 Ergan Li1,2†
Ergan Li1,2† Senlin Wang3†
Senlin Wang3† Youqin Li3Anke Liuli4Meifang Liang4
Youqin Li3Anke Liuli4Meifang Liang4 Jian Huang5,6,7
Jian Huang5,6,7 Yan Li5,7*Haifang Li8*
Yan Li5,7*Haifang Li8* Zhonghui Feng9*‡
Zhonghui Feng9*‡- 1The Third People's Hospital of Chengdu, Chengdu, Sichuan, China
- 2College of Animal Science and Veterinary Medicine, Southwest Minzu University, Chengdu, China
- 3College of Medicine, Southwest Jiaotong University, Chengdu, China
- 4College of Life Science and Engineering, Southwest Jiaotong University, Chengdu, China
- 5Key Laboratory of Animal Medicine, Southwest Minzu University, Chengdu, Sichuan, China
- 6Veterinary Teaching Hospital, Southwest Minzu University, Chengdu, China
- 7Department of Clinical Veterinary Medicine, College of Animal Science and Veterinary Medicine, Southwest Minzu University, Chengdu, Sichuan, China
- 8College of Life Science, Shandong Agricultural University, Tai'an, China
- 9The Center of Obesity and Metabolic Diseases, Department of General Surgery, The Third People's Hospital of Chengdu, The Affiliated Hospital of Southwest Jiaotong University, Chengdu, Sichuan, China
Purpose: With the evolution of dietary habits, obesity has emerged as a significant global health issue. Numerous studies have demonstrated a close association between obesity and gut microbiota; however, the specific contribution of gut microbiota to varying degrees of obesity remains inadequately understood. Consequently, this study aims to characterize the gut microbiota of individuals across different obesity severity levels.
Methods: We conducted a comprehensive characterization of the gut microbiome in Chinese obese patients and a healthy control group through the application of 16S rRNA gene sequencing, supplemented by metagenomic sequencing. The study cohort was stratified into five distinct categories based on body mass index (BMI): healthy, overweight, and obesity grades I, II, and III.
Results: In obese populations, the gut microbiome structure shifted significantly, with beneficial genera like Faecalibacterium, Roseburia, and Ruminococcus decreasing, and potentially harmful genera such as Blautia, Collinsella, and Streptococcus increasing. These changes impacted host metabolic pathways, including ribosome synthesis, RNA polymerase activity, and DNA repair. Clinical analyses also revealed strong links between specific genera and metabolic markers like lipid metabolism and insulin resistance.
Conclusion: Populations with different obesity traits show unique changes in gut flora. The level of dysbiosis, or imbalance in intestinal microbiota, rises with obesity. These microbial changes are linked to host metabolism, indicating that targeting harmful bacteria and supplementing with beneficial ones from normal-weight populations could effectively reduce obesity.
1 Introduction
The transformation in dietary patterns, particularly the increasing prevalence of high-calorie, high-fat, and high-sugar diets, has contributed to the escalating issue of obesity, which has reached epidemic levels within the past decade and poses a significant challenge to global public health (Isolauri and Laitinen, 2025). According to the World Health Organization (WHO), over 1.9 billion adults worldwide are classified as overweight, with 650 million of these individuals meeting the criteria for obesity (World Health Organization, 2000; Liu et al., 2025). Obesity not only adversely affects individual quality of life but is also strongly linked to a range of chronic diseases, including type 2 diabetes (T2D), cardiovascular disease (CVD), hypertension (HTN), neurological disorders, chronic respiratory disorders, digestive disorders, and various cancers (Maxim et al., 2025; Mayer et al., 2021). These conditions are frequently associated with dysregulation of the gut microbial community.
In China, the body mass index (BMI) is extensively utilized for the classification and evaluation of obesity (Gulati and Murphy, 2025). Nonetheless, various criteria and research findings have indicated that the applicability and accuracy of BMI across different populations remain contentious. A particular study highlighted discrepancies between the BMI criteria employed for assessing overweight and obesity in Chinese populations and those established by the World Health Organization (WHO). Specifically, the WHO defines overweight as a BMI ≥ 25 kg/m2 and obesity as a BMI ≥ 30 kg/m2, whereas the Chinese working group's criteria categorize overweight as a BMI ≥ 24 kg/m2 and obesity as a BMI ≥ 28 kg/m2. These divergent criteria significantly influence the prediction of non-communicable diseases (NCDs) and multimorbidity, with the application of the more stringent WHO criteria potentially offering enhanced benefits in terms of cardiovascular metabolism (Fan et al., 2025). Consequently, obesity is classified into grades I, II, and III according to the WHO's BMI standards (World Health Organization, 2000). Grade I obesity represents the initial phase of obesity, while Grade II obesity (moderate obesity) can exacerbate or precipitate obesity-related comorbidities, including diabetes mellitus, hypertension, and other chronic conditions. Grade III obesity (severe obesity) is associated with the onset of multiple chronic diseases and an elevated risk of mortality. A comprehensive understanding of these obesity classifications and gradations enables individuals to evaluate their own obesity status and implement timely interventions to prevent the progression of obesity and its detrimental impact on health and quality of life.
In recent years, a growing body of research has demonstrated the significant role of the gut microbiota in both the development and reversal of obesity (De Wit et al., 2023). The gut microbiota contributes to the onset and progression of obesity through mechanisms that affect inflammatory responses, appetite regulation, and metabolic processes (Ma and Lee, 2025; Deng et al., 2025; Mishra et al., 2023). Notably, the gut microbiota in obese individuals is characterized by reduced diversity and an imbalance in the proportions of specific microbial communities. Furthermore, the gut microbiota can modulate the host's energy balance and fat storage by producing metabolites such as short-chain fatty acids (SCFAs) (Aja et al., 2025). As a complex ecosystem within the human body, the gut microbiota participates in a variety of physiological processes, including nutrient absorption, metabolite synthesis, immune system regulation, and energy metabolism, thereby exerting a profound influence on the development of various diseases (including obesity) (Doghish et al., 2025; Zhang and Wang, 2024). Consequently, it is imperative to investigate the characteristics of the gut microbiota as a marker for the progression of obesity across individuals with varying degrees of obesity.
While numerous studies have investigated the relationship between obesity and the gut microbiota, there is a paucity of research focusing on the characteristics of the gut microbiota across varying degrees of obesity. Consequently, this study aims to compare the composition and diversity of the gut microbiota among individuals with different obesity levels using 16S rRNA gene sequencing and metagenomic sequencing. The objective is to systematically characterize the gut microbiota of Chinese patients with varying obesity levels, alongside healthy control cohorts, and to explore the potential mechanisms by which the gut microbiota may influence the progression of obesity. This research seeks to provide a novel scientific basis for obesity prevention and treatment, thereby laying the groundwork for the development of personalized medical strategies.
2 Materials and methods
2.1 Research participants and design
In accordance with the World Health Organization (WHO) criteria, the 36 study participants were categorized into five distinct groups based on their Body Mass Index (BMI): normal weight (N = 8, 18.5 ≤ BMI < 24.9 kg/m2), overweight (N = 7, 25.0 ≤ BMI < 29.9 kg/m2), grade 1 obesity (N = 8, 30.0 ≤ BMI < 34.9 kg/m2), grade 2 obesity (N = 8, 35.0 ≤ BMI < 39.9 kg/m2), and grade 3 obesity (N = 5, BMI ≥ 40.0 kg/m2). The obese participants were selected from patients scheduled for bariatric surgery between September 2019 and October 2020 at the Third People's Hospital of Chengdu City, while the control group consisted of healthy volunteers. A majority of the obese patients presented with comorbid conditions such as hypertension, hyperglycemia, fatty liver, type 2 diabetes mellitus, or obstructive sleep apnea syndrome. Furthermore, the study collected clinical indicators, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), high-density lipoprotein (HDL), low-density lipoprotein (LDL), total cholesterol (TC), fasting C-peptide (FCP), insulin, fasting blood glucose (FBG), and triglycerides (TG) from the hospital database, to examine the relationship between obesity and gut microbiota.
2.2 Fecal DNA extraction, PCR amplification, and 16S rRNA sequencing
Following storage of the fecal samples at −80°C, microbial genomic DNA was extracted utilizing the FastPure Stool DNA Isolation Kit (MJYH) and subsequently assessed for quality via 1% agarose gel electrophoresis. The V3-V4 region of the 16S rRNA gene was amplified using the primers 338F/806R. Post library construction with the NEXTFLEX Rapid DNA-Seq Kit, sequencing was conducted on the Illumina NextSeq 2000 PE300 platform (MJYH).
2.3 Statistical analysis
All data analyses were conducted using the Meggie BioCloud platform (https://cloud.majorbio.com). For alpha diversity, indices such as Chao1 and Shannon were calculated utilizing mothur (version 1.30.2), and inter-group differences were assessed using the Wilcoxon rank-sum test. Regarding beta diversity, Principal Coordinates Analysis (PCoA) was performed based on the Bray-Curtis distance, and differences in microbial community structure between groups were evaluated using PERMANOVA. Species Venn diagram analyzed using Python 2.7.10 software. For differential species analysis, the LEfSe method (with LDA > 4 and P < 0.05) was employed to identify significant differences in flora from the phylum to genus levels. Figure 1 clinical factor differences were analyzed using Prism version 10. The table of baseline patient characteristics by obesity category (Table 1) was generated using R software.
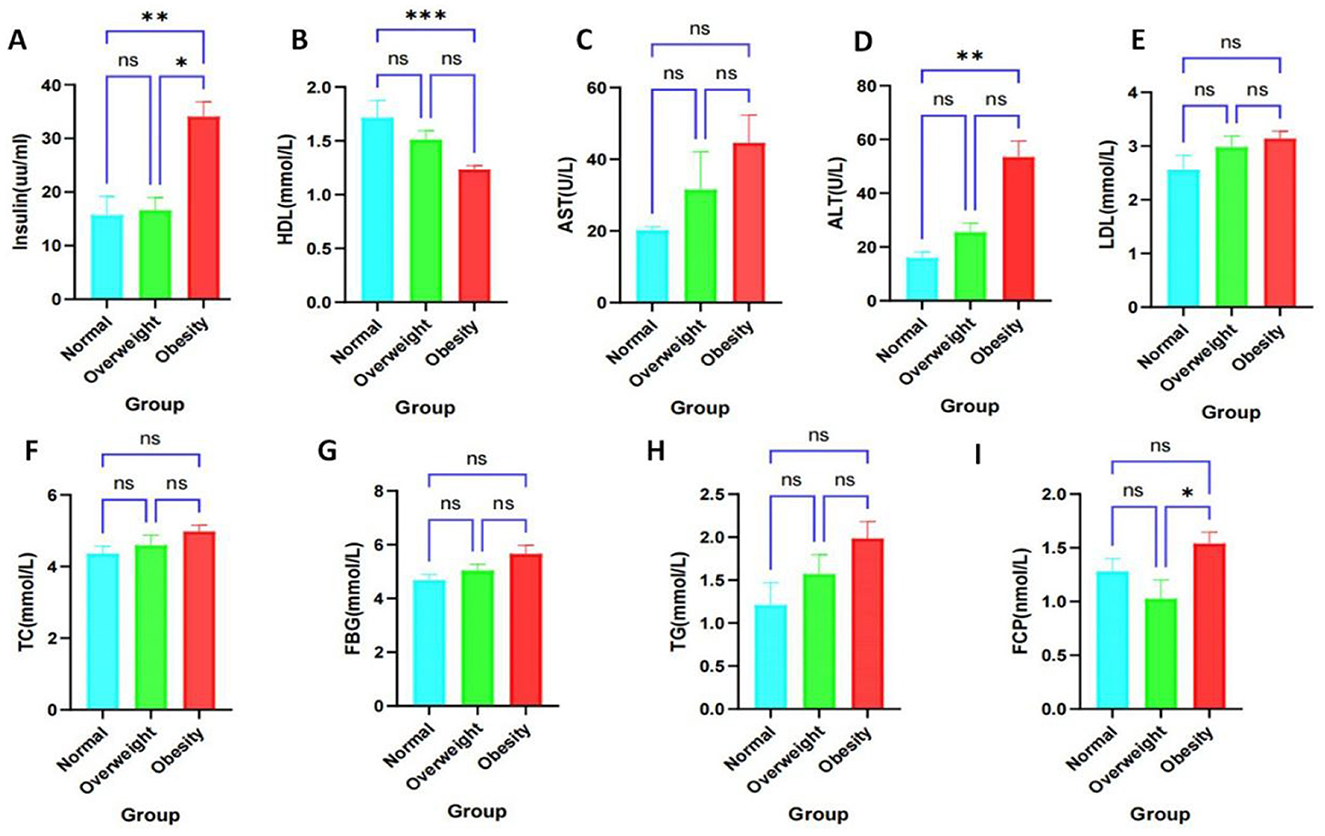
Figure 1. Clinical factor analysis of healthy, overweight and obese populations. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significantly different. (A) High-density lipoprotein. (B) Fasting insulin levels. (C) Aspartate aminotransferase. (D) Alanine aminotransferase. (E) Low-density lipoprotein. (F) Total cholesterol. (G) Fasting glucose. (H) Triglycerides. (I) Fasting C-peptide.
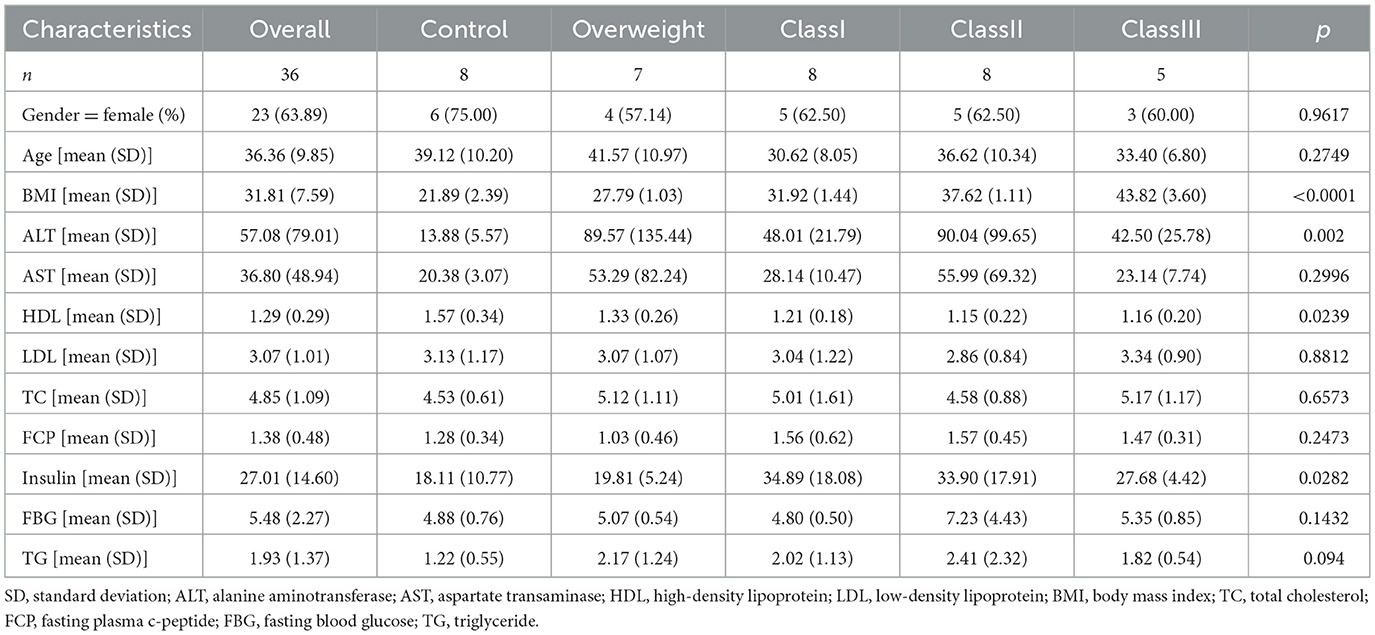
Table 1. Baseline patient characteristics by obesity category.
2.4 DNA extraction, library construction, and macro-genomics sequencing of fecal samples
Prior to the extraction and analysis of deoxyribonucleic acid (DNA), all fecal samples were frozen at −80 C. The methods employed for DNA extraction and the quality control procedures were identical to those utilized for 16S rRNA sequencing. The DNA was fragmented using the Covaris M220 (Genetics, China), and following screening of fragments measuring approximately 350 base pairs, libraries were constructed using the NEXTFLEX Rapid DNA-Seq (Bioo Scientific, USA). Subsequent to this, sequencing was performed using the Illumina NovaSeq™ X Plus (Illumina, USA) sequencing platform. The macrogenome sequencing procedure was carried out by Shanghai Meiji Biomedical Technology Co., Ltd.
2.5 Macrogenomics sequence quality control, genome assembly, and gene prediction
Before the extraction and analysis of DNA, all fecal samples were preserved by freezing at −80 C. The methods for DNA extraction and quality control adhered to established protocols. Quality control of DNA sequences was conducted using fastp (version 0.20.0), and sequence alignment was performed with BWA (version 0.7.17). MEGAHIT (version 1.1.2) facilitated sequence assembly, with contigs of ≥300 base pairs being selected as the final output. Open reading frames (ORFs) were predicted using Prodigal (version 2.6.3), with genes of ≥100 base pairs being retained and subsequently translated into amino acid sequences. To construct non-redundant gene sets, CD-HIT (version 4.7) was employed, while SOAPaligner (version 2.21) was utilized to quantify gene abundance.
2.6 Species and KEGG functional annotation
Sequence alignment analysis was conducted utilizing Diamond version 2.0.13. Amino acid sequences from non-redundant gene sets were aligned to the NR database using BLASTP with an e-value threshold of 1e-5, facilitating the calculation of species abundance in conjunction with taxonomic annotations. Similarly, these sequences were aligned to the KEGG database using BLASTP with the same e-value threshold, enabling the determination of functional class abundance based on KO, Pathway, EC, and Module annotations.
3 Results
3.1 Baseline characterization of patients in different obesity categories
We conducted an analysis of the biochemical profiles of obese individuals using collected clinical data to enhance diagnostic accuracy and clinical support. Specifically, we examined notable differences in levels of AST, ALT, HDL, LDL, TC, FCP, Insulin, FBG, and TG. Our findings indicated that HDL levels were significantly lower (Figure 1A), while Insulin levels were significantly higher (Figure 1B) in obese individuals compared to healthy controls. No significant differences were observed in AST, ALT, LDL, TC, FCP, FBG, and TG levels; however, there was a trend toward increased levels of AST, ALT, TC, FBG, and TG (Figures 1C–H). Additionally, FCP levels were significantly elevated in obese subjects compared to overweight subjects, but not significantly different from healthy individuals (Figure 1I). The baseline characteristics of patients, categorized by obesity status (Table 1), revealed significant differences in BMI, ALT, HDL, and Insulin among obese subgroups (P < 0.05). These findings suggest that obese patients are at an increased risk for insulin resistance, metabolic disorders, and obesity-related complications.
3.2 Structural changes in the gut microbiota of obese patients
The alterations in gut microbial composition were initially examined between normal controls and obese patients. It was observed that microbial abundance was significantly diminished in the obese cohort (Figure 2A). However, the Shannon index showed no significant difference (Supplementary Figure S1). Furthermore, Principal Coordinates Analysis (PCoA) at the Amplicon Sequence Variant (ASV) level indicated a divergence in the microbiota structure of obese individuals compared to normal controls (Figure 2B). Differential analyses of bacterial flora at the genus level, utilizing 16S rRNA and metagenomic sequencing, identified several microorganisms that exhibited significant differences between healthy and obese groups. These differential bacteria, particularly Subdoligranulum, Klebsiella, g__Roseburia, and g__Ruminococcus, may serve as potential biomarkers for distinguishing obesity status (Figures 2C, D). Metagenomic analysis further examined species differences among the top 10 species by abundance at the genus level (Supplementary Figure S2). It was found that s__Faecalibacterium_prausnitzii, s__Ruminococcus_bromii, s__Roseburia_faecis, s__Alistipes_putredinis were predominantly distributed in the normal and grade 1 obesity groups. Notably, s__Alistipes_putredinis was not observed in other groups. s__Bacteroides_stercoris was almost exclusively enriched in grade 1 obesity and may represent a potential biomarker. Ruminococcus gnavus exhibited higher abundance in the grade 2 obesity group. The species s__Collinsella_aerofaciens showed elevated abundance in overweight and grade 3 obesity groups.
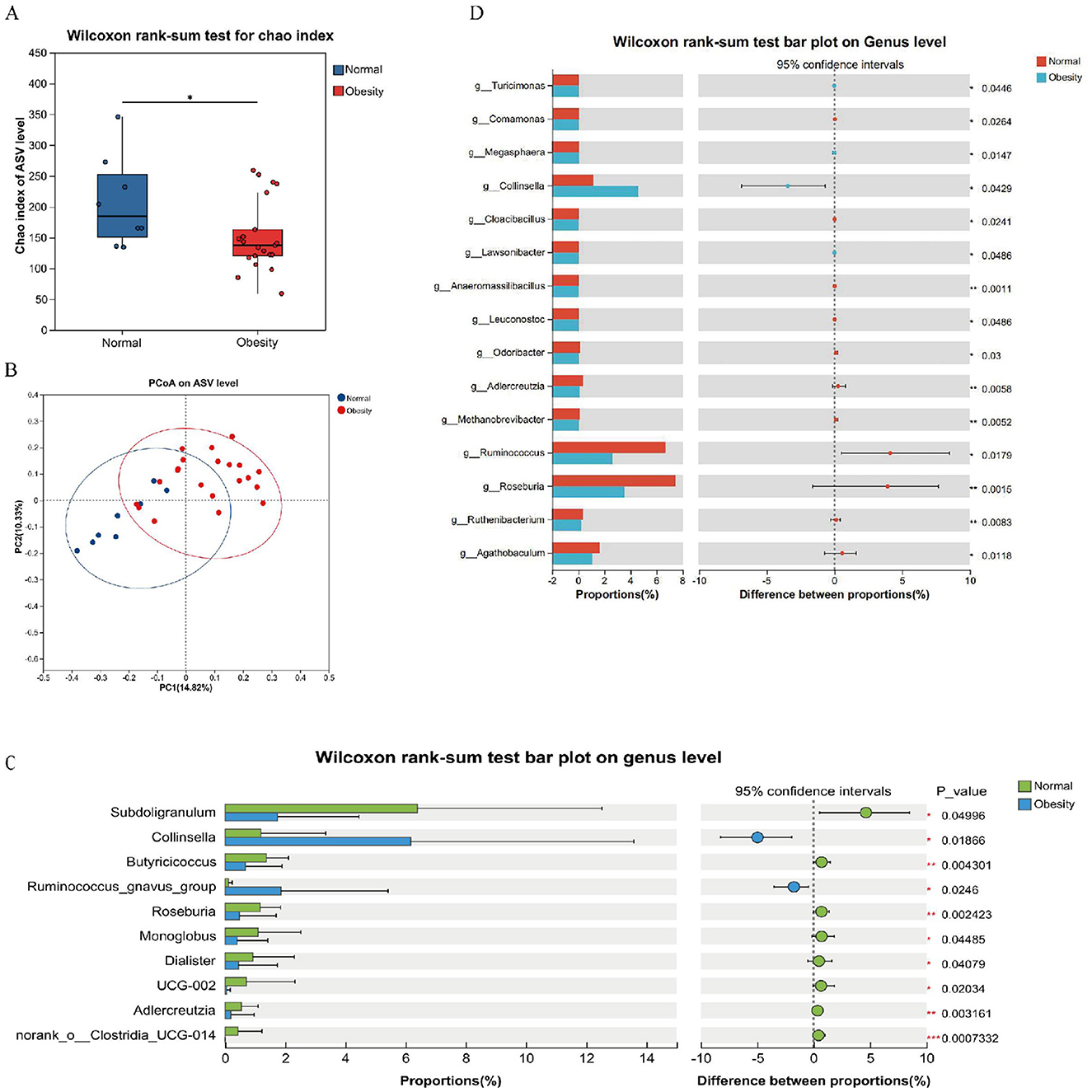
Figure 2. Structural analysis of the intestinal flora. (A) Alpha diversity chao index. (B) PCoA based on unweighted Unifrac distance used to show beta diversity between groups. (C) Analysis of the top 10 significantly different species from 16s rRNA abundance. (D) Analysis of significantly different species in the top 15 of abundance from macrogenomes. *p < 0.05, **p < 0.01, ***p < 0.001.
3.3 Biomarker analysis of health and obesity
To investigate the microorganisms potentially influencing obesity progression and to identify biomarkers for distinguishing obesity, a species Venn analysis at the Amplicon Sequence Variant (ASV) level was conducted. This analysis revealed that 730 ASVs were unique to healthy controls, 1,246 ASVs were unique to obese individuals, and 350 ASVs were common to both groups (Figure 3A). Further analysis of species composition at the ASV level, along with Linear Discriminant Analysis (LDA) Effect Size (LEfSe) for the top ten species based on genus-level richness, demonstrated an increase in the genera g__Blautia (70%), g__Streptococcus (67%), g__Bacteroides (67%), and g__Bifidobacterium (64%) in obese subjects. Conversely, there was a decrease in the genera g__Ruminococcus (28%), g__Roseburia (32%), g__Lachnospiraceae_unclassified (19%), g__Eubacterium (34%), and g__Faecalibacterium (41%). Notably, the genus g__Klebsiella was specific to obesity (Figure 3B). Consistent findings from 16S rRNA and metagenomic LEfSe analyses identified g__Collinsella as an obesity marker, whereas g__Roseburia and g__Ruminococcus were identified as markers of health (Figures 3C, D).
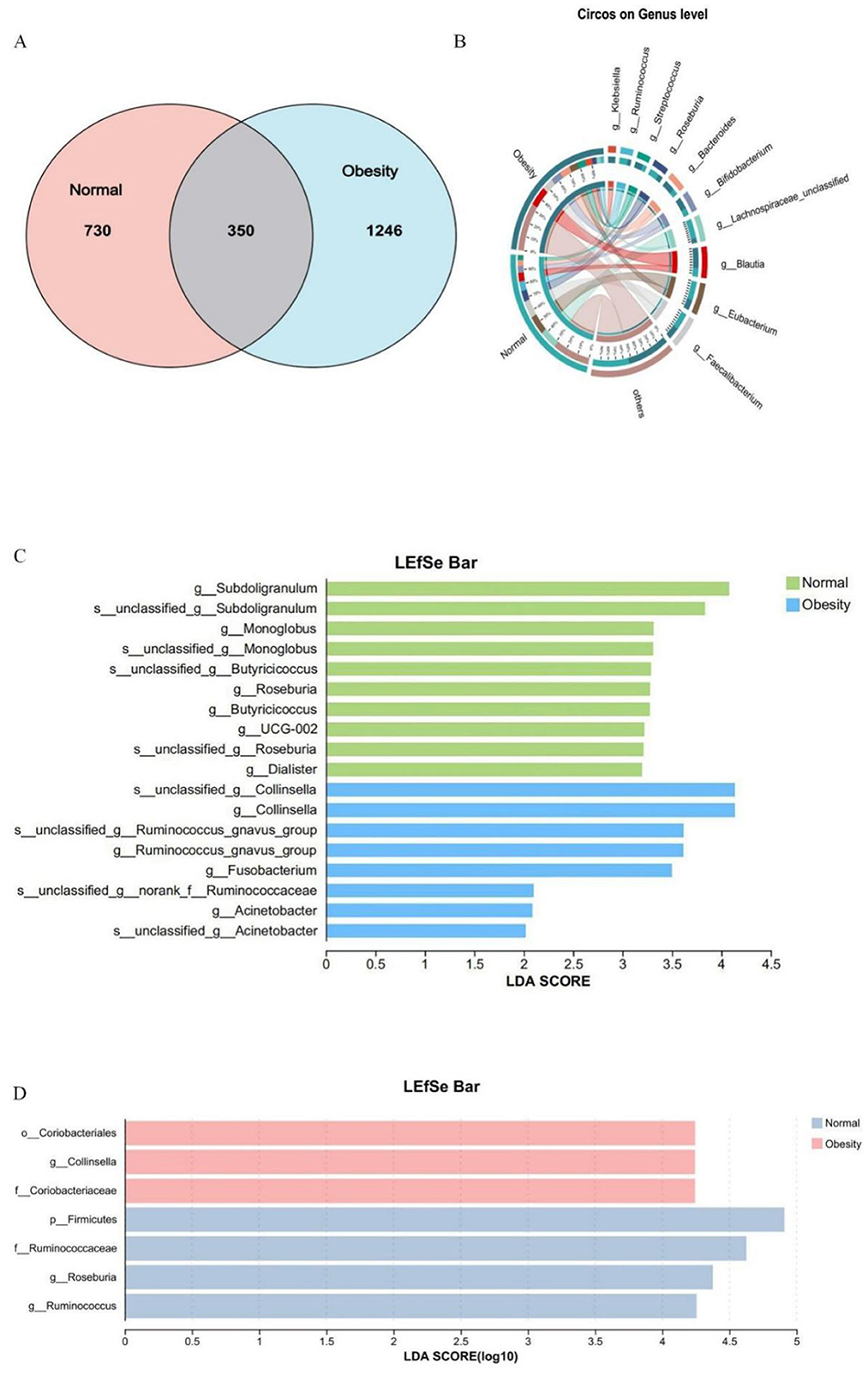
Figure 3. Normal weight and obesity biomarker analysis. (A) Venn diagram of shared or unique ASVs between healthy controls and obese patients. (B) Heatmap of species composition at the genus level in healthy controls and obese patients. (C) Linear discriminant analysis of 16SrRNA LEfSe, LDA score > 2.0. (D) Linear discriminant analysis of LEfSe in the macrogenome, LDA score > 4.0.
3.4 Gut microbial characteristics of patients with different levels of obesity
To comprehensively examine the intestinal microbiota composition across varying degrees of obesity, subjects were stratified into categories of overweight, and Obesity Classes I, II, and III. Analysis of alpha diversity using the Chao index, along with Principal Coordinates Analysis (PCoA) at the species abundance value (SAV) level, demonstrated significant alterations in microbiota composition among the different groups (Figures 4A, B). Furthermore, a Venn diagram at the genus level indicated a decline in the number of endemic species as obesity severity increased (Figure 4C). The microbial dysbiosis index (MDI) progressively rose, indicating a worsening of microbial community structure disorder (Figure 4D). Notably, the Class I obesity group did not exhibit significant differences. To elucidate species-specific alterations, the top ten species in terms of genus-level abundance were analyzed in relation to obesity levels. The analysis revealed that Collinsella, Streptococcus, Blautia, Romboutsia, and Klebsiella exhibited an increasing trend with obesity, whereas Faecalibacterium demonstrated a decreasing trend. Notably, the microbial structure of the Class I obesity group appeared to revert toward a normal configuration (Figure 4E and Supplementary Figure S3), indicating that the microbial community structure may undergo compensatory regulation during the progression of obesity. Further investigation into potential biomarkers of obesity severity among the top ten species revealed that Ruminococcus served as a marker for the normal group; Collinsella and Parvimonas were indicative of the overweight group; Faecalibacterium and Bacteroides were associated with the Class I obesity group; Ruminococcus_gnavus_group was identified as a marker for Class II obesity; and Acinetobacter was linked to Class III obesity (Figure 4F).
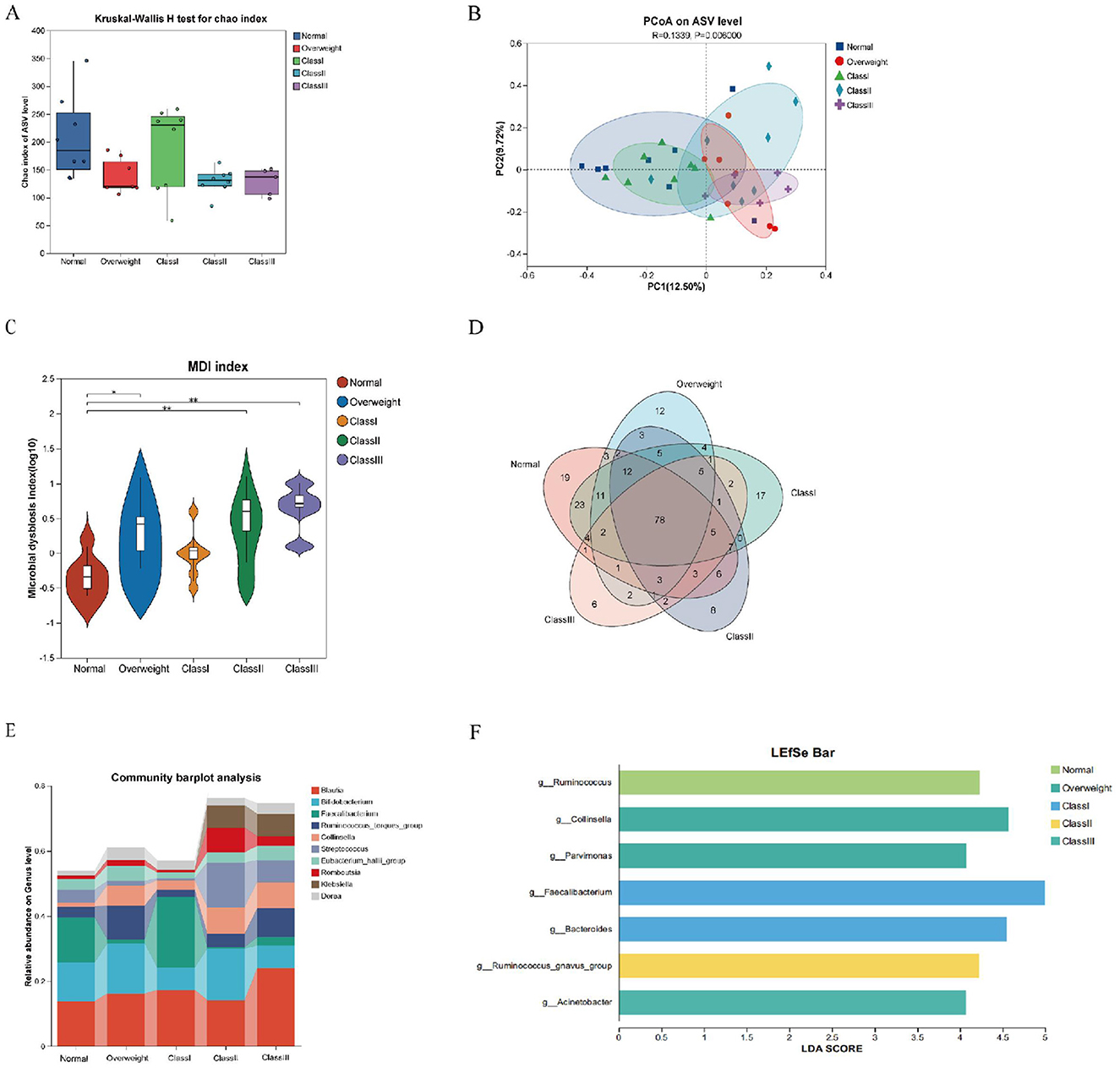
Figure 4. Characterization of gut flora in healthy, overweight and class I, II and III obese patients. (A) Alpha diversity chao index for the five groups. (B) PCoA analysis at the ASV level, demonstrating the degree of similarity and difference in the microbial communities of different groups. (C) Venn diagram at the genus level, reflecting species shared or endemic. Different colors represent different subgroups, overlapping parts indicate species common to multiple subgroups, parts without overlap indicate species endemic to the subgroup, and numbers indicate the corresponding number of species. (D) Genus-level colony structure disorder index (MDI). (E) Genus-level species composition analysis. (F) Genus-level Lefse species difference discriminant analysis with LDA score > 4.0. *p < 0.05; **p < 0.01.
3.5 Functional predictive analysis
The KEGG functional annotation predominantly enriched the Global and Overview Maps, Carbohydrate Metabolism, and Amino Acid Metabolism functions within the Metabolic Processes pathway. It also enriched the Translation function within the Genetic Information Processing pathway, the Membrane Transport function within the Environmental Information Processing pathway, and the Cellular Community - Prokaryotes function within the Cellular Processes pathway. Notably, the enrichment was more pronounced in the Metabolic Processes pathway (Figure 5).To predict functional differences attributable to colony contributions across varying levels of obesity, a functional difference analysis was conducted on the top five species abundances. Additionally, an analysis of species contributions to these top five abundances was performed at the KEGG Orthology (KO) functional level. The findings indicated significant variations in K02913 (L33 ribosomal protein), K03088 (σ70 RNA polymerase), K02914 (L34 ribosomal protein), K07473 (DNA damage repair protein J), and K02961 (S17 ribosomal protein) across different stages of obesity (Figure 6A).The L33 protein (K02913) analysis revealed that the normal and grade I obesity groups were predominantly influenced by Faecalibacterium, whereas Blautia was the principal contributor in the overweight and moderately severe obesity groups (grade II/III) (Figure 6B). In the case of the σ70 RNA polymerase (K03088), Collinsella and Streptococcus were the primary contributors in grade II/III obesity, with Collinsella also being predominant in the overweight group (Figure 6C). For the L34 protein (K02914), Bacteroides was the major contributor in the normal, class I, and class III obesity groups, while class II obesity was primarily influenced by Streptococcus (Figure 6D). The DNA damage repair protein J (K07473) analysis indicated that Roseburia was dominant in the normal group, Blautia in the overweight and moderate obesity groups, and Bacteroides in the primary obesity group (Figure 6E). Lastly, the S17 ribosomal proteins (K02961) analysis showed that Faecalibacterium was the main contributor in the normal and primary obesity groups, Bifidobacterium was predominant in the overweight and secondary obesity groups, and Collinsella was most prevalent in the tertiary obesity group (Figure 6F).
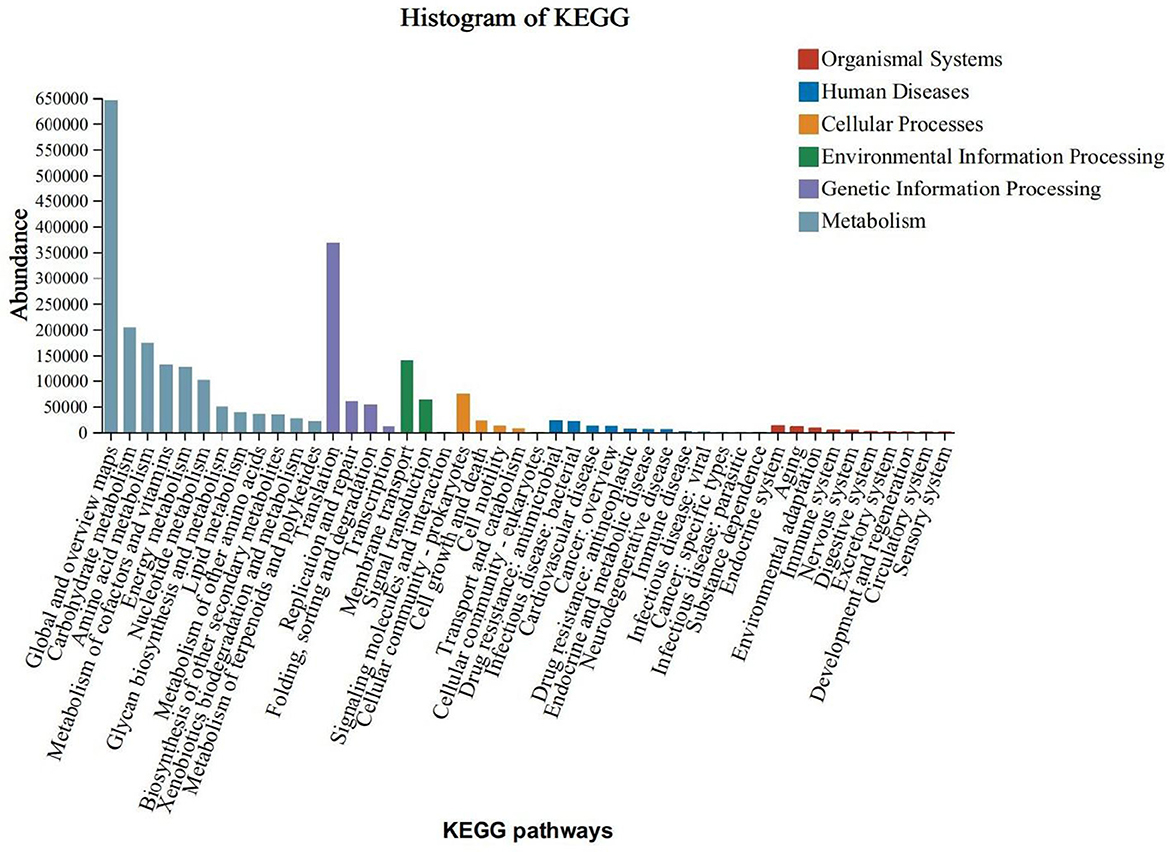
Figure 5. KEGG Functional Annotation Analysis.
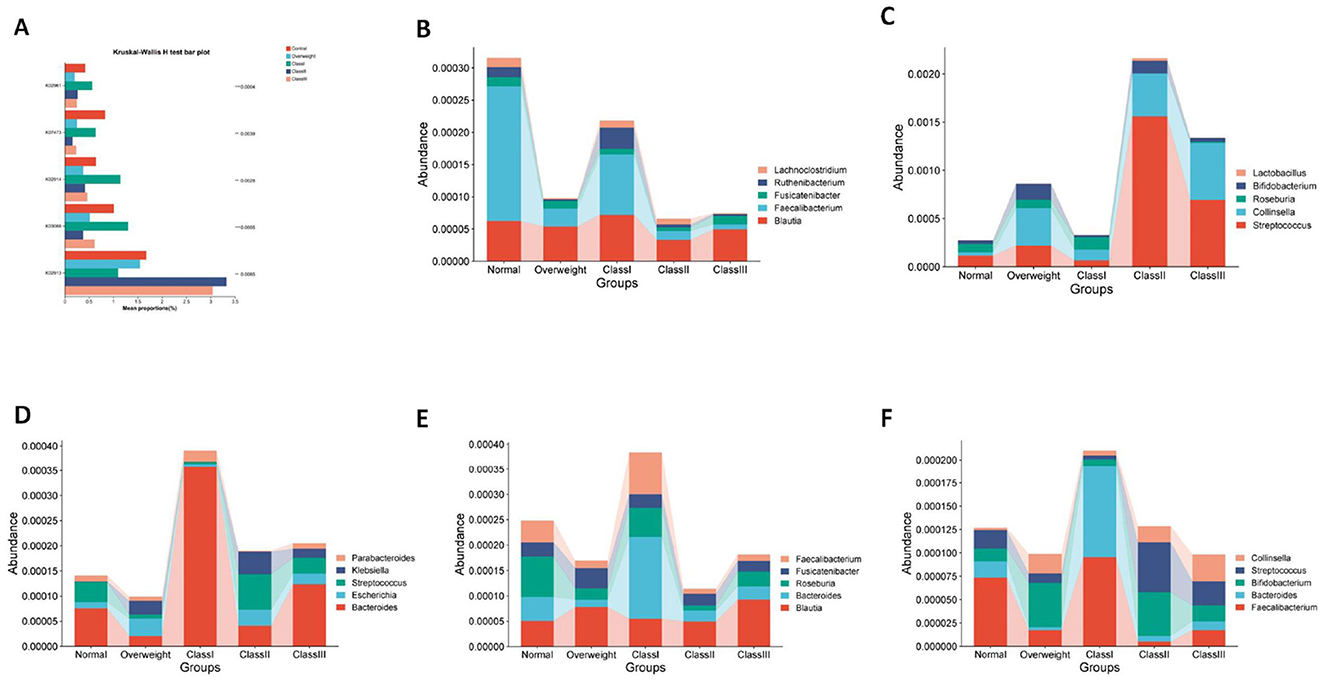
Figure 6. Functional prediction analysis. (A) Differences in the top five species/functional abundances KO. (B) Large subunit ribosomal protein L33, (C) RNA polymerase sigma-70 factor, ECF subfamily. (D) Large subunit ribosomal protein L34. (E) DNA-damage-inducible protein J. (F) Small subunit ribosomal protein S17.
In conclusion, the study identifies Blautia as frequently exhibiting dominant functions in individuals with overweight and moderate-to-severe obesity, such as the L33 protein and DNA repair protein J, suggesting its adaptation to obesity-related metabolic stresses. Conversely, Faecalibacterium is predominantly found in individuals with normal weight and mild obesity, associated with proteins like L33 and S17, and appears to play a role in maintaining metabolic homeostasis; its reduction may correlate with the progression of obesity. Additionally, Collinsella and Streptococcus are significantly implicated in RNA polymerase and ribosomal functions in cases of severe obesity, potentially linking them to metabolic disorders or inflammation. Roseburia, associated with DNA repair in the normal weight group, may offer protective benefits, with its reduced abundance potentially linked to obesity progression. Overall, the grade of obesity is intricately connected to the functional contributions of intestinal flora. Genera such as Blautia, Collinsella, and Streptococcus may adapt to host metabolic changes by modulating ribosome synthesis and stress responses, positioning them as potential microbial markers for obesity classification or as targets for therapeutic intervention.
3.6 Correlation between intestinal flora and biochemical indicators in patients with different degrees of obesity
Given the observed variations and trends in biochemical indicators among overweight and obese patients, we aimed to establish correlations between these indicators and intestinal flora. To achieve this, we analyzed the top 15 species based on species abundance for their correlation with biochemical indicators. Our findings revealed that these indicators were influenced by one or multiple intestinal flora, with the primary focus on identifying flora that could serve as potential biomarkers. Specifically, ALT, FCP, and FBG showed significant negative correlations with Ruminococcus, a biomarker of health, and significant positive correlations with HDL. Additionally, ALT and FBG were significantly negatively correlated with Faecalibacterium, identified as a biomarker for Class I obesity. Although Collinsella was not significantly correlated with these clinical factors as a marker of overweight, AST and HDL exhibited a tendency for negative correlation with Collinsella, while other factors showed a tendency for positive correlation. Furthermore, HDL demonstrated a highly significant negative correlation with Blautia, and FCP showed a highly significant positive correlation with Blautia (Figure 7).
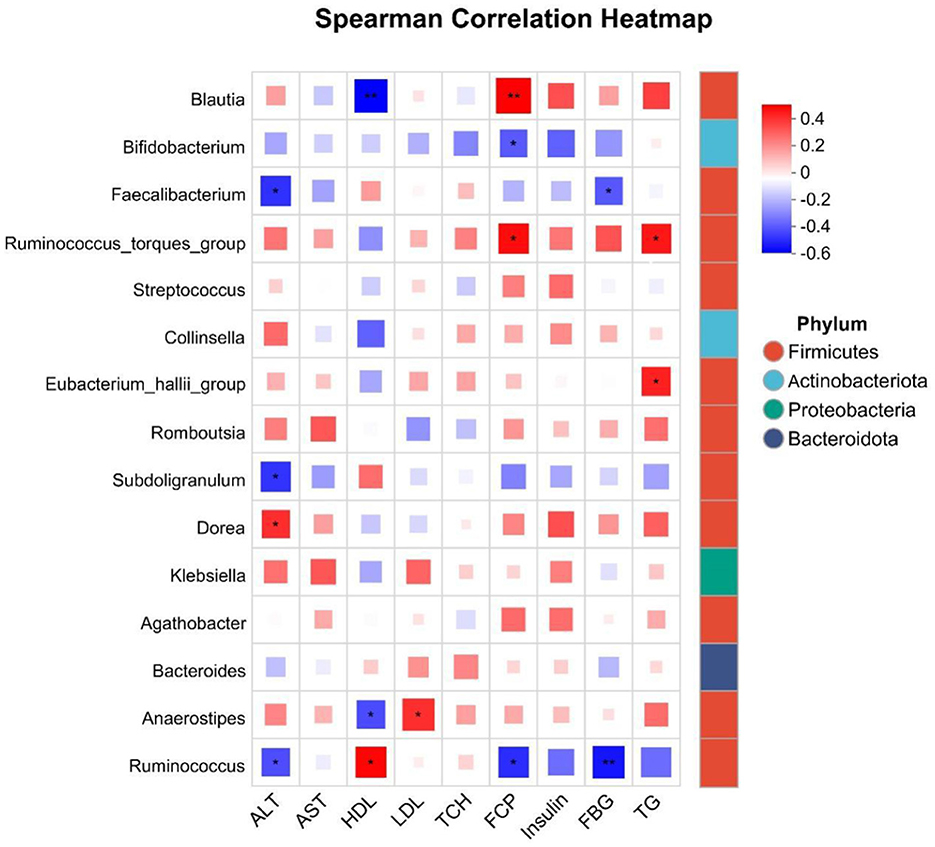
Figure 7. Correlation analysis of the top 20 species abundance with biochemical indicators. *P < 0.05, **P < 0.01.
4 Discussions
As the severity of obesity progresses from overweight to tertiary obesity, there is a notable decrease in the diversity of intestinal microbiota, accompanied by a gradual increase in the dysbiosis index (MDI). Interestingly, the alterations in microbiota composition observed in individuals with tertiary obesity closely resemble those found in healthy individuals, suggesting the potential presence of compensatory regulatory mechanisms during the early stages of obesity. Previous studies have demonstrated a significant reduction in intestinal microbiota diversity with increasing obesity, which may be linked to obesity-related metabolic disorders (Florêncio et al., 2025; Xie et al., 2025). It is hypothesized that in the initial phases of obesity, compensatory regulation of the intestinal microbiota may occur as an adaptive response to changes in the internal environment (Maqoud et al., 2024). Specifically, the gut microbiota of obese individuals is typically characterized by a higher proportion of Firmicutes and a lower proportion of Bacteroidetes, a shift that is believed to be associated with enhanced energy uptake and storage (Enache et al., 2024; Mamun et al., 2025). Furthermore, the presence of certain pro-inflammatory bacteria within the gut microbiota of obese individuals may exacerbate metabolic complications associated with obesity by compromising intestinal barrier integrity and promoting systemic inflammation (Maqoud et al., 2024; Custers et al., 2024).
During the initial stages of obesity, alterations in the gut microbiota may be subtle or even resemble those observed in healthy individuals. This phenomenon could be attributed to the body's ability to maintain gut microbiota equilibrium through compensatory mechanisms in the early phases of obesity (Deng et al., 2025). However, as obesity progresses, these compensatory mechanisms may become insufficient, resulting in reduced microbial diversity and an elevated dysbiosis index (MDI) (Xie et al., 2025). In summary, alterations in gut microbiota are pivotal in the pathogenesis of obesity, and elucidating these changes is crucial for the development of novel therapeutic strategies for obesity management.
A decline in beneficial bacteria, such as Faecalibacterium and Roseburia, alongside an increase in pathogenic bacteria, including Blautia and Collinsella, is correlated with the progression of obesity. Faecalibacterium and Roseburia are recognized for their production of short-chain fatty acids (SCFAs), particularly butyrate, which is crucial for maintaining gut barrier integrity and exerting anti-inflammatory effects. A reduction in these bacteria may compromise intestinal barrier function, potentially leading to chronic inflammation, a key pathophysiological mechanism underlying obesity and metabolic syndrome (Duan et al., 2021). Furthermore, the proliferation of pathogenic bacteria like Blautia and Collinsella is linked to metabolic abnormalities associated with obesity. The expansion of these microbial populations may disrupt intestinal microecological balance, thereby influencing host energy and lipid metabolism, and promoting adiposity and weight gain (Companys et al., 2021). Additionally, increased levels of Collinsella have been positively correlated with body fat and low-density lipoprotein (LDL) levels in obese individuals, underscoring its potential contribution to the pathogenesis of obesity (Zeng et al., 2019). In conclusion, alterations in the composition of the intestinal microbiota, particularly the reduction of beneficial bacteria and the proliferation of pathogenic bacteria, constitute a significant factor in the development and progression of obesity.
The functions of the microbiota in obese individuals are skewed toward ribosome synthesis and stress response pathways (e.g., K02913, K03088), indicating potential mechanisms of metabolic adaptation. This phenomenon suggests a significant role in metabolic adaptation, as the gut microbiota in an obese state may adjust to the host's metabolic demands by upregulating the ribosome synthesis pathway. Such an adaptation could be associated with the increased protein synthesis needs of cells in response to external environmental changes. In the context of stress response, bacteria typically modulate ribosomal functions to cope with environmental stresses. For instance, under heat stress conditions, bacterial ribosomes may selectively bind specific mRNAs, thereby regulating protein synthesis (Fisunov et al., 2017). This selective binding enables bacteria to swiftly modify their metabolic activities, ensuring survival and functionality under adverse conditions. Furthermore, the gut microbiota of obese individuals may influence the host's metabolic status by modulating ribosome-related pathways. Increased ribosome synthesis has been implicated in the development of obesity-associated metabolic disorders, potentially due to disruptions in protein synthesis that can adversely affect normal cellular functions and metabolic processes (Schöttl et al., 2020). Consequently, a comprehensive investigation into the role of gut microbiota in ribosome synthesis and stress response among obese individuals could offer novel insights into the mechanisms underlying obesity-related metabolic diseases.
Specific bacterial genera, such as g__Faecalibacterium and g__Bacteroides (class I), g__Ruminococcus_gnavus_group (class II), and Acinetobacter (class III), have been identified as markers indicative of obesity severity. Research indicates a strong association between these genera and metabolic abnormalities linked to obesity. For instance, there is a notable negative correlation between Faecalibacterium, prausnitzii and obesity-related metabolic markers, suggesting that this bacterium may play a crucial role in maintaining gut health and metabolic homeostasis (Brahe et al., 2015). Certain species within the genus Bacteroides exhibit varying abundance in obese individuals, potentially correlating with the host's metabolic status (Zeng et al., 2019).
Significant alterations in the composition and functionality of the gut microbiota can manifest at various stages of obesity. Research indicates that the gut microbiota of obese individuals is characterized by an increased proportion of Firmicutes and a decreased proportion of Bacteroidetes, a shift linked to enhanced energy absorption and fat storage (Krznarić et al., 2012). Furthermore, specific alterations in the genus Acinetobacter have been observed in individuals with grade III obesity, potentially correlating with more severe metabolic disturbances (Kong et al., 2013). The progression of obesity is also associated with modifications in other microbial populations. Studies have demonstrated that obese individuals generally exhibit reduced gut microbial diversity, which may contribute to impaired metabolic function (De la Cuesta-Zuluaga et al., 2018). Additionally, alterations in certain microbial taxa may be linked to obesity-related inflammatory responses, further exacerbating metabolic disturbances (Moreno-Indias et al., 2016). Moreover, the genus Blautia has been positively associated with insulin resistance, while Faecalibacterium has been positively correlated with high-density lipoprotein (HDL) levels, suggesting their potential as biomarkers for obesity diagnosis (Naderpoor et al., 2019). Firstly, concerning the association between Blautia and insulin resistance, research demonstrates a positive correlation between the abundance of Blautia and insulin resistance. One study found a significant association between Blautia abundance and insulin resistance in obese children, which was also correlated with increased levels of inflammatory markers, including TNF-α and IL-6 (Benítez-Páez et al., 2020; Zhu et al., 2020). Additionally, another study suggested that a reduction in Blautia abundance might be linked to obesity-related metabolic inflammation, thereby reinforcing the connection between Blautia and insulin resistance (Benítez-Páez et al., 2020). Secondly, the positive correlation between Faecalibacterium and high-density lipoprotein (HDL) levels has been corroborated by multiple studies. Faecalibacterium prausnitzii, a butyrate-producing bacterium, shows a positive correlation between its abundance and elevated HDL levels (Gruneck et al., 2020; Kashtanova et al., 2021). Research indicates that increased Faecalibacterium abundance is associated with improved metabolic health, potentially due to its production of short-chain fatty acids, such as butyrate, in the gut, which may enhance lipid metabolism (Gruneck et al., 2020). Finally, the positive correlation between Collinsella species and LDL cholesterol levels has been substantiated by multiple studies. One study reported a higher abundance of Collinsella aerofaciens in overweight and obese individuals, which was positively correlated with LDL levels (Companys et al., 2021). Furthermore, another study proposed that an increase in Collinsella may be linked to metabolic disorders, possibly through its influence on cholesterol metabolic pathways (Reimer et al., 2021). These genera exhibit distinct alterations at various stages of obesity, which may be intricately linked to the severity of obesity and its associated metabolic abnormalities (Chakaroun et al., 2021; Hu et al., 2024; Huang et al., 2023; Jie et al., 2021; Mohammadzadeh et al., 2024; Schwenger et al., 2025). In conclusion, the significance of the gut microbiota in metabolic health is garnering heightened scholarly interest. The correlation between specific bacterial taxa, including Blautia, Faecalibacterium, and Collinsella, and various metabolic markers provides novel insights into the involvement of gut microbiota in metabolic disorders. These findings contribute to a deeper understanding of the intricate relationship between gut microbiota and metabolic health and identify potential targets for future therapeutic interventions.
In summary, the present study conducted a systematic analysis of the gut microbiota characteristics among individuals with varying obesity levels (healthy, overweight, Grade I, Grade II, and Grade III obesity) in Southwest China, utilizing 16S rRNA gene sequencing and metagenomic sequencing technologies. This analysis has identified potential biomarkers for the microbiological diagnosis of obesity and has suggested novel approaches for the treatment of obesity and related metabolic disorders through the modulation of the intestinal microbiota. Nonetheless, this study is subject to several limitations. the most significant of which is the relatively small sample size and no discussion of gender differences was conducted. This limitation primarily arises from challenges in recruiting severely obese patients and healthy controls who fulfill stringent inclusion criteria, as well as the high costs associated with metagenomic sequencing analysis for each sample. The restricted sample size may compromise statistical power, heighten the risk of Type II errors, and potentially broaden the confidence intervals for effect estimates. To enhance the robustness of our findings, we conducted a review of the relevant literature and performed comparative studies, which revealed a reduction in microbial diversity within the obese population. Notably, we observed specific alterations in certain genera, including an increase in Blautia and Collinsella among obese individuals, alongside a decrease in Faecalibacterium and Roseburia (Supplementary Tables 1, 2). This study distinguishes itself by further refining specific biomarkers associated with varying levels of obesity—namely, Ruminococcus for normal weight, Collinsella for overweight, Faecalibacterium for Class I obesity, Ruminococcus_gnavus_group for Class II obesity, and Acinetobacter for Class III obesity. These findings elucidate a close relationship between the identified biomarkers and host metabolic functions, such as ribosome synthesis and RNA polymerase activity, offering a more in-depth analysis than previous research. Consequently, this study should be regarded as preliminary and exploratory, as it provides valuable targets and directions for future mechanistic investigations. However, the generalizability of these findings requires validation through studies involving larger and more representative population cohorts.
5 Conclusion
Populations exhibiting varying obesity profiles demonstrate distinct microbiota compositions, with flora adapted to obesity, and an escalation in dysregulation of these microbial communities is observed with increasing levels of obesity. There exists a correlation between alterations in microbial functionality and host metabolic processes, including RNA polymerase activity, DNA damage repair, and metabolic pathways such as ribosome synthesis, insulin resistance, glucose metabolism, and lipoprotein regulation. Targeted interventions aimed at modulating microbiota that are significantly elevated in obesity (e.g., Collinsella) and augmenting those significantly reduced in non-obese individuals (e.g., Faecalibacterium) may represent an effective strategy for mitigating obesity.
Data availability statement
The data presented in this study have been deposited in the figshare repository: 10.6084/m9.figshare.30529985.
Ethics statement
The studies involving humans originated from the China Western Weight Loss Surgery Longitudinal Study (ChiCTR2300073353). The studies have been reviewed and approved by the Ethics Committee of Chengdu Third People's Hospital (Approval No.: 2022-S-62). Written informed consent was obtained from the participants prior to enrollment.
Author contributions
ZF: Funding acquisition, Resources, Supervision, Writing – review & editing. EL: Conceptualization, Data curation, Visualization, Writing – original draft. SW: Conceptualization, Methodology, Writing – review & editing. YoL: Conceptualization, Writing – review & editing. AL: Conceptualization, Writing – review & editing. ML: Conceptualization, Writing – review & editing. JH: Supervision, Writing – review & editing. YaL: Supervision, Writing – review & editing. HL: Data curation, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study received support from the following funding sources: The National Natural Science Foundation of China (NSFC, 82202007, 82370884, 82170887), The Third People's Hospital of Chengdu Clinical Research Program (2023PI22, CSY-YN-01-2023-039), and the Foundation of Chengdu Science and Technology Bureau 2024-YF05-01311-SN, the Natural Science Foundation of Sichuan Province (2023NSFSC0739), Sichuan Provincial Department of Science and Technology Project, Sichuan-Chongqing Science and Technology Innovation Cooperation Plan: 2024YFHZ0066.
Acknowledgments
We thank the Third People's Hospital of Chengdu for providing the sequencing samples, and the Laboratory of Obesity and Metabolism Medical-Industrial Integration, School of Medicine, Southwest Jiaotong University, for providing the research platform.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1679119/full#supplementary-material
References
Aja, E., Zeng, A., Gray, W., Connelley, K., Chaganti, A., and Jacobs, J. P. (2025). Health effects and therapeutic potential of the gut microbe Akkermansia muciniphila. Nutrients 17:562. doi: 10.3390/nu17030562
Benítez-Páez, A., Gómez Del Pugar, E. M., López-Almela, I., Moya-Pérez, Á., Codoñer-Franch, P., and Sanz, Y. (2020). Depletion of Blautia species in the microbiota of obese children relates to intestinal inflammation and metabolic phenotype worsening. mSystems 5:e00857–e00819. doi: 10.1128/mSystems.00857-19
Brahe, L. K., Le Chatelier, E., Prifti, E., Pons, N., Kennedy, S., Hansen, T., et al. (2015). Specific gut microbiota features and metabolic markers in postmenopausal women with obesity. Nutr. Diabetes 5:e159. doi: 10.1038/nutd.2015.9
Chakaroun, R. M., Massier, L., Heintz-Buschart, A., Said, N., Fallmann, J., Crane, A., et al. (2021). Circulating bacterial signature is linked to metabolic disease and shifts with metabolic alleviation after bariatric surgery. Genome Med. 13:105. doi: 10.1186/s13073-021-00919-6
Companys, J., Gosalbes, M. J., Pla-Pagà, L., Calderón-Pérez, L., Llauradó, E., Pedret, A., et al. (2021). Gut microbiota profile and its association with clinical variables and dietary intake in overweight/obese and lean subjects: a cross-sectional study. Nutrients 13:2032. doi: 10.3390/nu13062032
Custers, E., Vreeken, D., Schuren, F., van den Broek, T. J., van Dongen, L., Geenen, B., et al. (2024). Impact of microbiota and metabolites on intestinal integrity and inflammation in severe obesity. Pharmaceuticals 17:918. doi: 10.3390/ph17070918
De la Cuesta-Zuluaga, J., Corrales-Agudelo, V., Carmona, J. A., Abad, J. M., and Escobar, J. S. (2018). Body size phenotypes comprehensively assess cardiometabolic risk and refine the association between obesity and gut microbiota. Int. J. Obes. 42, 424–432. doi: 10.1038/ijo.2017.281
De Wit, D. F., Hanssen, N. M. J., Wortelboer, K., Herrema, H., Rampanelli, E., and Nieuwdorp, M. (2023). Evidence for the contribution of the gut microbiome to obesity and its reversal. Sci. Transl. Med. 15:eadg2773. doi: 10.1126/scitranslmed.adg2773
Deng, M., Tang, F., and Zhu, Z. (2025). Altered cognitive function in obese patients: relationship to gut flora. Mol. Cell. Biochem. doi: 10.1007/s11010-024-05201-y. [Epub ahead of print].
Doghish, A. S., Elazazy, O., Mohamed, H. H., Mansour, R. M., Ghanem, A., Faraag, A. H. I., et al. (2025). A review on miRNAs in enteric bacteria-mediated host pathophysiology: mechanisms and implications. J. Biochem. Mol. Toxicol. 39:e70160. doi: 10.1002/jbt.70160
Duan, M., Wang, Y., Zhang, Q., Zou, R., Guo, M., and Zheng, H. (2021). Characteristics of gut microbiota in people with obesity. PLoS ONE 16:e0255446. doi: 10.1371/journal.pone.0255446
Enache, R. M., Profir, M., Roşu, O. A., Cretoiu, S. M., and Gaspar, B. S. (2024). The role of gut microbiota in the onset and progression of obesity and associated comorbidities. Int. J. Mol. Sci. 25:12321. doi: 10.3390/ijms252212321
Fan, H., Kouvari, M., Guo, C., Liu, Z., Zhang, X., Wang, H., et al. (2025). A comprehensive comparison of two commonly used BMI thresholds for non-communicable diseases and multimorbidity in the Chinese population. Clin. Nutr. 48, 70–79. doi: 10.1016/j.clnu.2025.03.016
Fisunov, G. Y., Evsyutina, D. V., Garanina, I. A., Arzamasov, A. A., Butenko, I. O., Altukhov, I. A., et al. (2017). Ribosome profiling reveals an adaptation strategy of reduced bacterium to acute stress. Biochimie 132, 66–74. doi: 10.1016/j.biochi.2016.10.015
Florêncio, G. P., Xavier, A. R., Natal, A. C. C., Sadoyama, L. P., Röder, D. V. D. M., Menezes, R. P., et al. (2025). Synergistic effects of probiotics and lifestyle interventions on intestinal microbiota composition and clinical outcomes in obese adults. Metabolites 15:70. doi: 10.3390/metabo15020070
Gruneck, L., Kullawong, N., Kespechara, K., and Popluechai, S. (2020). Gut microbiota of obese and diabetic Thai subjects and interplay with dietary habits and blood profiles. PeerJ 8:e9622. doi: 10.7717/peerj.9622
Gulati, S., and Murphy, W. J. (2025). Defining obesity in the context of cancer: thinking beyond body mass index. Trends Cancer 11, 441–447. doi: 10.1016/j.trecan.2025.01.009
Hu, M., Xiang, Q., Mei, Z., Gong, C., Pan, D., Liu, Y., et al. (2024). Bacterial and clinical metabolic signatures and their interactions in obese patients post-bariatric surgery. BMC Gastroenterol. 24:363. doi: 10.1186/s12876-024-03450-1
Huang, S., Zou, Y., Tang, H., Zhuang, J., Ye, Z., Wei, T., et al. (2023). Cordyceps militaris polysaccharides modulate gut microbiota and improve metabolic disorders in mice with diet-induced obesity. J. Sci. Food Agric. 103, 1885–1894. doi: 10.1002/jsfa.12409
Isolauri, E., and Laitinen, K. (2025). Resilience to global health challenges through nutritional gut microbiome modulation. Nutrients 17:741. doi: 10.3390/nu17030396
Jie, Z., Yu, X., Liu, Y., Sun, L., Chen, P., Ding, Q., et al. (2021). The baseline gut microbiota directs dieting-induced weight loss trajectories. Gastroenterology 160, 2029–2042.e16. doi: 10.1053/j.gastro.2021.01.029
Kashtanova, D. A., Klimenko, N. S., Tkacheva, O. N., Strazhesko, I. D., Metelskaya, V. A., Gomyranova, N. V., et al. (2021). Subfractional spectrum of serum lipoproteins and gut microbiota composition in healthy individuals. Microorganisms 9:1461. doi: 10.3390/microorganisms9071461
Kong, L. C., Tap, J., Aron-Wisnewsky, J., Pelloux, V., Basdevant, A., Bouillot, J. L., et al. (2013). Gut microbiota after gastric bypass in human obesity: increased richness and associations of bacterial genera with adipose tissue genes. Am. J. Clin. Nutr. 98, 16–24. doi: 10.3945/ajcn.113.058743
Krznarić, Z., Vranešić Bender, D., Kunović, A., Kekez, D., and Stimac, D. (2012). Gut microbiota and obesity. Dig. Dis. 30, 196–200. doi: 10.1159/000336965
Liu, H., Kang, J., Liu, W., and Shen, Y. (2025). Association between a body shape index and colorectal cancer in US population: a cross-sectional study based on NHANES. Front. Nutr. 12:1535655. doi: 10.3389/fnut.2025.1535655
Ma, Z. F., and Lee, Y. Y. (2025). The role of the gut microbiota in health, diet, and disease with a focus on obesity. Foods 14:320. doi: 10.3390/foods14030492
Mamun, M. A. A., Rakib, A., Mandal, M., and Singh, U. P. (2025). Impact of a high-fat diet on the gut microbiome: a comprehensive study of microbial and metabolite shifts during obesity. Cells 14:463. doi: 10.3390/cells14060463
Maqoud, F., Calabrese, F. M., Celano, G., Mallardi, D., Goscilo, F., D'Attoma, B., et al. (2024). Role of increasing body mass index in gut barrier dysfunction, systemic inflammation, and metabolic dysregulation in obesity. Nutrients 17:72. doi: 10.3390/nu17010072
Maxim, M., Soroceanu, R. P., Vlăsceanu, V. I., Platon, R. L., Toader, M., Miler, A. A., et al. (2025). Dietary habits, obesity, and bariatric surgery: a review of impact and interventions. Nutrients 17:785. doi: 10.3390/nu17030474
Mayer, S. B., Graybill, S., Raffa, S. D., Tracy, C., Gaar, E., Wisbach, G., et al. (2021). Synopsis of the 2020 U.S. VA/DoD clinical practice guideline for the management of adult overweight and obesity. Mil. Med. 186, 884–896. doi: 10.1093/milmed/usab114
Mishra, S. P., Wang, B., Jain, S., Ding, J., Rejeski, J., Furdui, C. M., et al. (2023). A mechanism by which gut microbiota elevates permeability and inflammation in obese/diabetic mice and human gut. Gut 72, 1848–1865. doi: 10.1136/gutjnl-2022-327365
Mohammadzadeh, N., Razavi, S., and Ebrahimipour, G. (2024). Impact of bariatric surgery on gut microbiota composition in obese patients compared to healthy controls. AMB Express 14:115. doi: 10.1186/s13568-024-01769-2
Moreno-Indias, I., Sánchez-Alcoholado, L., García-Fuentes, E., Cardona, F., Queipo-Ortuño, M. I., and Tinahones, F. J. (2016). Insulin resistance is associated with specific gut microbiota in appendix samples from morbidly obese patients. Am. J. Transl. Res. 8, 5672–5684.
Naderpoor, N., Mousa, A., Gomez-Arango, L. F., Barrett, H. L., Dekker Nitert, M., and de Courten, B. (2019). Faecal microbiota are related to insulin sensitivity and secretion in overweight or obese adults. J. Clin. Med. 8:452. doi: 10.3390/jcm8040452
Reimer, R. A., Wharton, S., Green, T. J., Manjoo, P., Ramay, H. R., Lyon, M. R., et al. (2021). Effect of a functional fibre supplement on glycemic control when added to a year-long medically supervised weight management program in adults with type 2 diabetes. Eur. J. Nutr. 60, 1237–1251. doi: 10.1007/s00394-020-02328-8
Schöttl, T., Pachl, F., Giesbertz, P., Daniel, H., Kuster, B., Fromme, T., et al. (2020). Proteomic and metabolite profiling reveals profound structural and metabolic reorganization of adipocyte mitochondria in obesity. Obesity 28, 590–600. doi: 10.1002/oby.22737
Schwenger, K. J. P., Copeland, J. K., Ghorbani, Y., Chen, L., Comelli, E. M., Guttman, D. S., et al. (2025). Characterization of liver, adipose, and fecal microbiome in obese patients with MASLD: links with disease severity and metabolic dysfunction parameters. Microbiome 13:9. doi: 10.1186/s40168-024-02004-7
World Health Organization (2000). Obesity: preventing and managing the global epidemic: report of a WHO consultation. World Health Organ. Tech. Rep. Ser. 894, i–xii, 1–253. Available online at: https://apps.who.int/iris/handle/10665/42330
Xie, Q. Y., Granato, A., Wong, A., Yau, C., Noseworthy, R., Chen, T., et al. (2025). Metabolic dysfunction associated with alterations in gut microbiota in adolescents with obesity. Diabetes 74, 720–733. doi: 10.2337/db24-0866
Zeng, Q., Li, D., He, Y., Li, Y., Yang, Z., Zhao, X., et al. (2019). Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 9:13424. doi: 10.1038/s41598-019-49462-w
Zhang, Y., and Wang, W. (2024). Probiotics in reducing obesity by reconfiguring the gut microbiota. Zhong Nan Da Xue Xue Bao Yi Xue Ban 49, 1042–1051. doi: 10.11817/j.issn.1672-7347.2024.240361
Keywords: degree of obesity, fecal microbiota, 16S rRNA sequencing, metagenomics, body mass index
Citation: Li E, Wang S, Li Y, Liuli A, Liang M, Huang J, Li Y, Li H and Feng Z (2025) Characterization of the gut microbiota in people with different levels of obesity. Front. Microbiol. 16:1679119. doi: 10.3389/fmicb.2025.1679119
Received: 05 August 2025; Accepted: 31 October 2025;
Published: 03 December 2025.
Edited by:
Shanshan Hu, Anhui Agricultural University, ChinaReviewed by:
Debojyoti De, National Institute of Technology, Durgapur, IndiaSiman Liu, University of Connecticut, United States
Copyright © 2025 Li, Wang, Li, Liuli, Liang, Huang, Li, Li and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haifang Li, aGFpZmFuZ2xpQHNkYXUuZWR1LmNu; Yan Li, bHl2ZXQyMDA0QDE2My5jb20=; Zhonghui Feng, ZmVuZ3poLjIwMDhAdHNpbmdodWEub3JnLmNu
†These authors have contributed equally to this work
‡ORCID: Zhonghui Feng orcid.org/0009-0007-5533-7144