 Bo Wang
Bo Wang Yuchu Xiang
Yuchu Xiang Xudong Liu1
Xudong Liu1- 1Institute of Medical Microbiology and Hygiene, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Department of Dermatology, West China Hospital, Sichuan University, Chengdu, China
- 3Laboratory of Dermatology, Clinical Institute of Inflammation and Immunology, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, Chengdu, China
- 4Department of Clinical Medicine, Shanghai Medical College, Fudan University, Shanghai, China
Renal cell carcinoma (RCC), particularly the clear-cell subtype (ccRCC), accounts for 75-85% of kidney cancers and exhibits distinct genetic and biological heterogeneity. While surgical resection remains the mainstay of treatment for localized ccRCC, the persistence of recurrence rates underscores the significant unmet need for effective adjuvant therapies. Recent advancements in immunotherapy and targeted therapies have revolutionized the management of RCC. Immune checkpoint inhibitors have significantly enhanced antitumor immune responses, whereas tyrosine kinase inhibitors (TKIs) and mammalian target of rapamycin (mTOR) inhibitors effectively disrupt angiogenesis and proliferation signaling pathways, respectively. However, non-clear cell RCC subtypes remain understudied due to their rarity and exclusion from major clinical trials. Consequently, this review primarily focuses on ccRCC, aiming to provide a comprehensive and up-to-date overview of the latest advancements in immunotherapy and targeted therapies. By synthesizing current evidence, this review seeks to elucidate the mechanisms of action, clinical efficacy, and limitations of these treatments, while also identifying gaps in knowledge and future research directions. Ultimately, the goal is to offer valuable insights for clinicians and researchers, facilitating the development of optimized, personalized treatment approaches to improve outcomes for ccRCC patients.
1 Introduction
Renal cancer remains a common cancer globally, with an estimated 431,288 new cases in 2020 worldwide (1). Recent studies suggest that new favorable subsets of cancers of unknown primary (CUP), including renal cell carcinoma (RCC) CUP, have emerged. This newly recognized clinical entity is treated similarly to RCC and contributes to the currently increasing incidence of RCC (2).The preponderance of kidney cancers comprises RCC, predominantly clear-cell RCC (ccRCC) occupying 75-85% of instances, trailed by papillary, chromophobe, medullary, and collecting duct subtypes. These subgroups of RCCs differ from each other in genetics, biology, and behavior (3). Localized ccRCC is conventionally managed through surgical excision, encompassing both radical and partial nephrectomy approaches, aimed at achieving definitive cure (4). However, the recurrence rate of RCC at 5-years is significant, varying from 2.2% to 58.1% depending on risk factors such as tumor size, histology, and other clinical features (5). Therefore, the necessity for adjuvant and/or neoadjuvant therapies to mitigate recurrence risks and enhance survival outcomes underscores a crucial, as yet unfulfilled, clinical requirement. Apart from surgical interventions, a diverse array of therapeutic modalities has emerged recently to arrest tumor progression and hinder RCC metastases, embracing radiotherapy, chemotherapy, immunological therapies, as well as precision-guided targeted therapies, among others (6).
The profound comprehension of the immune system and cancer’s molecular underpinnings has spawned immunotherapy and targeted therapy, respectively, revolutionizing contemporary therapeutic strategies for renal cancer (7). These innovative approaches offer more treatment options and better outcomes for kidney cancer patients.
Immunotherapy has significantly progressed, utilizing immune checkpoint inhibitors such as Programmed Cell Death Protein-1/Programmed Death-Ligand 1(PD-1/PD-L1) and Cytotoxic T-lymphocyte-associated protein 4(CTLA-4) antagonists, among others, to amplify the immune system’s capacity to counteract tumor growth (8). In addition, targeted therapies further complement this arsenal. For example, Sunitinib, a tyrosine kinase inhibitor (TKI), impedes tumor angiogenesis and expansion through Vascular Endothelial Growth Factor Receptor(VEGFR) disruption (9), while Everolimus and Temsirolimus, mammalian target of rapamycin (mTOR) inhibitors, hinder tumor growth and proliferation by modulating the mTOR signaling cascade (10–12). It should be noted that TKI monotherapy remains a suitable first-line option for patients ineligible for immunotherapy, as supported by the STAR trial. It also offers benefits as a second-line treatment. Although treatment breaks may be considered, caution is warranted, as these patients typically experience shorter progression-free survival compared to those receiving first-line TKI therapy (13).
Given the heterogeneous histology and the rarity of each individual subtype within non-clear cell RCCs (nccRCCs), these tumors have traditionally been under represented in clinical trials, resulting in a lack of well-studied treatment options specifically tailored to them (14). Consequently, this review primarily focuses on ccRCC, provides a comprehensive overview of the immunotherapy and targeted therapies for this specific subtype of kidney cancer.
2 Immunotherapy in RCC
2.1 Immunotherapy origins
The origins of immunotherapy date back to the late 19th century, when William Coley discovered that bacterial infections (Coley’s toxins) helped treat cancer. The patient’s immune system is provoked by these toxins, facilitating its ability to assault and eradicate cancerous tumor cells (15, 16). The development of modern immunotherapy is based on in-depth research into T cell function and immune checkpoint molecule (17). And immunotherapy works by activating the patient’s own immune system to attack tumor cells, primarily involving cytokine therapy and immune checkpoint inhibitors(ICIs) (Figure 1). In addition, with the continuous deepening of research, adoptive cell therapies-including non-gene-modified and gene-modified adoptive cell therapies- along with dendritic cells(DCs) and vaccines have gradually come into the spotlight.
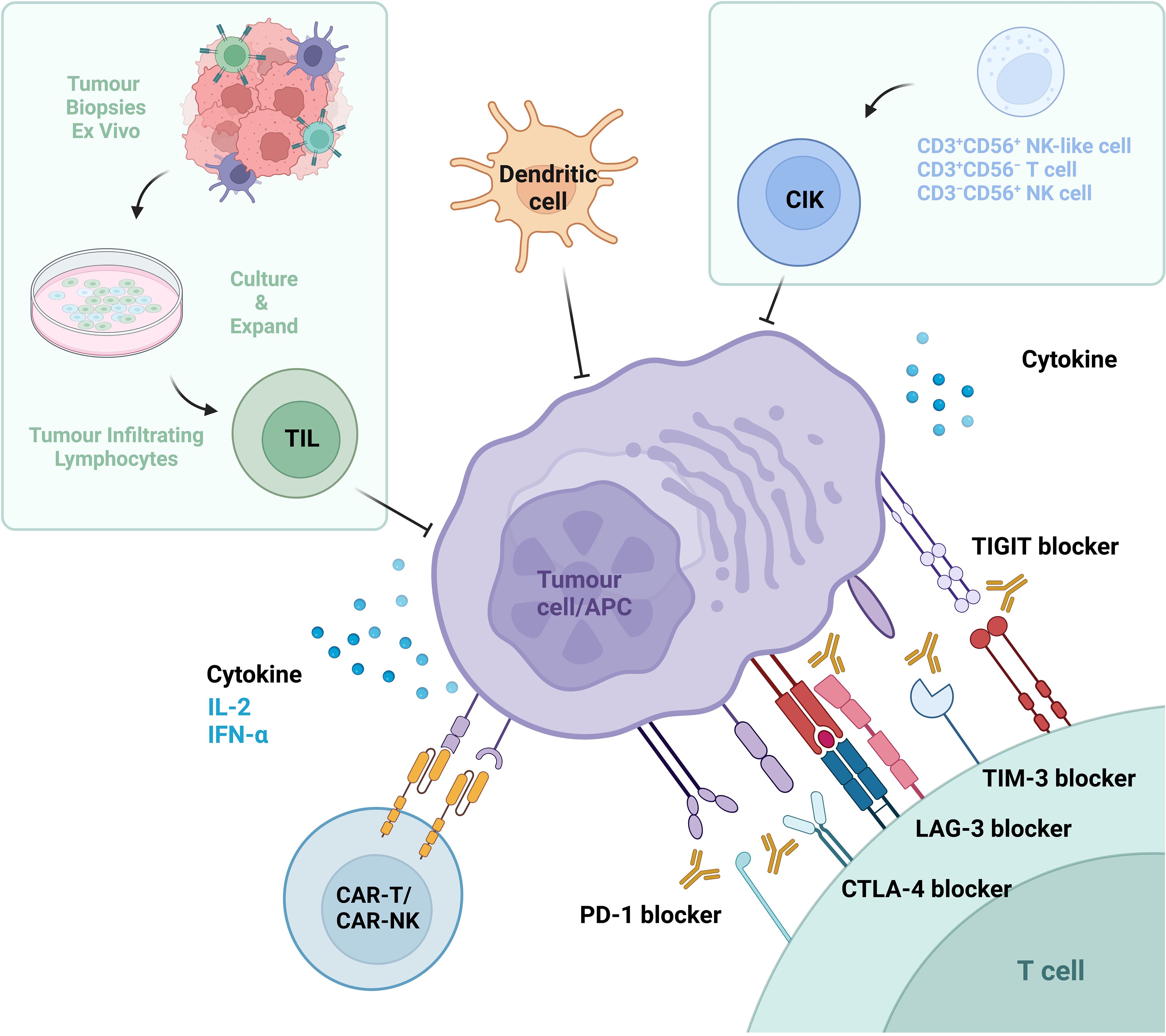
Figure 1. Immunotherapeutic strategies in renal cell carcinoma (RCC). Current immunotherapeutic approaches for RCC encompass three primary categories: (1) Cytokine therapy, involving interleukin-2 (IL-2) and interferon-alpha (IFN-α); (2) Immune checkpoint inhibitors (ICIs), including antibodies targeting coinhibitory receptors such as programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT); (3) Adoptive cell therapies, including ex vivo-expanded cytokine-induced killer (CIK) cells, tumor-infiltrating lymphocytes (TILs), genetically engineered chimeric antigen receptor T-cells (CAR-Ts) and natural killer cells (CAR-NKs), as well as dendritic cells (DCs). These modalities collectively aim to enhance intrinsic antitumor immune responses through distinct mechanistic frameworks.
2.1.1 Cytokines
Cytokine therapy, utilizing molecules like interleukin-2 (IL-2) and interferon-alpha (IFN-α), have surfaced as a validated therapeutic strategy for managing metastatic RCC (18–20). Historically, before the advent of targeted therapies, advanced RCC treatment heavily relied on cytokine immunotherapy using either interferon or IL-2 (21). Interferon, an early adopted protein, was administered for RCC treatment, potentiating the immune system’s efficacy in combating neoplastic diseases. Although some patients showed some tumor shrinkage after interferon treatment, the overall response rate was low (approximately 15-20%) and treatment was associated with severe side effects such as flu-like symptoms, fatigue, and depression (22). IL-2, a type of cytokine, stimulates T-lymphocytes and natural killer (NK) cells, enhancing their capacity to eliminate tumor cells. In RCC, select patients have responded favorably to high-dose IL-2 therapy, with some experiencing complete tumor regression (23). However, IL-2 administration is plagued by significant toxicity, particularly severe vascular leak syndrome, thus impeding its broad clinical application (24).
2.1.2 Immune checkpoint inhibitors
Immune checkpoint inhibitors (ICIs), a class of immunotherapy drugs, block checkpoint protein signaling pathways to enhance anti-tumor immune responses by relieving immune suppression (25). Their primary function is to block inhibitory signals, thereby enabling immune cells to mount a more effective tumor-specific cytotoxic response and eliminate cancer cells. Recent advancements highlight the pivotal role of this therapy in the management of ccRCC, particularly in advanced stages (Table 1).
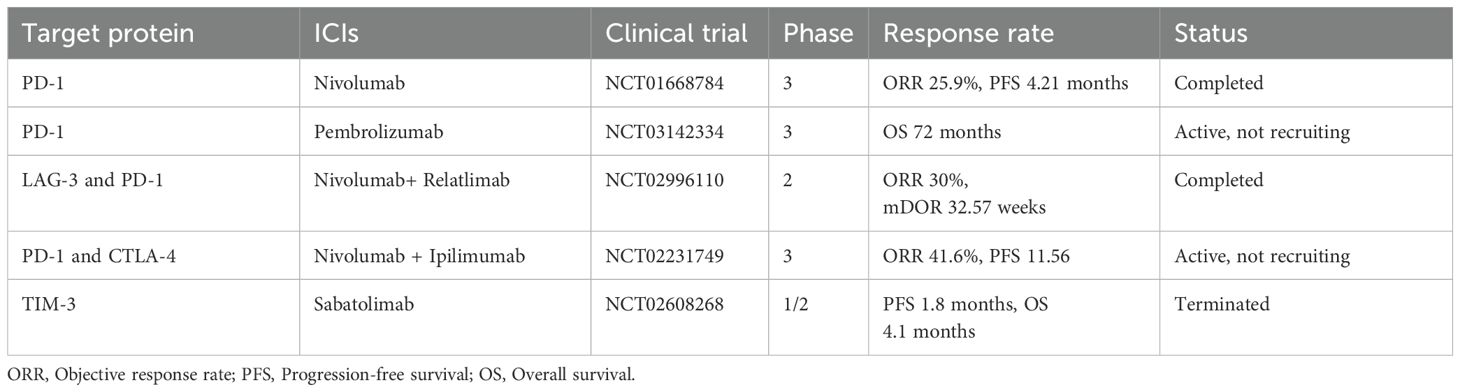
Table 1. Selected pivotal ICIs involved in RCC management.
2.1.2.1 PD-1 inhibitors
In 1992, T. Honjo identified PD-1, also designated as Cluster of Differentiation(CD)279. This transmembrane protein is encoded by the Programmed Cell Death 1 (PDCD1) gene (26). T. Honjo and J. Allison made groundbreaking discoveries regarding the role of PD-1 and CTLA-4 inhibition in cancer therapy, which earned them the 2018 Nobel Prize in Physiology or Medicine (27). PD-1 is prominently expressed on tumor-directed T-cells (CD8+ cytotoxic T-cells and CD4+ T helper1 cells), NK cells, B-cells, monocytes, and DCs. Upon engagement with its ligands, PD-L1 or Programmed Death-Ligand 2(PD-L2), PD-1 activates tyrosine phosphatases (such as Src Homology 2 Domain-Containing Phosphatase-1/2), which dephosphorylate key signaling molecules downstream of the T-cell receptor (TCR), thus inhibiting TCR signaling pathways crucial for T-cell activation. This results in attenuated cell survival signals, impaired cytokine release, and contributes to T-cell exhaustion, reducing immune cell responsiveness and promoting immune evasion. Notably, PD-1 ligands are expressed on both immune cells and tumor cells, allowing tumors to suppress immune responses and evade immune detection (28–30).
PD-1 inhibitors, a class of monoclonal antibodies, serve as immune checkpoint modulators by disrupting inhibitory signals mediated by PD-1 transmembrane proteins on effector immune cells. Currently, Pembrolizumab (Keytruda) and Nivolumab (Opdivo), two anti-PD-1 agents, are utilized in the treatment of ccRCC. Pembrolizumab, the first humanized IgG4 (S228P) monoclonal antibody, received Food and Drug Administration(FDA) approval in 2021 for various cancer indications, including RCC. Nivolumab, another human IgG4 antibody, preceded Pembrolizumab in 2015 as the first FDA-approved checkpoint inhibitor for advanced RCC. Both drugs can elicit adverse effects, such as gastrointestinal distress, muscular discomfort, and fatigue, likely by perturbing physiological immune processes (31–33). Notably, in ccRCC patients, anti-PD-1 therapies exhibit constrained efficacy against central nervous system metastases due to limited blood-brain barrier permeation (34). To mitigate these limitations, research has intensified on combinatorial immunotherapy strategies. In 2018, the FDA endorsed a PD-1/CTLA-4 (Ipilimumab) combination to enhance ccRCC treatment outcomes. Additionally, the potential synergy between Pembrolizumab and the anti-VEGF agent Pazopanib is still under investigation (25).
2.1.2.2 CTLA-4 inhibitors
CTLA-4 (CD152), a pivotal immune checkpoint, modulates antitumor immunity by inhibiting immune responses to cancer. Encoded by the CTLA-4 gene, it acts as a key regulator of immune tolerance. Its role was first discerned in 1987 and further elucidated in the 1990s (28). CTLA-4 is composed of 223 amino acids (35) and is upregulated in tumor-targeting T cells. Upon binding to CD80 (B7-1) or CD86 (B7-2) on antigen-presenting cells or tumor cells, it functions as an inhibitory switch. This interaction triggers the phosphorylation of its cytoplasmic YVKM motif, which recruits Src Homology 2 Domain-Containing Phosphatase-2 and activates Serine/Threonine Protein Phosphatase 2A. This cascade suppresses TCR/CD28-mediated activation of the Phosphatidylinositol 3-kinase (PI3K)/Protein Kinase B (Akt) signaling pathway, thereby limiting TCR signaling that is essential for T-cell activation and cytokine production (27, 36).
Ipilimumab (Yervoy) is an FDA-approved human Immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that specifically targets CTLA-4. While CTLA-4 inhibitors can elicit adverse reactions such as exhaustion and gastrointestinal disturbances, blocking CTLA-4 in melanoma therapy has been associated with severe Immune-Related Adverse Events(AE), including autoimmune complications in visceral organs such as the colon, liver, and endocrine glands (28). To enhance efficacy in resistant patients, the FDA approved the combination of Ipilimumab with Nivolumab as a first-line therapeutic regimen for advanced RCC in adults (37). This synergy arises from their complementary mechanisms of action: Anti-PD-1 therapy revitalizes exhausted effector T cells, thereby enhancing immune responses, while anti-CTLA-4 promotes antigen-specific T-cell activation and priming (38).
2.1.2.3 Lymphocyte-Activation Gene 3 inhibitors
Lymphocyte-Activation Gene 3 (LAG-3), also known as CD223, was first identified in 1990 by Triebel’s team. This 498-amino acid transmembrane protein, featuring four immunoglobulin (Ig) superfamily domains, is encoded by the LAG3 gene (39). It is expressed on T cells, NK cells, and B cells (40), binding to major histocompatibility complex II (MHC-II), liver and lymph node sinusoidal endothelial cell C-type lectin, and fibrinogen-like protein 1, all of which are found on antigen-presenting cells and tumor cells. Liver and lymph node sinusoidal endothelial cell C-type lectin, a C-lectin receptor on the cell surface, and MHC-II display the highest affinity for LAG-3 (27). Like other immune checkpoints, LAG-3 inhibits T cell proliferation, cytotoxicity, and homeostasis, hindering immune responses against tumor cells and foreign invaders (41–43). Elevated expression of LAG-3 alongside PD-1 is associated with a poor prognosis for patients with ccRCC (44).
Relatlimab, a humanized IgG4 monoclonal antibody, specifically targets the LAG-3 protein and is currently undergoing Phase II clinical trials for ccRCC. Recent findings suggest that monotherapy with anti-LAG-3 or its combination with anti-PD-1 shows promise for patients with LAG-3+ ccRCC who are refractory to anti-PD-1 therapy. Administered intravenously, Relatlimab may cause adverse reactions such as fatigue, rash, and arthralgia. Additionally, its combination with other immune checkpoint inhibitors, such as Nivolumab, is being investigated to improve therapeutic efficacy against RCC (44).
2.1.2.4 T cell immunoglobulin and Mucin domain 3 inhibitors
T cell Immunoglobulin and Mucin domain 3 (TIM-3), also known as Hepatitis A Virus Cellular Receptor 2 (HAVCR2), is a transmembrane immune checkpoint receptor involved in tumor-driven immune suppression. First identified by Kuchroo et al. in 2002, the HAVCR2 gene encodes this protein (45), initially observed on CD4+ and CD8+ T cells. Subsequent studies confirmed its expression on T helper 17 cells (46), Regulatory T cells (47), and innate immune cells such as DCs, NK cells, and monocytes (48). Structurally, TIM-3 comprises a membrane-distal single variable Ig domain, a glycosylated mucin domain, and an intracellular stem (49). TIM-3 interacts with multiple ligands, including galectin-9, high mobility group box 1 protein, phosphatidylserine, and carcinoembryonic antigen-related cell adhesion molecule 1. Binding between TIM-3 and its ligands results in NK and T cell dysfunction, ultimately leading to immune suppression in cancer and viral infections (50, 51). TIM-3 has been implicated in several malignancies, such as RCC, melanoma, and gastric cancer (52). However, its precise role in ccRCC remains unclear (53). TIM-3 blockade alone shows limited efficacy, necessitating its combination with other immune checkpoint inhibitors, such as anti-PD-1 antibodies, to enhance therapeutic outcomes (54).
In hematological malignancies, TIM-3 is found on CD8+ T cells in myelodysplastic syndrome, indicating its broader involvement beyond solid tumor immunity exhaustion (55). Sabatolimab, a pioneering immuno-myeloid agent targeting the TIM-3 transmembrane protein, is a humanized monoclonal antibody (IgG4). It is currently undergoing clinical trials for treating immunogenic cancers (56, 57).
2.1.2.5 T cell immunoreceptor with immunoglobulin and ITIM domains inhibitors
T cell Immunoreceptor with Immunoglobulin and ITIM domains (TIGIT), also known as WUCAM or VSTM3, is a transmembrane protein discovered in 2009. It belongs to the CD28 immunoglobulin superfamily and is encoded by the TIGIT gene. This 244-amino-acid protein (58) consists of an immunoglobulin domain, a transmembrane region, and an inhibitory cytoplasmic tail (59, 60). TIGIT is expressed on tumor-reactive T cells, NK cells, DCs, and macrophages, where it impedes antitumor activation and immune functionality (52, 61, 62). TIGIT binds to ligands such as CD155, CD112, and CD113. Ligand engagement triggers the phosphorylation of intracellular domains, leading to the recruitment of protein Growth factor receptor-bound protein 2 and inhibition of immune active SH2 domain-containing inositol 5′-phosphatase 1, Phosphatidylinositol 3-kinase (PI3K), and Mitogen-Activated Protein Kinase (MAPK) pathways (52, 60, 63). TIGIT expression varies across cancers and is generally low in RCC. However, tumor-infiltrating NK cells from metastatic lymph node patients exhibit elevated TIGIT expression, indicating a role in immune suppression (52).
Tiragolumab, a human IgG4κ monoclonal antibody targeting TIGIT, is currently under clinical evaluation (Phase II) for metastatic RCC. TIGIT has emerged as a promising therapeutic target for cancer immunomodulation, particularly when combined with PD-1 inhibitors. Preclinical studies have demonstrated that blocking both TIGIT and PD-1 significantly enhances tumor-specific T cell proliferation, degranulation, and cytokine production (64).The adverse reactions to Tiragolumab are similar to those of other ICIs, including fatigue, chills, and nausea (59, 65).
2.1.3 Adoptive cell therapy
2.1.3.1 Non-gene-modified cell therapies
2.1.3.1.1 Cytokine-Induced Killer cells
Cytokine-Induced Killer (CIK) cells are a heterogeneous population derived from peripheral or umbilical cord blood mononuclear precursors after stimulation with Interferon-gamma (IFN-γ), anti-CD3 monoclonal antibodies, and IL-2. This cell subset includes CD3+CD56+ NK-like cells, CD3+CD56− T lymphocytes, and CD3−CD56+ NK cells (66). CIK cells exhibit robust preclinical anti-tumor activity independent of major histocompatibility complex (MHC) or T cell receptor (TCR) specificity (66, 67). They express a diverse TCR repertoire and NK-associated markers such as Natural Killer Group 2 Member D, DNAX Accessory Molecule-1, and Natural Killer Cell p30-Activating Receptor (67), with Natural Killer Group 2 Member D engagement playing a pivotal role in their cytotoxic response against neoplastic cells (68).
In a prospective study comparing autologous CIK therapy to subcutaneous IL-2/Interferon-alpha (IFNα)-2a in 148 patients with metastatic ccRCC (69). CIK-treated patients demonstrated superior outcomes, including higher objective response rate (ORR) (53% vs. 27%), improved 3-year progression-free survival (PFS) (18% vs. 12%, p=0.031), and extended median overall survival (OS) (46 vs. 19 months, p<0.001). Subsequently, a randomized study involving 20 post-radical nephrectomy stage I/II patients contrasted autologous CIKs with investigator-selected treatments. Two weeks post-infusion, CIK-treated patients showed enhanced CD3+, CD3+CD8+, and CD3+CD56+ populations in peripheral blood (70). The CIK group also demonstrated significantly prolonged median PFS (32.2 vs. 21.6 months, p=0.032). In a separate study of 29 metastatic RCC (mRCC) patients, CIK therapy achieved a modest ORR of 13.8%, with elevated circulating myeloid-derived suppressor cells (MDSCs) correlating with poor outcomes, emphasizing the role of the tumor microenvironment (TME) in disease prognosis (71). Other mRCC CIK trials documented low toxicity and CD3+CD56+ cell expansion in vivo, yet definitive clinical benefits were obscured by study limitations including small sample sizes and heterogeneity (72, 73).
Incorporating DC vaccination to present tumor antigens, secrete cytokines, and engage in direct cell contact has shown promise in enhancing CIK activation and cytotoxicity (74). A retrospective study of 410 post-surgical mRCC patients revealed that DC-CIK therapy significantly outperformed IFNα, achieving a higher 3-year OS (96% vs. 83%, p<0.01) (75). Another randomized trial comparing autologous DC-CIK+IFNα vs. no adjuvant post-RCC surgery showed reduced recurrence and metastasis rates (p<0.01) (76). Similar findings were observed in a study where DC-CIK therapy led to a marked decline in post-surgery relapse (p = 0.0418) and superior 3-year DFS rates (96.7% vs. 57.7%) compared to no adjuvant therapy (75).
CIK cells are attractive therapeutic candidates due to their manufacturability and low toxicity. Future strategies focus on combining CIK cells with DC vaccines, TKIs, and ICIs to overcome the immunosuppressive TME and enhance therapeutic efficacy. A global registry for CIK cells is envisioned to establish benchmarks for future research endeavors (77).
2.1.3.1.2 Tumor infiltrating lymphocytes
Tumor-infiltrating lymphocytes (TILs) represent a promising adoptive cell therapy, especially for immunogenic tumors like metastatic melanoma, where clinical studies report an ORR of 49-72% and complete response (CR) rates of 10-20%, with approximately 40% achieving durable responses (78, 79). TILs are polyclonal T cells propagated ex vivo from patient tumor samples and re-infused after lymphodepleting chemotherapy, typically cyclophosphamide and fludarabine, to enhance in vivo expansion (78, 79).
Although promising in melanoma, TILs therapies have shown limited success in RCC. An early phase 3 clinical trial comparing CD8+ TILs combined with IL2 versus IL2 monotherapy in post-radical nephrectomy mRCC patients reported a modest ORR of 9.9%, compared to 11.4% in the IL2-only group (80). The trial was prematurely terminated due to insufficient efficacy, possibly attributed to diminished TIL viability and functional defects resulting from prolonged ex vivo culturing.
Recent advancements in TILs manufacturing, particularly the Rapid Expansion Protocol (REP) initially developed for melanoma, are being tested in mRCC. Despite successful TIL expansion using anti-CD3, high-dose IL-2, and irradiated allogeneic feeder cells, the products often exhibit functional defects, including poor cytotoxicity and reduced cytokine secretion (81). This may result from the limited T-cell recognition of tumor-specific antigens or manufacturing-induced quality decline (82).
Single-cell RNA sequencing analysis of RCC-derived TILs before and after REP revealed preferential expansion of CD4+ T cells, reduced T-cell diversity, and stagnation of exhausted T-cell clones (83). Emerging methods, such as Dynabeads-based manufacturing, have demonstrated improved TIL expansion and functionality (84). Achieving polyclonal, tumor-specific T-cell diversity and reactivity remains pivotal for advancing TIL therapies targeting RCC.
2.1.3.2 Gene-modified adoptive cell therapies
Chimeric Antigen Receptor T-Cells (CAR-Ts) and NK Cells (CAR-NKs).
Chimeric Antigen Receptor T-Cells (CAR-Ts) are T lymphocytes genetically reprogrammed to recognize and kill tumor cells independently of HLA presentation. This contrasts with endogenous T cells, which require peptide-HLA complexes for activation. By circumventing HLA restriction, CAR-T cells offer broader applicability and can target a wide range of surface antigens. Structurally, CARs are modular receptors composed of an extracellular antigen-binding domain, a hinge/spacer, a transmembrane domain, and intracellular signaling motifs (85). The antigen-binding domain typically derives from a single-chain variable fragment of tumor-specific antibodies, though nanobodies and ligands have also shown efficacy (86, 87).
The hinge and transmembrane domains, sourced from proteins like CD8 or CD28, modulate flexibility and anchoring. Intracellular domains usually consist of CD3ζ and one or more co-stimulatory motifs, enabling potent T cell activation and cytokine secretion. Unlike natural T cells, CAR-Ts integrate signal 1 and signal 2 into a single receptor, facilitating autonomous tumor engagement (85).
Five CAR generations have evolved since their 1987 inception (88). First-generation CARs included only CD3ζ and lacked persistence. Second- and third-generation CARs introduced co-stimulatory domains, improving cytotoxicity and expansion (89). More advanced fourth-generation CARs, known as T cell Redirected for Universal Cytokine Killing, are engineered to secrete immunostimulatory cytokines like IL-12, IL-15, or IL-18, thereby enhancing anti-tumor efficacy and modulating the tumor microenvironment (90). Fifth-generation CARs incorporate cytokine receptor signaling to promote persistence and mimic physiological T cell signaling (91).
To enhance safety, “suicide switches” (e.g., inducible caspase-9) enable pharmacologic CAR-T ablation in AE (92). CAR-T production involves leukapheresis, gene modification (commonly via viral vectors), and expansion. While viral vectors ensure stable gene integration, they carry rare risks of insertional mutagenesis and secondary malignancies (93–95). Alternatives such as CRISPR/Cas9, TALENs, and transposon systems offer non-viral solutions with enhanced safety profiles (96).
While CAR-T cells have shown significant success in hematologic malignancies, their application in solid tumors like RCC faces unique challenges (97). Solid tumors are shielded by physical and immunosuppressive barriers (e.g., hypoxia, acidic pH, Tregs), limiting CAR-T infiltration and function. Tumor antigen heterogeneity and off-tumor toxicity (due to shared antigen expression with healthy tissues) further complicate treatment.
Unlike T cells, NK cells naturally eliminate tumor cells without HLA recognition (98). Allogeneic NK cell therapies have been explored, though clinical outcomes have been modest thus far. Chimeric Antigen Receptor NK Cells (CAR-NK) are now under early-stage clinical evaluation (99). Their inherent lack of HLA restriction makes them attractive candidates for off-the-shelf manufacturing, though protocols for efficient expansion and gene modification still require optimization. Similarly, macrophages, which are often recruited into tumor tissues and play key roles in shaping the immune microenvironment, have emerged as novel platforms for CAR engineering (100, 101).
In RCC, multiple tumor-associated antigens have been identified for CAR-targeting, including Carboxy-Anhydrase IX (CAIX), CD70, AXL receptor tyrosine kinase (AXL), Receptor tyrosine kinase-like orphan receptor 2 (ROR2), DnaJ heat shock protein family (Hsp40) member B8 (DNAJB8), Mucin 1, C-Mesenchymal-Epithelial Transition Factor (c-Met), and Epidermal Growth Factor Receptor (EGFR). CAR-Ts and CAR-NKs therapies against these targets are under various stages of development, with some already in clinical trials (Table 2).
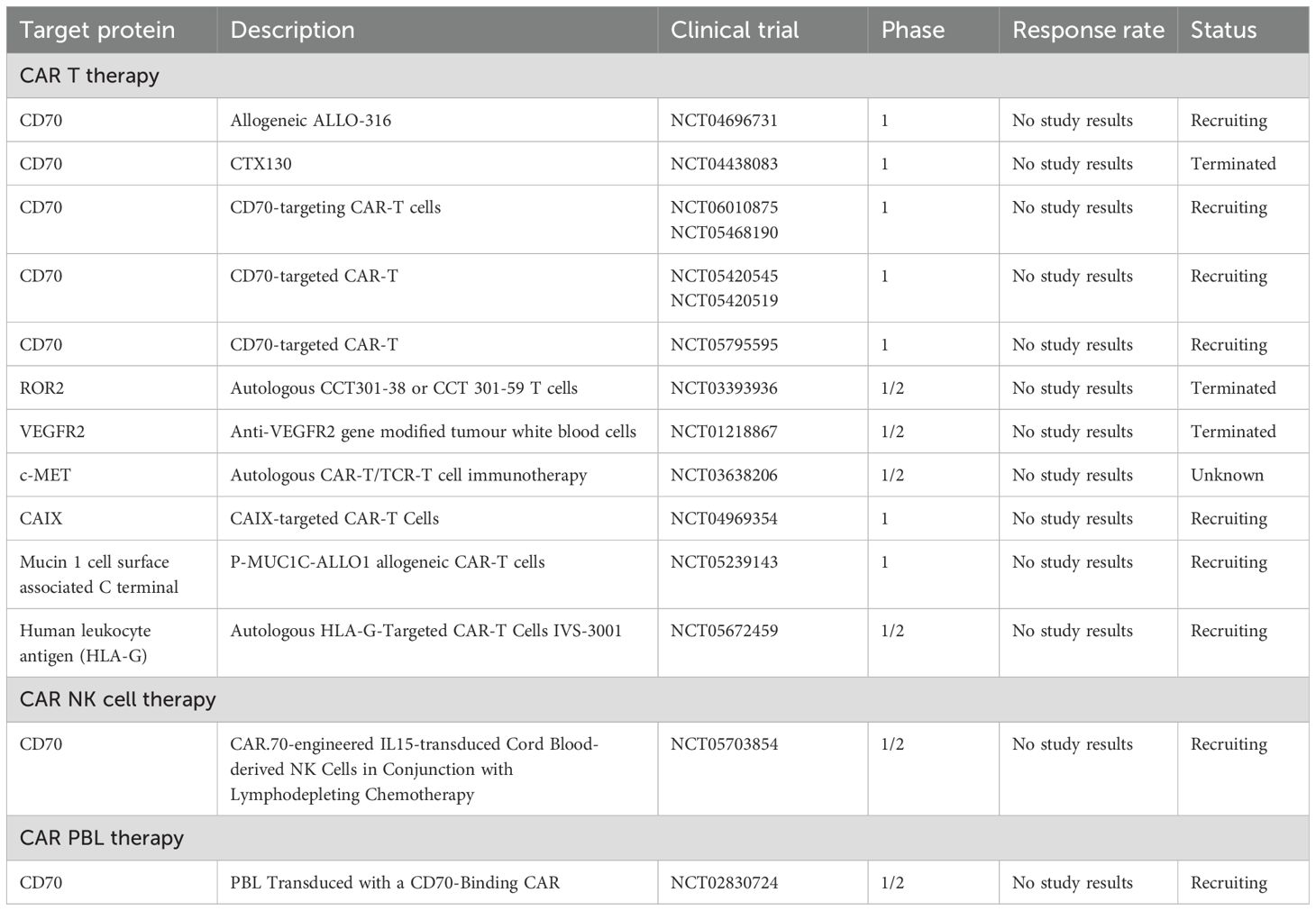
Table 2. Selected pivotal CAR Therapy Clinical Trials for RCC.
2.1.3.2.1 CAIX
CAIX is an enzyme frequently upregulated in hypoxic solid tumors, notably mRCC, making it an early target for CAR-T therapy in these malignancies (102). In a Phase I trial involving 12 patients, first-generation autologous anti-CAIX CAR-T cells were administered without lymphodepletion preconditioning. No clinical responses were observed, and significant toxicities, including liver enzyme disturbances, were reported (103).
Subsequent research has focused on enhancing the efficacy and safety of anti-CAIX CAR-T therapies. A self-inactivating bi-cistronic CAR-T targeting CAIX, fused with a CD28ζ endodomain and designed to locally release anti-PD-L1 antibodies at RCC sites, demonstrated heightened antitumor efficacy and reduced T-cell exhaustion markers in a humanized mouse model of CAIX+PD-L1+RCC (104). This suggests that modulating the TME is pivotal in mRCC prognosis (105).
To address antigenic heterogeneity, dual-targeting strategies have been explored. A dual-targeted fine-tuned immune-restoring CAR-T for ccRCC, aimed at both CAIX and CD70, and engineered to co-release immune checkpoint inhibitors, is currently under preclinical assessment (106).
Combining anti-CAIX CAR-T products with TKIs like sunitinib has shown promise (107). Sunitinib is known to augment IFNγ-producing T-cells while diminishing regulatory T cells and MDSCs (108, 109). In a humanized RCC mouse model, the combination of sunitinib and anti-CAIX CAR-Ts significantly reduced tumor load, outperforming either monotherapy (107).
Regarding CAR-modified NK cells, third-generation anti-CAIX CAR-modified NK92 cells, combined with bortezomib, reduced tumor burden in immunocompromised RCC xenograft mice (110). Bortezomib is believed to augment NK-mediated antitumor responses (111).
These findings underscore the potential of targeting CAIX in RCC using CAR-T and CAR-NK cell therapies, while highlighting the importance of addressing associated toxicities and the immunosuppressive tumor microenvironment.
2.1.3.2.2 CD70
CD70 is a transmembrane glycoprotein that interacts with CD27 receptors on T cells, promoting the generation of effector and memory T cells (112). Its expression is notable in activated T and B lymphocytes, as well as mature dendritic cells (113). Importantly, CD70 is prominently expressed in RCC, making it a compelling target for CAR-T therapy (114).
The CTX130 Phase I multicenter trial evaluated an allogeneic CD70-targeting CAR-T therapy in 16 patients with relapsed or refractory ccRCC. Preclinical studies demonstrated favorable proliferation and cytotoxicity profiles, with complete regression of RCC xenograft tumors. In the clinical setting, patients received escalating doses of CTX130 without encountering dose-limiting toxicities. Disease control was achieved in 81.3% of patients, and one patient remained in a durable complete response at three years. The study also introduced CTX131, a next-generation CAR-T construct with synergistic potency enhancements, which demonstrated improved expansion and efficacy in preclinical models. These findings establish proof of concept for CD70-targeted allogeneic CAR-T therapies in ccRCC and other CD70-expressing malignancies (115).
In contrast, the TRAVERSE study assessed ALLO316, an allogeneic Transcription Activator–Like Effector Nuclease gene-edited anti-CD70 CAR-T product, in 17 patients with pretreated advanced or metastatic ccRCC. To mitigate the risk of graft-versus-host disease, the T-cell receptor alpha constant gene was deleted, and CD52 was ablated to enhance lymphodepletion using ALLO647 (a humanized anti-CD52 antibody) in combination with fludarabine and cyclophosphamide. Cytokine Release Syndrome occurred in 65% of patients, including one case of grade 3 severity. The disease control rate reached 71%, peaking at 100% in CD70-positive subsets.
Additionally, CRISPR/Cas9-edited CAT-248 CAR-NK cells have been developed to target CD70. These cells are engineered to prevent self-targeting by deleting endogenous CD70 and secrete interleukin-15 (IL-15) to enhance persistence. In preclinical studies, CAT-248 demonstrated efficacy in vitro and in xenograft RCC models, significantly reducing tumor burden by more than 99%.
These findings underscore the potential of CD70-targeted CAR-T and CAR-NK therapies in treating CD70-expressing malignancies, particularly RCC.
2.1.3.2.3 AXL & ROR2
AXL, a member of the TAM family tyrosine kinase receptors, is frequently overexpressed in various solid tumors, including RCC (116), where it interacts with its high-affinity ligand growth arrest-specific protein 6 to promote proliferation, survival, angiogenesis, and invasion (117). Similarly, ROR2, an orphan tyrosine kinase receptor critical during embryogenesis, exhibits limited expression in adult tissues but is aberrantly upregulated in RCC, where it contributes to enhanced tumor growth, migration, and invasiveness (118). Given their roles in tumor progression, both AXL and ROR2 have emerged as promising targets for CAR-T therapies. A Phase I/II clinical trial investigated the safety and efficacy of ROR2-targeted CAR-T (CCT301-59) and AXL-targeted CAR-T (CCT301-38) therapies in patients with refractory or relapsed metastatic RCC.
2.1.3.2.4 DNAJB8
DNAJB8, a cancer-testis antigen, has emerged as a potential therapeutic target due to its expression in cancer stem/initiating cells and implication in tumorigenicity in RCC and osteosarcoma. Recent developments in CAR-T cell therapy have focused on second-generation constructs utilizing the B10 binder (B10-CAR), specifically targeting Human Leukocyte Antigen(HLA)-A*24:02/DNAJB8 peptide complexes on RCC cells (119). These engineered cells demonstrated robust antigen-specific and HLA-mediated activation, inducing IFN-γ secretion in vitro when exposed to RCC cell lines and significantly reducing tumor burden in vivo.
Although these findings highlight the therapeutic potential of targeting cancer stem-like cell antigens, further investigations are required to validate this approach and optimize its clinical efficacy.
2.1.3.2.5 Mucin 1
Mucin 1, a transmembrane glycoprotein normally expressed on epithelial surfaces, plays a protective role in healthy tissues but frequently undergoes aberrant glycosylation and overexpression in various malignancies, including RCC (120). An allogeneic CAR-T therapy targeting the Mucin 1 C-terminal antigen utilizes Cas-CLOVER gene editing to eliminate TCR and MHC-I expression, thereby mitigating the risk of graft-versus-host disease and immune rejection. This therapy is currently being evaluated in a Phase I clinical trial involving patients with advanced or metastatic epithelial tumors, including RCC. Further investigations are required to assess its efficacy and safety in this patient population.
2.1.3.2.6 c-met
c-Met, a receptor tyrosine kinase involved in tumor cell migration, proliferation, and invasion, is overexpressed in approximately 97% of papillary RCC cases while being largely absent in healthy renal tissue (121). A third-generation CAR-T cell therapy targeting c-Met, incorporating CD28, 4-1BB, and CD3ζ co-stimulatory domains, demonstrated antitumor efficacy in an orthotopic RCC mouse model, where tumor growth inhibition was observed in 60% of cases. Histological analysis confirmed the infiltration of CAR+ and CD8+ T cells into the tumor microenvironment. Furthermore, when combined with Axitinib, a tyrosine kinase inhibitor, the therapy produced a synergistic antitumor effect, warranting further investigation in clinical studies (121).
2.1.3.2.7 EGFR
EGFR Specific CAR-NK92 construct has been evaluated in combination with Cabozantinib, a tyrosine kinase inhibitor, demonstrating significant antigen-specific activation and enhanced antitumor efficacy in both in vitro and in vivo preclinical models (122). Cabozantinib potentiates CAR-NK92 activity by upregulating EGFR expression and downregulating PD-L1 on tumor cells, thereby improving immune cell infiltration and reducing immune suppression within the tumor microenvironment (122). This synergistic interaction highlights the potential of combining CAR-NK92 therapy with Cabozantinib for targeting EGFR-positive solid tumors, particularly in RCC.
2.1.4 DCs
DCs, as the most potent antigen-presenting cells, play a pivotal role in initiating T-cell activation by cross-presenting tumor antigens via MHC-I and MHC-II molecules, thereby inducing durable immunological memory against pathogens and tumors (123, 124). Harnessing DC-mediated antitumor responses has been a central focus of cell-based cancer immunotherapies, with strategies ranging from loading DCs with tumor lysates, proteins, and peptides to genetically engineered vectors encoding tumor-specific neoantigens (125–129). Clinical studies have demonstrated the safety and partial efficacy of antigen-loaded DC vaccines, albeit with limited response rates (10-20% in most trials), highlighting the need for improved therapeutic strategies (130–133). Significant barriers, including tumor-mediated immune suppression (e.g., via IL-10 and TGF-β), downregulation of antigen expression, regulatory T-cell dominance, and robust checkpoint pathways (e.g., PD-1/PD-L1), impede the full potential of DC-based therapies (134, 135). To overcome these challenges, researchers are exploring combinatorial approaches, such as integrating DC vaccines with immune checkpoint inhibitors, cytokine modulation (e.g., IL-12 and IFN-α), and neoantigen-specific targeting, aiming to achieve more sustained clinical outcomes and broader patient benefits (136).
2.1.5 Vaccines
Cancer vaccines represent a promising immunotherapeutic strategy designed to stimulate the immune system to recognize and eliminate malignant cells (137, 138). Unlike traditional prophylactic vaccines that prevent virus-associated cancers, therapeutic cancer vaccines aim to enhance anti-tumor immunity in patients with established malignancies (139, 140). These vaccines introduce tumor-associated antigens or neoantigens to activate DCs, which in turn prime cytotoxic T lymphocytes to mount a targeted immune response against tumor cells (141, 142). While initial attempts using autologous tumor cell vaccines did not demonstrate significant clinical efficacy in phase III trials (143, 144), recent advances have focused on combining vaccines with ICIs to amplify immune responses. This synergistic approach aims to overcome tumor-induced immune suppression and improve therapeutic outcomes.
One promising strategy is NeoVax, a personalized neoantigen vaccine derived from tumor DNA sequencing. By incorporating synthetic neoantigens alongside the toll-like receptor agonist polyinosinic-polycytidylic acid stabilized with poly-L-lysine and carboxymethylcellulose, NeoVax has been shown to elicit durable immune responses (145). A phase I study in high-risk melanoma patients demonstrated its ability to induce sustained T-cell immunity (146). Building on these findings, a phase I trial (NCT02950766) is currently evaluating the combination of NeoVax and ipilimumab in RCC.
Despite encouraging preliminary results, challenges remain in fully harnessing the therapeutic potential of cancer vaccines for RCC. The immunosuppressive tumor microenvironment and tumor heterogeneity present significant hurdles to achieving sustained clinical benefit. While no RCC-specific vaccines have received regulatory approval, ongoing research is focused on improving vaccine formulations, identifying optimal target antigens, and integrating vaccines with existing treatment strategies to enhance efficacy. Continued clinical trials will be essential in defining the role of vaccine-based immunotherapy in RCC management.
2.1.6 Summary of immunotherapy for RCC
The landscape of RCC treatment has been transformed by immunotherapy, with ICIs, adoptive cell therapies, and cancer vaccines demonstrating significant clinical potential. By disrupting inhibitory pathways and enhancing immune activation, these approaches have improved patient outcomes, particularly in advanced RCC. However, despite these advancements, immunotherapy alone is often insufficient to achieve durable tumor control. Given the molecular complexity of RCC, targeted therapies remain essential, directly inhibiting oncogenic signaling pathways that drive tumor progression. The integration of immunotherapy with targeted agents represents a promising strategy, leveraging the strengths of both modalities to optimize treatment efficacy and overcome resistance mechanisms.
3 Targeted therapy in RCC
3.1 Targeted therapy origins
The advent of targeted therapy emerged from breakthroughs in understanding cancer molecular mechanisms in the late 20th century. The approval of imatinib (Gleevec) in the late 1990s, the first successful TKI, revolutionized cancer treatment by dramatically improving outcomes for chronic myeloid leukemia through the specific inhibition of Breakpoint Cluster Region-Abelson (147). This milestone spurred the development of targeted therapies across various cancers, including RCC. In 2005, the approval of sorafenib marked the introduction of targeted treatment for RCC, paving the way for multi-targeted TKIs (148). Currently, the targeted therapies for RCC mainly focus on key oncogenic drivers, as is shown in Figure 2.
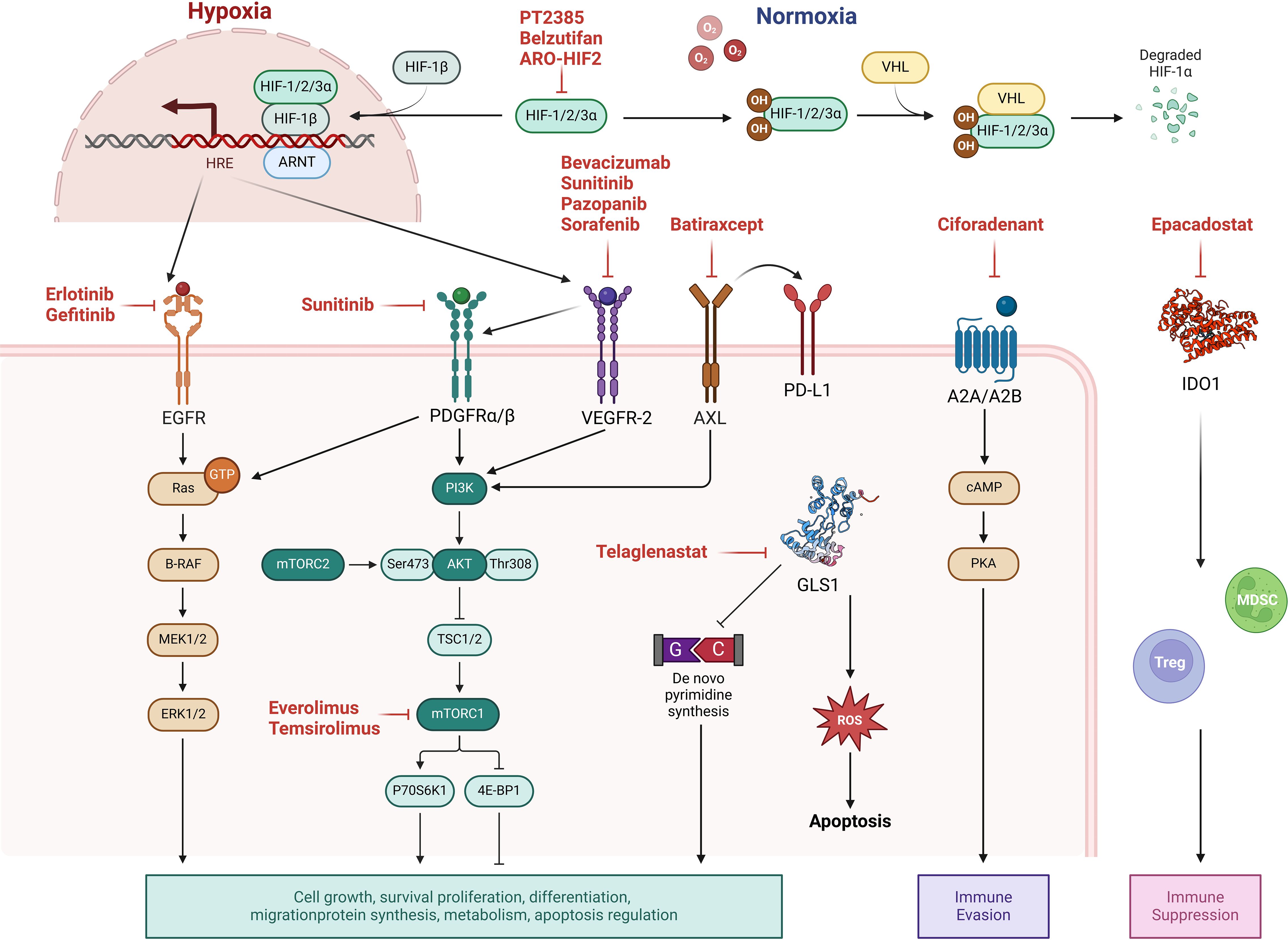
Figure 2. Molecular targets and pathway modulations in renal cell carcinoma (RCC) therapy. Current targeted therapies for RCC focus on key oncogenic drivers: (1) Hypoxia-inducible factor 2α (HIF2α) inhibition in clear cell RCC (ccRCC) to disrupt hypoxia response signaling; (2) Growth factor signaling blockade targeting vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF) pathways; (3) mTOR pathway modulation through inhibition of mechanistic target of rapamycin (mTOR); (4) Metabolic reprogramming interventions including Inhibition of glutaminase 1 (GLS1); (5) Receptor tyrosine kinase inhibition (e.g., AXL kinase blockade); and (6) Adenosine receptor antagonism to counteract immunosuppressive tumor microenvironments. These strategies collectively aim to interfere with tumor survival, proliferation, and microenvironmental adaptation.
ccRCC, the most prevalent RCC subtype, frequently harbors mutations in the tumor suppressor gene von Hippel-Lindau (VHL). VHL loss leads to aberrant stabilization and overexpression of hypoxia-inducible factor 2 alpha (HIF2α), which dysregulates multiple tumorigenic pathways (149). HIF2α promotes angiogenesis by upregulating vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), enhances cell proliferation via cyclin D1 and glucose metabolism through glucose transporter 1, and drives tumor invasion and metastasis via stromal cell-derived factor 1 and its receptor C-X-C motif chemokine receptor 4 (150). Additionally, HIF2α attenuates EGFR endocytosis, sustaining EGFR-mediated signaling and contributing to tumor growth and progression (151, 152).
In RCC, the mTOR pathway is frequently hyperactivated, often due to VHL mutations or dysregulated upstream signaling (9). This aberrant activation promotes downstream targets, including S6 kinase and eukaryotic translation initiation factor 4E-binding protein 1, enhancing protein synthesis and cellular proliferation (10). The intricate interconnections between the VHL-HIF2α pathway and mTOR signaling underscore their collective role in RCC pathogenesis, offering multiple potential therapeutic targets. Consequently, therapeutic interventions targeting these pathways continue to be a focus of ongoing research and clinical trials (11, 12) (Table 3).
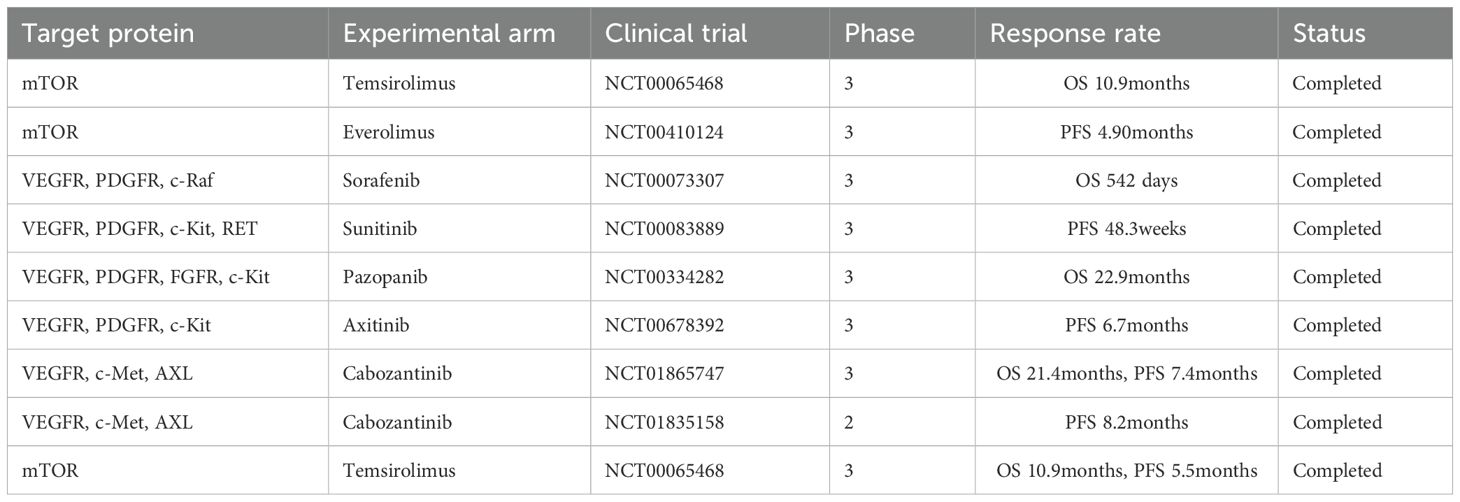
Table 3. Selected pivotal studies of targeted therapies approved as monotherapies for the treatment of RCC.
3.1.1 HIF2α inhibition in ccRCC
The VHL tumor suppressor gene, located on chromosome 3p, plays a central role in the pathogenesis of ccRCC. Inactivating mutations in both alleles of VHL represent the most prevalent genetic alterations in ccRCC, observed in both sporadic and hereditary cases (153). VHL disease, an autosomal dominant hereditary cancer syndrome, stems from germline VHL mutations, conferring a 70% lifetime risk for RCC, along with increased susceptibility to tumors such as hemangioblastomas and paragangliomas (154).
The VHL protein (pVHL) functions as a key component of the E3 ubiquitin ligase complex, which regulates the degradation of hypoxia-inducible factors (HIFs), including hypoxia-inducible factor 1 alpha (HIF1α), hypoxia-inducible factor 2 alpha (HIF2α), and hypoxia-inducible factor 3 alpha (HIF3α) (155). Among these, HIF2α has emerged as a critical driver of ccRCC progression (153). Under normoxic conditions, the pVHL/E3 ubiquitin ligase complex targets HIF1α and HIF2α for proteasomal degradation (156). VHL inactivation leads to the accumulation of HIFα subunits, which dimerize with HIFβ to form an active transcriptional complex. This complex induces the expression of hypoxia-responsive genes such as VEGF, PDGF-β, and erythropoietin, contributing to ccRCC’s characteristic hypervascularity and paraneoplastic erythrocytosis (154). Clinical trials have been conducted to evaluate the efficacy and safety of enrolled HIF-2α inhibitors for RCC (Table 4).
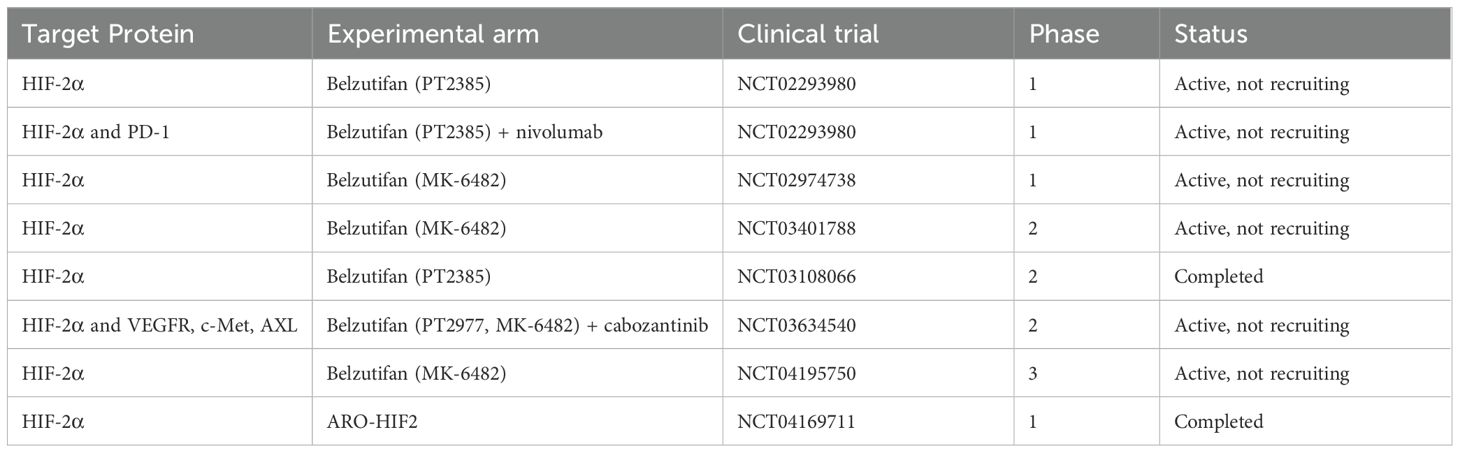
Table 4. Selected pivotal HIF-2α inhibitors Clinical Trials for RCC.
Initially considered an undruggable target due to the absence of a clear ligand-binding domain, HIF2α became a viable therapeutic target with the identification of a small binding site within its Per-Arnt-Sim B domain. This discovery led to the development of PT2385, the first HIF2α-specific inhibitor (157, 158).
A Phase I dose-escalation trial involving 51 pretreated metastatic ccRCC patients demonstrated PT2385’s favorable safety profile, with anemia, peripheral edema, and fatigue as the most common adverse effects. Partial and complete responses were observed in 12% and 2% of patients, respectively, while 52% achieved disease stabilization (159). Pharmacokinetic and pharmacodynamic analyses identified 800 mg twice daily as the optimal dose for Phase II trials. Notably, PT2385 rapidly suppressed HIF2α-mediated erythropoietin expression, confirming target engagement and biological activity (160).
Despite these promising findings, PT2385 exhibited variable pharmacokinetics, prompting the development of Belzutifan (MK-6482, formerly PT2977), a second-generation HIF2α inhibitor with improved pharmacokinetic properties (161). Structural modifications, including the replacement of a geminal difluoro group with a cis-vicinal difluoro group, enhanced Belzutifan’s serum availability and binding affinity for HIF2α (162).
The first-in-human Phase I/II study (NCT02974738) evaluated Belzutifan in patients with advanced ccRCC and other solid tumors. Belzutifan achieved an ORR of 24%, with 67% of patients experiencing tumor size reduction. The median PFS was 11.0 months. Anemia was the most common adverse event, primarily managed with EPO replacement without necessitating dose reductions. Hypoxia occurred in 26% of patients but was generally manageable with supplemental oxygen.
Further studies have demonstrated Belzutifan’s efficacy in VHL disease-associated RCC. A Phase II trial (NCT03401788) involving 61 patients reported a 49% ORR and a 98% 12-month PFS. Belzutifan also showed efficacy in non-RCC manifestations, including pancreatic neuroendocrine tumors (77% ORR) and central nervous system hemangioblastomas (30% ORR) (163). Based on these findings, the FDA approved Belzutifan in August 2021 for VHL-associated RCC and other tumors not requiring immediate surgical intervention.
Notably, a recent phase III trial showed that belzutifan significantly improved progression-free survival and objective response rates compared to everolimus in patients with advanced ccRCC previously treated with immune checkpoint inhibitors and antiangiogenic therapies (NCT04195750) (164). The ongoing LITESPARK-001 Phase I study has highlighted Belzutifan’s potential in treating metastatic ccRCC, achieving a 25% ORR over a median 3-year follow-up (165). Combination trials are underway, including Belzutifan with cabozantinib (NCT03634540) (166) and pembrolizumab (NCT05239728) (167), aiming to enhance therapeutic outcomes.
RNA interference (RNAi)-based therapies targeting HIF2α are also emerging as promising strategies (168). ARO-HIF2, an RNAi therapy, demonstrated encouraging interim results in a Phase Ib trial (NCT04169711), with one partial response and stable disease in several patients (169). Several Phase II/III trials assessing HIF2α inhibitors, either solo or combined with ICIs, TKIs, and novel analogs, are ongoing. These findings underscore the potential of novel therapeutic approaches to overcome resistance mechanisms associated with HIF2α inhibition.
The development of HIF2α inhibitors, particularly Belzutifan, marks a significant advancement in ccRCC therapy. Ongoing clinical trials and preclinical studies continue to explore combination strategies and novel therapeutic approaches. As understanding of resistance mechanisms and biomarker development evolves, HIF2α inhibition is poised to remain a cornerstone of targeted therapy for ccRCC.
3.1.2 VEGF signaling pathway in RCC
VEGF is a key driver of angiogenesis and is produced by various cell types, including tumor cells, stromal fibroblasts, inflammatory cells, and endothelial cells (160). VEGF exerts its pro-angiogenic effects primarily by binding to specific receptors on vascular endothelial cells: VEGFR-1, VEGFR-2, and VEGFR-3. Among these, VEGFR-2 is particularly critical for angiogenesis (170). Upon VEGF-A binding, VEGFR-2 undergoes dimerization and activates tyrosine kinases, triggering downstream signaling pathways such as PI3K/Akt and MAPK. This activation promotes endothelial cell survival, proliferation, and neovascularization (171).
In the context of RCC, VEGF and its receptors are markedly upregulated, often in response to hypoxia-induced HIF activation within the tumor microenvironment. The loss of VHL protein, a hallmark of ccRCC, leads to constitutive stabilization of HIF, driving the overexpression of VEGF-A. This upregulated VEGF-A binds to VEGFR-2, accelerating angiogenesis and providing essential nutrients and oxygen to the tumor. Consequently, the VEGF-A/VEGFR-2 axis has emerged as a critical therapeutic target in RCC due to its profound role in stimulating angiogenesis (172).
Moreover, VEGF contributes to immune evasion in RCC by inhibiting the maturation of DCs, impairing T-cell activation, and promoting the recruitment of immunosuppressive regulatory T cells. The interaction between VEGF and immune checkpoints, such as the PD-1/PD-L1 axis, further enhances tumor immune evasion. These insights have prompted the development of combination therapies targeting both angiogenesis and immune checkpoints (173).
A variety of VEGF inhibitors, including monoclonal antibodies and TKIs, have been approved for the treatment of RCC. These agents disrupt VEGF-VEGFR interactions or inhibit receptor tyrosine kinase activity to block angiogenesis and tumor growth. Bevacizumab, a monoclonal antibody targeting VEGF-A, demonstrated improved PFS in phase II clinical trials, although long-term survival benefits remain limited (174). Among TKIs, sunitinib and pazopanib are well-established first-line therapies for mRCC, consistently reducing tumor progression in clinical studies (175). Another TKIs, sorafenib, initially developed as a B-Raf inhibitor, was later found to inhibit VEGFR2 (KDR) in ccRCC due to the pivotal role of pVHL loss. Cabozantinib and lenvatinib are primarily used as second-line treatments; cabozantinib inhibits VEGFRs as well as MET and AXL receptors (176), targeting resistance mechanisms, while lenvatinib concurrently blocks VEGFR and Fibroblast Growth Factor Receptor (FGFR) (177). Recent phase III trials have demonstrated the superiority of both drugs in PFS compared to everolimus in pre-treated patients. Additionally, tivozanib, a selective TKI targeting VEGFR1, VEGFR2, and VEGFR3, has demonstrated significant improvements in median PFS compared to sorafenib in two pivotal phase III clinical trials, underscoring its potential as a promising therapeutic option for advanced RCC (178, 179). Cabozantinib was found to be superior to sunitinib in the front-line setting (180) and was also superior to everolimus in patients previously treated with other TKIs that target VEGF (181, 182). Lenvatinib (in combination with everolimus) was also superior to everolimus alone in previously treated patients with RCC (183). It is important to note that although the perceived greater clinical benefit with cabozantinib or lenvatinib, compared with that of earlier TKIs, has been largely considered to be due to their targeting of resistance pathways to VEGF, the benefit may simply be due to more-potent inhibition of KDR (184).
Complete responses to VEGF inhibitors are rare, with tumors often activating angiogenic escape pathways to restore perfusion (185). Recent TKIs such as cabozantinib and lenvatinib provide dual targeting capabilities to overcome resistance (186). Cabozantinib has shown superiority to sunitinib as a front-line treatment and to everolimus in pre-treated patients. Lenvatinib, combined with everolimus, demonstrated enhanced efficacy compared to everolimus monotherapy (187, 188).
Attempts to target other angiogenic pathways, such as the angiopoietin-Tie axis and the Transforming Growth Factor-beta (TGFβ)-Activin Receptor-Like Kinase 1 (ALK1)-endoglin pathway, have largely been unsuccessful (189, 190). For example, combining trebananib (a peptibody neutralizing Angiopoietin-1 and Angiopoietin-2) with sorafenib did not improve PFS compared to sorafenib alone (191). Similarly, the endoglin inhibitor carotuximab and the ALK1 inhibitor dalantercept failed to show superior efficacy when combined with axitinib compared to axitinib monotherapy (192).
However, it should be noted that inhibition of multiple targets by TKIs can result in various AE, necessitating careful monitoring. Hematologic and hepatic toxicities are particularly significant, and VEGF receptor blockade is associated with hypertension and other cardiovascular complications. Additionally, pazopanib may induce side effects such as hair depigmentation, diarrhea, nausea, anorexia, vomiting, and, in rare cases, posterior reversible encephalopathy syndrome (193).
3.1.3 PDGF signaling pathway in RCC
PDGF comprises four isoforms: PDGF-A, PDGF-B, PDGF-C, and PDGF-D. These isoforms form functional dimers that bind to PDGF receptors (PDGFR), which have two subtypes: PDGFRα and PDGFRβ (194). Upon ligand binding, these receptors dimerize and activate downstream signaling cascades, including the PI3K-AKT and Ras-MAPK pathways (195). This activation promotes processes such as tumor growth, angiogenesis, metastasis, and modulation of the tumor microenvironment in RCC. Therapeutic strategies targeting the PDGF-PDGFR axis have shown promise in inhibiting angiogenesis and cell proliferation, making it a potential avenue for RCC treatment (194).
A key finding from recent studies is the role of VEGF165b, an inhibitory isoform of VEGF-A, in modulating PDGFRβ phosphorylation. This interaction suppresses endothelial cell proliferation and migration, potentially contributing to resistance against antiangiogenic therapies. Evidence from clinical and preclinical studies indicates that concurrent activation of PDGF-B/PDGFRβ and VEGF165b pathways may lead to complex vascular phenotypes and therapeutic resistance (196).
A comprehensive study analyzed the immunohistochemical expression of PDGF-B, and its receptor PDGFRβ in 1,423 prospectively collected tumor samples from patients undergoing radical or partial nephrectomy at a tertiary referral center. Among the 1,091 patients (mean age: 54 years), the majority (88.7%) had ccRCC, followed by papillary (7.5%), chromophobe (2.8%), unclassified (0.4%), and other rare types (0.5%). The findings revealed that PDGFRβ expression was highest in ccRCC, whereas PDGF-B expression were most prominent in papillary RCC. After adjusting for T stage and Fuhrman nuclear grade, multivariate logistic regression analysis demonstrated that PDGF-B (OR = 2.46, P = 0.019) expression were significantly higher in papillary RCC compared to clear cell type. These findings underscore the distinct biological characteristics and angiogenic profiles of RCC subtypes (197).
Another study evaluated the prognostic significance of PDGF-BB expression and differentiated microvascular density in 100 patients with ccRCC using immunohistochemistry on tissue microarrays. The results indicated that higher-grade and more advanced-stage ccRCC tumors exhibited significantly lower PDGF-BB expression and differentiated MVD (P < 0.05). Interestingly, elevated PDGF-BB expression emerged as an independent prognostic factor for improved survival. Incorporating PDGF-BB expression into a clinicopathological model significantly enhanced the predictive accuracy for disease-free survival, increasing Harrell’s concordance index from 0.695 to 0.707. A strong positive correlation was observed between PDGF-BB expression and differentiated microvascular density (r = 0.634, P < 0.001), suggesting that PDGF-BB may play a role in vascular differentiation. These insights suggest that targeting PDGF-BB and its associated pathways may provide therapeutic benefits and enhance current anti-angiogenic strategies for RCC management (198).
PDGF-C plays an essential role in developmental and physiological processes as well as in human diseases. A novel splice variant of PDGF-C, termed PDGF-Cb, encodes an N-terminally truncated protein lacking the signal peptide and Complement C1r/C1s, Uegf, and Bmp1 domain. This variant is co-expressed with PDGF-C in normal tissues. PDGF-Cb is produced as a cytoplasmic protein with a similar intracellular localization to PDGF-C but is not secreted from cells. PDGF-Cb can form heterodimers (PDGF-CCb) with PDGF-C, retaining it within the cytoplasm and targeting it for degradation. In primary RCC tumors, expression of the full-length PDGF-C transcript was elevated, while PDGF-Cb expression remained unchanged. These findings suggest that deregulation of PDGF-C may contribute to RCC tumorigenesis and that PDGF-Cb acts as a dominant-negative molecule modulating PDGF-C secretion (199).
A real-world analysis assessed the effectiveness and safety profile of sunitinib in 702 advanced or metastatic RCC (mRCC) patients from 116 German sites (STAR-TOR registry, NCT00700258). Between 2010 and 2020, sunitinib was administered as first-line (83.5%), second-line (11.7%), or third-line (4.8%) therapy. Clear-cell RCC was the predominant subtype (81.6%). Drug-related AEs were reported in 66.3% of patients, with gastrointestinal disorders being the most common (39.7%). Serious AEs occurred in 13.9% of patients. This study provides critical insights into the real-world outcomes and AE profile of sunitinib in a/mRCC patients (200).
Ball et al. (2007) demonstrated that VEGF-A not only stimulates the expression of PDGFRα and PDGFRβ but also binds directly to both receptor types. This interaction positions VEGF-A as a key regulator for recruiting both endothelial cells and perivascular smooth muscle cells, highlighting its multifaceted role in vascular development. The interplay between PDGFRβ and VEGF pathways promotes endothelial cell migration and proliferation while facilitating the recruitment of perivascular cells, which are essential for stabilizing and maturing blood vessels (201).
In conclusion, targeting the PDGF-B/PDGFRβ axis in combination with VEGF inhibitors may offer a novel therapeutic approach for managing resistance to conventional anti-angiogenic therapies in RCC. Further studies are warranted to elucidate the precise mechanisms underlying these interactions and their clinical implications.
3.1.4 Epidermal Growth Factor signaling pathway in RCC
The Epidermal Growth Factor (EGF) signaling pathway plays a crucial role in the development and progression of RCC. Upon binding to the EGFR, EGF triggers receptor dimerization and autophosphorylation, leading to the activation of downstream pathways such as PI3K/Akt, Ras/Raf/MAPK/ERK Kinase (MEK)/Extracellular Signal-Regulated Kinase (ERK), and Janus Kinase (JAK)/Signal Transducer and Activator of Transcription (STAT). These pathways regulate essential cellular processes, including proliferation, survival, angiogenesis, and metastasis (202). In RCC, particularly the clear-cell subtype, the loss of the VHL gene contributes to upregulated HIF2α activity, which enhances EGFR expression. This upregulation promotes tumor growth and survival, making the EGF/EGFR axis a potential therapeutic target (203).
Despite the established role of EGFR-targeting agents in other cancers, such as non-small cell lung cancer and colorectal cancer, their efficacy in RCC has been limited. Early clinical trials using first-generation EGFR inhibitors, such as erlotinib and gefitinib, showed only modest efficacy in unselected RCC populations (203). This limited success can be attributed to the intrinsic molecular heterogeneity of RCC and compensatory activation of alternative pathways, notably EGF-driven angiogenesis. Efforts to overcome these limitations have led to the investigation of combination therapies targeting both EGF and VEGF pathways.
A notable example involved a phase II trial exploring the combination of erlotinib and bevacizumab, a VEGF inhibitor, in patients with metastatic RCC. The combination demonstrated a modest improvement in progression-free survival compared to erlotinib monotherapy but was associated with significant toxicities, including skin rash and gastrointestinal disturbances (204). Another study evaluated gefitinib in combination with sorafenib, a VEGF-targeting TKI, in refractory RCC patients. Although the combination showed limited clinical benefit, it underscored the importance of patient selection and the need for novel therapeutic strategies (205).
Given the limited efficacy of EGFR inhibitors alone, recent studies have explored their combination with ICIs such as pembrolizumab. Preclinical models suggest that EGFR inhibition may enhance tumor immunogenicity, thereby improving responses to ICIs. Additionally, multi-kinase inhibitors like cabozantinib, which target VEGFR, MET, and AXL, have shown promise in overcoming resistance mechanisms associated with EGFR inhibition (206). The dual inhibition of angiogenesis and EGF signaling offers a promising approach to enhance antitumor efficacy by disrupting key survival pathways in RCC (207).
The use of EGFR inhibitors is associated with various adverse effects. Dermatologic toxicities, particularly acneiform rash, are among the most common side effects and may require corticosteroid treatment. Gastrointestinal disturbances, such as diarrhea and mucositis, are also frequently reported. Hepatotoxicity, characterized by elevated liver enzymes, may necessitate dose adjustments. In rare cases, interstitial lung disease has been observed, requiring immediate intervention. Despite these challenges, the integration of EGFR inhibitors with other targeted therapies holds promise for addressing the complex tumor microenvironment in RCC (208).
Future research efforts are focused on identifying predictive biomarkers to guide patient selection and optimizing combination therapies to maximize therapeutic efficacy. The development of next-generation EGFR inhibitors with improved selectivity and safety profiles is also underway. Understanding the intricate interplay between EGF signaling and other oncogenic pathways remains crucial for developing more effective and durable treatment options for RCC patients.
3.1.5 mTOR signaling pathway in RCC
The mammalian mTOR is an atypical serine/threonine protein kinase that plays a critical role within the PI3K/Akt/mTOR signaling cascade. Activation of this pathway drives the proliferation and invasiveness of renal cancer cells by phosphorylating key downstream targets such as ribosomal protein S6 kinase and eukaryotic translation initiation factor 4E-binding protein 1. These phosphorylation events facilitate protein synthesis and promote cell cycle progression, thereby enhancing the proliferative potential of renal cancer cells (209).
Beyond its role in cell proliferation, mTOR signaling also regulates the epithelial-mesenchymal transition process, granting renal cancer cells enhanced invasive and migratory capacities. Additionally, mTOR signaling protects these cancer cells from apoptotic signals by inhibiting apoptosis-related proteins, thereby promoting their survival and sustained proliferation. Another crucial function of the mTOR pathway is the promotion of angiogenesis within renal cancer tissues through the upregulation of VEGF (210). This neovascularization provides cancer cells with essential nutrients and oxygen, contributing to tumor growth and dissemination.
Given its central role in RCC pathogenesis, inhibiting mTOR activity has emerged as a promising therapeutic strategy. Currently, two mTOR inhibitors, everolimus and temsirolimus, have been approved for the treatment of advanced RCC. Temsirolimus is approved as a first-line therapy, particularly for poor-risk patients, while everolimus is commonly used as a second-line option following the failure of VEGFR inhibitor-based treatments. In a pivotal phase III trial, everolimus demonstrated a trend toward improved recurrence-free survival compared to placebo; however, this benefit was accompanied by an increase in AE, including stomatitis, hypertriglyceridemia, and hyperglycemia (211).
At the molecular level, some ccRCC tumors harbor mutations in genes encoding components of the mTOR pathway, further underscoring the importance of targeting this signaling cascade (212). mTOR functions within two distinct multiprotein complexes, mTORC1 and mTORC2 (213). Rapamycin analogs (‘rapalogs’) such as everolimus and temsirolimus selectively inhibit mTORC1, leading to reduced translation of mRNAs involved in cell survival, proliferation, and angiogenesis (214). Despite relatively low response rates, randomized controlled trials have demonstrated the superiority of everolimus and temsirolimus over interferon-α and placebo, respectively, leading to their regulatory approval for advanced RCC treatment (215, 216).
However, the inhibition of mTORC1 has been associated with the unintended consequence of relieving negative feedback inhibition on mTORC2 (217). This can stabilize hypoxia-inducible factor 2 alpha (HIF2α), thereby enhancing PI3K and Akt-mediated cell survival and proliferation (218–220). Preclinical studies in ccRCC cell lines have suggested that novel agents capable of inhibiting both mTORC1 and mTORC2, as well as PI3K, may be more effective than rapalogs (221, 222). Unfortunately, clinical trials evaluating dual mTORC1/mTORC2 inhibitors (e.g., AZD2014 and sapanisertib) and mTOR/PI3K inhibitors (e.g., apitolisib and BEZ235) encountered significant challenges due to considerable on-target toxicity, such as hyperglycemia (223–225). These adverse effects often necessitated dose reductions, which may have compromised their therapeutic efficacy.
Despite these setbacks, ongoing research continues to explore innovative approaches to improve the efficacy and safety of mTOR-targeted therapies in RCC. Combining mTOR inhibitors with other targeted agents or immune checkpoint inhibitors is being investigated to overcome resistance mechanisms and enhance therapeutic outcomes (210). Recent studies have further revealed that down-regulation of lactotransferrin, a critical protein involved in the innate immune system, promotes metastasis in RCC. Interestingly, this down-regulation has also been associated with increased sensitivity of RCC tumor cells to mTOR inhibitors, suggesting that lactotransferrin expression may serve as a predictive biomarker for therapeutic efficacy (226). Understanding the intricate interactions between mTOR signaling and other oncogenic pathways remains critical for the development of more effective and durable treatments for RCC.
3.1.6 Glutaminase inhibition in RCC
In RCC, particularly in cells deficient in the VHL tumor suppressor gene, there is a pronounced reliance on glutamine for metabolic processes. Glutamine serves as a critical substrate for producing key molecules such as citrate and lipids through metabolic pathways that support cellular growth and survival. Inhibition of glutaminase 1 disrupts these pathways by reducing intracellular levels of aspartic acid, thereby inhibiting de novo pyrimidine synthesis, which impairs DNA synthesis and cellular proliferation. Moreover, glutaminase 1 inhibition leads to the accumulation of reactive oxygen species, inducing oxidative stress and triggering apoptosis in RCC cells (227).
Telaglenastat (CB-839) is a first-in-class, selective, reversible, and orally active GLS1 inhibitor that has been investigated in several clinical trials for its potential to enhance therapeutic outcomes in RCC. The ENTRATA trial, a randomized, double-blind, phase II study, evaluated the combination of telaglenastat and everolimus (T + E) versus placebo plus everolimus (P + E) in 69 patients with mRCC who had progressed after two or more prior systemic therapies. The trial showed a trend toward improved median PFS with T + E (3.8 months vs. 1.9 months, HR 0.64, p = 0.079). However, grade ≥3 AE (AEs) were more frequent in the T + E group (80%) compared to the P + E group (60%). The most common AEs included anemia (17% vs. 17%), pneumonia (7% vs. 4%), abdominal pain (7% vs. 0%), thrombocytopenia (7% vs. 0%), and fatigue (4% vs. 9%). Despite these toxicities, discontinuation due to AEs was similar between groups (28% vs. 30%) (228).
The subsequent phase II CANTATA trial evaluated telaglenastat in combination with cabozantinib (T + C) versus placebo plus cabozantinib (P + C) in patients with advanced ccRCC who had progressed on prior first- or second-line therapies, including anti-angiogenic or ICI-based regimens. Among 444 randomized patients, no statistically significant difference in PFS was observed between the T + C and P + C groups (median PFS 9.2 months vs. 9.3 months, HR 0.94, p = 0.65). ORR were similar (31% vs. 28%, respectively), and OS data remained immature at the time of analysis. In a prespecified subgroup analysis, patients who had previously received ICI-based therapy showed a trend toward PFS benefit with T + C (11.1 months vs. 9.2 months, HR 0.77) (229).
AE were prevalent in both treatment arms, with grade 3–4 AEs occurring in 71% of patients on T + C and 79% in those on P + C. The most common grade 3–4 AEs included hypertension (17% vs. 18%) and diarrhea (15% vs. 13%). These findings highlight the challenges of combining glutaminase inhibition with other targeted therapies in RCC and underscore the need for better patient selection and toxicity management strategies.
While glutaminase inhibition holds promise as a therapeutic approach in RCC, the modest clinical outcomes observed in current trials suggest that further optimization is necessary. Potential strategies include the development of more potent glutaminase 1 inhibitors, improved combination regimens, and the identification of predictive biomarkers to guide patient selection.
3.1.7 AXL in RCC
AXL is a receptor tyrosine kinase that plays a crucial role in immune regulation and tumor progression. Normally expressed in both immune and non-immune cells, AXL is upregulated in ccRCC and is associated with poor prognosis (230). Elevated AXL levels contribute to immune evasion by increasing PD-L1 expression and promoting the clearance of tumor antigens, thereby limiting effective anti-tumor immune responses (230). Additionally, AXL activation is essential for the PI3K/AKT signaling pathway via VEGF, which may underlie resistance to anti-angiogenic therapies (185).
Targeting AXL has emerged as a promising therapeutic approach in ccRCC. Batiraxcept, a recombinant fusion protein that inhibits AXL by binding to its activating ligand Growth Arrest-Specific 6, is currently being evaluated in a phase 1/2b study in combination with cabozantinib for patients with advanced ccRCC. Interim analysis showed an acceptable safety profile, with the most common AEs being decreased appetite, diarrhea, and fatigue. The ORR was 46%, and a baseline serum soluble AXL (sAXL)/Growth Arrest-Specific 6 ratio of 2.3 or greater was associated with a higher ORR of 67%, suggesting its potential as a predictive biomarker (231).
3.1.8 Adenosine receptor inhibition
Metabolic reprogramming within the TME is another critical mechanism contributing to immune evasion and resistance to ICIs (232). One key pathway involves the breakdown of extracellular Adenosine Triphosphate(ATP) into immunosuppressive adenosine (233). ATP, a metabolite released during pro-inflammatory stimuli, promotes immune responses by activating T cells and inducing cytokine production. However, regulatory mechanisms exist to protect tissues from excessive immune activation. This includes the enzymatic conversion of ATP to adenosine by ectonucleotidases CD39 and CD73 (234).
In the TME, hypoxic conditions upregulate the expression of CD39 and CD73 (235), accelerating the breakdown of ATP into adenosine and leading to its accumulation. Adenosine exerts potent immunosuppressive effects by engaging the adenosine receptors A2A and A2B, which are expressed on various immune cell subsets (236). Hypoxia further increases the expression of A2A and A2B receptors, enhancing cell responsiveness to adenosine and promoting immune evasion (234). Compared to other solid tumors, RCC exhibits higher levels of A2AR and CD73 expression, underscoring the relevance of this pathway in disease progression (237).
Targeting the adenosine pathway has shown promise in clinical trials. Ciforadenant (formerly known as CPI-444), a small molecule A2AR antagonist, has been evaluated in a phase I clinical trial involving patients with advanced refractory RCC. The trial demonstrated clinical responses both as monotherapy and in combination with atezolizumab, an anti-PD-L1 antibody. Importantly, higher baseline levels of adenosine-induced genes in tumor biopsies were associated with tumor regression, suggesting their potential as predictive biomarkers for identifying patients who are likely to benefit from adenosine pathway inhibition (237).
The combination of AXL and A2AR inhibition, alongside ICIs and anti-angiogenic therapies, holds promise for overcoming immune resistance and enhancing treatment outcomes in RCC. Continued research and clinical validation are needed to optimize therapeutic strategies and identify predictive biomarkers for patient selection.
3.1.9 Tryptophan catabolism pathway in RCC
The tryptophan catabolism pathway plays a pivotal role in modulating immune responses within TME. Indoleamine 2,3-dioxygenase 1 (IDO1) is a key catabolic enzyme that degrades tryptophan, a critical amino acid for T-cell proliferation. The depletion of tryptophan in the TME promotes immunosuppression by activating regulatory T cells and MDSCs, fostering an immune-tolerant environment that supports tumor progression (238).
Inhibiting IDO1 has emerged as a promising therapeutic strategy to restore anti-tumor immunity. Epacadostat, a potent and selective IDO1 inhibitor, reduces tryptophan catabolism, thereby enhancing immune surveillance (239). By increasing the proliferation of effector T cells and NK cells while suppressing the expansion of Tregs, epacadostat reactivates anti-tumor immune responses. The combination of epacadostat with pembrolizumab, an anti-PD-1 immune checkpoint inhibitor, has been investigated in a phase I trial involving patients with advanced solid tumors, including RCC. Among 33 patients in the advanced RCC cohort, the ORR was 47% for those with zero-to-one prior treatments and 37% for patients who had undergone two or more prior therapies. The most common treatment-related AE included fatigue, rash, arthralgia, and diarrhea (240).
Building on these promising early-phase results, the combination of epacadostat and pembrolizumab is currently being evaluated as a first-line therapy in a phase III trial (NCT03260894) against the previous standard of care (sunitinib or pazopanib). This ongoing trial aims to establish whether dual IDO1 and PD-1 blockade can provide superior clinical outcomes in advanced RCC patients (241).
Additionally, the RENAVIV randomized phase III trial (NCT03592472) is underway, comparing pazopanib plus abexinostat versus pazopanib plus placebo in first- or second-line settings for patients with advanced RCC. Abexinostat, a histone deacetylase inhibitor, is believed to exert synergistic effects by modulating immune responses and enhancing the efficacy of VEGF inhibitors such as pazopanib. The outcomes of this trial are expected to shed light on the potential benefits of combining metabolic and epigenetic modulation in RCC treatment.
While targeting the tryptophan catabolism pathway offers a novel therapeutic avenue, further studies are required to optimize treatment regimens and identify predictive biomarkers for patient selection. Understanding the complex interactions between metabolic pathways and immune checkpoints is essential for the development of effective combination therapies in RCC.
3.1.10 Exosome-based biomarkers in RCC
In recent years, advances in medical diagnostic technology have significantly improved early detection rates and clinical outcomes in RCC. Exosomes, small extracellular vesicles with a unique bilayer membrane structure, have emerged as a promising non-invasive source of tumor biomarkers. This bilayer provides protection against external Ribonucleases and proteases, thereby preserving the integrity of enclosed messenger RNAs, microRNAs, and functional proteins, and enhancing the stability and sensitivity of these markers for disease diagnosis. Tumor-derived exosomes, particularly their miRNA cargo, have shown potential as biomarkers in the serum and urine of RCC patients, offering valuable targets for early detection, disease monitoring, and therapeutic stratification (242).
4 Integration of radiotherapy, chemotherapy, and immunotherapy/targeted therapy in RCC
RCC has traditionally been considered resistant to radiotherapy and chemotherapy. However, recent advances in immunotherapy and targeted therapy have led to renewed interest in combining these modalities to enhance treatment efficacy (243). And we summarized the pivotal studies of combination therapies with immunotherapies and targeted Therapies for the treatment of RCC in Table 5. Radiotherapy, while not conventionally used as a primary treatment for RCC, has been shown to induce immunogenic cell death, releasing tumor-associated antigens that promote dendritic cell activation and enhance T-cell priming. Additionally, radiotherapy can modulate the TME by increasing the expression of immune checkpoint molecules such as PD-L1, thereby sensitizing tumors to ICIs (244–246). Clinical studies, including the NIVES trial, have demonstrated that stereotactic body radiotherapy combined with nivolumab results in increased immune activation and durable responses in selected patients (247). Similarly, the RADVAX-RCC trial investigated stereotactic body radiotherapy in combination with dual checkpoint blockade using nivolumab and ipilimumab, showing enhanced T-cell activation and improved response rates. These findings suggest that radiotherapy may act as an immunomodulatory agent that enhances the efficacy of ICIs in RCC (248).
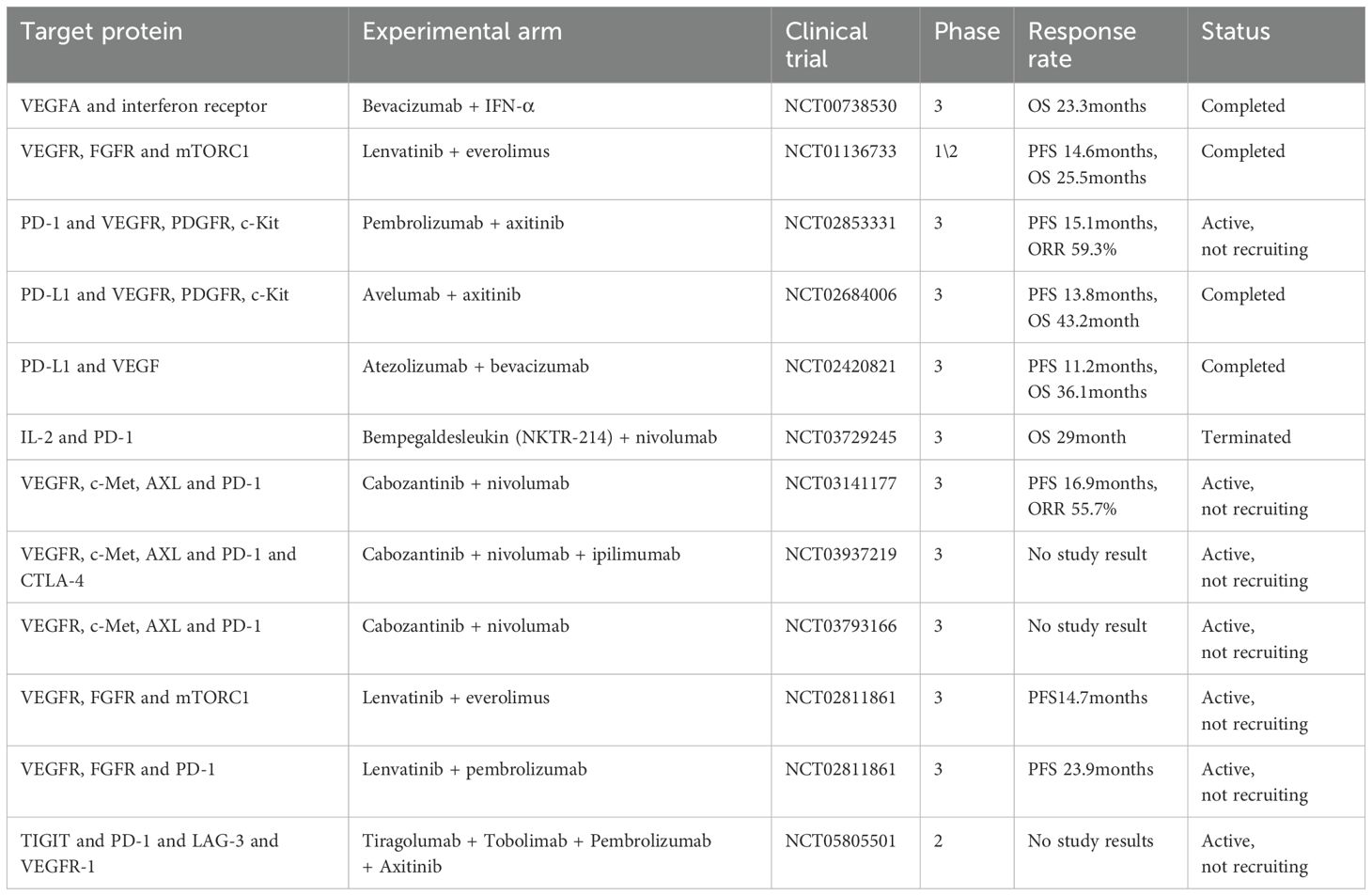
Table 5. Selected pivotal studies of combination therapies with Immunotherapies and Targeted Therapies for the treatment of RCC.
Beyond its synergy with immunotherapy, radiotherapy has also been investigated in combination with targeted therapy, particularly VEGF-targeted TKIs (249–251). Preclinical and clinical studies have demonstrated that radiotherapy can disrupt tumor vasculature, leading to increased sensitivity to anti-angiogenic agents. For example, stereotactic body radiotherapy combined with sunitinib has been shown to improve local tumor control compared to sunitinib alone, while a combination of stereotactic body radiotherapy and axitinib demonstrated prolonged progression-free survival in advanced RCC patients (252). The ability of VEGF inhibitors to reduce tumor-induced immunosuppression further suggests that their combination with radiotherapy may provide dual benefits of angiogenesis inhibition and enhanced immune priming (253).
Although RCC has historically exhibited low sensitivity to conventional cytotoxic chemotherapy, emerging evidence suggests that specific subtypes, such as nccRCC and sarcomatoid RCC, may benefit from chemotherapy in combination with immunotherapy or targeted therapy (254). Chemotherapy-induced immunogenic effects, including increased antigen presentation and modulation of T-cell responses, may enhance the efficacy of ICIs (254). For example, albumin-bound paclitaxel (nab-paclitaxel) combined with PD-1 inhibitors has shown promising responses in sarcomatoid RCC (255). Additionally, gemcitabine combined with VEGF-TKIs has demonstrated potential in high-risk RCC patients by simultaneously reducing tumor burden and enhancing the anti-angiogenic effects of VEGFR inhibition (256). These findings indicate that while chemotherapy remains a secondary option in RCC, its integration into combination treatment strategies may provide additional therapeutic benefits.
Despite these promising advancements, challenges remain in optimizing the integration of radiotherapy and chemotherapy with immunotherapy and targeted therapy. One of the key challenges is determining the optimal sequencing and dosing of these treatments to maximize efficacy while minimizing toxicity. For instance, excessive immune activation following radiotherapy could lead to increased immune-related AE when combined with ICIs (257, 258). Additionally, the identification of biomarkers predictive of response to combination therapy remains a critical area of investigation. Advances in molecular profiling may help stratify patients who are most likely to benefit from these treatment combinations (259). Future research is also focusing on the incorporation of novel radiosensitizers, DNA damage repair inhibitors such as Poly(ADP-ribose) polymerase inhibitors, and other immunomodulatory agents to further enhance therapeutic outcomes.
In conclusion, the integration of radiotherapy and chemotherapy with immunotherapy and targeted therapy represents a promising avenue for RCC treatment. While additional clinical trials are needed to refine these strategies, current evidence suggests that multi-modal approaches can improve survival outcomes and overcome resistance mechanisms in RCC. The continued exploration of synergistic combinations and biomarker-driven patient selection will be critical in optimizing these treatment paradigms.
5 Discussion
The landscape of RCC treatment has evolved considerably, with immunotherapy and targeted therapy emerging as the two most effective approaches for advanced disease (260). While immunotherapy have demonstrated durable responses in a subset of patients, primary and acquired resistance mechanisms, such as immune exclusion, upregulation of alternative immune checkpoints, and metabolic adaptations, continue to limit their efficacy. Similarly, targeted therapies, particularly VEGF and mTOR inhibitors, disrupt tumor angiogenesis and key oncogenic pathways but often lead to adaptive resistance, necessitating combination strategies (261).
Combination approaches have shown promise in overcoming these limitations, particularly the use of ICIs with VEGF-targeted TKIs. This dual strategy aims to normalize tumor vasculature, enhance immune infiltration, and reduce immunosuppressive factors within the tumor microenvironment. Clinical trials have validated the efficacy of combinations such as pembrolizumab plus axitinib and nivolumab plus cabozantinib, demonstrating improved PFS and OS compared to monotherapies. However, these regimens are associated with increased toxicity, raising the need for better patient stratification and biomarker-driven treatment selection (262, 263).
Beyond ICIs and TKIs, adoptive cell therapies, including CAR-T and CAR-NK cells, are being explored in RCC. Despite promising preclinical evidence, challenges such as the immunosuppressive tumor microenvironment, antigen heterogeneity, and limited CAR persistence remain significant hurdles. Advances in gene-modified cell therapies and the development of novel tumor antigens may enhance the feasibility of these approaches. Additionally, other combination modalities, including radiotherapy, chemotherapy, and metabolic inhibitors, hold potential for reshaping the tumor microenvironment to enhance immune response. Radiotherapy, for instance, has been shown to induce immunogenic cell death, which may synergize with ICIs to improve therapeutic efficacy (264).
Moving forward, the integration of multi-omic approaches, including genomics, transcriptomics, and single-cell sequencing, is critical for identifying predictive biomarkers and understanding resistance mechanisms. Precision medicine strategies will play a pivotal role in refining patient selection for specific combination therapies, optimizing treatment sequencing, and minimizing toxicity. Finally, we summarized the FDA-Approved pivotal Immunotherapies and Targeted Therapies for RCC in Table 6.
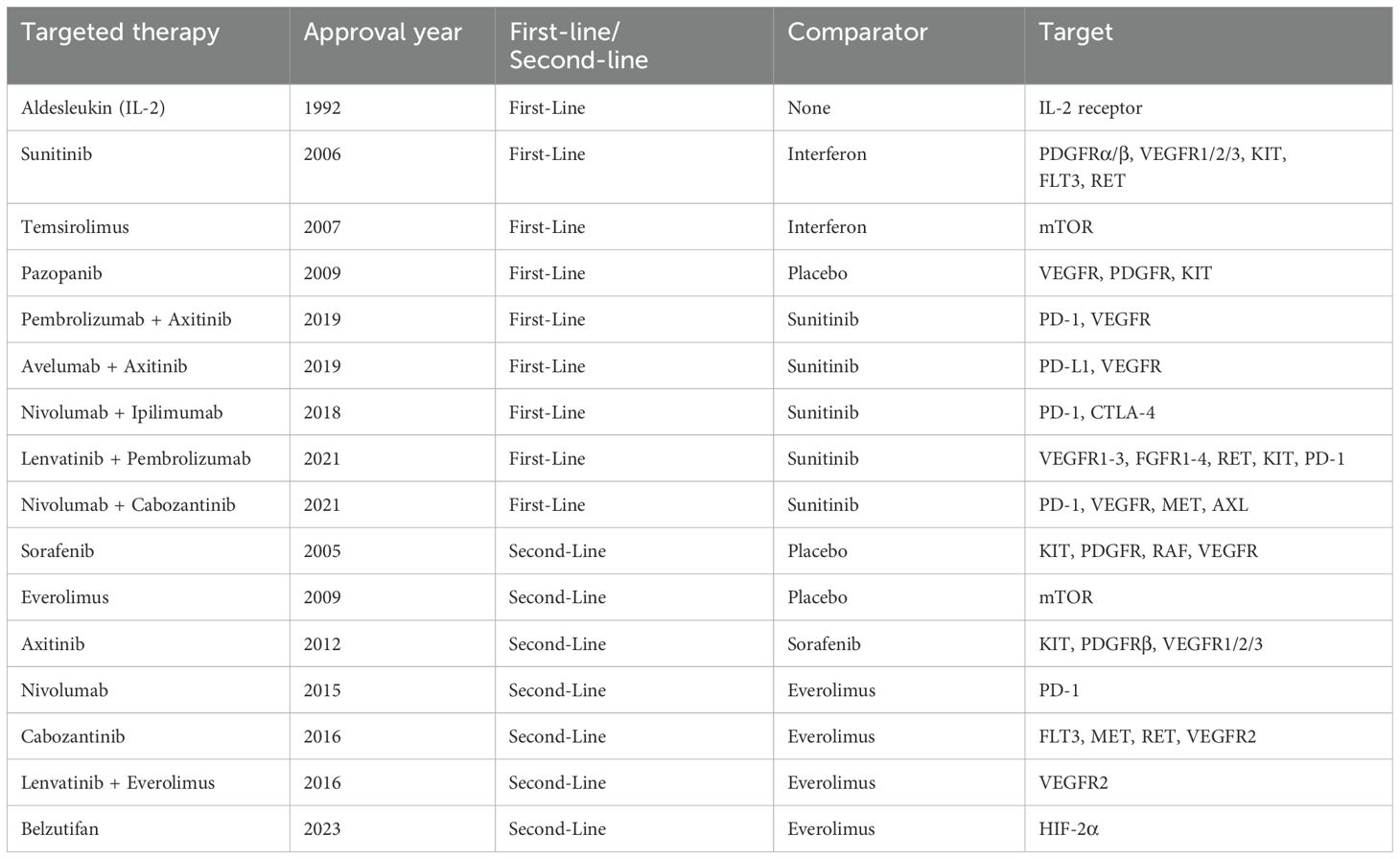
Table 6. FDA-Approved pivotal Immunotherapies and Targeted Therapies for RCC.
6 Conclusion
Immunotherapy and targeted therapy have significantly improved outcomes for RCC patients, yet resistance and toxicity remain key challenges. Emerging strategies, such as adoptive cell therapies and metabolic inhibitors, may further enhance treatment efficacy, but their clinical translation requires overcoming substantial biological and technical barriers. Future efforts should focus on identifying robust biomarkers, developing rational combination regimens, and optimizing treatment sequencing to maximize patient benefit. With ongoing advancements in immuno-oncology and precision medicine, a more personalized and durable approach to RCC treatment is on the horizon.
Author contributions
BW: Writing – original draft, Writing – review & editing. YX: Writing – original draft, Writing – review & editing. XL: Writing – original draft. XP: Writing – original draft. LP: Writing – original draft. JM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. JM and XL were supported by China Scholarship Council (CSC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1582887/full#supplementary-material
Glossary
RCC: Renal cell carcinoma
ccRCC: clear-cell renal cell carcinoma
TKI/TKIs: tyrosine kinase inhibitor/tyrosine kinase inhibitors
mTOR: mammalian target of rapamycin
CUP: cancers of unknown primary
PD-1: Programmed Cell Death Protein-1
PD-L1: Programmed Death-Ligand 1
CTLA-4: Cytotoxic T-lymphocyte-associated protein 4
VEGF/VEGFR: Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor Receptor
nccRCC/nccRCCs: non-clear cell renal cell carcinoma/non-clear cell renal cell carcinomas
ICIs: immune checkpoint inhibitors
DC/DCs: dendritic cell/dendritic cells
IL-2: Interleukin-2
IFN-α: Interferon-alpha
NK cells: natural killer cells
CD: Cluster of Differentiation 279 (或)
PDCD1: Programmed Cell Death 1 (gene)
TCR: T-cell receptor
FDA: Food and Drug Administration
PP2A: Serine/Threonine Protein Phosphatase 2A
PI3K: Phosphatidylinositol 3-kinase
Akt: Protein kinase B
IgG1κ: Immunoglobulin G1 kappa
AE: Adverse Events
LAG-3: Lymphocyte-Activation Gene 3
Ig: immunoglobulin
MHC-II: major histocompatibility complex II
TIM-3: T cell Immunoglobulin and Mucin domain 3
HAVCR2: Hepatitis A Virus Cellular Receptor 2
TIGIT: T cell Immunoreceptor with Immunoglobulin and ITIM domains
PI3K: Phosphatidylinositol 3-kinase
MAPK: Mitogen-Activated Protein Kinase
CIK cells: Cytokine-Induced Killer cells
IFN-γ: Interferon-gamma
MHC: major histocompatibility complex
TCR: T cell receptor
IFNα: Interferon-alpha
ORR: objective response rate
PFS: progression-free survival
OS: overall survival
mRCC: metastatic RCC
MDSCs: myeloid-derived suppressor cells
TME: tumor microenvironment
TILs: Tumor-infiltrating lymphocytes
CR: complete response
REP: Rapid Expansion Protocol
CAR-Ts: Chimeric Antigen Receptor T-Cells
CAR-NKs: Chimeric Antigen Receptor NK Cells
CAR: chimeric antigen receptor
CAIX: Carboxy-anhydrase-IX
AXL: AXL receptor tyrosine kinase
ROR2: Receptor tyrosine kinase-like orphan receptor 2
DNAJB8: DnaJ heat shock protein family (Hsp40) member B8
MUC1: Mucin 1, cell surface associated
c-Met: C-Mesenchymal-Epithelial Transition Factor
EGF/EGFR: Epidermal Growth Factor/Epidermal Growth Factor Receptor
HLA: Human Leukocyte Antigen
VHL: von Hippel-Lindau
HIF2α: hypoxia-inducible factor 2 alpha
PDGF/PDGFR: platelet-derived growth factor/platelet-derived growth factor receptors
pVHL: VHL protein
HIFs: hypoxia-inducible factors
HIF1α: hypoxia-inducible factor 1 alpha
HIF2α: hypoxia-inducible factor 2 alpha
HIF3α: hypoxia-inducible factor 3 alpha
RNAi: RNA interference
FGFR: Fibroblast Growth Factor Receptor
TGFβ: Transforming Growth Factor-beta
ALK1: Activin Receptor-Like Kinase 1
aRCC: advanced RCC
MEK: ERK Kinase
ERK: Extracellular Signal-Regulated Kinase
JAK: Janus Kinase
STAT: Signal Transducer and Activator of Transcription
T: telaglenastat
E: everolimus
P: placebo
C: cabozantinib
ATP: Adenosine Triphosphate
IDO1: Indoleamine 2,3-dioxygenase 1.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Rassy E, Parent P, Lefort F, Boussios S, Baciarello G, and Pavlidis N. New rising entities in cancer of unknown primary: Is there a real therapeutic benefit? Crit Rev Oncol Hematol. (2020) 147:102882. doi: 10.1016/j.critrevonc.2020.102882
3. Rini BI, Campbell SC, and Escudier B. Renal cell carcinoma. Lancet. (2009) 373:1119–32. doi: 10.1016/S0140-6736(09)60229-4
4. Jonasch E, Walker CL, and Rathmell WK. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol. (2021) 17:245–61. doi: 10.1038/s41581-020-00359-2
5. Speed JM, Trinh Q-D, Choueiri TK, and Sun M. Recurrence in localized renal cell carcinoma: a systematic review of contemporary data. Curr Urol Rep. (2017) 18:15. doi: 10.1007/s11934-017-0661-3
6. Yang J, Wang K, and Yang Z. Treatment strategies for clear cell renal cell carcinoma: Past, present and future. Front Oncol. (2023) 13:1133832. doi: 10.3389/fonc.2023.1133832
7. Motzer RJ, Banchereau R, Hamidi H, Powles T, McDermott D, Atkins MB, et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell. (2020) 38:803–817.e4. doi: 10.1016/j.ccell.2020.10.011
8. Davidson G, Helleux A, Vano YA, Lindner V, Fattori A, Cerciat M, et al. Mesenchymal-like tumor cells and myofibroblastic cancer-associated fibroblasts are associated with progression and immunotherapy response of clear cell renal cell carcinoma. Cancer Res. (2023) 83:2952–69. doi: 10.1158/0008-5472.CAN-22-3034
9. Jin J, Xie Y, Zhang J-S, Wang J-Q, Dai S-J, He W-F, et al. Sunitinib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Drug Resist Update. (2023) 67:100929. doi: 10.1016/j.drup.2023.100929
11. Goudarzi Z, Mostafavi M, Salesi M, Jafari M, Mirian I, Hashemi Meshkini A, et al. Everolimus and temsirolimus are not the same second-line in metastatic renal cell carcinoma: a systematic review and meta-analysis. Cost Eff Resour Alloc. (2023) 21:10. doi: 10.1186/s12962-023-00420-4
12. Bukowski RM. Temsirolimus: a safety and efficacy review. Expert Opin Drug Saf. (2012) 11:861–79. doi: 10.1517/14740338.2012.713344
13. Brown JE, Royle K-L, Gregory W, Ralph C, Maraveyas A, Din O, et al. Temporary treatment cessation versus continuation of first-line tyrosine kinase inhibitor in patients with advanced clear cell renal cell carcinoma (STAR): an open-label, non-inferiority, randomised, controlled, phase 2/3 trial. Lancet Oncol. (2023) 24:213–27. doi: 10.1016/S1470-2045(22)00793-8
14. de Velasco G, McKay RR, Lin X, Moreira RB, Simantov R, and Choueiri TK. Comprehensive analysis of survival outcomes in non-clear cell renal cell carcinoma patients treated in clinical trials. Clin Genitourin Cancer. (2017) 15:652–660.e1. doi: 10.1016/j.clgc.2017.03.004
16. Carlson RD, Flickinger JC, and Snook AE. Talkin’ Toxins: from coley’s to modern cancer immunotherapy. Toxins (Basel). (2020) 12:241. doi: 10.3390/toxins12040241
17. Liu K, Zhao S, Li J, Zheng Y, Wu H, Kong J, et al. Knowledge mapping and research hotspots of immunotherapy in renal cell carcinoma: A text-mining study from 2002 to 2021. Front Immunol. (2022) 13:969217. doi: 10.3389/fimmu.2022.969217
18. Vuky J and Motzer RJ. Cytokine therapy in renal cell cancer. Urol Oncol. (2000) 5:249–57. doi: 10.1016/s1078-1439(00)00068-5
19. Johannsen M, Brinkmann OA, Bergmann L, Heinzer H, Steiner T, Ringsdorf M, et al. The role of cytokine therapy in metastatic renal cell cancer. Eur Urol Suppl. (2007) 6:658–64. doi: 10.1016/j.eursup.2007.03.001
20. Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, and Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. (1995) 13:688–96. doi: 10.1200/JCO.1995.13.3.688
21. Papaetis GS, Karapanagiotou LM, Pandha H, and Syrigos KN. Targeted therapy for advanced renal cell cancer: cytokines and beyond. Curr Pharm Des. (2008) 14:2229–51. doi: 10.2174/138161208785740153
22. Bonetti E, Jenzer M, Nientiedt C, Kaczorowski A, Geisler C, Zschäbitz S, et al. Interleukin-2 and interferon-α for advanced renal cell carcinoma: patient outcomes, sexual dimorphism of responses, and multimodal treatment approaches over a 30-year period. Urol Int. (2022) 106:1158–67. doi: 10.1159/000524097
23. Achkar T, Arjunan A, Wang H, Saul M, Davar D, Appleman LJ, et al. High-dose interleukin 2 in patients with metastatic renal cell carcinoma with sarcomatoid features. PloS One. (2017) 12:e0190084. doi: 10.1371/journal.pone.0190084
24. Garcia JA and Rini BI. Recent progress in the management of advanced renal cell carcinoma. CA Cancer J Clin. (2007) 57:112–25. doi: 10.3322/canjclin.57.2.112
25. Parikh M and Bajwa P. Immune checkpoint inhibitors in the treatment of renal cell carcinoma. Semin Nephrol. (2020) 40:76–85. doi: 10.1016/j.semnephrol.2019.12.009
26. Kamimura N, Wolf AM, and Iwai Y. Development of cancer immunotherapy targeting the PD-1 pathway. J Nippon Med Sch. (2019) 86:10–4. doi: 10.1272/jnms.JNMS.2019_86-2
27. Qin S, Xu L, Yi M, Yu S, Wu K, and Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. (2019) 18:155. doi: 10.1186/s12943-019-1091-2
28. Fuertes Marraco SA, Neubert NJ, Verdeil G, and Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol. (2015) 6:310. doi: 10.3389/fimmu.2015.00310
29. Schildberg FA, Klein SR, Freeman GJ, and Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. (2016) 44:955–72. doi: 10.1016/j.immuni.2016.05.002
30. Bardhan K, Anagnostou T, and Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. (2016) 7:550. doi: 10.3389/fimmu.2016.00550
31. Wojtukiewicz MZ, Rek MM, Karpowicz K, Górska M, Polityńska B, Wojtukiewicz AM, et al. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. (2021) 40:949–82. doi: 10.1007/s10555-021-09976-0
32. Deutsch JS, Lipson EJ, Danilova L, Topalian SL, Jedrych J, Baraban E, et al. Combinatorial biomarker for predicting outcomes to anti-PD-1 therapy in patients with metastatic clear cell renal cell carcinoma. Cell Rep Med. (2023) 4:100947. doi: 10.1016/j.xcrm.2023.100947
33. Xu JX, Maher VE, Zhang L, Tang S, Sridhara R, Ibrahim A, et al. FDA approval summary: nivolumab in advanced renal cell carcinoma after anti-angiogenic therapy and exploratory predictive biomarker analysis. Oncologist. (2017) 22:311–7. doi: 10.1634/theoncologist.2016-0476
34. Chatzkel J, Schell MJ, Chahoud J, Zhang J, Jain R, Swank J, et al. Coordinated pembrolizumab and high dose IL-2 (5-in-a-row schedule) for therapy of metastatic clear cell renal cancer. Clin Genitourin Cancer. (2022) 20:252–9. doi: 10.1016/j.clgc.2022.01.010
35. Xu J, Zhang Y, Shen L, Du L, Xue H, Wu B, et al. Mechanistic insights into the inhibition of a common CTLA-4 gene mutation in the cytoplasmic domain. Molecules. (2024) 29:1330. doi: 10.3390/molecules29061330
36. Zhou X, Li C, Chen T, Li W, Wang X, and Yang Q. Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy. Mol Cancer. (2023) 22:36. doi: 10.1186/s12943-023-01746-6
37. Wu B, Zhang Q, and Sun J. Cost-effectiveness of nivolumab plus ipilimumab as first-line therapy in advanced renal-cell carcinoma. J Immunother Cancer. (2018) 6:124. doi: 10.1186/s40425-018-0440-9
38. Sammarco E, Manfredi F, Nuzzo A, Ferrari M, Bonato A, Salfi A, et al. Immune checkpoint inhibitor rechallenge in renal cell carcinoma: current evidence and future directions. Cancers (Basel). (2023) 15:3172. doi: 10.3390/cancers15123172
39. Andrews LP, Marciscano AE, Drake CG, and Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. (2017) 276:80–96. doi: 10.1111/imr.12519
40. Kisielow M, Kisielow J, Capoferri-Sollami G, and Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. (2005) 35:2081–8. doi: 10.1002/eji.200526090
41. Ma J, Yan S, Zhao Y, Yan H, Zhang Q, and Li X. Blockade of PD-1 and LAG-3 expression on CD8+ T cells promotes the tumoricidal effects of CD8+ T cells. Front Immunol. (2023) 14:1265255. doi: 10.3389/fimmu.2023.1265255
42. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. (2018) 9:176–89. doi: 10.18632/genesandcancer.180
43. Chen N, Liu Y, Guo Y, Chen Y, Liu X, and Liu M. Lymphocyte activation gene 3 negatively regulates the function of intrahepatic hepatitis C virus-specific CD8+ T cells. J Gastroenterol Hepatol. (2015) 30:1788–95. doi: 10.1111/jgh.13017
44. Lee CH, Jung SJ, Seo WI, Chung JI, Lee DS, Jeong DH, et al. Coexpression of lymphocyte-activation gene 3 and programmed death ligand-1 in tumor infiltrating immune cells predicts worse outcome in renal cell carcinoma. Int J Immunopathol Pharmacol. (2022) 36:3946320221125588. doi: 10.1177/03946320221125588
45. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
46. Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. (2009) 39:2492–501. doi: 10.1002/eji.200939274
47. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One. (2012) 7:e30676. doi: 10.1371/journal.pone.0030676
48. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. (2012) 119:3064–72. doi: 10.1182/blood-2011-06-360321
49. Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. (2007) 26:311–21. doi: 10.1016/j.immuni.2007.01.016
50. Acharya N, Sabatos-Peyton C, and Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. (2020) 8:e000911. doi: 10.1136/jitc-2020-000911
51. Zhao L, Cheng S, Fan L, Zhang B, and Xu S. TIM-3: An update on immunotherapy. Int Immunopharmacol. (2021) 99:107933. doi: 10.1016/j.intimp.2021.107933
52. Sun H and Sun C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front Immunol. (2019) 10:2354. doi: 10.3389/fimmu.2019.02354
53. Cong Y, Liu J, Chen G, and Qiao G. The emerging role of T-cell immunoglobulin mucin-3 in breast cancer: A promising target for immunotherapy. Front Oncol. (2021) 11:723238. doi: 10.3389/fonc.2021.723238
54. Donini C, D’Ambrosio L, Grignani G, Aglietta M, and Sangiolo D. Next generation immune-checkpoints for cancer therapy. J Thorac Dis. (2018) 10:S1581–601. doi: 10.21037/jtd.2018.02.79
55. Tao J, Li L, Wang Y, Fu R, Wang H, and Shao Z. Increased TIM3+CD8+T cells in Myelodysplastic Syndrome patients displayed less perforin and granzyme B secretion and higher CD95 expression. Leuk Res. (2016) 51:49–55. doi: 10.1016/j.leukres.2016.11.003
56. Xu S, Zhang N, Rinne ML, Sun H, and Stein AM. Sabatolimab (MBG453) model-informed drug development for dose selection in patients with myelodysplastic syndrome/acute myeloid leukemia and solid tumors. CPT Pharmacometr Syst Pharmacol. (2023) 12:1653–65. doi: 10.1002/psp4.12962
57. Schwartz S, Patel N, Longmire T, Jayaraman P, Jiang X, Lu H, et al. Characterization of sabatolimab, a novel immunotherapy with immuno-myeloid activity directed against TIM-3 receptor. Immunother Adv. (2022) 2:ltac019. doi: 10.1093/immadv/ltac019
58. Zheng Q, Xu J, Gu X, Wu F, Deng J, Cai X, et al. Immune checkpoint targeting TIGIT in hepatocellular carcinoma. Am J Transl Res. (2020) 12:3212–24.
59. Chauvin J-M and Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. (2020) 8:e000957. doi: 10.1136/jitc-2020-000957
60. Liu L, You X, Han S, Sun Y, Zhang J, and Zhang Y. CD155/TIGIT, a novel immune checkpoint in human cancers (Review). Oncol Rep. (2021) 45:835–45. doi: 10.3892/or.2021.7943
61. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. (2011) 41:902–15. doi: 10.1002/eji.201041136
62. Xiong F, Yu M, Xu H, Zhong Z, Li Z, Guo Y, et al. Discovery of TIGIT inhibitors based on DEL and machine learning. Front Chem. (2022) 10:982539. doi: 10.3389/fchem.2022.982539
63. Zhou X, Ren T, Zan H, Hua C, and Guo X. Novel immune checkpoints in esophageal cancer: from biomarkers to therapeutic targets. Front Immunol. (2022) 13:864202. doi: 10.3389/fimmu.2022.864202
64. Chauvin J-M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
65. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. (2022) 23:781–92. doi: 10.1016/S1470-2045(22)00226-1
66. Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. (2011) 118:3301–10. doi: 10.1182/blood-2011-02-336321
67. Zhang Y and Schmidt-Wolf IGH. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J Cell Physiol. (2020) 235:9291–303. doi: 10.1002/jcp.29827
68. Verneris MR, Karimi M, Baker J, Jayaswal A, and Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. (2004) 103:3065–72. doi: 10.1182/blood-2003-06-2125
69. Liu L, Zhang W, Qi X, Li H, Yu J, Wei S, et al. Randomized study of autologous cytokine-induced killer cell immunotherapy in metastatic renal carcinoma. Clin Cancer Res. (2012) 18:1751–9. doi: 10.1158/1078-0432.CCR-11-2442
70. Zhang Y, Wang J, Wang Y, Lu X-C, Fan H, Liu Y, et al. Autologous CIK cell immunotherapy in patients with renal cell carcinoma after radical nephrectomy. Clin Dev Immunol. (2013) 2013:195691. doi: 10.1155/2013/195691
71. Kasherman L, Siu DHW, Woodford R, and Harris CA. Angiogenesis inhibitors and immunomodulation in renal cell cancers: the past, present, and future. Cancers (Basel). (2022) 14:1406. doi: 10.3390/cancers14061406
72. Zhang J, Zhu L, Wei J, Liu L, Yin Y, Gu Y, et al. The effects of cytokine-induced killer cells for the treatment of patients with solid tumors: a clinical retrospective study. J Cancer Res Clin Oncol. (2012) 138:1057–62. doi: 10.1007/s00432-012-1179-1
73. Su X, Zhang L, Jin L, Ye J, Guan Z, Chen R, et al. Immunotherapy with cytokine-induced killer cells in metastatic renal cell carcinoma. Cancer Biother Radiopharm. (2010) 25:465–70. doi: 10.1089/cbr.2010.0762
74. Märten A, Ziske C, Schöttker B, Renoth S, Weineck S, Buttgereit P, et al. Interactions between dendritic cells and cytokine-induced killer cells lead to an activation of both populations. J Immunother. (2001) 24:502–10. doi: 10.1097/00002371-200111000-00007
75. Zhao X, Zhang Z, Li H, Huang J, Yang S, Xie T, et al. Cytokine induced killer cell-based immunotherapies in patients with different stages of renal cell carcinoma. Cancer Lett. (2015) 362:192–8. doi: 10.1016/j.canlet.2015.03.043
76. Zhan H-L, Gao X, Pu X-Y, Li W, Li Z-J, Zhou X-F, et al. A randomized controlled trial of postoperative tumor lysate-pulsed dendritic cells and cytokine-induced killer cells immunotherapy in patients with localized and locally advanced renal cell carcinoma. Chin Med J (Engl). (2012) 125:3771–7.
77. Schmeel LC, Schmeel FC, Coch C, and Schmidt-Wolf IGH. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol. (2015) 141:839–49. doi: 10.1007/s00432-014-1864-3
78. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. (2013) 19:4792–800. doi: 10.1158/1078-0432.CCR-13-0380
79. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
80. Figlin RA, Thompson JA, Bukowski RM, Vogelzang NJ, Novick AC, Lange P, et al. Multicenter, randomized, phase III trial of CD8(+) tumor-infiltrating lymphocytes in combination with recombinant interleukin-2 in metastatic renal cell carcinoma. J Clin Oncol. (1999) 17:2521–9. doi: 10.1200/JCO.1999.17.8.2521
81. Markel G, Cohen-Sinai T, Besser MJ, Oved K, Itzhaki O, Seidman R, et al. Preclinical evaluation of adoptive cell therapy for patients with metastatic renal cell carcinoma. Anticancer Res. (2009) 29:145–54.
82. Andersen R, Westergaard MCW, Kjeldsen JW, Müller A, Pedersen NW, Hadrup SR, et al. T-cell responses in the microenvironment of primary renal cell carcinoma-implications for adoptive cell therapy. Cancer Immunol Res. (2018) 6:222–35. doi: 10.1158/2326-6066.CIR-17-0467
83. Lee MH, Theodoropoulos J, Huuhtanen J, Bhattacharya D, Järvinen P, Tornberg S, et al. Immunologic characterization and T cell receptor repertoires of expanded tumor-infiltrating lymphocytes in patients with renal cell carcinoma. Cancer Res Commun. (2023) 3:1260–76. doi: 10.1158/2767-9764.CRC-22-0514
84. Baldan V, Griffiths R, Hawkins RE, and Gilham DE. Efficient and reproducible generation of tumour-infiltrating lymphocytes for renal cell carcinoma. Br J Cancer. (2015) 112:1510–8. doi: 10.1038/bjc.2015.96
85. Roselli E, Faramand R, and Davila ML. Insight into next-generation CAR therapeutics: designing CAR T cells to improve clinical outcomes. J Clin Invest. (2021) 131:e142030, 142030. doi: 10.1172/JCI142030
86. Mandriani B, Pellè E, Mannavola F, Palazzo A, Marsano RM, Ingravallo G, et al. Development of anti-somatostatin receptors CAR T cells for treatment of neuroendocrine tumors. J Immunother Cancer. (2022) 10:e004854. doi: 10.1136/jitc-2022-004854
87. Hanssens H, Meeus F, De Veirman K, Breckpot K, and Devoogdt N. The antigen-binding moiety in the driver’s seat of CARs. Med Res Rev. (2022) 42:306–42. doi: 10.1002/med.21818
88. Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. (1987) 149:960–8. doi: 10.1016/0006-291x(87)90502-x
89. Abate-Daga D and Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncol. (2016) 3:16014. doi: 10.1038/mto.2016.14
90. Tang L, Pan S, Wei X, Xu X, and Wei Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol Ther. (2023) 31:3146–62. doi: 10.1016/j.ymthe.2023.09.021
91. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang C-H, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
92. Del Bufalo F, De Angelis B, Caruana I, Del Baldo G, De Ioris MA, Serra A, et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N Engl J Med. (2023) 388:1284–95. doi: 10.1056/NEJMoa2210859
93. Elsallab M, Ellithi M, Lunning MA, D’Angelo C, Ma J, Perales M-A, et al. Second primary Malignancies after commercial CAR T-cell therapy: analysis of the FDA Adverse Events Reporting System. Blood. (2024) 143:2099–105. doi: 10.1182/blood.2024024166
94. Verdun N and Marks P. Secondary cancers after chimeric antigen receptor T-cell therapy. N Engl J Med. (2024) 390:584–6. doi: 10.1056/NEJMp2400209
95. Levine BL, Pasquini MC, Connolly JE, Porter DL, Gustafson MP, Boelens JJ, et al. Unanswered questions following reports of secondary Malignancies after CAR-T cell therapy. Nat Med. (2024) 30:338–41. doi: 10.1038/s41591-023-02767-w
96. Morgan MA, Büning H, Sauer M, and Schambach A. Use of cell and genome modification technologies to generate improved “Off-the-shelf” CAR T and CAR NK cells. Front Immunol. (2020) 11:1965. doi: 10.3389/fimmu.2020.01965
97. Albelda SM. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat Rev Clin Oncol. (2024) 21:47–66. doi: 10.1038/s41571-023-00832-4
98. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
99. Li T, Niu M, Zhang W, Qin S, Zhou J, and Yi M. CAR-NK cells for cancer immunotherapy: recent advances and future directions. Front Immunol. (2024) 15:1361194. doi: 10.3389/fimmu.2024.1361194
100. Lei A, Yu H, Lu S, Lu H, Ding X, Tan T, et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat Immunol. (2024) 25:102–16. doi: 10.1038/s41590-023-01687-8
101. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
102. Stillebroer AB, Mulders PFA, Boerman OC, Oyen WJG, and Oosterwijk E. Carbonic anhydrase IX in renal cell carcinoma: implications for prognosis, diagnosis, and therapy. Eur Urol. (2010) 58:75–83. doi: 10.1016/j.eururo.2010.03.015
103. Lamers CHJ, Klaver Y, Gratama JW, Sleijfer S, and Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. (2016) 44:951–9. doi: 10.1042/BST20160037
104. Suarez ER, Chang DK, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
105. Wang Y, Suarez ER, Kastrunes G, de Campos NSP, Abbas R, Pivetta RS, et al. Evolution of cell therapy for renal cell carcinoma. Mol Cancer. (2024) 23:8. doi: 10.1186/s12943-023-01911-x
106. Wang Y, Buck A, Grimaud M, Culhane AC, Kodangattil S, Razimbaud C, et al. Anti-CAIX BBζ CAR4/8 T cells exhibit superior efficacy in a ccRCC mouse model. Mol Ther Oncol. (2022) 24:385–99. doi: 10.1016/j.omto.2021.12.019
107. Li H, Ding J, Lu M, Liu H, Miao Y, Li L, et al. CAIX-specific CAR-T cells and sunitinib show synergistic effects against metastatic renal cancer models. J Immunother. (2020) 43:16–28. doi: 10.1097/CJI.0000000000000301
108. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. (2009) 15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332
109. Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. (2008) 14:6674–82. doi: 10.1158/1078-0432.CCR-07-5212
110. Zhang Q, Xu J, Ding J, Liu H, Li H, Li H, et al. Bortezomib improves adoptive carbonic anhydrase IX−specific chimeric antigen receptor−modified NK92 cell therapy in mouse models of human renal cell carcinoma. Oncol Rep. (2018) 40:3714–24. doi: 10.3892/or.2018.6731
111. Lundqvist A, Yokoyama H, Smith A, Berg M, and Childs R. Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood. (2009) 113:6120–7. doi: 10.1182/blood-2008-11-190421
112. Denoeud J and Moser M. Role of CD27/CD70 pathway of activation in immunity and tolerance. J Leukoc Biol. (2011) 89:195–203. doi: 10.1189/jlb.0610351
113. Borst J, Hendriks J, and Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol. (2005) 17:275–81. doi: 10.1016/j.coi.2005.04.004
114. Law C-L, Gordon KA, Toki BE, Yamane AK, Hering MA, Cerveny CG, et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. (2006) 66:2328–37. doi: 10.1158/0008-5472.CAN-05-2883
115. Pal SK, Tran B, Haanen JBAG, Hurwitz ME, Sacher A, Tannir NM, et al. CD70-targeted allogeneic CAR T-cell therapy for advanced clear cell renal cell carcinoma. Cancer Discov. (2024) 14:1176–89. doi: 10.1158/2159-8290.CD-24-0102
116. Yu H, Liu R, Ma B, Li X, Yen H-Y, Zhou Y, et al. Axl receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br J Cancer. (2015) 113:616–25. doi: 10.1038/bjc.2015.237
117. Tanaka M and Siemann DW. Gas6/axl signaling pathway in the tumor immune microenvironment. Cancers (Basel). (2020) 12:1850. doi: 10.3390/cancers12071850
118. Wright TM, Brannon AR, Gordan JD, Mikels AJ, Mitchell C, Chen S, et al. Ror2, a developmentally regulated kinase, promotes tumor growth potential in renal cell carcinoma. Oncogene. (2009) 28:2513–23. doi: 10.1038/onc.2009.116
119. Watanabe Y, Tsukahara T, Murata K, Hamada S, Kubo T, Kanaseki T, et al. Development of CAR-T cells specifically targeting cancer stem cell antigen DNAJB8 against solid tumours. Br J Cancer. (2023) 128:886–95. doi: 10.1038/s41416-022-02100-1
120. Milella M, Rutigliano M, Lasorsa F, Ferro M, Bianchi R, Fallara G, et al. The role of MUC1 in renal cell carcinoma. Biomolecules. (2024) 14:315. doi: 10.3390/biom14030315
121. Mori J-I, Adachi K, Sakoda Y, Sasaki T, Goto S, Matsumoto H, et al. Anti-tumor efficacy of human anti-c-met CAR-T cells against papillary renal cell carcinoma in an orthotopic model. Cancer Sci. (2021) 112:1417–28. doi: 10.1111/cas.14835
122. Zhang Q, Tian K, Xu J, Zhang H, Li L, Fu Q, et al. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res. (2017) 2017:6915912. doi: 10.1155/2017/6915912
123. Mastelic-Gavillet B, Balint K, Boudousquie C, Gannon PO, and Kandalaft LE. Personalized dendritic cell vaccines-recent breakthroughs and encouraging clinical results. Front Immunol. (2019) 10:766. doi: 10.3389/fimmu.2019.00766
124. Gardner A, de Mingo Pulido Á, and Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol. (2020) 11:924. doi: 10.3389/fimmu.2020.00924
125. Chung DJ, Sharma S, Rangesa M, DeWolf S, Elhanati Y, Perica K, et al. Langerhans dendritic cell vaccine bearing mRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv. (2022) 6:1547–58. doi: 10.1182/bloodadvances.2021005941
126. De Keersmaecker B, Claerhout S, Carrasco J, Bar I, Corthals J, Wilgenhof S, et al. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J Immunother Cancer. (2020) 8:e000329. doi: 10.1136/jitc-2019-000329
127. Dörrie J, Schaft N, Schuler G, and Schuler-Thurner B. Therapeutic cancer vaccination with ex vivo RNA-transfected dendritic cells-an update. Pharmaceutics. (2020) 12:92. doi: 10.3390/pharmaceutics12020092
128. Cozzi S, Najafi M, Gomar M, Ciammella P, Iotti C, Iaccarino C, et al. Delayed effect of dendritic cells vaccination on survival in glioblastoma: A systematic review and meta-analysis. Curr Oncol. (2022) 29:881–91. doi: 10.3390/curroncol29020075
129. Jansen Y, Kruse V, Corthals J, Schats K, van Dam P-J, Seremet T, et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol Immunother. (2020) 69:2589–98. doi: 10.1007/s00262-020-02618-4
130. Filin IY, Kitaeva KV, Rutland CS, Rizvanov AA, and Solovyeva VV. Recent advances in experimental dendritic cell vaccines for cancer. Front Oncol. (2021) 11:730824. doi: 10.3389/fonc.2021.730824
131. Batich KA, Mitchell DA, Healy P, Herndon JE, and Sampson JH. Once, twice, three times a finding: reproducibility of dendritic cell vaccine trials targeting cytomegalovirus in glioblastoma. Clin Cancer Res. (2020) 26:5297–303. doi: 10.1158/1078-0432.CCR-20-1082
132. Laureano RS, Sprooten J, Vanmeerbeerk I, Borras DM, Govaerts J, Naulaerts S, et al. Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology. (2022) 11:2096363. doi: 10.1080/2162402X.2022.2096363
133. Jeng L-B, Liao L-Y, Shih F-Y, and Teng C-F. Dendritic-cell-vaccine-based immunotherapy for hepatocellular carcinoma: clinical trials and recent preclinical studies. Cancers (Basel). (2022) 14:4380. doi: 10.3390/cancers14184380
134. Grenier JM, Yeung ST, and Khanna KM. Combination immunotherapy: taking cancer vaccines to the next level. Front Immunol. (2018) 9:610. doi: 10.3389/fimmu.2018.00610
135. Joyce JA and Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
136. Ghasemi A, Martinez-Usatorre A, Li L, Hicham M, Guichard A, Marcone R, et al. Cytokine-armed dendritic cell progenitors for antigen-agnostic cancer immunotherapy. Nat Cancer. (2024) 5:240–61. doi: 10.1038/s43018-023-00668-y
137. Han M-H and Kim CH. Current immunotherapeutic approaches for Malignant gliomas. Brain Tumor Res Treat. (2022) 10:1–11. doi: 10.14791/btrt.2022.10.e25
138. Siemens DR and Ratliff TL. Vaccines in urologic Malignancies. Urol Res. (2001) 29:152–62. doi: 10.1007/s002400100178
139. Cho H-I, Jung S-H, Sohn H-J, Celis E, and Kim T-G. An optimized peptide vaccine strategy capable of inducing multivalent CD8+ T cell responses with potent antitumor effects. Oncoimmunology. (2015) 4:e1043504. doi: 10.1080/2162402X.2015.1043504
140. Jugniot N, Dahl JJ, and Paulmurugan R. Immunotheranostic microbubbles (iMBs) - a modular platform for dendritic cell vaccine delivery applied to breast cancer immunotherapy. J Exp Clin Cancer Res. (2022) 41:299. doi: 10.1186/s13046-022-02501-3
141. Durgeau A, Virk Y, Corgnac S, and Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. (2018) 9:14. doi: 10.3389/fimmu.2018.00014
142. Chen M-H, Jiang J, Chen H, Wu R-H, Xie W, Dai S-Z, et al. Reinforcing cancer immunotherapy with engineered porous hollow mycobacterium tuberculosis loaded with tumor neoantigens. J Immunother Cancer. (2025) 13:e010150. doi: 10.1136/jitc-2024-010150
143. Jocham D, Richter A, Hoffmann L, Iwig K, Fahlenkamp D, Zakrzewski G, et al. Adjuvant autologous renal tumour cell vaccine and risk of tumour progression in patients with renal-cell carcinoma after radical nephrectomy: phase III, randomised controlled trial. Lancet. (2004) 363:594–9. doi: 10.1016/S0140-6736(04)15590-6
144. Wood C, Srivastava P, Bukowski R, Lacombe L, Gorelov AI, Gorelov S, et al. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet. (2008) 372:145–54. doi: 10.1016/S0140-6736(08)60697-2
145. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
146. Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. (2021) 27:515–25. doi: 10.1038/s41591-020-01206-4
147. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. (2017) 376:917–27. doi: 10.1056/NEJMoa1609324
148. Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. (2006) 12:7271–8. doi: 10.1158/1078-0432.CCR-06-1249
149. Tu R, Ma J, Chen Y, Kang Y, Ren D, Cai Z, et al. USP7 depletion potentiates HIF2α degradation and inhibits clear cell renal cell carcinoma progression. Cell Death Dis. (2024) 15:749. doi: 10.1038/s41419-024-07136-0
150. Dengler VL, Galbraith M, and Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. (2014) 49:1–15. doi: 10.3109/10409238.2013.838205
151. Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature. (2012) 486:126–9. doi: 10.1038/nature11055
152. Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med. (2009) 15:319–24. doi: 10.1038/nm.1922
153. Choueiri TK and Kaelin WG. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat Med. (2020) 26:1519–30. doi: 10.1038/s41591-020-1093-z
154. Kaelin WG. HIF2 inhibitor joins the kidney cancer armamentarium. J Clin Oncol. (2018) 36:908–10. doi: 10.1200/JCO.2017.77.5254
155. Gossage L, Eisen T, and Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. (2015) 15:55–64. doi: 10.1038/nrc3844
156. Choueiri TK and Motzer RJ. Systemic therapy for metastatic renal-cell carcinoma. N Engl J Med. (2017) 376:354–66. doi: 10.1056/NEJMra1601333
157. Wehn PM, Rizzi JP, Dixon DD, Grina JA, Schlachter ST, Wang B, et al. Design and activity of specific hypoxia-inducible factor-2α (HIF-2α) inhibitors for the treatment of clear cell renal cell carcinoma: discovery of clinical candidate (S)-3-((2,2-difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1 H-inden-4-yl)oxy)-5-fluorobenzonitrile (PT2385). J Med Chem. (2018) 61:9691–721. doi: 10.1021/acs.jmedchem.8b01196
158. Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, and Gardner KH. Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci U.S.A. (2009) 106:450–5. doi: 10.1073/pnas.0808092106
159. Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α Antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. (2018) 36:867–74. doi: 10.1200/JCO.2017.74.2627
160. Strowd R, Ellingson B, Raymond C, Yao J, Wen PY, Ahluwalia M, et al. Activity of a first-in-class oral HIF2-alpha inhibitor, PT2385, in patients with first recurrence of glioblastoma. J Neurooncol. (2023) 165:101–12. doi: 10.1007/s11060-023-04456-7
161. Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, et al. 3-[(1S,2S,3R)-2,3-difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell renal cell carcinoma. J Med Chem. (2019) 62:6876–93. doi: 10.1021/acs.jmedchem.9b00719
162. Yang G, Ge S, Singh R, Basu S, Shatzer K, Zen M, et al. Glucuronidation: driving factors and their impact on glucuronide disposition. Drug Metab Rev. (2017) 49:105–38. doi: 10.1080/03602532.2017.1293682
163. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von hippel-lindau disease. N Engl J Med. (2021) 385:2036–46. doi: 10.1056/NEJMoa2103425
164. Choueiri TK, Powles T, Peltola K, de Velasco G, Burotto M, Suarez C, et al. Belzutifan versus everolimus for advanced renal-cell carcinoma. N Engl J Med. (2024) 391:710–21. doi: 10.1056/NEJMoa2313906
165. Jonasch E, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, et al. Phase 1 LITESPARK-001 (MK-6482-001) study of belzutifan in advanced solid tumors: Update of the clear cell renal cell carcinoma (ccRCC) cohort with more than 3 years of total follow-up. J Clin Oncol. (2022) 40(16_suppl):4509. doi: 10.1200/JCO.2022.40.16_suppl.4509
166. Choueiri TK, Bauer TM, McDermott DF, Arrowsmith E, Roy A, Perini RF, et al. Phase 2 study of the oral hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 in combination with cabozantinib in patients with advanced clear cell renal cell carcinoma (ccRCC). JCO. (2021) 39:272–2. doi: 10.1200/JCO.2021.39.6_suppl.272
167. Choueiri TK, Tomczak P, Park SH, Venugopal B, Ferguson T, Chang Y-H, et al. Adjuvant pembrolizumab after nephrectomy in renal-cell Carcinoma. N Engl J Med. (2021) 385:683–94. doi: 10.1056/NEJMoa2106391
168. Gartel AL and Kandel ES. RNA interference in cancer. Biomol Eng. (2006) 23:17–34. doi: 10.1016/j.bioeng.2006.01.002
169. Arrowhead Announces Positive Interim Results from Phase 1b Study of ARO-HIF2 for Treatment of Clear Cell Renal Cell Carcinoma | Business Wire . Available online at: https://www.businesswire.com/news/home/20210706005215/en/Arrowhead-Announces-Positive-Interim-Results-from-Phase-1b-Study-of-ARO-HIF2-for-Treatment-of-Clear-Cell-Renal-Cell-Carcinoma (Accessed August 3, 2024).
170. Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C, Istrate M, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. (2018) 59:455–67.
171. Liu X, He H, Zhang F, Hu X, Bi F, Li K, et al. m6A methylated EphA2 and VEGFA through IGF2BP2/3 regulation promotes vasculogenic mimicry in colorectal cancer via PI3K/AKT and ERK1/2 signaling. Cell Death Dis. (2022) 13:483. doi: 10.1038/s41419-022-04950-2
172. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers. (2017) 3:17009. doi: 10.1038/nrdp.2017.9
173. Rini BI, Battle D, Figlin RA, George DJ, Hammers H, Hutson T, et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of advanced renal cell carcinoma (RCC). J Immunother Cancer. (2019) 7:354. doi: 10.1186/s40425-019-0813-8
174. Pal K, Madamsetty VS, Dutta SK, Wang E, Angom RS, and Mukhopadhyay D. Synchronous inhibition of mTOR and VEGF/NRP1 axis impedes tumor growth and metastasis in renal cancer. NPJ Precis Oncol. (2019) 3:31. doi: 10.1038/s41698-019-0105-2
175. Casper J, Schumann-Binarsch S, and Köhne C-H. Pazopanib versus sunitinib in renal cancer. N Engl J Med. (2013) 369:1969. doi: 10.1056/NEJMc1311795
176. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. (2011) 10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264
177. Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. (2014) 6:18. doi: 10.1186/2045-824X-6-18
178. Motzer RJ, Nosov D, Eisen T, Bondarenko I, Lesovoy V, Lipatov O, et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: results from a phase III trial. J Clin Oncol. (2013) 31:3791–9. doi: 10.1200/JCO.2012.47.4940
179. Molina AM, Hutson TE, Nosov D, Tomczak P, Lipatov O, Sternberg CN, et al. Efficacy of tivozanib treatment after sorafenib in patients with advanced renal cell carcinoma: crossover of a phase 3 study. Eur J Cancer. (2018) 94:87–94. doi: 10.1016/j.ejca.2018.02.009
180. Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: the alliance A031203 CABOSUN trial. J Clin Oncol. (2017) 35:591–7. doi: 10.1200/JCO.2016.70.7398
181. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med. (2015) 373:1814–23. doi: 10.1056/NEJMoa1510016
182. Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. (2016) 17:917–27. doi: 10.1016/S1470-2045(16)30107-3
183. Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. (2015) 16:1473–82. doi: 10.1016/S1470-2045(15)00290-9
184. Bhargava P and Robinson MO. Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr Oncol Rep. (2011) 13:103–11. doi: 10.1007/s11912-011-0154-3
185. Zhou L, Liu X-D, Sun M, Zhang X, German P, Bai S, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. (2016) 35:2687–97. doi: 10.1038/onc.2015.343
186. Larroquette M, Lefort F, Heraudet L, Bernhard J-C, Ravaud A, Domblides C, et al. Therapeutic management of metastatic clear cell renal cell carcinoma: A revolution in every decade. Cancers (Basel). (2022) 14:6230. doi: 10.3390/cancers14246230
187. Študentová H, Vitásková D, and Melichar B. Lenvatinib for the treatment of kidney cancer. Expert Rev Anticancer Ther. (2018) 18:511–8. doi: 10.1080/14737140.2018.1470506
188. O’Reilly A and Larkin J. Lenvatinib for use in combination with everolimus for the treatment of patients with advanced renal cell carcinoma following one prior anti-angiogenic therapy. Expert Rev Clin Pharmacol. (2017) 10:251–62. doi: 10.1080/17512433.2017.1289840
189. Jonker L. TGF-β & BMP receptors endoglin and ALK1: overview of their functional role and status as antiangiogenic targets. Microcirculation. (2014) 21:93–103. doi: 10.1111/micc.12099
190. Huang H, Bhat A, Woodnutt G, and Lappe R. Targeting the ANGPT-TIE2 pathway in Malignancy. Nat Rev Cancer. (2010) 10:575–85. doi: 10.1038/nrc2894
191. Rini B, Szczylik C, Tannir NM, Koralewski P, Tomczak P, Deptala A, et al. AMG 386 in combination with sorafenib in patients with metastatic clear cell carcinoma of the kidney: a randomized, double-blind, placebo-controlled, phase 2 study. Cancer. (2012) 118:6152–61. doi: 10.1002/cncr.27632
192. Voss MH, Bhatt RS, Plimack ER, Rini BI, Alter RS, Beck JT, et al. The DART study: results from the dose-escalation and expansion cohorts evaluating the combination of dalantercept plus axitinib in advanced renal cell carcinoma. Clin Cancer Res. (2017) 23:3557–65. doi: 10.1158/1078-0432.CCR-16-2395
193. Savaliya M, Surati D, Surati R, Padmani S, and Boussios S. Posterior reversible encephalopathy syndrome after pazopanib therapy. Diseases. (2023) 11:76. doi: 10.3390/diseases11020076
194. Jung S-C, Kang D, and Ko E-A. Roles of PDGF/PDGFR signaling in various organs. Korean J Physiol Pharmacol. (2024) 29(2):139–55. doi: 10.4196/kjpp.24.309
195. Wee P and Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel). (2017) 9:52. doi: 10.3390/cancers9050052
196. Cumpănas AA, Cimpean AM, Ferician O, Ceausu RA, Sarb S, Barbos V, et al. The involvement of PDGF-B/PDGFRβ Axis in the resistance to antiangiogenic and antivascular therapy in renal cancer. Anticancer Res. (2016) 36:2291–5.
197. Song SH, Jeong IG, You D, Hong JH, Hong B, Song C, et al. VEGF/VEGFR2 and PDGF-B/PDGFR-β expression in non-metastatic renal cell carcinoma: a retrospective study in 1,091 consecutive patients. Int J Clin Exp Pathol. (2014) 7:7681–9.
198. Qi L, Du J, Zhang Z, Diao L, Chen X, and Yao X. Low differentiated microvascular density and low expression of platelet-derived growth factor-BB (PDGF-BB) predict distant metastasis and poor prognosis in clear cell renal cell carcinoma. BJU Int. (2013) 112:E415–423. doi: 10.1111/bju.12191
199. Zhao J, Liu Z, Liu T, Nilsson S, and Nistér M. Identification and expression analysis of an N-terminally truncated isoform of human PDGF-C. Exp Cell Res. (2008) 314:2529–43. doi: 10.1016/j.yexcr.2008.06.007
200. Uhlig A, Bergmann L, Bögemann M, Fischer T, Goebell PJ, Leitsmann M, et al. Sunitinib for metastatic renal cell carcinoma: real-world data from the STAR-TOR registry and detailed literature review. Urol Int. (2024) 108:198–210. doi: 10.1159/000536563
201. Ball SG, Shuttleworth CA, and Kielty CM. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol. (2007) 177:489–500. doi: 10.1083/jcb.200608093
202. Li Y, Leng Q, Pang X, Shi H, Liu Y, Xiao S, et al. Therapeutic effects of EGF-modified curcumin/chitosan nano-spray on wound healing. Regener Biomater. (2021) 8:rbab009. doi: 10.1093/rb/rbab009
203. Masoud GN and Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
204. Hainsworth JD, Sosman JA, Spigel DR, Edwards DL, Baughman C, and Greco A. Treatment of metastatic renal cell carcinoma with a combination of bevacizumab and erlotinib. J Clin Oncol. (2005) 23:7889–96. doi: 10.1200/JCO.2005.01.8234
205. Takimoto CH and Awada A. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother Pharmacol. (2008) 61:535–48. doi: 10.1007/s00280-007-0639-9
206. Ma X, Wu S, Li B, Zhang Q, Zhang J, Liu W, et al. EGFR blockade confers sensitivity to cabozantinib in hepatocellular carcinoma. Cell Discov. (2022) 8:82. doi: 10.1038/s41421-022-00425-y
207. Zhong H and Bowen JP. Recent advances in small molecule inhibitors of VEGFR and EGFR signaling pathways. Curr Top Med Chem. (2011) 11:1571–90. doi: 10.2174/156802611795860924
208. Izzedine H and Perazella MA. Adverse kidney effects of epidermal growth factor receptor inhibitors. Nephrol Dial Transplant. (2017) 32:1089–97. doi: 10.1093/ndt/gfw467
209. Salles DC, Asrani K, Woo J, Vidotto T, Liu HB, Vidal I, et al. GPNMB expression identifies TSC1/2/mTOR-associated and MiT family translocation-driven renal neoplasms. J Pathol. (2022) 257:158–71. doi: 10.1002/path.5875
210. Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, and Kolenko VM. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol Cancer Ther. (2018) 17:1355–64. doi: 10.1158/1535-7163.MCT-17-1299
211. Ryan CW, Tangen C, Heath EI, Stein MN, Meng M, Alva AS, et al. EVEREST: Everolimus for renal cancer ensuing surgical therapy—A phase III study (SWOG S0931, NCT01120249). J Clin Oncol. (2022) 40(17_suppl):LBA4500. doi: 10.1200/JCO.2022.40.17_suppl.LBA4500
212. Chan J, Bayliss PE, Wood JM, and Roberts TM. Dissection of angiogenic signaling in zebrafish using a chemical genetic approach. Cancer Cell. (2002) 1:257–67. doi: 10.1016/s1535-6108(02)00042-9
213. Saxton RA and Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
214. Hua H, Kong Q, Zhang H, Wang J, Luo T, and Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. (2019) 12:71. doi: 10.1186/s13045-019-0754-1
215. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. New Engl J Med. (2007) 356:2271–81. doi: 10.1056/NEJMoa066838
216. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. (2008) 372:449–56. doi: 10.1016/S0140-6736(08)61039-9
217. Santoni M, Pantano F, Amantini C, Nabissi M, Conti A, Burattini L, et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim Biophys Acta. (2014) 1845:221–31. doi: 10.1016/j.bbcan.2014.01.007
218. O’Reilly KE, Rojo F, She Q-B, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. (2006) 66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925
219. Dibble CC, Asara JM, and Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. (2009) 29:5657–70. doi: 10.1128/MCB.00735-09
220. Toschi A, Lee E, Gadir N, Ohh M, and Foster DA. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem. (2008) 283:34495–9. doi: 10.1074/jbc.C800170200
221. Zheng B, Mao J-H, Qian L, Zhu H, Gu D, Pan X, et al. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. (2015) 357:468–75. doi: 10.1016/j.canlet.2014.11.012
222. Cho DC, Cohen MB, Panka DJ, Collins M, Ghebremichael M, Atkins MB, et al. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clin Cancer Res. (2010) 16:3628–38. doi: 10.1158/1078-0432.CCR-09-3022
223. Carlo MI, Molina AM, Lakhman Y, Patil S, Woo K, DeLuca J, et al. A phase ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist. (2016) 21:787–8. doi: 10.1634/theoncologist.2016-0145
224. Powles T, Wheater M, Din O, Geldart T, Boleti E, Stockdale A, et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur Urol. (2016) 69:450–6. doi: 10.1016/j.eururo.2015.08.035
225. Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. (2016) 34:1660–8. doi: 10.1200/JCO.2015.64.8808
226. Aweys H, Lewis D, Sheriff M, Rabbani RD, Lapitan P, Sanchez E, et al. Renal cell cancer - insights in drug resistance mechanisms. Anticancer Res. (2023) 43:4781–92. doi: 10.21873/anticanres.16675
227. Contento G, Wilson J-AA, Selvarajah B, Platé M, Guillotin D, Morales V, et al. Pyruvate metabolism dictates fibroblast sensitivity to GLS1 inhibition during fibrogenesis. JCI Insight. (2024) 9:e178453. doi: 10.1172/jci.insight.178453
228. Home Page: Annals of Oncology . Available online at: https://www.annalsofoncology.org/ (Accessed August 3, 2024).
229. Tannir NM, Agarwal N, Porta C, Lawrence NJ, Motzer RJ, Lee RJ, et al. CANTATA: Primary analysis of a global, randomized, placebo (Pbo)-controlled, double-blind trial of telaglenastat (CB-839) + cabozantinib versus Pbo + cabozantinib in advanced/metastatic renal cell carcinoma (mRCC) patients (pts) who progressed on immune checkpoint inhibitor (ICI) or anti-angiogenic therapies. J Clin Oncol. (2021) 39(15_suppl):4501. doi: 10.1200/JCO.2021.39.15_suppl.4501
230. Hahn AW, George DJ, and Agarwal N. An evolving role for AXL in metastatic renal cell carcinoma. Clin Cancer Res. (2021) 27:6619–21. doi: 10.1158/1078-0432.CCR-21-3029
231. Beckermann K, Shah NJ, Vogelzang NJ, Mao SS, Ornstein MC, Hammers HJ, et al. A phase 1b/2 study of batiraxcept (AVB-S6-500) in combination with cabozantinib in patients with advanced or metastatic clear cell renal cell (ccRCC) carcinoma who have received front-line treatment (NCT04300140). J Clin Oncol. (2022) 40(16_suppl):TPS4599. doi: 10.1200/JCO.2022.40.16_suppl.TPS4599
232. Chang C-H and Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. (2016) 17:364–8. doi: 10.1038/ni.3415
233. Allard B, Allard D, Buisseret L, and Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
234. Allard B, Beavis PA, Darcy PK, and Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. (2016) 29:7–16. doi: 10.1016/j.coph.2016.04.001
235. Vijayan D, Young A, Teng MWL, and Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:765. doi: 10.1038/nrc.2017.110
236. Young A, Mittal D, Stagg J, and Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov. (2014) 4:879–88. doi: 10.1158/2159-8290.CD-14-0341
237. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. (2020) 10:40–53. doi: 10.1158/2159-8290.CD-19-0980
238. Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. (2018) 11:100. doi: 10.1186/s13045-018-0644-y
239. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. (2010) 115:3520–30. doi: 10.1182/blood-2009-09-246124
240. Mitchell TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J Clin Oncol. (2018) 36:3223–30. doi: 10.1200/JCO.2018.78.9602
241. Aggarwal RR, Thomas S, Pawlowska N, Grabowsky JA, Calabrese S, Lam P, et al. Exceptional responders to abexinostat (ABX) plus pazopanib (PAZ) in pretreated renal cell carcinoma (RCC) and other solid tumors: Long-term follow-up of a phase 1b study. J Clin Oncol. (2019) 37(15_suppl):3022. doi: 10.1200/JCO.2019.37.15_suppl.3022
242. Boussios S, Devo P, Goodall ICA, Sirlantzis K, Ghose A, Shinde SD, et al. Exosomes in the diagnosis and treatment of renal cell cancer. Int J Mol Sci. (2023) 24:14356. doi: 10.3390/ijms241814356
243. Gouttefangeas C, Stenzl A, Stevanović S, and Rammensee H-G. Immunotherapy of renal cell carcinoma. Cancer Immunol Immunother. (2007) 56:117–28. doi: 10.1007/s00262-006-0172-4
244. Kong Y, Ma Y, Zhao X, Pan J, Xu Z, and Zhang L. Optimizing the treatment schedule of radiotherapy combined with anti-PD-1/PD-L1 immunotherapy in metastatic cancers. Front Oncol. (2021) 11:638873. doi: 10.3389/fonc.2021.638873
245. Wang N-H, Lei Z, Yang H-N, Tang Z, Yang M-Q, Wang Y, et al. Radiation-induced PD-L1 expression in tumor and its microenvironment facilitates cancer-immune escape: a narrative review. Ann Transl Med. (2022) 10:1406. doi: 10.21037/atm-22-6049
246. Huang KC-Y, Chiang S-F, Ke T-W, Chen T-W, Hu C-H, Yang P-C, et al. DNMT1 constrains IFNβ-mediated anti-tumor immunity and PD-L1 expression to reduce the efficacy of radiotherapy and immunotherapy. Oncoimmunology. (2021) 10:1989790. doi: 10.1080/2162402X.2021.1989790
247. Masini C, Iotti C, Giorgi UD, Bellia RS, Buti S, Salaroli F, et al. Nivolumab in combination with stereotactic body radiotherapy in pretreated patients with metastatic renal cell carcinoma. Results of the phase II NIVES study. Eur Urol. (2022) 81:274–82. doi: 10.1016/j.eururo.2021.09.016
248. ASCO GU 2020: Combination of Dual Immune Checkpoint Inhibition with Stereotactic Radiation in Metastatic Renal Cell Carcinoma (RADVAX RCC) . Available online at: https://www.urotoday.com/conference-highlights/asco-gu-2020/asco-gu-2020-kidney-cancer/119275-asco-gu-2020-combination-of-dual-immune-checkpoint-inhibition-with-stereotactic-radiation-in-metastatic-renal-cell-carcinoma-radvax-rcc.html?utm_source=chatgpt.com (Accessed February 21, 2025).
249. Berro A, Assi A, Farhat M, Hatoum L, Saad J-P, Mohanna R, et al. Unlocking Hope: Anti-VEGFR inhibitors and their potential in glioblastoma treatment. Crit Rev Oncol Hematol. (2024) 198:104365. doi: 10.1016/j.critrevonc.2024.104365
250. Heine A, Held S, Bringmann A, Holderried T, and Brossart P. Immunomodulatory effects of anti-angiogenic drugs. Leukemia. (2011) 25:899–905. doi: 10.1038/leu.2011.24
251. Somasundaram A, Socinski MA, and Villaruz LC. Immune checkpoint blockade in oncogene-driven non-small-cell lung cancer. Drugs. (2020) 80:883–92. doi: 10.1007/s40265-020-01320-0
252. Liu Y, Wei W, Zhang Z, Liu R, Gao J, Guo S, et al. Preliminary results from a phase II study comparing sunitinib alone or with stereotactic body radiotherapy (SBRT) for newly diagnosed oligometastatic renal cell carcinoma. JCO. (2023) 41:4533–3. doi: 10.1200/JCO.2023.41.16_suppl.4533
253. Hirsch L, Flippot R, Escudier B, and Albiges L. Immunomodulatory roles of VEGF pathway inhibitors in renal cell carcinoma. Drugs. (2020) 80:1169–81. doi: 10.1007/s40265-020-01327-7
254. Ahrens M, Scheich S, Hartmann A, Bergmann L, and IAG-N Interdisciplinary Working Group Kidney Cancer of the German Cancer Society. Non-clear cell renal cell carcinoma - pathology and treatment options. Oncol Res Treat. (2019) 42:128–35. doi: 10.1159/000495366
255. Tian Z, Dong S, Yang Y, Gao S, Yang Y, Yang J, et al. Nanoparticle albumin-bound paclitaxel and PD-1 inhibitor (sintilimab) combination therapy for soft tissue sarcoma: a retrospective study. BMC Cancer. (2022) 22:56. doi: 10.1186/s12885-022-09176-1
256. Chen Y-W, Wang L, Panian J, Dhanji S, Derweesh I, Rose B, et al. Treatment landscape of renal cell carcinoma. Curr Treat Options Oncol. (2023) 24:1889–916. doi: 10.1007/s11864-023-01161-5
257. Wang Q, Yu M, and Zhang S. Peptic ulcer induced by immune checkpoint inhibitors successfully treated with glucocorticoids: A report of three cases and a literature review. Exp Ther Med. (2024) 28:410. doi: 10.3892/etm.2024.12699
258. Hu X, Ren J, Xue Q, Luan R, Ding D, Tan J, et al. Anti-PD-1/PD-L1 and anti-CTLA-4 associated checkpoint inhibitor pneumonitis in non-small cell lung cancer: Occurrence, pathogenesis and risk factors (Review). Int J Oncol. (2023) 63:122. doi: 10.3892/ijo.2023.5570
259. Kohsaka S, Tada Y, Ando M, Nakaguro M, Shirai Y, Ueno T, et al. Identification of novel prognostic and predictive biomarkers in salivary duct carcinoma via comprehensive molecular profiling. NPJ Precis Oncol. (2022) 6:82. doi: 10.1038/s41698-022-00324-1
260. Moreira M, Pobel C, Epaillard N, Simonaggio A, Oudard S, and Vano Y-A. Resistance to cancer immunotherapy in metastatic renal cell carcinoma. cdr. (2020) 3:454–71. doi: 10.20517/cdr.2020.16
261. Sweeney PL, Suri Y, Basu A, Koshkin VS, and Desai A. Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. cdr. (2023) 6:858–73. doi: 10.20517/cdr.2023.89
262. ASCO GU. Nivolumab plus Cabozantinib vs Sunitinib for Previously Untreated Advanced Renal Cell Carcinoma: Final Follow-up Results from the CheckMate 9ER Trial (2025). Available online at: https://www.urotoday.com/conference-highlights/asco-gu-2025/asco-gu-2025-kidney-cancer/158352-asco-gu-2025-nivolumab-plus-cabozantinib-n-c-vs-sunitinib-s-for-previously-untreated-advanced-renal-cell-carcinoma-arcc-final-follow-up-results-from-the-checkmate-9er-trial.html?utm_source=chatgpt.com (Accessed February 21, 2025).
263. ASCO GU. Phase 1b/2 Study of Combination 177Lu Girentuximab + Cabozantinib and Nivolumab in Treatment Naive Patients With Advanced Clear Cell RCC (2025). Available online at: https://www.urotoday.com/conference-highlights/asco-gu-2025/asco-gu-2025-kidney-cancer/158314-asco-gu-2025-phase-1b-2-study-of-combination-177lu-girentuximab-cabozantinib-and-nivolumab-in-treatment-naive-patients-with-advanced-clear-cell-rcc.html (Accessed February 21, 2025).
Keywords: renal cell carcinoma, immunotherapy, targeted therapy, current strategies, future directions
Citation: Wang B, Xiang Y, Liu X, Pan X, Peng L and Ma J (2025) Advances in immunotherapy and targeted therapy for advanced clear-cell renal cell carcinoma: current strategies and future directions. Front. Immunol. 16:1582887. doi: 10.3389/fimmu.2025.1582887
Received: 25 February 2025; Accepted: 06 June 2025;
Published: 03 July 2025.
Edited by:
Meng Zhang, First Affiliated Hospital of Anhui Medical University, ChinaReviewed by:
Karthik Dhatchinamoorthy, University of Massachusetts Medical School, United StatesSawkar Vijay Pramod, Padjadjaran University, Indonesia
Copyright © 2025 Wang, Xiang, Liu, Pan, Peng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Wang, Ym93YW5nNTgxQGdtYWlsLmNvbQ==; Jiajia Ma, amlhamlhLm1hQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==
†These authors have contributed equally to this work