 Jesús Avila
1,2*
Jesús Avila
1,2*
- 1 Centro de Biología Molecular “Severo Ochoa”, Campus Cantoblanco, Universidad Autónoma de Madrid, Madrid, Spain
- 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, Universidad Complutense, Madrid, Spain
In this short review is described the toxicity of modified intracellular tau and that of extracellular tau that could be released into the extracellular space after neuron death.
Introduction
The tau protein was initially identified from isolated brain microtubules as a microtubule-associated protein (MAP) (Weingarten et al., 1975 ). Subsequent in vitro analyses and studies in cultured cells then suggested that the tau protein facilitates in microtubule assembly and the stabilization of microtubule polymers (Cleveland et al., 1977 ; Drubin and Kirschner, 1986 ; Caceres and Kosik, 1990 ), indicating that tau could suppress microtubule dynamics (Panda et al., 1995 ). This function could also be performed by other MAPs, which would explain the viability of tau-deficient mice produced by gene targeting (Harada et al., 1994 ; Dawson et al., 2001 ), although some differences have been identified with respect to the wild-type mouse (see below).
It has been suggested that alterations in the amount or conformation of tau, as well as other modifications to this protein could have pathological effects. This modifications provoke disorders known as tauopathies (Hernandez and Avila, 2007 ), of which Alzheimer’s disease (AD) is the most prevalent. Tauopathies are neurodegenerative disease in which neurodegeneration is associated with the presence of phosphorylated or/and aggregated tau (Hernandez and Avila, 2007 ).
Alzheimer’s disease is characterized by the presence of two specific structures in the brain of patients, senile plaques (SP), and neurofibrillary tangles (NFT), which are accompanied by more evident neuron death. The main component of SP is the beta amyloid peptide (Masters et al., 1985 ) whereas tau protein, in its phosphorylated form, is the main constituent of NFT (Grundke-Iqbal et al., 1986a ,b ).
The development of a tau pathology in AD correlates with the neurodegeneration found during the progression of the disease (Braak and Braak, 1991 ), which also correlates with the appearance of phosphorylated tau (Delacourte et al., 1999 ). In addition, an inverse relationship has been found in damaged regions between the number of extracellular NFT ghost tangles and the number of surviving cells (Bondareff et al., 1989 ; Cras et al., 1995 ). This observation suggests that neurons containing NFT could degenerate and release their intracellular NFT into the extracellular environment (Gomez-Ramos et al., 2006 ), which may then be toxic for the surrounding neurons (Gomez-Ramos et al., 2006 , 2008 ). Indeed, all cytoplasmic proteins are released into the extracellular space after neuron death and some of these proteins could be toxic in this milieu. We hypothesized that it is extracellular tau that is indeed toxic to neurons (Gomez-Ramos et al., 2006 ) and in this short review, I will describe some of the studies that have been carried out to test this hypothesis. However, in testing this hypothesis, we must take care to distinguish between the possible toxic role for intracellular tau and that of extracellular tau.
Intracellular Tau
It has been suggested that as well as an increase in the amount of tau (Andorfer et al., 2005 ), structural changes to this protein, modifications by phosphorylation, or its aggregation could produce toxic effects in cells (Avila et al., 2004 ; Duff and Planel, 2005 ; Bretteville and Planel, 2008 ; Takashima, 2008 ). An excess of tau protein could inhibit the trafficking of vesicles and organelles in neurons (Stamer et al., 2002 ). Indeed, an increase in the amount of tau protein could impair microtubule dependent axonal transport (Dixit et al., 2008 ), although this remains to be confirmed (Yuan et al., 2008 ). Nevertheless, reducing the amount of endogenous tau ameliorates amyloid-beta toxicity (Rapoport et al., 2002 ; Roberson et al., 2007 ) and tau suppression in a mouse model improves memory function (Santacruz et al., 2005 ).
There is more data available about the toxicity of phosphorylated intracellular tau and it has been suggested that phosphotau can sequester some other brain MAPs, producing a disorganization of the microtubule network that might be toxic to a neuron (Alonso et al., 1997 ; Alonso Adel et al., 2006 ). Indeed, phosphotau appears to be toxic to neurons in a Drosophila model (Steinhilb et al., 2007 ; Feuillette et al., 2010 ), and inhibition of tau phosphorylation by altering GSK3 activity is correlated with reduced degeneration in vivo (Noble et al., 2005 ; Gomez de Barreda et al., 2010b ). Moreover, phosphorylated tau impairs learning in aged mice expressing human tau (Kimura et al., 2007 ).
Toxicity in neurons has also been related with conformational changes in the tau protein (Garcia-Sierra et al., 2008 ), which could provoke the appearance of aggregated truncated tau (Park and Ferreira, 2005 ; Zilka et al., 2009 ). However, the toxicity of aggregated tau still remains under debate (Duff and Planel, 2005 ), since neurons may live for decades with NFT in humans (Morsch et al., 1999 ). Also, in hibernating squirrels, tau accumulates in the somato-dendritic compartment and becomes hyperphosphorylated without causing neuronal cell death (Arendt et al., 2003 ). Thus it has been suggested that phosphorylation at specific sites, but not at others, could be needed for the toxic effect of tau (Alonso et al., 2008 ).
Extracellular Tau
The addition of recombinant tau protein to cultured neuronal cells produces an increase in intracellular calcium that could lead to neuron degeneration (Gomez-Ramos et al., 2006 ). As indicated above, it has been suggested that endogenous intracellular tau may be released to the extracellular space upon neuron degeneration, where it could be toxic to other neurons (Gomez-Ramos et al., 2006 ). The concentration of Tau in a neuron is about 2 µM (Gamblin et al., 2003 ; Reynolds et al., 2005 ) and concentrations as low as 35 pM of extracellular tau might produce an increase in the intracellular calcium concentration in neurons (Gomez-Ramos et al., 2009 ).
Although, it has been postulated that the presence of tau in the extracellular milieu is due to neuron degeneration, and that it can then be found in the cerebrospinal fluid (Iqbal et al., 2005 ), the presence of extracellular tau could be due to other causes. For example, it cannot be ruled out that tau might be exocytosed by cells, as indicated for the prion protein (Fevrier et al., 2004 ). Indeed, intraneuronal transfer of tau protein between neurons in situ has been recently described, suggesting that N-terminal region of tau is required for tau secretion (Kim et al., 2010 ).
The increase in intracellular calcium induced by Tau is caused by its interaction with specific cell receptors on neurons. After studying the different possible mechanisms for the calcium mobilization provoked by tau, it was found to promote an increase in intracellular calcium through its interaction with the M1 and M3 muscarinic receptors expressed by neurons (Gomez-Ramos et al., 2008 ). Monomeric tau interacts with cell receptors with a higher affinity than aggregated tau (Gomez-Ramos et al., 2006 ), and unmodified tau more readily than phosphorylated tau (Gomez-Ramos et al., 2006 ). However, the tau present in the filaments that make up NFT is both phosphorylated and aggregated.
Nevertheless, using a fraction of enriched aggregated tau protein it was found that tau aggregates can enter cells of neuronal origin (Frost et al., 2009 ) and that exogenous aggregated tau induces the aggregation of intracellular tau (Frost et al., 2009 ). In this process, it is not known if the tau aggregates enter the cells by endocytosis or by another mechanism (Frost et al., 2009 ).
In a similar study in vivo (Clavaguera et al., 2009 ), the injection of a brain extract containing tau aggregates provoked the transmission and spreading of the tau pathology in a transgenic mouse that overexpresses human tau with a mutation (P301S) that is found in some patients with frontotemporal dementia (another tauopathy). Again, little is known about the possible mechanism underlying the spreading of tau from cell to cell, although in this case the influence of monomeric tau cannot be excluded since the brain extract used in the experiments could have contained both monomeric and aggregated tau.
A summary of the possible role of intracellular and extracellular tau is shown in Figure 1 .
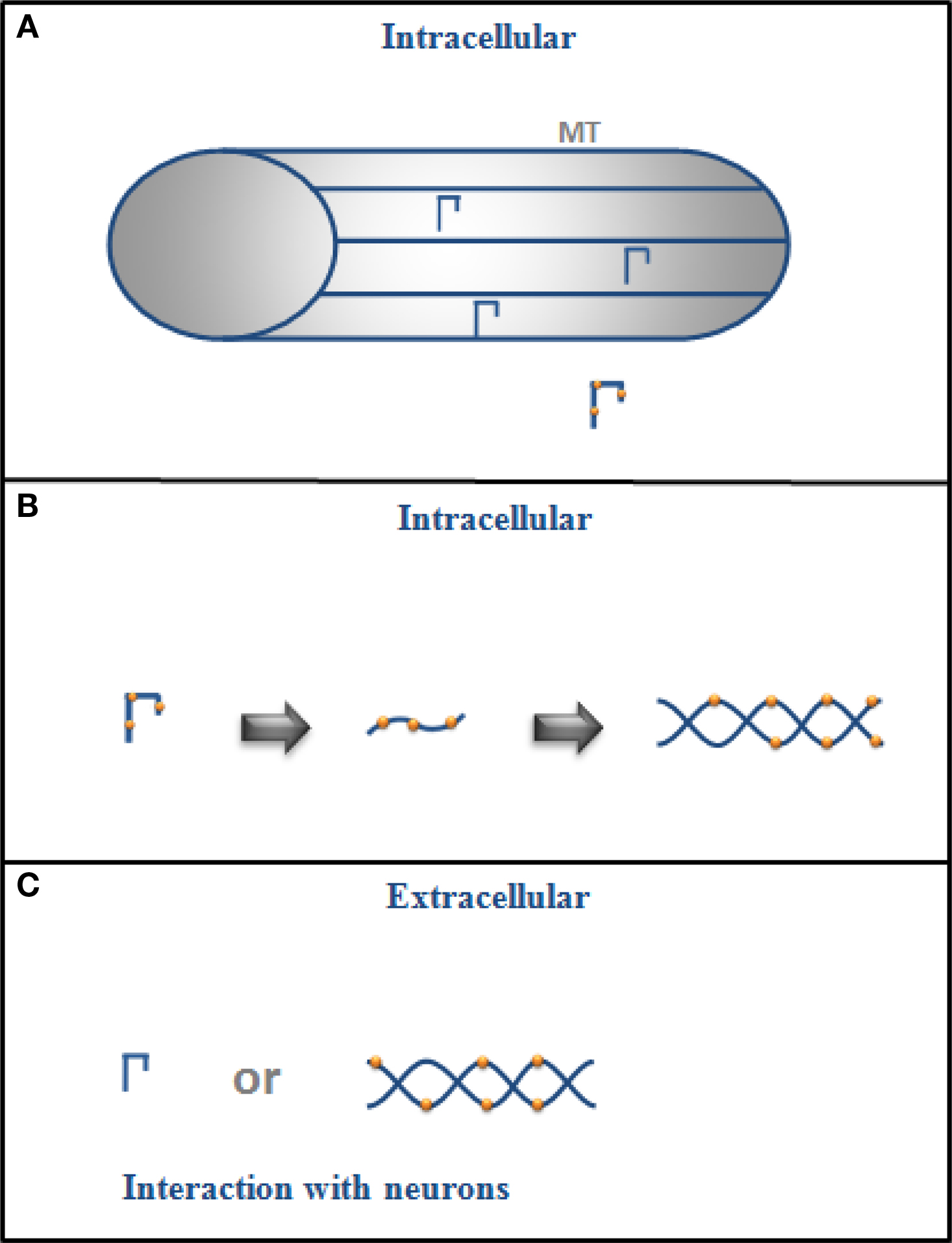
Figure 1. (A) Intracellular tau is mainly bound to microtubules (MT) in the cytoplasm of a neuron but if it is phosphorylated (drawing with dots), its binding to MT decreases, and (B) it could also change its conformation and aggregate into filaments. (C) In its extracellular form, tau could exist as monomers or in filaments (aggregated form), and it could interact with neurons.
Tau as a Target
It is known that tau-deficient mice produced by gene targeting are viable (Harada et al., 1994 ; Dawson et al., 2001 ) and can reproduce, probably since some tau functions can be compensated for by other proteins, such as its role on microtubule dynamics. Accordingly, it is possible to consider the tau protein as a suitable target for the treatment of tauopathies. Nevertheless, the absence of tau could produce some defects due to its role in processes unrelated to its other activities as a MAP. Indeed, different defects have been described for tau null mice, like muscle weakness, some behavioral deficits (Ikegami et al., 2000 ), and hyperactivity (Cantero et al., 2010b ).
The tau protein has also been described as an inhibitor of Histone deacetylase 6 (HDAC6) (Perez et al., 2009 ). Tubulin acetylation is regulated by HDAC6 and acetylated tubulin enhances the recruitment of molecular motors like kinesin to microtubules (Kazantsev and Thompson, 2008 ). Thus, the acetylation of tubulin in microtubules enhances the binding of motor proteins that play a crucial function in axonal transport. This observation could explain the fact that an excess of tau may be beneficial in maintaining tubulin in an acetylated form that will facilitate axonal transport, but it may also be detrimental to the competition of tau protein for the same binding site as that of kinesin on the tubulin molecule. This possibility could help explain the discrepancies regarding the effect of the amount of tau on axonal transport.
In addition, the lack of tau could result in a delay in neuron differentiation during adult neurogenesis in the dentate gyrus (Gomez de Barreda et al., 2010a ; Sirerol-Piquer et al., 2010 ). Moreover, a role for tau protein has been proposed in the sleep/wake cycle and indeed, tau-deficient mice sleep significantly less (Cantero et al., 2010a ), which could also result in an increase in the expression of Aβ peptide aggregates (Kang et al., 2009 ).
Together, these observations suggest that the tau protein may not be the most suitable target for the treatment of tauopathies.
Conclusion
In summary, intracellular tau not bound to microtubules and in its phosphorylated form may be toxic within a neuron, while tau protein may also promote neurodegeneration in its extracellular form. However, the depletion of tau, that might prevent its toxic function, could result in collateral effects that might also be detrimental to neurons. However, depletion of extracellular tau could be advisable and the use of a vaccine using tau protein as potential target for immunotherapy has been proposed (Kayed and Jackson, 2009 ).
References
Alonso, A. C., Li, B., Grundke-Iqbal, I., and Iqbal, K. (2008). Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 5, 375–384.
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303.
Alonso Adel, C., Li, B., Grundke-Iqbal, I., and Iqbal, K. (2006). Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. U.S.A. 103, 8864–8869.
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454.
Arendt, T., Stieler, J., Strijkstra, A. M., Hut, R. A., Rudiger, J., Van der Zee, E. A., Harkany, T., Holzer, M., and Hartig, W. (2003). Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 23, 6972–6981.
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384.
Bondareff, W., Mountjoy, C. Q., Roth, M., and Hauser, D. L. (1989). Neurofibrillary degeneration and neuronal loss in Alzheimer’s disease. Neurobiol. Aging 10, 709–715.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Bretteville, A., and Planel, E. (2008). Tau aggregates: toxic, inert, or protective species? J. Alzheimer’s Dis. 14, 431–436.
Caceres, A., and Kosik, K. S. (1990). Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 343, 461–463.
Cantero, J. L., Hita-Yañez, E., Moreno-Lopez, B., Portillo, F., Rubio, A., and Avila, J. (2010a). Tau protein role in sleep-wake cycle. J. Alzheimer’s Dis. (in press).
Cantero, J. L., Moreno-Lopez, B., Portillo, F., Rubio, A., Hita-Yañez, E., and Avila, J. (2010b). Role of tau protein on neocortical and hippocampal oscillatory patterns. Hippocampus. (in press).
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., Fraser, G., Stalder, A. K., Beibel, M., Staufenbiel, M., Jucker, M., Goedert, M., and Tolnay, M. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913.
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225.
Cras, P., Smith, M. A., Richey, P. L., Siedlak, S. L., Mulvihill, P., and Perry, G. (1995). Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease. Acta Neuropathol. 89, 291–295.
Dawson, H. N., Ferreira, A., Eyster, M. V., Ghoshal, N., Binder, L. I., and Vitek, M. P. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187.
Delacourte, A., David, J. P., Sergeant, N., Buee, L., Wattez, A., Vermersch, P., Ghozali, F., Fallet-Bianco, C., Pasquier, F., Lebert, F., Petit, H., and Di Menza, C. (1999). The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 52, 1158–1165.
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089.
Drubin, D. G., and Kirschner, M. W. (1986). Tau protein function in living cells. J. Cell Biol. 103, 2739–2746.
Feuillette, S., Miguel, L., Frebourg, T., Campion, D., and Lecourtois, M. (2010). Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J. Neurochem. 113, 895–903.
Fevrier, B., Vilette, D., Archer, F., Loew, D., Faigle, W., Vidal, M., Laude, H., and Raposo, G. (2004). Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688.
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852.
Gamblin, T. C., Berry, R. W., and Binder, L. I. (2003). Modeling tau polymerization in vitro: a review and synthesis. Biochemistry 42, 15009–15017.
Garcia-Sierra, F., Mondragon-Rodriguez, S., and Basurto-Islas, G. (2008). Truncation of tau protein and its pathological significance in Alzheimer’s disease. J. Alzheimer’s Dis. 14, 401–409.
Gomez-Ramos, A., Diaz-Hernandez, M., Cuadros, R., Hernandez, F., and Avila, J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850.
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Diaz-Hernandez, J. I., Miras-Portugal, M. T., and Avila, J. (2009). Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 19, 708–717.
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell Neurosci. 37, 673–681.
Gomez de Barreda, E., Dawson, H. N., Vitek, M. P., and Avila, J. (2010a). Tau deficiency leads to the upregulation of BAF-57, a protein involved in neuron-specific gene repression. FEBS Lett. 584, 2265–2270.
Gomez de Barreda, E., Perez, M., Gomez Ramos, P., de Cristobal, J., Martin-Maestro, P., Moran, A., Dawson, H. N., Vitek, M. P., Lucas, J. J., Hernandez, F., and Avila, J. (2010b). Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 37, 622–629.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986a). Abnormal phosphorylation of the microtubule- associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917.
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986b). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Harada, A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Ohshima, T., Sato-Yoshitake, R., Takei, Y., Noda, T., and Hirokawa, N. (1994). Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491.
Ikegami, S., Harada, A., and Hirokawa, N. (2000). Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 279, 129–132.
Iqbal, K., Flory, M., Khatoon, S., Soininen, H., Pirttila, T., Lehtovirta, M., Alafuzoff, I., Blennow, K., Andreasen, N., Vanmechelen, E., and Grundke-Iqbal, I. (2005). Subgroups of Alzheimer’s disease based on cerebrospinal fluid molecular markers. Ann. Neurol. 58, 748–757.
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., Fujiki, N., Nishino, S., and Holtzman, D. M. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007.
Kayed, R., and Jackson, G. R. (2009). Prefilament tau species as potential targets for immunotherapy for Alzheimer disease and related disorders. Curr. Opin. Immunol. 21, 359–363.
Kazantsev, A. G., and Thompson, L. M. (2008). Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7, 854–868.
Kim, W., Lee, S., Jung, C., Ahmed, A., Lee, G., and Hall, G. F. (2010). Interneuronal transfer of human tau between Lamprey central neurons in situ. J. Alzheimer’s Dis. 19, 647–664.
Kimura, T., Yamashita, S., Fukuda, T., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2007). Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 26, 5143–5152.
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249.
Morsch, R., Simon, W., and Coleman, P. D. (1999). Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197.
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., Gaynor, K., Wang, L., LaFrancois, J., Feinstein, B., Burns, M., Krishnamurthy, P., Wen, Y., Bhat, R., Lewis, J., Dickson, D., and Duff, K. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995.
Panda, D., Goode, B. L., Feinstein, S. C., and Wilson, L. (1995). Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 34, 11117–11127.
Park, S. Y., and Ferreira, A. (2005). The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J. Neurosci. 25, 5365–5375.
Perez, M., Santa-Maria, I., Gomez de Barreda, E., Zhu, X., Cuadros, R., Cabrero, J. R., Sanchez-Madrid, F., Dawson, H. N., Vitek, M. P., Perry, G., Smith, M. A., and Avila, J. (2009). Tau – an inhibitor of deacetylase HDAC6 function. J. Neurochem. 109, 1756–1766.
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tau is essential to beta-amyloid- induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 6364–6369.
Reynolds, M. R., Berry, R. W., and Binder, L. I. (2005). Site-specific nitration differentially influences tau assembly in vitro. Biochemistry 44, 13997–14009.
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., Gerstein, H., Yu, G. Q., and Mucke, L. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754.
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., DeTure, M., Ramsden, M., McGowan, E., Forster, C., Yue, M., Orne, J., Janus, C., Mariash, A., Kuskowski, M., Hyman, B., Hutton, M., and Ashe, K. H. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481.
Sirerol-Piquer, M. S., Gomez-Ramos, P., Hernandez, F., Perez, M., Moran, M. A., Fuster-Matanzo, A., Lucas, J. J., Avila, J., and Garcia-Verdugo, J. M. (2010). GSK3β overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus (in press).
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063.
Steinhilb, M. L., Dias-Santagata, D., Fulga, T. A., Felch, D. L., and Feany, M. B. (2007). Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 18, 5060–5068.
Takashima, A. (2008). Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J. Alzheimer’s Dis. 14, 371–375.
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862.
Yuan, A., Kumar, A., Peterhoff, C., Duff, K., and Nixon, R. A. (2008). Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 28, 1682–1687.
Keywords: tau, toxicity, Alzheimer disease, tauopathies
Citation: Avila J (2010) Intracellular and extracellular tau. Front. Neurosci. 4:49. doi: 10.3389/fnins.2010.00049
This article was submitted to Frontiers in Neurodegenerative Diseases, a specialty of Frontiers in Neuroscience.
Received: 08 April 2010;
Paper pending published: 26 April 2010;
Accepted: 09 June 2010;
Published online: 28 June 2010
Edited by:
Emmanuel Planel, Centre Hospitalier de l’université Laval, CanadaColumbia University Medical Center, USA
Reviewed by:
Sebastien S. Hebert, Université Laval, CanadaNicole Leclerc, Université de Montréal, Québec, Canada
Emmanuel Planel, Columbia University Medical Center, USA; Centre Hospitalier de l’université Laval, Canada
Copyright: © 2010 Avila. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jesús Avila, Centro de Biología Molecular “Severo Ochoa”, Campus Cantoblanco, Universidad Autónoma de Madrid, Nicolás Cabrera, 3, 28050 Madrid, Spain. e-mail:amF2aWxhQGNibS51YW0uZXM=