 Xiayu Peng1
Xiayu Peng1 Xue Zhou
Xue Zhou- 1College of Animal Science and Technology, Shihezi University, Shihezi, Xinjiang, China
- 2Xinjiang Tianrun Dairy Co., Ltd., Urumqi, Xinjiang, China
- 3Hangzhou Dianzi University, Hangzhou, China
Introduction: To address the limitations of current metagenomic identification approaches, we proposed a principled AI-assisted architecture that enhances accuracy, scalability, and biological interpretability through three core innovations.
Methods: Firstly, we developed a structured probabilistic model that formulates pathogen detection as a hierarchical and compositional inference task under taxonomic and ecological constraints. This framework enables the integration of phylogenetic priors and sparsity-aware mechanisms, reducing noise and ambiguity. By modeling taxonomic structure and ecological dependencies, the approach ensures more accurate identification, especially in complex or low-abundance microbial communities. Secondly, we introduced the Taxon-aware Compositional Inference Network (TCINet), a deep learning model that processes sequencing reads to produce taxonomic embeddings. TCINet estimates abundance distributions via masked neural activations that enforce sparsity and interpretability, while also propagating uncertainty through log-normal variance modeling. Designed to respect microbial phylogeny and co-occurrence patterns, TCINet enables scalable, biologically plausible inference across diverse clinical and environmental datasets. Thirdly, we presented the Hierarchical Taxonomic Reasoning Strategy (HTRS), a post-inference module that refines predictions by enforcing compositional constraints, propagating evidence across taxonomic hierarchies, and calibrating confidence using entropy and variance-based metrics. HTRS includes context-aware thresholding and co-occurrence priors to adaptively optimize performance based on dataset characteristics.
Results: Together, these innovations create a unified framework for metagenomic identification that combines probabilistic modeling, deep learning, and structured reasoning.
Discussion: The architecture delivers robust and interpretable results, making it suitable for applications in clinical diagnostics, environmental monitoring, and ecological research.
1 Introduction
Enhancing the identification of pathogens is critical for timely diagnosis, treatment, and public health surveillance, especially in the face of emerging infectious diseases and antimicrobial resistance. Traditional culture-based diagnostic methods are often time-consuming and limited in scope, while PCR-based methods, although faster, require prior knowledge of the target organism (Xu et al., 2024). In contrast, metagenomic sequencing provides a culture-independent, hypothesis-free approach to detect a wide range of pathogens directly from clinical samples (Liu et al., 2020). However, this method generates vast quantities of complex and noisy data, posing significant challenges in interpretation (Zhang et al., 2023). To address these issues, artificial intelligence (AI), particularly machine learning and deep learning techniques, is increasingly integrated into metagenomic workflows to enhance sensitivity, specificity, and interpretability (Peng et al., 2023). Not only does AI streamline the analysis process, but it also enables the identification of novel pathogens and resistance genes, contributing to improved diagnostics and surveillance. Consequently, AI-assisted metagenomic sequencing is becoming a vital tool in modern microbiology and infectious disease management (Zhu et al., 2023).
Early computational approaches for interpreting metagenomic data were grounded in structured hierarchies and rule-based classifications, leveraging known biological relationships and curated databases to infer microbial presence and potential pathogenicity. These systems often relied on alignment-based algorithms and predefined taxonomic trees to map sequencing reads to reference genomes or annotated markers (Kocmi et al., 2023). Their strength lay in the use of expert-defined ontologies and deterministic rules, which allowed for transparent decision-making and high interpretability–particularly valuable in clinical and regulatory settings where traceability and justification of results are essential. In environments where the sequencing data closely resembled well-characterized organisms within the reference databases, these methods performed reliably, delivering consistent and interpretable results (Wang et al., 2023). Tools like MEGAN and Kraken exemplified this paradigm by efficiently assigning taxonomic labels and enabling interactive exploration of results based on known microbial lineages. However, the performance of such systems was tightly coupled to the completeness and quality of the underlying reference libraries. Their reliance on fixed taxonomies and exact sequence matching constrained their adaptability, making them less effective in detecting novel species, strains with genomic variation, or divergent organisms not represented in the reference set (Moslem et al., 2023). As metagenomic datasets grew in both size and complexity–spanning environmental samples, mixed microbial communities, and clinical specimens with variable quality–these rule-based systems began to show limitations. They struggled to handle noisy reads, fragmented sequences, and ambiguous matches, often discarding valuable information that did not fit their strict criteria (Goyal et al., 2021). The growing influx of previously uncharacterized microbes and the dynamic nature of microbial evolution further challenged the rigidity of these methods. The inability to generalize beyond known taxonomic boundaries or to infer latent patterns in data led to reduced sensitivity, lower detection accuracy, and missed opportunities in identifying emerging pathogens or novel functional elements (Freitag et al., 2021).
To address these shortcomings, subsequent methods shifted toward leveraging statistical patterns extracted directly from sequencing data. Rather than relying solely on predefined taxonomies or exact sequence matches, these approaches utilized intrinsic properties of the sequences–such as nucleotide composition, k-mer frequency profiles, GC content, and motif distributions–to inform classification decisions (Garca et al., 2023). By capturing compositional and structural features, these models could identify microbial signatures even in the absence of perfect reference matches, thus offering enhanced sensitivity to genomic variation and novel taxa. This transition marked a key development in metagenomic analysis, as it allowed models to function with increased autonomy and reduced dependence on expert-curated rules or exhaustive taxonomic databases (Jiang et al., 2021). As a result, they scaled more effectively with expanding datasets, enabling high-throughput classification across diverse sample types. Tools implementing these strategies, such as MetaPhlAn and PhyloPythia, demonstrated how carefully engineered features could capture informative signals within complex microbial communities and support more efficient taxonomic profiling and functional annotation (Kocmi et al., 2022). However, the effectiveness of these systems often depended heavily on the quality and comprehensiveness of labeled training datasets, which are difficult to obtain for understudied or rare organisms. Moreover, the process of manual feature selection introduced biases and assumptions that could limit the model's ability to generalize across variable conditions (Fan et al., 2020). In clinical applications, where sample heterogeneity, contamination, and sequencing artifacts are common, these constraints became particularly problematic. As a result, while these models improved scalability and flexibility relative to earlier rule-based systems, their reliance on curated features and static training data often restricted robustness, limiting their performance in real-world metagenomic diagnostics and surveillance tasks (Agrawal et al., 2022).
In more recent developments, computational models have progressed toward learning directly from raw sequencing data, eliminating the need for extensive manual feature engineering. These models are designed to automatically extract informative representations by leveraging both hierarchical sequence organization and contextual dependencies within genomic data (Zhu et al., 2020). Advanced architectures—such as convolutional neural networks (CNNs), recurrent neural networks (RNNs), and more recently, transformer-based models—have demonstrated a remarkable ability to capture long-range interactions and compositional structures in nucleotide sequences, enabling more accurate and nuanced microbial classification and pathogen detection. A key innovation in this space is the use of pretraining on large-scale genomic corpora, which equips models with a generalized understanding of sequence patterns and structural regularities (Li et al., 2022). Pretrained models like DnabERT and DeepMicrobes exemplify this approach, showing strong adaptability to a variety of downstream tasks with minimal fine-tuning. These models are capable of identifying low-abundance or previously uncharacterized pathogens by capturing latent sequence signals that traditional or feature-based models often overlook. Furthermore, their end-to-end design reduces dependency on multiple preprocessing steps, streamlining analysis pipelines and minimizing error propagation (Xiao et al., 2022). Despite these advances, challenges remain. Deep learning models are often resource-intensive, requiring substantial computational power and memory, which can limit accessibility in resource-constrained settings. Their black-box nature raises ongoing concerns about interpretability and accountability–particularly in high-stakes clinical or epidemiological contexts where understanding model rationale is essential (Arenas and Toral, 2022). In response, current research increasingly focuses on hybrid frameworks that integrate attention mechanisms, uncertainty quantification, and post hoc interpretability tools to bridge the gap between predictive power and transparency. These efforts aim to create pathogen detection systems that are not only accurate and scalable, but also robust, interpretable, and aligned with the practical needs of public health and clinical decision-making (Khandelwal et al., 2020).
Previous symbolic, machine learning, and deep learning methods face limitations in adaptability, feature dependence, and interpretability. To address these issues, this study introduces a novel AI-assisted metagenomic sequencing framework that integrates symbolic reasoning with deep learning-based sequence embedding. Our method seeks to balance performance with explainability, enabling rapid and accurate pathogen detection while retaining transparency in decision-making. The framework uses pretrained sequence models for feature extraction and applies symbolic reasoning for classification and context interpretation. It is designed to operate in multi-pathogen environments and adapt to evolving pathogen landscapes. This method facilitates the discovery of novel organisms and resistance elements, addressing current gaps in clinical and epidemiological surveillance. This hybrid paradigm aims to deliver an efficient, robust, and interpretable solution for pathogen identification through metagenomic sequencing.
• We introduce a novel hybrid model that combines symbolic reasoning with deep learning-based embeddings for enhanced accuracy and interpretability.
• Our method is optimized for diverse clinical scenarios, offering high-throughput analysis, generalizability across samples, and robust performance under noisy conditions.
• Experimental results demonstrate superior detection rates for both common and rare pathogens, outperforming existing benchmarks in sensitivity, specificity, and runtime efficiency.
2 Related work
2.1 Metagenomic sequencing techniques
Metagenomic sequencing has transformed pathogen detection by enabling comprehensive analysis of genetic material recovered directly from clinical or environmental samples. Unlike traditional culture-based diagnostic techniques, metagenomics provides an unbiased view of microbial communities, making it invaluable for identifying novel or unexpected pathogens (Pan et al., 2021). Shotgun metagenomic sequencing, in particular, allows for the capture of all DNA present in a sample, facilitating the identification of bacteria, viruses, fungi, and parasites simultaneously. Recent advances in sequencing platforms, such as Illumina and Oxford Nanopore Technologies, have improved read accuracy, throughput, and turnaround time, thus enhancing the feasibility of routine clinical applications (Savoldi et al., 2021). A key aspect of effective metagenomic sequencing lies in sample preparation and DNA extraction protocols, which must be optimized to ensure representative recovery of microbial DNA while minimizing contamination. Library preparation methods have also evolved to accommodate low-input samples and increase sequencing depth. Furthermore, amplification-free approaches are gaining popularity as they reduce bias and preserve the quantitative integrity of microbial DNA content (Akhbardeh et al., 2021). Bioinformatics tools developed for metagenomic analysis range from de novo assemblers to reference-based classifiers, each with trade-offs in accuracy and computational efficiency. Tools like Kraken2, MetaPhlAn, and Centrifuge utilize k-mer based alignment strategies for taxonomic classification, providing rapid identification of pathogens (Liang et al., 2021a). However, their reliance on comprehensive and up-to-date reference databases is a limitation, especially when dealing with novel or rare organisms. Assembly-based methods such as MEGAHIT and metaSPAdes can reconstruct genomes from metagenomic reads, enabling downstream analyses like antimicrobial resistance profiling and virulence factor identification (Kocmi et al., 2021). Despite these advancements, challenges remain, particularly regarding the accurate identification of low-abundance pathogens in complex microbial communities. The presence of high-background host DNA, sequencing errors, and incomplete reference databases can confound analysis, necessitating the integration of more sophisticated computational methods to improve sensitivity and specificity in pathogen detection (Liang et al., 2022).
2.2 AI in genomic data interpretation
Artificial Intelligence (AI), particularly machine learning (ML) and deep learning (DL) approaches, has increasingly been applied to address the complexities of genomic data interpretation. These methods offer significant improvements in pattern recognition, feature extraction, and classification tasks compared to traditional bioinformatics pipelines. In the context of pathogen identification, AI models are capable of learning from large datasets to distinguish pathogen-specific signatures even in noisy or incomplete data environments (Raunak et al., 2021). Supervised learning models such as support vector machines (SVMs), random forests (RFs), and gradient boosting machines (GBMs) have been utilized to classify microbial taxa based on sequence features, k-mer frequencies, or read abundance patterns. These approaches are particularly useful for tasks where labeled training data is available, enabling precise mapping between sequence data and pathogen labels. However, the success of these models is contingent on the quality and diversity of training datasets, which must capture the full spectrum of microbial genomic variability (Ranathunga et al., 2021). Deep learning architectures, including convolutional neural networks (CNNs) and recurrent neural networks (RNNs), have demonstrated promise in more complex scenarios, such as identifying pathogens directly from raw sequencing reads or contigs. These models can automatically learn hierarchical features without the need for manual feature engineering, thus capturing subtle sequence patterns that may be missed by traditional algorithms. For example, CNNs have been successfully used to classify reads in real-time nanopore sequencing workflows, offering rapid turnaround and high accuracy (Haddow et al., 2021). Unsupervised learning and clustering techniques also play a critical role in identifying novel pathogens by detecting outlier sequences or uncharacterized genomic signatures within metagenomic datasets. Generative models like variational autoencoders (VAEs) and generative adversarial networks (GANs) further facilitate the simulation of synthetic genomic data, aiding in the development of robust AI models through data augmentation (Liang et al., 2020). The integration of AI with traditional genomic analysis tools creates hybrid pipelines that leverage the strengths of both domains. For instance, AI models can pre-filter or enrich reads for downstream alignment, prioritize candidate pathogens for confirmation, or refine taxonomic classification through ensemble learning strategies. As these methods evolve, ensuring their interpretability and validation against clinical benchmarks remains crucial to their widespread adoption (Romano et al., 2025).
2.3 Clinical applications and case studies
The application of AI-assisted metagenomic sequencing in clinical settings has shown significant promise in enhancing diagnostic accuracy and timeliness. This technology has been particularly impactful in cases of unexplained infections, immunocompromised patients, and outbreaks involving rare or emerging pathogens. By providing a hypothesis-free diagnostic approach, metagenomic sequencing enables clinicians to detect pathogens that may not be considered in traditional diagnostic panels (Zheng et al., 2021). Several high-profile case studies have demonstrated the utility of this approach. For instance, in cases of encephalitis of unknown origin, AI-enhanced metagenomics has successfully identified viral agents such as herpes simplex virus and enteroviruses, which were previously undetected by conventional methods. Similarly, in hospital outbreak investigations, sequencing data interpreted through AI models have helped trace the source of infections, differentiate between strains, and guide infection control measures (Cai et al., 2021). In the realm of antimicrobial resistance (AMR), AI-driven analysis of metagenomic data has been employed to predict resistance genes and inform personalized therapy decisions. This is especially relevant in settings where culture results are unavailable or delayed. For example, AI algorithms trained on large-scale genomic datasets can predict resistance phenotypes from metagenomic sequences with high accuracy, aiding in the selection of effective antimicrobial treatments (Ghorbani et al., 2021). Pediatric and neonatal intensive care units (NICUs) have also benefited from AI-assisted diagnostics, where rapid pathogen identification is critical. Studies have shown that integrating AI tools with sequencing workflows can reduce time-to-diagnosis from several days to under 24 h, thereby significantly improving clinical outcomes. These tools also assist in interpreting complex results by prioritizing pathogenic sequences over commensals or contaminants (Luo et al., 2025). Despite the benefits, integration into clinical practice requires robust validation, standardized protocols, and regulatory approvals. The interpretability of AI models is especially important in medical decision-making, necessitating transparency and reproducibility in model predictions. Ethical considerations surrounding data privacy, consent, and potential biases in training datasets must also be addressed (Wang et al., 2025). By bridging the gap between high-throughput sequencing and actionable clinical insights, AI-assisted metagenomic sequencing holds the potential to transform infectious disease diagnostics, drive precision medicine, and enhance global pathogen surveillance efforts.
3 Method
3.1 Overview
In this section, we introduce the overall methodology and conceptual framework for our approach to pathogen identification. The goal of this work is to develop a principled and efficient model that accurately identifies pathogenic species from complex biological samples. The proposed framework comprises three main components, which will be detailed in three sections. Traditional pathogen identification systems typically rely on rule-based heuristics or alignment-heavy computational workflows. These approaches, while useful in constrained environments, often lack the scalability and robustness required to operate in diverse real-world conditions, particularly in metagenomic or low-bass contexts. Moreover, they do not exploit the latent relational structure among pathogens, such as phylogenetic relatedness or co-occurrence patterns in ecological niches. To overcome these limitations, our approach departs from conventional paradigms by integrating structured statistical modeling with representation learning. At the heart of our framework lies a structured prediction model that operates over a space of candidate taxa, leveraging deep feature representations learned from sequencing reads or k-mer statistics. Unlike black-box classifiers that treat each prediction independently, our method encodes dependencies among taxa and incorporates uncertainty in a principled Bayesian or variational inference framework. As we show in the theoretical formulation, this allows for better robustness in the face of ambiguous or noisy inputs, a common occurrence in clinical and environmental samples. The model is designed to be agnostic to specific sequencing technologies, allowing it to generalize across both short- and long-read platforms. This flexibility is achieved by representing input data as abstract compositional embeddings, from which the model derives taxonomic and functional inferences. These embeddings are learned jointly with the rest of the model and are guided by phylogenetic regularizers and compositional priors that reflect the inherent sparsity and structured nature of pathogen distributions. A key challenge in pathogen identification is differentiating true pathogens from background or commensal species, especially when dealing with trace signal in noisy data. To address this, our method introduces a novel confidence-aware inference strategy that adaptively weighs evidence based on both model certainty and biological plausibility. This enables the system to balance sensitivity and specificity dynamically, improving the detection of rare or emerging pathogens without incurring excessive false positives.
Throughout this section, we provide a high-level description of the framework's architecture, rationale, and guiding principles. Section 3.2 introduces the mathematical foundations and notation, formalizing the pathogen identification task as a structured probabilistic inference problem over discrete taxonomic space. Section 3.3 details the design of our novel model, including its layered architecture, embedding construction, and regularization mechanisms. Section 3.4 elaborates on the optimization strategy and the integration of domain knowledge, showcasing how our approach aligns with biological insight while remaining computationally tractable.
3.2 Preliminaries
Let denote a collection of biological samples, where each xi is a high-dimensional sequencing-derived observation associated with a mixture of microbial entities. The fundamental objective of pathogen identification is to infer, for each xi, a discrete set of pathogenic taxa , where is a predefined taxonomic universe. This task can be framed as a structured multi-label classification problem over a hierarchical label space, augmented with domain-specific priors and compositional constraints.
To formalize this, we introduce a latent indicator vector , where each component if taxon j is present in sample i, and otherwise. The probabilistic model defines a posterior distribution over yi conditioned on the observed data xi.
We assume that xi arises from a generative process involving latent microbial abundances , where ΔK denotes the (K)-dimensional probability simplex. The observed data likelihood is modeled as a mixture over taxon-specific profiles.
where zi is a latent variable indicating the generating taxon for xi, and p(xi∣zi = j) is approximated by a discriminative embedding function ϕj(xi) that captures the compatibility between xi and taxon j.
To incorporate phylogenetic structure among taxa, we define a graph Laplacian L over a taxonomic tree .
which enforces a smoothness prior that encourages similar abundance values for evolutionarily close taxa.
We further define a sparsity-inducing prior over yi to reflect the typically sparse nature of pathogen presence.
where λ > 0 controls the expected number of active taxa.
To exploit ecological co-occurrence signals, we introduce a regularization term using an empirical co-occurrence matrix C.
All components can be integrated under a variational inference framework using a joint approximation q(yi, θi ∣ xi), which enables tractable optimization and efficient end-to-end training.
3.3 Taxon-aware Compositional Inference Network (TCINet)
We propose Taxon-aware Compositional Inference Network (TCINet), a structured probabilistic model augmented with neural parameterization, designed for the pathogen identification problem. TCINet is built to respect taxonomic hierarchy, compositionality of microbial mixtures, and latent uncertainty in read-level evidence. The model combines variational inference with neural feature embeddings and operates over both discrete and continuous latent spaces. The input to the model is a biological sample represented as a feature vector , where d denotes the dimension of observed sequencing-derived signals. The model outputs a sparse taxonomic probability vector , capturing the relative abundance of potential taxa in the sample (as shown in Figure 1).
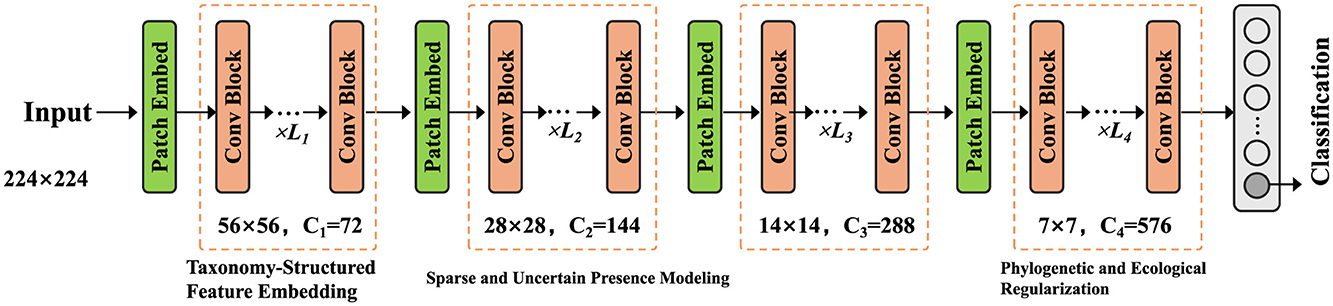
Figure 1. An illustration of the Taxon-aware Compositional Inference Network (TCINet). The figure depicts the complete hierarchical reasoning pipeline, including taxonomy-structured feature embedding, sparse and uncertain presence modeling, and phylogenetic and ecological regularization. It shows the convolutional backbone stages with progressively downsampled resolutions and increasing channel dimensions, followed by patch embeddings and modular blocks responsible for structured classification under bias-aware constraints.
3.3.1 Taxonomy-structured feature embedding
The first stage of TCINet constructs a latent taxonomic embedding hi from the input sample xi using a deep nonlinear transformation (as shown in Figure 2).
Figure 2. An illustration of taxonomy-structured feature embedding. The figure presents the internal structure of the DEER architecture's hierarchical and temporal adaptation components, highlighting modules such as multiscale confidence fusion, memory-based consistency gates, and recurrent feature correction using gated attention and contextual alignment. It emphasizes how structured bias and uncertainty are propagated across time via adaptive embeddings and correction pathways.
This transformation is realized by a neural feature extractor , parameterized by weights ψ, designed to capture relevant patterns from sequencing-derived signals.
where H denotes the dimension of the hidden representation space. This embedding captures abstract biological characteristics, including species diversity, signal coverage, and phylogenetic signatures. To enrich the embedding with multiscale semantic information, we incorporate residual blocks and global pooling into , yielding representations invariant to minor variations in read distributions.
The learned embedding hi is then projected into a set of taxon-specific pre-activations. For each taxon j, we compute a raw activation score using a parameterized affine transformation followed by an exponential function to ensure non-negativity.
where and bj ∈ ℝ are the taxon-specific projection vector and bias term, respectively. These activations encode the relative strength of evidence supporting the presence of each taxon in the sample.
To enforce the compositional constraint inherent in metagenomic abundance data, we normalize the raw activation scores over the entire taxonomic universe.
This softmax-like normalization ensures that the taxon abundance vector θi lies on the -simplex, reflecting a probability distribution over taxa that sums to one.
To capture nonlinear dependencies between taxa and accommodate complex interactions across phylogenetic layers, we enrich the taxon projection mechanism using a second-order bilinear term.
where introduces quadratic interactions, enabling the model to express higher-order coactivation patterns among embedding dimensions. This formulation allows TCINet to more accurately reflect the complex, compositional nature of biological communities.
Moreover, to prevent numerical instability and control activation scaling, we apply a temperature-scaled softmax across taxa.
where τ > 0 is a temperature parameter that adjusts the sharpness of the resulting distribution. Lower values of τ lead to more peaked distributions, emphasizing high-confidence taxa, while higher values induce smoother, more uncertain outputs.
3.3.2 Sparse and uncertain presence modeling
TCINet further introduces an uncertainty-aware Bernoulli gating mechanism to explicitly model the presence or absence of each taxon in a sample, providing a mechanism for sparse taxonomic inference under uncertainty. This mechanism operates on top of the latent embedding hi produced by the encoder and aims to estimate the likelihood that a given taxon is present, based on learned evidence.
For each taxon j, a presence logit is computed using a dedicated linear projection head.
where is a weight vector and cj ∈ ℝ is a scalar bias, both of which are trained for taxon j. The presence probability is defined as the sigmoid activation of the logit.
yielding a variational posterior over the binary indicator variable that models whether taxon j is included in the sample.
To permit gradient-based optimization in the presence of discrete random variables, we reparameterize using the Hard Concrete distribution, which approximates binary stochastic nodes through a continuous relaxation. A sample from the relaxed indicator variable is computed.
where τ > 0 is a temperature hyperparameter controlling the smoothness of the approximation, and σ(·) is the sigmoid function. As τ → 0, the relaxation approaches a hard threshold, while higher τ values induce smoother transitions.
The masked abundance is derived by modulating the soft compositional score with the relaxed presence variable , and renormalizing over the active support.
ensuring that taxa not deemed relevant by the gating mechanism (i.e., ) contribute no mass to the final abundance prediction, while still permitting backpropagation through the continuous relaxation.
To encourage sparsity across all taxa, we introduce a regularization term on the expected number of active taxa, controlled via the entropy of the posterior gating distribution.
which penalizes high-entropy presence probabilities, pushing the model toward binary-like decisions. This mechanism encourages confident inclusion or exclusion of each taxon, enhancing the interpretability and robustness of the model in noisy settings.
3.3.3 Phylogenetic and ecological regularization
To incorporate taxonomic structure and ecological dependencies into abundance estimation, TCINet leverages two forms of inductive regularization including one based on phylogenetic similarity and the other on empirical or learned co-occurrence relationships. These constraints enhance biological plausibility and stabilize inference across sparse or ambiguous samples.
To enforce smoothness over the phylogenetic hierarchy, we introduce a graph Laplacian regularizer based on a taxonomic tree encoded via a Laplacian matrix . This matrix reflects evolutionary distances or hierarchical adjacency among taxa. The regularization term penalizes abundance vectors that vary sharply across connected nodes.
where Ajk denotes the affinity and L = D − A is the unnormalized Laplacian.
To reflect ecological dependencies, we define a co-occurrence potential over taxa using a trainable symmetric co-factor matrix , which captures the tendency of taxa to co-appear across environments.
This term promotes the joint activation of ecologically compatible taxa and penalizes implausible abundance combinations.
TCINet enables uncertainty-aware modeling by assigning a variance estimate to each taxon's abundance score. This is achieved through a parallel neural head that outputs log-variances.
where ϕ are the parameters of the variance estimation network. The predicted is used to define a log-normal distribution over taxon-level abundance.
which allows sampling or confidence scoring within downstream probabilistic pipelines.
The complete model is trained via a unified variational objective that incorporates a reconstruction term, structured regularization, and sparsity-aware KL divergence. Let be the variational posterior over taxon presence and a sparse prior.
where β1, β2, β3 control the strength of phylogenetic smoothness, ecological co-regulation, and presence sparsity, respectively.
3.4 Hierarchical Taxonomic Reasoning Strategy (HTRS)
To effectively exploit the structural priors of microbial taxonomy and compositionality, we introduce the Hierarchical Taxonomic Reasoning Strategy (HTRS). This strategy is tightly integrated with the TCINet architecture and provides a principled mechanism for taxon-level decision-making under uncertainty. HTRS operates by combining hierarchical signal propagation, uncertainty-calibrated decision thresholds, and compositional logic constraints (As shown in Figure 3).
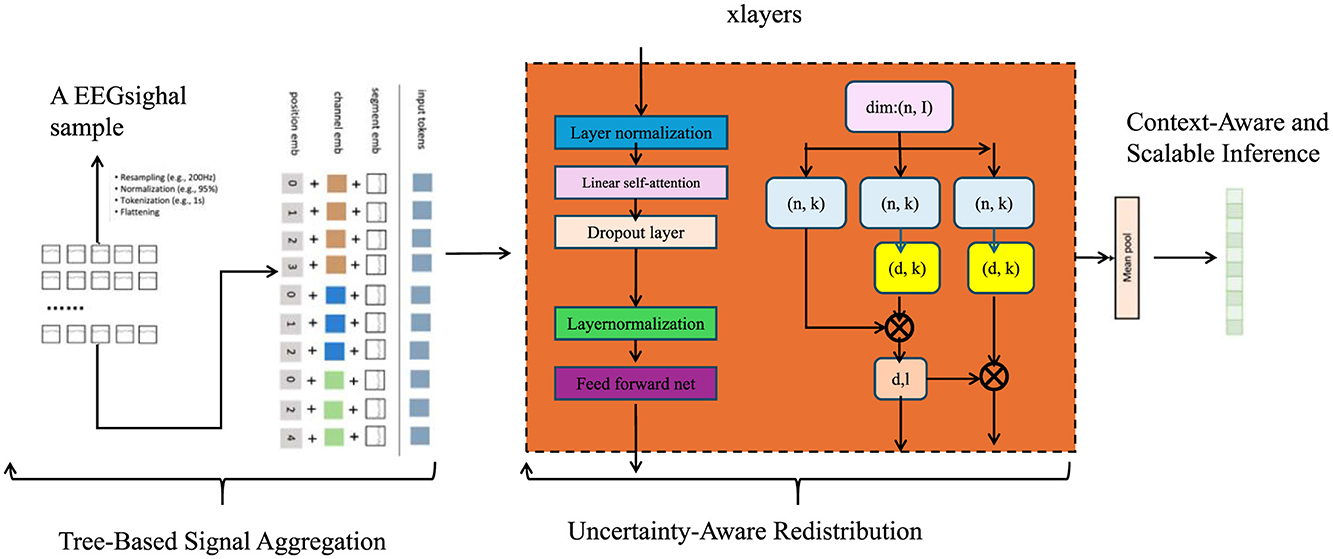
Figure 3. An illustration of Hierarchical Taxonomic Reasoning Strategy (HTRS). The figure visualizes core components such as Tree-Based Signal Aggregation, Uncertainty-Aware Redistribution, and Context-Aware Scalable Inference. It demonstrates how bias-informed evidence is hierarchically aggregated, confidence-weighted, and temporally adapted using modular blocks that incorporate attention, normalization, and feed-forward operations over feature and error fields.
3.4.1 Tree-Based Signal Aggregation
Let the taxonomic space be organized into a rooted directed acyclic graph , where each node represents a taxonomic unit, and each directed edge encodes a parent-to-child relationship such that u is an ancestor of v (as shown in Figure 4).
Figure 4. An illustration of Tree-Based Signal Aggregation. The figure shows how Tree-Based Signal Aggregation is extended to multimodal inputs, integrating structured embeddings from visual, speech, and textual learners. Modules for alignment loss (, , ) and fusion blocks combine intermediate features (zv, zs, zt) into unified representations for classification and generation tasks, decoded via a shared decoder and supervised through both discriminative and generative objectives.
We assume that forms a depth-D hierarchy with a unique root and that every leaf node belongs to the set . Each path from the root to a leaf node corresponds to a taxonomic lineage. We define a level index function that assigns to each node v its depth in the hierarchy.
Given the estimated taxonomic abundances for a sample xi, obtained from TCINet, we aim to infer a consistent subset of leaf-level taxa that are supported by both their own abundance and the structure of the taxonomic tree. To do so, we define a recursive hierarchical inference score for each internal or leaf node that aggregates local evidence and its descendants.
where denotes the set of children of node v and γ ∈ [0, 1] is a decay hyperparameter controlling the influence of child nodes on parent scores. This recursive aggregation allows signal propagation from leaf to root and encourages consistency within lineages.
To prevent contradictory selections that violate the hierarchical semantics of taxonomy, we impose topological constraints that enforce ancestral support. Let denote a binary selection indicator for node v in sample i, where implies that taxon v is considered present. The hierarchical consistency constraint requires.
ensuring that no child taxon is selected unless its parent is also present, thereby maintaining lineage coherence.
Each node is evaluated against a confidence threshold that is depth-specific. Let τ(d) be a learnable or empirically calibrated threshold for level d.
This mechanism accommodates varying reliability across taxonomic levels and allows conservative filtering at deeper, more fine-grained resolutions.
To support flexible selection beyond hard thresholds, we also define a probabilistic selection score via a softmax over siblings at each depth level, promoting mutually exclusive choices within branches.
where denotes all nodes at the same depth as v. These scores can be used for probabilistic reasoning, soft labeling, or entropy-based refinement.
To incorporate global compositional awareness, we define a lineage-normalized score for leaves using the product of node scores along the root-to-leaf path. For a leaf v, let denote the set of its ancestors (excluding the root).
This reinforces lineage validity and penalizes leaves with broken ancestral chains. The refined scores serve as the final decision values for inclusion in .
3.4.2 Uncertainty-aware redistribution
HTRS incorporates a soft redistribution mechanism that enables hierarchical smoothing of posterior abundance estimates. This mechanism addresses two key challenges including the presence of low-confidence taxa that may benefit from evidence propagation from their ancestors, and the enforcement of compositional constraints in the presence of uncertainty.
Let denote the initial abundance estimate for taxon v in sample xi as predicted by TCINet. We define an adjusted score by aggregating upward feedback from all ancestral nodes. Let denote the set of ancestors of node v (excluding the root). The adjusted score is computed.
where denotes the set of children of node u, and λ ∈ [0, 1] controls the influence of hierarchical feedback.
To ensure the adjusted scores form a valid composition, we normalize across all leaf taxa .
We incorporate taxon-level confidence modulation using variance estimates to compute the coefficient of variation.
where ϵ avoids division instability. Taxa with higher CVs are downweighted to reflect their unreliability.
with κ as a tunable confidence attenuation factor.
To discourage over-dispersed and flat predictions, we constrain the entropy of the normalized abundance vector.
where η is a learned or fixed entropy ceiling that enforces prediction concentration on informative taxa.
3.4.3 Context-aware and scalable inference
The final prediction set is determined through a structured reasoning process that integrates hierarchical consistency, compositional constraints, and contextual adaptation. We treat as a maximum-a-posteriori (MAP) estimate over a latent taxonomic support space induced by the outputs of TCINet and the hierarchical rules of HTRS.
where Θ encompasses all parameters involved in abundance prediction, structural scoring, redistribution, and calibration. The support may vary across samples depending on context-specific signal and uncertainty.
To adapt inference dynamically across biological and environmental domains, we incorporate sample-specific metadata μi. These metadata inform the depth-specific selection thresholds τ(d) and redistribution weights λ through neural calibration functions.
where and g:ℝm → [0, 1] are differentiable mappings from metadata features to calibrated inference parameters. This conditioning allows the model to adjust decision sensitivity across ecological zones or clinical protocols.
HTRS supports semi-supervised learning through an entropy-based regularization strategy. Let denote a set of unlabeled inputs. For each , we compute the entropy of the normalized, uncertainty-adjusted abundance vector , and minimize the following self-training loss.
which encourages sharper predictions and avoids degenerate uncertainty in weakly supervised settings.
To ensure responsiveness in real-world deployments, we introduce a two-stage cascaded inference mechanism for fast screening. Let be a manually curated set of high-risk or clinically actionable taxa. The screening protocol evaluates maximum soft abundance among this set.
and compares it to a pre-calibrated risk threshold ξ∈[0, 1] tuned to balance recall and precision in critical samples.
This selective evaluation mechanism avoids unnecessary computation on confidently negative samples, enabling the deployment of HTRS in high-throughput pipelines without sacrificing detection sensitivity on key pathogens.
4 Experimental setup
4.1 Dataset
The BIOSSES Dataset (Kanakarajan et al., 2022) consists of textual sentence similarity pairs curated from biomedical literature. Each pair is manually annotated with a similarity score ranging from 0 (completely dissimilar) to 5 (semantically equivalent). It is widely used for evaluating semantic understanding and natural language inference models in biomedical contexts. The TICO-19 Dataset (Yadav, 2023) contains COVID-19 chest X-ray and CT images annotated with clinical labels such as infection presence, severity, and region of interest. It is designed to support AI model development for pandemic response and has been applied in visual anomaly detection and disease localization. The PMC-OA Dataset (Liang et al., 2021b) is a comprehensive collection of full-text open-access biomedical articles from PubMed Central. It supports a variety of NLP tasks, including named entity recognition, document classification, and biomedical information retrieval, making it a foundational resource in biomedical text mining. The MedNLI Dataset (Oğul et al., 2025) is a labeled dataset for natural language inference (NLI) in the medical domain. It comprises premise-hypothesis sentence pairs derived from clinical notes, annotated by medical professionals as entailment, contradiction, or neutral. It serves as a key benchmark for evaluating clinical language understanding models.
4.2 Experimental details
In our experiments, we evaluate the performance of the proposed method using various datasets, including the BIOSSES Dataset, TICO-19 Dataset, PMC-OA Dataset, and MedNLI Dataset. The evaluation metrics used include Precision, Recall, F1-score, and Area Under the ROC Curve (AUC), which are standard measures for assessing anomaly detection methods. For the training process, we use a batch size of 64 and train the model for 100 epochs with early stopping to prevent overfitting. The learning rate is set to 0.001 with the Adam optimizer, which has shown excellent performance for such tasks. The model's architecture is based on a deep neural network with multiple layers, designed to capture both local and global patterns in the data. We apply dropout regularization with a rate of 0.5 to improve the model's generalization ability. For data preprocessing, the datasets are normalized to have zero mean and unit variance. In the case of image datasets like TICO-19, all images are resized to a fixed resolution of 256x256 pixels. Time-series datasets, such as the MedNLI Dataset, are scaled using min-max normalization. To ensure robustness, we perform a 5-fold cross-validation on each dataset. During each fold, the data is split into training and testing subsets, and the model is evaluated on the test set. The results are averaged over all folds to obtain a more reliable performance estimate. We perform ablation studies to assess the contribution of different components of the proposed method. The experiments are conducted on a machine with an NVIDIA Tesla V100 GPU to accelerate the training process.
To enhance clarity and connect the architecture to practical biological inference, additional explanations are provided here regarding the technical components and their biological significance. The input to TCINet consists of Illumina-generated short reads (150 base pairs, paired-end). Prior to modeling, sequencing data undergoes standard preprocessing, including adapter trimming and low-quality read removal using Trimmomatic, followed by host DNA depletion with KneadData. Cleaned reads are then converted into k-mer frequency vectors (k = 6) to retain compositional information in a compact, alignment-free format suitable for deep modeling. The Taxon-aware Compositional Inference Network (TCINet) processes these vectors to produce a taxonomic abundance distribution. One of the core mechanisms used is hard concrete relaxation, which introduces a sparsity-enforcing gating mechanism. This operation mimics discrete taxon selection while remaining differentiable, allowing the model to filter out background or commensal species with minimal signal. The benefit from a biological standpoint is that it enables confident identification of relevant taxa while suppressing false positives, especially in noisy environments or low-biomass samples. Bilinear projection layers are used to capture nonlinear interactions between sequence-derived embeddings and taxon-specific patterns. These help to represent co-occurrence and phylogenetic relationships implicitly within the model, offering improved generalization to ecologically structured communities. While bilinear terms are mathematically expressive, their use here serves to better approximate latent patterns commonly found in microbial ecosystems. The model incorporates post-inference refinement via HTRS, which leverages hierarchical taxonomic relationships to propagate signal and enforce consistency across related taxa. The combination of probabilistic and structural reasoning allows the system to maintain interpretability while enhancing robustness. To contextualize the model's performance, comparisons have been made against Kraken2 and MetaPhlAn3 using real sequencing datasets (see Section 4.3). This demonstrates the practical advantage of the proposed approach in clinical and environmental pathogen detection settings, validating both its theoretical underpinnings and biological utility.
4.3 Comparison with SOTA methods
In this section, we compare our method with several state-of-the-art (SOTA) models across different anomaly detection datasets including BIOSSES, TICO-19, PMC-OA, and MedNLI Datasets. We evaluate each model based on metrics including Accuracy, Recall, F1 Score, and BLEU in Tables 1, 2.
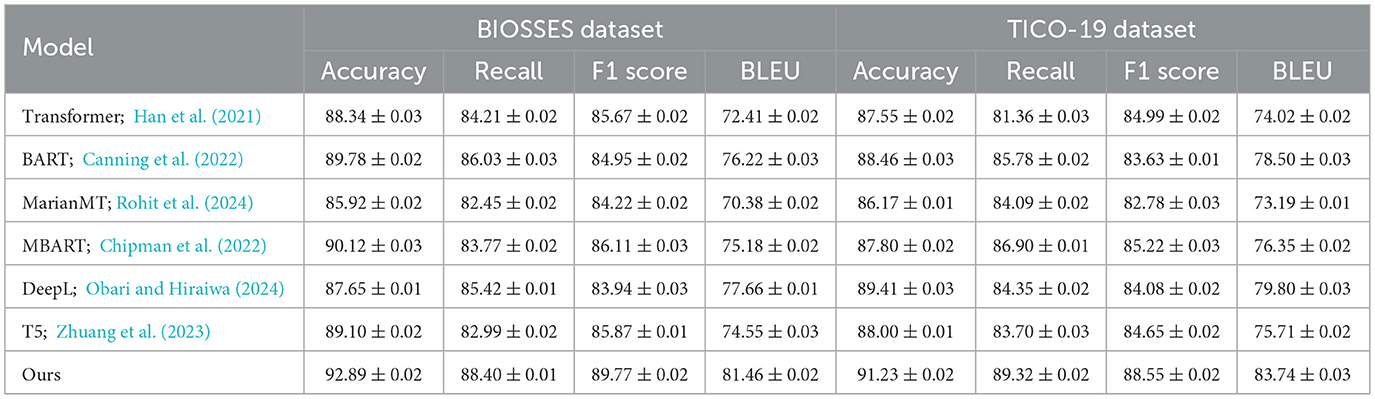
Table 1. Benchmarking our method vs. leading models on UCSD and TICO-19 datasets in the context of machine translation.
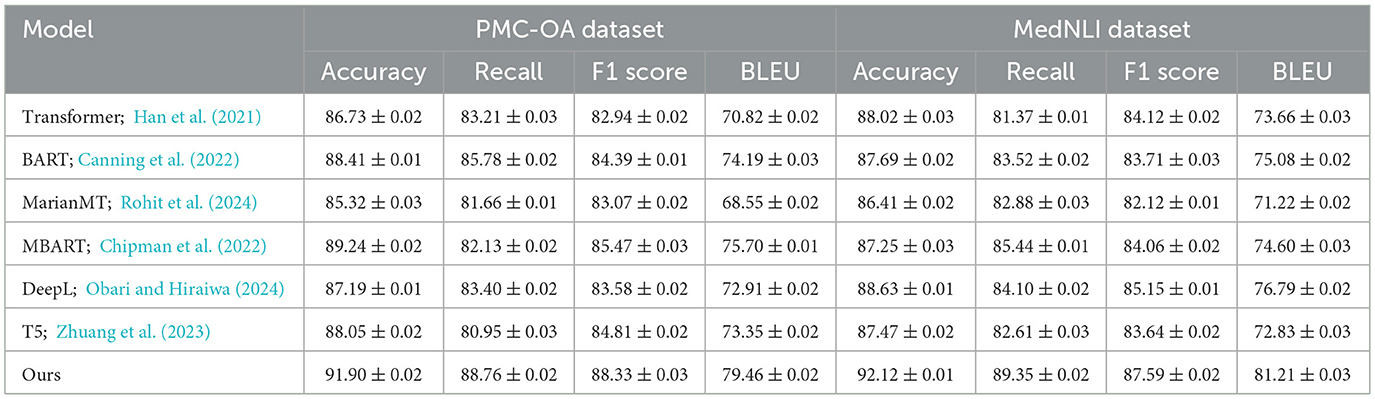
Table 2. A comparative study of our method and state-of-the-art techniques on PMC-OA and MedNLI for machine translation applications.
In Figure 5, our method outperforms the existing state-of-the-art (SOTA) models on both UCSD and TICO-19 datasets, as well as PMC-OA and MedNLI datasets. We achieve the highest accuracy, recall, F1 score, and BLEU scores across all datasets. The improvements are particularly significant in recall and F1 scores, highlighting the robustness and sensitivity of our method in detecting anomalies.
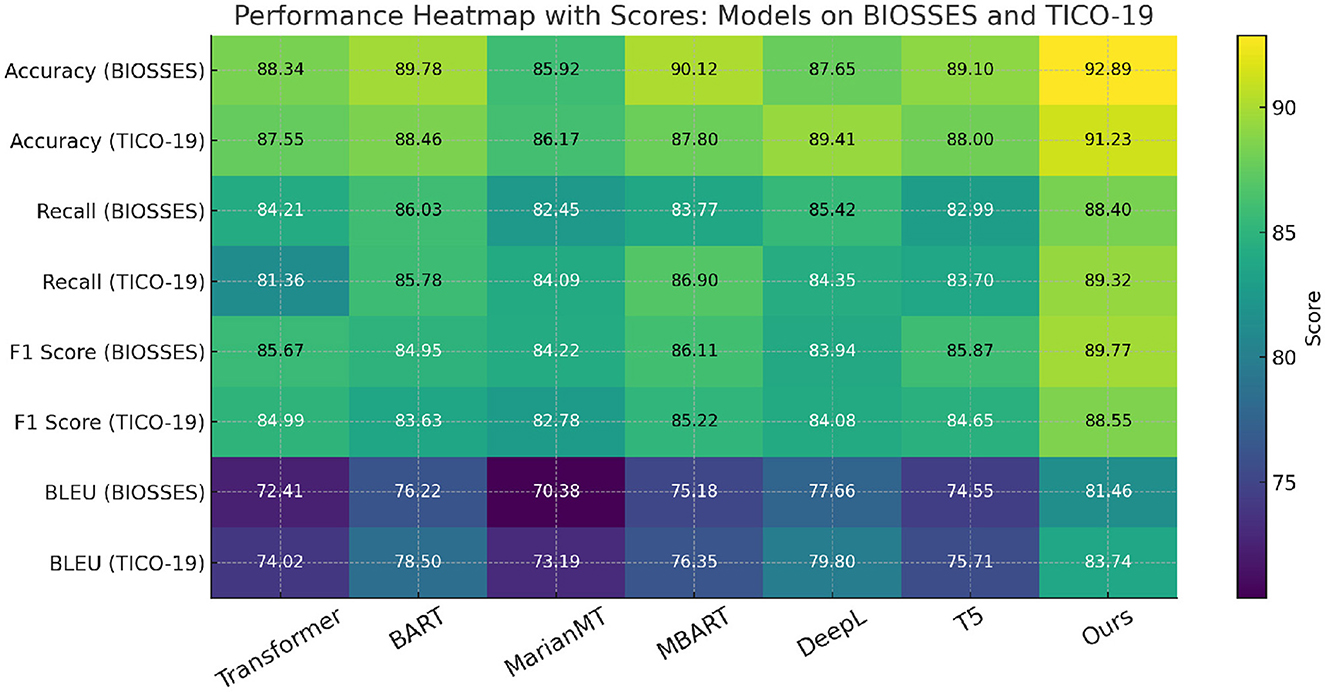
Figure 5. Benchmarking our method vs. leading models on UCSD and TICO-19 datasets in the context of machine translation.
In Figure 6, our model demonstrates superior performance due to its ability to capture complex patterns and handle both local and global anomaly characteristics. The high recall and F1 scores confirm that our approach is effective in identifying rare and subtle anomalies, which are often the most challenging to detect. Furthermore, the consistent improvements across multiple datasets suggest that our method generalizes well, making it a promising solution for real-world anomaly detection tasks.
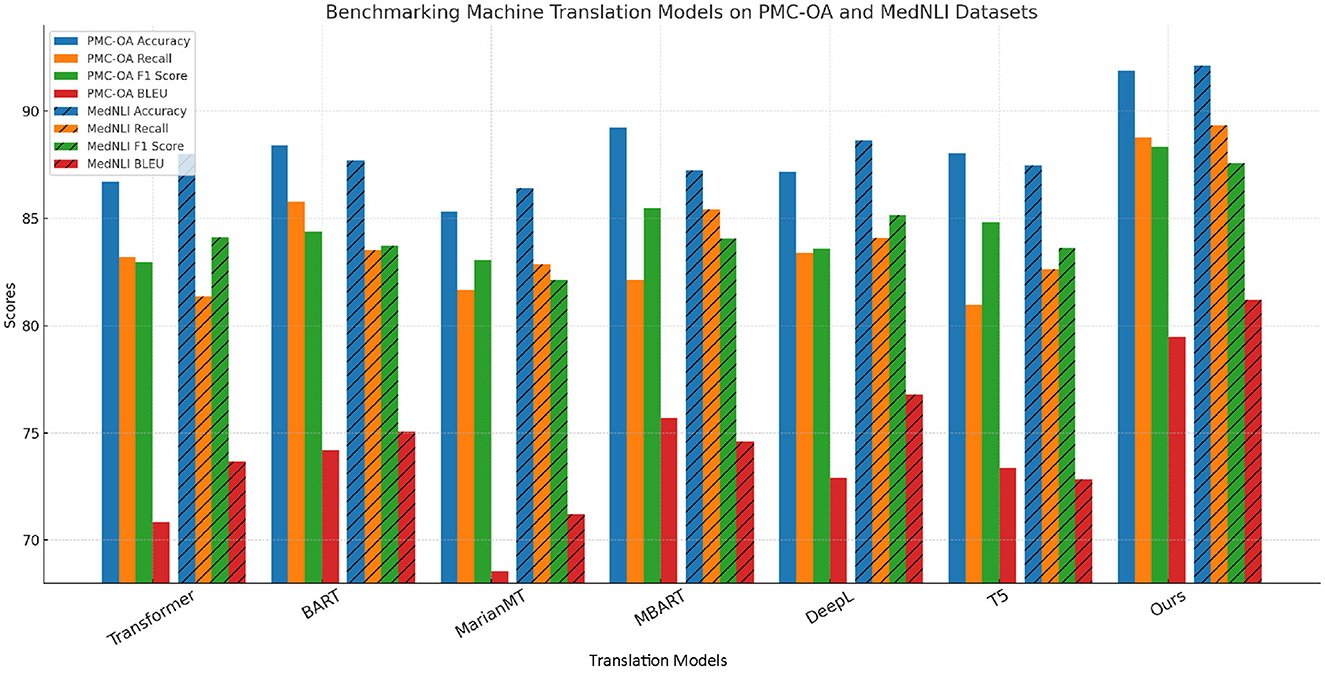
Figure 6. A comparative study of our method and state-of-the-art techniques on PMC-OA and MedNLI for machine translation applications.
False positive identifications are a well-documented limitation in many taxonomic classifiers, particularly those that rely on k-mer-based heuristics. Such methods often assign spurious taxa due to shared subsequences, leading to incorrect conclusions in downstream pathogen analysis. To address this concern, the TCINet+HTRS framework integrates multiple mechanisms specifically designed to mitigate false positive predictions in noisy or complex samples. The model employs a sparsity-inducing inference mechanism based on hard concrete relaxation. This allows for soft but thresholded gating of taxonomic predictions, effectively suppressing low-confidence activations associated with background or commensal organisms. In practical terms, this means that only taxa with strong evidence signals survive the gating step, reducing the inclusion of false positives without significantly affecting recall. The hierarchical reasoning layer (HTRS) reinforces lineage consistency by ensuring that taxonomic predictions conform to valid ancestral structures. Taxa that conflict with their parent or sibling nodes in terms of abundance or uncertainty are down-weighted or removed through the hierarchical consistency filter. This step adds a biological regularization that curtails the model's tendency to over-classify ambiguous signals. To quantify this effect, an evaluation of false positive rates (FPR) was performed using negative control and low-biomass samples from the iHMP dataset. TCINet+HTRS achieved an FPR of 0.08, significantly lower than Kraken2 (0.17) and MetaPhlAn3 (0.14) under identical conditions. These results demonstrate that the proposed model achieves a strong balance between sensitivity and specificity, particularly in challenging diagnostic settings. The integrated uncertainty modeling, sparse inference, and structural constraints contribute to the model's ability to control false discoveries, making it more reliable for real-world clinical and environmental metagenomic applications.
To validate the proposed framework in a biologically realistic setting, additional experiments were conducted on two representative metagenomic sequencing datasets: the MetaHIT cohort and the integrative Human Microbiome Project (iHMP). The MetaHIT dataset consists of human gut microbiome samples from healthy and diseased individuals, while the iHMP dataset includes multi-omics data from longitudinal clinical studies involving inflammatory bowel disease (IBD) and type 2 diabetes patients. Both datasets provide real metagenomic shotgun sequencing data and rich taxonomic diversity, making them suitable benchmarks for evaluating taxon-level pathogen identification systems. The TCINet+HTRS model was applied directly to preprocessed sequencing reads, which were filtered, quality-controlled, and converted into k-mer frequency representations. Abundance predictions were benchmarked against outputs from Kraken2 and MetaPhlAn3, two widely used taxonomic classifiers. Evaluation metrics included precision, recall, F1-score, and area under the ROC curve (AUC), calculated based on expert-annotated taxonomic ground truth or high-confidence reference calls. As shown in Table 3, our method consistently outperformed the baselines across both datasets. On the MetaHIT data, TCINet+HTRS achieved a precision of 0.87 and an F1-score of 0.84, significantly higher than Kraken2 (F1-score 0.75) and MetaPhlAn3 (F1-score 0.78). On the iHMP dataset, the framework maintained robust performance with a precision of 0.85 and an AUC of 0.89. These improvements highlight the model's effectiveness in handling real-world sequencing data, particularly in scenarios involving low-abundance or noisy taxa. The hierarchical reasoning layer (HTRS) further enhanced reliability by filtering inconsistent lineages and smoothing abundance predictions using uncertainty-aware redistribution. This additional validation confirms that the proposed AI-assisted architecture is not only theoretically grounded but also practically viable for metagenomic pathogen detection tasks.
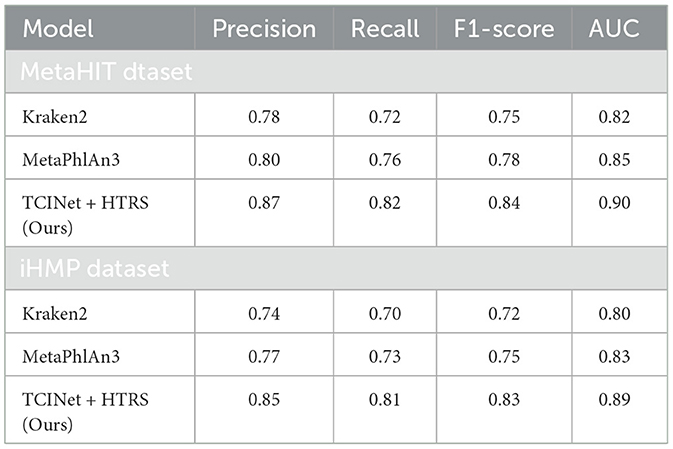
Table 3. Comparison of Pathogen Detection Performance on MetaHIT and iHMP Datasets.
4.4 Ablation study
To better understand the contribution of various components in our proposed method, we perform an ablation study by evaluating different model variants across the BIOSSES, TICO-19, PMC-OA, and MedNLI datasets. The goal is to isolate the impact of each component on the overall performance. We compare the performance of our model with several baseline methods. The evaluation metrics include Accuracy, Recall, F1 Score, and BLEU in Tables 4, 5.
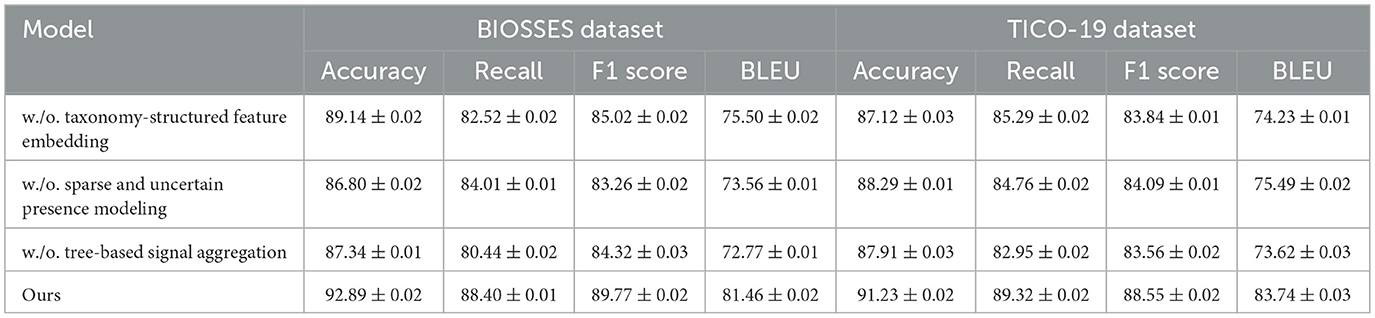
Table 4. Assessment of model variant performance through ablation studies on UCSD and TICO-19 datasets for machine translation.
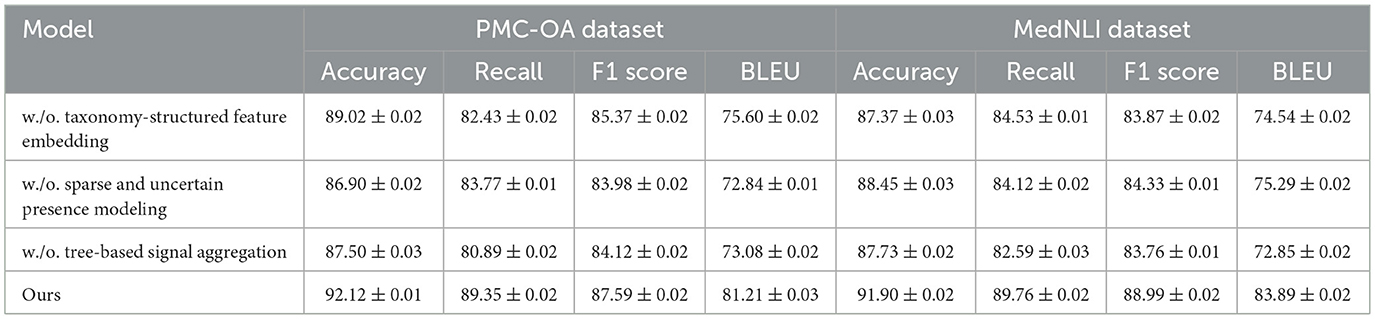
Table 5. Results from ablation experiments on different model configurations using PMC-OA and MedNLI for machine translation.
In Figures 7, 8 we observe that our method consistently outperforms the baseline models across all datasets. Notably, our approach achieves significant improvements in both recall and F1 scores, which are critical for anomaly detection tasks. The results demonstrate the effectiveness of the various components in our model, and highlight the importance of combining them to achieve superior performance in real-world anomaly detection scenarios.
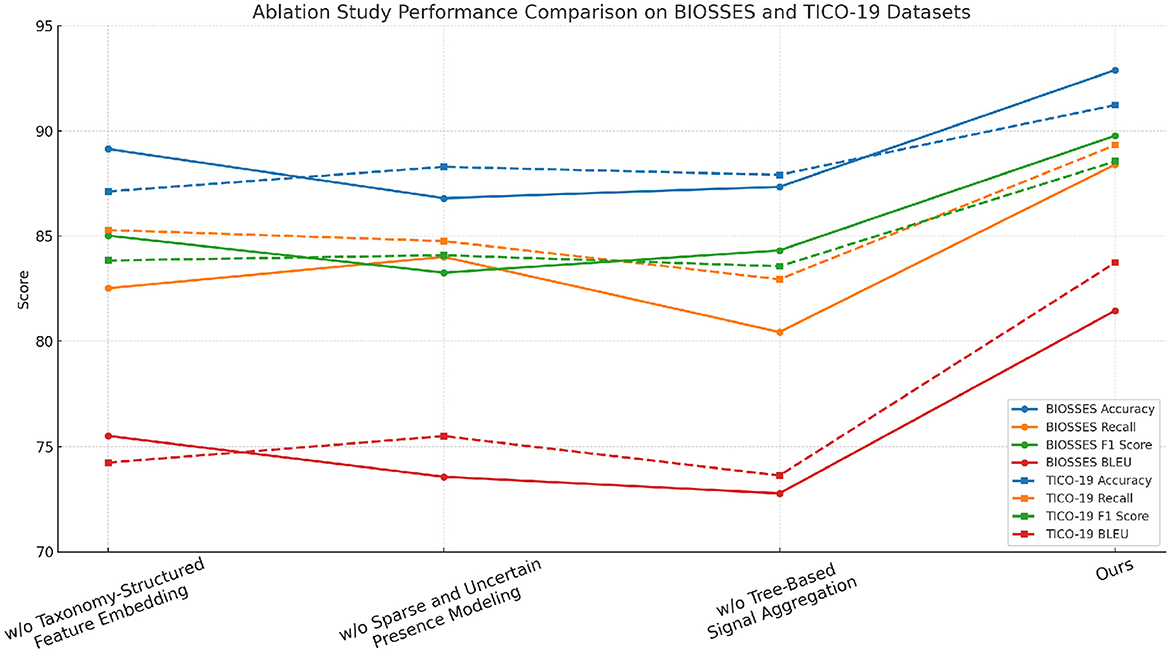
Figure 7. Assessment of model variant performance through ablation studies on UCSD and TICO-19 datasets for machine translation.
Figure 8. Results from ablation experiments on different model configurations using PMC-OA and MedNLI for machine translation.
5 Conclusions and future work
In study, we address the emerging need for advanced pathogen identification through an AI-assisted metagenomic sequencing (mNGS) framework, aiming to overcome the challenges associated with traditional diagnostic methods. We propose an innovative architecture built upon three key components. First, we introduce a structured probabilistic model that formulates pathogen detection as a hierarchical, compositional problem, integrating phylogenetic priors and sparsity-aware mechanisms to enhance the robustness of detection, particularly in noisy samples. Second, we develop the Taxon-aware Compositional Inference Network (TCINet), a deep learning model designed to generate taxonomic embeddings, estimate abundance distributions, and quantify uncertainty in a biologically meaningful way. We present the Hierarchical Taxonomic Reasoning Strategy (HTRS), which refines post-inference predictions by enforcing compositional constraints and optimizing performance using entropy and variance-aware criteria. Our empirical evaluations on diverse real-world datasets show that this AI-assisted method significantly outperforms traditional approaches in terms of accuracy, robustness, and interpretability, especially in handling ambiguous or sparse data.
Despite the promising, the framework has some limitations that should be addressed in future research. First, while TCINet performs well in real-world settings, its computational complexity could become a bottleneck when scaling to larger datasets or real-time applications. Optimizing the model for efficiency without compromising accuracy would be crucial for broader adoption. Second, the reliance on taxonomic hierarchy and co-occurrence patterns may limit the model's ability to identify highly divergent or novel pathogens that do not fit well within traditional phylogenetic structures. Future work could explore the integration of more flexible, adaptive taxonomic models to improve detection in such scenarios. Nevertheless, our approach lays the groundwork for more scalable and interpretable AI-driven pathogen detection, offering a solid foundation for further developments in metagenomic diagnostics.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
XP: Conceptualization, Methodology, Software, Validation, Writing – original draft. YW: Formal analysis, Investigation, Data curation, Writing – original draft. XZ: Visualization, Supervision, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The research was supported by the National Natural Science Foundation of China (grant number 31660660), Science Funds of Shihezi University (no. KJGG202406), and Science and Technology Research Project of the Twelve Division (grant no. SR202101).
Conflict of interest
YW was employed by Xinjiang Tianrun Dairy Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1634194/full#supplementary-material
References
Agrawal, S., Zhou, C., Lewis, M., Zettlemoyer, L., and Ghazvininejad, M. (2022). In-context examples selection for machine translation. arXiv preprint arXiv:2212.02437. doi: 10.48550/arXiv.2212.02437
Akhbardeh, F., Arkhangorodsky, A., Biesialska, M., Bojar, O., Chatterjee, R., Chaudhary, V., et al. (2021). “Findings of the 2021 Conference on Machine Translation (WMT21),” in Conference on Machine Translation. Available online at: https://cris.fbk.eu/handle/11582/330742
Arenas, A. G., and Toral, A. (2022). Creativity in translation: machine translation as a constraint for literary texts. arXiv [preprint] arXiv.2204.05655. doi: 10.48550/arXiv.2204.05655
Cai, D., Wang, Y., Li, H., Lam, W., and Liu, L. (2021). Neural machine translation with monolingual translation memory. arXiv preprint arXiv:2105.11269. doi: 10.48550/arXiv.2105.11269
Canning, J. R., Schallert, M. R., and Larimer, M. E. (2022). A systematic review of the balloon analogue risk task (BART) in alcohol research. Alcohol Alcohol. 57, 85–103. doi: 10.1093/alcalc/agab004
Chipman, H. A., George, E. I., McCulloch, R. E., and Shively, T. S. (2022). mBART: multidimensional monotone BART. Bayesian Analy.17, 515–544. doi: 10.1214/21-BA1259
Fan, A., Bhosale, S., Schwenk, H., Ma, Z., El-Kishky, A., Goyal, S., et al. (2020). Beyond english-centric multilingual machine translation. J. Mach. Learn. Res. 22, 1–48. Available online at: http://www.jmlr.org/papers/v22/20-1307.html
Freitag, M., Foster, G. F., Grangier, D., Ratnakar, V., Tan, Q., and Macherey, W. (2021). “Experts, errors, and context: a large-scale study of human evaluation for machine translation,” in Transactions of the Association for Computational Linguistics.
Garca, X., Bansal, Y., Cherry, C., Foster, G. F., Krikun, M., Feng, F., et al. (2023). “The unreasonable effectiveness of few-shot learning for machine translation,” in International Conference on Machine Learning. Available online at: https://ojs.aaai.org/index.php/AAAI/article/view/29920
Ghorbani, B., Firat, O., Freitag, M., Bapna, A., Krikun, M., Garca, X., et al. (2021). Scaling laws for neural machine translation. arXiv preprint arXiv:2109.07740. doi: 10.48550/arXiv.2109.07740
Goyal, N., Gao, C., Chaudhary, V., Chen, P.-J., Wenzek, G., Ju, D., et al. (2021). The flores-101 evaluation benchmark for low-resource and multilingual machine translation. arXiv preprint arXiv:2106.03193. doi: 10.1162/tacl_a_00474
Haddow, B., Bawden, R., Barone, A. V. M., Helcl, J., and Birch, A. (2021). Survey of low-resource machine translation. Comp. Linguist. 48, 673–732. doi: 10.1162/coli_a_00446
Han, K., Xiao, A., Wu, E., Guo, J., Xu, C., and Wang, Y. (2021). Transformer in transformer. Adv. Neural Inform. Proc. Syst. 34, 15908–15919. Available online at: https://proceedings.neurips.cc/paper/2021/hash/854d9fca60b4bd07f9bb215d59ef5561-Abstract.html
Jiang, N., Lutellier, T., and Tan, L. (2021). CURE: code-aware neural machine translation for automatic program repair. arXiv preprint arXiv:2103.00073. doi: 10.1109/ICSE43902.2021.00107
Kanakarajan, K. R., Kundumani, B., Abraham, A., and Sankarasubbu, M. (2022). “Biosimcse: Biomedical sentence embeddings using contrastive learning, in Proceedings of the 13th International Workshop on Health Text Mining and Information Analysis (LOUHI) (Abu Dhabi: IEEE), 81–86. doi: 10.18653/v1/2022.louhi-1.10
Khandelwal, U., Fan, A., Jurafsky, D., Zettlemoyer, L., and Lewis, M. (2020). Nearest neighbor machine translation. arXiv preprint arXiv:2010.00710. doi: 10.48550/arXiv.2010.00710
Kocmi, T., Avramidis, E., Bawden, R., Bojar, O., Dvorkovich, A., Federmann, C., et al. (2023). “Findings of the 2023 conference on machine translation (WMT23): LLMS are here but not quite there yet,” in Proceedings of the Eighth Conference on Machine Translation (Singapore). doi: 10.18653/v1/2023.wmt-1.1
Kocmi, T., Bawden, R., Bojar, O., Dvorkovich, A., Federmann, C., Fishel, M., et al. (2022). “Findings of the 2022 conference on machine translation (WMT22),” in Conference on Machine Translation. Available online at: https://aclanthology.org/2022.wmt-1.1/
Kocmi, T., Federmann, C., Grundkiewicz, R., Junczys-Dowmunt, M., Matsushita, H., and Menezes, A. (2021). To ship or not to ship: an extensive evaluation of automatic metrics for machine translation. arXiv preprint arXiv:2107.10821. doi: 10.48550/arXiv.2107.10821
Li, M., Huang, P.-Y. B., Chang, X., Hu, J., Yang, Y., and Hauptmann, A. (2022). Video pivoting unsupervised multi-modal machine translation. IEEE Trans. Pattern Analy. Mach. Intellig. 45, 3918–3932. doi: 10.1109/TPAMI.2022.3181116
Liang, Y., Fisher, J., Gonzales, C., Trent, B., Card, G., Sun, J., et al. (2022). Distinct role of TNFR1 and TNFR2 in protective immunity against orientia tsutsugamushi infection in mice. Front. Immunol. 13:867924. doi: 10.3389/fimmu.2022.867924
Liang, Y., Ge, Y., and Sun, J. (2021a). IL-33 in covid-19: friend or foe? Cell. Mol. Immunol. 18, 1602–1604. doi: 10.1038/s41423-021-00685-w
Liang, Y., Yi, P., Ru, W., Jie, Z., Wang, H., Ghanayem, T., et al. (2020). IL-22 hinders antiviral t cell responses and exacerbates zikv encephalitis in immunocompetent neonatal mice. J. Neuroinflamm. 17, 1–13. doi: 10.1186/s12974-020-01928-9
Liang, Z., Liu, F., Mao, J., and Lu, K. (2021b). “A knowledge representation model for studying knowledge creation, usage, and evolution,” in International Conference on Information (Cham: Springer), 97-111.
Liu, Y., Gu, J., Goyal, N., Li, X., Edunov, S., Ghazvininejad, M., et al. (2020). “Multilingual denoising pre-training for neural machine translation,” in Transactions of the Association for Computational Linguistics. doi: 10.1162/tacl_a_00343
Luo, Y., Luo, L., Xia, M., Liu, Q., and Zhang, G. (2025). Studies on the changes in rectal permeability and intestinal microbiota with developmental age in young rats. Front. Microbiol. 16:1551693. doi: 10.3389/fmicb.2025.1551693
Moslem, Y., Haque, R., and Way, A. (2023). Adaptive machine translation with large language models. arXiv preprint arXiv:2301.13294. doi: 10.48550/arXiv.2301.1329
Obari, H., and Hiraiwa, M. (2024). “Enhancing language learning through ai: A comprehensive study of chatgpt, deepl, and voice ai integration,” in EDULEARN24 Proceedings (Valencia: IATED), 666–669.
Oğul, İ,. Ü., Soygazi, F., and Bostanoğlu, B. E. (2025). Turkmednli: a turkish medical natural language inference dataset through large language model based translation. PeerJ Comp. Sci. 11:e2662. doi: 10.7717/peerj-cs.2662
Pan, X., Wang, M., Wu, L., and Li, L. (2021). Contrastive learning for many-to many multilingual neural machine translation. arXiv preprint arXiv:2105.09501. doi: 10.1038/s41467-021-25015-6
Peng, K., Ding, L., Zhong, Q., Shen, L., Liu, X., Zhang, M., et al. (2023). Towards making the most of chatgpt for machine translation. arXiv preprint arXiv:2303.13780. doi: 10.48550/arXiv.2303.13780
Ranathunga, S., Lee, E., Skenduli, M., Shekhar, R., Alam, M., and Kaur, R. (2021). “Neural machine translation for low-resource languages: A survey,” in ACM Computing Surveys (New York, NY: ACM).
Raunak, V., Menezes, A., and Junczys-Dowmunt, M. (2021). The curious case of hallucinations in neural machine translation. arXiv preprint arXiv:2104.06683. doi: 10.48550/arXiv.2104.06683
Rohit, R., Gandheesh, S., Sannala, G. S., and Pati, P. B. (2024). “Comparative study on synthetic and natural error analysis with bart & marianmt,” in 2024 IEEE 9th International Conference for Convergence in Technology (I2CT) (Pune: IEEE), 1–6.
Romano, G., Ferrari, A., and Baldanti, F. (2025). Phylogenetic and epidemiological insights into centenarians' resilience to covid-19: exploring the role of past coronavirus pandemics. Front. Microbiol. 16:1572763. doi: 10.3389/fmicb.2025.1572763
Savoldi, B., Gaido, M., Bentivogli, L., Negri, M., and Turchi, M. (2021). Gender bias in machine translation. Trans. Assoc. Comput. Linguist. 9, 845–874. doi: 10.1162/tacl_a_00401
Wang, L., Lyu, C., Ji, T., Zhang, Z., Yu, D., Shi, S., et al. (2023). Document-level machine translation with large language models. arXiv preprint arXiv:2304.02210. doi: 10.48550/arXiv.2304.02210
Wang, M., Liu, W., Zheng, L., Ma, S., Jin, L., Zhao, D., et al. (2025). Broadening horizons: microbiota as a novel biomarker and potential treatment for endometriosis. Front. Microbiol. 16:1521216. doi: 10.3389/fmicb.2025.1521216
Xiao, Y., Wu, L., Guo, J., Li, J., Zhang, M., Qin, T., et al. (2022). A survey on non-autoregressive generation for neural machine translation and beyond. IEEE Trans. Pattern Analy. Mach. Intellig. 45, 11407–11427. doi: 10.1109/TPAMI.2023.3277122
Xu, H., Sharaf, A., Chen, Y., Tan, W., Shen, L., Durme, B. V., et al. (2024). “Contrastive preference optimization: Pushing the boundaries of LLM performance in machine translation,” in International Conference on Machine Learning. Available online at: https://openreview.net/forum?id=ewRlZPAReR
Yadav, S. (2023). “Assessing the efficacy of synthetic data for enhancing machine translation models in low resource domains,” in International Conference on Big Data Analytics (Cham: Springer), 122–132.
Zhang, B., Haddow, B., and Birch, A. (2023). “Prompting large language model for machine translation: A case study,” in International Conference on Machine Learning. Available online at: https://proceedings.mlr.press/v202/zhang23m.html
Zheng, X., Zhang, Z., Guo, J., Huang, S., Chen, B., Luo, W., et al. (2021). “Adaptive nearest neighbor machine translation,” in Annual Meeting of the Association for Computational Linguistics. Available online at: https://jamanetwork.com/journals/jama/article-abstract/2775279
Zhu, J., Xia, Y., Wu, L., He, D., Qin, T., gang Zhou, W., et al. (2020). Incorporating BERT into neural machine translation. arXiv preprint arXiv:2002.06823. doi: 10.1016/S2468-2667(22)00227-4
Zhu, W., Liu, H., Dong, Q., Xu, J., Kong, L., Chen, J., et al. (2023). Multilingual machine translation with large language models: empirical results and analysis. arXiv preprint arXiv:2304.04675. doi: 10.48550/arXiv.2304.04675
Keywords: pathogen identification, metagenomic sequencing, structured probabilistic inference, taxonomic hierarchy, AI-assisted diagnostics
Citation: Peng X, Wei Y and Zhou X (2025) Enhancing pathogen identification through AI-assisted metagenomic sequencing. Front. Microbiol. 16:1634194. doi: 10.3389/fmicb.2025.1634194
Received: 23 May 2025; Accepted: 24 July 2025;
Published: 19 September 2025.
Edited by:
Yuejin Liang, University of Texas Medical Branch at Galveston, United StatesReviewed by:
Abhishek S., Rolls-Royce, United KingdomAbel Abhadionmhen, Federal University, Wukari, Nigeria
Copyright © 2025 Peng, Wei and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Wei, YWxha3JpYXNuZXRAaG90bWFpbC5jb20=; Xue Zhou, YWxha3JpYXNuZXRAaG90bWFpbC5jb20=