 Ting Zhang
Ting Zhang Shuai Zhao
Shuai Zhao Jia-Xun Feng
Jia-Xun Feng- State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangxi Research Center for Microbial and Enzyme Engineering Technology, College of Life Science and Technology, Guangxi University, Nanning, China
Limited information on transcription factor (TF)-mediated regulation exists for most filamentous fungi, specifically for regulation of the production of plant-biomass-degrading enzymes (PBDEs). The filamentous fungus, Talaromyces pinophilus, can secrete integrative cellulolytic and amylolytic enzymes, suggesting a promising application in biotechnology. In the present study, the regulatory roles of a Zn2Cys6 protein, TP05746, were investigated in T. pinophilus through the use of biochemical, microbiological and omics techniques. Deletion of the gene TP05746 in T. pinophilus led to a 149.6% increase in soluble-starch-degrading enzyme (SSDE) production at day one of soluble starch induction but an approximately 30% decrease at days 2 to 4 compared with the parental strain ΔTpKu70. In contrast, the T. pinophilus mutant ΔTP05746 exhibited a 136.8–240.0% increase in raw-starch-degrading enzyme (RSDE) production, as well as a 90.3 to 519.1% increase in cellulase and xylanase production following induction by culturing on wheat bran plus Avicel, relative to that exhibited by ΔTpKu70. Additionally, the mutant ΔTP05746 exhibited accelerated mycelial growth at the early stage of cultivation and decreased conidiation. Transcriptomic profiling and real-time quantitative reverse transcription-PCR (RT-qPCR) analyses revealed that TP05746 dynamically regulated the expression of genes encoding major PBDEs and their regulatory genes, as well as fungal development-regulated genes. Furthermore, in vitro binding experiments confirmed that TP05746 bound to the promoter regions of the genes described above. These results will contribute to our understanding of the regulatory mechanism of PBDE genes and provide a promising target for genetic engineering for improved PBDE production in filamentous fungi.
Introduction
Soil filamentous fungi play crucial roles in terrestrial carbon cycling because they can decay organic matter, including plant biomass (Gougoulias et al., 2014) via secretion of plant-biomass-degrading enzymes (PBDEs), such as cellulase, hemicellulose, and amylase (Bornscheuer et al., 2014). Most of the PBDEs are produced in nature by Trichoderma, Penicillium and Aspergillus, but they generate only low yields (Passos et al., 2018). Therefore, genetic improvement of natural fungal producers such as enzymes needs to be further explored, with an aim of developing strains capable of high production of PBDEs.
In the enzyme market, cellulases and amylases account for the majority of PBDEs. Cellulases are the complexes comprising cellobiohydrolases (EC 3.2.1.91; CBHs), endo-β-1,4-glucanases (EC 3.2.1.4; EGs), and β-1,4-glucosidases (EC 3.2.1.21; BGLs)1. Synergistic action among these enzymes is required for cellulose hydrolysis into glucose. During the process, EGs randomly break internal β-1,4-glycosidic bonds in glucan chains to release glucooligosaccharides, resulting in new chain ends. CBHs act on both ends of glucooligosaccharide chains to produce mainly cellobiose. Both soluble glucooligosaccharide and cellobiose are hydrolyzed by BGLs to yield glucose (Bornscheuer et al., 2014).
Comparably, amylases are classified broadly into glucoamylases (EC 3.2.1.3; GLAs), α-amylases (EC 3.2.1.1; AMYs), α-glucosidases (EC 3.2.1.20; AGAs) and 1,4-α-glucan-branching enzymes (EC 2.4.1.18) according to the action principle (see text footnote 1). Among them, GLAs attack α-1,4- or α-1,6-glucosidic bonds at the non-reducing ends of starch chains to release glucose, while AMYs break internal α-1,4-glycosidic bonds of starch chains to generate straight and branched oligosaccharides or maltose. AGAs digest maltose into glucose (Marín-Navarro and Polaina, 2011). Moreover, amylolytic enzymes can show activity toward soluble starch, known as soluble starch-degrading enzymes (SSDEs). Some amylolytic enzymes, known as raw starch-degrading enzymes (RSDEs), can directly hydrolyze raw starch granules below the starch gelatinization temperature (Sun et al., 2010). SSDEs are commonly used for conventional starch processing, including two-step liquefaction and saccharification. During liquefaction, raw starch is first gelatinized at a high temperature (95–105°C) and then liquefied into dextrin via the thermophilic AMYs. Subsequently, GLAs are applied in the saccharification step to hydrolyze the cooled dextrin into glucose at 60–65°C (Sánchez and Cardona, 2008; Görgens et al., 2015). However, the conventional liquefaction step requires high energy input and extra equipment, resulting in a high cost of the starch-derived products.
RSDEs contain the specific starch-binding domain (SBD) that enables them to bind onto the surface of raw starch granules (Sorimachi et al., 1997; Machovic and Janecek, 2006; Xu et al., 2016). RSDE application can reduce the process cost since the liquefaction step would not be needed (Sun et al., 2010; Lee et al., 2012). It was estimated that the application of RSDE in ethanol production, using starch as feedstock, could reduce the fuel cost of the total ethanol product by about 10–20% (Robertson et al., 2006).
Genetic engineering of fungal strains, based on the regulatory network of transcription factors (TFs) and their target genes, is an efficient strategy by which to improve functional enzyme yields. TFs are proteins that control transcription of target genes through binding to specific DNA sequence elements such as promoters, enhancers, etc. TF-mediated transcriptional control is a central regulatory mechanism in all eukaryotic organisms (Weirauch and Hughes, 2011). More than 90 types of TFs in eukaryotes have been identified, including zinc finger (Zn2Cys6, C2H2, GATA, CCHC, DHHC, etc.), helix-turn-helix (HTH), basic leucine zipper (bZIP), APSES and winged helix repressor DNA-binding domain (Weirauch and Hughes, 2011; Carrillo et al., 2017). The regulatory roles of individual TFs vary under different conditions.
Transcriptional expression of genes encoding PBDEs is strictly controlled by specific TFs in filamentous fungi. Since XlnR was first identified to be involved in regulating the expression of genes encoding enzymes involved in degrading plant cell walls in Aspergillus (van Peij et al., 1998), several TFs have been found to be expressed in the presence of recalcitrant carbon sources, such as the key activators CLR-2 (Coradetti et al., 2012; Zhao et al., 2016), AmyR (Li et al., 2015), and PoxCxrA (Yan et al., 2017). Among them, CLR-2 and PoxCxrA encode Gal4-like Zn2Cys6 proteins, and play essential roles in the regulation of fungal cellulase gene expression in the presence of cellulose. In Neurospora crassa, the expression of clr-2 is induced by the TF CLR-1 (Coradetti et al., 2012), while it is induced by PoxCxrA in Penicillium oxalicum (Liao et al., 2019). The CLR-1 homolog ClrA is less involved in the regulation of cellulase gene expression in P. oxalicum (Liao et al., 2019), as well as in Aspergillus nidulans (Coradetti et al., 2012; Tani et al., 2014).
The Zn2Cys6 proteins AmyR and/or COL-26 are necessary for amylolytic gene expression in filamentous fungi such as P. oxalicum, N. crassa, T. pinophilus, Aspergillus spp., and are required for starch and maltose utilization (Li et al., 2015; Xiong et al., 2017; Zhang et al., 2017). Additionally, AmyR inhibits the expression of cellulase genes and its expression is regulated by ClrB in P. oxalicum (Li et al., 2015). COL-26 regulates cellulase gene expression and enzyme production, synergistically functioning with the carbon catabolite repressor, CRE-1 (Xiong et al., 2014).
CRE-1 mediates carbon catabolite repression (CCR) that represses cellulase gene expression in the presence of favorable carbon source such as glucose. CRE-1 could directly bind to the promoter regions of major cellulase and xylanase genes and their regulatory genes, such as clr-2, resulting in reduction of enzyme yields (Li et al., 2015; Huberman et al., 2016).
The soil fungus T. pinophilus, formerly called Penicillium pinophilus, produces useful PBDEs such as α-amylase, glucoamylase, cellulase, xylanase, and laccase (Pol et al., 2012; Visser et al., 2013; Kusum et al., 2014; Xian et al., 2015) and medical metabolites such as 3-O-methylfunicone and talaromycolides 1–3, 5, and 11. The metabolite 3-O-methylfunicone is able to repress mesothelioma cell motility, while talaromycolide kills human-pathogenic methicillin-resistant Staphylococcus aureus (Buommino et al., 2012; Zhai et al., 2015). T. pinophilus strain 1–95 was isolated in China (Xian et al., 2015), and shown to produce several PBDEs (Li et al., 2017), albeit at low yields.
Comparative transcriptomic profiling and genetic analyses of T. pinophilus strain 1–95 identified seven novel regulatory genes that regulate SSDE production. Among them, TpRfx1 (TP06128), positively regulated SSDE production of T. pinophilus via binding to the promoter regions of major amylase genes (Liao et al., 2018). Intriguingly, deletion of another candidate regulatory gene TP05746, encoding a Zn2Cys6 protein, resulted in a 51.4% increase in SSDE production, compared with the parental strain ΔTpKu70 when cultured directly on medium containing soluble corn starch (SCS) for 5 days (Liao et al., 2018), but its detailed biological roles are unknown.
In this study, we found that TP05746 regulated the production of various PBDEs including SSDE, RSDE, cellulase and xylanase of T. pinophilus, as well as mycelial growth and conidiation. Experiments further confirmed that TP05746 could bind to the promoter regions of major PBDE genes and their key regulatory genes, and to growth- and development-associated regulatory genes.
Materials and Methods
Fungal Strains and Culture Conditions
Talaromyces pinophilus wild-type strain 1–95 (#2645, China General Microbiological Culture Collection, Beijing, China; CGMCC) was isolated from a dry ploughed field in China (Xian et al., 2015). Deletion mutants ΔTpKu70 and ΔTP05746 were constructed by knocking out the TpKu70 gene in strain 1–95 and the TP05746 gene in ΔTpKu70, respectively (Zhang et al., 2017; Liao et al., 2018). The ΔTpKu70 has no apparent difference in vegetative growth and enzyme production when compared with wild-type strain 1–95 (Zhang et al., 2017). All T. pinophilus strains were maintained on potato-dextrose agar (PDA) plates at 4°C or stored in 25% glycerol at -80°C. Cultivation of Penicillium oxalicum strains was consistent with the T. pinophillus strains. Mutant ΔPoxKu70 (#3.15650, CGMCC) was derived from the wild-type HP7-1 via deleting gene PoxKu70 (Zhao et al., 2016).
Asexual spores (conidia) were collected from fungal cells cultured on PDA plates at 28°C for 6 days, resuspended in aqueous 0.2% (v/v) Tween-80, and adjusted to a concentration of 1 × 108 spores mL–1. For mycelial growth for DNA extraction, the fungus was cultured on liquid complete medium (LCM; Liao et al., 2018). Culture conditions for enzyme activity analysis, RNA sequencing, and real-time quantitative reverse transcription-PCR (RT-qPCR) analysis were as described previously (Liao et al., 2018). For RNA sequencing, total RNA was extracted from mycelium harvested after 12-h culture following transfer from glucose into medium containing 1% (w/v) soluble corn starch (SCS; Sigma-Aldrich, St. Louis, MO, United States).
For mycelial growth and observation by light microscopy, standard liquid medium (SLM; Liao et al., 2018) containing 1% (w/v) D-glucose, 1% (w/v) SCS, 2% (w/v) wheat bran plus Avicel [WA; wheat bran: Avicel = 1: 1; (w/w); Sigma-Aldrich, St.] or 2% (w/v) Avicel and/or 0.2% (w/v) 2-deoxy-glucose (2-DG) was inoculated with T. pinophilus spores and cultured at 28°C for 12 to 120 h. For phenotypic analyses, 1% (w/v) D-glucose, 1% (w/v) SCS or 2% (w/v) WA were added to solid low-salt minimal medium (LsMM) plates, with PDA being used as a positive control. For measurement of enzymatic activity, P. oxalicum strains were cultivated in Avicel medium according to the descried by Yan et al. (2017).
Extraction of Total DNA and RNA From Talaromyces pinophilus
Extraction of total DNA and RNA from mycelia of T. pinophilus strains was carried out by the method described previously (Yan et al., 2017) with some modifications. In brief, for total DNA extraction, the harvested mycelia were ground in liquid nitrogen and lysate reagent (pH 8.0) was added immediately at a ratio of 10:1 (v: w). Subsequently, an equal volume of a phenol-chloroform mixture was added to the extract to remove proteins, followed by centrifuging at 11,300 × g at 4°C for 10 min. Finally, DNA was precipitated using isopropanol at a ratio of 1:1 (v: v).
Total RNA was extracted using TRIzol RNA Kit (Life Technologies, Carlsbad, CA, United States), according to the manufacturer’s instructions.
Southern Hybridization Analysis
The only one-site mutation in the mutant ΔTP05746 was confirmed using Southern hybridization analysis, according to the protocols of the DIG (digoxigenin)-High Prime DNA Labeling & Detection Starter Kit (Life Technologies, Carlsbad, CA, United States). Briefly, genomic DNA from each of the ΔTpKu70 and the ΔTP05746 mutants was extracted and digested by HindIII (TaKaRa Bio Inc., Dalian, China), and then transferred onto a Hybond-N+ Nylon membrane (GE Healthcare Limited, Amersham, United Kingdom). PCR was used to amplify the hybridization probe, using a specific primer pair TP05746-T-F/TP05746-T-R (Supplementary Table S1).
Measurement of PBDE Production and Intracellular Protein Concentration Assays
Plant-biomass-degrading enzymes activities, including SSDE, raw starch-digesting enzymes (RSDE), cellulase and xylanase activities, were assayed as previously described (Liao et al., 2018; Zhang et al., 2019). Briefly, 50 μL of appropriately diluted crude extract produced from the parental strain ΔTpKu70 or its deletion mutant ΔTP05746, were added to 450 μL of 100 mM citrate-phosphate buffer (pH 5.0) containing 1% SCS (Sigma-Aldrich), 1% raw cassava flour (farmer’s market, Nanning, China), 1% CMC-Na (Sigma-Aldrich, Darmstadt, Germany), or 1% beechwood xylan (Megazyme International Ireland, Bray, Ireland), and 1 mL of 100 mM citrate-phosphate buffer pH 5.0, containing Whatman No. 1 filter paper (50 mg, 1.0 cm × 6.0 cm; GE Healthcare Limited, Little Chalfont, United Kingdom). The mixture was incubated at 50–60°C for 10–60 min. The reducing sugars released were determined using the 3,5-dinitrosalicyclic acid method, measuring A540 (Miller, 1959). One unit of enzymatic activity (U) was defined as the required amount of enzyme required to produce 1 μmol reducing sugar per minute from the reaction substrates.
The substrates p-nitrophenyl-β-D-cellobioside (pNPC) and p-nitrophenyl-β-D-glucopyranoside (pNPG) (both from Sigma-Aldrich) were used for measurement of pNPCase and pNPGase activities, respectively. An aliquot (10 μL or 68 μL) of appropriately diluted crude extract from ΔTpKu70 or ΔTP05746, was added into 130 or 72 μL of a mixture containing 14 μL 25 mM pNPG or pNPC, and incubated at 50°C for 15 min. Sodium carbonate (70 μL, 0.4 M) was added to stop the reaction. The p-nitrophenol liberated was measured at wavelength of 410 nm, with one unit of enzymatic activity (U) being defined as the amount of enzyme that produced 1 μmol of p-nitrophenol per minute from an appropriate substrate. All the assays were performed in three biological replicates.
Protein concentration in extracts of T. pinophilus strains was measured using the Bradford assay kit (Pierce Biotechnology, Rockford, IL, United States).
Determination of Mycelial Dry Weight
Mycelial dry weight of T. pinophilus strains cultured in SLM containing 1% D-glucose or 1% SCS as the sole carbon source was determined gravimetrically as previously described (Liao et al., 2018). In brief, 1.0 × 108 fresh spores were inoculated into 100 mL of the above medium and shake-cultured at 28°C and 180 rpm for 12 to 84 h. The hyphae were harvested by vacuum suction filtration every 12 h and then dried at 50°C to a constant weight. All the assays were performed in three biological replicates and the mean ± SD of the three replicates is presented.
Light Microscopy Observation
Light microscopy observation of T. pinophilus mycelia was performed according to previously described methods (Xiong et al., 2018). The harvested hyphae from T. pinophilus strains grown on SLM containing 1% D-glucose, 1% SCS, or 2% WA for 12–36 h was transferred onto microscope slides. The slides were observed under a light microscope (OLYMPUS DP480; Olympus, Tokyo, Japan), and photomicrographs were analyzed using cellSence Dimension digital imaging software (Olympus).
Real-Time Quantitative Reverse Transcription-PCR (RT-qPCR) Analysis
The PrimeScriptTM RT Reagent kit (TaKaRa Bio Inc.) was used to synthesize the first-strand cDNA from total RNA of mutant ΔTpKu70 as the template, according to the manufacturer’s instruction. The PCR reaction mixture (20 μL) was composed of 10 μL of SYBR Premix ExTaq II (TaKaRa Bio Inc), 1.6 μL of 10 μM each primer (Supplementary Table S1), 2.0 μL of first-strand cDNA and 6.4 μL of sterile water. All the reaction cycles were run as follows: initial denaturation for 3 min at 96°C, followed by 40 cycles of 10 s each at 96°C, and 60°C for 30 s. The fluorescence signals were observed at the end of each extension step at 80°C. The relative expression levels of the tested genes were calculated as described previously (Zhang et al., 2017).
RNA Sequencing
RNA sequencing of T. pinophilus strains was performed by BGI, Shenzhen, China, as described by He et al. (2018). A cDNA library was constructed with an average length of 100 bp for each sample and assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States) and an ABI StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA, United States), and then sequenced using an Illumina HiSeq 4000 system. Clean reads generated were mapped onto the genome of T. pinophilus strain 1–95 (Li et al., 2017) for functional annotation using BWA v0.7.10-r789 (Li and Durbin, 2009) and Bowtie2 v2.1.0 (Langmead and Salzberg, 2012). The gene expression levels (fragments per kilobase of exon per million mapped reads, FPKM) and differentially expressed genes (DEGs) were calculated and screened for, using the software RAEM v1.2.12 (Li and Dewey, 2011) and DESeq (Love et al., 2014), respectively. Genes with | log2 (ΔTP05746_FPKM/ΔTpKu70_FPKM)| ≥ 1 and p-value ≤ 0.05 were defined as DEGs.
Overexpression of Gene TP05746 in Penicillium oxalicum
Overexpression of gene TP05746 was performed in filamentous fungus P. oxalicum according to the method for complementary strain construction described by Yan et al. (2017). Briefly, the overexpression cassette comprised of four fragments including approximately 2 kb of left- and right-flanking sequences of gene POX05007 encoding a aspartic protease (Wang et al., 2018), G418 resistance gene, and gene TP05746 sequence with a strong promoter pPoxEgCel5B (Wang et al., 2018) and gene TP05586 terminator, was constructed via fusion PCR. These fragments were amplified using PCR with specific primer pairs (Supplementary Table S1). The constructed overexpression cassette was introduced into the fresh protoplasts of the parental strain ΔPoxKu70 based on the methods previously described (Zhao et al., 2016). The overexpressed transformants were isolated and confirmed via corresponding antibiotics G418 and hygromycin, and PCR with specific confirmation primers (Supplementary Table S1).
Recombinant Expression of DNA Fragment Encoding TP05746
A DNA fragment (1002 bp) of TP05746 was amplified with PCR using primer pairs TP05746-F2/TP05746-R2 (Supplementary Table S1). The DNA fragment was cloned into vector pET32a (+) digested with SacI, using ClonExpress® II One Step Cloning Kit (Vazyme Biotech, Nanjing, China). The recombinant plasmid pET32a-TP05746 was introduced into E. coli Trans-DE3 cells, and then induced with 0.5 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) and cultured at 16°C for 24 h. The recombinant protein, rTP05746, labeled with Trx-His-S tags, was purified using Ni-nitrilotriacetic acid (Ni-NTA) resin. Trx-His-S fusion protein and bovine serum albumin (BSA) were used as negative controls.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed as previously described (Yan et al., 2017). Briefly, DNA fragments approximately 1,000-bp upstream from the ATG start codons of the tested genes were amplified by PCR, using specific primers labeled with 6-carboxyfluorescein (FAM) at the 3′ terminus (Supplementary Table S1) as EMSA probes, and a 500-bp DNA sequence upstream from the ATG start codon of TP10751 encoding β-tubulin (with a FAM label) was used as a control. DNA fragments of the same length but without the FAM label were used as competitive EMSA probes. Approximately 50 ng of DNA probes were mixed with 0, 0.5, 1.0, 1.5, or 2.0 μg of rTP05746 in 2 μL of binding buffer (Liao et al., 2018) and incubated at 25°C for 20 min. In each EMSA reaction, non-specific sheared salmon sperm DNA was added, in order to prevent non-specific binding between protein and probes. For the control, 2.0 μg of BSA or Trx-His-S fusion protein were used. For the competitive EMSA, 2.0 μg of rTP05746 were mixed with 50 ng of DNA probe with the FAM label and 250–2500 ng of competitive EMSA probes. DNA-protein complexes were separated by 4% polyacrylamide Tris–acetic acid–EDTA gel electrophoresis and detected with the Bio-Rad ChemiDocTM MP Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, United States) at 489–506 nm.
Phylogenetic Analysis
Proteins homologous to TP05746 were downloaded from NCBI website using BLASTP2. The neighbor-joining method and a Poisson correction model were used to construct a phylogenetic tree using software MEGA7.0 (Kumar et al., 2016), with 1000 replicates being used to calculate bootstrap values and gaps, and to handle missing data.
Statistical Analysis
All experimental data associated with enzyme production, intracellular protein concentration, counts by microscopy, gene transcription and biomass were statistically analyzed using Microsoft Excel (Office 2016; Microsoft, Redmond, WA, United States). Summary statistics presented are mean ± SD. Significance analyses (p ≤ 0.05 or p ≤ 0.01) among samples were performed using Student’s t test.
Accession Numbers
The DNA sequence of TP05746 is available from the GenBank database under accession number MH447996. The transcriptomic data of T. pinophilus strains have been deposited in Gene Expression Omnibus (GEO) on NCBI (accession No. GSE131872).
Results
TP05746 Regulates the Production of PBDEs in Talaromyces pinophilus
Previous work had initially found that a deletion mutant ΔTP05746 of T. pinophilus showed a 51.4% increase in SSDE production, compared with the parental strain ΔTpKu70 cultured directly in medium containing SCS as the sole carbon source (Liao et al., 2018). Protein TP05746, composed of 333 amino acids, contains a conserved Gal4-like domain, also known as a Zn2Cys6 zinc finger domain, at its C-terminus, as identified through NCBI BlastP query (Figure 1A). TP05746 shared 87% and 43% of identities with putative C6 transcription factor PMAA_081800 in Talaromyces marneffei ATCC 18224 (XP_002147682.1) and AN8177.2 in Aspergillus nidulans FGSC A4 (XP 022399897.1), respectively, with the coverage of 100% and 83%, respectively, whereas most of the aligned proteins were selected based on the conserved Zn2Cys6 zinc finger domain at the C-terminus. Additionally, phylogenetic analyses demonstrated that the TP05746 and its homologs were specific to Talaromyces, and phylogenetically close to those in the Ajellomycetaceae (Figure 1B).
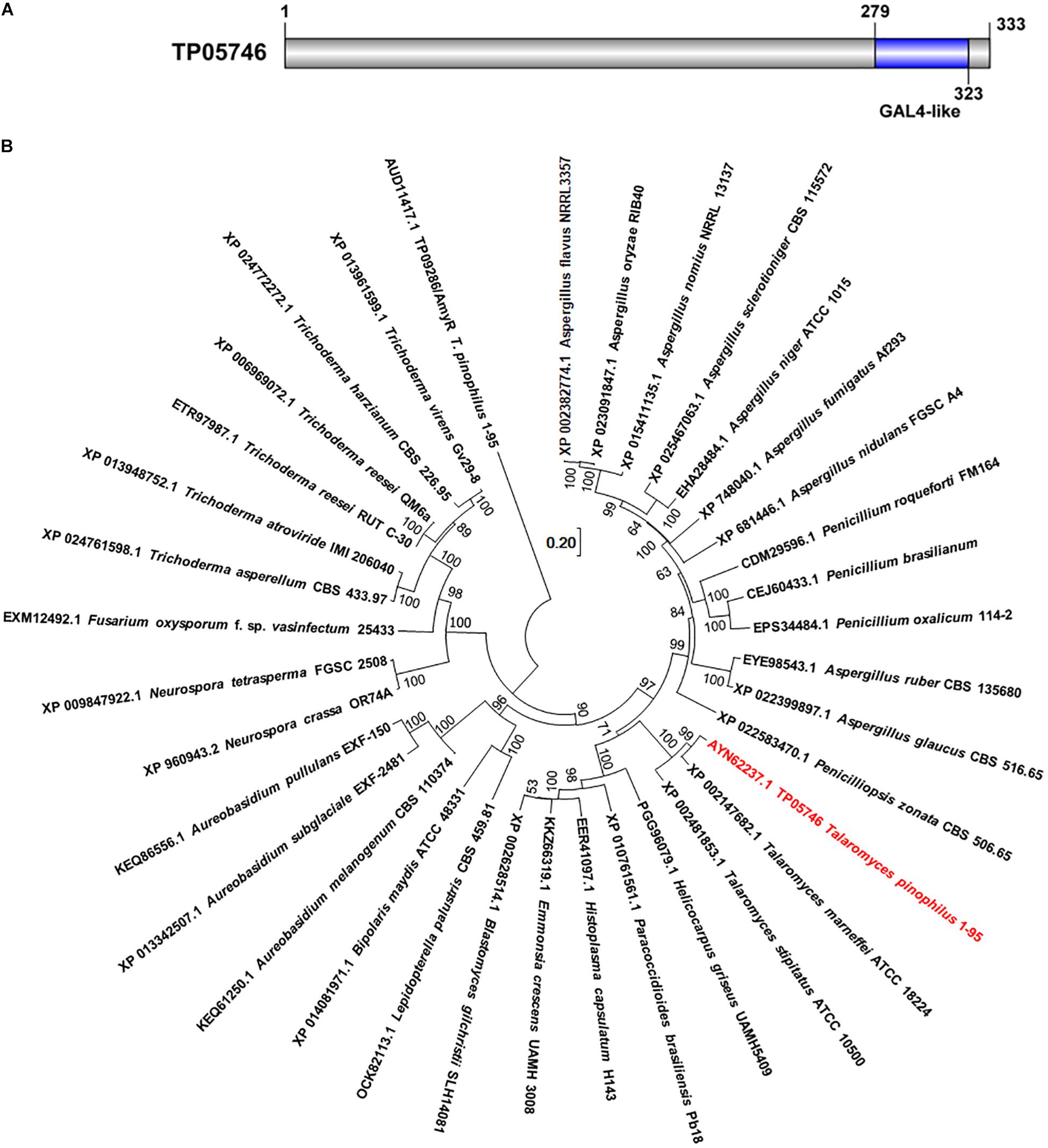
Figure 1. Characterization of TP05746 from Talaromyces pinophilus. (A) Conserved domain in TP05746. (B) Phylogenetic analyses of TP05746 and its homologous proteins. The cladogram was constructed by software MEGA7 based on the neighbor-joining method and a Poisson correction model. Scale bar indicates branch lengths.
Southern hybridization analysis was employed to confirm the identity of the mutant ΔTP05746. The results showed that an expected 6.3-kb band appeared in the transformants of ΔTP05746, compared with a 3.2-kb band for ΔTpKu70 (Supplementary Figure S1), confirming that a TP05746 deletion cassette inserted into only the one correct site in the fungal genome. PCR analysis also confirmed that the deletion cassette replaced the TP05746 locus (Liao et al., 2018).
Talaromyces pinophilus mutant strain ΔTP05746 and its parental strain ΔTpKu70 were then precultured in glucose medium for 24 h, and the same amounts of mycelia were transferred into fresh SLM containing SCS as carbon source and cultured for 1–4 days. Enzyme activity tests indicated that ΔTP05746 exhibited 149.6% higher SSDE production at day one of SCS induction but then 27.6–33.1% decrease at days 2–4, relative to that exhibited by ΔTpKu70 (p < 0.01, Student’s t-test; Figure 2A). Intriguingly, the RSDE production of ΔTP05746 increased by 136.8–240.0% over the entire culture period (p < 0.01, Student’s t-test; Figure 2B).
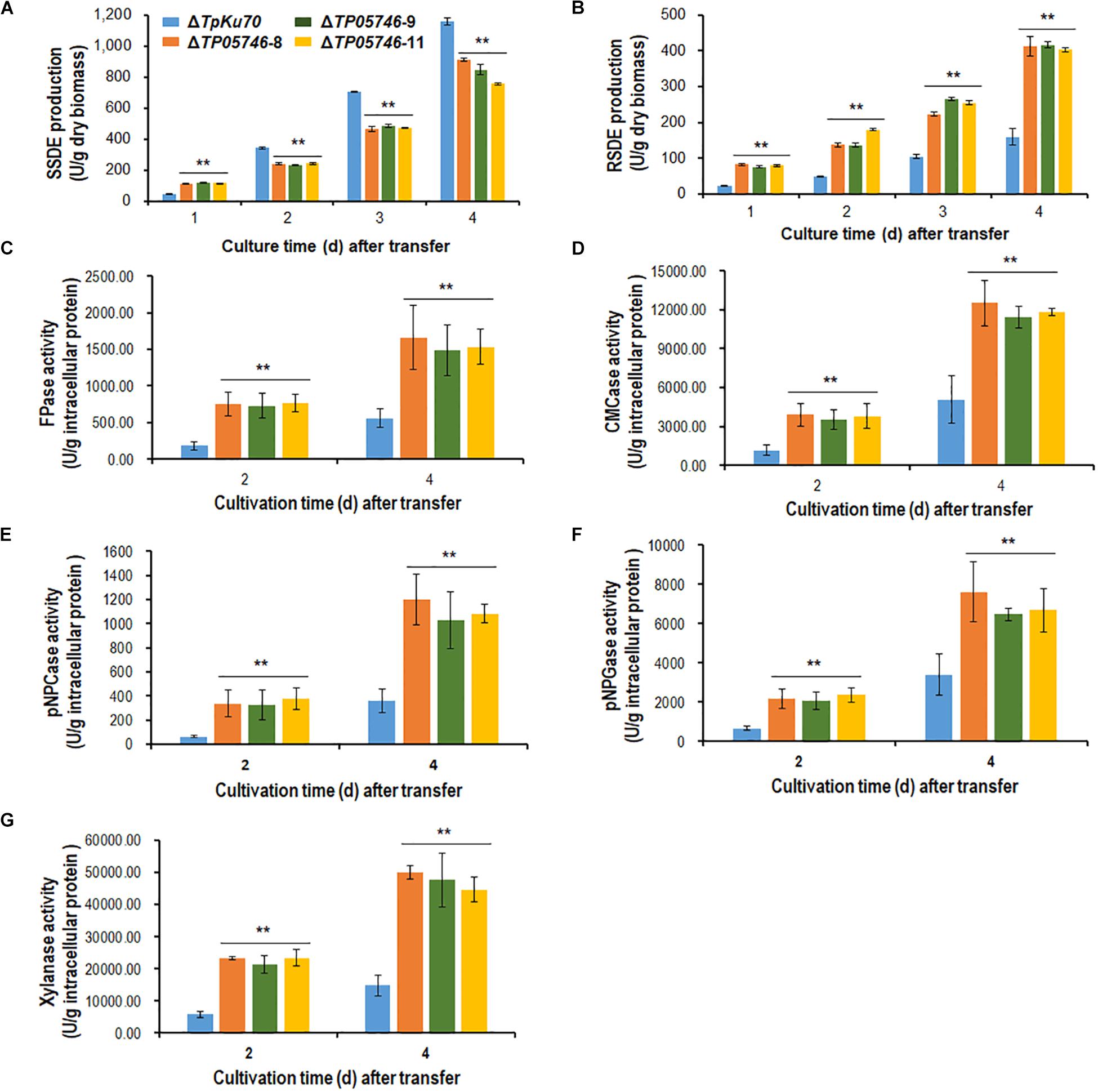
Figure 2. Plant-biomass-degrading enzymes production by T. pinophilus mutant ΔTP05746 and the parental strain ΔTpKu70. Crude extracts were produced from T. pinophilus strains cultured after a transfer from glucose medium to SCS medium (A,B) or WA medium (C–G) for 1–4 days. (A) SSDE production. (B) RSDE production. (C) FPase production. (D) CMCase production. (E) pNPCase production. (F) pNPGase production. (G) Xylanase production. All experiments were performed with three independent biological replicates. Each data point is mean ± SD. ∗∗p ≤ 0.01 indicates differences between the deletion mutant ΔTP05746 and the parental strain ΔTpKu70 by Student’s t-test. PBDE, plant-biomass-degrading enzymes; SSDE, soluble-starch-degrading enzymes; SCS, soluble corn starch; RSDE, raw-starch-degrading enzymes; FPase, filter paper cellulase; CMCase, carboxymethylcellulase; pNPCase, p-nitrophenyl-β-cellobiosidase; pNPGase, p-nitrophenyl-β-glucopyranosidase.
In addition to amylase production, cellulase and xylanase production of the ΔTP05746 were also tested on WA for 2–4 days, using the parental strain ΔTpKu70 as a control. The results indicated that the production of cellulase, including FPase, CMCase, pNPCase, pNPGase, and xylanase in ΔTP05746 increased to various degrees, ranging from 90.1 to 519.1%, depending on the enzyme class, compared with those in ΔTpKu70 (p < 0.01, Student’s t-test; Figures 2C–G).
TP05746 Controls Conidiation of Talaromyces pinophilus
When both strains, ΔTP05746 and ΔTpKu70, were inoculated onto solid LsMM containing D-glucose, SCS or WA, with PDA being used as a control, colony phenotypes were observed. As shown in Figure 3A, no significant difference was found in the colony size between ΔTP05746 and ΔTpKu70 on any of the above-mentioned plates, while colony color of ΔTP05746 was slightly different from that of ΔTpKu70 on all plates.
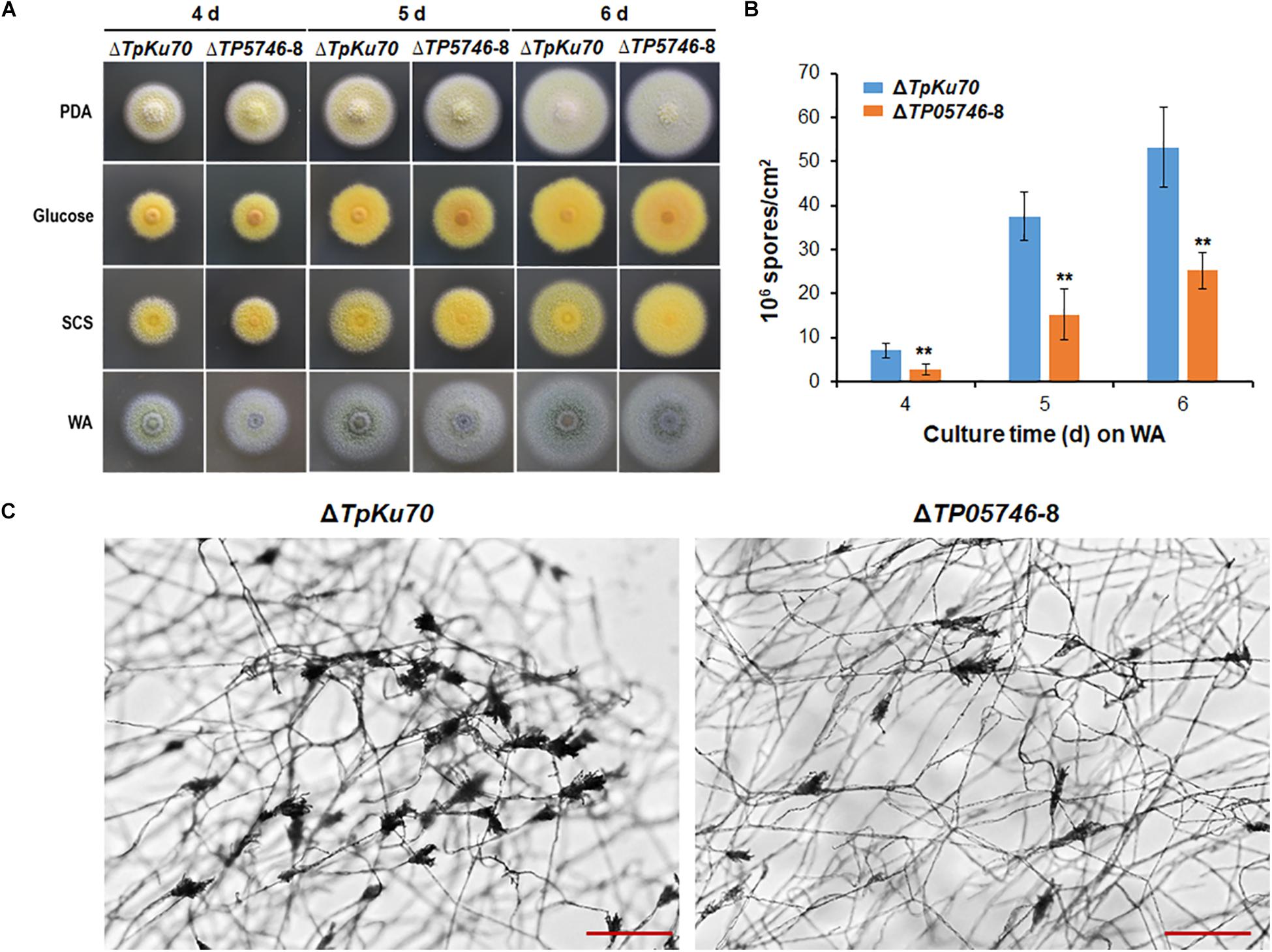
Figure 3. Phenotypic analyses of T. pinophilus mutant ΔTP05746 and the parental strain ΔTpKu70. (A) Colonies on solid LsMM plates containing glucose, SCS, or WA, with PDA being used for a positive control. Fungal strains were incubated at 28°C for 4–6 days. (B) Numbers of asexual spores on WA. Each data point is mean ± SD. ∗∗p < 0.01 indicates differences between the ΔTP05746 and parental strain ΔTpKu70 by Student’s t test. (C) Microscopic investigation of conidiophores. Fungal strains were cultivated on WA at 28°C for 36 h. The mutant ΔTP05746 exhibited fewer phialides in comparison with the parental strain ΔTpKu70. LsMM, low-salt minimal medium; SCS, soluble corn starch; WA, wheat bran plus Avicel; PDA, potato dextrose agar.
Asexual spores (conidia) produced by ΔTP05746 were then counted following inoculation onto WA plates, using the hemocytometer method (Christensen et al., 2012). The results revealed that ΔTP05746 produced 52.6–61.9% of (p < 0.01, Student’s t-test) spores relative to ΔTpKu70 at 4–6 days (Figure 3B).
As shown in Figure 3C, microscopic investigation showed that the mutant ΔTP05746 exhibited delayed phialide development compared with the parental strain ΔTpKu70 on WA-supplemented solid LsMM plates.
TP05746 Accelerates Growth of Talaromyces pinophilus at Early Stage of SCS or WA Induction but Delays Growth at Later Stages
To investigate whether TP05746 affected the growth of Talaromyces pinophilus, real-time quantitative growth curves in the presence of glucose, SCS or WA of both ΔTP05746 and ΔTpKu70 were determined and then compared. The results revealed that ΔTP05746 produced mycelial dry weights similar to ΔTpKu70 in the glucose medium (Figure 4A). When using SCS or WA instead of glucose, however, ΔTP05746 grew faster (p < 0.01, Student’s t-test) than ΔTpKu70 at the early induction period (i.e., 24–36 h for WA, and between 24–60 h for SCS) but the growth rate of ΔTP05746, compared with ΔTpKu70, fell sharply at the later stages (p < 0.05, Student’s t-test; Figures 4B,C).
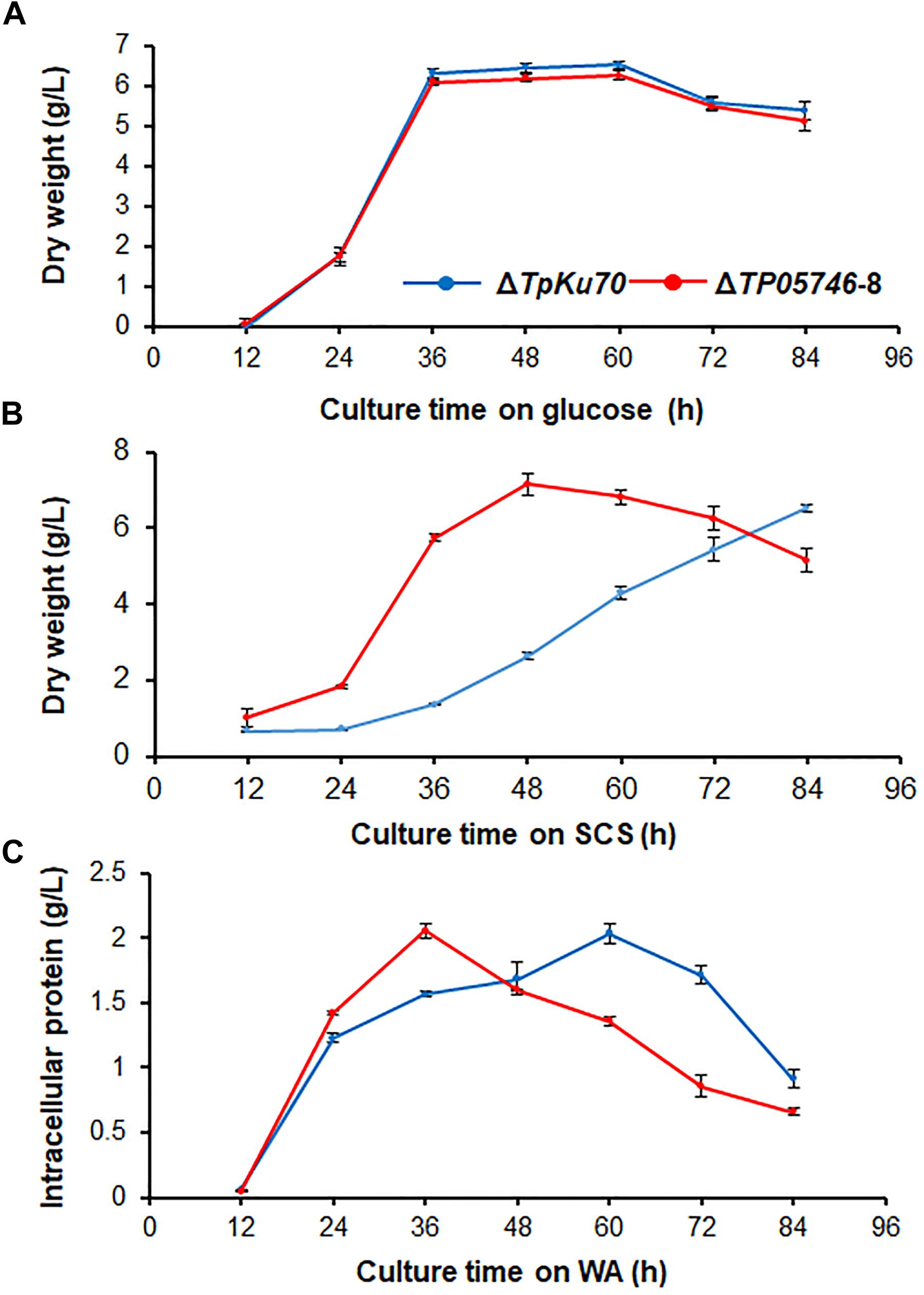
Figure 4. Growth curves of T. pinophilus mutant ΔTP05746 and the parental strain ΔTpKu70 in SLM containing glucose (A), SCS (B), or WA (C) as the sole carbon source for 12–84 h at 28°C, respectively. Three independent experiments were performed as biological replicates. SLM, standard liquid medium; SCS, soluble corn starch; WA, wheat bran plus Avicel.
Further microscopic observations revealed that the mutant ΔTP05746 exhibited significantly more hyphal branching when compared with the parental strain ΔTpKu70 at 24 and 36 h in both liquid SCS and WA medium, although there was no significant difference at 12 h (Figure 5). Moreover, mycelial development of the ΔTP05746 was the same as that of the ΔTpKu70 when cultivated on liquid glucose medium (Supplementary Figure S2).
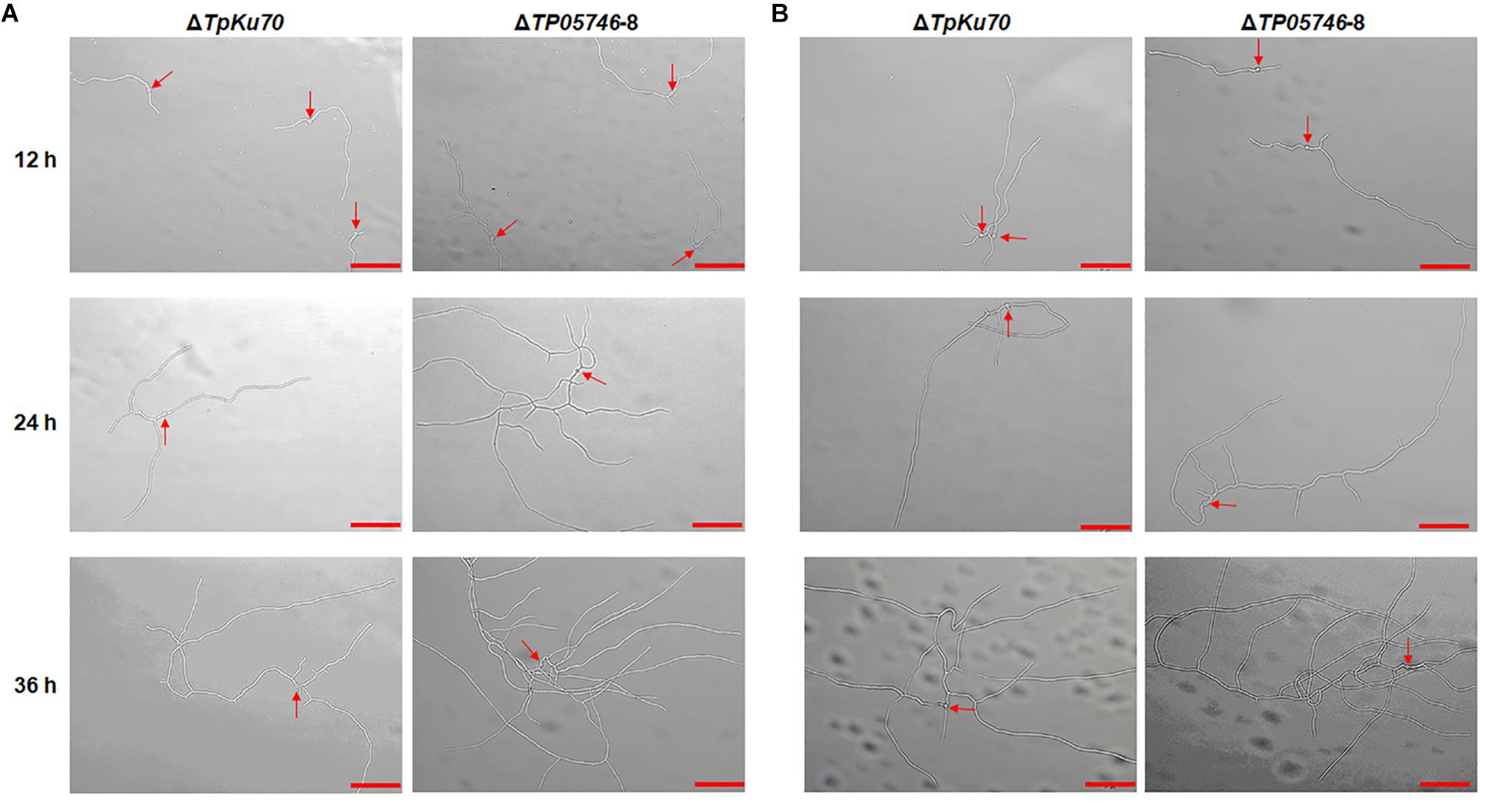
Figure 5. Microscopic investigation of growth of T. pinophilus mutant ΔTP05746 and the parental strain ΔTpKu70 on SLM containing SCS (A) or WA (B). Fungal strains were cultured at 28°C for 12–36 h. The mutant ΔTP05746 cultured on SCS or WA for 24–36 h showed more hyphal branches than that of the parental strain ΔTpKu70. Red arrows indicate the developing conidia. Bar is 50 μm. SLM, standard liquid medium; SCS, soluble corn starch; WA, wheat bran plus Avicel.
Transcriptomic Analyses Reveal That TP05746 Regulates the Expression of Genes Encoding PBDEs and Their Regulators, and Hyphal Development-Associated Genes in Talaromyces pinophilus
The genome-wide mRNA abundance of both the parental strain ΔTpKu70 and its mutant ΔTP05746 were measured in cultures grown in SLM containing SCS as the sole carbon source for 12 h. Comparative transcriptomics identified 4429 differentially expressed genes (DEGs) in mutant ΔTP05746, using a | log2 (fold change)| ≥ 1 and p-value ≤ 0.05 as thresholds relative to expression in the parental strain ΔTpKu70 (Supplementary Table S2). Among them, the transcripts of 2470 genes were up-regulated and 1959 were down-regulated. Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation revealed that the DEGs were mainly involved in metabolism (47%), with the others being involved in human diseases (15%), organismal systems (15%), environmental information processing (8%), genetic information processing (8%), and cellular processes (7%) (Figure 6A). Moreover, the number of up-regulated genes involved in metabolism was greater than that of down-regulated genes (Supplementary Figure S3).
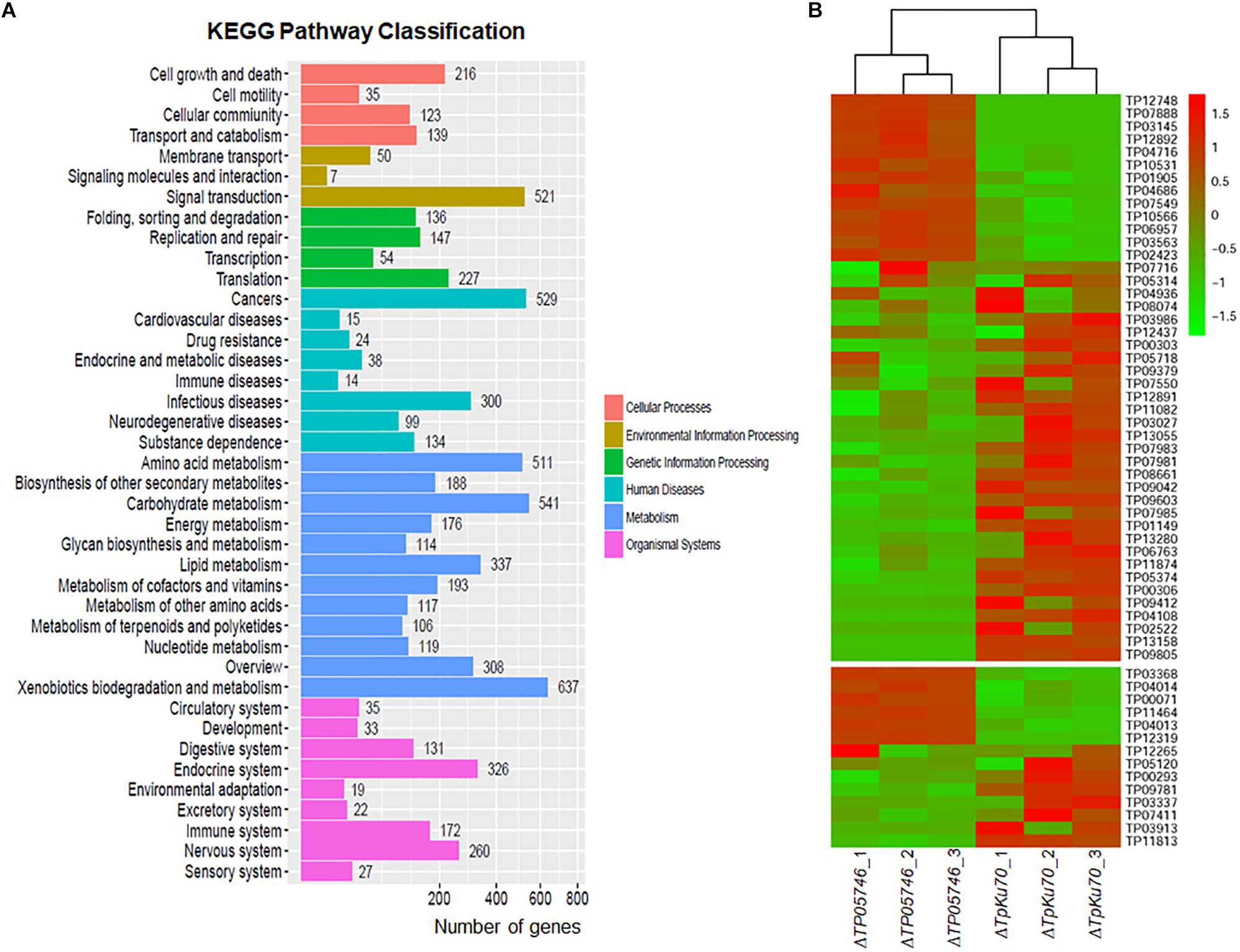
Figure 6. Transcriptomic analysis of T. pinophilus mutant ΔTP05746 and the parental strain ΔTpKu70 cultured in SLM containing SCS as the sole carbon source for 12 h. (A) KEGG pathway classification annotation of 4429 DEGs in the ΔTP05746 [a | log2(fold change)| ≥ 1 and a p-value ≤ 0.05 as thresholds]. (B) Plant cell-wall-degrading enzymes and amylase genes regulated by TP05746. SLM, standard liquid medium; SCS, soluble corn starch; KEGG, kyoto encyclopedia of genes and genomes; DEGs: differently expressed genes.
Of the DEGs, 14 were involved in starch degradation, including three amy genes TP03368, TP04014/Amy13A and TP07411, one gla gene TP12319, and ten aga genes TP00071, TP00293, TP03337, TP03913, TP04013, TP05120, TP09781, TP11813, TP11464, and TP12265. Comparative analyses revealed that the transcripts of six of these 14 starch-degradation-associated DEGs (TP03368, TP04014/Amy13A, TP12319, TP00071, TP04013, and TP11464) were upregulated in ΔTP05746 in comparison with the expression level in ΔTpKu70, with a log2 fold change ranging from 1.27 to 5.90. The other eight DEGs were downregulated, with a log2 fold change ranging from −5.46 to −1.42 (Figure 6B).
Interestingly, 44 of the DEGs included in the TP05746 regulon encoded plant cell wall-degrading enzymes (CWDEs), including one cbh gene (TP09412/cbh1), two eg genes (TP04686 and TP06957), and 13 bgl genes (TP01149, TP02423, TP04716, TP05374, TP07549, TP07716, TP07981, TP07983, TP08074, TP09042, TP09603, TP11082, and TP12437). Of these 44 CWDE-encoding DEGs, 13 genes (TP01905, TP02423, TP03145, TP03563, TP04686, TP04716, TP06957, TP07549, TP07888, TP10531, TP10566, TP12748, and TP12892) were upregulated in ΔTP05746 compared with ΔTpKu70, with a log2 fold change ranging from 1.51 to 5.93, whereas the other 31 genes were down-regulated, with a log2 fold change ranging from −7.83 to −1.13 (Figure 6B).
In addition to genes encoding carbohydrate-degrading enzymes, the 377 DEGs encoding putative TFs were found, most of which contained Zn2Cys6, C2H2, bZIP, winged helix repressor or homeodomain-like domains, consisting of 159 upregulated (1.03 < log2 fold change < 9.81) and 218 down-regulated (−10.68 < log2 fold change < −1.00) DEGs (Supplementary Table S2). Notably, six regulatory genes, known to be involved in controlling the expression of genes encoding PBDEs, were detected, including TP08849/AreA (log2 fold change = 3.45), TP09286/AmyR (log2 fold change = 1.10), TP10486/ClrB (log2 fold change = 5.52), TP02627/XlnR (log2 fold change = −1.53), TP00292/BglR (log2 fold change = 3.89), and TP06351/Vib1 (log2 fold change = −3.22) (Li et al., 2017).
Previous work had shown that deletion of TP05746 resulted in a reduction in asexual spore production, suggesting that TP05746 controls conidiation of T. pinophilus. In the DEGs, 13 genes (TP04427/Bud4, TP10226/Asp, TP10901/VosA, TP03893/FadA, TP04237/NimX, TP05733/WetA, TP11170/FlbA-like gene, TP10098/FlbA, TP03987/VeA, TP05897/RodA, TP05092/ArpA, TP13250/ArpA-like gene, and TP06809/ArpA-like gene) were observed, which were previously reported to be involved in fungal conidiogenesis (Qin et al., 2013), of which five (TP04427/Bud4, TP10226/Asp, TP10901/VosA, TP03893/FadA, and TP04237/NimX) were upregulated (1.67 < log2 fold change < 4.73) and eight were downregulated (−6.55 < log2 fold change < −1.19) in ΔTP05746 (Supplementary Table S2).
RT-qPCR Shows That TP05746 Dynamically Controls the Expression of Genes Encoding Major PBDE Genes and Their Regulators, and Hyphal Development-Associated Genes in Talaromyces pinophilus
RT-qPCR was employed to investigate regulatory dynamics by TP05746 of the expression of genes involved in the degradation of plant biomass in T. pinophilus. On the basis of comparative transcriptomic data, the target genes were identified, including three amy genes TP03368, TP04014/Amy13A and TP07411, one gla gene TP12319, and five aga genes TP09781, TP11464, TP12265, TP00071 and TP04013, as well as their known regulatory genes TP09286/AmyR (Zhang et al., 2017), TP06128/Rfx1 (Liao et al., 2018), and TP00292/BglR (Xiong et al., 2017), and their expression (relative to the levels in the parental strain ΔTpKu70) was measured at 12, 24, and 48 h after SCS induction in the mutant ΔTP05746. The results showed that the transcript abundance of these 12 genes was significantly altered in ΔTP05746 relative to the parental strain. At 12 h after induction, the expression of genes TP03368, TP04014/Amy13A, TP12319, TP11464, TP00071, TP04013, TP09286/AmyR, and TP06128/Rfx1 in the mutant ΔTP05746 exhibited significant up-regulation, ranging from 1.17- to 87.77-fold, whereas the expression of four genes, TP07411, TP09781, TP12265, and TP00292/BglR, was down-regulated by 59.7–81.9% in the mutant ΔTP05746 (p < 0.05, Student’s t-test). At 24 h, all these targeted genes, except for TP07411, TP12265, TP09286/AmyR, TP06128/Rfx1, and TP00292/BglR, were significantly upregulated in ΔTP05746, the increase ranging from 614.8 to 10108.2% (p < 0.05, Student’s t test). The transcript abundance of TP09286/AmyR, TP06128/Rfx1, and TP07411 decreased by 43.7 to 84.0%. By contrast, the transcript abundance of only three genes, namely TP12319, TP09781, and TP00071, continued to increase, by 190.0 to 2142.8% in ΔTP05746 at 48 h. Six genes, TP04014, TP11464, TP12265, TP04013, TP09286/AmyR, and TP06128/Rfx1 showed a 44.2–70.2% decrease in transcript abundance (p < 0.05, Student’s t test; Figure 7A).
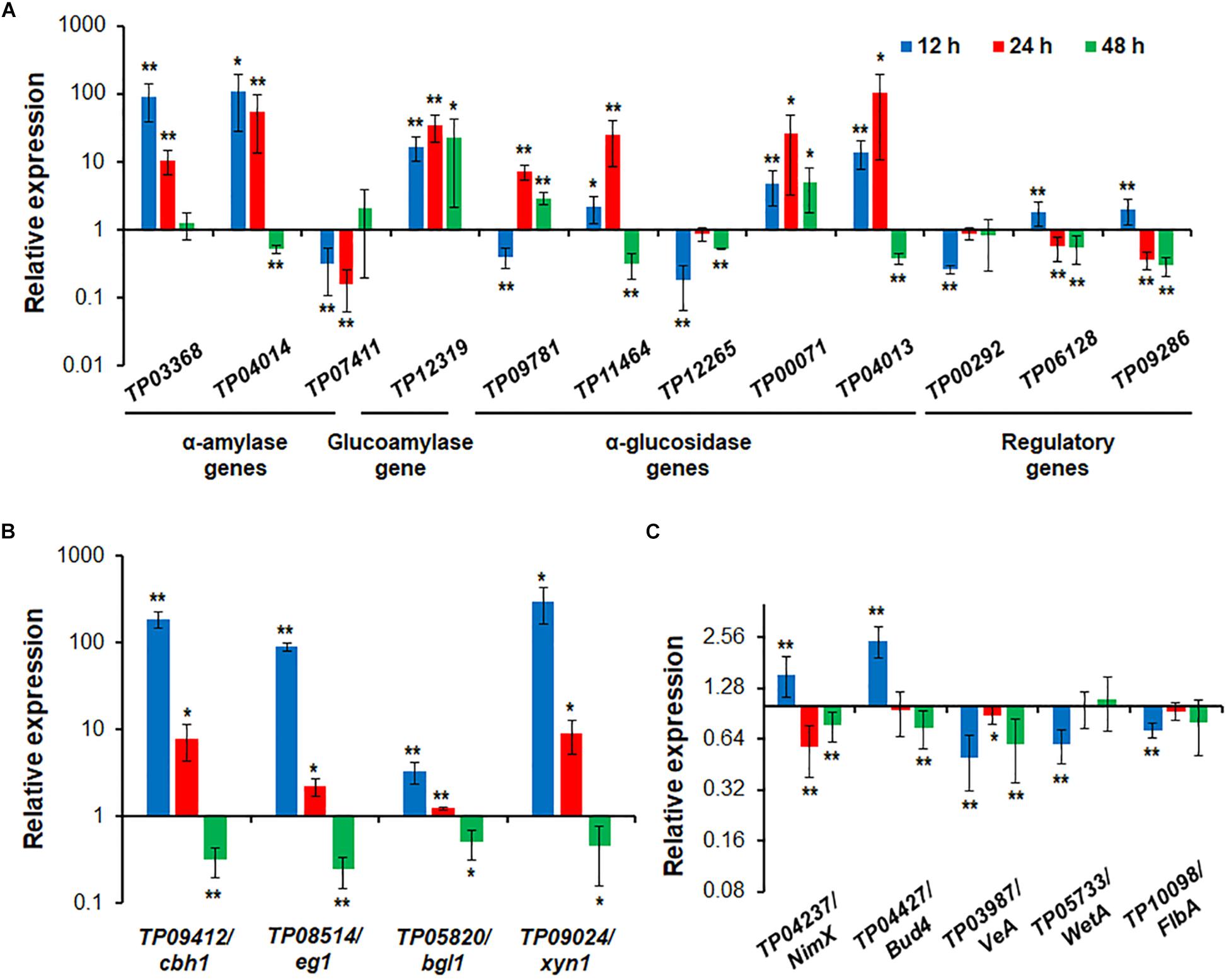
Figure 7. Expressional regulation of enzyme genes and regulatory genes by TP05746 in T. pinophilus as demonstrated by RT-qPCR. (A) Amylase genes and their regulatory genes. Total RNA was extracted from fungal strains cultured in medium containing SCS as the carbon source for 12, 24, and 48 h after transfer from glucose. (B) Cellulase and xylanase genes. (C) Genes involved in conidiogenesis. Fungal cells were cultured on WA for 12, 24, and 48 h after transfer from glucose. Expression levels of the genes tested in the ΔTP05746 mutant were normalized against the parental strain ΔTpKu70. ∗∗p ≤ 0.01 and ∗p ≤ 0.05 indicate differences between ΔTP05746 and ΔTpKu70 by Student’s t test. Each experiment contained three biological replicates. RT-qPCR, real-time quantitative reverse transcription PCR; WA, wheat bran plus Avicel.
In addition to starch-degrading enzyme genes, four cellulase and xylanase genes, TP09412/cbh1, TP08514/eg1, TP05820/bgl1 and TP09024/xyn1, were also selected and their real-time transcription investigated in both ΔTP05746 and ΔTpKu70 cultured on WA. The results indicated that the expression of all the cellulase and xylanase genes tested was upregulated to various degrees in ΔTP05746 relative to ΔTpKu70 in the early induction period (before 24 h) but downregulated in the later induction period. For example, the transcription levels of all four genes increased by between 228.6% and 29565.0% at 12 h but by between 22.4% and 802.8% at 24 h (p < 0.05, Student’s t-test) in ΔTP05746 (Figure 7B). Conversely, their expression was consistently downregulated by 50.4–75.6% in the mutant at 48 h (p < 0.05, Student’s t-test) (Figure 7B).
Five key genes involved in fungal conidiogenesis, namely TP04427/Bud4, TP04237/NimX, TP05733/WetA, TP10098/FlbA, and TP03987/VeA, were selected for RT-qPCR. The results indicated that, at 12 h, the transcripts of both TP04237/NimX, and TP04427/Bud4 were significantly upregulated, by 54.8% and 143.7%, respectively, while expression of the others was downregulated by 27.5 to 50.0% (p < 0.01, Student’s t-test). At 24 h, the expression of two genes, TP04237/NimX, and TP03987/VeA, increased by 42.7% and 11.4%, whereas that of TP04237/NimX, TP03987/VeA, and TP04427/Bud4 decreased by 23.1 to 40.7% (p < 0.05, Student’s t-test; Figure 7C).
TP05746 Binds to the Promoter Regions of Genes Encoding Major PBDEs and Their Regulators in vitro, as Well as to Conidiogenesis-Involved Genes
Electrophoretic mobility shift assay was used to confirm whether TP05746 directly or indirectly regulated the expression of target genes. The cDNA of TP05746 was fused to a DNA fragment encoding Trx-His-S-tags and recombinantly expressed in Escherichia coli. The 6-carboxyflurescein (FAM)-tagged DNA probes (∼1,000 bp) in the promoter regions of two α-amylase genes TP03368, TP04014/Amy13A, one glucoamylase gene TP12319, three α-glucosidase genes TP11464, TP00071, TP04013, three cellulase genes TP09412/cbh1, TP08514/eg1, TP05820/bgl1, one xylanase gene TP09024/xyn1, and three regulatory genes TP00292/BglR, TP06128/Rfx1, and TP09286/AmyR, as well as four conidiogenesis-involved genes TP04427/Bud4, TP04237/NimX, TP05733/WetA, and TP10098/FlbA, were amplified using specific primer pairs (Supplementary Table S1). The promoter region of the β-tubulin gene TP10751, and either bovine serum albumin (BSA) or the Trx-His-S fusion protein was used as controls. DNA fragments of the same length but without the FAM label, were used as competitive probes. As shown in Figures 8, 9, shifted bands representing rTP05746-DNA complexes in all mixtures of the recombinant protein rTP05746 and EMSA probes of the target genes were observed to various degrees. Moreover, the band size gradually increased along with an increase in rTP05746 amounts (0–2.0 μg), whereas shifted bands did not occur between EMSA probes and either BSA or the Trx-His-S fusion protein, or between rTP05746 and the promoter region of the β-tubulin gene TP10751.
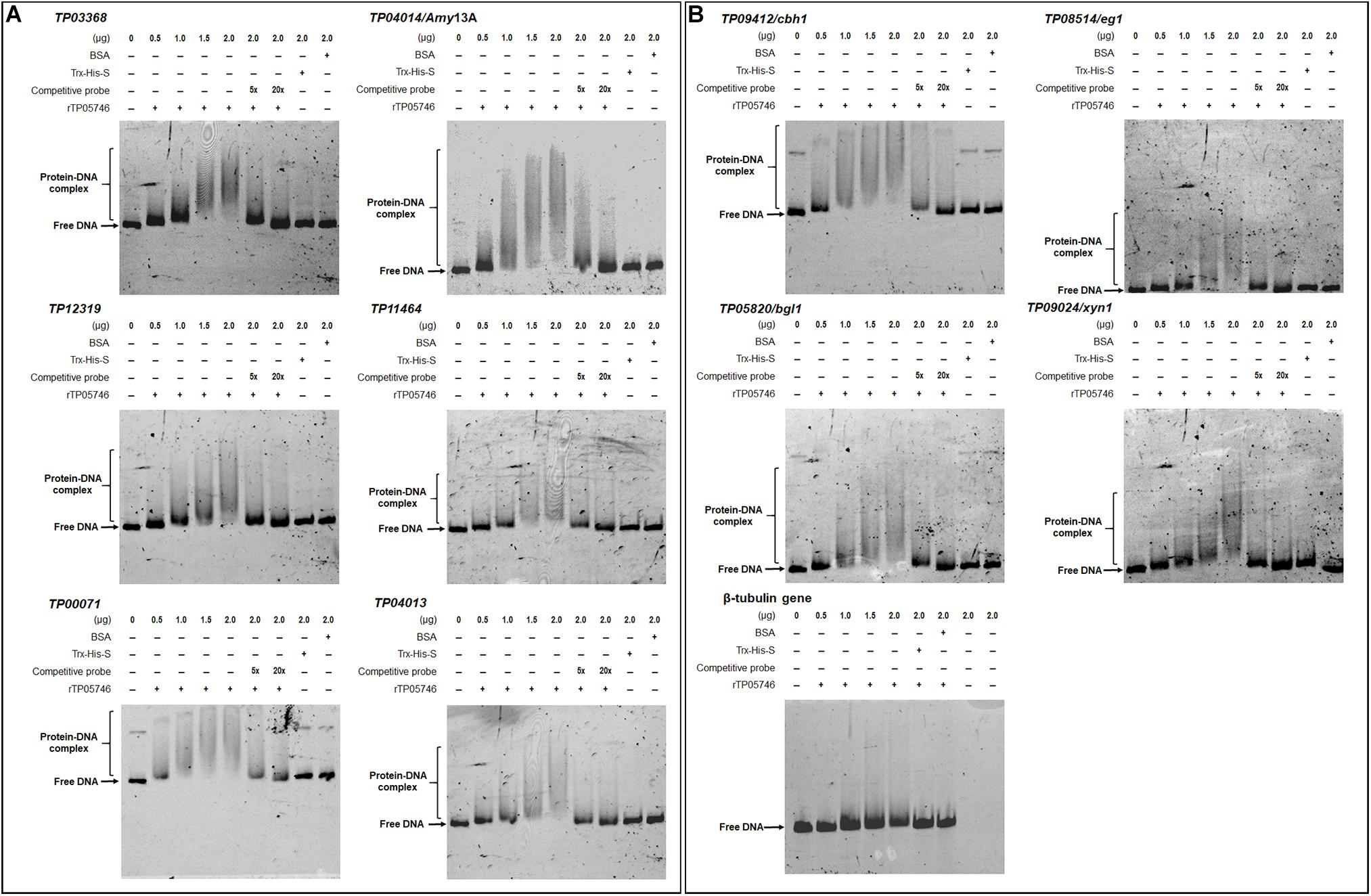
Figure 8. Interaction between TP05746 and genes encoding enzymes as revealed by electrophoretic mobility shift assay (EMSA). (A) Amylase genes. (B) Cellulase and xylanase genes. The recombinant protein rTP05746 (0–2.0 μg) was mixed with approximately 50 ng of FAM-labeled EMSA probes. EMSA probes lacking the FAM label were used for competitive EMSA. BSA, Trx-His-S fusion protein or the promoter region of the β-tubulin gene alone were used as controls. FAM, 6-carboxyfluorescein; EMSA, electrophoretic mobility shift assay; BSA, bovine serum albumin. In each EMSA reaction, non-specific sheared salmon sperm DNA was added, in order to prevent non-specific binding between protein and probes.
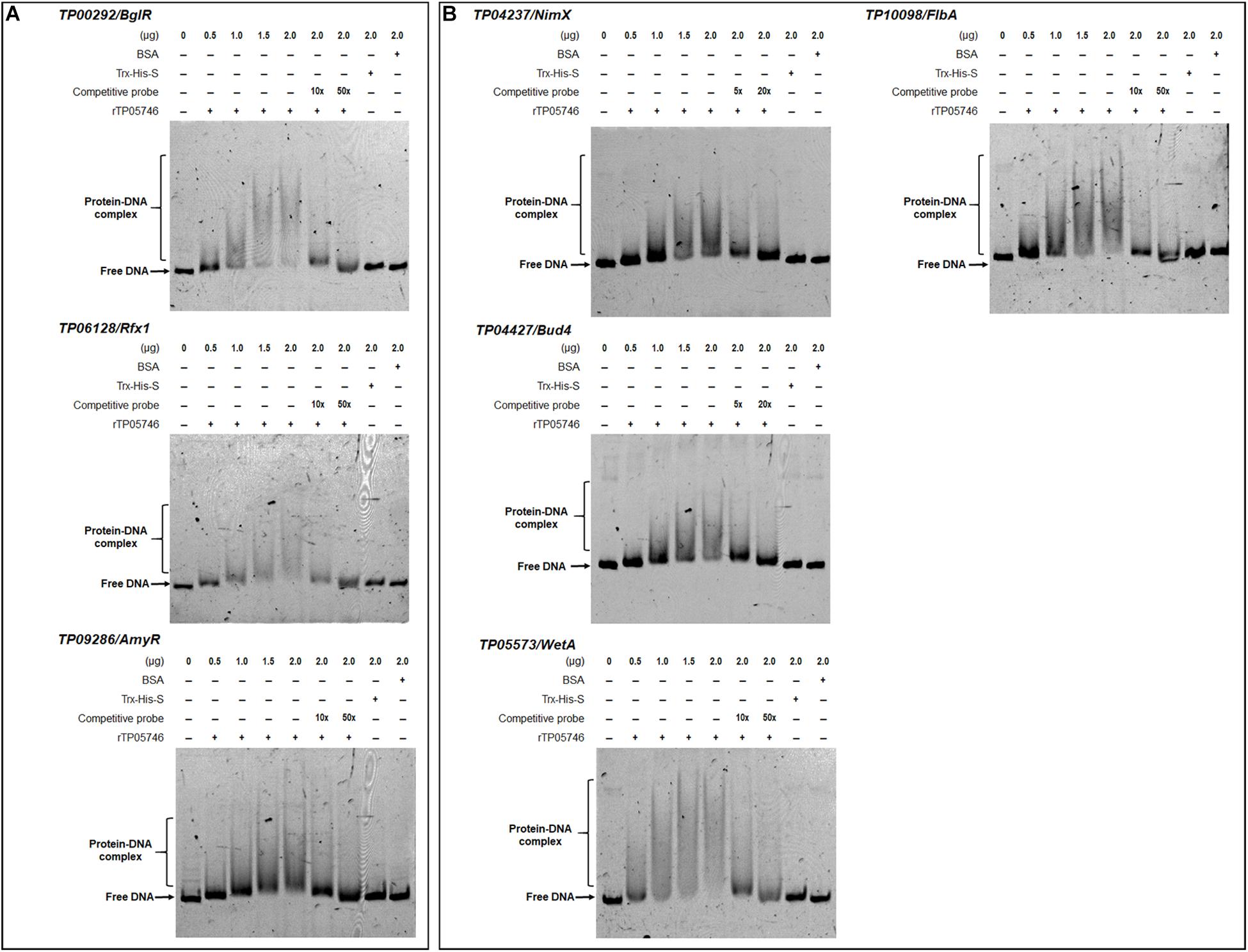
Figure 9. Interaction between TP05746 and regulatory genes revealed by electrophoretic mobility shift assay (EMSA). (A) Genes involved in the regulation of PBDE gene expression. (B) Genes involved in conidiogenesis. The recombinant protein rTP05746 (0–2.0 μg) was mixed with approximately 50 ng of FAM-labeled EMSA probes. EMSA probes lacking the FAM label were used for competitive EMSA. BSA, Trx-His-S fusion protein or the promoter region of the β-tubulin gene alone were used as controls. FAM, 6-carboxyfluorescein; EMSA, electrophoretic mobility shift assay; BSA: bovine serum albumin; PBDE, plant-biomass-degrading enzymes. In each EMSA reaction, non-specific sheared salmon sperm DNA was added, in order to prevent non-specific binding between protein and probes.
Simultaneously, competitive EMSA was performed using the competitive probes, and the results revealed that the concentration of the shifted bands gradually decreased in response to increasing amounts of the competitive probes without the FAM label (Figures 8, 9). However, in fact how interact with these promoters by TP05746 in vivo still awaits to be performed.
TP05746 Inhibits the Production of PBDEs in Filamentous Fungus Penicillium oxalicum
To further confirm the regulatory roles and potential application of gene TP05746 in genetic engineering, gene TP05746 was over-expressed in filamentous fungus P. oxalicum parental strain ΔPoxKu70 (Supplementary Figure S4) derived from the wild-type HP7-1 via deleting gene PoxKu70 (Zhao et al., 2016), and then its production of PBDEs was measured when cultivated on Avicel for 1–3 days. The results indicated the obtained overexpressed strain OXTP05746_POX lost 54.4–84.6% of cellulase (FPase, pNPCase and CMCase) and xylanase production, 15.6–49.9% of SSDE and RSDE production, while increased 131.1–275.1% of pNPGase production, compared with the parental strain ΔPoxKu70 (p < 0.01, Student’s t test; Figures 10A–G). Intracellular proteins of strain OXTP05746_POX had no significant difference from that of the ΔPoxKu70 when cultivated on WA for 1 day, but decreased by approximately 70% when for 3 days (p < 0.01, Student’s t test; Figure 10H).
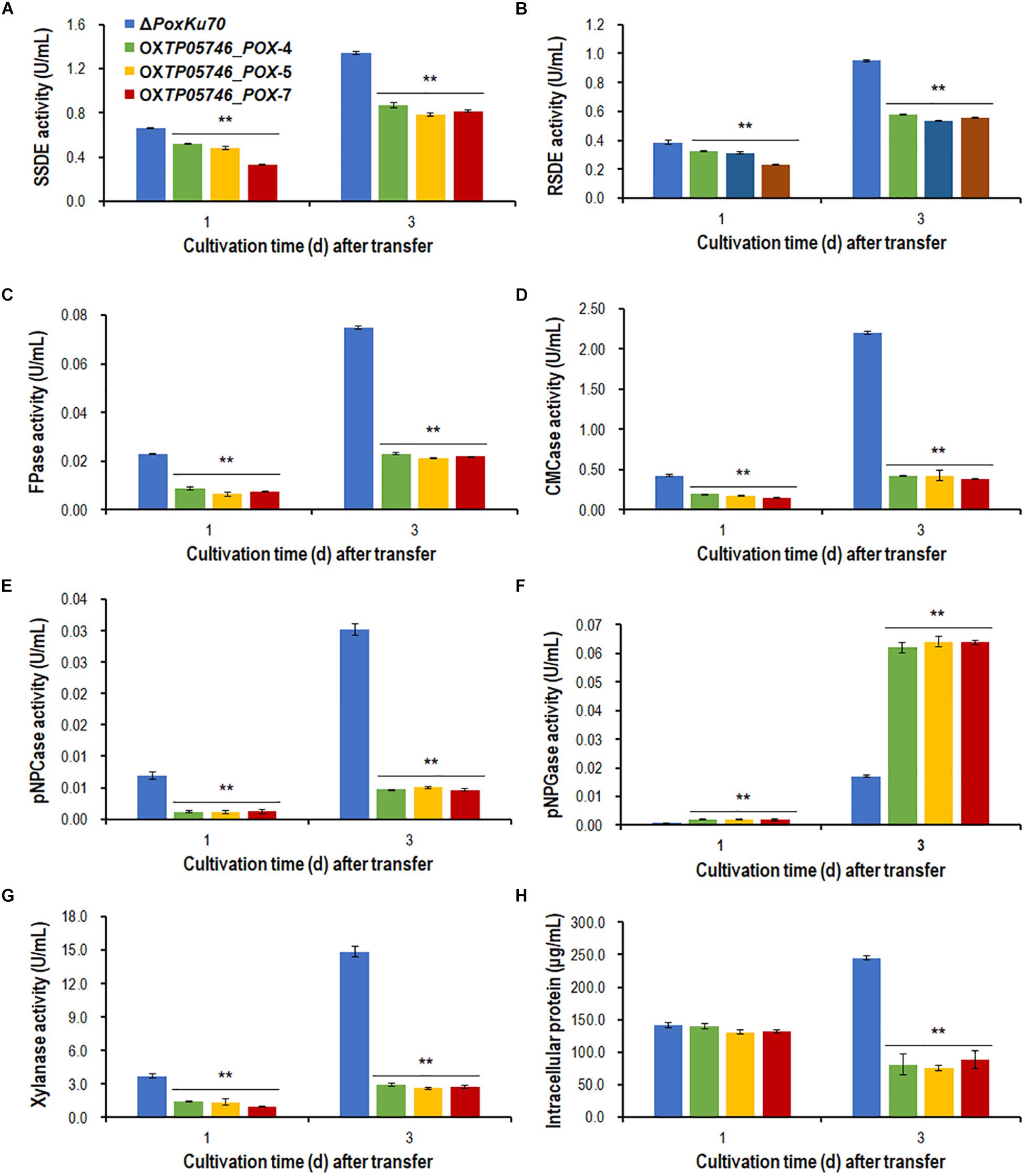
Figure 10. Plant-biomass-degrading enzymes production by overexpression strain OXTP05746_POX and the parental strain ΔPoxKu70. Crude extracts were produced from Penicillium oxalicum strains cultured after a transfer from glucose medium to Avicel medium for 1–3 days. (A) SSDE production. (B) RSDE production. (C) FPase production. (D) CMCase production. (E) pNPCase production. (F) pNPGase production. (G) Xylanase production. (H) intracellular protein. All experiments were performed independently for three biological replicates. Each data point is mean ± SD. ∗∗p ≤ 0.01 indicates differences between the deletion mutant ΔTP05746 and the parental strain ΔTpKu70 by Student’s t test. PBDE, plant-biomass-degrading enzyme; SSDE, soluble-starch-degrading enzyme; SCS, soluble corn starch; RSDE, raw-starch-degrading enzyme; FPase, filter paper cellulase; CMCase, carboxymethylcellulase; pNPCase, p-nitrophenyl-β-cellobiosidase; pNPGase, p-nitrophenyl-β-glucopyranosidase.
Discussion
In this study, we explored regulatory roles of a novel Zn2Cys6 protein, TP05746, that regulated PBDE (i.e., SSDE, RSDE, cellulase, and xylanase) production as well as growth and positively regulated conidiation of T. pinophilus through regulating the expression of the associated genes and their regulatory genes (Figure 11). TP05746 specifically belonged to Talaromyces, and plays an essential regulatory role via a molecular mechanism distinct from that of the known CreA-mediated CCR.
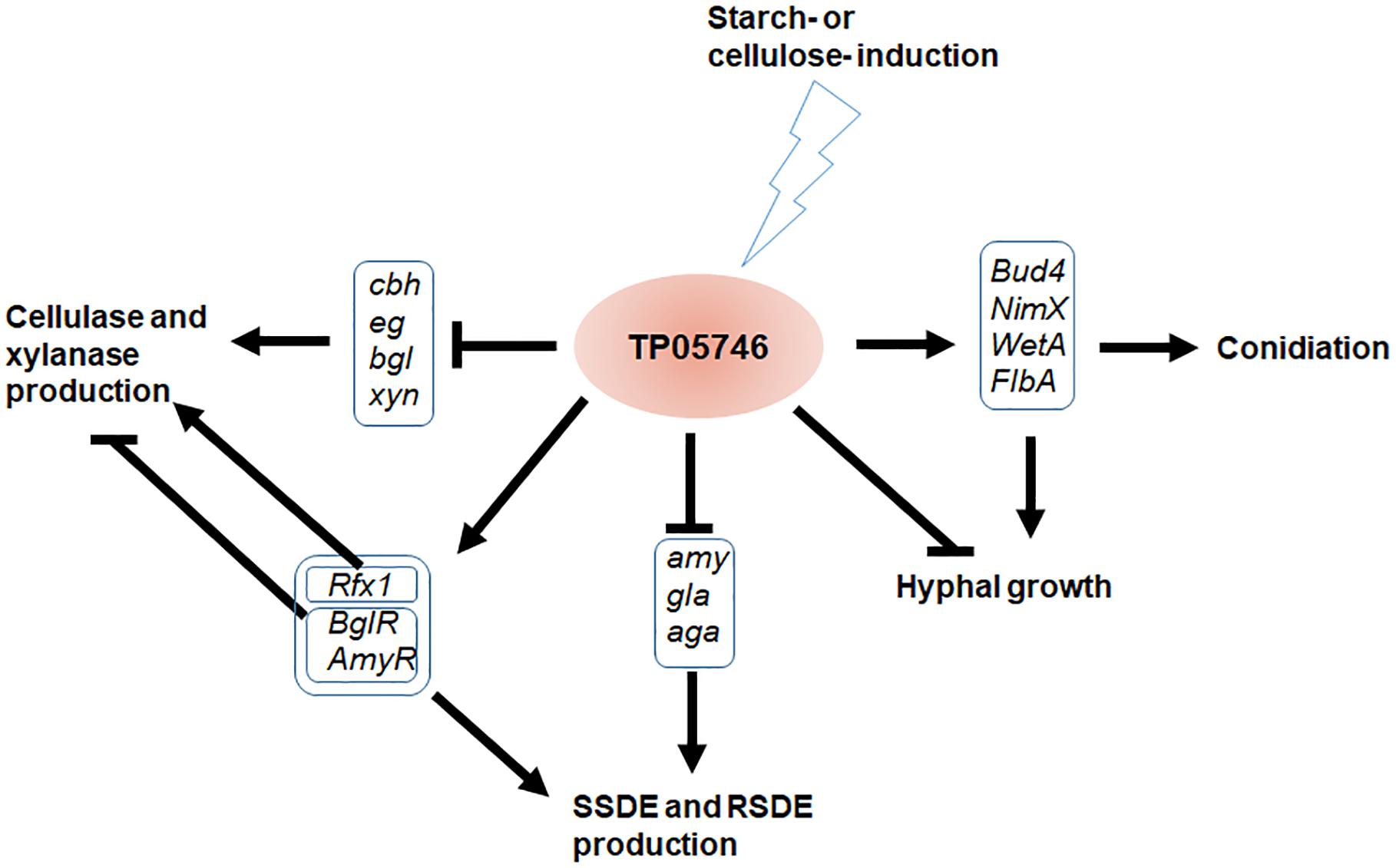
Figure 11. Proposed model of TP05746 regulation in T. pinophilus. cbh, cellobiohydrolase gene; eg, endo-β-1,4-glucanase gene; bgl, β-1,4-glucosidase gene; gla, glucoamylase gene; amy, α-amylase gene; aga, α-glucosidase gene.
The regulation of TP05746 on PBDE production is time-dependent, as was expression of their encoded genes. For example, deletion of TP05746 resulted in an increase in RSDE, cellulase and xylanase activity over the entire induction period, while the level of increasing production tended to fall over time, corresponding to dynamic changes in expression of major PBDE genes. The transcripts of TP03368-, and TP04014/Amy13A-encoding proteins that included SBDs in the mutant ΔTP05746 first increased and then decreased, while the expression of TP00071 continued to increase over time. Cellulase and xylanase genes TP09412/cbh1, TP08514/eg1, TP05820/bgl1, and TP09024/xyn1 were repressed at the early induction stage but activated at later stages.
These observed phenomena are closely associated with substrate induction and a complex regulatory network. Plant biomass is degraded into single sugars via the synergistic action of several PBDEs working at appropriate respective rates, with the most-efficient enzyme mixture being secreted by the fungal cells based on which enzymes are needed in response to specific substrates. To the best of our knowledge, specific inducing substrates are low molecular weight sugars, in-process products of biomass degradation, such as cellobiose, xylobiose and maltose (Amore et al., 2013), but with unstable contents. Additionally, a complex regulatory network, consisting of various TFs and the target genes, are prerequisites to respond to the in-process products. As a note of this complex regulatory network, TP05746 regulates not only the expression of genes encoding specific enzymes, but also that of other TF genes, such as AmyR, TpRfx1, and BglR. AmyR and BglR positively regulate fungal amylase production and negatively control cellulase and xylanase production (Nitta et al., 2012; Li et al., 2015; Xiong et al., 2017; Zhang et al., 2017), whereas TpRfx1 positively mediates PBDE production, including amylase, cellulase and xylanase, in T. pinopholus (Liao et al., 2018). However, the actual regulatory mechanism of TP05746 in fungal cells still needs to be further elucidated.
In addition to fungal PBDE production, TP05746 also controls conidiation. Fungal conidiation is governed by the BrlA → AbaA → WetA regulatory cascade in concert with other genes, such as genes encoding FLBs (fluffy low BrlA expression) and the velvet family of proteins (Park and Yu, 2016). Here, this study found that deletion of TP05746 affected the expression of several key conidiation-associated regulatory genes, such as TP04427/Bud4, TP04237/NimX, TP05733/WetA, TP10098/FlbA and TP03987/VeA. The gene Bud4 is involved in spectrum formation in hyphal growth and the development of conidiophores in Aspergillus, deletion of which resulted in non-production of conidia (Si et al., 2012), as well as FlbA (Wieser et al., 1994). NimX, also called cdc2, encoding the cyclin-dependent kinase, accelerates hyphal branching and produces abnormal conidiophores at restrictive temperatures in A. nidulans (Lin and Momany, 2004). WetA is required for the middle to late phases of conidiation, to complete conidiation, and functions in the germination of these asexual spores and the early phase of mycelial growth (Tao and Yu, 2011). The velvet gene VeA is essential for proper asexual spore development but exhibits the opposite pattern of conidiation in Aspergillus fumigatus according to previous work (Dhingra et al., 2012; Park et al., 2012). Unfortunately, at this point it is not known how TP03987/VeA function in T. pinophilus. The expression of the above genes regulated by TP05746 is time-dependent, and reaches a balance, resulting in the phenotypes observed in the mutant ΔTP05746. It should be noted that these conidiation-related genes might contribute to the regulation of cellulase and xylanase genes by TP05746. For example, the VeA ortholog Vel1 of T. reesei positively regulates the expression of key cellulase and xylanase genes (Karimi Aghcheh et al., 2014).
In addition, TP05746 represses hyphal growth of T. pinophilus, which might affect the utilization of nutrients. For example, under SCS induction, the expression of TP05746 is activated, thereby inhibiting the transcription of major amylase genes via a complex regulatory network, resulting in low yields of extracellular amylases. Starch could not be digested into small sugars, including glucose and maltose, to provide energy for the fungal cells.
To the best of our knowledge, few negative TFs have been identified in filamentous fungi to date, deletion of which results in an increase in PBDE production. The best-known repressor is CreA/Cre1/Cre-1, which inhibits almost all fungal genes involved in the degradation of plant biomass in the presence of the preferred carbon source, glucose, including both structural enzyme genes and regulatory genes (Portnoy et al., 2011; Sun and Glass, 2011; Li et al., 2015). Further experimental data revealed that, in the presence of D-glucose, both SSDE and RSDE production by ΔTP05746 increased approximately 1.3- to 2.2-fold relative to that of the parental ΔTpKu70. In addition, 2-DG could activate CCR in the mutant ΔTP05746, leading to insufficient cellulase production, as in ΔTpKu70 (Supplementary Figure S5). These data suggested that TP05746 was not involved in CreA-mediated CCR, suggesting that the mode of action of TP05746 is different from that of CreA.
We tried to construct the complementary strain of the mutant ΔTP05746 at least four times but failed for unknown reasons. Three randomly chosen knock-out transformants for the gene TP05746 in T. pinophilus were confirmed by both PCR with specific primers and by Southern hybridization analysis. Furthermore, in the enzymatic activity assay, all three randomly chosen transformants showed similar and consistent results, confirming that the obtained phenotype of each transformant was specifically generated by the deletion of the gene TP05746. Fortunately, gene TP05746 was heterologously overexpressed in filamentous fungus P. oxalicum. Overexpression of TP05746 resulted in the reduction of plant-biomass-degrading enzyme production, thereby leading to less accumulation of fungal hyphae. These data also confirmed that TP05746 plays a negative role in the production of PBDEs.
It should be noted that negatively regulatory genes are potential targets for genetic engineering to achieve increased enzyme production. Deletion of TP05746 led to a several-fold increase in RSDE production. RSDE application to starch biorefineries to generate biofuels or other biochemical products efficiently saves costs compared with traditional starch processing (Robertson et al., 2006; Görgens et al., 2015).
Conclusion
In conclusion, the present study explored the regulatory roles of the novel TF TP05746 in T. pinophilus with respect to the control of PBDE production (SSDE, RSDE, cellulase and xylanase), as well as growth and conidiation. Further studies indicated that TP05746 dynamically regulated the expression of the associated genes described above, thereby affecting the corresponding fungal phenotypes. These findings provided novel insights into the regulatory mechanism of fungal PBDE gene expression and identify a potential target for genetic engineering for industrial application.
Data Availability Statement
The datasets generated for this study can be found in the DNA sequence of TP05746 is available from the GenBank database under the accession number MH447996. The transcriptomic data of T. pinophilus strains have been deposited in Gene Expression Omnibus (GEO) on NCBI (accession no. GSE131872).
Author Contributions
J-XF designed and supervised the study, and involved in data analysis and manuscript revision. SZ co-supervised all the experiments and revised the manuscript. TZ carried out the enzyme activity assay, phenotypic analysis, Southern hybridization analysis, RT-qPCR, and EMSA, and drafted the manuscript. L-SL involved in genomic DNA, RNA, and protein extraction and transcriptomic analysis. C-XL carried out the bioinformatic analysis. G-YL, XL, and X-ML took part in the preparation of experimental materials and data analysis. All authors read and approved the final manuscript.
Funding
This work was financially supported by the grants from the Guangxi Natural Science Foundation (Grant No. 2018GXNSFAA281103), the State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources (SKLCUSA-a201902; SKLCUSA-a201923), Training Program for 1000 Young and Middle-aged Key Teachers in Guangxi at 2019, and the “One Hundred Person” Project of Guangxi.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02875/full#supplementary-material
Footnotes
References
Amore, A., Giacobbe, S., and Faraco, V. (2013). Regulation of cellulase and hemicellulase gene expression in fungi. Curr. Genomics 14, 230–249.
Bornscheuer, U., Buchholz, K., and Seibel, J. (2014). Enzymatic degradation of (Ligno)cellulose. Angew. Chem. Int. Ed. 53, 10876–10893. doi: 10.1002/anie.201309953
Buommino, E., Filippis, A. D., Nicoletti, R., Menegozzo, M., Menegozzo, S., Ciavatta, M. L., et al. (2012). Cell-growth and migration inhibition of human mesothelioma cells induced by 3-O-Methylfunicone from Penicillium pinophilum and cisplatin. Invest. New Drug 30, 1343–1351. doi: 10.1007/s10637-011-9698-1
Carrillo, A. J., Schacht, P., Cabrera, I. E., Blahut, J., Prudhomme, L., Dietrich, S., et al. (2017). Functional profiling of transcription factor genes in Neurospora crassa. G3 7, 2945–2956.
Christensen, S., Borrego, E., Shim, W. B., Isakeit, T., and Kolomiets, M. (2012). Quantification of fungal colonization, sporogenesis, and production of mycotoxins using kernel bioassays. J. Vis. Exp. 62:3727. doi: 10.3791/3727
Coradetti, S. T., Craig, J. P., Xiong, Y., Shock, T., Tian, C. G., and Glass, N. L. (2012). Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 7397–7402. doi: 10.1073/pnas.1200785109
Dhingra, S., Andes, D., and Calvo, A. M. (2012). VeA regulates conidiation, gliotoxin production, and protease activity in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot. Cell 11, 1531–1543. doi: 10.1128/EC.00222-12
Görgens, J. F., Bressler, D. C., and van Rensburg, E. (2015). Engineering Saccharomyces cerevisiae for direct conversion of raw, uncooked or granular starch to ethanol. Crit. Rev. Biotechnol. 35, 369–391. doi: 10.3109/07388551.2014.888048
Gougoulias, C., Clark, J. M., and Shaw, L. J. (2014). The role of soil microbes in the global carbon cycle: tracking the below-ground microbial processing of plant-derived carbon for manipulating carbon dynamics in agricultural systems. J. Sci. Food Agric. 94, 2362–2371. doi: 10.1002/jsfa.6577
He, Q. P., Zhao, S., Wang, J. X., Li, C. X., Yan, Y. S., Wang, L., et al. (2018). Transcription factor NsdD regulates the expression of genes involved in plant biomass-degrading enzymes, conidiation, and pigment biosynthesis in Penicillium oxalicum. Appl. Environ. Microbiol. 84:e01039-18. doi: 10.1128/AEM.01039-18
Huberman, L. B., Liu, J., Qin, L. N., and Glass, N. L. (2016). Regulation of the lignocellulolytic response in filamentous fungi. Fungal Biol. Rev. 30, 101–111. doi: 10.1016/j.fbr.2016.06.001
Karimi Aghcheh, R., Németh, Z., Atanasova, L., Fekete, E., Paholcsek, M., Sándor, E., et al. (2014). The VELVET A orthologue VEL1 of Trichoderma reesei regulates fungal development and is essential for cellulase gene expression. PLoS One 9:e112799. doi: 10.1371/journal.pone.0112799
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kusum, D., Rahul, J., Sushma, T., and Anita, P. (2014). Prolonged laccase production by a cold and pH tolerant strain of Penicillium pinophilum (MCC 1049) isolated from a low temperature environment. Enzyme Res. 2014:120708. doi: 10.1155/2014/120708
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, W. S., Chen, I. C., Chang, C. H., and Yang, S. S. (2012). Bioethanol production from sweet potato by co-immobilization of saccharolytic molds and Saccharomyces cerevisiae. Renew. Energ. 39, 216–222. doi: 10.1016/j.renene.2011.08.024
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, C. X., Zhao, S., Zhang, T., Xian, L., Liao, L. S., Liu, J. L., et al. (2017). Genome sequencing and analysis of Talaromyces pinophilus provide insights into biotechnological applications. Sci. Rep. 7:490. doi: 10.1038/s41598-017-00567-0
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Z. H., Yao, G. S., Wu, R. M., Gao, L. W., Kan, Q. B., Liu, M., et al. (2015). Synergistic and dose-controlled regulation of cellulase gene expression in Pencillium oxalicum. PLoS Genet. 11:e1005509. doi: 10.1371/journal.pgen.1005509
Liao, G. Y., Zhao, S., Zhang, T., Li, C. X., Liao, L. S., Zhang, F. F., et al. (2018). The transcription factor TpRfx1 is an essential regulator of amylase and cellulase gene expression in Talaromyces pinophilus. Biotechnol. Biofuels 11:276. doi: 10.1186/s13068-018-1276-8
Liao, L. S., Li, C. X., Zhang, F. F., Yan, Y. S., Luo, X. M., Zhao, S., et al. (2019). How an essential Zn2Cys6 transcription factor PoxCxrA regulates cellulase gene expression in ascomycete fungi? Biotechnol. Biofuels 12:105. doi: 10.1186/s13068-019-1444-5
Lin, X. R., and Momany, M. (2004). Identification and complementation of abnormal hyphal branch mutants ahbA1 and ahbB1 in Aspergillus nidulans. Fungal Genet. Biol. 41, 998–1006. doi: 10.1016/j.fgb.2004.07.005
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA seq data with DESeq2. Genome Biol. 15:550.
Machovic, M., and Janecek, S. (2006). Starch-binding domains in the post-genome era. Cell Mol. Life Sci. 63, 2710–2724. doi: 10.1007/s00018-006-6246-9
Marín-Navarro, J., and Polaina, J. (2011). Glucoamylases: structural and biotechnological aspects. Appl. Microbiol. Biotechnol. 89, 1267–1273. doi: 10.1007/s00253-010-3034-0
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428. doi: 10.1021/ac60147a030
Nitta, M., Furukawa, T., Shida, Y., Mori, K., Kuhara, S., Morikawa, Y., et al. (2012). A new Zn(II)2Cys6-type transcription factor BglR regulates β-glucosidase expression in Trichoderma reesei. Fungal Genet. Biol. 49, 388–397. doi: 10.1016/j.fgb.2012.02.009
Park, H. S., Bayram, O., Braus, G. H., Kim, S. C., and Yu, J. H. (2012). Characterization of the velvet regulators in Aspergillus fumigatus. Mol. Microbiol. 86, 937–953. doi: 10.1111/mmi.12032
Park, H. S., and Yu, J. H. (2016). Developmental regulators in Aspergillus fumigatus. J. Microbiol. 54, 223–231.
Passos, D. D. F., Pereira, N., and Castro, A. M. D. (2018). A comparative review of recent advances in cellulases production by Aspergillus, Penicillium and Trichoderma strains and their use for lignocellulose deconstruction. Curr. Opin. Green Sustain. Chem. 14, 60–66.
Pol, D., Laxman, R. S., and Rao, M. (2012). Purifcation and biochemical characterization of endoglucanase from Penicillium pinophilum MS. Indian J. Biochem. Biophys. 49, 189–194.
Portnoy, T., Margeot, A., Linke, R., Atanasova, L., Fekete, E., Sandor, E., et al. (2011). The CRE1 carbon catabolite repressor of the fungus Trichoderma reesei: a master regulator of carbon assimilation. BMC Genomics 12:269. doi: 10.1186/1471-2164-12-269
Qin, Y., Bao, L., Gao, M., Chen, M., Lei, Y., Liu, G., et al. (2013). Penicillium decumbens BrlA extensively regulates secondary metabolism and functionally associates with the expression of cellulase genes. Appl. Microbiol. Biotechnol. 97, 10453–10467. doi: 10.1007/s00253-013-5273-3
Robertson, G. H., Wong, D. W., Lee, C. C., Wagschal, K., Smith, M. R., and Orts, W. J. (2006). Native or raw starch digestion: a key step in energy efficient biorefining of grain. J. Agric. Food Chem. 54, 353–365. doi: 10.1021/jf051883m
Sánchez, O. J., and Cardona, C. A. (2008). Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresour. Technol. 99, 5270–5295. doi: 10.1016/j.biortech.2007.11.013
Si, H. Y., Rittenour, W. R., Xu, K., Nicksarlian, M., Calvo, A. M., and Harris, S. D. (2012). Morphogenetic and developmental functions of the Aspergillus nidulans homologues of the yeast bud site selection proteins Bud4 and Axl2. Mol. Microbiol. 85, 252–270. doi: 10.1111/j.1365-2958.2012.08108.x
Sorimachi, K., Le Gal-Coëffet, M. F., Williamson, G., Archer, D. B., and Williamson, M. P. (1997). Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to β-cyclodextrin. Structure 5, 647–661. doi: 10.1016/s0969-2126(97)00220-7
Sun, H., Zhao, P., Ge, X., Xia, Y., Hao, Z., Liu, J., et al. (2010). Recent advances in microbial raw starch degrading enzymes. Appl. Biochem. Biotechnol. 160, 988–1003. doi: 10.1007/s12010-009-8579-y
Sun, J., and Glass, N. L. (2011). Identification of the CRE-1 cellulolytic regulon in Neurospora crassa. PLoS One 6:e25654. doi: 10.1371/journal.pone.0025654
Tani, S., Kawaguchi, T., and Kobayashi, T. (2014). Complex regulation of hydrolytic enzyme genes for cellulosic biomass degradation in filamentous fungi. Appl. Microbiol. Biotechnol. 98, 4829–4837. doi: 10.1007/s00253-014-5707-6
Tao, L., and Yu, J. H. (2011). AbaA and WetA govern distinct stages of Aspergillus fumigatus development. Microbiology 157, 313–326. doi: 10.1099/mic.0.044271-0
van Peij, N. N., Gielkens, M. M., de Vries, R. P., Visser, J., and de Graaff, L. H. (1998). The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl. Environ. Microbiol. 64, 3615–3619.
Visser, E. M., Falkoski, D. L., de Almeida, M. N., Maitan-Alfenas, G. P., and Guimarães, V. M. (2013). Production and application of an enzyme blend from Chrysoporthe cubensis and Penicillium pinophilum with potential for hydrolysis of sugarcane bagasse. Bioresour. Technol. 144, 587–594. doi: 10.1016/j.biortech.2013.07.015
Wang, L., Zhao, S., Chen, X. X., Deng, Q. P., Li, C. X., and Feng, J. X. (2018). Secretory overproduction of a raw starch-degrading glucoamylase in Penicillium oxalicum using strong promoter and signal peptide. Appl. Microbiol. Biotechnol. 102, 9291–9301. doi: 10.1007/s00253-018-9307-8
Weirauch, M. T., and Hughes, T. R. (2011). A catalogue of eukaryotic transcription factor types, their evolutionary origin, and species distribution. Subcell. Biochem. 52, 25–73. doi: 10.1007/978-90-481-9069-0_3
Wieser, J., Lee, B. N., Fondon, J. W. III, and Adams, T. H. (1994). Genetic requirements for initiating asexual development in Aspergillus nidulans. Curr. Genet. 27, 62–69.
Xian, L., Wang, F., Luo, X., Feng, Y. L., and Feng, J. X. (2015). Purification and characterization of a highly efficient calcium-independent alpha-amylase from Talaromyces pinophilus 1-95. PLoS One 10:e121531. doi: 10.1371/journal.pone.0121531
Xiong, Y., Sun, J. P., and Glass, N. L. (2014). VIB1, a link between glucose signaling and carbon catabolite repression, is essential for plant cell wall degradation by Neurospora crassa. PLoS Genet. 10:e1004500. doi: 10.1371/journal.pgen.1004500
Xiong, Y., Wu, V. W., Lubbe, A., Qin, L., Deng, S., Kennedy, M., et al. (2017). A fungal transcription factor essential for starch degradation affects integration of carbon and nitrogen metabolism. PLoS Genet. 13:e1006737. doi: 10.1371/journal.pgen.1006737
Xiong, Y. R., Zhao, S., Fu, L. H., Liao, X. Z., Li, C. X., Yan, Y. S., et al. (2018). Characterization of novel roles of a HMG-box protein PoxHmbB in biomass-degrading enzyme production by Penicillium oxalicum. Appl. Microbiol. Biotechnol. 102, 3739–3753. doi: 10.1007/s00253-018-8867-y
Xu, Q. S., Yan, Y. S., and Feng, J. X. (2016). Efficient hydrolysis of raw starch and ethanol fermentation: a novel raw starch-digesting glucoamylase from Penicillium oxalicum. Biotechnol. Biofuels 9:216.
Yan, Y. S., Zhao, S., Liao, L. S., He, Q. P., Xiong, Y. R., Wang, L., et al. (2017). Transcriptomic profiling and genetic analyses reveal novel key regulators of cellulase and xylanase gene expression in Penicillium oxalicum. Biotechnol. Biofuels 10:279. doi: 10.1186/s13068-017-0966-y
Zhai, M. M., Niu, H. T., Li, J., Xiao, H., Shi, Y. P., Di, D. L., et al. (2015). Talaromycolides A-C, novel phenyl-substituted phthalides isolated from the green chinese onion-derived fungus Talaromyces pinophilus AF-02. J. Agric. Food Chem. 63, 9558–9564. doi: 10.1021/acs.jafc.5b04296
Zhang, M. Y., Zhao, S., Ning, Y. N., Fu, L. H., Li, C. X., Wang, Q., et al. (2019). Identification of an essential regulator controlling the production of raw-starch-digesting glucoamylase in Penicillium oxalicum. Biotechnol. Biofuels 12:7. doi: 10.1186/s13068-018-1345-z
Zhang, T., Zhao, S., Liao, L. S., Li, C. X., Liao, G. Y., and Feng, J. X. (2017). Deletion of TpKu70 facilitates gene targeting in Talaromyces pinophilus and identification of TpAmyR involvement in amylase production. World J. Microbiol. Biotechnol. 33:171. doi: 10.1007/s11274-017-2331-5
Zhao, S., Yan, Y. S., He, Q. P., Yang, L., Yin, X., Li, C. X., et al. (2016). Comparative genomic, transcriptomic and secretomic profiling of Penicillium oxalicum HP7-1 and its cellulase and xylanase hyper-producing mutant EU2106, and identification of two novel regulatory genes of cellulase and xylanase gene expression. Biotechnol. Biofuels 9:203. doi: 10.1186/s13068-016-0616-9
Keywords: transcription factor, plant-biomass-degrading enzymes, mycelial development, conidiation, Talaromyces pinophilus
Citation: Zhang T, Liao L-S, Li C-X, Liao G-Y, Lin X, Luo X-M, Zhao S and Feng J-X (2019) Identification of a Novel Transcription Factor TP05746 Involved in Regulating the Production of Plant-Biomass-Degrading Enzymes in Talaromyces pinophilus. Front. Microbiol. 10:2875. doi: 10.3389/fmicb.2019.02875
Received: 04 October 2019; Accepted: 28 November 2019;
Published: 13 December 2019.
Edited by:
Guodong Liu, Shandong University, ChinaCopyright © 2019 Zhang, Liao, Li, Liao, Lin, Luo, Zhao and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuai Zhao, c2h1YWl6aGFvMDIyN0BneHUuZWR1LmNu; Jia-Xun Feng, amlheHVuZmVuZ0Bzb2h1LmNvbQ==