 Gabriela N. Tenea*
Gabriela N. Tenea* Evelyn Angamarca
Evelyn Angamarca- Biofood and Nutraceutics Research and Development Group, Faculty of Engineering in Agricultural and Environmental Sciences, Universidad Técnica del Norte, Ibarra, Ecuador
Introduction: Foodborne bacterial infections remain a critical global health challenge, exacerbated by the increasing prevalence of antimicrobial resistance (AMR). Misuse of antimicrobials in agriculture and inadequate food handling practices facilitate the spread of resistant bacteria across the human–animal–environment interface, a central concern of the One Health approach. Comprehensive understanding of microbial threats in food systems is vital for effective risk assessment and control. In this study, we report the first complete genome of a multidrug-resistant Staphylococcus xylosus strain, FFCShyA4, isolated from commercially sold avocados.
Methods: Whole-genome sequencing and comparative genomics were employed for taxonomic classification and phylogenetic analysis. In silico tools identified antibiotic resistance genes (ARGs), virulence factors, mobile genetic elements (MGEs), CRISPR loci, genomic islands, and biosynthetic gene clusters (BGCs). In vitro assays assessed hemolysis, gelatinase activity, antibiotic susceptibility, and PCR-based gene detection.
Results: The FFCShyA4 genome spans 3.09 Mb with a 32.63% GC content and includes a 32 kb plasmid. It shares 99.97% average nucleotide identity with S. xylosus NBRC 109770 yet displays extensive structural rearrangements indicative of niche-specific adaptation. The genome encodes 2,720 protein-coding genes, including ARGs for β-lactams, macrolides, fluoroquinolones, tetracyclines, and lincosamides. The presence of 133 MGEs, CRISPR systems, an intact prophage, and 138 genomic islands reflects a strong potential for horizontal gene transfer. Virulence profiling identified 121 genes across 34 families, with a predicted human pathogenicity of 98.2%. BGCs linked to bacteriocins, siderophores, and staphylopine biosynthesis were also detected. In vitro assays confirmed multidrug resistance and pathogenicity.
Conclusion: These results emphasize the critical need for integrated One Health surveillance of antimicrobial resistance within food production and commercial environments to facilitate early detection and reduce dissemination of resistance determinants across interconnected human, animal, and environmental reservoirs.
1 Introduction
Staphylococcus xylosus is a Gram-positive, coagulase-negative bacterium that primarily acts as a commensal on the skin and mucous membranes of mammals. It also adapts well to diverse environmental niches, such as soil, water, and surfaces linked to animal husbandry and food processing environments (Schiffer et al., 2023). Highly adaptable, S. xylosus can persist in challenging conditions, such as biofilms, high-salt environments, and low oxygen levels (Condas et al., 2017). It plays a significant role in food fermentation, contributing to the production of fermented meat products and specific cheeses by enhancing flavor development, stabilizing color, and ensuring safety through enzymatic activity and the production of antimicrobial metabolites (Leroy et al., 2017). Although primarily considered non-pathogenic, it has been associated with opportunistic infections like bovine mastitis and is recognized as a reservoir for antibiotic resistance genes and virulence factors, raising concerns about HGT to more pathogenic species such as S. aureus (Condas et al., 2017). Furthermore, growing evidence underscores the potential of coagulase-negative staphylococci (CNS) to act as reservoirs for virulence-associated factors (Marincola et al., 2021). Besides, S. xylosus is a rare colonizer of human skin, but it is more frequently found on the skin of individuals who have regular contact with animals (Battaglia and Garrett-Sinha, 2023). When human skin is transplanted onto nude mice, S. xylosus can be isolated from the grafts, though it colonizes a smaller proportion of grafts compared to the host mouse skin. This suggests that certain characteristics of murine skin support the colonization of S. xylosus, whereas human skin traits may prevent it (Kearney et al., 1982). While the most common staphylococcal species on human skin are part of the Epidermidis–Aureus group, S. xylosus, which is less prevalent, falls within the Saprophyticus sub-group (Lamers et al., 2012). Early genomic and in situ analyses of S. xylosus in a meat model reveal its adaptation to meat substrates, supported by genes enabling the utilization of diverse carbon, energy, and nitrogen sources (Dordet-Frisoni et al., 2007; Leroy et al., 2017). Its genome encodes pathways for osmotic, oxidative/nitrosative, and acidic stress responses, nitrate reductase activity for cured meat coloration, and enzymes for pyruvate and amino acid catabolism, contributing to odorous compounds (Labrie et al., 2014). In another study, a multidrug-resistant strain of S. xylosus NM36 was isolated from a cow with chronic mastitis (Al-Tameemi et al., 2023). This strain exhibited resistance to methicillin, ampicillin, cefoxitin, oxacillin, and tetracycline but remained susceptible to vancomycin, alongside a strong biofilm-forming capacity (Al-Tameemi et al., 2023).
While existing literature emphasizes the ecological specialization of S. xylosus in animal-related environments, a recent study revealed the presence of S. xylosus in fermented soybean foods in Korea (Kong et al., 2022). Most strains were susceptible to multiple antibiotics, but 23 showed potential acquired resistance to erythromycin, lincomycin, and tetracycline. Recently, we detected several Staphylococcus species on the exocarp of avocado fruits sold in open markets in Ecuador (Angamarca et al., 2023). Among these, S. xylosus FFCShyA4 exhibited antibiotic resistance. The presence of S. xylosus on the fruit exocarp can be explained by its ability to thrive in diverse environments, including plant surfaces, soil, and food-processing settings. This bacterium is known for its resilience, forming biofilms and utilizing various carbohydrates, which may facilitate its survival on the fruit surface. Thus, the presence of antibiotic-resistant bacteria in ready to eat fruits raises public health concerns (Rahman et al., 2021). Open markets are environments where both human and animal interactions are common, often facilitating the spread of bacteria between different sources (Tenea et al., 2023). However, it highlights the potential for these bacteria to spread from surfaces contaminated by animal products or human handling (Angamarca et al., 2023). These bacteria can then be transferred to consumers through direct contact with the food or improper handling during preparation. This scenario underscores the importance of proper hygiene and food safety measures to limit the spread of antibiotic-resistant bacteria and prevent potential health risks to consumers. To date, there are no reports on the genome of S. xylosus isolated from sources other than animals. In this study, we performed a comprehensive genomic analysis of the FFCShyA4 strain. The isolate was taxonomically classified, and its phylogenetic relationship with closely related strains was established. Functional genome mining was conducted to predict antibiotic resistance genes, virulence factors, CRISPR systems, MGE, and secondary metabolite biosynthesis. Additionally, in vitro assays were carried out to assess hemolytic activity, gelatinase production, and susceptibility to various antibiotics. These findings enhance our understanding of the genomic diversity, evolutionary dynamics, and public health risks associated with S. xylosus. By highlighting its potential role in foodborne contamination and cross-contamination, this study supports the development of targeted interventions to strengthen food safety systems and protect public health.
2 Materials and methods
2.1 Bacterial strain cultivation and DNA extraction
S. xylosus FFCShyA4 was isolated from the exocarp of avocado (Persea nubigena var. guatemalensis) as described by Angamarca et al. (2023). The strain was cultured in Brain Heart Infusion (BHI) broth (Merck Millipore, MA, USA). Genomic DNA and total RNA were extracted using the Illumina DNA Prep Kit (Illumina Inc., San Diego, CA, USA), following quality control procedures. DNA concentration was measured using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, USA), and libraries were prepared according to the manufacturer’s protocol.
2.2 De novo assembly and workflow sequencing
Genome sequencing was conducted using the Illumina HiSeq X Ten platform (Macrogen Inc., Seoul, Korea). Libraries were prepared by fragmenting DNA or cDNA samples and ligating 5′ and 3′ adapters, as per the manufacturer’s protocol. Adapter-ligated fragments were PCR-amplified, gel-purified, and loaded onto a flow cell for cluster generation via bridge amplification or ExAmp cluster amplification on patterned flow cells. Sequencing was performed using Illumina SBS technology, a reversible terminator-based method that minimizes incorporation bias and reduces sequence errors, even in repetitive or homopolymer regions, by ensuring natural competition among all four dNTPs. Raw sequencing data were analyzed, and FastQC (v0.11.5) was used to assess quality, base count, GC content, and other metrics. De novo assembly was carried out with SPAdes v3.15.5 using multiple k-mers, and the best assembly was chosen based on parameters like contig number, total contig bases, and N50 (Bankevich et al., 2012). Assembly completeness was evaluated using BUSCO v5.1.3 against the bacterial or eukaryotic database, based on near-universal single-copy orthologs. BLAST analysis was used to identify the species corresponding to each scaffold.
2.3 Typing and species relatedness
ANI analysis was performed by aligning the reference sequence (Taxon ID: GCF_007992815.1_S. xylosus_NBRC_109,770) with contig 1 using BLASTN (custom assay project, Macrogen Inc., Seoul, Korea). A circular genome map was generated via the PROKSEE server1 (Grant et al., 2023). Genome sequence data underwent further analysis through the Type (Strain) Genome Server (TYGS) for a comprehensive genome-based taxonomic study (Meier-Kolthoff and Göker, 2019). The closest related strain genomes were identified using MASH distance comparisons, with precise distances calculated by the Genome BLAST Distance Phylogeny (GBDP) method under the “coverage” algorithm (Meier-Kolthoff et al., 2013). Phylogroups were classified using in silico Clermont Phylotyper EzClermont (Waters et al., 2020). Synteny analysis was conducted via multiple genome alignment using Mauve with default parameters (minimum Localized Collinear Blocks (LCBs) of 1,000, island size of 50, backbone size of 50, and maximum gap of 50). The draft genome of FFCShyA4 was aligned relative to the S. xylosus CCM2738 and S. xylosus NBRC 109770, reference genomes—originally isolated from human skin (Darling et al., 2004).
2.4 General genome features, gene prediction, and functional annotation
The identification of coding sequences (CDS), rRNA, tRNA/tmRNA, signal leader peptides, and noncoding RNA was conducted following the methodology outlined by Molina et al. (2024). Gene annotation was performed using Prokka v1.14.6 (Seemann, 2014), while plasmid annotation was carried out with pLannotate (McGuffie and Barrick, 2021). Functional annotation was achieved using InterProScan v5.0 (Jones et al., 2014), which evaluates sequence similarity at the family level and matches sequences against member databases within InterPro, including Pfam, the Conserved Domain Database (CDD), and TIGRFAM, a curated database focusing on prokaryotic protein families (Selengut et al., 2007). Furthermore, the EggNOG database (Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups) was utilized for additional annotations (Huerta-Cepas et al., 2019), with psi-BLAST applied to align predicted protein sequences to the EggNOG database.
2.5 In silico genome analysis
2.5.1 Prediction of CRISPR sequences, prophage, antibiotic resistant genes (ARGs), virulence factors (VFs), genomic island (GIs), genetic mobile elements (MGEs), and pathogenicity
To identify CRISPR, Cas sequences, and prophage regions in bacterial genomes, CRISPRCasFinder (Crispr-Cas++1.1.2) and the PHAge Search Tool Enhanced Release (PHASTER) were employed (Arndt et al., 2016). The CARD (Comprehensive Antibiotic Resistance Database) tool and the RGI (Resistance Gene Identifier) were used to detect ARGs, applying Perfect hit, Rigorous hit, and Loose hit criteria (Jia et al., 2017; Arndt et al., 2016). To identify acquired ARGs, the ResFinder 4.1 server was utilized with a 90% ID threshold and a minimum length of 60% (Zankari et al., 2012), alongside the detection of chromosomal mutations (Bortolaia et al., 2020). Putative virulence factors were predicted using the VFDB database (Liu et al., 2019). Genomic islands were predicted using the IslandViewer 4 server (Bertelli et al., 2017) and bacterial pathogenicity was assessed via the PathogenFinder web server (Cosentino et al., 2013). MGEs were predicted using mobileOG-db tool (Brown et al., 2022) incorporated in the PROKSEE server (see text footnote 1).
2.5.2 Prediction of bacteriocin encoding genes clusters and secondary metabolites
For the detection of biosynthetic gene clusters (BGCs) of antimicrobial compounds, the web tool BAGEL 4 (van Heel et al., 2018) was used. The secondary metabolites prediction was assessed using antiSMASH version 4 webtool (Blin et al., 2023).
2.6 In vitro analysis
2.6.1 Hemolysis and gelatinase production
Hemolysin production in the FFCShyA4 isolate was assessed by inoculating the strain onto 5% human blood agar plates, followed by incubation at 37°C for 24 h, as described by Tabasi et al. (2015). Hemolytic activity was evaluated based on the presence of partial or complete zones of erythrocyte lysis surrounding the bacterial colonies. Gelatinase production was determined using gelatin nutrient agar plates, following the protocol established by Mittal et al. (2014). An overnight culture of the isolate was inoculated onto the plates, and after visible growth, the medium was treated with a mercuric chloride solution. A clearing zone surrounding the bacterial colonies, resulting from gelatin hydrolysis, indicated a positive gelatinase activity. S. aureus ATCC43300 and S. aureus ATCC1026 were included as controls in both assays.
2.6.2 Antibiotic susceptibility evaluation
Antibiotic susceptibility was determined using the Muller-Hilton (MH) agar disk diffusion procedure, and according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (Clinical and Laboratory Standards Institute, 2021). Briefly, 100 μL of inoculum (107–108 CFU/mL) was streaked onto MH plates. The commercial antibiotic disks (Merck, USA) chosen are listed in Supplementary Table S1A. The disks were plated on MH agar plates, and incubated at 37°C for 48 h. The diameter of each clear zone was measured in millimeters by scanning the plates with a microplate reader (SCAN500, Interscience, Fr). As controls, S. aureus ATCC43300 and S. aureus ATCC1026 were included in the experiments. The microbiological breakpoints reported by Clinical and Laboratory Standards Institute (2021) and the European Committee on Antimicrobial Susceptibility Testing (2021) standards were used to categorize Staphylococcus as susceptible (S), intermediate (I), or resistant (R) (Supplementary Table S1B). The MAR index was also determined as a ration between the total number of antibiotics to which the isolate is resistant and the total number of antibiotics tested.
2.6.3 Virulence and antibiotic genes detection by PCR
Primers targeting virulence genes thermonuclease (nuc), intracellular adhesin (icaA), putative adhesin (sdrE), hemolysin (hlg), meticillinA (mecA), and meticillinC (mecC) (Stegger et al., 2012) were prepared at a concentration of 0.3 μM (Supplementary Table S2). Genomic DNA was extracted using the Wizard® Genomic DNA Purification Kit (#1120, Promega, USA). DNA concentration and purity were measured using a NanoDrop™ spectrophotometer (Thermo Fisher Scientific, USA) at absorbance wavelengths of 230, 260, and 280 nm. PCR amplifications were carried out in 25 μL reaction volumes, each containing 2X GoTaq® Green Master Mix (#7132, Promega, USA). Reactions were performed using a Genemax Thermal Cycler (IQM, Oslo, Norway). The amplification conditions are shown in Supplementary Table S2. PCR products were separated on 1% agarose gels in 1X Tris-Borate-EDTA (TBE, pH 8.0) buffer (Sigma-Aldrich, USA). The gels were stained with TBE buffer containing 0.5 μg/mL ethidium bromide. Results were recorded as positive or negative based on the presence or absence of the expected amplicons.
3 Results and discussion
3.1 General genome characteristics, taxonomy, and phylo-genetic relationship
For the FFCShyA4 strain, a total of 2,972,650,628 bases were generated, producing 19,686,428 pair-end reads. The total number of contigs was 2,856,035 bp. The GC content was calculated at 32.63%, with a Q30 value of 92.75%. The estimated genome size was 3,093,530 bp. In addition, the strain harbored one plasmid of 32,035 bp. The general genome features of FFCShyA4 are shown in Supplementary Table S3. The circular genome and plasmid maps are shown in Figures 1A,B. Following assembly of the complete or draft genome, BLAST analysis was conducted to identify species-level similarities for each scaffold. Genus-level classification revealed that 90.91% of the scaffolds matched Staphylococcus, while 9.09% aligned with Mammaliicoccus. ANI analysis revealed a 99.97% nucleotide identity and 94.19% alignment coverage with S. xylosus NBRC109770 (human skin) (Lamers et al., 2012). The five most closely related genomes identified based on ANI results are presented in Supplementary Table S4. Hierarchical clustering of ANI data was visualized as two-dimensional dendrograms, using simple linkage for both ANI percentage identity and ANI alignment coverage (Supplementary Figures S1A,B). BLASTN analysis revealed that plasmid pFFCShyA4 shares 99.26% sequence identity with plasmid pDMSX03-1 from S. xylosus strain DMSX03. Phylogenetic analysis identified S. xylosus strain CCM2738 and S. pseudoxylosus strain S044009 as the closest related strains as shown by TYGS analysis (Supplementary Figure S2). The Mauve alignment of FFCShyA4, and the two references S. xylosus NBRC109770 and S. xylosus CCM2738 reveals a mix of conserved and variable regions, highlighting both evolutionary stability and divergence (Figure 2). The conserved regions, mainly found at the genomes terminal ends and encompassing essential housekeeping genes, suggest a high degree of preservation among the three strains (González et al., 2010). However, extensive genomic rearrangements, including inversions and translocations, are observed between 1,000,000–1,800,000 bp, suggesting evolutionary divergence (Shukla et al., 2009). Notable insertion/deletion (indel) events occur within 600,000–1,000,000 bp and 2,200,000–2,600,000 bp, with FFCShyA4 displaying unique genomic islands absent in the reference strains, potentially linked to adaptation in food-related environments (Vermassen et al., 2016). Additionally, the high density of intersecting lines observed in the 1,200,000–1,600,000 bp region indicates potential hotspots for horizontal gene transfer (HGT), likely involving MGEs (Ghaly et al., 2024). These regions frequently harbor genes linked to antibiotic resistance, responses to metal and oxidative stress (e.g., copper and arsenic resistance genes, superoxide dismutase), as well as carbohydrate metabolism and fermentation. The presence of HGT-associated markers, including transposases and prophage-related genes, underscores the strain’s metabolic versatility and genomic plasticity (Coll et al., 2025). However, the alignment highlights conserved core regions while also identifying unique genomic segments in FFCShyA4 that may contribute to its distinct functional capabilities, including potential virulence and resistance traits (Zhou et al., 2020). These results support the hypothesis that FFCShyA4 shares a common lineage with NBRC 109770 and CCM2738 but has undergone genetic modifications that could enhance its survival in food-associated environments or human-related settings, warranting further investigation into its role in public health and food safety (Vermassen et al., 2014). These findings suggest that the FFCShyA4 strain may possess unique traits enhancing its environmental adaptability and resilience, particularly in response to stressful or competitive conditions such fruit environment.
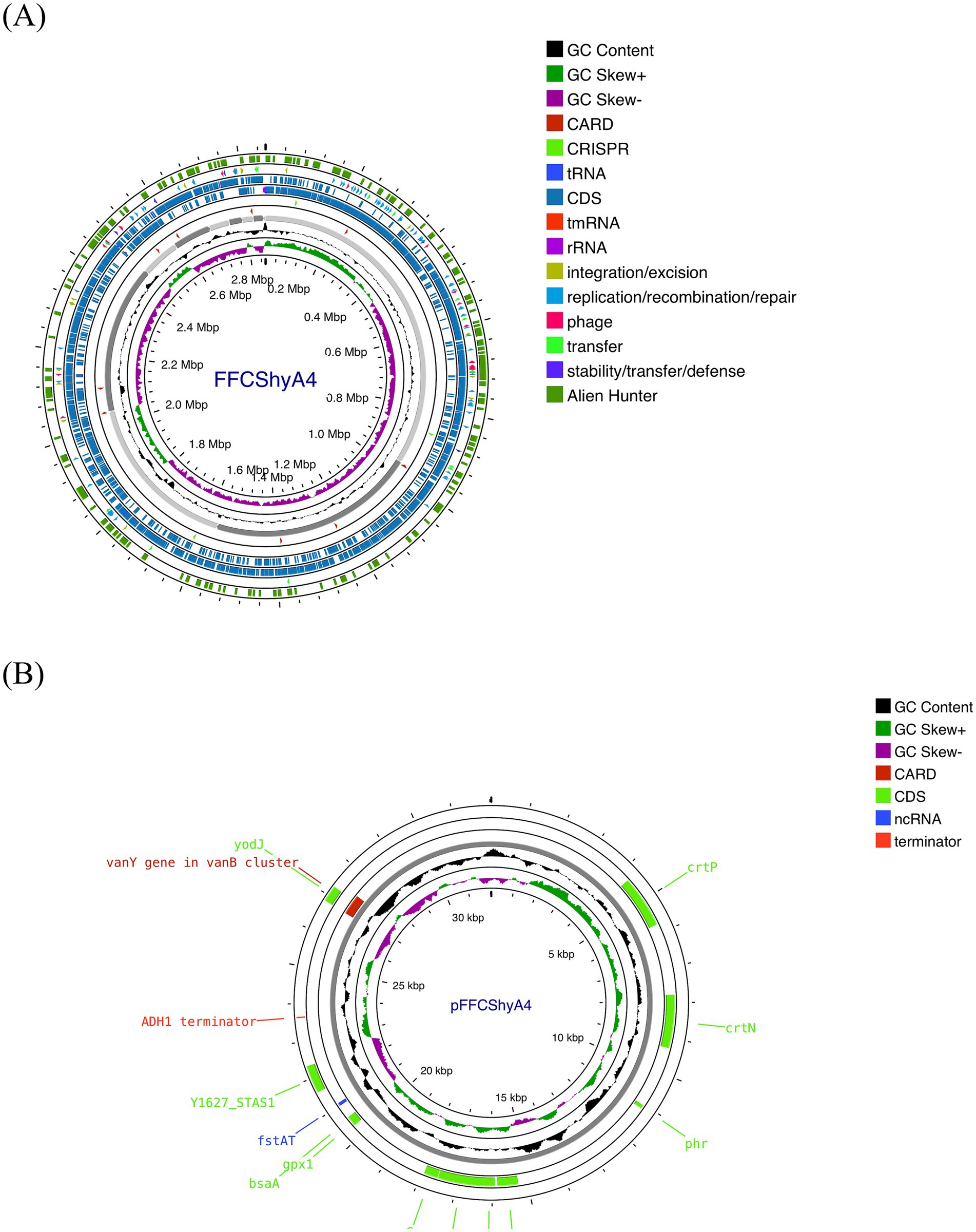
Figure 1. (A) Genome map of S. xylosus FFCShyA4 predicted with the PROKSEE viewer (Accessed on January 20, 2025). (B) Plasmid map as predicted by pLannotate. The contents are arranged in feature rings. Starting with the outermost ring: ring 1, mobile genetic elements (MGE) annotation with Alien Hunter predicting HGT (Horizontal Genetic Transfer) events (in red color); ring 2, MGE annotation with Mobile OG DB marking the hsdR gene involved in stability/transfer/defense; rings 3–4 show the CDS (protein-coding genes) with Prokka annotation (blue color), tRNA, rRNA, and tmRNA are marked; ring 5 displays the CRISPR-Cas annotation; ring 6, CARD annotation, ring 7 GC content plot (black); ring 8 displays G/C skew information in the (+) strand (green color) and (−) strand (purple color).
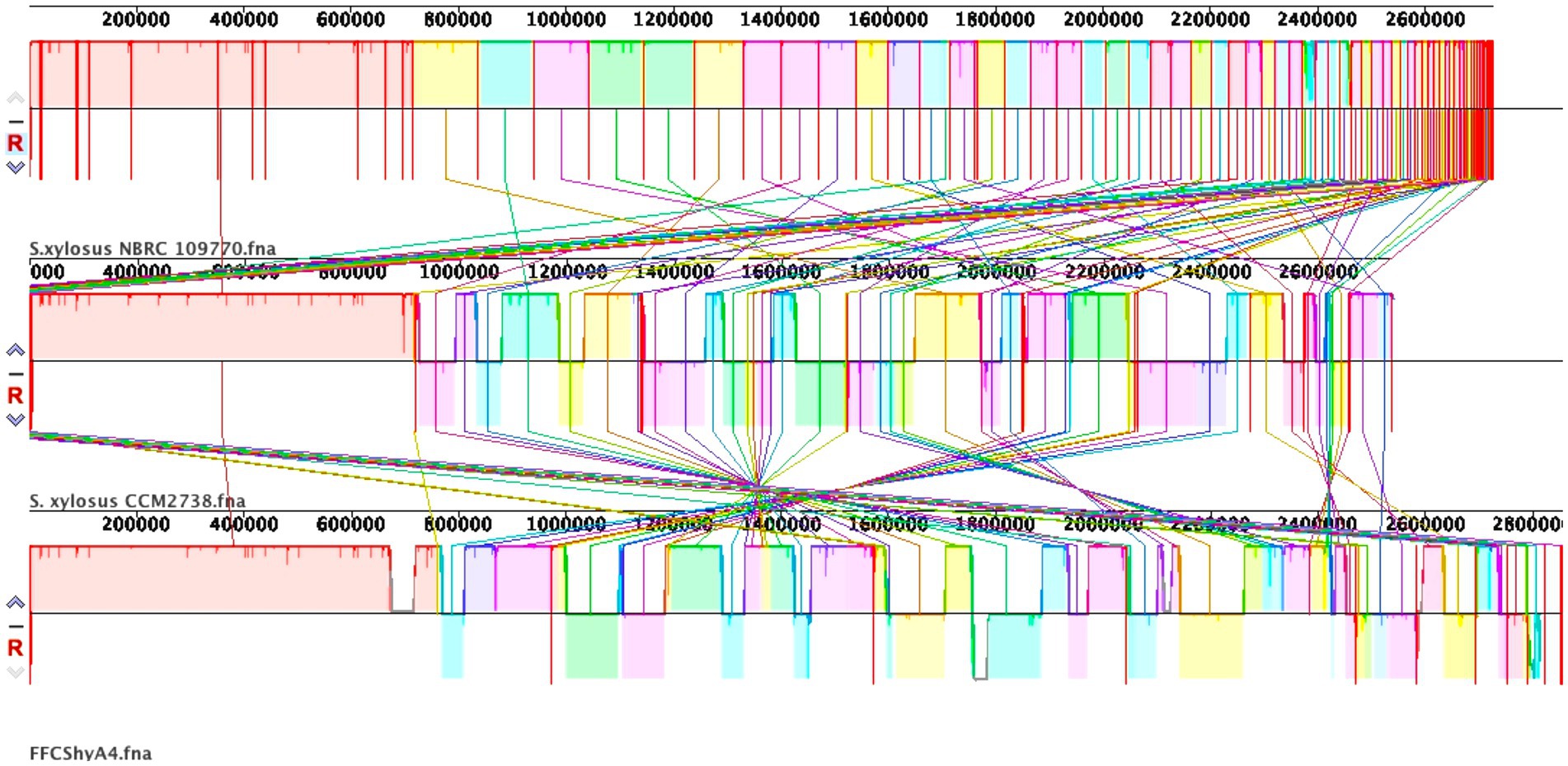
Figure 2. Whole-genome alignment of FFCShyA4 and references strains S. xylosus CCM2738 and S. xylosus NBRC109770 using Mauve Contig Mover. Colored blocks represent local collinear blocks (LCBs) with highly conserved regions between the genomes. Crossing lines indicate genomic rearrangements, while the absence of LCBs in specific regions suggests strain-specific sequences.
3.2 Gene prediction and functional annotation
The genome comprises 2,720 genes, including 2,665 coding sequences (CDS), 47 tRNAs, 7 rRNAs, and 1 tmRNA (Supplementary Table S3). Gene locations were predicted using Prokka, while BLAST analysis was performed to determine the functions and identify the assembled sequences by comparing them against nucleotide and protein sequence databases. The predicted genes were subsequently aligned with multiple databases to obtain functional annotations using specific aligners, as summarized in Table 1. Among the total proteins, 2,576 matched EggNOG DB proteins (2,538 Single EggNOG and 38 Multi EggNOG proteins) and 89 proteins with no hit (Figure 3A). Similarly, 2,476 proteins matched COG DB (2,210 Single COG category and 266 Multi COG category) while 189 showed no hit (Figure 3B). The number of genes associated with KEGG (2,580 genes) functional annotation categories is shown in Supplementary Figure S3. The plasmid annotation (Figure 1B) includes several key features: arsB (100% identity) from S. xylosus, involved in arsenical resistance and likely forming the channel of an arsenite pump; arsR (99% identity) from S. aureus, a transcriptional repressor for the ars operon, which regulates its own expression and is derepressed by oxyanions of arsenic, antimony, and bismuth in the +III oxidation state, as well as arsenate (As(V)); arsC (95.4% identity) from S. aureus strain N315, encoding an enzyme that reduces arsenate [As(V)] to arsenite [As(III)]; bin3 (83.2% identity) from S. aureus, a potential DNA invertase; crtN (66.5% identity) and crtP (65% identity) from S. aureus, involved in staphyloxanthin biosynthesis, a carotenoid contributing to virulence by protecting against oxidative stress; and ADH1 terminator (100% identity), a transcription terminator for the S. cerevisiae alcohol dehydrogenase 1 (ADH1) gene. These features highlight roles in resistance, enzymatic activity, and oxidative stress protection. ADH1 is not naturally found in Staphylococcus species (Makhlin et al., 2007). In the context of plasmid annotation, the ADH1 terminator is used as a transcription terminator, and possible serves as a genetic element for controlling gene expression.
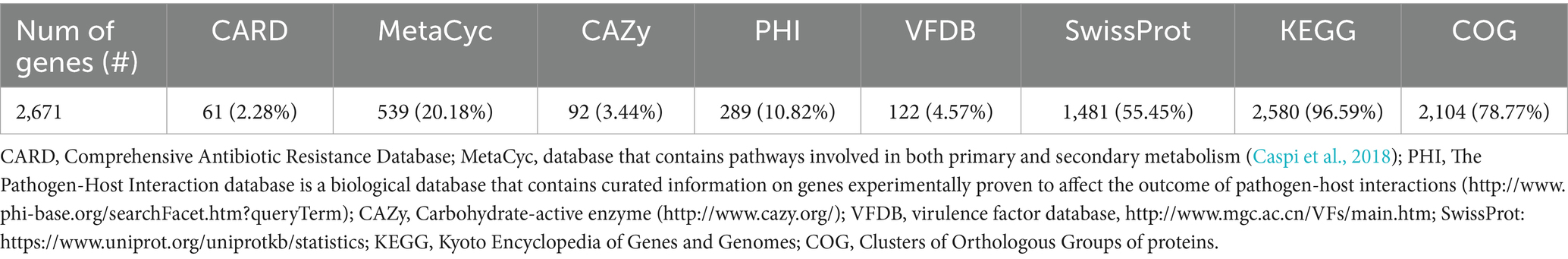
Table 1. Gene annotation summary.
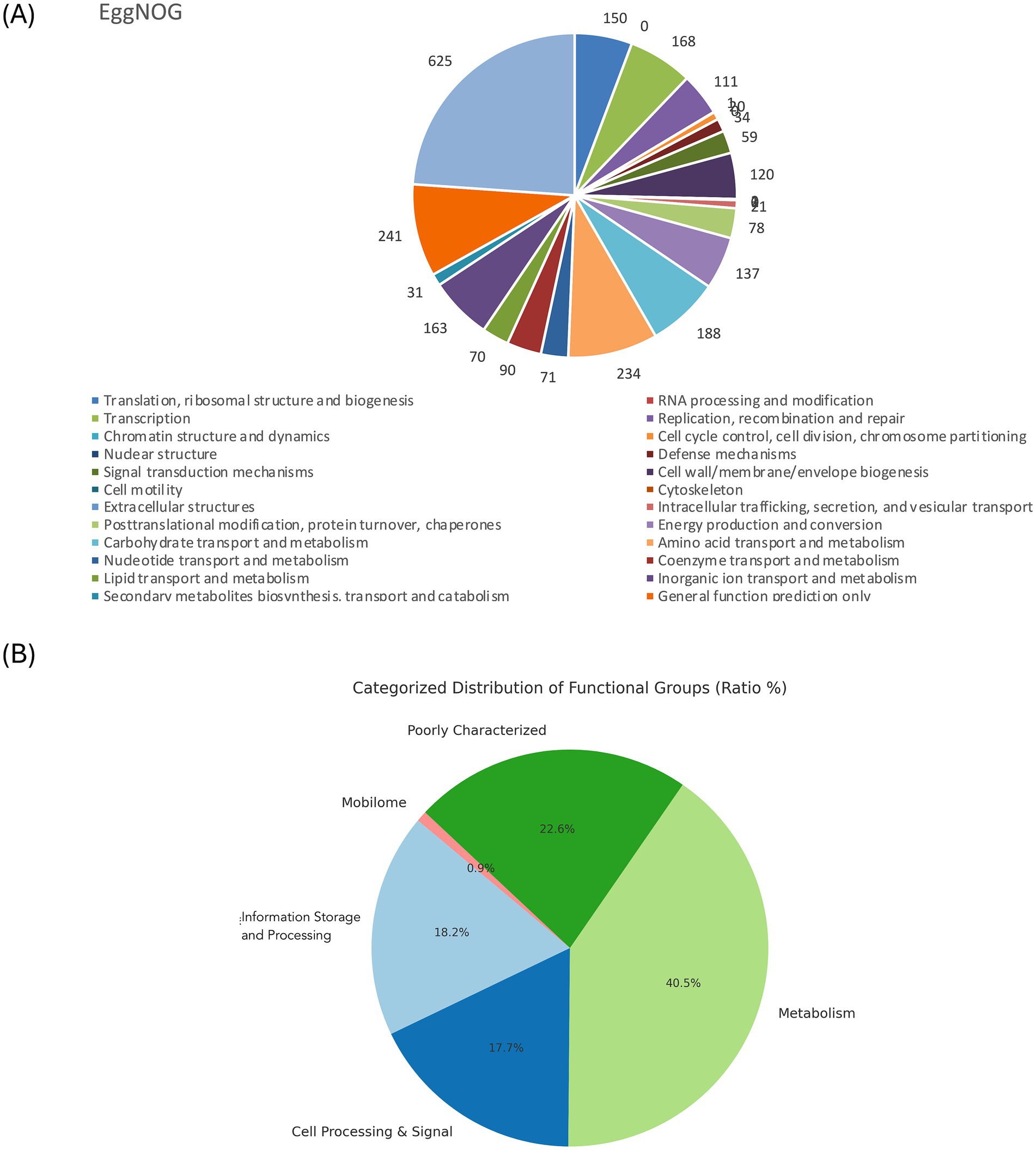
Figure 3. (A). EggNOG category distribution of functional annotation result. (B). Pie chart showing the COG distribution of the overall categories.
3.3 Prediction of CRISPR sequences, prophage, ARGs, VFs, GIs, MGEs, and pathogenicity
Three CRISPR sequences were identified in the FFCShyA4 genome, located at positions 79,658 to 79,743, 868,840 to 868,928, and 47,184 to 47,284, respectively. Each sequence includes a short spacer sequence flanked by degenerate repeats (consensus DRs), with repeat conservation of 96.68, 96.65, and 100%, and spacer conservation of 100%. No Cas genes were found. One intact prophage was found in the genome (Supplementary Table S5). The detection of Phage Staphy_47 in the FFCShyA4 genome is significant as it highlights the potential influence of prophages on the genetic and functional diversity of the host bacterium. This phage can contribute to HGT, providing the host with adaptive advantages, including virulence factors, resistance genes, or enhanced survival mechanisms under stress conditions (Deghorain and Van Melderen, 2012). Additionally, a total of 138 GIs of 126,044 bp were predicted with IslandViewer using as a reference genome of non-coagulase S. xylosus strain DMSX03 isolated from fermented soybean (Heo et al., 2021). Many hypothetical proteins and enzymes, such as proteases, and virulence factors (mazE, PemK/MazF), were identified in the GIs. These genomic regions are crucial for the rapid evolution, diversification, and adaptation of Staphylococcus due to their frequent rearrangements, excisions, transfers, and the acquisition of additional DNA (Supplementary Table S6). Among the 121 genes identified from the virulence database, the thermonuclease gene (nuc), the intercellular adhesion protein C involved in polysaccharide intercellular adhesin (PIA) synthesis (icaC), and the ATP-dependent Clp protease proteolytic subunit (clpP) were detected, although they exhibited imperfect matches (identity < 85%, with the input sequence length shorter than the full virulence gene length, and 60% alignment coverage). In addition, the elongation factor Tu (tufA) and phosphopyruvate hydratase (eno) were annotated showing 74 and 79.8% sequence identity, respectively. The detected virulence factors are summarized in Supplementary Table S7. Interesting, the presence of nuc but not icaC, sdrE, and hlg genes was confirmed by PCR (Supplementary Figure S4), as these genes typically are associated with S. aureus. The detection of the nuc gene in CNS strain may be attributed to HGT, genomic variability within CNS, or the presence of a divergent gene variant with partial homology, highlighting the potential for genetic exchange and adaptation in diverse environments (Canning et al., 2020). Besides, several virulence factors such as surface-displayed alpha-enolase (eno), adaptor protein MecA (mecA), type II toxin-antitoxin system PemK/MazF family toxin, a virulence associated protein E (virE), were annotated with EggNOG. Moreover, within FFCShyA4 genome, a total of 133 MGEs were identified, including 17 related to insertion/excision, 74 associated with replication/recombination/repair, 21 linked to phages, 6 involved in stability/transfer/defense, and 15 related to transfer. Analysis of the 34 matched pathogenic families (accounting for 1.28% of the proteome) indicated that the FFCShyA4 isolate is a human pathogen with a likelihood of 0.982. This finding was further supported by the presence of an inhibition zone on blood agar, indicative of hemolytic activity (Supplementary Figure S5). Additionally, the gelatinase test for FFCShyA4 was positive, consistent with previous research showing that gelatinase is a common virulence factor among Staphylococcus spp. (Bertelloni et al., 2021). Using CARD protein IDs, 61 predicted genes were classified by drug class, resistance mechanism, and AMR gene family, with the distribution of genes shown in Supplementary Figure S6. Besides, 11 ARGs including vancomycin (vanY gene in vanA cluster), lincosamide (salD), and fosfomycin (fosBx1) resistance were detected according to the RGI tool criteria. The presence of acquired ARGs indicates that this strain was likely exposed to selective pressures, such as environmental antimicrobial use, which may have promoted the acquisition of these resistance genes through horizontal gene transfer (HGT) or other mechanisms (Chen et al., 2022). Based on EggNOG annotation, we confirmed the presence of several key antibiotic resistance-related genes in the FFCShyA4 genome, including fusA, pbp4, and ileS. The fusA gene encodes elongation factor G (EF-G), which is essential for protein synthesis and serves as a target for antibiotics like fusidic acid, suggesting a potential mechanism for resistance to this antibiotic (Resch et al., 2008). The pbp4 gene encodes penicillin-binding protein 4 (PBP4), which plays a critical role in bacterial cell wall synthesis and is a key target of β-lactam antibiotics like penicillin (Satishkumar et al., 2021). Alterations in PBP4 can reduce the binding affinity of these antibiotics, contributing to resistance. However, its role in S. xylosus was not yet identified. Additionally, ileS, which encodes isoleucine-tRNA ligase, is involved in protein synthesis and may influence resistance indirectly by impacting bacterial growth and response to translation-targeting antibiotics (Grundy et al., 1997). These findings indicate that the FFCShyA4 strain exhibits a high level of adaptability to antibiotic pressure, its genomic features suggest it may serve as a reservoir of ARGs across environmental interface. These genomic features suggest that FFCShyA4 possesses ecological adaptability and may function as a reservoir of ARGs, with potential relevance to One Health pathways of AMR dissemination across human, animal, and environmental interfaces (Robinson et al., 2016).
3.4 BGCs organization predicted from genome study
Two bacteriocin clusters (Area of interest, AOI) were annotated within the FFCShyA4 genome as follows: contig 5.2 (AOI_01) (started at 18217, end at 38349) of auto-inducing peptide III (AIP III) class, and contig 5.2 (AOI_02) (started at 85346, end at 105367) of the sactipeptides class (ribosomally synthesized peptides) (Figure 4). BLASTP analysis revealed that AIP III shares 100% amino acid sequence identity with the cyclic lactone autoinducer peptide AgrD found in several Staphylococcus species, including S. xylosus. The agr locus was originally characterized in S. aureus as a regulatory element that controls the production of exoproteins involved in virulence (Dufour et al., 2002). Besides, AIP III plays a critical role in quorum sensing within Staphylococcus species, particularly S. aureus (Tamai et al., 2023). By regulating gene expression in response to population density, AIP III influences virulence factors, biofilm formation, and antibiotic resistance. This peptide signals the bacteria to produce toxins and surface proteins that aid in immune evasion and infection persistence, while biofilms protect the bacteria from antimicrobials (Tamai et al., 2023). Disrupting AIP III-mediated quorum sensing presents a potential therapeutic strategy to reduce bacterial virulence and biofilm formation (Gonzales et al., 2024). In addition, the sactipeptides, a class of ribosomally synthesized and post-translationally modified peptides (RiPPs), exhibit diverse biological activities, including antibacterial, spermicidal, and hemolytic properties (Chen et al., 2021). Analysis of the FFCShyA4 genome using the antiSMASH web tool identified nine distinct biosynthetic regions (Table 2). These include a terpene biosynthetic cluster (region 1.1) with heme D1 as the closest known cluster (11% similarity), an NI-siderophore cluster (region 2.1) with 100% similarity to a known siderophore cluster, and regions encoding Type III polyketide synthase (T3PKS) and non-ribosomal peptide synthase (NRPS) on contigs 2 and 3. Based on gene similarity analysis with the MiBIG database, a 54% sequence similarity was observed with staphylopherrin A found in S. aureus subsp. aureus NCTC 8325 (Supplementary Table S8), which plays a vital role in iron acquisition (Battaglia and Garrett-Sinha, 2023). Staphylopherrin A aids the bacteria by binding to iron (Fe3+) in the environment and facilitating its transport into the bacterial cell (Battaglia and Garrett-Sinha, 2023). Early genomic analysis of the S. xylosus C2a strain reveals the presence of genes encoding staphylopherrin A (Vermassen et al., 2014). This iron acquisition mechanism enables S. aureus to survive and thrive within the host, enhancing its virulence and ability to cause infections. This suggests that S. xylosus may utilize similar mechanisms as S. aureus to acquire iron from the environment. Notably, one NRPS cluster predicted within FFCShyA4 genome shared 8% similarity with surfactin. Additionally, an opine-like metallophore cluster located on contig 3 displayed 100% similarity to the staphylopine biosynthesis cluster. Finally, a cyclic lactone autoinducer biosynthetic region (region 5) showed 4% similarity to the kijanimicin cluster. Staphylopine, a versatile metallophore produced by the prominent human pathogen Staphylococcus aureus, is essential for the uptake of transition metals and contributes significantly to bacterial virulence (Song et al., 2018). These findings suggest that S. xylosus may employ virulence-like strategies like S. aureus, underscoring the need to assess its role in antimicrobial resistance dissemination and cross-domain adaptation (Battaglia and Garrett-Sinha, 2023). Further research should explore the functional roles of the AIP III and sactipeptide bacteriocin clusters in FFCShyA4, particularly their involvement in quorum sensing, biofilm formation, and microbial competition within foodborne ecosystems.
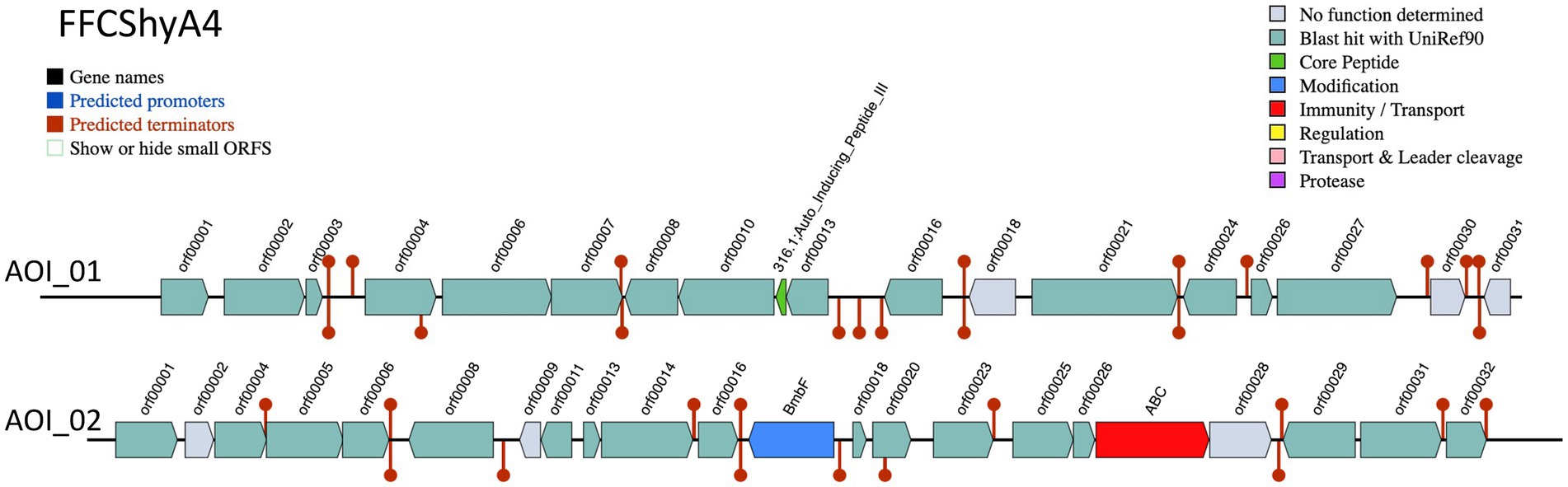
Figure 4. Bacteriocin cluster genes organization. Area of Interest (AOI) of FFCShyA4. Legend: red blocks: immunity and transport; green arrow: core peptide; blue block: peptide modifications; grey blocks: no function determined.
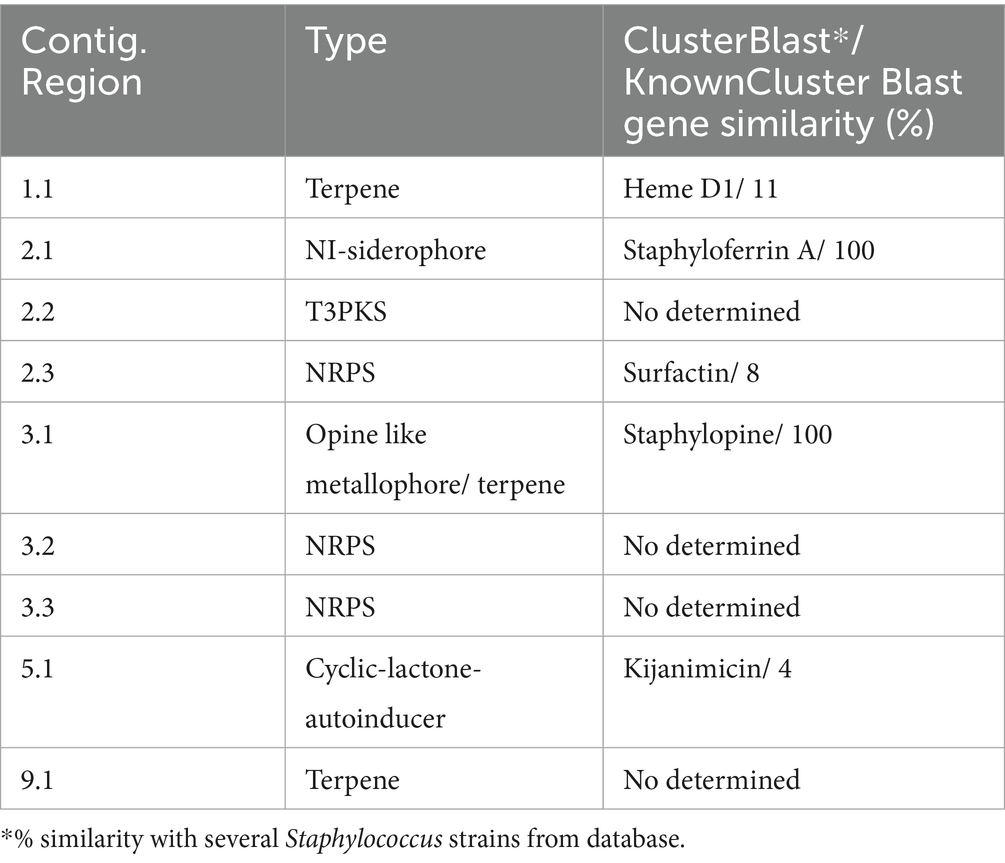
Table 2. Identified secondary metabolite biosynthetic gene clusters with antiSMASH using strictness “strict.”
3.5 Hemolysis, gelatinase, and antibiotic susceptibility tests in vitro
S. xylosus FFCShyA4 exhibited β-hemolytic activity and tested positive for gelatinase, like the S. aureus reference strains ATCC1026 and ATCC43300, indicating a potential adaptive trait rather than a direct virulence marker (Supplementary Figure S5). Genome annotation revealed the presence of a hemolysin III family protein in FFCShyA4; however, the failure to amplify hemolysin genes via PCR suggests potential sequence variations leading to primer mismatches or regulatory mechanisms influencing gene expression. Alternatively, hemolytic activity may be mediated by proteases or phospholipases rather than classical hemolysins (Vandenesch et al., 2012). It is also possible that homologous hemolysin genes or functional analogs were acquired through horizontal gene transfer (HGT) or retained for ecological adaptation. Unlike S. aureus, where hemolysins are key virulence factors, S. xylosus likely utilizes β-hemolysis for ecological fitness, particularly in food-associated environments such as plant surfaces. While this activity may enhance colonization potential, it does not necessarily indicate pathogenicity in healthy individuals, underscoring the phenotypic diversity and evolutionary plasticity of staphylococcal species (Eltwisy et al., 2020). The FFCShyA4 strain contained several putative resistance genes linked to resistance to lincosamide (1), tetracycline (4), fosfomycin (1), fluoroquinolone (16), macrolide (3), mupirocin (1), and salicylic acid (1) antimicrobials (Supplementary Figure S7). The antibiotic resistance profile of FFCShyA4 was compared with S. aureus ATCC1026 and S. aureus ATCC43300, highlighting key differences in their susceptibility patterns (Figure 5). FFCShyA4 shows resistance to several key antibiotics, particularly TE30 (tetracycline), MET5 (methicillin), CXM30 (cefuroxime), VA30 (Vancomycin), AX25 (amoxycillin), AM10 (ampicillin), OX1 (oxacillin), and LZD30 (linezolid), while displaying intermediate resistance to CIP5 (ciprofloxacin), C30 (chloramphenicol), and DA2 (clindamycin) (Supplementary Table S9). The MAR index was 0.67 for FFCShyA4, 0.70 for S. aureus ATCC 1026, and 0.62 for S. aureus ATCC 43300. Typically, a MAR index above 0.2 is considered significant, as it suggests exposure to antimicrobials in environments where misuse or overuse occurs, such as hospitals or livestock settings (Tang et al., 2023). The high MAR index and resistance to critical antibiotics, including methicillin and vancomycin, reflect possible exposure to antimicrobials in agricultural or post-harvest settings, raising concerns over antimicrobial misuse beyond clinical boundaries (Iwu et al., 2020). From a One Health perspective, the detection of multidrug-resistant S. xylosus on plant-based food products underscores the interconnectedness of human, animal, and environmental health, emphasizing the need for integrated surveillance of AMR across non-clinical reservoirs such as agricultural commodities, food supply chains, and market environments (Robinson et al., 2016). Compared to the reference S. aureus strains, FFCShyA4 exhibited phenotypic resistance to tetracycline, corroborating the resistance determinants identified through genomic analysis. The EggNOG annotation of FFShyA4 revealed the presence of tpiA and bla genes, indicating its potential for methicillin and β-lactam resistance, aligning with the observed multidrug resistance in the antibiogram. The bla gene suggests β-lactamase production, leading to resistance to penicillin and related antibiotics. Although the FFCShyA4 strain exhibited phenotypic resistance to methicillin, complementary PCR analysis using mecA and mecC primers failed to detect these genes. This discrepancy may be attributed to primer mismatches arising from sequence variations or the genomic location of mecA, potentially affecting primer binding efficiency. Besides, the presence of tpiA confirms the species identity, as it is a conserved housekeeping gene in S. xylosus. Early investigations into methicillin-resistant coagulase-negative staphylococci revealed that resistance to most β-lactam antibiotics arises primarily from the acquisition of the mecA or mecC genes, which encode alternative penicillin-binding proteins (PBPs) with reduced β-lactam binding affinity (Schwarz et al., 2018). The absence of mecA/mecC genes, common in livestock-associated Staphylococcus, does not exclude an animal origin but may indicate a less direct or recent exposure. Besides, the multidrug efflux pumps such as NorA which was annotated in the FFCShyA4 genome may contribute to methicillin resistance by actively exporting β-lactam antibiotics, reducing their intracellular concentration (Dashtbani-Roozbehani and Brown, 2021). Further comparative genomic and epidemiological studies would be necessary to confirm the specific origin of this strain. Another common resistance mechanism against penicillin involves enzymatic hydrolysis of the antibiotic by β-lactamases encoded by the blaZ or blaARL genes (Schwarz et al., 2018). The absence of mecA/mecC alongside the detection of alternative resistance determinants (bla, NorA) supports the idea that non-traditional or environmental pathways may be driving resistance evolution in underregulated sectors, such as local markets. Early studies indicate that antimicrobial resistance in Staphylococcus spp. isolated from various food products varies significantly depending on the species and source of isolation (Resch et al., 2008). Among S. xylosus strains, resistance rates ranged from 22% for tetracycline to 69% for penicillin, with 93% of isolates originating from meat starter cultures. Notably, all coagulase-negative staphylococci (CNS) strains were fully susceptible to clinically relevant antibiotics such as chloramphenicol, clindamycin, cotrimoxazole, gentamicin, kanamycin, linezolid, neomycin, streptomycin, and vancomycin (Resch et al., 2008). Additionally, S. xylosus strains isolated from fermented soybean products were reported to be susceptible to chloramphenicol, erythromycin, gentamicin, kanamycin, lincomycin, oxacillin, tetracycline, and trimethoprim (Kong et al., 2022). However, 23 strains exhibited acquired phenotypic resistance to erythromycin, lincomycin, and tetracycline. Minimum inhibitory concentration (MIC) testing showed continuous or unimodal distribution patterns for these antibiotics, which did not align with the resistance profiles, suggesting possible discrepancies. These findings indicate that the current CLSI breakpoint values for ampicillin and penicillin G in S. xylosus may need to be re-evaluated. Moreover, the presence of acquired resistance to erythromycin, lincomycin, and tetracycline underscores the importance of performing antibiotic susceptibility testing before utilizing these strains in food-related applications (Kong et al., 2022). These observations underscore the necessity for coordinated, cross-sectoral surveillance strategies encompassing food production and environmental interfaces to monitor the emergence and dissemination of antimicrobial resistance, particularly in regions with insufficient regulation of antimicrobials use in agri-food systems.
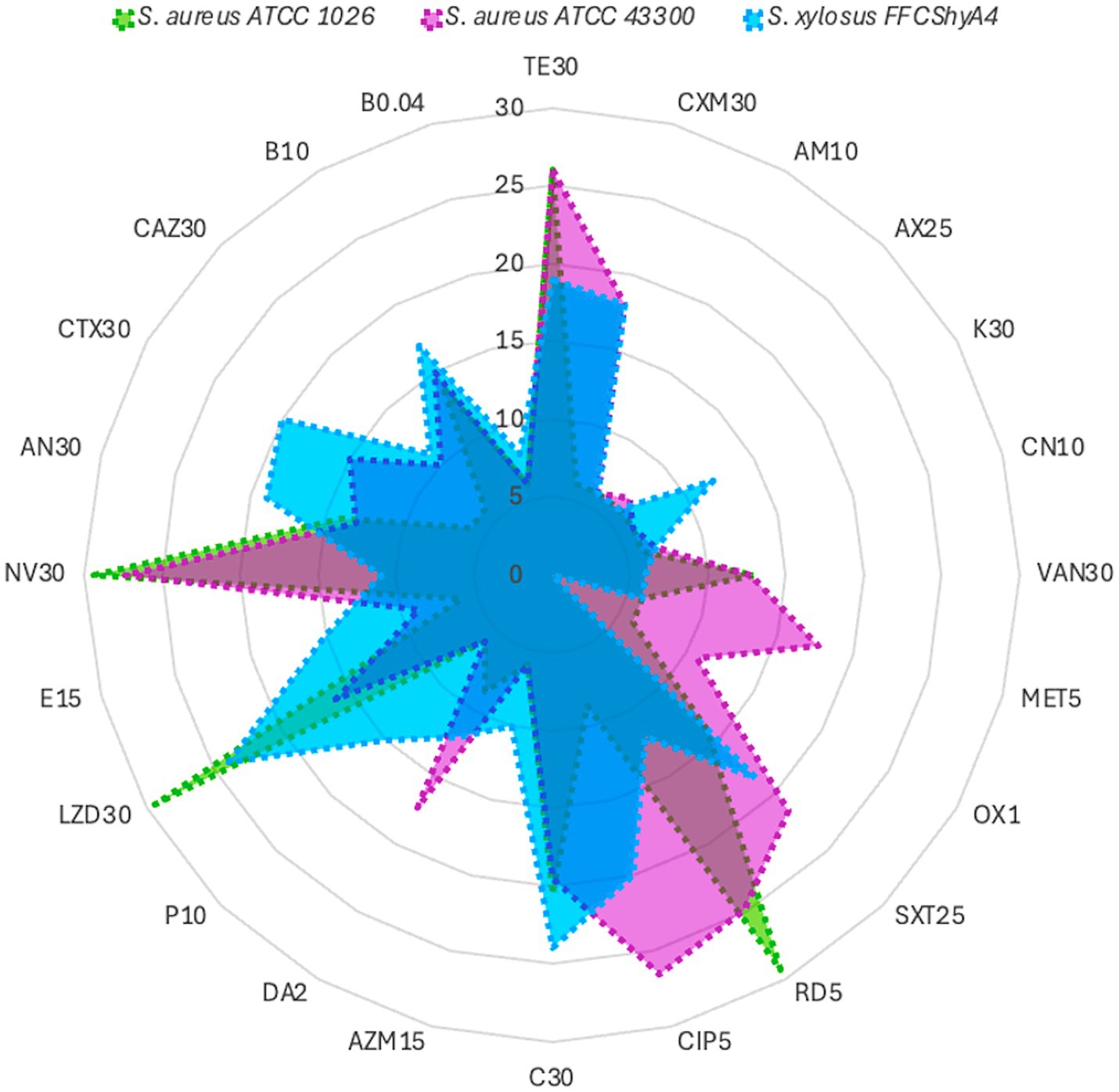
Figure 5. Antibiotic profile of S. xylosus FFCShyA4 compared with S. aureus ATCC1026 and S. aureus ATCC43300.
4 Conclusion
This study highlights a multidrug-resistant S. xylosus strain FFCShyA4 from avocados as a potential foodborne threat, with genomic and phenotypic features suggesting cross-domain antimicrobial resistance and adaptation, reinforcing the need for integrated One Health surveillance in food systems. Genomic evidence suggests an animal-associated origin, potentially resulting from cross-contamination via human handling, market environments, or contact with animal-derived materials. The genome analysis reveals extensive structural rearrangements, and a wide array of genes linked to antibiotic resistance, virulence, and environmental adaptation. However, resistance genes (e.g., fusA, pbp4, ileS, bla, and tpiA), stress response elements, quorum sensing systems, and bacteriocin clusters, together with MGEs, indicate high potential for horizontal gene transfer and adaptation across niches. These features underscore the role of mobile produce surfaces as reservoirs and potential vectors for resistance genes, bridging ecosystems across the food production and consumption continuum. In vitro assays further confirmed hemolytic activity and resistance to multiple antimicrobial classes, highlighting its potential as an opportunistic pathogen. These findings highlight the critical need to extend antimicrobial resistance surveillance beyond clinical and veterinary contexts to include plant-derived food matrices. The comprehensive genomic analysis of FFCShyA4 illustrates how a single, well-characterized foodborne isolate can provide valuable insights into resistance mechanisms and their potential transmission across ecological boundaries. As such, this case underscores the importance of incorporating microbial genomics into food safety and public health strategies. Integrating such data into One Health surveillance systems is essential to guide effective interventions and evidence-based policymaking.
Data availability statement
The genome assembly data of S. xylosus FFCShyA4 are publicly available. This data can be found here: NCBI Sequence Read Archive, BioProject ID PRJNA1249250, BioSample accession SAMN47884644.
Author contributions
GT: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. EA: Investigation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by Universidad Tecnica del Norte, No. 9674/2024 awarded to Dr. Tenea.
Acknowledgments
The authors would like to express their gratitude to interim students B. Carlosama and A. Gordillo for their assistance with in vitro analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1629139/full#supplementary-material
Footnotes
References
Al-Tameemi, H. M., Al-Hraishawi, H., Al-Hejjaj, M. Y., Abdulah, N. S., Alrafas, H. R., and Dawood, Y. A. (2023). Whole genome sequence and comparative genomics analysis of multidrug-resistant Staphylococcus xylosus NM36 isolated from a cow with mastitis in Basrah city. J. Genet. Eng. Biotechnol. 21:163. doi: 10.1186/s43141-023-00606-6
Angamarca, E., Castillejo, P., and Tenea, G. N. (2023). Microbiota and its antibiotic resistance profile in avocado Guatemalan fruits (Persea nubigena var. guatemalensis) sold at retail markets of Ibarra city, northern Ecuador. Front. Microbiol. 14:1228079. doi: 10.3389/fmicb.2023.1228079
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Battaglia, M., and Garrett-Sinha, L. A. (2023). Staphylococcus xylosus and Staphylococcus aureus as commensals and pathogens on murine skin. Lab. Anim. Res. 39:18. doi: 10.1186/s42826-023-00169-0
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing GroupLau, B. Y., Hoad, G., et al. (2017). Islandviewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bertelloni, F., Cagnoli, G., and Ebani, V. V. (2021). Virulence and antimicrobial resistance in canine Staphylococcus spp. isolates. Microorganisms 9:515. doi: 10.3390/microorganisms9030515
Blin, K., Shaw, S., Augustijn, H. E., Reitz, Z. L., Biermann, F., Alanjary, M., et al. (2023). antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucl. Acids Res. 51, W46–W50. doi: 10.1093/nar/gkad344
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Brown, C. L., Mullet, J., Hindi, F., Stoll, J. E., Gupta, S., Choi, M., et al. (2022). mobileOG-db: a manually curated database of protein families mediating the life cycle of bacterial Mobile genetic elements. Appl. Environ. Microbiol. 88:e0099122. doi: 10.1128/aem.00991-22
Canning, B., Mohamed, I., Wickramasinghe, N., Swindells, J., and O'Shea, M. K. (2020). Thermonuclease test accuracy is preserved in methicillin-resistant Staphylococcus aureus isolates. J. Med. Microbiol. 69, 548–551. doi: 10.1099/jmm.0.001166
Caspi, R., Billington, R., Fulcher, C. A., Keseler, I. M., Kothari, A., Krummenacker, M., et al. (2018). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46:D633. doi: 10.1093/nar/gkx935
Chen, M., Li, Y., Li, S., Cui, W., Zhou, Y., Qu, Q., et al. (2022). Molecular mechanism of Staphylococcus xylosus resistance against Tylosin and Florfenicol. Infect. Drug Resist. 15, 6165–6176. doi: 10.2147/IDR.S379264
Chen, Y., Wang, J., Li, G., Yang, Y., and Ding, W. (2021). Current advancements in Sactipeptide natural products. Front. Chem. 9:595991. doi: 10.3389/fchem.2021.595991
Clinical and Laboratory Standards Institute, Performance standards for antimicrobial susceptibility testing, 31st edition. M100-Ed31. CLSI; Wayne, PA: (2021). Available online at: https://clsi.org/standards/products/microbiology/documents (Accessed September 6, 2024).
Coll, F., Blane, B., Bellis, K. L., Matuszewska, M., Wonfor, T., Jamrozy, D., et al. (2025). The mutational landscape of Staphylococcus aureus during colonisation. Nat. Commun. 16:302. doi: 10.1038/s41467-024-55186-x
Condas, L. A. Z., de Buck, J., Nobrega, D. B., Carson, D. A., Roy, J.-P., Keefe, G. P., et al. (2017). Distribution of non-aureus staphylococci species in udder quarters with low and high somatic cell count, and clinical mastitis. J. Dairy Sci. 100, 5613–5627. doi: 10.3168/jds.2016-12479
Cosentino, S., Larsen, M. V., Aarestrup, F. M., and Lund, O. (2013). Pathogenfinder - distinguishing friend from foe using bacterial whole genome sequence data. PLoS One 8:e77302. doi: 10.1371/journal.pone.0077302
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dashtbani-Roozbehani, A., and Brown, M. H. (2021). Efflux pump mediated antimicrobial resistance by staphylococci in health-related environments: challenges and the quest for inhibition. Antibiotics 10:1502. doi: 10.3390/antibiotics10121502
Deghorain, M., and Van Melderen, L. (2012). The staphylococci phages family: an overview. Viruses 4, 3316–3335. doi: 10.3390/v4123316
Dordet-Frisoni, E., Dorchies, G., De Araujo, C., Talon, R., and Leroy, S. (2007). Genomic diversity in Staphylococcus xylosus. Appl. Environ. Microbiol. 73, 7199–7209. doi: 10.1128/AEM.01629-07
Dufour, P., Jarraud, S., Vandenesch, F., Greenland, T., Novick, R. P., Bes, M., et al. (2002). High genetic variability of the agr locus in Staphylococcus species. J. Bacteriol. 184, 1180–1186. doi: 10.1128/jb.184.4.1180-1186.2002
Eltwisy, H. O., Abdel-Fattah, M., Elsisi, A. M., Omar, M. M., Abdelmoteleb, A. A., and El-Mokhtar, M. A. (2020). Pathogenesis of Staphylococcus haemolyticus on primary human skin fibroblast cells. Virulence 11, 1142–1157. doi: 10.1080/21505594.2020.1809962
European Committee on Antimicrobial Susceptibility Testing, Breakpoint tables for interpretation of MICs and zone diameters, Version 11.0. (2021). Available online at https://eucast.org/clinical_breakpoints/ (Accessed September 6, 2024).
Ghaly, T. M., Gillings, M. R., Rajabal, V., Paulsen, I. T., and Tetu, S. G. (2024). Horizontal gene transfer in plant microbiomes: integrons as hotspots for cross-species gene exchange. Front. Microbiol. 15:1338026. doi: 10.3389/fmicb.2024.1338026
Gonzales, M., Kergaravat, B., Jacquet, P., Billot, R., Grizard, D., Chabrière, É., et al. (2024). Disrupting quorum sensing as a strategy to inhibit bacterial virulence in human, animal, and plant pathogens. Pathog. Dis. 82:ftae009. doi: 10.1093/femspd/ftae009
González, V., Acosta, J. L., Santamaría, R. I., Bustos, P., Fernández, J. L., Hernández González, I. L., et al. (2010). Conserved symbiotic plasmid DNA sequences in the multireplicon pangenomic structure of Rhizobium etli. Appl. Environ. Mcrobiol. 76, 1604–1614. doi: 10.1128/AEM.02039-09
Grant, J. R., Enns, E., Marinier, E., Mandal, A., Herman, E. K., Chen, C. Y., et al. (2023). Proksee: in-depth characterization and visualization of bacterial genomes. Nucl. Acids Res. 51, W484–W492. doi: 10.1093/nar/gkad326
Grundy, F. J., Haldeman, M. T., Hornblow, G. M., Ward, J. M., Chalker, A. F., and Henkin, T. M. (1997). The Staphylococcus aureus ileS gene, encoding isoleucyl-tRNA synthetase, is a member of the T-box family. J. Bacteriol. 179, 3767–3772. doi: 10.1128/jb.179.11.3767-3772.1997
Heo, S., Lee, J., and Jeong, D. (2021). Complete genome sequence of Staphylococcus xylosus strain DMSX03 from fermented soybean, meju. Korean J. Microbiol. 57, 52–54. doi: 10.7845/kjm.2021.0120
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucl. Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Iwu, C. D., Korsten, L., and Okoh, A. I. (2020). The incidence of antibiotic resistance within and beyond the agricultural ecosystem: a concern for public health. Microbiology Open 9:e1035. doi: 10.1002/mbo3.1035
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kearney, J. N., Gowland, G., Holland, K. T., and Cunliffe, W. J. (1982). Maintenance of the normal flora of human skin grafts transplanted to mice. J. Gen. Microbiol. 128, 2431–2437. doi: 10.1099/00221287-128-10-2431
Kong, H., Jeong, D. W., Kim, N., Lee, S., Sul, S., and Lee, J. H. (2022). Safety and technological characterization of Staphylococcus xylosus and Staphylococcus pseudoxylosus isolates from fermented soybean foods of Korea. J. Microbiol. Biotechnol. 32, 458–463. doi: 10.4014/jmb.2111.11040
Labrie, S. J., El Haddad, L., Tremblay, D. M., Plante, P. L., Wasserscheid, J., Dumaresq, J., et al. (2014). First complete genome sequence of Staphylococcus xylosus, a meat starter culture and a host to propagate Staphylococcus aureus phages. Genome Announc. 2:e00671-14. doi: 10.1128/genomeA.00671-14
Lamers, R. P., Muthukrishnan, G., Castoe, T. A., Tafur, S., Cole, A. M., and Parkinson, C. L. (2012). Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC Evol. Biol. 12:171. doi: 10.1186/1471-2148-12-171
Leroy, S., Vermassen, A., Ras, G., and Talon, R. (2017). Insight into the genome of Staphylococcus xylosus, a ubiquitous species well adapted to meat products. Microorganisms 5:52. doi: 10.3390/microorganisms5030052
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Makhlin, J., Kofman, T., Borovok, I., Kohler, C., Engelmann, S., Cohen, G., et al. (2007). Staphylococcus aureus ArcR controls expression of the arginine deiminase operon. J. Bacteriol. 189, 5976–5986. doi: 10.1128/JB.00592-07
Marincola, G., Liong, O., Schoen, C., Abouelfetouh, A., Hamdy, A., Wencker, F. D. R., et al. (2021). Antimicrobial resistance profiles of coagulase-negative staphylococci in community-based healthy individuals in Germany. Front. Public Health 9:684456. doi: 10.3389/fpubh.2021.684456
McGuffie, M. J., and Barrick, J. E. (2021). pLannotate: engineered plasmid annotation. Nucleic Acids Res. 49, W516–W522. doi: 10.1093/nar/gkab374
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Mittal, S., Sharma, M., and Chaudhary, U. (2014). Study of virulence factors of uropathogenic Escherichia coli and its antibiotic susceptibility pattern. Indian J. Pathol. Microbiol. 57:61. doi: 10.4103/0377-4929.130899
Molina, D., Carrión-Olmedo, J. C., Jarrín-V, P., and Tenea, G. N. (2024). Genome characterization of a multi-drug resistant Escherichia coli strain, L1PEag1, isolated from commercial cape gooseberry fruits (Physalis peruviana L.). Front. Microbiol. 15:1392333. doi: 10.3389/fmicb.2024.1392333
Rahman, M., Alam, M. U., Luies, S. K., Kamal, A., Ferdous, S., Lin, A., et al. (2021). Contamination of fresh produce with antibiotic-resistant Bacteria and associated risks to human health: a scoping review. Int. J. Environ. Publ. Health. 19:360. doi: 10.3390/ijerph19010360
Resch, M., Nagel, V., and Hertel, C. (2008). Antibiotic resistance of coagulase-negative staphylococci associated with food and used in starter cultures. Int. J. Food Microbiol. 127, 99–104. doi: 10.1016/j.ijfoodmicro.2008.06.013
Robinson, T. P., Bu, D. P., Carrique-Mas, J., Fèvre, E. M., Gilbert, M., Grace, D., et al. (2016). Antibiotic resistance is the quintessential one health issue. Trans. R. Soc. Trop. Med. Hyg. 110, 377–380. doi: 10.1093/trstmh/trw048
Satishkumar, N., Alexander, J. A. N., Poon, R., Buggeln, E., Argudín, M. A., Strynadka, N. C. J., et al. (2021). PBP4-mediated β-lactam resistance among clinical strains of Staphylococcus aureus. J. Antimicrob. Chemother. 76, 2268–2272. doi: 10.1093/jac/dkab201
Schiffer, C. J., Grätz, C., Pfaffl, M. W., Vogel, R. F., and Ehrmann, M. A. (2023). Characterization of the Staphylococcus xylosus methylome reveals a new variant of type I restriction modification system in staphylococci. Front. Microbiol. 14:946189. doi: 10.3389/fmicb.2023.946189
Schwarz, S., Feßler, A. T., Loncaric, I., Wu, C., Kadlec, K., Wang, Y., et al. (2018). Antimicrobial resistance among staphylococci of animal origin. Microbiol. Spectr. 6. doi: 10.1128/microbiolspec.arba-0010-2017
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Selengut, J. D., Haft, D. H., Davidsen, T., Ganapathy, A., Gwinn-Giglio, M., Nelson, W. C., et al. (2007). TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucl. Acids Res. 35, D260–D264. doi: 10.1093/nar/gkl1043
Shukla, S. K., Kislow, J., Briska, A., Henkhaus, J., and Dykes, C. (2009). Optical mapping reveals a large genetic inversion between two methicillin-resistant Staphylococcus aureus strains. J. Bacteriol. 191, 5717–5723. doi: 10.1128/JB.00325-09
Song, L., Zhang, Y., Chen, W., Gu, T., Zhang, S. Y., and Ji, Q. (2018). Mechanistic insights into staphylopine-mediated metal acquisition. Proc. Natl. Acad. Sci. USA 115, 3942–3947. doi: 10.1073/pnas.1718382115
Stegger, M., Andersen, P. S., Kearns, A., Pichon, B., Holmes, M. A., Edwards, G., et al. (2012). Rapid detection, differentiation and typing of methicillin-resistant Staphylococcus Aureus Harbouring either MecA or the new MecA homologue MecALGA251. Clin. Microbiol. Infect. 18, 395–400. doi: 10.1111/j.1469-0691.2011.03715.x
Tabasi, M., Karam, M. R. A., Habibi, M., Yekaninejad, M. S., and Bouzari, S. (2015). Phenotypic assays to determine virulence factors of Uropathogenic Escherichia coli (UPEC) isolates and their correlation with antibiotic resistance pattern. Osong Public Heal. Res. Perspect. 6, 261–268. doi: 10.1016/j.phrp.2015.08.002
Tamai, M., Yamazaki, Y., Ito, T., Nakagawa, S., and Nakamura, Y. (2023). Pathogenic role of the staphylococcal accessory gene regulator quorum sensing system in atopic dermatitis. Front. Cell. Infect. Microbiol. 13:1178650. doi: 10.3389/fcimb.2023.1178650
Tang, K. W. K., Millar, B. C., and Moore, J. E. (2023). Antimicrobial Resistance (AMR). Br. J. Biomed. Sci. 80:11387. doi: 10.3389/bjbs.2023.11387
Tenea, G. N., Reyes, P., Molina, D., and Ortega, C. (2023). Pathogenic microorganisms linked to fresh fruits and juices purchased at low-cost Markets in Ecuador, potential carriers of antibiotic resistance. Antibiotics 12:236. doi: 10.3390/antibiotics12020236
van Heel, A. J., de Jong, A., Song, C., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucl. Acids Res. 46, W278–W281. doi: 10.1093/nar/gky383
Vandenesch, F., Lina, G., and Henry, T. (2012). Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front. Cell. Infect. Microbiol. 2:12. doi: 10.3389/fcimb.2012.00012
Vermassen, A., de la Foye, A., Loux, V., Talon, R., and Leroy, S. (2014). Transcriptomic analysis of Staphylococcus xylosus in the presence of nitrate and nitrite in meat reveals its response to nitrosative stress. Front. Microbiol. 5:691. doi: 10.3389/fmicb.2014.00691
Vermassen, A., Dordet-Frisoni, E., de La Foye, A., Micheau, P., Laroute, V., Leroy, S., et al. (2016). Adaptation of Staphylococcus xylosus to nutrients and osmotic stress in a salted meat model. Front. Microbiol. 7:87. doi: 10.3389/fmicb.2016.00087
Waters, N. R., Abram, F., Brennan, F., Holmes, A., and Pritchard, L. (2020). Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2:acmi000143. doi: 10.1099/acmi.0.000143
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Staphylococcus xylosus , genomic analysis, virulence factors, antibiotic susceptibility, food safety, avocado
Citation: Tenea GN and Angamarca E (2025) Genomic characterization of a multidrug-resistant Staphylococcus xylosus from Ecuadorian open market avocados: food safety and public health implications. Front. Microbiol. 16:1629139. doi: 10.3389/fmicb.2025.1629139
Edited by:
Federica Savini, University of Bologna, ItalyReviewed by:
Marta Laranjo, University of Evora, PortugalElisabetta Chiarini, University of Turin, Italy
Copyright © 2025 Tenea and Angamarca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela N. Tenea, Z250ZW5lYUB1dG4uZWR1LmVj