Z, Ouaras N, Achour N, Bouchair N, Boudiaf H, Bekkat-Berkani D, Maouche H, Bouzrar Z, Aissat L, Ibsaine O, Bioud B, Kedji L, Dahlouk D, Bensmina M, Radoui A, Bessahraoui M, Bensaadi N, Mekki A, Zeroual Z, Chan K-W, Leung D, Tebaibia A, Ayoub S, Mekideche D, Gharnaout M, Casanova JL, Puel A, Lau YL, Cherif N, Ladj S, Smati L, Boukari R, Benhalla N and Djidjik R (2022) Inborn Errors of Immunity in Algerian Children and Adults: A Single-Center Experience Over a Period of 13 Years (2008–2021). Front. Immunol. 13:900091. doi: 10.3389/fimmu.2022.900091
Received: 19 March 2022; Accepted: 25 March 2022;
Published: 21 April 2022.
Edited by:
Waleed Al-Herz, Kuwait University, KuwaitReviewed by:
Kathleen Sullivan, Children’s Hospital of Philadelphia, United StatesJavier Chinen, Baylor College of Medicine, United States
Copyright © 2022 Belaid, Lamara Mahammed, Drali, Oussaid, Touri, Melzi, Dehimi, Berkani, Merah, Larab, Allam, Khemici, Kirane, Boutaba, Belbouab, Bekkakcha, Guedouar, Chelali, Baamara, Noui, Baaziz, Rezak, Azzouz, Aichaoui, Moktefi, Benhatchi, Oussalah, Benaissa, Laredj, Bouchetara, Adria, Habireche, Tounsi, Dahmoun, Touati, Boucenna, Bouferoua, Sekfali, Bouhafs, Aboura, Kherra, Inouri, Dib, Medouri, Khelfaoui, Redjedal, Zelaci, Yahiaoui, Medjadj, Touhami, Kadi, Amireche, Frada, Houasnia, Benarab, Boubidi, Ferhani, Benalioua, Sokhal, Benamar, Aggoune, Hadji, Bellouti, Rahmoune, Boutrid, Okka, Ammour, Saadoune, Amroun, Belhadj, Ghanem, Abbaz, Boudrioua, Zebiche, Ayad, Hamadache, Ouaras, Achour, Bouchair, Boudiaf, Bekkat-Berkani, Maouche, Bouzrar, Aissat, Ibsaine, Bioud, Kedji, Dahlouk, Bensmina, Radoui, Bessahraoui, Bensaadi, Mekki, Zeroual, Chan, Leung, Tebaibia, Ayoub, Mekideche, Gharnaout, Casanova, Puel, Lau, Cherif, Ladj, Smati, Boukari, Benhalla and Djidjik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reda Djidjik, b3VydGlsYW5lQHlhaG9vLmZy