 Dan Sexton
Dan Sexton Anton Kichev
Anton Kichev Salomé Juethner3
Salomé Juethner3 Dave Yeung
Dave Yeung Ezequiel Anokian
Ezequiel Anokian Bin Li
Bin Li- 1Takeda Development Center Americas, Inc., Cambridge, MA, United States
- 2Clarivate PLC, Barcelona, Spain
- 3Takeda Pharmaceuticals USA Inc., Lexington, MA, United States
Introduction: Plasma proteomics analyses were performed to identify novel disease state biomarkers of hereditary angioedema due to C1 inhibitor deficiency (HAE-C1INH) and investigate the biological consequences of specific plasma kallikrein inhibition with lanadelumab.
Methods: Affinity proteomic analyses were performed using plasma from healthy controls (n=30) and patients with HAE-C1INH before (baseline, n=125) and after 6 months of treatment with lanadelumab (300 mg every 2 weeks, n=112) using the SomaScan platform.
Results: Relative plasma levels for several proteins differed significantly between controls and patients with HAE-C1INH, and between matched baseline and post-treatment samples from patients with HAE-C1INH. As expected, C1 inhibitor and complement C4 were significantly lower (P<1.10e-39 false discovery rate [fdr], P<6.6e-25 fdr, respectively) in HAE-C1INH baseline plasma versus controls. Cleaved high-molecular-weight kininogen, a biomarker of excess kallikrein-kinin system (KKS) activation, was higher in HAE-C1INH baseline plasma versus controls (P<6.7e-6 fdr) and was reduced in HAE-C1INH plasma after lanadelumab treatment. Of 1041 identified proteins that differed significantly (P<0.05) from controls and HAE-C1INH baseline plasma, 120 proteins were no longer different between controls and patients with HAE-C1INH after 6 months of lanadelumab treatment. Canonical pathway and local network analyses of HAE-C1INH plasma proteomics suggest dysregulation in KKS, coagulation, cell adhesion, and connective tissue degradation that approach that of healthy controls following treatment with lanadelumab.
Conclusion: Proteomic analyses of plasma from patients with HAE-C1INH before and after treatment with lanadelumab compared with healthy controls confirmed known HAE-C1INH biomarkers and identified additional potential biomarkers of plasma kallikrein dysregulation for further investigation.
Introduction
Hereditary angioedema due to C1INH deficiency (HAE-C1INH) is an autosomal dominant genetic disease mediated by a dysregulated plasma kallikrein-kinin system (KKS), which generates excess bradykinin in the vascular compartment; pathophysiology that causes episodic attacks of angioedema (1). KKS activation occurs upon FXII activation to FXIIa, which converts prekallikrein to active plasma kallikrein (PKa), that activates additional FXII and cleaves high-molecular-weight kininogen (HK) to generate cleaved HK (HKa) and bradykinin (1, 2).
Despite the well described pathophysiological role of the KKS and the multitude of approved treatments for HAE-C1INH, the search for novel biomarkers (3) remains in order to elucidate further biological consequences of excess KKS activation, develop improved therapies, and reduce diagnosis time for patients, which for many patients can take 4–9 years on average (4), if the identified biomarkers can be developed as diagnostic assays.
Lanadelumab (Takhzyro, TAK-743, SHP643, DX-2930) is a specific antibody inhibitor of PKa approved to prevent attacks in patients with HAE (5). We performed plasma proteomics with samples from HAE-C1INH patients before and after treatment with lanadelumab to assess the impact of specific PKa inhibition on the plasma proteome.
Methods
Plasma was collected from patients with HAE-C1INH enrolled in the phase III HELP study for lanadelumab as well as from patients who only enrolled into the open-label extension portion of the HELP study (ClinicalTrials.gov ID: NCT02586805 and NCT02741596, respectively). Plasma was collected from age- and gender-matched healthy controls (n = 30) by BioIVT (Westbury, NY). All participants provided written informed consent for blood samples to be used for the investigation of exploratory biomarkers of contact system activation. To minimize ex vivo activation of the contact pathway system during blood collection, plasma was collected from patients with HAE-C1INH and healthy controls by means of a clean venipuncture with a butterfly needle/catheter kit (BD Biosciences part number 367296, San Jose, CA, USA) and removal of the tourniquet upon blood flow to decrease stasis. The first tube of blood was discarded and blood was collected into polypropylene evacuated tubes containing 3.2% sodium citrate (BD Biosciences). Blood samples were centrifuged within 1 hour, and plasma was aliquoted and stored at –80°C until processing.
Plasma proteomic analyses were performed using a multiplex approach that compares relative levels of >7000 proteins (SomaScan, Somalogic, Denver, CO, USA) (6) on the following 3 groups: 1) healthy control plasma (n = 30); 2) HAE-C1INH plasma before lanadelumab treatment (baseline, n = 125); and 3) HAE-C1INH plasma after 26 weeks of lanadelumab treatment (300 mg Q2W, after treatment, n = 114). Of the 114 HAE-C1INH patients for which baseline plasma was available, matched post-treatment samples were available for 112 patients.
Results
We compared plasma proteomics of HAE-C1INH plasma at baseline (i.e., before treatment with lanadelumab) to that of healthy control plasma in a volcano plot of P-value versus fold change (Figure 1). Out of the proteins detected, 1041 proteins were statistically different between HAE-C1INH and healthy controls (Supplementary Table 1).
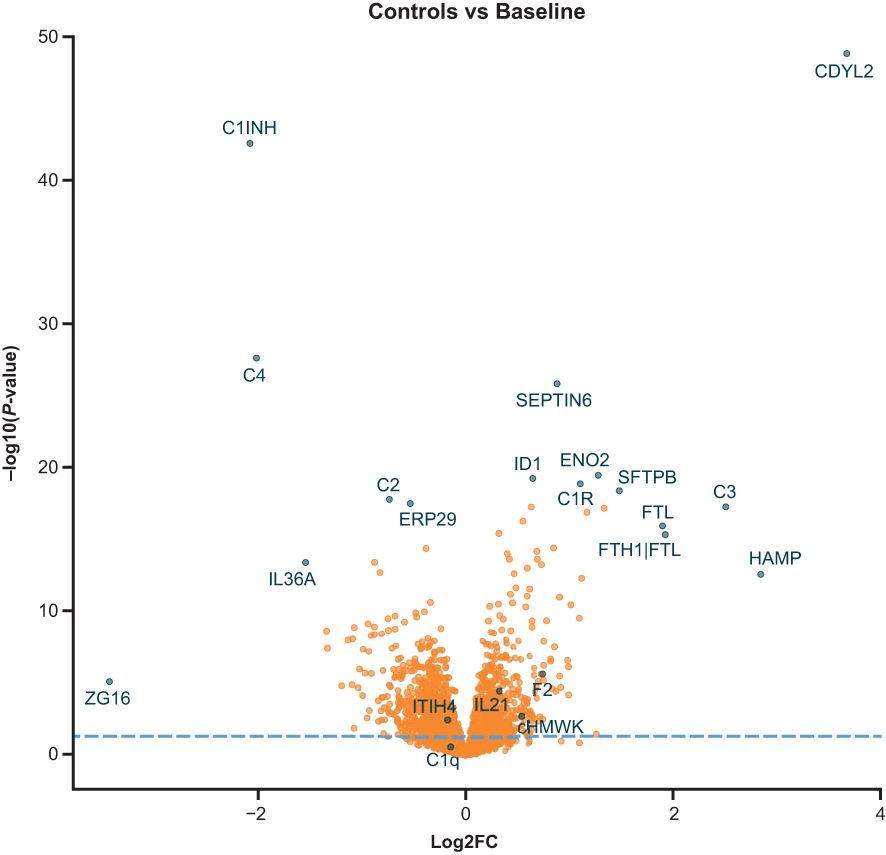
Figure 1. Volcano plot comparison of protein levels from SomaScan analyses in plasma from patients with HAE-C1INH that was collected at baseline to levels present in plasma from age- and gender-matched healthy controls. The x-axis is the fold change (log2FC) in signal for each protein in HAE-C1INH plasma as compared to healthy control plasma. The y-axis is the calculated P-value (Log10) for the difference in signal for each protein between HAE-C1INH patients and healthy controls plasma. HAE-C1INH, hereditary angioedema due to C1 inhibitor deficiency.
Of the 1041 proteins, C4 and C1INH were lower in HAE-C1INH baseline plasma than healthy control plasma (Figure 2), which was expected since low levels of both proteins are used in the diagnosis of HAE-C1INH (7). HAE-C1INH may be associated with C1INH plasma levels that are <50% of a healthy control standard at 1 U/mL (~0.24 g/L) and C4 plasma concentrations below that of healthy controls (8, 9).
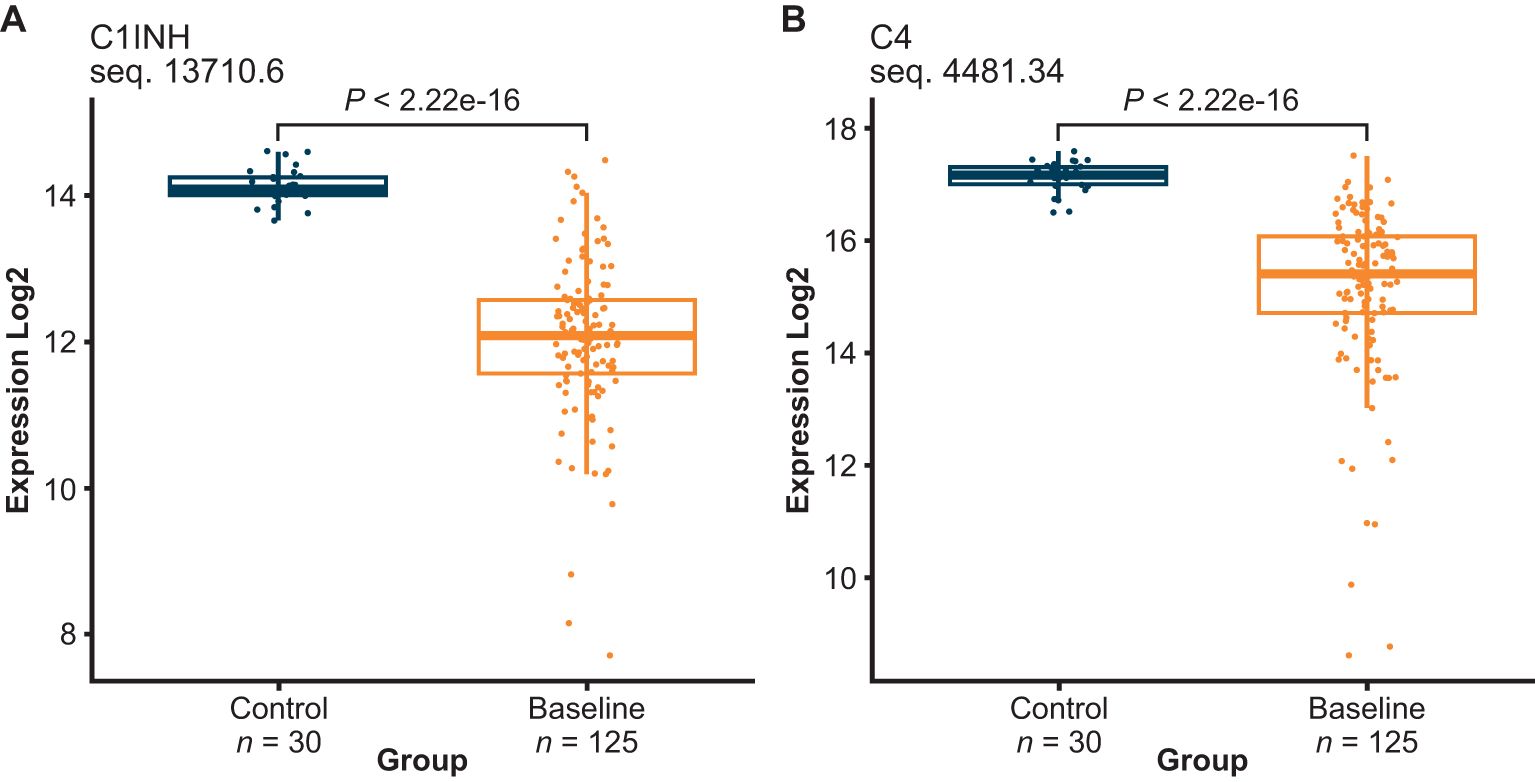
Figure 2. C1INH (A) and complement 4 (C4) (B) plasma levels were both decreased in HAE-C1INH samples collected at baseline as compared to healthy control plasma. HAE-C1INH, hereditary angioedema due to C1 inhibitor deficiency.
We also compared plasma proteomics between healthy controls, baseline HAE-C1INH, and HAE-C1INH after 26 weeks of lanadelumab treatment. Out of the 1041 proteins that were different between healthy controls and HAE-C1INH baseline, 120 proteins were observed to be no longer different from healthy controls after HAE-C1INH patients were treated with lanadelumab for 26 weeks. The list of the 120 proteins is provided in Supplementary Table 1 and includes HKa, thrombin, tissue kallikrein 14, interleukin-21, α-2-macroglobulin (A2M), and apolipoprotein B (Supplementary Figure 1).
The first one of these 120 potential biomarkers to highlight is HKa, a protein previously shown to be elevated in plasma of patients with HAE-C1INH and a pharmacodynamic biomarker of lanadelumab bioactivity (10–12). Figure 3 shows the HKa signal measured using SOMAmer sequence 19631-13, which shows that HKa levels were elevated in HAE-C1INH baseline plasma, but were no longer different from healthy control plasma levels after HAE-C1INH patients received lanadelumab for 26 weeks. HKa levels measured using sequence 19631-13 positively correlated to the %HKa measured by Western blot analyses performed during the clinical study with lanadelumab (13). The SomaScan panel contains 3 other SOMAmers against kininogen (15343-337, 7784-1, and 4918-21). The profile for 15343-337 appeared similar to 19631-13, whereas the profile of 7784-1 suggests preferential binding to intact HK, and the profile of 4918-21 suggests that it may bind both HK and HKa (Supplementary Figures 2, 3). Furthermore, the observed reduction in HKa signal using SOMAmers 19631-13 and 15343-337 in paired HAE-C1INH patient samples collected at baseline and after 26 weeks of lanadelumab treatment is consistent with the pharmacodynamic activity previously reported for this PKa inhibitor (Supplementary Figure 4) (14).
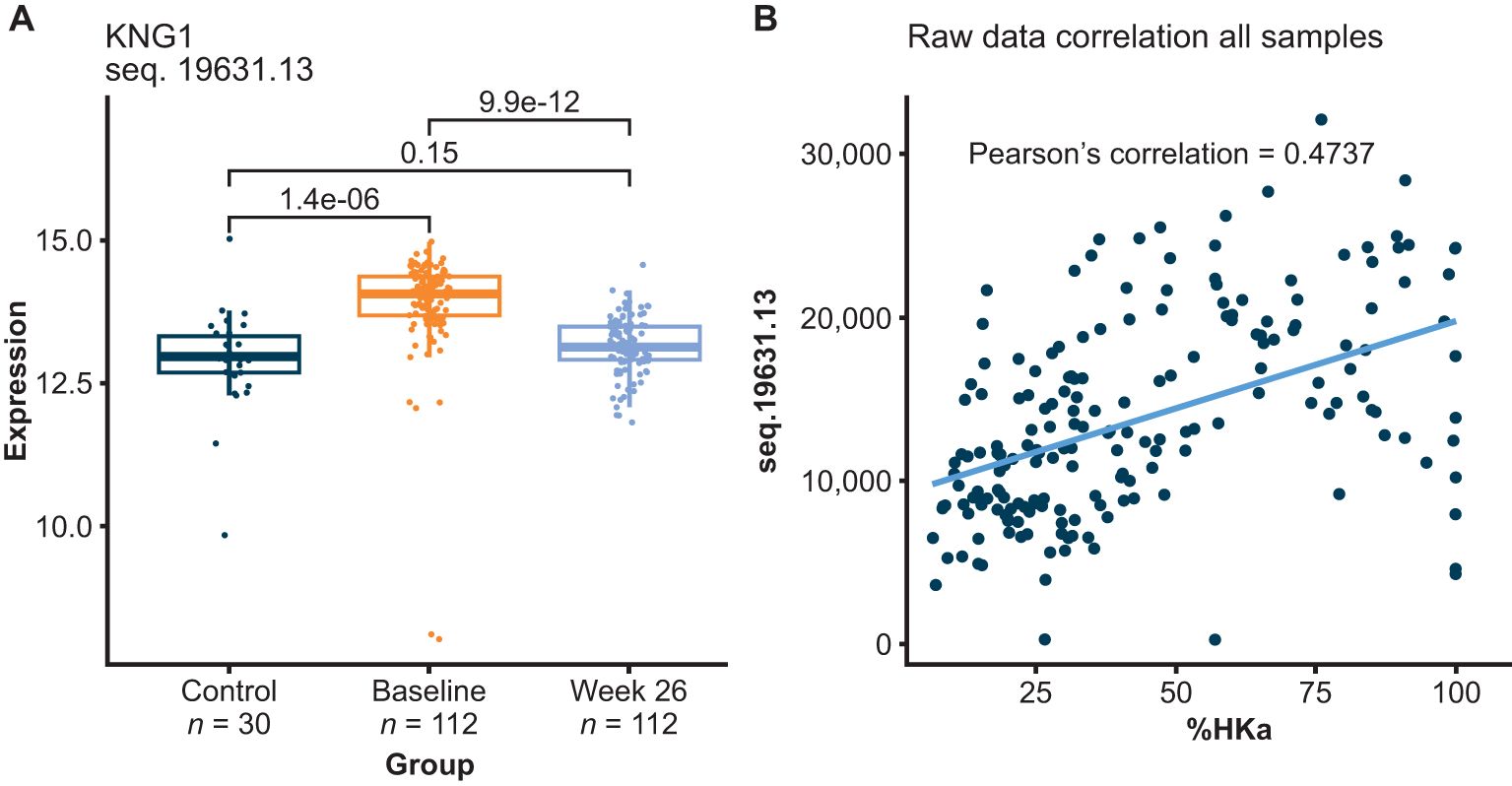
Figure 3. HKa relative levels measured using SOMAmer 19631-13 increased in plasma from HAE-C1INH patients prior to receiving lanadelumab (baseline), relative to healthy controls (Control) and HKa decreased following lanadelumab treatment (A). Correlation between HKa levels as measured using SOMAmer 19631-13 and Western blot analyses of HK (B). SOMAmer 19631-13 was reported by the manufacturer to be raised against kininostatin (domain 5 of HK) with binding observed to HKa (although with~10-folder weaker affinity than for kininostatin) and no binding observed with intact HK or intact LK. HAE-C1INH, hereditary angioedema due to C1 inhibitor deficiency; HK, high-molecular-weight kininogen; HKa, cleaved HK; LK, low-molecular-weight kininogen.
We next investigated whether additional potential biomarkers from the 120 proteins discovered previously would show measurable protein differences with commercially available ELISA kits. To test this, additional plasma samples (from different HAE-C1INH patients from the same clinical study with lanadelumab and different healthy controls) were analyzed using commercially available ELISA kits (RayBiotech, Peachtree Corners, GA) for interleukin-21, A2M, and apolipoprotein B. We observed no differences between healthy controls and HAE-C1INH plasma for these 3 proteins (data not shown). Differences between results obtained using affinity proteomic methods, including SomaScan, and immunoassays have been previously reported and potentially attributed to differences in the epitopes or protein complexes targeted between the different assays (15). Furthermore, targeted immunoassays often disagree with each other, so disagreement with a proteomic platform does not definitively imply which result is superior. Consequently, the lack of concordance between methods does not demonstrate that the results from the affinity proteomic analyses are necessarily incorrect and require further validation (e.g., using assays based on the specific SOMAmers used for each protein).
We next performed pathway enrichment analysis on these 120 altered proteins using Metacore (Clarivate). As shown in Table 1, the KKS is the most significant enriched pathway, which is consistent with the fact that this is a study on patients with HAE-C1INH and the effects of their treatment. Other enriched pathways include blood coagulation, cell adhesion, and connective tissue degradation. These 120 proteins were then analyzed using a known knowledge network approach called Causal-ASsociational NETwork (CASNET), which refers to consistently active subnetworks of known protein-protein interactions (16). Finding active subnetworks in diseases can be a way of generating biological insights, including novel feedback loops, using data with differentially expressed genes or in our case different protein expression levels from proteomic analyses (Supplementary Figure 5). Proteins identified by CASnet analyses out of the list of 120 proteins include proteases (thrombin, tissue kallikreins, cathepsin K, plasminogen) protease inhibitors (inter-alpha trypsin inhibitor heavy chain 4, A2M), apolipoproteins, and complement system proteins (Supplementary Table 2, Supplementary Figure 5).
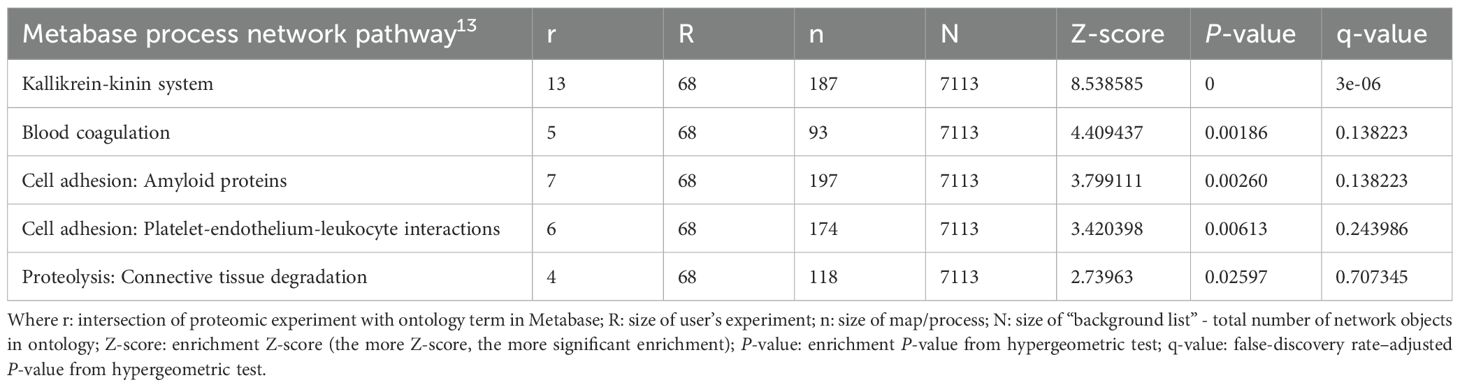
Table 1. Pathway analysis using the 120 proteins.
Discussion
Biomarker discovery in HAE-C1INH remains an active area of research that can provide novel insights into pathophysiology and may identify novel biomarkers to potentially improve diagnosis (3, 17–20). We used an affinity proteomic platform capable of measuring relative amounts of more than 7000 different human proteins to compare the plasma proteome of patients with HAE-C1INH to that of healthy controls. In addition, we investigated the effect of PKa inhibition using a highly specific antibody inhibitor (lanadelumab) on the plasma proteome of patients with HAE-C1INH.
SomaScan proteomics includes the known diagnostic biomarkers of HAE-C1INH, C1INH and C4, both of which were lower than healthy controls as expected. In addition, SomaScan proteomics measures HKa, a previously identified disease state and pharmacodynamic biomarker of KKS activation (10–12). C1INH, C4, and HKa levels in HAE-C1INH plasma measured using SomaScan proteomics indicate that this technology could be considered for investigations of HAE-C1INH plasma biomarkers and possibly as a pharmacodynamic assay for the investigation of novel therapies targeting the KKS.
By comparing the HAE-C1INH plasma proteome before and after 26 weeks of treatment with lanadelumab to that of healthy controls, we were able to investigate the effect of specific PKa inhibition on the 1041 proteins with levels that differed between HAE-C1INH plasma baseline and healthy controls. These 1041 proteins are listed in the Supplementary Table 1 and may find use in further studies into the biological consequences of excess KKS activation in HAE-C1INH and other diseases potentially mediated by the KKS, including comorbidities associated with HAE-C1INH (21–23). From this analysis, we identified 120 out of the 1041 proteins that were no longer different from that of healthy controls after 26 weeks of lanadelumab treatment.
A2M, one of these 120 proteins, is a broad-spectrum covalent protein inhibitor of proteases, including PKa, which has multiple functions, including binding and regulating pro-inflammatory cytokines and hormones (24). A2M has been investigated as a biomarker for a number of different diseases, including diabetes mellitus (25, 26). We observed that A2M was elevated in HAE-C1INH plasma relative to that of healthy controls and returned to approximate healthy control levels after 26 weeks of treatment with lanadelumab (Supplementary Figure 1). Further studies could elucidate whether the SOMAmer against A2M also binds the A2M-PKa covalent complex, which has been shown to be elevated during an attack in plasma from patients with HAE-C1INH (27).
Complement protein C3 is another one of 120 altered proteins that has been implicated in the pathophysiology of many diseases including diabetic macular edema (28). C3 is a central protein component of the complement system (29). The observation here of increased C3 cleavage fragments (C3b, C3a, and C3d) in baseline HAE-C1INH plasma as compared with that of healthy controls, followed by a reduction upon lanadelumab treatment is consistent with previous reports of C3 activation by PKa (Supplementary Figure 6) (30).
Apolipoprotein B, a protein involved in cholesterol deposition in the arterial wall and a marker of cardiovascular risk (31), was among the 120 altered proteins that were elevated in HAE-C1INH baseline plasma and reduced after 26 weeks of treatment with lanadelumab to levels comparable with that of healthy controls. It was previously reported that apolipoprotein B is cleaved by PKa (32). Consequently, it could be useful in future studies to determine whether the SOMAmer against apolipoprotein B exhibits differential binding to intact protein compared with the PKa-cleaved protein. Further investigation into a role for PKa in cholesterol metabolism is supported by the recent observation that prekallikrein binds the low-density lipoprotein receptor and could be a therapeutic target to decrease cholesterol (33).
Another protein among the 120 altered proteins worthy of further investigation is interleukin-21, which is a pleiotropic cytokine involved in T helper 17 cell expansion that was previously identified as being elevated in patients with HAE (34). Inflammation and elevated cytokine levels have been suggested as potentially contributing to attack onset triggers (35).
The pathways identified from comparing pre-dose HAE-C1INH plasma proteomics to that of healthy controls are supported by previous observations. For example, biomarkers of blood coagulation and fibrinolysis have previously been shown to be associated with HAE-C1INH, especially upon attack onset (36). Even though patients with HAE-C1INH may not have an increased risk of thrombosis (37), recent preclinical and clinical studies indicate that high plasma levels of C1 INH reduce the risk of venous thromboembolism (38). Pathway analyses using this proteomic data also identified differences in cell adhesion pathways based on plasma proteomics in HAE-C1INH that appeared to approach levels in healthy controls after treatment with lanadelumab. Elevated levels of adhesion proteins such as VE-cadherin have also been previously shown in plasma from HAE-C1INH patients (39, 40).
In summary, we used plasma proteomic analyses to identify potential HAE disease state biomarkers for further examination with additional patient samples and orthogonal methods for confirmation. Combining proteomic analyses with matched samples from patients before and after treatment with a highly specific antibody therapeutic can help identify active subnetworks in the disease that are mediated by the target.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Western Institutional Review Board (WIRB), Puyallup, WA 98374-2115, USA; IRB Services, Aurora, Ontario, L4G 0A5, Canada; Program for the Protection of Human Subjects, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Penn State Milton S. Hershey Medical, Center/Penn State College of Medicine, Human Subjects Protection Office/Institutional Review Board, Hershey, PA 17033, USA; Human Research Protection Office, Washington University School of Medicine, St. Louis, MO 63110, USA; Human Subjects Committee, University of Kansas Medical Center, City, KS 66160, USA; Cleveland Clinic Foundation, Cleveland, OH 44195, USA; University of Michigan Medical School Institutional Review Board (IRBMED), Ann Arbor, MI 48109-2800, USA; Duke University Health System Institutional Review Board, Durham, NC 27705, USA; Children’s Hospital Institutional Review Board, Wauwatosa, WI 53226, USA; Health Research Authority, Manchester, M1 3DZ, UK; Ethik-Kommission des Landes Berlin, 10707 Berlin, Germany; Ethik-Kommission der Landesärztekammer Rheinland-Pfalz, 55116 Mainz, Germany; Comitato Etico Interaziendale Milano Area A, Segreteria Centrale, c/o Ospedale L. Sacco COMITATO ETICO, 20157 Milano, Italy; Ethikkommission der Landesärztekammer Hessen, 60488 Frankfurt am Main, Germany; Ethik-Kommission des Fachbereichs Medizin der Johann Wolfgang Goethe-Universität, 60590 Frankfurt am Main, Germany; University of Alberta Research Ethics, 308 Campus Tower, Edmonton, AB T6G 1K8, Canada; Triumpharma Institutional Review Board, Amman 11941, Jordan; BioIVT, Westbury, NY, USA. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
DS: Writing – original draft, Writing – review & editing. AK: Writing – review & editing, Writing – original draft. SJ: Writing – review & editing, Writing – original draft. DY: Writing – review & editing, Writing – original draft. AM: Writing – review & editing, Writing – original draft. EA: Writing – review & editing, Writing – original draft. BL: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was sponsored by Takeda Development Center Americas, Inc. Takeda Pharmaceuticals USA, Inc., provided funding to Excel Scientific Solutions, Inc., for support in editing this manuscript. The intepretation of the data was made by the authors independently.
Conflict of interest
AK and EA are employees of Clarivate PLC. DS is an employee of Sexton Bio Consulting, LLC, former employee of Takeda Development Center Americas, Inc., and holds stock and/or share options in Takeda Pharmaceutical Company Limited. AM is a former employee of Takeda Development Center Americas, Inc., and holds stock and/or share options in Takeda Pharmaceutical Company Limited. SJ is an employee of Takeda Pharmaceuticals USA Inc., and holds stock and/or share options in Takeda Pharmaceutical Company Limited. DY and BL are employees of Takeda Development Center Americas, Inc., and hold stock and/or share options in Takeda Pharmaceutical Company Limited.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1471168/full#supplementary-material
References
1. Kaplan AP, Joseph K. Pathogenic mechanisms of bradykinin mediated diseases: dysregulation of an innate inflammatory pathway. Adv Immunol. (2014) 121:41–89. doi: 10.1016/B978-0-12-800100-4.00002-7
2. Zuraw BL, Christiansen SC. HAE pathophysiology and underlying mechanisms. Clin Rev Allergy Immunol. (2016) 51:216–29. doi: 10.1007/s12016-016-8561-8
3. Porebski G, Kwitniewski M, Reshef A. Biomarkers in hereditary angioedema. Clin Rev Allergy Immunol. (2021) 60:404–15. doi: 10.1007/s12016-021-08845-6
4. Isono M, Kokado M, Kato K. Why does it take so long for rare disease patients to get an accurate diagnosis?-A qualitative investigation of patient experiences of hereditary angioedema. PLoS One. (2022) 17:e0265847. doi: 10.1371/journal.pone.0265847
5. Zuraw BL, Maurer M, Sexton DJ, Cicardi M. Therapeutic monoclonal antibodies with a focus on hereditary angioedema. Allergol Int. (2023) 72:54–62. doi: 10.1016/j.alit.2022.06.001
6. Candia J, Daya GN, Tanaka T, Ferrucci L, Walker KA. Assessment of variability in the plasma 7k SomaScan proteomics assay. Sci Rep. (2022) 12:17147. doi: 10.1038/s41598-022-22116-0
7. Sinnathamby ES, Issa PP, Roberts L, Norwood H, Malone K, Vemulapalli H, et al. Hereditary angioedema: diagnosis, clinical implications, and pathophysiology. Adv Ther. (2023) 40:814–27. doi: 10.1007/s12325-022-02401-0
8. Davis AE 3rd, Lu F, Mejia P. C1 inhibitor, a multi-functional serine protease inhibitor. Thromb Haemost. (2010) 104:886–93. doi: 10.1160/TH10-01-0073
9. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygoren-Pursun E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema - The 2021 revision and update. World Allergy Organ J. (2022) 15:100627. doi: 10.1016/j.waojou.2022.100627
10. Suffritti C, Zanichelli A, Maggioni L, Bonanni E, Cugno M, Cicardi M. High-molecular-weight kininogen cleavage correlates with disease states in the bradykinin-mediated angioedema due to hereditary C1-inhibitor deficiency. Clin Exp Allergy. (2014) 44:1503–14. doi: 10.1111/cea.12293
11. Cugno M, Zanichelli A, Bellatorre AG, Griffini S, Cicardi M. Plasma biomarkers of acute attacks in patients with angioedema due to C1-inhibitor deficiency. Allergy. (2009) 64:254–7. doi: 10.1111/j.1398-9995.2008.01859.x
12. Christiansen SC, Zuraw BL. Laboratory approaches for assessing contact system activation. Immunol Allergy Clin North Am. (2017) 37:527–39. doi: 10.1016/j.iac.2017.04.008
13. Banerji A, Riedl MA, Bernstein JA, Cicardi M, Longhurst HJ, Zuraw BL, et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. (2018) 320:2108–21. doi: 10.1001/jama.2018.16773
14. Banerji A, Busse P, Shennak M, Lumry W, Davis-Lorton M, Wedner HJ, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med. (2017) 376:717–28. doi: 10.1056/nejmoa1605767
15. Rooney MR, Chen J, Ballantyne CM, Hoogeveen RC, Tang O, Grams ME, et al. Comparison of proteomic measurements across platforms in the Atherosclerosis Risk in Communities (ARIC) Study. Clin Chem. (2023) 69:68–79. doi: 10.1093/clinchem/hvac186
16. Gaire RK, Smith L, Humbert P, Bailey J, Stuckey PJ, Haviv I. Discovery and analysis of consistent active sub-networks in cancers. BMC Bioinf. (2013) 14 Suppl 2:S7. doi: 10.1186/1471-2105-14-S2-S7
17. Farkas H, Veszeli N, Kajdacsi E, Cervenak L, Varga L. "Nuts and bolts" of laboratory evaluation of angioedema. Clin Rev Allergy Immunol. (2016) 51:140–51. doi: 10.1007/s12016-016-8539-6
18. Singh U, Bernstein JA. Determining biomarkers for evaluation and diagnosis of hereditary angioedema. Clin Transl Allergy. (2022) 12:e12202. doi: 10.1002/clt2.12202
19. Parsopoulou F, Loules G, Zamanakou M, Csuka D, Szilagyi A, Kompoti M, et al. Searching for genetic biomarkers for hereditary angioedema due to C1-inhibitor deficiency (C1-INH-HAE). Front Allergy. (2022) 3:868185. doi: 10.3389/falgy.2022.868185
20. Zhang Z, Wang X, Gu J, Wu J, Cao Y, Xu Y, et al. Validation of diagnostic and predictive biomarkers for hereditary angioedema via plasma N-glycomics. Clin Transl Allergy. (2021) 11:e12090. doi: 10.1002/clt2.12090
21. Christiansen SC, Wilmot J, Castaldo AJ, Zuraw BL. The US Hereditary Angioedema Association Scientific Registry: hereditary angioedema demographics, disease severity, and comorbidities. Ann Allergy Asthma Immunol. (2023) 131:766–74.e8. doi: 10.1016/j.anai.2023.08.012
22. Sundler Bjorkman L, Persson B, Aronsson D, Skattum L, Nordenfelt P, Egesten A. Comorbidities in hereditary angioedema-A population-based cohort study. Clin Transl Allergy. (2022) 12:e12135. doi: 10.1002/clt2.12135
23. Zanichelli A, Senter R, Merlo A, Gidaro A, Popescu Janu V, Cogliati CB, et al. Comorbidities in angioedema due to C1-inhibitor deficiency: an Italian survey. J Allergy Clin Immunol Pract. (2024) 12:1029–36. doi: 10.1016/j.jaip.2023.12.046
24. Vandooren J, Itoh Y. Alpha-2-macroglobulin in inflammation, immunity and infections. Front Immunol. (2021) 12:803244. doi: 10.3389/fimmu.2021.803244
25. Rehman AA, Ahsan H, Khan FH. Alpha-2-macroglobulin: a physiological guardian. J Cell Physiol. (2013) 228:1665–75. doi: 10.1002/jcp.24266
26. de Paula Silva L, de Moraes Rego FG, Picheth G, Muller-Santos M, Alberton D. Prospection of plasma proteins as biomarkers for diabetes mellitus monitoring. J Diabetes Metab Disord. (2021) 20:611–20. doi: 10.1007/s40200-021-00788-1
27. Kaufman N, Page JD, Pixley RA, Schein R, Schmaier AH, Colman RW. Alpha 2-macroglobulin-kallikrein complexes detect contact system activation in hereditary angioedema and human sepsis. Blood. (1991) 77:2660–7. doi: 10.1182/blood.v77.12.2660.bloodjournal77122660
28. Honoré B, Hajari JN, Pedersen TT, Ilginis T, Al-Abaiji HA, Lønkvist CS, et al. Proteomic analysis of diabetic retinopathy identifies potential plasma-protein biomarkers for diagnosis and prognosis. Clin Chem Lab Med. (2024) 62:1177–97. doi: 10.1515/cclm-2023-1128
29. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
30. Irmscher S, Döring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
31. Glavinovic T, Thanassoulis G, de Graaf J, Couture P, Hegele RA, Sniderman AD. Physiological bases for the superiority of apolipoprotein B over low-density lipoprotein cholesterol and non-high-density lipoprotein cholesterol as a marker of cardiovascular risk. J Am Heart Assoc. (2022) 11:e025858. doi: 10.1161/JAHA.122.025858
32. Hardman DA, Gustafson A, Schilling JW, Donaldson VH, Kane JP. Scission of human apolipoprotein B-100 by kallikrein: Characterization of the cleavage site. Biochem Biophys Res Commun. (1986) 137:821–5. doi: 10.1016/0006-291x(86)91153-8
33. Wang JK, Li Y, Zhao XL, Liu YB, Tan J, Xing YY, et al. Ablation of plasma prekallikrein decreases low-density lipoprotein cholesterol by stabilizing low-density lipoprotein receptor and protects against atherosclerosis. Circulation. (2022) 145:675–87. doi: 10.1161/CIRCULATIONAHA.121.056491
34. Arcoleo F, Lo Pizzo M, Misiano G, Milano S, Romano GC, Muggeo V, et al. The complex alteration in the network of IL-17-type cytokines in patients with hereditary angioedema. Clin Exp Med. (2018) 18:355–61. doi: 10.1007/s10238-018-0499-0
35. Hofman ZL, Relan A, Zeerleder S, Drouet C, Zuraw B, Hack CE. Angioedema attacks in patients with hereditary angioedema: Local manifestations of a systemic activation process. J Allergy Clin Immunol. (2016) 138:359–66. doi: 10.1016/j.jaci.2016.02.041
36. Cugno M, Cicardi M, Bottasso B, Coppola R, Paonessa R, Mannucci PM, et al. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood. (1997) 89:3213–8. doi: 10.1182/blood.v89.9.3213
37. Cugno M, Asero R, Tedeschi A, Lazzari R, Marzano AV. Inflammation and coagulation in urticaria and angioedema. Curr Vasc Pharmacol. (2012) 10:653–8. doi: 10.2174/157016112801784558
38. Grover SP, Snir O, Hindberg K, Englebert TM, Braekkan SK, Morelli VM, et al. High plasma levels of C1-inhibitor are associated with lower risk of future venous thromboembolism. J Thromb Haemost. (2023) 21:1849–60. doi: 10.1016/j.jtha.2023.03.024
39. Bindke G, Gehring M, Wieczorek D, Kapp A, Buhl T, Wedi B. Identification of novel biomarkers to distinguish bradykinin-mediated angioedema from mast cell-/histamine-mediated angioedema. Allergy. (2022) 77:946–55. doi: 10.1111/all.15013
Keywords: kallikrein-kinin system, lanadelumab, bradykinin, antibody inhibitor of protease, hereditary angioedema
Citation: Sexton D, Kichev A, Juethner S, Yeung D, MacDonald A, Anokian E and Li B (2025) Hereditary angioedema plasma proteomics following specific plasma kallikrein inhibition with lanadelumab. Front. Immunol. 15:1471168. doi: 10.3389/fimmu.2024.1471168
Received: 31 July 2024; Accepted: 26 November 2024;
Published: 09 May 2025.
Edited by:
Michael Rudenko, London Allergy and Immunology Centre, United KingdomReviewed by:
Thomas Buttgereit, Charité University Medicine Berlin, GermanyNatasa Teovska Mitrevska, Remedika General Hospital, North Macedonia
Regis Campos, Federal University of Bahia (UFBA), Brazil
This work is authored by Sexton, Kichev, Juethner, Yeung, MacDonald, Anokian and Li. © 2025 Takeda Pharmaceuticals USA Inc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Li, QmluLkxpMkB0YWtlZGEuY29t
†Present address: Dan Sexton, Sexton Bio Consulting, LLC, Melrose, MA, United States