 Stefano Raimondi1†
Stefano Raimondi1† Francesco Candeliere1†
Francesco Candeliere1† Alberto Amaretti1,2
Alberto Amaretti1,2 Stefania Costa3
Stefania Costa3 Silvia Vertuani4
Silvia Vertuani4 Gloria Spampinato1
Gloria Spampinato1 Maddalena Rossi1,2*
Maddalena Rossi1,2*- 1Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
- 2Biogest Siteia, University of Modena and Reggio Emilia, Reggio Emilia, Italy
- 3Department of Chemical, Pharmaceutical and Agricultural Sciences—DOCPAS, University of Ferrara, Ferrara, Italy
- 4Department of Life Sciences and Biotechnology, University of Ferrara, Ferrara, Italy
Leuconostoc is a genus of saccharolytic heterofermentative lactic acid bacteria that inhabit plant-derived matrices and a variety of fermented foods (dairy products, dough, milk, vegetables, and meats), contributing to desired fermentation processes or playing a role in food spoilage. At present, the genus encompasses 17 recognized species. In total, 216 deposited genome sequences of Leuconostoc were analyzed, to check the delineation of species and to infer their evolutive genealogy utilizing a minimum evolution tree of Average Nucleotide Identity (ANI) and the core genome alignment. Phylogenomic relationships were compared to those obtained from the analysis of 16S rRNA, pheS, and rpoA genes. All the phylograms were subjected to split decomposition analysis and their topologies were compared to check the ambiguities in the inferred phylogenesis. The minimum evolution ANI tree exhibited the most similar topology with the core genome tree, while single gene trees were less adherent and provided a weaker phylogenetic signal. In particular, the 16S rRNA gene failed to resolve several bifurcations and Leuconostoc species. Based on an ANI threshold of 95%, the organization of the genus Leuconostoc could be amended, redefining the boundaries of the species L. inhae, L. falkenbergense, L. gelidum, L. lactis, L. mesenteroides, and L. pseudomesenteroides. Two strains currently recognized as L. mesenteroides were split into a separate lineage representing a putative species (G16), phylogenetically related to both L. mesenteroides (G18) and L. suionicum (G17). Differences among the four subspecies of L. mesenteroides were not pinpointed by ANI or by the conserved genes. The strains of L. pseudomesenteroides were ascribed to two putative species, G13 and G14, the former including also all the strains presently belonging to L. falkenbergense. L. lactis was split into two phylogenetically related lineages, G9 and G10, putatively corresponding to separate species and both including subgroups that may correspond to subspecies. The species L. gelidum and L. gasicomitatum were closely related but separated into different species, the latter including also L. inhae strains. These results, integrating information of ANI, core genome, and housekeeping genes, complemented the taxonomic delineation with solid information on the phylogenetic lineages evolved within the genus Leuconostoc.
Introduction
The present project aims to investigate the evolutionary relationships within the genus Leuconostoc through a phylogenomic approach and to verify the consistency of phylogenetic relationships with the current taxonomy. This genus holds a group of Lactic Acid Bacteria (LAB) belonging, together with the genera Convivina, Fructobacillus, Oenococcus, and Weissella, to the family of Leuconostocaceae (www.bacterio.net; Nieminen et al., 2014; Bello et al., 2022), in its turn included in the order Lactobacillales, based on the average amino acid identity of core proteins (cAAI) (Zheng et al., 2020). The genus Leuconostoc encompasses 17 species according to the List of Prokaryotic names with Standing in Nomenclature (https://lpsn.dsmz.de/genus/leuconostoc) (Parte et al., 2020). They are saccharolytic bacteria that catabolize carbohydrates to lactic acid through heterolactic fermentation and inhabit a variety of niches where carbohydrate-based substrates are available, such as plants, plant-derived matrices, silage, fermented foods (e.g., dairy products, fermented dough, milk, vegetables, and meats), spoiled foods, and sewage (Dellaglio et al., 1995). In some cases, species are associated with a specific habitat, such as vegetables (Yu et al., 2020), meats (Candeliere et al., 2021), or other foods (Vedamuthu, 1994). The genus is considered safe products and has been accorded the status of “generally recognized as safe” (GRAS) (Ogier et al., 2008). Fermentation of suitable substrates has attracted attention in recent years to produce antimicrobial extracts (Venegas-Ortega et al., 2019; Ahmadi-Ashtiani et al., 2020; Costa et al., 2021). Strains of Leuconostoc spp. have found promising industrial applications in the preparation of biopreservative systems by fermentation of different substrates. Leuconostoc are used as starters in food and beverage fermentation to improve the nutritional and sensorial properties and to extend the shelf life (Shin and Han, 2015). Moreover, bioactive extracts obtained from vegetables fermented with Leuconostoc spp. found application in the formulation of innovative cosmetics (INCI Name: Leuconostoc/Radish Root Ferment Filtrate) (Ahmadi-Ashtiani et al., 2020).
The early taxonomy of LAB was based on phenotypic and morphological features, then chemotaxonomic criteria such as DNA–DNA hybridization and G+C content became the reference for species assignment (Vandamme et al., 1996). With the advent of phylogenetic taxonomy (Woese and Fox, 1977), the sequence of the gene encoding 16S rRNA turned into the gold standard for taxonomic and phylogenetic analysis (Lane et al., 1985; Stackebrandt and Goebel, 1994; Stackebrandt and Ebers, 2006), and allowed the systematic study of evolutionary relationships among prokaryotes. A phylogenetic tree of a conserved gene that is assumed to be vertically inherited is expected to produce the statistical trend or the real genealogy of evolving entities. However, due to the limited size of individual genes, multiple substitutions, parallel, convergent, or reversal events, and horizontal transfer of DNA, the strength of the phylogenetic signal in single molecules is often too low to infer the evolutionary relationships underlying species differentiation. Therefore, multiple phylogenetic markers have been applied to obtain a well-resolved and informative tree, either by the concatenation of genes, aiming to average their phylogenetic information, or by the corroboration of individual phylogenetic signals (Whelan and Morrison, 2017).
With the advent of high-throughput sequencing, an increasing amount of whole bacterial genomes had been accumulated, thus plenty of information became available to improve the resolution of bacterial diversity and the accuracy of phylogenetic reconstruction. In this context, core genome phylogenesis is widely used to infer phyletic lines in the evolutionary history of prokaryotic species (Stott and Bobay, 2020). The average nucleotide identity (ANI) of the genes shared between two genomes was introduced as the gold standard for the delineation of bacterial species (Richter and Rosselló-Móra, 2009; Chun and Rainey, 2014) and has been recently proposed as a tool to determine statistically supported phylogenies (Gosselin et al., 2022).
In the present study, the taxonomy of 221 Leuconostoc genome sequences belonging to 17 species has been preliminarily investigated based on ANI to measure genome similarity. ANI, which provides an average measure of similarity across homologous regions shared by a pair of genomes, is a major metric used for this purpose (Palmer et al., 2020), considering the ANI threshold of 95% for species delineation (Richter and Rosselló-Móra, 2009). The subspecies delimitations were also investigated, considering a threshold of 98% (Minias et al., 2020; Pearce et al., 2021), although it is not recognized as a standard in the taxonomy of prokaryotes. For the strains presenting low ANI values, the Average Aminoacidic Identity (AAI) was utilized to confirm the inclusion in the genus Leuconostoc and the different putative species, considering the threshold of 55–60% and 85–90%, respectively (Rodriguez-R and Konstantinidis, 2014). The evolutive genealogy of the genomes was reconstructed utilizing a minimum evolution tree of ANI, computed according to Gosselin et al. (2022), and the core genome alignment.
Phylogenetic relationships were also inferred using the housekeeping genes encoding phenylalanyl-tRNA synthase alpha-subunit (pheS) and RNA polymerase alpha-subunit (rpoA) (Das et al., 2014), in addition to the 16S rRNA gene. rpoA was previously exploited for the identification of L. falkenbergense strains at the species level (Wu and Gu, 2021a), while pheS is one of the targets in the Multilocus Sequence Analysis (MLSA), successfully applied for differentiation of species of the genus Leuconostoc (Rahkila et al., 2014). As a whole, multiple phylogenomic approaches have been applied to investigate evolutionary relationships within the genus Leuconostoc, following a preliminary reclassification of the strains in putative species according to ANI comparison. Split decomposition analysis has been carried out to check the ambiguities in the inferred phylogenesis, evaluate consistency among the output of the diverse analysis, and determine the approach that provides the strongest phylogenetic signal (Whelan and Morrison, 2017).
Methods
In total, 221 Leuconostoc genome sequences available on 1 October 2021 were retrieved from the NCBI database. The accession numbers are reported in Supplementary Table S1. Only genomes obtained from pure culture sequencing were used, while metagenome-assembled genomes were discarded. The genomes were inspected for completeness and contamination with CheckM (Parks et al., 2015). Genomes were also checked for the presence of the molecular signatures characterizing the genus Leuconostoc (Bello et al., 2022). The genomes were annotated with Prokka (Seemann, 2014) to calculate the pangenome using Roary (Page et al., 2015), with the minimum percentage identity parameter set at 80%.
The genes encoding rpoA, pheS, and 16S rRNA were extracted from each genome and aligned with Clustal Omega on EMBL-EBI website (Sievers et al., 2011; Madeira et al., 2019). The Total Average Nucleotide Identity (ANI) was calculated following the method described by Gosselin et al. (2022). This method allowed us to calculate the bootstrap values from 100 ANI distance matrices replicates, thanks to a process of genome segmentation and random segment selection utilized to calculate the desired number of replicates. The AAI between all the genomes was calculated with AAI Calculator (http://enve-omics.ce.gatech.edu/aai/) by all-against-all approach (Rodriguez-R and Konstantinidis, 2016).
The alignments of 16S rRNA, rpoA, and pheS genes and the 247 core genes identified by Roary out of a total of 25,377 genes (Supplementary Spreadsheet 1) were utilized to generate phylogenetic trees constructed with the maximum likelihood method using the RAxML tool with 100 bootstrap replicates (Stamatakis, 2014). The ANI distance matrix was used to build a tree using the script provided by Gosselin et al. (2022). In this script, the balanced minimum evolution algorithm implemented in the FastME function of the R package APE (Paradis et al., 2004) was applied to generate phylogenies for each distance matrix (Desper and Gascuel, 2002), whereas the function plotBS of the R package Phangorn (Schliep, 2011) was exploited to map support values onto the tree. The AAI distance matrix was used to compute an Unweighted Pair Group Method with the Arithmetic mean (UPGMA) unrooted phylogenetic tree with the DendroUPGMA tool (Garcia-Vallvé et al., 1999). The phylogeny was further inferred with SplitsTree v. 4.18.2 (Huson and Bryant, 2006) with a neighbor net drawing and Jukes–Cantor correction for alignment-derived trees (Bandelt and Dress, 1992; Huson and Bryant, 2006).
The tree of ANI was compared with those constructed from the alignment of 16S rRNA, pheS, rpoA genes, or of the core genome, utilizing the following “generalized” Robinson–Foulds metrics: the Jaccard–Robinson–Foulds (JRF), computed with k = 1 (Nye et al., 2006), and the information-based measures of Mutual Clustering Information (MCI) and Shared Phylogenetic Information (SPI) (Smith, 2020). JRF, MCI, and SPI were computed with the R package “TreeDist,” archived at https://dx.doi.org/10.5281/zenodo.3528123. Comparison relied on matching each informative split within a tree (i.e., an internal branch of the tree, with at least two leaves at each extremity) with an informative split within the other tree. For each metric, the comparison yielded a normalized similarity score in the range 0–1 between two trees and a score for each match of paired tree splits.
Results
ANI phylogenomic analysis
All the genome sequences available for species of Leuconostoc on the date 1 October 2021, containing all the type strains, were included in the analysis. The majority of the sequences presented low or no contamination (≤ 5%) after CheckM analysis, except for six genomes. Five of them were discarded, while the sequence of L. inhae KCTC 3774 (contamination = 6.1%) was retained, being a type strain. As a whole, 216 genomes were analyzed. They were ascribed to 17 correctly named species (17 L. carnosum, 29 L. citreum, three L. falkenbergense, two L. fallax, three L. gelidum, seven L. gasicomitatum, one L. holzapfelii, five L. inhae, two L. kimchii, 19 L. lactis, one L. litchii, one L. miyukkimchii, 83 L. mesenteroides, one L. palmae, 34 L. pseudomesenteroides, one L. rapi, and six L. suionicum), to the not validly published species “L. garlicum” (one strain), and broadly assigned to the genus Leuconostoc (five strains) (Supplementary Table S1). All the strains, including those without an assigned species, were coherent with the indel signatures of Leuconostoc genus.
Pairwise ANI values were calculated for the data set (Figure 1, Supplementary Spreadsheet S2). Considering the species threshold of 95%, the 216 strains nominally ascribed to 17 species were redistributed in 18 groups (referred to as G1–G18), which were consistent with the taxonomy of the Genome Taxonomy Database web server (GTDB, https://gtdb.ecogenomic.org; Supplementary Table S1). Pairwise ANI between strains of different groups was always <95%, while it was always >6% between strains belonging to the same group, thus ascribable to the same putative species (Figures 1, 2A). The mean pairwise ANI similarities between the strains of each group are presented in Figure 2B.

Figure 1. Heatmap of the ANI similarities matrix reporting pairwise ANI values of 216 Leuconostoc genomes. Blue, ANI > 98%; green, 95% < ANI <98%; gray shades, ANI <95% (77–94.9%). Strain labels are colored according to the groups.
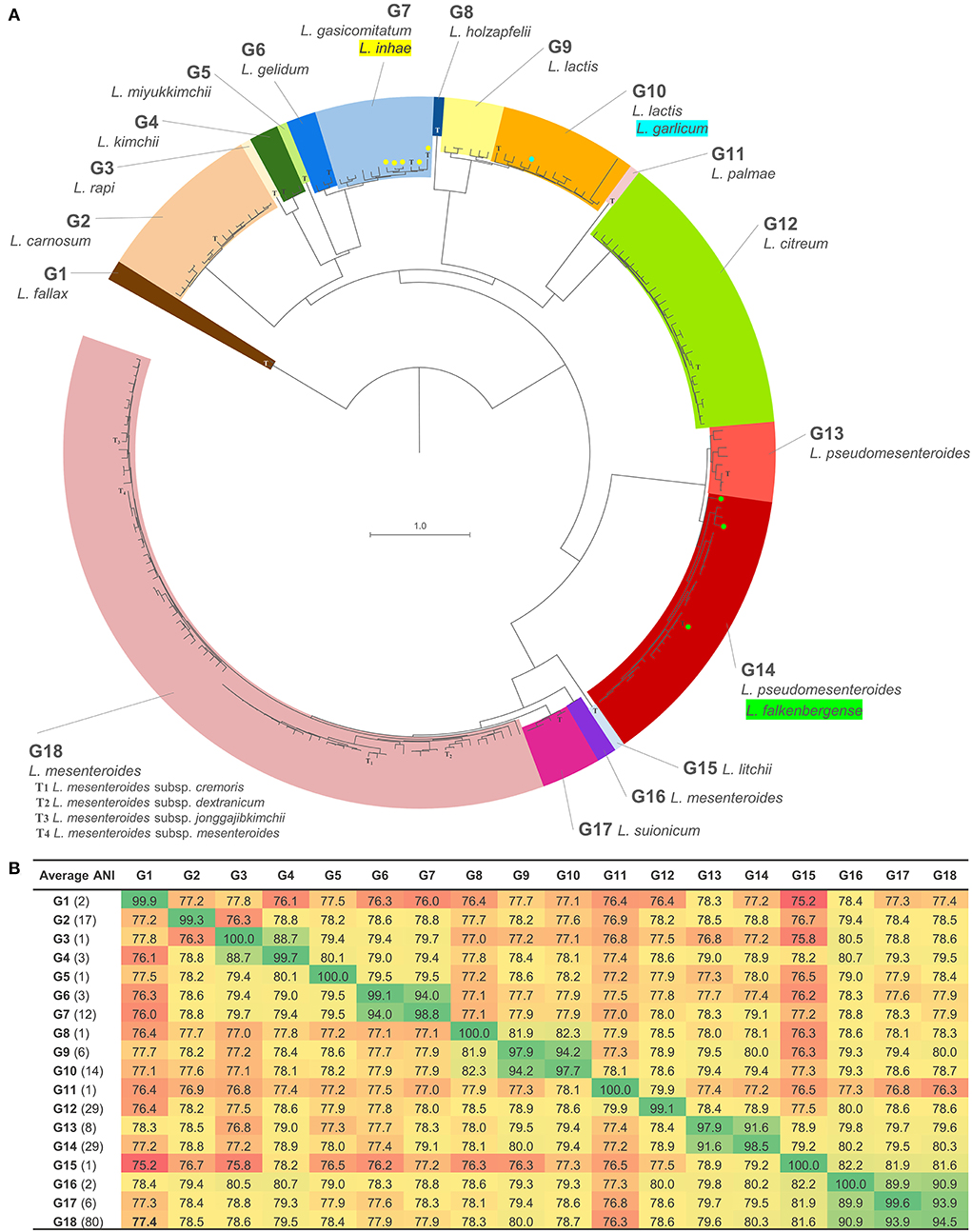
Figure 2. Phylogenomic analysis of Leuconostoc species: (A) Minimum evolution tree of ANI, reporting the 18 proposed clades with values >95%; T, type strain; strains currently ascribed to the species of L. falkenbergense, L. garlicum, and L. inhae are indicated in green, cyan, and yellow, respectively. (B) Heatmap of the mean ANI values between the strains of the groups.
The species L. fallax, L. carnosum, L. rapi, L. kimchii, L. miyukkimchii, L. holzapfelii, L. citreum, L. palmae, L. litchii, and L. suionicum were consistent with the current taxonomy and coincided with groups G1, G2, G3, G4, G5, G8, G11, G12, G15, and G17, respectively. Except for a sole strain of L. citreum, none of the strains attributed to these species was included in a different group. On the other hand, a bijective relationship between nominal species and ANI groups (i.e., one species–one group, and vice versa) was not observed for the strains currently belonging to L. mesenteroides, L. pseudomesenteroides, L. falkenbergense, L. lactis, L. inhae, and L. gelidum. The five strains lacking a nominal species designation were placed in groups G4, G12, G13, G14, and G18 (Supplementary Table S1).
Most of the strains assigned to the species L. mesenteroides (80) clustered in G18. This group encompassed all the strains belonging to the four validly published L. mesenteroides subspecies (L. mesenteroides subsp. cremoris, L. mesenteroides subsp. dextranicum, L. mesenteroides subsp. mesenteroides, and L. mesenteroides subsp. jonggajibkimchii), including the corresponding type strains (ATCC 19254T, DSM 20484T, ATCC 8293T, and DRC1506T, respectively), and to the not valid subspecies L. mesenteroides subsp. sake. Within G18, the strains were very similar and, despite the existence of different nominal subspecies, they presented on average ANI values of 99.1%. Pairwise ANI values between members of the different subspecies of L. mesenteroides were always >98.6%. Likely evolving as a separate lineage, only L. mesenteroides subsp. cremoris seemed fully resolved at the subspecies level (except for L. mesenteroides subsp. cremoris LbT16, likely misclassified). Another strain of L. mesenteroides and L. mesenteroides subsp. mesenteroides clustered in a diverse clade, G16, which are closely related to both G17 and G18, the latter grouping the L. suionicum strains.
The strains attributed to the species L. pseudomesenteroides were distributed in the closely related groups G13 and G14. The mean value of pairwise ANI between strains from the two distinct groups was 91.8%. The type strain L. pseudomesenteroides NCDO 768T, seven other L. pseudomesenteroides, and a Leuconostoc sp. were comprehended in G13, with a mean ANI value of 98.4%. G14 held 26 L. pseudomesenteroides, three strains currently ascribed to L. falkenbergense, including the type strain L. falkenbergense LMG 10779T, and an additional Leuconostoc sp. strain, with a mean ANI value of 99.4%.
The strains belonging to the species L. lactis were split into two phylogenetically related groups, G9 and G10. G9 included six L. lactis strains, among which L. lactis KCTC 3773, which used to be the type strain of the species L. argentinum until it was merged with L. lactis. G10 encompassed the type strain of L. lactis (strain JCM 6123T), the putative “L. garlicum” KFRI01 (species currently not validly published), L. citreum 1300_LCIT, and other 12 L. lactis. Pairwise ANI between strains of G9 and G10 was always <94.5%, while it was always >96.9% within the two groups. Within G9, two subgroups of strains with ANI values >98% within them and in the range of 95–98% between the subgroups could be delineated. Likewise, pairs of strains presenting ANI values >98% were identified within G10, even though a clear delineation of subgroups could not be accomplished.
The strains of L. gelidum and L. gasicomitatum clustered in two separated but closely related clades, corresponding to G6 and G7, the latter also including L. inhae (Figure 1). G6 comprised three L. gelidum strains, among which the type strain L. gelidum KCTC_3527T. G7 comprised all the L. gasicomitatum (7) and all the L. inhae (5), including the corresponding type strain. Within this group, the pairwise ANI values between strains of L. gasicomitatum and L. inhae were always > 98%.
Phylogenomic relationships highlighted by ANI joined G17 and G18, which were more distantly related to G16 and G15 (Figure 2). Another strict relationship is associated with G13 and G14, which were more remotely linked to the clade harboring G15, G16, G17, and G18. A close relationship connected G9 and G10, which were more distantly related to G8, with these three groups descending from a common branch that evolved independently also toward G11 and G12. Strict relationships were found between G3 and G4 and between G6 and G7, which lay in a clade harboring also G5, and with a more ancient bifurcation, G2. On the other side, group G1 resulted phylogenetically distant from all the other putative species, albeit belonging to the genus Leuconostoc according to ANI thresholds.
Comparison between ANI and phylogenetic genomic markers
Trees were computed utilizing ANI, the core genome alignment, AAI, and alignment of the genes rpoA, pheS, and 16S rRNA (Supplementary Figures 1,2). To evaluate possible conflicting phylogenetic signals, the trees were subjected to split decomposition analysis (Figure 3), which revealed that the core genome alignment yielded the less reticulated network, from which the evolutionary trajectories could be inferred with the lowest ambiguity. ANI, AAI, rpoA, and pheS yielded more reticulated networks, that corroborated the phylogenetic reconstruction, although with increasing uncertainty in the location of some bifurcations, particularly toward the origin of the main branches. The trees of ANI, AAI, rpoA, and pheS genes maintained the same general topology with respect to the most peripheral region of the trees, reflecting the most recent evolutionary derivations (Figure 4).
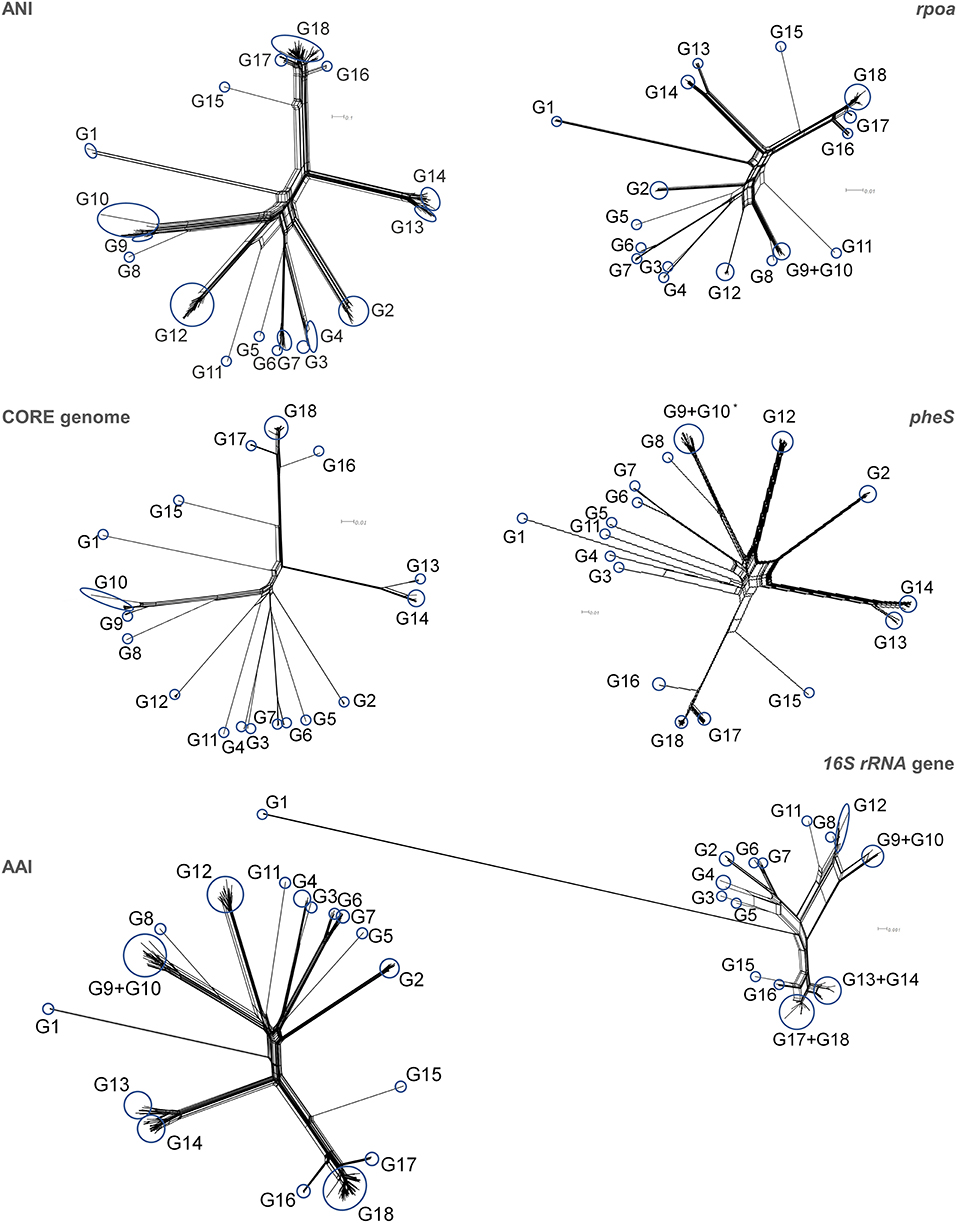
Figure 3. Split decomposition analysis of the phylogenetic trees of ANI, core genome, AAI, rpoA, pheS, and 16S rRNA genes. The analysis was performed utilizing gene alignments for the core genome, rpoA, pheS, and 16S rRNA genes, and distance matrix for ANI. Blue circles indicate the position of groups on split-decomposed trees. *For clarity of representation, the strain L. lactis AV1N that harbored a highly divergent pheS sequence, was not included.
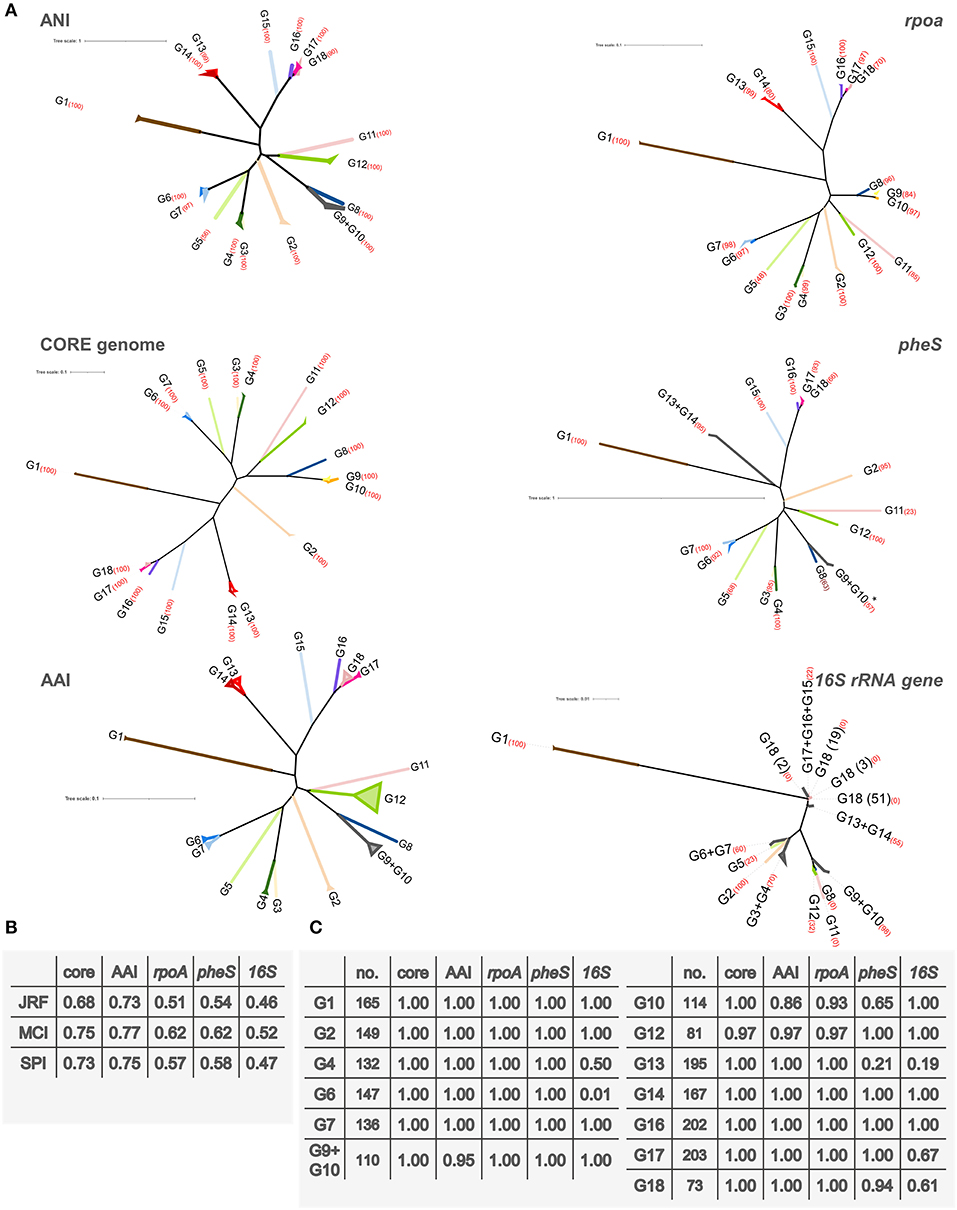
Figure 4. (A) Phylogenetic trees representing ANI distances and the alignment of the core genome, rpoA, pheS, and the 16S rRNA gene. For core genome, rpoA, pheS, and 16S rRNA genes trees, an alignment was produced with Clustal Omega, respectively, and it was used to infer a tree using RAxML. ANI tree was inferred from the ANI distance matrix with R package APE. AAI distance matrix was used to compute the UPGMA tree. Strains are collapsed into their corresponding group. The percentage bootstrap value of each clade is reported in red brackets. *For clarity of representation, the strain L. lactis AV1N that harbored a highly divergent pheS sequence, was not included. (B) Comparison of the ANI tree with the other phylogenetic trees. Normalized similarity score obtained with the generalized Robinson–Foulds metrics Jaccard–Robinson–Foulds (JRF), Mutual Clustering Information (MCI), and Shared Phylogenetic Information (SPI) are reported. (C) JRF scores of the 15 groups identified in the ANI tree.
The topology of the ANI phylogram was compared to the other trees, utilizing three “generalized” Robinson–Foulds metrics to establish the consistency of the splits (Figure 4, Supplementary Spreadsheet S3). This analysis indicated that the core genome, AAI, pheS, and rpoA, in order of decreasing adherence, delineated the same groups identified by ANI analysis. The informative splits corresponding to several groups defined by ANI (i.e., G1, G2, G4, G6, G7, G14, G16, and G17) scored JRF values of 1.00, indicating that the “leaves” of each branch generally coincided in ANI, core genome, AAI, rpoA, and pheS trees. The phylogeny reconstructed according to ANI, pheS, rpoA, the core genome, and AAI were in general agreement in the identification of three major branches: one harboring the groups from G2 to G7, another from G8 to G12, and a third from G13 to G18. The gene rpoA was not suitable for phylogenetic positioning of groups G9 and G10, including the strains currently assigned to the species L. lactis, whereas the nodes of speciation of the other groups, except for G18 (bootstrap 70%), were identified with very good resolution (bootstraps ≥80%). pheS was not able to detect with high confidence (bootstrap <80%) the common speciation of strains assigned to G9, G10, G13, and G18. On the other hand, the 16S rRNA gene presented the lowest phylogenetic signal, generating a highly reticulated network that failed to pinpoint phylogenetic relationships and a maximum likelihood tree where several ANI groups were not resolved. Most of the nodes of the 16S rRNA gene tree presented low bootstrap values, indicating that the confidence of this tree was low.
Phylogenomic relationships closely associated G17 and G18, which were more distantly related to G16, and then to G15 that lay closer to a common ancestor (Figure 2). The nodes from which G15 and G16 descend and separate from G17 and G18 have bootstrap confidence of 100% in the trees of ANI, core genome, rpoA, and pheS. In turn, G17 and G18 descended from a solid node with a bootstrap of 100% in both ANI and core genome, found also in rpoA and pheS trees with lower confidence. Coherently, JRF scores of 1.00 indicated that the branches leading to these ANI groups were highly conserved in all these trees. Within G18, a sole strain out of 80 lay on a separate branch, divided from the other 79 strains by a highly solid node (90 and 100% in ANI and the core genome trees, respectively). Most of the strains ascribed to the subspecies L. mesenteroides subsp. cremoris evolved from a common lineage. On the other side, the strains ascribed to the subspecies L. mesenteroides subsp. mesenteroides were shuffled within the tree. The clade harboring G13 and G14 was the closest to the one harboring G15, G16, G17, and G18, originating from a node with bootstrap confidence of 100 in the ANI, core genome, and rpoA trees. In these trees, G13 and G14 descended from a solid node (99 or 100%) and were highly conserved, presenting a JRF value of 1.00, although the amount of inferred evolutionary change was low. pheS failed to separate G13 and G14.
The strictly related species G9 and G10 were resolved in separate branches in the core genome tree by a node with a bootstrap of 100%. Despite pairwise ANI values between the strains of G9 and G10 suggesting their distribution in separate species, the separation was not displayed in the minimum evolution tree of ANI (Figure 2), where the two subgroups of G9 had different locations with respect to G10. In fact, in the ANI tree the subgroup of G9 harboring L. lactis KCTC 3773 was separated from G10 by a solid node (bootstrap of 100%), while the other subgroup was placed in a branch of G10 by a more uncertain node (bootstrap of 83%). However, all the trees consistently indicated that G9 and G10 are derived from a lineage that evolved separately also into G8 and that had a common ancestor with G11 and G12. Bootstrap values of 100% corroborated this descendance in both ANI and core genome trees. G11 evolved separately from G12, from which it diverged by a conspicuous evolutionary change.
Another major lineage that could be inferred in the trees of ANI, core genome, AAI, rpoA, and pheS encompassed G2, G3, G4, G5, G6, and G7. Bootstrap values of 100% in both the ANI and the core genome trees corroborated a pattern of bifurcations that was consistently found in the general traits also in the trees of AAI, rpoA, and pheS, although with lower bootstrap values. The first bifurcation in the inferred lineage was the branch leading to G2. Subsequently, a branch led to both G3 and G4, reciprocally characterized by a minor amount of inferred evolutionary change. G5 evolved separately from the branch leading to G6 and G7 that, in their turn, were very strictly related. For these groups, the high JRF values reflected the accordance of ANI classification with the core genome, AAI, rpoA, and pheS trees.
Group G1 was the less closely related species within the genus, presenting on average ANI scores with other groups <82.3%, and being separated from all the other species of Leuconostoc by a large amount of inferred evolutionary change. Nonetheless, the placement of L. fallax inside the genus was confirmed by AAI, since they presented an AAI value >66% with all the other Leuconostoc strains.
Discussion
Bacterial species can be defined within populations of similar strains on the basis of maximum likelihood methods that determine the point of transition of the evolutionary processes. As a result of common evolution, the strains belonging to the same species share genetic features and thus biological properties. Species delimitations constitute an important challenge in biodiversity studies, mostly for genera such as Leuconostoc that have an important role in several industrial and food fermentation processes (Buckenhüskes, 1993; Caplice and Fitzgerald, 1999; Steinkraus, 2002), to produce fermented sausages, fermented vegetables and cereals, and dairy products (e.g., butter, cream, fresh and raw milk, cheeses), and to obtain novel ingredients for cosmeceutical formulations (Ahmadi-Ashtiani et al., 2020).
Taxonomic classifications of the 216 Leuconostoc strains based on the core genome sequences indicated that many strains attributed to the same Leuconostoc species laying onto paraphyletic branches necessitate reclassification and that the taxonomy of the genus is not entirely resolved. During the revision of the manuscript, a study suggesting the reorganization of Leuconostoc taxonomy was published (Kumar et al., 2022). In that manuscript, presenting some overlapping with our survey also in terms of procedures, the proposal of a taxonomic update of the genus Leuconostoc was based on ANI and core genome analyses and is consistent with the outcome of our investigation. The phylogenomic reconstruction of the genus herein presented, integrating the information of ANI, core genome, and housekeeping genes complemented the taxonomic delineation with solid information on the phylogenetic lineages evolved within the genus Leuconostoc.
In this study, the phylogenomic trees built aligning housekeeping single genes corroborated ANI results. The topology of the phylogenetic trees consistently indicates the remote diversification of most of the species, with a few exceptions. Split decomposition analysis and comparison of the ANI tree with the other ones revealed robust consistency for most of the branches identified, with strong signals for the majority of the nodes identified by ANI, core genome, AAI, rpoA, and pheS genes, that generally presented very high bootstrap values. 16S rRNA gene yielded weak signals and thus was not suitable to define taxonomy and phylogeny within this genus. According to the consistent results of ANI and phylogenomic trees, the strains currently attributed to L. mesenteroides lay onto paraphyletic branches and need to be split into two separate species, corresponding to G16 and G18. Interestingly, these groups are already distinct in GTDB where they are referred to as Leuconostoc mesenteroides_B and Leuconostoc mesenteroides, respectively, and have been delineated also by Kumar et al. (2022). G18 encompasses most of the strains currently assigned to L. mesenteroides, including the type strain, and all the strains belonging to the subspecies L. mesenteroides subsp. cremoris, L. mesenteroides subsp. dextranicum, L. mesenteroides subsp. jonggajibkimchii, and L. mesenteroides subsp. mesenteroides. Nonetheless, the strains belonging to different L. mesenteroides subspecies present a genome similarity always >99.1%, higher than the threshold of 98% utilized to delineate subspecies in other bacterial taxa (Minias et al., 2020; Pearce et al., 2021). Accordingly, both in this study and in previous ones the conserved genes failed to differentiate between the subspecies of L. mesenteroides (Ricciardi et al., 2020).
G18 is closely related to G17, which includes strains of the species L. suionicum, previously recognized as subspecies L. mesenteroides subsp. suionicum (Jeon et al., 2017). The two strains belonging to G16, currently assigned to L. mesenteroides, should form a new species that would require characterization and formal definition (Das et al., 2014; Ramasamy et al., 2014). The strains currently assigned to L. pseudomesenteroides should be split into two species, G13 and G14. The former harbors the type strain L. pseudomesenteroides NCDO 768T, and the latter encompasses also the strains of L. falkenbergense, including the type strain L. falkenbergense LMG 10779T. L. falkenbergense has been already recognized as a different species phylogenetically related to L. pseudomesenteroides (Wu and Gu, 2021a) and recently added in GTDB.
The strains currently attributed to L. lactis should be split into two separate species, corresponding to the groups G9 and G10. Consistently, in GTDB the groups are already distinct and referred to as Leuconostoc lactis_A and Leuconostoc lactis. G10 encompasses the type strain L. lactis JCM 6123T, the strain KFRI01 named L. garlicum, a species that is not formally recognized, and L. citreum 1300_LCIT. G9 harbors L. lactis KCTC 3773, previously identified as the type strain of L. argentinum, species that is not anymore accepted (Dicks et al., 1993; Vancanneyt et al., 2006). Within both G10 and G9, subgroups of strains putatively ascribable to different subspecies can be identified, that present ANI scores between each other lower than 98%. Therefore, further phenotypic and genotypic studies could investigate whether subspecies have to be created within G10 and G9 groups. The strains of L. gelidum and L. gasicomitatum fall into G6 and G7, respectively, which include the type strains L. gelidum subsp. gelidum KCTC 3527T and L. gasicomitatum LMG_18811T. This classification confirms the observations by Wu and Gu (2021b) that rejected the proposal to recognize L. gasicomitatum as a subspecies of L. gelidum (L. gelidum subsp. gasicomitatum) (Rahkila et al., 2014). Moreover, G7 includes also the type strain L. inhae KCTC 3774T, suggesting that L. inhae and L. gasicomitatum could belong to the same species. No clear correlation could be established between the phylogenetic relationships and the source of isolation of the strains, except for G2 (L. carnosum) and G14 (L. falkenbergense) that encompassed mostly strains isolated from meat and dairy products, respectively (Supplementary Table S1) (Raimondi et al., 2018; Wu and Gu, 2021a). The evolutive force that shaped the speciation of Leuconostoc deserves deeper comparative genomics and functional analysis.
The results of this study confirmed and deepened the evidence on the evolutionary relationship among Leuconostoc species and may provide a basis for a possible future reorganization of the genus, as summarized in Supplementary Table S1. However, any update in the organization of Leuconostoc genus, such as the creation of new species and/or subspecies, would require, all the biochemical and physiological data based on strains survey that support the creation of new species and the reclassification of others.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
Conceptualization: SR, AA, and MR. Investigation: SR, FC, and GS. Data curation: FC, AA, SV, and SC. Formal Analysis: AA. Visualization: SR, FC, and AA. Writing—original draft: All authors. Funding acquisition: MR and SV. Supervision: MR. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.897656/full#supplementary-material
References
Ahmadi-Ashtiani, H. R., Baldisserotto, A., Cesa, E., Manfredini, S., Zadeh, H. S., Gorab, M. G., et al. (2020). Microbial biosurfactants as key multifunctional ingredients for sustainable cosmetics. Cosmetics 7, 46. doi: 10.3390/COSMETICS7020046
Bandelt, H. J., and Dress, A. W. (1992). Split decomposition: a new and useful approach to phylogenetic analysis of distance data. Mol. Phylogenet. Evol. 1, 242–252. doi: 10.1016/1055-7903(92)90021-8
Bello, S., Rudra, B., and Gupta, R. S. (2022). Phylogenomic and comparative genomic analyses of Leuconostocaceae species: identification of molecular signatures specific for the genera Leuconostoc, Fructobacillus and Oenococcus and proposal for a novel genus Periweissella gen. nov. Int. J. Syst. Evol. Microbiol. 72, 005284. doi: 10.1099/ijsem.0.005284
Buckenhüskes, H. J. (1993). Selection criteria for lactic acid bacteria to be used as starter cultures for various food commodities. FEMS Microbiol. Rev. 12, 253–271 doi: 10.1016/0168-6445(93)90067-J
Candeliere, F., Raimondi, S., Spampinato, G., Tay, M. Y. F., Amaretti, A., Schlundt, J., et al. (2021). Comparative genomics of Leuconostoc carnosum. Front. Microbiol. 11, 605127. doi: 10.3389/fmicb.2020.605127
Caplice, E., and Fitzgerald, G. F. (1999). Food fermentations: role of microorganisms in food production and preservation. Int. J. Food Microbiol. 50, 131–149. doi: 10.1016/s0168-1605(99)00082-3
Chun, J., and Rainey, F. A. (2014). Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 64, 316–324. doi: 10.1099/ijs.0.054171-0
Costa, S., Summa, D., Semeraro, B., Zappaterra, F., Rugiero, I., and Tamburini, E. (2021). Fermentation as a strategy for bio-transforming waste into resources: lactic acid production from agri-food residues. Fermentation 7, 3. doi: 10.3390/fermentation7010003
Das, S., Dash, H. R., Mangwani, N., Chakraborty, J., and Kumari, S. (2014). Understanding molecular identification and polyphasic taxonomic approaches for genetic relatedness and phylogenetic relationships of microorganisms. J. Microbiol. Methods 103, 80–100. doi: 10.1016/j.mimet.2014.05.013
Dellaglio, F., Dicks, L. M. T., and Torriani, S. (1995). “The Genus Leuconostoc,” in The Genera of Lactic Acid Bacteria, Vol. 2, eds B. J. B. Wood, and W. H. Holzapfel (Boston, MA: Springer), 235–278. doi: 10.1007/978-1-4615-5817-0_7
Desper, R., and Gascuel, O. (2002). Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J. Comput. Biol. 9, 687–705. doi: 10.1089/106652702761034136
Dicks, L. M. T., Fantuzzi, L., Gonzalez, F. C., Du Toit, M., and Dellaglio, F. (1993). Leuconostoc argentinum sp. nov., isolated from Argentine raw milk. Int. J. Syst. Bacteriol. 43, 347–351. doi: 10.1099/00207713-43-2-347
Garcia-Vallvé, S., Palau, J., and Romeu, A. (1999). Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Mol. Biol. Evol. 16, 1125–1134. doi: 10.1093/oxfordjournals.molbev.a026203
Gosselin, S., Fullmer, M. S., Feng, Y., and Gogarten, J. P. (2022). Improving phylogenies based on average nucleotide identity, incorporating saturation correction and nonparametric bootstrap support. Syst Biol. 71, 396–409. doi: 10.1093/sysbio/syab060
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Jeon, H. H., Kim, K. H., Chun, B. H., Ryu, B. H., Han, N. S., and Jeon, C. O. (2017). A proposal of Leuconostoc mesenteroides subsp. jonggajibkimchii subsp. nov. and reclassification of Leuconostoc mesenteroides subsp. suionicum (Gu et al., 2012) as Leuconostoc suionicum sp. nov. based on complete genome sequences, Int. J. Syst. Evol. Microbiol. 67, 2225–2230, doi: 10.1099/ijsem.0.001930
Kumar, S., Bansal, K., and Sethi, S. K. (2022). Comparative genomics analysis of genus Leuconostoc resolves its taxonomy and elucidates its biotechnological importance. Food Microbiol. 106, 104039. doi: 10.1016/j.fm.2022.104039
Lane, D. J., Pace, B., Olsen, G. J., Stahl, D. A., Sogin, M. L., and Pace, N. R. (1985). Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl Acad. Sci. USA. 82, 6955–6959. doi: 10.1073/pnas.82.20.6955
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Minias, A., Zukowska, L., Lach, J., Jagielski, T., Strapagiel, D., Kim, S. Y., et al. (2020). Subspecies-specific sequence detection for differentiation of Mycobacterium abscessus complex. Sci. Rep. 10, 16415. doi: 10.1038/s41598-020-73607-x
Nieminen, T. T., Säde, E., Endo, A., Johansson, P., and Björkroth, J. (2014). “The Family Leuconostocaceae,” in The Prokaryotes, 4th Edn, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin, Heidelberg: Springer), 215–240. doi: 10.1007/978-3-642-30120-9_208
Nye, T. M., Li,ò, P., and Gilks, W. R. (2006). A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics 22, 117–119. doi: 10.1093/bioinformatics/bti720
Ogier, J. C., Casalta, E., Farrokh, C., and Saihi, A. (2008). Safety assessment of dairy microorganisms: the Leuconostoc genus, Int. J. Food Microbiol. 126, 286–290, doi: 10.1016/j.ijfoodmicro.2007.08.012
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palmer, M., Steenkamp, E. T., Blom, J., Hedlund, B. P., and Venter, S. N. (2020). All ANIs are not created equal: implications for prokaryotic species boundaries and integration of ANIs into polyphasic taxonomy. Int. J. Syst. Evol. Microbiol. 70, 2937–2948. doi: 10.1099/ijsem.0.004124
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Pearce, M. E., Langridge, G. C., Lauer, A. C., Grant, K., Maiden, M., and Chattaway, M. A. (2021). An evaluation of the species and subspecies of the genus Salmonella with whole genome sequence data: proposal of type strains and epithets for novel S. enterica subspecies VII, VIII, IX, X and XI. Genomics 113, 3152–3162. doi: 10.1016/j.ygeno.2021.07.003
Rahkila, R., De Bruyne, K., Johansson, P., Vandamme, P., and Björkroth, J. (2014). Reclassification of Leuconostoc gasicomitatum as Leuconostoc gelidum subsp. gasicomitatum comb. nov., description of Leuconostoc gelidum subsp. aenigmaticum subsp. nov., designation of Leuconostoc gelidum subsp. gelidum subsp. nov. and emended description of Leuconostoc gelidum. Int. J. Syst. Evol. Microbiol. 64, 1290–1295. doi: 10.1099/ijs.0.058263-0
Raimondi, S., Nappi, M. R., Sirangelo, T. M., Leonardi, A., Amaretti, A., Ulrici, A., et al. (2018). Bacterial community of industrial raw sausage packaged in modified atmosphere throughout the shelf life. Int. J. Food Microbiol. 280, 78–86. doi: 10.1016/j.ijfoodmicro.2018.04.041
Ramasamy, D., Mishra, A. K., Lagier, J. C., Padhmanabhan, R., Rossi, M., Sentausa, E., et al. (2014). A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species. Int. J. Syst. Evol. Microbiol. 64, 384–391. doi: 10.1099/ijs.0.057091-0
Ricciardi, A., Storti, L. V., Zotta, T., Felis, G. E., and Parente, E. (2020). Analysis of rpoB polymorphism and PCR-based approaches for the identification of Leuconostoc mesenteroides at the species and subspecies level. Int. J. Food. Microbiol. 318, 108474. doi: 10.1016/j.ijfoodmicro.2019.108474
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodriguez-R, L. M., and Konstantinidis, K. T. (2014). Bypassing cultivation to identify bacterial species. Microbe 9, 111–118.
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J. Preprints 4, e1900ve1901. doi: 10.7287/peerj.preprints.1900v1
Schliep, K. P. (2011). Phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shin, S. Y., and Han, N. S. (2015). “Leuconostoc spp. as starters and their beneficial roles in fermented foods,” in Beneficial Microorganisms in Food and Nutraceuticals. Microbiology Monographs, Vol. 27, ed M. T. Liong (Cham: Springer), pp. 111–132. doi: 10.1007/978-3-319-23177-8_5
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Smith, M. R. (2020). Information theoretic generalized Robinson-Foulds metrics for comparing phylogenetic trees. Bioinformatics 36, 5007–5013. doi: 10.1093/bioinformatics/btaa614
Stackebrandt, E., and Ebers, J. (2006). Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today. 33, 152–155
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in Bacteriology. Int. J. Syst. Bacteriol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steinkraus, K. H. (2002). Fermentations in world food processing. Compr. Rev. Food Sci. Food Saf. 1, 23–32. doi: 10.1111/j.1541-4337.2002.tb00004.x
Stott, C. M., and Bobay, L. M. (2020). Impact of homologous recombination on core genome phylogenies. BMC Genom. 21, 829. doi: 10.1186/s12864-020-07262-x
Vancanneyt, M., Zamfir, M., De Wachter, M., Cleenwerck, I., Hoste, B., Rossi, F., et al. (2006). Reclassification of Leuconostoc argentinum as a later synonym of Leuconostoc lactis. Int. J. Syst. Evol. Microbiol. 56, 213–216. doi: 10.1099/ijs.0.63898-0
Vandamme, P., Pot, B., Gillis, M., de Vos, P., Kersters, K., and Swings, J. (1996). Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol. Rev. 60, 407–438. doi: 10.1128/mr.60.2.407-438.1996
Vedamuthu, E. R. (1994). The dairy leuconostoc: use in dairy products. J. Dairy Sci. 77, 2725–2737. doi: 10.3168/jds.S0022-0302(94)77215-5
Venegas-Ortega, M. G., Flores-Gallegos, A. C., Martínez-Hernández, J. L., Aguilar, C. N., and Nevárez-Moorillón, G. V. (2019). Production of bioactive peptides from lactic acid bacteria: a sustainable approach for healthier foods. Compr. Rev. Food Sc. Food Saf. 18, 1039–1051. doi: 10.1111/1541-4337.12455
Whelan, S., and Morrison, D. A. (2017). Inferring trees. Methods Mol. Biol. 1525, 349–377. doi: 10.1007/978-1-4939-6622-6_14
Woese, C. R., and Fox, G. E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc. Natl Acad. Sci. U. S. A. 74, 5088–5090. doi: 10.1073/pnas.74.11.5088
Wu, Y., and Gu, C. T. (2021a). Leuconostoc falkenbergense sp. nov., isolated from a lactic culture, fermentating string beans and traditional yogurt. Int. J. Syst. Evol. Microbiol. 71, 004602. doi: 10.1099/ijsem.0.004602
Wu, Y., and Gu, C. T. (2021b). Rejection of the reclassification of Leuconostoc gasicomitatum as Leuconostoc gelidum subsp. gasicomitatum based on whole genome analysis. Int J Syst Evol Microbiol. 71, 005027. doi: 10.1099/ijsem.0.005027
Yu, A. O., Leveau, J. H. J., and Marco, M. L. (2020). Abundance, diversity and plant-specific adaptations of plant-associated lactic acid bacteria. Environ. Microbiol. Rep. 12, 16–29. doi: 10.1111/1758-2229.12794
Zheng, J., Wittouck, S., Salvetti, E., Franz, C. M. A. P., Harris, H. M. B., Mattarelli, P., et al. (2020). A taxonomic note on the genus Lactobacillus: description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 70, 2782–2858. doi: 10.1099/ijsem.0.004107
Keywords: Leuconostoc, phylogenomics, average nucletide identity (ANI), 16S rRNA gene, cosmeceutics, biopreservatives
Citation: Raimondi S, Candeliere F, Amaretti A, Costa S, Vertuani S, Spampinato G and Rossi M (2022) Phylogenomic analysis of the genus Leuconostoc. Front. Microbiol. 13:897656. doi: 10.3389/fmicb.2022.897656
Received: 16 March 2022; Accepted: 28 June 2022;
Published: 25 July 2022.
Edited by:
Iain Sutcliffe, Northumbria University, United KingdomReviewed by:
Margarita Gomila, University of the Balearic Islands, SpainGiovanna E. Felis, University of Verona, Italy
Radhey S. Gupta, McMaster University, Canada
Copyright © 2022 Raimondi, Candeliere, Amaretti, Costa, Vertuani, Spampinato and Rossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maddalena Rossi, bWFkZGFsZW5hLnJvc3NpQHVuaW1vcmUuaXQ=
†These authors have contributed equally to this work