 Amanda Sierra*
Amanda Sierra* Juan M. Encinas and Mirjana Maletic-Savatic*
Juan M. Encinas and Mirjana Maletic-Savatic*
- Department of Pediatrics, Baylor College of Medicine, Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, Houston, TX, USA
Neural stem cells reside in well-defined areas of the adult human brain and are capable of generating new neurons throughout the life span. In rodents, it is well established that the new born neurons are involved in olfaction as well as in certain forms of memory and learning. In humans, the functional relevance of adult human neurogenesis is being investigated, in particular its implication in the etiopathology of a variety of brain disorders. Adult neurogenesis in the human brain was discovered by utilizing methodologies directly imported from the rodent research, such as immunohistological detection of proliferation and cell-type specific biomarkers in postmortem or biopsy tissue. However, in the vast majority of cases, these methods do not support longitudinal studies; thus, the capacity of the putative stem cells to form new neurons under different disease conditions cannot be tested. More recently, new technologies have been specifically developed for the detection and quantification of neural stem cells in the living human brain. These technologies rely on the use of magnetic resonance imaging, available in hospitals worldwide. Although they require further validation in rodents and primates, these new methods hold the potential to test the contribution of adult human neurogenesis to brain function in both health and disease. This review reports on the current knowledge on adult human neurogenesis. We first review the different methods available to assess human neurogenesis, both ex vivo and in vivo and then appraise the changes of adult neurogenesis in human diseases.
Introduction: A Brief History of the Adult Mammalian Neurogenesis Discovery
The discovery of adult neurogenesis crushed the century-old dogma that no new neurons are formed in the mammalian brain after birth. However, this finding and its acceptance by the scientific community did not happen without hurdles. At the beginning of the last century, based on detailed observations of the brain anatomy reported by Santiago Ramon y Cajal and others, it was established that the human nervous system develops in utero (Colucci-D’Amato et al., 2006). In adult brains, it was thought, no more neurons could be generated, as the brain is grossly incapable of regenerating after damage (for a more detailed historical report see Watts et al., 2005; Whitman and Greer, 2009). This dogma was deeply entrenched in the Neuroscience community, and Altman’s (1962) discovery of newborn cells in well-defined areas of the adult rodent brain was largely ignored. The phenomenon was reexamined in the 1970–1980s, when Michael Kaplan (Kaplan and Hinds, 1977) and Fernando Nottebohm (Goldman and Nottebohm, 1983) demonstrated the presence of newborn cells in the adult brain of mice and canaries, respectively, and showed that these cells had ultrastructural characteristics of neurons. However, such findings could not be repeated in adult rhesus monkeys, where proliferating cells appeared to be glial and endothelial cells and not neurons (Rakic, 1985; Eckenhoff and Rakic, 1988). Thus, neurogenesis seemed to be absent in adult primates (Eckenhoff and Rakic, 1988).
The field of adult neurogenesis finally took off in the 1990s with the development of new technologies. First, the use of 3H-thymidine, a radioactive nucleotide used to study proliferation when incorporated into the cells during the S phase of the cell cycle, was replaced by its analog, bromodeoxyuridine (BrdU), which could be detected by a specific antibody. Utilization of the BrdU for labeling of newborn cells via immunohistochemistry allowed their further studies by co-labeling with specific neuronal markers (Miller and Nowakowski, 1988). Further, it was shown that neuroprogenitor cells (NPCs), isolated from adult mouse brains, proliferated and differentiated into neurons and astrocytes in vitro (Reynolds and Weiss, 1992). In addition, NPCs labeled with viral vectors were able to migrate and differentiate into neurons in the adult mouse brain (Lois and Alvarez-Buylla, 1993), demonstrating that the adult neurogenesis was functional in rodents. Finally, the existence of adult neurogenesis in humans was firmly established when, in 1998, Gage and colleagues demonstrated for the first time that new neurons were produced in the adult hippocampus (Eriksson et al., 1998).
Currently, adult neurogenesis is one of the hot topics in Neuroscience especially because of the new opportunities it may bring for treatments of neurodegenerative diseases, either by harnessing resident progenitors to regenerate the lost tissue (Sohur et al., 2006) or by cell transplantation therapies (Goldman and Windrem, 2006). The field is currently on the rise, as shown by the exponential growth of publications with the key words “adult” AND “neurogenesis OR neural stem cells” (PubMed search up to December 31, 2010): a total of 6,437 papers have been published, of which 57% (3,695 papers) was published in the last 5 years (Figure 1). However, only 8% of published papers (530 papers) deal with human data (search including the term “human” in the title), suggesting that the research on adult neurogenesis in humans is still in its infancy. Thus, the actual knowledge on adult human neurogenesis is limited and in many cases, data is directly extrapolated from the rodent literature. Herein, we review the methodologies used to assess adult human neurogenesis and its status in several neuropsychiatric disorders.
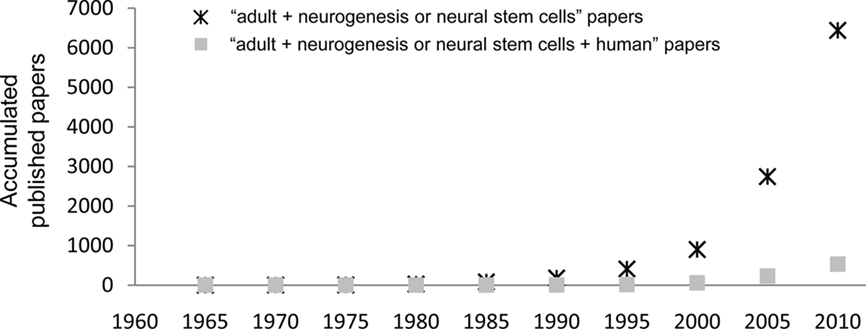
Figure 1. Published papers on adult neurogenesis per quinquennium. The graph shows the accumulated papers published from 1960 onward, searched in PubMed with the search terms “adult” AND (“neurogenesis” OR “stem cells”). The asterisks show the total number of papers, and the filled squares show those papers with the term “human” in their title.
Methods to Assess Neurogenesis in Humans
The extent of our knowledge on adult human neurogenesis directly correlates with the type of available techniques that can be applied to human brain tissue research. Several methodologies exist, but each method yields different sensitivity, specificity, and ultimately different units of quantification, thus rendering it difficult to compare different studies. In addition, some methodologies can assess only proliferation (NPCs or total proliferating cells) while others can provide the data on neurogenesis [neuroblasts (NBs, neuronal committed cells) or newborn neurons]. Herein, we review the advantages and disadvantages of methods used to assess adult human neurogenesis both ex vivo and in vivo.
Methods to Assess Neurogenesis on Postmortem and Biopsied Tissue
The majority of the methodologies used to study neurogenesis ex vivo have been inherited from the rodent literature, where they have been thoroughly validated (Figure 2). These techniques require brain tissue that is obtained postmortem, either frozen fresh immediately after death or, more frequently, fixed and stored in brain tissue banks. In both cases, it is important to note that the cause of death, presence of brain and/or systemic illnesses, age of death, and postmortem interval (the time from death to tissue fixation) may be confounding factors when interpreting the results (Boldrini et al., 2009). Alternatively, brain tissue can also be obtained from biopsies or surgical resections, such as in temporal lobectomy due to intractable epilepsy.
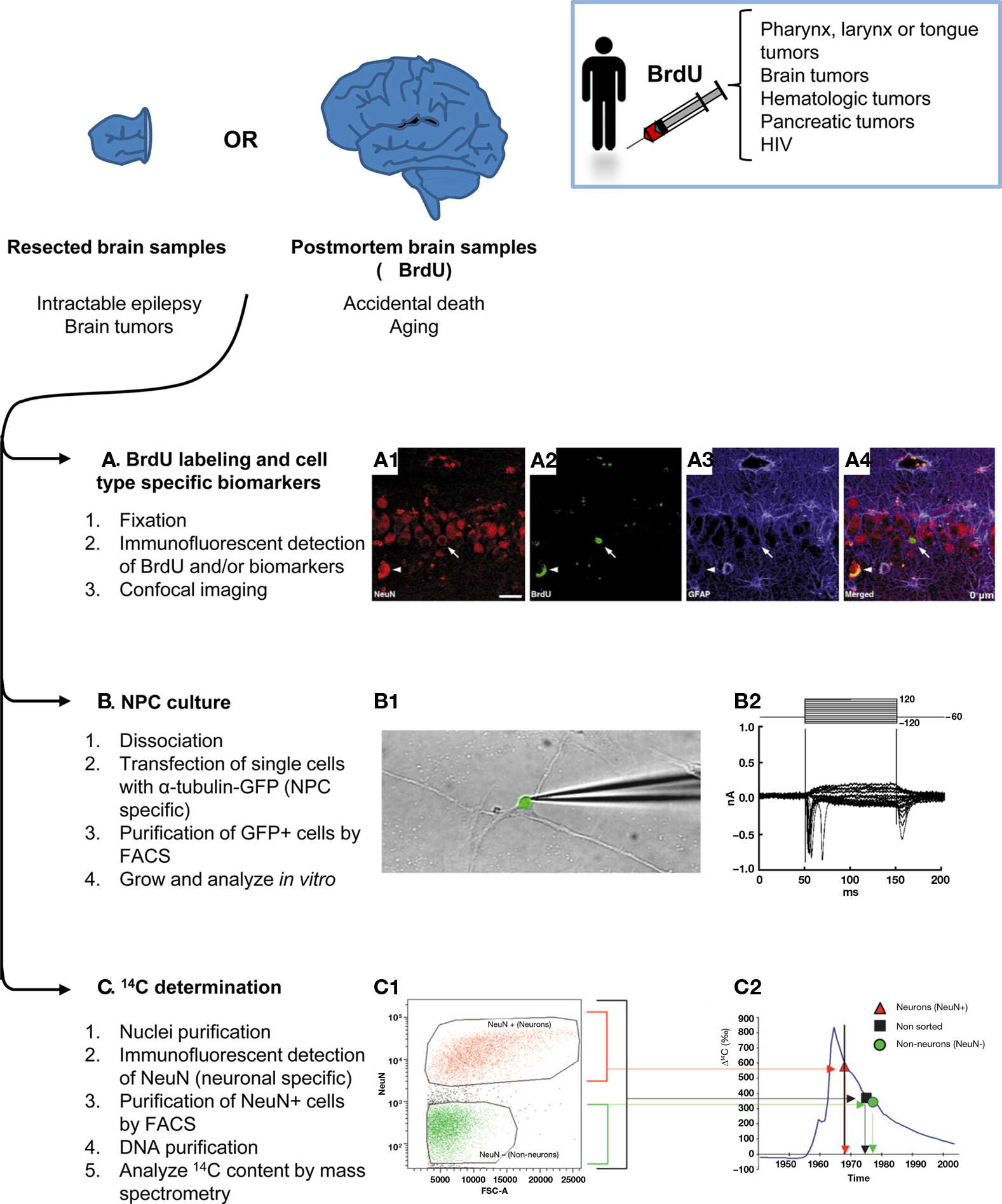
Figure 2. Methods to assess adult human neurogenesis in resected or postmortem brain samples. Brain samples from live individuals can be obtained by surgical resection of the focus of the seizures in intractable epilepsy or brain tumors. Alternatively, bank tissues store frozen or fixed brain samples from accidental deaths and aging individuals, some of which may have been treated with BrdU. Once the tissue is obtained, three main methods can be applied to study neurogenesis: (A) Immunodetection of BrdU and/or proliferation and cell-type specific biomarkers in fixed tissue, followed by analysis by confocal analysis. An example of new born neurons in the SGZ of the hippocampus of an aging patient is shown in (A1) NeuN+ neurons (red), (A2) BrdU labeling (green), (A3) GFAP+ astrocytes (blue), (A4) merged image showing one newborn neuron labeled with BrdU and NeuN, but not with GFAP (arrow); and non-specific fluorescence emission due to lipofuscin accumulation in old tissue (arrowhead). Scale bar is 25 μm. (B) Culture of NPCs. In this case, the fresh tissue is dissociated and transfected with a reporter gene (GFP) driven by a Tα1 tubulin promoter, an early neuronal regulatory gene. The transfected cells are then purified by FACS and cultured in vitro. The grown cells differentiate into fully functional neurons which when patch-clamped in a voltage-clamped configuration (B1) show typical depolarizations of the voltage-gated sodium currents measured in nanoAmperes (nA) over a time course of milliseconds (ms) (B2). (C) Determination of the 14C content. Cell nuclei from prefrontal cortex samples are purified by density gradients, labeled with NeuN and purified by FACS. Next, their DNA is purified and the 14C content is analyzed by mass spectrometry. (C1) FACS plot showing the expression of NeuN (vertical axis) versus the area of the forward scatter (FSC-A, which is considered a measure of the particle size). The neuronal nuclei (NeuN+, orange) are separated from the rest of the nuclei (NeuN−, green). (C2) 14C content of the sorted nuclei (Δ14C, given in relation to a universal standard and corrected for radioactive decay) compared to the atmosphere 14C content (blue line) and the age of the individual (vertical black line). Neuronal nuclei (orange triangles) have a 14C content that indicates that their age is coincident with that of the individual. Non-neuronal nuclei (green circles) are younger than the individual, making the non-sorted (black squares) cells younger than the individual in average as well. Figures (A1–A4) and (B1–B2) are reprinted by permission from Macmillan Publishers Ltd: Nature Medicine, Eriksson et al. (1998), copyright 1998; and Roy et al. (2000), copyright 2000, respectively. Figures (C1–C2) are reprinted by permission from Bhardwaj et al. (2006), copyright 2006, National Academy of Sciences, U.S.A.
Bromodeoxyuridine labeling
Bromodeoxyuridine is widely used in animal models to quantify the number of dividing cells in a tissue and to trace their progeny. When administered systemically, it is incorporated into the DNA during the S phase of the cell cycle and is transmitted to the daughter cells, as long as it is not diluted through many rounds of proliferation (Karpowicz et al., 2005). Using a variety of specific anti-BrdU antibodies, it can be detected by immunohistochemistry. Although it can be mutagenic (Rakic, 2002a), BrdU was approved in 1995 by the Food and Drug Administration (FDA) to be used in humans under the commercial name of Broxine/Neomark as a radiation sensitizer in the treatment of primary brain tumors1. Furthermore, BrdU is currently used in several clinical trials to measure cell cycle kinetics in patients with hematologic malignancies, to study white blood cell replication and survival in patients with human immunodeficiency virus, and to assess the degree of tumor proliferation in biopsies as well as to treat patients with pancreatic tumors as an antineoplastic agent2. It was the treatment with BrdU of patients who suffered from larynx, pharynx, or tongue cancers that enabled the detection of proliferating NPCs in the hippocampus (Eriksson et al., 1998), the discovery which paved the way for further investigations of adult neurogenesis in the human brain.
The major advantage of BrdU labeling is its sensitivity to detect proliferating cells compared to other immunohistological methods. For instance, neurogenesis in adult rhesus monkeys was only detected using BrdU (Kornack and Rakic, 1999) but not H3-thymidine, which also incorporates into the DNA of dividing cells (Rakic and Nowakowski, 1981). In addition, proliferation in the adult human hippocampus was found using BrdU (Eriksson et al., 1998), but not other markers of proliferation such as MiB-1 (Seress et al., 2001). Another advantage of BrdU labeling is utilization of pulse-and-chase analysis, which enables studies of both proliferation (hours after the BrdU injection) and differentiation (days or months after the BrdU injection). Such studies may ultimately be used to distinguish between neurogenesis and gliogenesis, a particularly significant feature in human brain diseases.
Despite its wide use, BrdU labeling has some drawbacks. For instance, BrdU does not diffuse freely through the blood–brain barrier (BBB), but rather, it likely uses the deoxythymidine transporters (Spector and Johanson, 2007). Therefore, conditions that disrupt the BBB, such as inflammation, irradiation, status epilepticus, trauma, etc., may lead to different BrdU availability which, in turn, leads to labeling of different number of cells without actual changes in proliferation (von Bohlen Und Halbach, 2007). To overcome this possible cause of misinterpretation of the data, it is particularly important to determine the integrity of the BBB when comparing neurogenesis in healthy and diseased individuals. Furthermore, BrdU can label phenomena other than proliferation, such as DNA turnover or repair, or abortive reentry in the cell cycle during apoptosis (Rakic, 2002a,b). Finally, some dividing cells may preferentially use de novo synthesis of deoxythymidine rather than the salvage enzymes which phosphorylate existing deoxynucleotides (including BrdU) to generate the deoxynucleotides for DNA synthesis during the S phase (Spector and Johanson, 2007). In such cases, BrdU labeling will not correlate with the proliferative activity of the cells. Thus, while it has revolutionized the studies of neurogenesis, BrdU labeling should be meticulously analyzed to avoid possible misinterpretations as noted above.
Expression of cell-specific biomarkers
Particular cell types and particular stages of the cell cycle of dividing cells can be assessed by specific antibodies. When utilizing these reagents for immunostaining of the human tissue, it is important to take into account antigenicity, which can be affected by the delayed fixation of the postmortem tissue (Boekhoorn et al., 2006; Liu et al., 2008) and the specificity of the antibodies, which may be related to a particular species (for example, antibodies which work for rodent tissue may not work for human tissue). Ideally, quantification of cells expressing the biomarker of interest should be obtained using unbiased stereology, such as the optical dissector method (Lemmens et al., 2010), although in human samples this goal can be difficult to achieve due to the low number of proliferating cells (Liu et al., 2008).
Proliferation biomarkers are expressed while cells are cycling (Table 1), such as Ki-67 (Hall and Woods, 1990; Yerushalmi et al., 2010) and MCM2 (Stoeber et al., 2001; Bailis and Forsburg, 2004). Ki-67 immunolabeling relies heavily on pH (Boekhoorn et al., 2006); thus other antibodies against the Ki67 antigen, such as MIB-1, have been developed for different tissue conditions (Cattoretti et al., 1992). Another widely used marker is PCNA (Takasaki et al., 1981), although it is also expressed by some non-proliferating cells (Rakic, 2002a). Compared to BrdU, these markers offer the advantage of not requiring a priori administration of the label, thus increasing the number of samples that can be studied. However, these markers only label proliferating cells and cannot be used to trace their fate or to assess actual neurogenesis. Importantly, proliferation biomarkers in neurogenic regions are usually assumed to label NPCs, but it is also possible that they label proliferating astrocytes, oligoprogenitors, or endothelial cells. Gliogenesis should be ruled out by double-labeling with specific glial biomarkers.
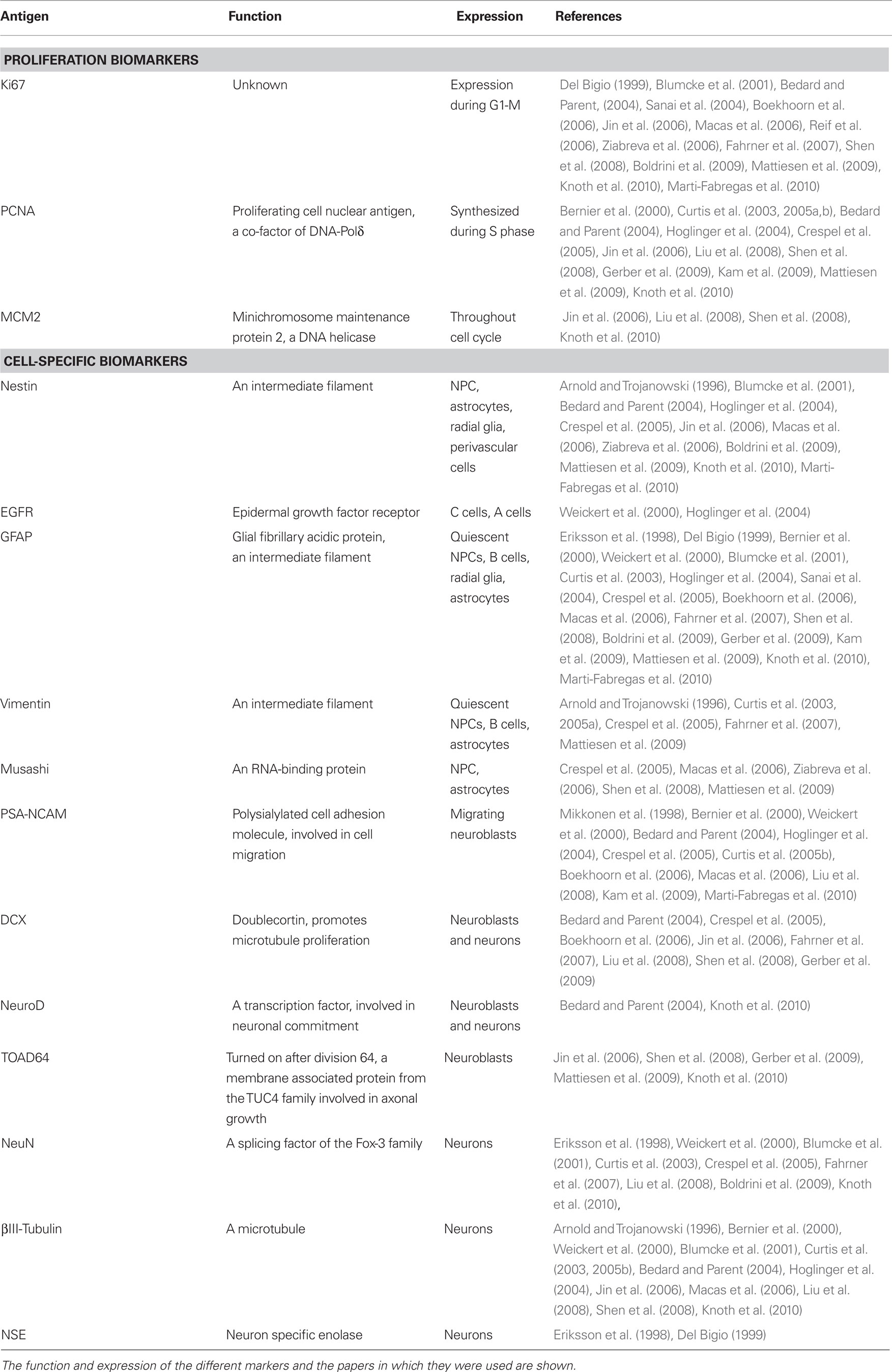
Table 1. Proliferation and cell-type specific biomarkers commonly used in human neurogenesis studies.
Cell-specific biomarkers can also be detected using specific antibodies (thoroughly reviewed by von Bohlen Und Halbach, 2007; Encinas and Enikolopov, 2008). The most common cell biomarkers used for studies of the human tissue are described in Table 1: Nestin (Lendahl et al., 1990), GFAP (Doetsch et al., 1997), Vimentin (Doetsch et al., 1997), EGFR (Danilov et al., 2009), Musashi (Sakakibara and Okano, 1997), PSA-NCAM (Doetsch et al., 1997), Doublecortin (DCX; des Portes et al., 1998), NeuroD (Miyata et al., 1999), TOAD-64 (Minturn et al., 1995), NeuN (Mullen et al., 1992; Kim et al., 2009), NSE (Kaiser et al., 1989), and βIII-Tubulin (Encinas and Enikolopov, 2008). However, caution must be observed when using these antibodies to assign the cell identity. For instance, apart from labeling migrating NBs, PSA-NCAM is also involved in synaptic plasticity (Dityatev et al., 2004) and is expressed in non-neurogenic areas in rodents (Nacher et al., 2002). In addition, the specificity of DCX as a marker for neurogenesis has been recently challenged, as it has been found in mature astrocytes in the human neocortex in patients suffering from neurodegeneration (Verwer et al., 2007). Finally, some authors (Boekhoorn et al., 2006), but not others (Liu et al., 2008), find that DCX immunolabeling is very sensitive to postmortem delay, an important confounding factor when comparing human postmortem samples.
NPC culture
Human NPCs have been isolated and cultured in vitro. Originally, Steindler and colleagues were able to grow NPCs from the temporal lobe tissue extracted from patients with intractable epilepsy. These NPCs proliferated in vitro in the form of neurospheres and differentiated into neurons and astrocytes (Johansson et al., 1999; Kukekov et al., 1999). Later on, Goldman and colleagues refined the technique and were able to specifically isolate the NPCs (Roy et al., 2000). They transfected cells from mixed brain cultures with viral constructs expressing a humanized green fluorescent protein (GFP) under regulatory elements of the nestin or the early neuronal Tα1 tubulin genes, and then purified the NPCs based on the expression of the transgene by fluorescence-activated cell sorting (FACS). The isolated NPCs divided in culture and gave rise to physiologically active neurons (Roy et al., 2000). This technique was not designed for quantification purposes and, thus, cannot be used to assess the degree of neurogenesis in different disorders or after different treatments. Nonetheless, it has generated a great excitement because it was seen as the first serious step toward cell replacement therapies for human neurological diseases by transplantation of either precursors or already differentiated cells (Antel et al., 2000).
14C retrospective labeling
Similar to the 14C-dating used in archeology, this method uses the 14C-content to assess the average age of the cells present in a particular tissue (Spalding et al., 2005; Bhardwaj et al., 2006). Due to the extensive testing of nuclear weapons in the 1950–1960s, large quantities of 14C were generated; since then, the 14C levels in the biosphere have decayed at a known rate. This 14C incorporates into the cells, matching the 14C levels in the atmosphere. The exception is genomic DNA because the molecular composition of DNA is stable after the last cell division (except in case of DNA repair). The DNA 14C-content reflects the age of the cell and not the atmospheric levels (Spalding et al., 2005; Bhardwaj et al., 2006). Thus, the strategy devised by the Jonas Frisen’s group was to isolate cell nuclei from fresh or frozen brain autopsy specimens and label them with NeuN. Next, neuronal nuclei (NeuN+) were sorted by FACS, their DNA purified and the 14C-content measured by accelerator mass spectrometry (Spalding et al., 2005; Bhardwaj et al., 2006). Using this technique, it was shown that neurogenesis in the adult neocortex is absent or very limited because the age of the neurons matched the age of the individual and had 14C levels similar to atmospheric levels at the time when the individual was born (Bhardwaj et al., 2006). On the contrary, NeuN-nuclei, including glial cells which are known to have a higher turnover rate, showed an average age several years younger than the age of the individual, suggesting that those were cells born during adulthood (Bhardwaj et al., 2006). This method yields high sensitivity as it has been estimated that it detects newborn cells down to 1% of the population (Bhardwaj et al., 2006). However, it only provides the average age of the neurons in the tissue and does not allow dating or tracing of individual cells. Surprisingly, this method has not yet been used to test adult neurogenesis in the hippocampus or the olfactory bulb (OB), the two major areas of adult neurogenesis.
Methods to Assess Neurogenesis in vivo
More recently, methods have been specifically designed to detect neurogenesis in live human brain by means of magnetic resonance imaging (MRI; Figure 3). In the MRI scanner, the subjects are exposed to a harmless magnetic field that aligns the magnetic spin of all the protons in the tissue in a low energy configuration; next, the subjects receive radiofrequency electrical stimulation, which excites the spins out of equilibrium. The spins then naturally relax back to their original conformation with time constants T1 (spin–lattice relaxation time, for longitudinal magnetization) and T2 (spin–spin relaxation time, for transversal magnetization; Maletic-Savatic et al., 2008). The difference in relaxation times of different molecules, such as water and fat, is used to generate detailed MRI images of the brain. In addition, further information can be extracted from these constants, and different MR modalities have been adapted to study neurogenesis (Modo and Bulte, 2011).
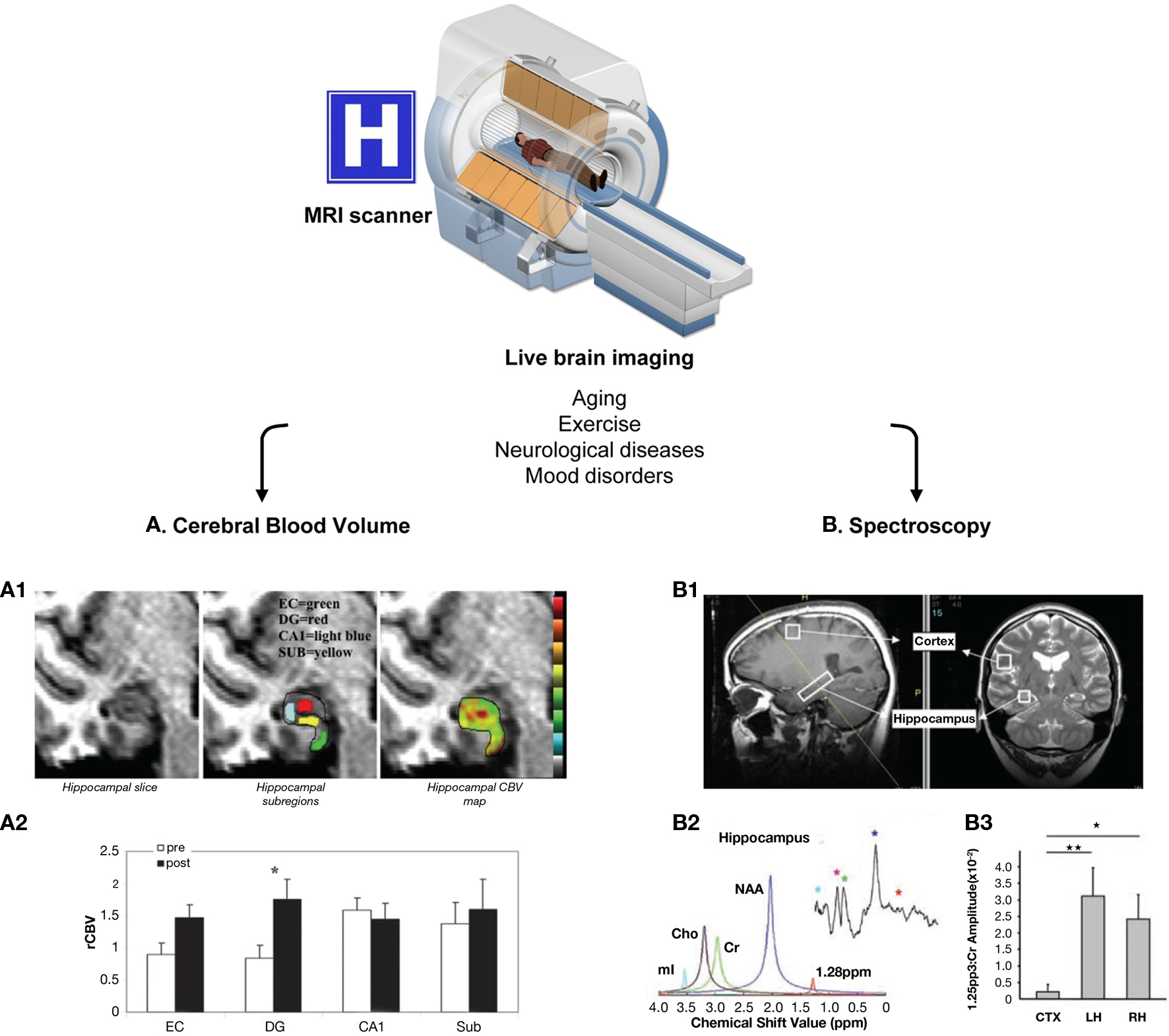
Figure 3. Live methods to assess adult human neurogenesis. These methods are based on magnetic resonance, using MRI scanners available in hospitals worldwide. Because there are no side effects, both healthy and diseased people can be re-scanned throughout aging, before and after exercise, to follow-up the effect of pharmacological interventions, etc. Two main methods have been developed to indirectly quantify adult human neurogenesis using different MR modalities: (A) CBV measurement. The dentate gyrus CBV is a proxy for neurogenesis in physical exercise paradigms. This method is based on the consecutive correlation of neurogenesis–angiogenesis, and angiogenesis–CBV. (A1) High resolution MRI slice of the adult human hippocampus (right panel), showing the different hippocampal subregions (entorhinal cortex, EC, green; dentate gyrus, DG, red; cornu ammonis 1, CA1, light blue; and subiculum, SUB, yellow; central panel) and a typical hippocampal CBV map (warmer colors indicate higher CBV). (A2) Quantification of the mean relative hippocampal CBV (rCBV), before (white bars) and after (black bars) exercise in healthy humans. As in mice, physical exercise resulted in a significant increase in CVB only in the DG (asterisk). (B) Spectroscopy. A lipidic metabolite resonating at 1.28 ppm was identified as a marker of rodent NPCs. (B1) Positioning of the voxel of interest in the cortex and the hippocampus of a healthy person. (B2) Spectroscopic analysis of the metabolite content in the hippocampal voxel using SVD and FFT (small upper insert). Identified metabolites are myoInositol (mI, light blue), choline (Cho, purple), creatine (green), N-acetylaspartate (NAA, dark blue), and the 1.28 ppm metabolite (red). (B3) Quantification of the abundance of the 1.28 ppm metabolite in the cortical (CTX) and hippocampal voxels (LH and RH for left and right hippocampus, respectively), normalized over the amplitude of the creatine peak. The hippocampi had much higher content of the 1.28 ppm metabolite than the cortex. The MRI cutaway is printed from permission from the National High Magnetic Field Lab website (http://www.magnet.fsu.edu/education /tutorials/magnetacademy/mri/). Figures (A1–A2) are reprinted by permission from Pereira et al. (2007), copyright 2007, National Academy of Sciences, U.S.A. Figures (B1–B3) are from Manganas et al. (2007) and are reprinted with permission from AAAS.
The major advantage of MR-based methods is that they are performed in live individuals with no side effects, supporting repeated measures and longitudinal studies. Thus, these methods allow a more controlled experimental design, and variables such as the cause or age of death no longer have to be taken into account. Nonetheless, these methods rely on correlations to indirectly quantify neurogenesis, and extensive validation in both rodents and humans is required to demonstrate that they are specific for neurogenesis. More importantly, it is essential to determine whether the data correlate with the number of NPCs, proliferating NPCs (versus other cell types that proliferate), or newborn neurons. Another major advantage of MRI-based methods is that MRI scanners are widely available in hospitals and research centers worldwide. Thus, these methods could be easily implemented in many labs and offer a unique research opportunity to increase our understanding of the role of adult neurogenesis in humans.
Cerebral blood volume measurements
Cerebral blood volume (CBV) can be measured by several methods, one of which is MRI. In MR-based CBV measurements, the contrast agent gadolinium is injected systemically. The chelated gadolinium used is a non-toxic highly lipophobic agent, thus restricted to the intravascular space when the BBB is not challenged (Zaharchuk, 2007). Due to its paramagnetic properties, it creates variations in the local magnetic field which lead to decreased T1 signal intensity. These changes can be used to generate maps of the CBV and cerebral blood flow (CBF) by an array of computational methods (reviewed in Zaharchuk, 2007). Among these, the steady-state T1 method is based on the assumption that the MRI signal derives from two separate compartments – intravascular (vessels) and extravascular (brain parenchyma; Lin et al., 1999). When gadolinium is administered, only the T1 signal from the intravascular compartment will decrease, assuming the BBB is intact. Then, the difference between pre-contrast and post-contrast images normalized over a voxel that contains only blood, such as the sagittal sinus, is used to generate the CBV map (Lin et al., 1999). The main advantage of the steady-state T1 method, compared to other methods such as bolus tracking (also called dynamic imaging), is that it renders absolute estimations of the CBV, supporting longitudinal studies of brain perfusion, and has high spatial resolution. This method has been validated through correlation with estimations of gray matter CBV using other imaging modalities. However, it requires longer acquisition time and has lower signal-to-noise ratio than bolus tracking (Lin et al., 1999). Nevertheless, the steady-state T1 method is well-established for determining CBV (Zaharchuk, 2007) and has been recently used by the group of Scott Small to indirectly assess changes in adult human neurogenesis (Pereira et al., 2007).
The basis for the CBV studies of neurogenesis is the correlation between angiogenesis and neurogenesis. Increased cortical CBV correlates with angiogenesis in ischemia (Lin et al., 2002; Seevinck et al., 2010) and gliomas (Aronen et al., 2000; Cha et al., 2003). In turn, angiogenesis occurs in the hippocampal neurogenic niche (Palmer et al., 2000), and both angiogenesis and neurogenesis are elevated in the hippocampus following physical exercise (van Praag et al., 2005; Van der Borght et al., 2009). Thus, because of the correlation of CBV–angiogenesis and angiogenesis–neurogenesis, the CBV might provide an indirect measure of neurogenesis in the adult human hippocampus (Pereira et al., 2007). In fact, the CBV increased selectively in the human dentate gyrus (DG, where NPCs reside) after a 12-week exercise paradigm, and this increase correlated with cognitive performance, such as declarative memory but not delayed recognition (Pereira et al., 2007). As a validation experiment, the authors showed that in running mice, the CBV increased in the DG and not in other hippocampal areas, and this increase correlated with the number of 1- to 3-week-old BrdU+ cells (Pereira et al., 2007). More recently, others have demonstrated increased number of micro-vessels occurring in parallel to increased proliferation (Ki67+ cells) and the number of newborn cells committed to the neuronal lineage (DCX+ cells) after 10 days of running (Van der Borght et al., 2009). However, it remains to be elucidated if the angiogenesis–neurogenesis coupling occurs in conditions other than exercise, which would render the CBV measurements for assessments of human neurogenesis more widely applicable.
Spectroscopic biomarker of NPCs
Here, the MRI modality used is proton magnetic resonance spectroscopy (1H-MRS), which exploits the magnetic properties of different protons to detect an array of small metabolites in the living tissue. Some protons of some metabolites are mobile in the magnetic field, and the relaxation of their spins can be detected by MR. This relaxation is observed as a sinusoid wave of decay in the time domain (free-induction decay, or FID), which is conventionally transformed into a function in the frequency domain (Fourier-fast transform, or FFT; Maletic-Savatic et al., 2008). The FFT is usually plotted as a graph of peaks representing the proton content of different metabolites. The x-axis represents the resonant frequency of each particular metabolite (in parts per million or ppm); and the y-axis represents the intensity of the signal, so that the integrated area under each peak is a readout of the amount of protons that contribute to that particular signal (Sibtain et al., 2007). Using this conventional signal analysis, a few relevant metabolites can be detected (Soares and Law, 2009): N-acetylaspartate (NAA), a marker of neurons whose major peak resonates at 2.02 ppm; Creatine, resonating at 3.02 ppm, an energy metabolite considered to be stable and thus used as a house-keeping metabolite for normalization; and Myoinositol, a marker of astrocytes which resonates at 3.56 ppm. Other metabolites commonly analyzed include choline, alanine, lactate, glutamate, glutamine, glucose, and some macromolecular proteins and lipids (Soares and Law, 2009). Using the 1H-MRS, our group recently identified a metabolite with main resonance at 1.28 ppm, which was enriched in rodent NPCs and was used to indirectly quantify adult human neurogenesis (Manganas et al., 2007).
This metabolite was initially discovered in rodent NPCs, grown as embryonically derived neurospheres, by high-field nuclear magnetic resonance (NMR; Manganas et al., 2007; Ma et al., 2011), although it had not been previously found in NPCs differentiated in culture from embryonic stem cells (ESCs; Jansen et al., 2006). Its association with neurogenesis in vivo was validated by microMR spectroscopy, which detected it in the rodent hippocampus, as well as in the cortex after NPC transplantation (Manganas et al., 2007). Furthermore, the amount of the metabolite detected by microMR spectroscopy correlated with the number of BrdU+ cells in the hippocampus following an electroconvulsive shock-induced increase in endogenous neurogenesis. These studies further led to imaging of the human hippocampus, where this metabolite was expressed in very small amounts and was not detectable using conventional signal processing. Thus, to extract it from the human 1H-MRS, Manganas et al. (2007) utilized singular value decomposition (SVD), a parametric method which models metabolite signals as decaying complex sinusoids in the time domain. The validity and reliability of SVD to quantify the 1.28 ppm signal encountered criticisms mostly due to potential overfitting of the data (Friedman, 2008; Hoch et al., 2008; Jansen et al., 2008; Dong et al., 2009). However, most of these issues were addressed using simulated and semi-simulated data showing that the rate of false positives was less than 5% for the range of signal-to-noise ratios found in the initial studies (under −20 dB), and thus that the estimations of human NPCs based on the 1.28 ppm peak were reliable (Djuric et al., 2008). Overall, these issues have been considered a matter of optimization of the technique rather than a fundamental problem in the methodology (Romer et al., 2008; Dong et al., 2009), with the agreement that more research is necessary to unequivocally establish the 1.28 ppm spectral peak as a marker of neurogenesis with clinical value (Djuric et al., 2008; Romer et al., 2008; Dong et al., 2009).
The identity of the 1.28 ppm metabolite remains unknown. Although it most likely contains a lipid component (Manganas et al., 2007), its exact molecular nature has not yet been determined and its functional significance for neurogenesis awaits further studies. Recent reports indicate that the 1.28 ppm and adjacent resonances may also be associated with apoptosis. A similar lipidic peak resonating at 1.30 ppm has been also reported in apoptotic rat gliomas in vivo (Liimatainen et al., 2008), and more recent studies have found that the 1.28 ppm peak in cultured NPC increased during conditions that favored quiescence and apoptosis (Ramm et al., 2009). Apoptosis is common in the hippocampal neurogenic niche, as vast amounts of newborn cells die (Sierra et al., 2010). Thus, whether the 1.28 ppm peak detected in living brains originates from living or apoptotic NPCs remains to be determined.
Sites of Neurogenesis in the Adult Human Brain
Nowadays, the consensus is that adult neurogenesis occurs in two main areas of the brain: the subgranular zone (SGZ) of the DG of the hippocampus, where new granule neurons are locally produced and have been associated with learning and memory, and mood disorders; and the subventricular zone (SVZ), from where newborn cells migrate through the rostral migratory stream (RMS) and give rise to neurons in the OB, related to olfaction (Ma et al., 2009). Additionally, there are recent reports of adult neurogenesis in the neocortex, as well as the striatum, amygdala, substantia nigra (SN), and a few other areas in the rodent brain, as reviewed by Gould (2007). In humans, multipotent progenitors have been isolated from the temporal and frontal cortex, as well as the amygdala from patients undergoing brain resection due to epilepsy, trauma, or dysplasia (Arsenijevic et al., 2001). However, studies in healthy humans have suggested that neocortical neurogenesis is restricted to the perinatal period or, at least, that the contribution of adult-born neurons to the total cortical population is extremely small and undetectable by 14C methods (Spalding et al., 2005; Bhardwaj et al., 2006).
Neurogenesis in the Adult Human Hippocampus
In rodents, the hippocampal neurogenic cascade starts with the quiescent neuroprogenitors (QNPs; type-1 cells; radial glia), which reside in the SGZ. QNPs proliferate, giving rise to a transient population, the amplifying neuroprogenitors (ANPs; type-2a cells) which in turn proliferate and differentiate into neuronal-committed NBs (type-2b and type-3 cells). Finally, at the end of a 4-week period, the surviving NBs become mature neurons integrated into the circuitry (reviewed by Kempermann et al., 2004; Encinas and Enikolopov, 2008).
In humans, adult hippocampal neurogenesis was demonstrated by analysis of postmortem tissue of cancer patients (Eriksson et al., 1998), and changes in it under different conditions such as physical exercise and aging have been observed indirectly using CBV (Pereira et al., 2007) and 1H-MRS (Manganas et al., 2007) in healthy adults in vivo. The presence of functional NPCs in the adult human hippocampus was further demonstrated by culture, expansion, and differentiation of human NPCs in vitro (Kukekov et al., 1999; Roy et al., 2000; Moe et al., 2005). A recent study has shown that the adult human SGZ contains DCX-expressing cells that co-localize both with markers of proliferation (MCM2, Ki67, PCNA) and mature neurons (NeuN, β-III-tubulin), supporting the existence of NBs throughout the human lifespan (Knoth et al., 2010).
Other studies, however, failed to detect NPCs or proliferating cells in the adult hippocampus of epileptic patients using immunohistochemical methods, such as expression of nestin, vimentin, or Ki67 (Arnold and Trojanowski, 1996; Del Bigio, 1999; Blumcke et al., 2001; Seress et al., 2001; Fahrner et al., 2007). This conflicting literature can be explained by different sensitivities of the particular method used in each study. Overall, future work is needed to determine all components of the hippocampal neurogenic niche and the cellular types that comprise human neurogenic cascade.
Neurogenesis in the Adult SVZ
In rodents, the stem cells of the SVZ are specialized astrocytes called B cells. These cells proliferate and give rise to C cells, the transient amplifying population of the system (Doetsch et al., 1999). These C cells generate NBs or A cells, which form chains of proliferating cells that migrate ensheathed by astrocytes forming the RMS toward the OB, where they differentiate into granule cells and periglomerular interneurons (Lois et al., 1996). In non-human primates, the structure is notably similar, with astrocytic-like precursors in the SVZ that generate chains and honeycomb-like structures of migrating NBs that reach the OB through the RMS (Kornack and Rakic, 2001; Pencea et al., 2001).
In humans, proliferating BrdU+ cells were found in the SVZ, but they did not co-localize with either GFAP or NeuN (Eriksson et al., 1998). Similarly to human SGZ NPCs, the functional NPCs from the human SVZ were grown in vitro (Kukekov et al., 1999) and, more recently, isolated from the healthy and diseased elderly SVZ (Leonard et al., 2009). Further studies showed the presence of proliferating putative NPCs, labeled with nestin and PCNA (Bernier et al., 2000), as well as putative NBs, labeled with PSA-NCAM, in the human SVZ (Weickert et al., 2000). However, only recently was the human SVZ niche thoroughly described. First, a well-defined astrocytic ribbon formed by the B cells was observed in the SVZ, similarly to rodent SVZ, but no evidence of cells migrating in an organized RMS was found (Sanai et al., 2004). The human RMS remained elusive, until a report that it was organized around the lateral ventricular extension which reached the OB (Curtis et al., 2007b). This report was criticized by Alvarez-Buylla and colleagues, who claimed that it had not unquestionably proved the existence of an olfactory ventricle and the migrating, proliferating NBs in the RMS (Sanai et al., 2007). Further study then showed scarce cells that co-expressed markers of proliferation (PCNA) and NBs (PSA-NCAM), but no clear evidence of migration (Kam et al., 2009). In addition, it appears that the human RMS consists of four layers, similar to the SVZ (Kam et al., 2009), which prompted others to suggest that the human RMS may be a rostral extension of the proliferative zone, rather than the migratory pathway as found in rodents and non-human primates (Whitman and Greer, 2009). Nonetheless, there is no consensus in the literature on whether the SVZ/RMS are actively providing a source of newborn neurons to the human OB throughout adulthood.
Finally, the human OB hosts NPCs, which have been isolated from patients undergoing neurosurgery and grown in culture, where they differentiated into neurons, astrocytes, and oligodendrocytes (Pagano et al., 2000). In agreement, new neurons seem to be produced locally in the human OB, as shown by co-localization with Ki67 and NeuroD (Bedard and Parent, 2004). However, the significance of local neurogenesis in the adult human OB is still debatable.
Relevance of Adult Neurogenesis to Human Disease
The majority of studies on human neurogenesis compare findings in healthy people to those in patients with a variety of neurological diseases. A summary comparing the alteration in neurogenesis in rodent models of disease and human patients is shown in Table 2. These studies use immunohistochemistry to detect biomarkers of proliferation or specific cell-types, and thus are only able to report differences in proliferation and putative NPCs and NBs (pNPCs, pNBs), but not actual neurogenesis (i.e., formation of new neurons). Thus, we label these detected cells “putative” because none of the studies demonstrated that proliferating cells differentiated into mature, functional neurons. To directly reach the conclusion that neurogenesis is occurring lineage tracing using BrdU or analogs is required.
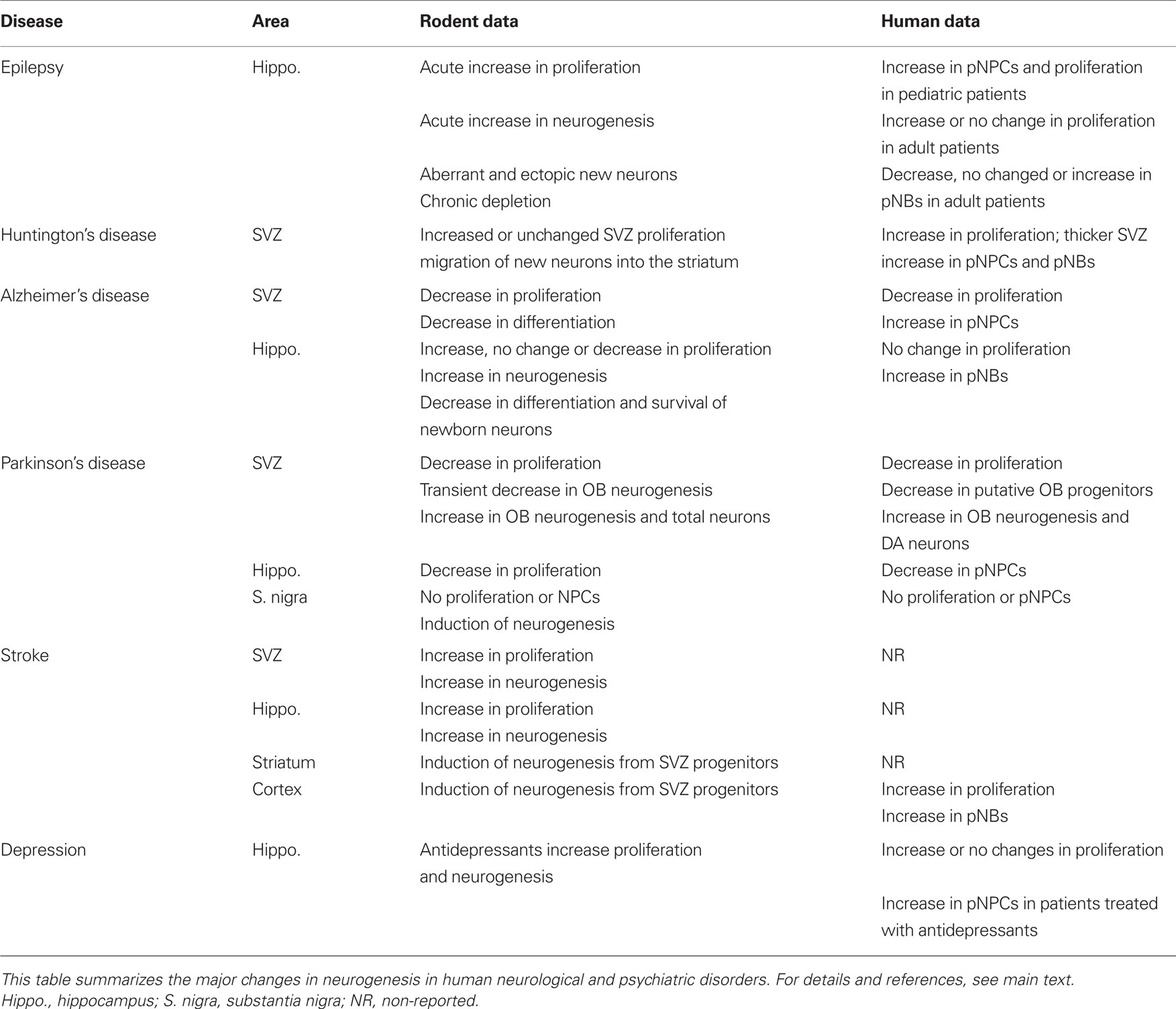
Table 2. Adult neurogenesis during disease.
Epilepsy
Epilepsy is a common human disease that affects more than 50 million people worldwide (Kuruba et al., 2009). One of the most intractable forms is temporal lobe epilepsy (TLE), characterized by altered electrical activity, aberrant synaptic reorganization and neurodegeneration in the hippocampus, as well as development of depression and impairments in learning and memory (Kuruba et al., 2009).
In rodents, there is abundant literature reporting that hippocampal neurogenesis increases in models of acute seizures, either by administration of glutamate receptors agonists such as kainic acid, or by electrical stimulation of the hippocampus or the piriform cortex. In these models an increment in the proliferation of the hippocampal NPCs results in a concomitant increase in the number of newborn neurons (reviewed by Parent, 2002; Curtis et al., 2007a; Kuruba et al., 2009). However, the newborn neurons are located ectopically (in the hilus and the molecular layer of the dentate gyrus) and display aberrant connectivity and morphology (Parent et al., 1997; Scharfman et al., 2000). The seizure-induced abnormal neurogenesis may have detrimental consequences and contribute to aberrant synaptic reorganization (Parent, 2002). In contrast, in models of chronic epilepsy, neurogenesis seems to return to basal levels or even to be downregulated as compared to control animals (Kuruba et al., 2009).
Several studies have reported changes in adult neurogenesis in patients with TLE, who were all pharmacoresistant and had to undergo temporal lobectomy, providing the source for studies of neurogenesis. Utilizing Ki67 labeling the initial study showed no evidence of proliferation in the adult epileptic SGZ, since the number of Ki67+ cells was not significantly larger compared to other DG regions (Del Bigio, 1999). In addition, the number of pNBs (PSA-NCAM+ cells) was smaller in TLE patients than in age-matched controls (Mikkonen et al., 1998). Further comparison of adult TLE patients and controls showed similar levels of proliferation, measured by the number of Ki67+ and MCM2+ cells, as well as DCX protein expression and the number of DCX+ cells (Fahrner et al., 2007). In contrast, pediatric (<19 months of age) TLE patients had increased number of proliferating pNPCs, labeled with nestin and Ki67 (Blumcke et al., 2001). More recent studies, however, contradict the previous literature. One study described a not quantified increase in proliferating cells (PCNA+ cells), and pNPCs (vimentin, musashi+ cells) in the SGZ and SVZ of adult TLE patients compared to controls (Crespel et al., 2005), while another reported augmented DCX protein and gene expression and number of pNBs (DCX+ cells) in TLE patients in the hippocampus and other temporal cortical regions (Liu et al., 2008). In these patients the DCX+ cells expressed markers of the neuronal lineage, such as PSA-NCAM or NeuN, but the authors did not provide the rationale for their findings contradicting previous literature and only noted that the effect of TLE on human neurogenesis was not as “dramatic” as in animal models of epilepsy (Liu et al., 2008). Finally, although many ex vivo studies showed the presence of functional NPCs in the epileptic hippocampus (Kukekov et al., 1999; Roy et al., 2000; Moe et al., 2005), it has been recently shown that their proliferative and multipotential properties in vitro depend on disease duration and were almost absent in epileptic patients with mesial temporal sclerosis, a late consequence of TLE (Paradisi et al., 2010). In conclusion, in infants, epilepsy is associated with increased proliferation in the hippocampus, but whether this proliferation leads to an increase of newborn neurons remains unknown. In adults, reports are controversial and most likely reflect a wide range of disease duration, which apparently affects NPCs (Paradisi et al., 2010). Whether changes in neurogenesis also relate to the changes in mood and cognitive performance observed in TLE patients remains to be determined.
Epilepsy also alters SVZ neurogenesis. In rats, pilocarpin-induced status epilepticus produced an increased proliferation of SVZ NPCs as well as an expansion of the RMS, which contained more proliferating cells and NBs (Parent et al., 2002). In addition, some cells in the RMS were found to migrate ectopically into the surrounding cortical parenchyma, although the majority of these ectopic cells did not survive (Parent et al., 2002). In human, increased proliferation, ectopic parenchymal migration, and neuronal differentiation were also found using organotypic slice preparations from tissue resected from patients with a variety of intractable cortical seizures (Gonzalez-Martinez et al., 2007). The functional significance of increased neurogenesis and ectopic migration of SVZ precursors induced by SE in rodents and humans remains to be established.
Huntington’s Disease
Huntington’s disease (HD) is caused by expanded CAG repeats in the huntingtin gene, which leads to protein accumulation and neurodegeneration in the striatum, a brain area that lies below the SVZ. In transgenic mice models of HD, in which little striatal neurodegeneration occurs, neurogenesis in the SVZ remains unaltered; whereas in rat models of striatal degeneration an increased SVZ proliferation is observed (reviewed by Curtis et al., 2007a). Interestingly, some of the newly generated cells are able to migrate into the damaged striatum, where they express neuronal markers (Tattersfield et al., 2004), although it is unknown whether they are functionally mature neurons. These results suggested an endogenous regeneration potential in HD because the new born neurons would take long time to develop huntingtin inclusions and in the meanwhile could contribute to maintaining the striatal circuitry (Curtis et al., 2007a).
Studies of HD patients have shown increased intensity of PCNA staining in the SVZ lining of the striatum compared to age-matched controls, suggesting an increase in SVZ proliferation. Additionally, the expression levels of PCNA correlated with the number of CAG repeats in these patients. The proliferating cells expressed markers of glia (GFAP) and neurons (β-III-Tubulin), suggesting the presence of putative B and A cells, respectively (Curtis et al., 2003). Further, the SVZ was thicker due to an increase in the number of B cells, identified by cellular morphology, and the number of dividing cells, labeled with PCNA, in HD patients compared to age-matched controls (Curtis et al., 2005a,b). Unexpectedly, some of the proliferating PCNA+ cells expressed markers of mature neurons such as βIII-Tubulin and this was interpreted as increased neurogenesis in HD patients (Curtis et al., 2005a). However, it takes days, if not weeks, for newborn cells to express markers of mature neurons. Therefore, an aberrant expression of PCNA in mature neurons or an aberrant expression of βIII-Tubulin in dividing cells in the SVZ of HD patients cannot be ruled out. Finally, migration of the newborn cells from the SVZ into the damaged human striatum and their neuronal differentiation has not been described yet and, thus, it remains unknown if the endogenous SVZ cells can be harnessed for repair in HD patients.
Alzheimer’s Disease
Alzheimer’s disease (AD) is characterized by accumulation of β-amyloid and neurofibrillary tangles containing hyperphosphorylated tau protein throughout the cortex and the hippocampus, resulting in progressive dementia (Curtis et al., 2007a). Some pathological features of AD can be modeled in transgenic mice overexpressing amyloid precursor protein and presenilin 1 (APP/PS1). In these mice, memory impairment and increased hippocampal proliferation and neurogenesis were observed at 9, but not 3 months of age (Yu et al., 2009). However, earlier works showed that 6-month-old APP/PSE1 mice have unaltered proliferation and short-term survival (1–13 days), whereas they have a significant reduction of long-term survival (30–42 days) and differentiation (Verret et al., 2007). In addition, other transgenic mouse models of AD have shown otherwise. For instance, in triple transgenic mice (APP/PSE1/Tau) there is a gradual decrease in SGZ proliferation starting at 6 months of age (Rodriguez et al., 2008). On the other hand, 3-month-old mice expressing mutated APP have increased proliferation (Jin et al., 2004a) although this increase was reverted to control levels in older animals (Lopez-Toledano and Shelanski, 2007). Finally, in 6-week-old transgenic mouse expressing human APP showed decreased proliferation in control housing conditions as well as a decreased 4-week survival in enriched environment conditions (Naumann et al., 2010). In postmortem hippocampal samples from patients with advanced AD, an increased expression of NB proteins (DCX, PSA-NCAM, and NeuroD) compared to age-matched controls was reported, suggesting increased neurogenesis perhaps as a compensatory mechanism to cope with the AD-related cognitive impairment (Jin et al., 2004b). However, a more recent study of presenile AD patients failed to demonstrate increased proliferation in the DG, whereas it showed an increased proliferation (Ki67+ cells) associated with gliogenesis and angiogenesis in other hippocampal regions. Further, the same study attributed changes in DCX immunolabeling to postmortem breakdown (Boekhoorn et al., 2006). Thus, it is clear that more comprehensive studies are needed to clarify the changes in SGZ neurogenesis in AD. Furthermore, the relation between potentially altered neurogenesis and the cognitive impairments observed in AD remains to be elucidated (Lazarov et al., 2010).
Subventricular zone neurogenesis is also altered in mouse AD-models. For instance, transgenic APP or PSE1 mice as well as wild-type mice infused in the lateral ventricles with βA peptide had reduced SVZ proliferation compared to control mice (Haughey et al., 2002; Rodriguez et al., 2009; Veeraraghavalu et al., 2010). Decreased proliferation and neuronal differentiation were also observed in cultured NPCs isolated from PSE1 mutant SVZ (Veeraraghavalu et al., 2010) and from APP/PS1 mutant SVZ (Demars et al., 2010). In human AD patients, decreased proliferation (Ki67+ cells) accompanied by a puzzling increase in nestin expression was observed in postmortem sections (Ziabreva et al., 2006). In agreement, cultured embryonic human NPC had decreased proliferation and increased apoptosis when treated with Aβ peptide compared to control NPCs treated with vehicle (Haughey et al., 2002). Thus, there are consistently lower levels in SVZ neurogenesis in AD patients as well as in and rodent AD models, prompting the suggestion that impaired SVZ neurogenesis may have functional consequences in AD (Curtis et al., 2007a). For instance, olfactory deficits significantly predict development of AD in patients with mild cognitive impairment (Devanand et al., 2000), although whether these olfactory deficits are related to decreased SVZ neurogenesis remains unknown.
Parkinson’s Disease
The major hallmark of the Parkinson’s disease (PD) is the death of dopaminergic neurons in the SN, with consequent impairment of movement control, mood, and motivation (Hoglinger et al., 2004). In animal models of PD, a reduced proliferation in the SVZ overlaying the striatum is observed. C and A cells in the SVZ receive dopaminergic fibers from the SN (Hoglinger et al., 2004). In rodents, this dopaminergic innervation controls the proliferation of these two cell types, because the injection of a toxic dopamine analog (6-hydroxydopamine) in the nigrostriatal pathway dennervates both the striatum and the SVZ and results in decreased SVZ proliferation (Hoglinger et al., 2004), resulting in a transient decrease of newborn neurons in the OB granule cell layer (Hoglinger et al., 2004; Winner et al., 2006). In the lesioned mice, administration of a dopaminergic precursor (levodopa) partially recovered SVZ proliferation close to control levels (Hoglinger et al., 2004). Similar results were obtained in PD patients. The number of SVZ proliferating cells, labeled with PCNA, as well as OB pNPCs (nestin+ cells) was reduced compared to age-matched controls (Hoglinger et al., 2004), although no changes in the number of cells in the OB granule cell layer have been reported.
Interestingly, in the OB glomerule cell layer there is an increase in the number of newborn as well as total dopaminergic cells, expressing the synthesizing enzyme tyrosine hydroxylase, both in rodents whose nigrostriatal pathway was lesioned with 6-hydroxydopamine (Winner et al., 2006) and in PD patients (Huisman et al., 2004). This increase in dopaminergic cells remains unexplained and it is unexpected in the light of the decreased SVZ proliferation. Nonetheless, because dopamine inhibits olfactory transmission, it has been suggested that the increase in OB dopaminergic neurons could explain the hyposmia (Huisman et al., 2004) that occurs in up to 95% of PD patients prior to the onset of other clinical symptoms (Haehner et al., 2009). Dopaminergic innervation may also regulate neurogenesis in the hippocampus. In rodents, lesions of the nigrostriatal pathway decrease hippocampal proliferation (Hoglinger et al., 2004; Suzuki et al., 2010), and in the hippocampus of PD patients, less pNPCs (nestin+ cells) are observed compared to age-matched controls (Hoglinger et al., 2004). While initial reports suggested the neurogenesis was induced in the SN in rodent models of PD (Zhao et al., 2003), later studies, however, showed no evidences of proliferation or NPCs in the SN either in rodent models of PD (Frielingsdorf et al., 2004; Yoshimi et al., 2005) nor in PD patients (Yoshimi et al., 2005).
Stroke/Ischemia
A stroke, or cerebrovascular accident, results from occlusion of cerebral arteries leading to decreased local blood flow (ischemia) or from a hemorrhage. In the stroked tissue, two areas of injury can be discriminated: the core infarcted area, where neurons die of necrosis and very little, if any, regeneration is possible; and the penumbra area, which surrounds the infarcted area, is perfused by collateral arteries, and is not irreversibly damaged. Given that the ischemic stroke is the third most frequent cause of mortality in industrialized countries, major scientific efforts have been directed toward discoveries of therapies to facilitate recovery from the insult.
In rodent and non-human primate models of stroke, such as occlusion of the medial cerebral artery occlusion (MCAO), adult neurogenesis is up-regulated both in the SVZ–RMS–OB and the hippocampus (Jin et al., 2001; Zhang et al., 2001; Koketsu et al., 2006; Lledo et al., 2006). In addition, stroke also induces ectopic neurogenesis in penumbra areas, such as the striatum, due to atypical migration of SVZ newborn cells (Arvidsson et al., 2002). Cortical neurogenesis in the penumbra area in rodent models of stroke has been found by some (Gu et al., 2000; Jin et al., 2003) but not by others (Arvidsson et al., 2002). Interestingly, the newborn cells differentiated into striatal neurons and acquired the same phenotype of the neurons which had died as a consequence of the stroke, suggesting that neuronal replacement can occur in the stroked striatum (Arvidsson et al., 2002). Although the vast majority of the striatal newborn cells died, possibly due to an unfavorable environment (Arvidsson et al., 2002), stroke-induced striatal neurogenesis seems to have functional consequences in rodents, since it has been shown that the transgenic ablation of the NB protein DCX prevented stroke-induced neurogenesis and worsened the sensorimotor and behavioral deficits after MCAO (Jin et al., 2010).
This research indicated that harnessing aberrant striatal neurogenesis in stroke may be useful to reduce the neurological deficits in patients (reviewed in Zhang and Chopp, 2009). The patients who suffered the ischemic, middle cerebral artery stroke showed increased proliferation of putative B cells (Ki67, GFAP+ cells) and putative C cells (PSA-NCAM+ cells), in the ipsilateral SVZ compared to the contralateral side of the stroke (Marti-Fabregas et al., 2010). In addition, there were traces of ectopic neurogenesis not in the striatum, but in the cortex. A significant increase in proliferating Ki67+ cells and pNBs (PSA-NCAM+ cells) was found in the cortical penumbra region of ischemic stroke patients compared to age-matched controls (Jin et al., 2006; Macas et al., 2006) as well as in perihematomal regions in patients with intracerebral hemorrhage (Shen et al., 2008). The relevance of this increase in cortical neurogenesis in stroke patients remains to be investigated, but the phenomena certainly raise the hope that neurogenesis might be harnessed as a possible treatment for stroke patients.
Mood Disorders
Depression, or major depressive disorder (MDD), is characterized by anhedonia and the absence of positive affect (Craske et al., 2009). MDD is thought to be caused by an imbalance in the levels of monoamines, such as serotonin and noradrenalin, as formulated in the “monoamine hypothesis of depression” (Duman et al., 2000; Wong and Licinio, 2001). This hypothesis is strongly supported by the ability of antidepressant drugs, such as selective serotonin reuptake inhibitors (SSRIs) or tricyclic antidepressants (TCAs), to improve the symptomatology by increasing synaptic levels of monoamines (Wong and Licinio, 2001). However, while antidepressant drugs modify monoamine levels within hours, it takes weeks of daily treatments to observe the behavioral effects, indicating that long-term changes underlie the effects of antidepressant therapies (Wong and Licinio, 2001). Structural changes in the cortex and hippocampus, such as alterations in neuronal morphology, synaptic plasticity, and cell survival, may also be part of the disease (Duman et al., 2000). For instance, MDD patients may have hippocampal atrophy (Sheline et al., 1996), which correlates with poor cognitive performance in these patients (Frodl et al., 2006). In addition, MDD patients have decreased pyramidal neuronal soma size and higher density of glial, pyramidal, and granule cells in the hippocampus compared to age-matched controls (Stockmeier et al., 2004). Together, these results suggest a significant reduction in the neuropil (glial and neuronal processes) in MDD patients (Stockmeier et al., 2004). While the absolute numbers of glia and neurons in the hippocampus of MDD patients are unknown (Stockmeier et al., 2004), small increases in hippocampal apoptosis in MDD patients have been reported (Lucassen et al., 2001).
The hippocampal pathology in MDD has led to the “neurogenesis hypothesis of depression.” Postulated by Drew and Hen (2007), the hypothesis states that altered levels of adult hippocampal neurogenesis may underlie the pathology of depression as well as the behavioral effects of antidepressants. This hypothesis is based on several lines of evidence showing that in rodents, chronic antidepressant treatments increase hippocampal neurogenesis (Malberg et al., 2000) and that, conversely, hippocampal neurogenesis is necessary for the behavioral effects of antidepressant drugs (Santarelli et al., 2003). The main caveat of this hypothesis is that neurogenesis seems not to be sufficient to cause depression, because decreased neurogenesis does not always induce depressive behaviors, and vice versa, depressive behaviors can be induced in experimental paradigms that do not affect neurogenesis (Vollmayr et al., 2007). To further complicate the interpretation of the data, the depression-induced morphological alterations, including those in neurogenesis, have generally been blamed on depression-related stress (Duman et al., 2000).
Stress, or more properly, the failure to adapt to stressful situations, is a shared symptom of depression and other mood disorders, such as anxiety and fear disorders (Craske et al., 2009), although is still under debate whether it is a concurrent epiphenomenon or an actual pathological state (Wong and Licinio, 2001). The connection of stress, anxiety, and MDD is not trivial, because the most commonly utilized rodent models of depression, such as learned helplessness, chronic mild stress, and chronic psychosocial stress induce depressive behaviors by increasing stress levels (Vollmayr et al., 2007; Pryce and Seifritz, 2011). Stress, through the actions of glucocorticoids, reduces adult hippocampal neurogenesis (Mirescu and Gould, 2006), produces dendritic atrophy (McEwen, 2001), and leads to decreased hippocampal volume (Tata and Anderson, 2010). From the rodent literature, it is therefore difficult to extract a clear picture of whether the alterations in adult hippocampal neurogenesis are due to stress, anxiety, depression, or a combination of these factors.
In MDD patients, recent studies have shown alterations in adult hippocampal neurogenesis (Reif et al., 2006; Boldrini et al., 2009). An initial study on postmortem samples of a cohort of 15 MDD patients showed no alterations in proliferation (Ki67+ cells) compared to age-matched controls (Reif et al., 2006). In contrast, a more recent study of a cohort of 19 MDD patients treated with TCAs such as nortriptyline, showed a significant increase in proliferation (Ki67+ cells) compared to the untreated MDD patients. In addition, a significant increase in the number of pNPCs (nestin+ cells) was found in patients treated with TCAs or SSRIs such as fluoxetine, compared to the untreated MDD patients (Boldrini et al., 2009). Interestingly, and similar to the observation that decreased neurogenesis is not required to induce depression-like behavior in rodents (Vollmayr et al., 2007), there were no significant differences in the number of proliferative cells or pNPCs in MDD patients compared to age-matched controls (Boldrini et al., 2009). Several limitations where identified, such as the small sample size, the possibility that treated MDD patients had more severe symptoms than untreated patients (hence, the prescription of pharmacological treatment), and the high incidence of suicide in the untreated patients (Boldrini et al., 2009). Taken together, these data suggest that decreased neurogenesis is not causative for depressive symptoms; however, treatment with antidepressants does improve the symptoms while increasing neurogenesis in the adult hippocampus. Further research needs to test the correlations between neurogenesis, MDD, stress, and antidepressant drugs. In particular, methods to assess human neurogenesis in vivo are of particular importance for longitudinal studies in MDD patients, to quantify neurogenesis before and after the antidepressant treatment, and in correlation with the onset and/or improvement of depressive symptoms.
Miscellanea
Finally, there is also sporadic evidence that adult neurogenesis may be altered in other brain diseases. For instance, in traumatic brain injury, increased hippocampal proliferation has been reported in humans (Gerber et al., 2009) and mice (Yu et al., 2008). In patients with hypoxic-ischemic encephalopathy, which results in neuronal loss in the cortex and the hippocampus, there is a non-significant increase in PCNA labeled cells (Mattiesen et al., 2009). During bacterial meningitis an increase in hippocampal proliferation as well as the number of pNBs was observed (Gerber et al., 2009), although in mice inflammation results in decreased proliferation and neurogenesis (Monje et al., 2003). In patients with subarachnoid hemorrhage, increased mRNA and protein levels of vimentin, musashi, and nestin were observed in the frontal lobe compared to control brains, while proliferating pNPCs (Ki67, nestin+ cells) were also found in the damaged frontal lobes (Sgubin et al., 2007). In autism, it has recently been suggested that neurogenesis may be altered because of the found increased thickness and dysplasia of the SVZ (Wegiel et al., 2010). In schizophrenia, decreased hippocampal proliferation (Ki67+ cells) has been reported (Reif et al., 2006), in agreement with the findings of decreased NPC proliferation (Mao et al., 2009) and aberrant morphology and excitability of the newborn neurons in mice with lower expression of disrupted-in-schizophrenia 1 (DISC1), a schizophrenia susceptibility gene (Duan et al., 2007). In alcohol abuse patients, no significant differences in hippocampal proliferation have been found (Reif et al., 2006), although chronic alcohol exposure decreases neurogenesis in rats (He et al., 2005).
Conclusion
Overall, studies of adult human neurogenesis, even though hampered by limitations of the available methodologies for both ex vivo and in vivo assessments, are promising. Development of new antibodies targeted to human antigens will certainly improve immunohistochemical data, but even then, such labeling will provide only putative information. It is in combination with BrdU labeling that the production of new neurons can be assessed and quantified. As more BrdU labeled tissue is generated, the changes in the neurogenic cascade that accompany brain disorders will be elucidated. However, several considerations need to be taken into account when studying postmortem human tissue, in particular the postmortem delay, the cause of death, and the age at the time of death. Thus, the future of adult human neurogenesis research and the prospects of harnessing its potential for treatments of brain disorders will heavily depend on the development and thorough validation of methods for in vivo assessments, as those offer unique opportunity for both cross-sectional and longitudinal studies of the neurogenic niches while they are intact within the living brain tissue.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Juan J.P. Deudero, William T. Choi, and Fatih Semerci for their help during the preparation of this manuscript. This work was supported by the NIH Intellectual and Developmental Disabilities Research Grant (P30HD024064), the McKnight Endowment Fund, the DANA Foundation, and the Farish Foundation (Mirjana Maletic-Savatic).
Footnotes
References
Antel, J. P., Nalbantoglu, J., and Olivier, A. (2000). Neuronal progenitors-learning from the hippocampus. Nat. Med. 6, 249–250.
Arnold, S. E., and Trojanowski, J. Q. (1996). Human fetal hippocampal development: II. The neuronal cytoskeleton. J. Comp. Neurol. 367, 293–307.
Aronen, H. J., Pardo, F. S., Kennedy, D. N., Belliveau, J. W., Packard, S. D., Hsu, D. W., Hochberg, F. H., Fischman, A. J., and Rosen, B. R. (2000). High microvascular blood volume is associated with high glucose uptake and tumor angiogenesis in human gliomas. Clin. Cancer Res. 6, 2189–2200.
Arsenijevic, Y., Villemure, J. G., Brunet, J. F., Bloch, J. J., Deglon, N., Kostic, C., Zurn, A., and Aebischer, P. (2001). Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Exp. Neurol. 170, 48–62.
Arvidsson, A., Collin, T., Kirik, D., Kokaia, Z., and Lindvall, O. (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8, 963–970.
Bailis, J. M., and Forsburg, S. L. (2004). MCM proteins: DNA damage, mutagenesis and repair. Curr. Opin. Genet. Dev. 14, 17–21.
Bedard, A., and Parent, A. (2004). Evidence of newly generated neurons in the human olfactory bulb. Brain Res. Dev. Brain Res. 151, 159–168.
Bernier, P. J., Vinet, J., Cossette, M., and Parent, A. (2000). Characterization of the subventricular zone of the adult human brain: evidence for the involvement of Bcl-2. Neurosci. Res. 37, 67–78.
Bhardwaj, R. D., Curtis, M. A., Spalding, K. L., Buchholz, B. A., Fink, D., Bjork-Eriksson, T., Nordborg, C., Gage, F. H., Druid, H., Eriksson, P. S., and Frisén, J. (2006). Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. U.S.A. 103, 12564–12568.
Blumcke, I., Schewe, J. C., Normann, S., Brustle, O., Schramm, J., Elger, C. E., and Wiestler, O. D. (2001). Increase of nestin-immunoreactive neural precursor cells in the dentate gyrus of pediatric patients with early-onset temporal lobe epilepsy. Hippocampus 11, 311–321.
Boekhoorn, K., Joels, M., and Lucassen, P. J. (2006). Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis. 24, 1–14.
Boldrini, M., Underwood, M. D., Hen, R., Rosoklija, G. B., Dwork, A. J., John Mann, J., and Arango, V. (2009). Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34, 2376–2389.
Cattoretti, G., Becker, M. H., Key, G., Duchrow, M., Schluter, C., Galle, J., and Gerdes, J. (1992). Monoclonal antibodies against recombinant parts of the Ki-67 antigen (MIB 1 and MIB 3) detect proliferating cells in microwave-processed formalin-fixed paraffin sections. J. Pathol. 168, 357–363.
Cha, S., Johnson, G., Wadghiri, Y. Z., Jin, O., Babb, J., Zagzag, D., and Turnbull, D. H. (2003). Dynamic, contrast-enhanced perfusion MRI in mouse gliomas: correlation with histopathology. Magn. Reson. Med. 49, 848–855.
Colucci-D’Amato, L., Bonavita, V., and di Porzio, U. (2006). The end of the central dogma of neurobiology: stem cells and neurogenesis in adult CNS. Neurol. Sci. 27, 266–270.
Craske, M. G., Rose, R. D., Lang, A., Welch, S. S., Campbell-Sills, L., Sullivan, G., Sherbourne, C., Bystritsky, A., Stein, M. B., and Roy-Byrne, P. P. (2009). Computer-assisted delivery of cognitive behavioral therapy for anxiety disorders in primary-care settings. Depress. Anxiety 26, 235–242.
Crespel, A., Rigau, V., Coubes, P., Rousset, M. C., de Bock, F., Okano, H., Baldy-Moulinier, M., Bockaert, J., and Lerner-Natoli, M. (2005). Increased number of neural progenitors in human temporal lobe epilepsy. Neurobiol. Dis. 19, 436–450.
Curtis, M. A., Faull, R. L., and Eriksson, P. S. (2007a). The effect of neurodegenerative diseases on the subventricular zone. Nat. Rev. Neurosci. 8, 712–723.
Curtis, M. A., Kam, M., Nannmark, U., Anderson, M. F., Axell, M. Z., Wikkelso, C., Holtas, S., van Roon-Mom, W. M., Bjork-Eriksson, T., Nordborg, C., Frisén, J., Dragunow, M., Faull, R. L., and Eriksson, P. S. (2007b). Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science 315, 1243–1249.
Curtis, M. A., Penney, E. B., Pearson, A. G., van Roon-Mom, W. M., Butterworth, N. J., Dragunow, M., Connor, B., and Faull, R. L. (2003). Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc. Natl. Acad. Sci. U.S.A. 100, 9023–9027.
Curtis, M. A., Penney, E. B., Pearson, J., Dragunow, M., Connor, B., and Faull, R. L. (2005a). The distribution of progenitor cells in the subependymal layer of the lateral ventricle in the normal and Huntington’s disease human brain. Neuroscience 132, 777–788.
Curtis, M. A., Waldvogel, H. J., Synek, B., and Faull, R. L. (2005b). A histochemical and immunohistochemical analysis of the subependymal layer in the normal and Huntington’s disease brain. J. Chem. Neuroanat. 30, 55–66.
Danilov, A. I., Gomes-Leal, W., Ahlenius, H., Kokaia, Z., Carlemalm, E., and Lindvall, O. (2009). Ultrastructural and antigenic properties of neural stem cells and their progeny in adult rat subventricular zone. Glia 57, 136–152.
Del Bigio, M. R. (1999). Proliferative status of cells in adult human dentate gyrus. Microsc. Res. Tech. 45, 353–358.
Demars, M., Hu, Y. S., Gadadhar, A., and Lazarov, O. (2010). Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 88, 2103–2117.
des Portes, V., Pinard, J. M., Billuart, P., Vinet, M. C., Koulakoff, A., Carrie, A., Gelot, A., Dupuis, E., Motte, J., Berwald-Netter, Y., Catala, M., Kahn, A., Beldjord, C., and Chelly, J. (1998). A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92, 51–61.
Devanand, D. P., Michaels-Marston, K. S., Liu, X., Pelton, G. H., Padilla, M., Marder, K., Bell, K., Stern, Y., and Mayeux, R. (2000). Olfactory deficits in patients with mild cognitive impairment predict Alzheimer’s disease at follow-up. Am. J. Psychiatry 157, 1399–1405.
Dityatev, A., Dityateva, G., Sytnyk, V., Delling, M., Toni, N., Nikonenko, I., Muller, D., and Schachner, M. (2004). Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses. J. Neurosci. 24, 9372–9382.
Djuric, P. M., Benveniste, H., Wagshul, M. E., Henn, F., Enikolopov, G., and Maletic-Savatic, M. (2008). Response to comments on “magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321, 640e.
Doetsch, F., Caille, I., Lim, D. A., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716.
Doetsch, F., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1997). Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. 17, 5046–5061.
Dong, Z., Dreher, W., Leibfritz, D., and Peterson, B. S. (2009). Challenges of using MR spectroscopy to detect neural progenitor cells in vivo. AJNR Am. J. Neuroradiol. 30, 1096–1101.
Drew, M. R., and Hen, R. (2007). Adult hippocampal neurogenesis as target for the treatment of depression. CNS Neurol Disord Drug Targets 6, 205–218.
Duan, X., Chang, J. H., Ge, S., Faulkner, R. L., Kim, J. Y., Kitabatake, Y., Liu, X. B., Yang, C. H., Jordan, J. D., Ma, D. K., Liu, C. Y., Ganesan, S., Cheng, H. J., Ming, G. L., Lu, B., and Song, H. (2007). Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130, 1146–1158.
Duman, R. S., Malberg, J., Nakagawa, S., and D’Sa, C. (2000). Neuronal plasticity and survival in mood disorders. Biol. Psychiatry 48, 732–739.
Eckenhoff, M. F., and Rakic, P. (1988). Nature and fate of proliferative cells in the hippocampal dentate gyrus during the life span of the rhesus monkey. J. Neurosci. 8, 2729–2747.
Encinas, J. M., and Enikolopov, G. (2008). Identifying and quantitating neural stem and progenitor cells in the adult brain. Methods Cell Biol. 85, 243–272.
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A., and Gage, F. H. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Fahrner, A., Kann, G., Flubacher, A., Heinrich, C., Freiman, T. M., Zentner, J., Frotscher, M., and Haas, C. A. (2007). Granule cell dispersion is not accompanied by enhanced neurogenesis in temporal lobe epilepsy patients. Exp. Neurol. 203, 320–332.
Friedman, S. D. (2008). Comment on “magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321, 640; author reply 640.
Frielingsdorf, H., Schwarz, K., Brundin, P., and Mohapel, P. (2004). No evidence for new dopaminergic neurons in the adult mammalian substantia nigra. Proc. Natl. Acad. Sci. U.S.A. 101, 10177–10182.
Frodl, T., Schaub, A., Banac, S., Charypar, M., Jager, M., Kummler, P., Bottlender, R., Zetzsche, T., Born, C., Leinsinger, G., Reiser, M., Möller, H. J., and Meisenzahl, E. M. (2006). Reduced hippocampal volume correlates with executive dysfunctioning in major depression. J. Psychiatry Neurosci. 31, 316–323.
Gerber, J., Tauber, S. C., Armbrecht, I., Schmidt, H., Bruck, W., and Nau, R. (2009). Increased neuronal proliferation in human bacterial meningitis. Neurology 73, 1026–1032.
Goldman, S. A., and Nottebohm, F. (1983). Neuronal production, migration, and differentiation in a vocal control nucleus of the adult female canary brain. Proc. Natl. Acad. Sci. U.S.A. 80, 2390–2394.
Goldman, S. A., and Windrem, M. S. (2006). Cell replacement therapy in neurological disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1463–1475.
Gonzalez-Martinez, J. A., Bingaman, W. E., Toms, S. A., and Najm, I. M. (2007). Neurogenesis in the postnatal human epileptic brain. J. Neurosurg. 107, 628–635.
Gu, W., Brannstrom, T., and Wester, P. (2000). Cortical neurogenesis in adult rats after reversible photothrombotic stroke. J. Cereb. Blood Flow Metab. 20, 1166–1173.
Haehner, A., Boesveldt, S., Berendse, H. W., Mackay-Sim, A., Fleischmann, J., Silburn, P. A., Johnston, A. N., Mellick, G. D., Herting, B., Reichmann, H., and Hummel, T. (2009). Prevalence of smell loss in Parkinson’s disease – a multicenter study. Parkinsonism Relat. Disord. 15, 490–494.
Hall, P. A., and Woods, A. L. (1990). Immunohistochemical markers of cellular proliferation: achievements, problems and prospects. Cell Tissue Kinet. 23, 505–522.
Haughey, N. J., Liu, D., Nath, A., Borchard, A. C., and Mattson, M. P. (2002). Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromol. Med. 1, 125–135.
He, J., Nixon, K., Shetty, A. K., and Crews, F. T. (2005). Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Eur. J. Neurosci. 21, 2711–2720.
Hoch, J. C., Maciejewski, M. W., and Gryk, M. R. (2008). Comment on “magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321, 640; author reply 640.
Hoglinger, G. U., Rizk, P., Muriel, M. P., Duyckaerts, C., Oertel, W. H., Caille, I., and Hirsch, E. C. (2004). Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 7, 726–735.
Huisman, E., Uylings, H. B., and Hoogland, P. V. (2004). A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson’s disease. Mov. Disord. 19, 687–692.
Jansen, J. F., Gearhart, J. D., and Bulte, J. W. (2008). Comment on “magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321, 640; author reply 640.
Jansen, J. F., Shamblott, M. J., van Zijl, P. C., Lehtimaki, K. K., Bulte, J. W., Gearhart, J. D., and Hakumaki, J. M. (2006). Stem cell profiling by nuclear magnetic resonance spectroscopy. Magn. Reson. Med. 56, 666–670.
Jin, K., Galvan, V., Xie, L., Mao, X. O., Gorostiza, O. F., Bredesen, D. E., and Greenberg, D. A. (2004a). Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc. Natl. Acad. Sci. U.S.A. 101, 13363–13367.
Jin, K., Peel, A. L., Mao, X. O., Xie, L., Cottrell, B. A., Henshall, D. C., and Greenberg, D. A. (2004b). Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 343–347.
Jin, K., Minami, M., Lan, J. Q., Mao, X. O., Batteur, S., Simon, R. P., and Greenberg, D. A. (2001). Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. U.S.A. 98, 4710–4715.
Jin, K., Sun, Y., Xie, L., Peel, A., Mao, X. O., Batteur, S., and Greenberg, D. A. (2003). Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol. Cell. Neurosci. 24, 171–189.
Jin, K., Wang, X., Xie, L., Mao, X. O., and Greenberg, D. A. (2010). Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc. Natl. Acad. Sci. U.S.A. 107, 7993–7998.
Jin, K., Wang, X., Xie, L., Mao, X. O., Zhu, W., Wang, Y., Shen, J., Mao, Y., Banwait, S., and Greenberg, D. A. (2006). Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. U.S.A. 103, 13198–13202.
Johansson, C. B., Svensson, M., Wallstedt, L., Janson, A. M., and Frisen, J. (1999). Neural stem cells in the adult human brain. Exp. Cell Res. 253, 733–736.
Kaiser, E., Kuzmits, R., Pregant, P., Burghuber, O., and Worofka, W. (1989). Clinical biochemistry of neuron specific enolase. Clin. Chim. Acta 183, 13–31.
Kam, M., Curtis, M. A., McGlashan, S. R., Connor, B., Nannmark, U., and Faull, R. L. (2009). The cellular composition and morphological organization of the rostral migratory stream in the adult human brain. J. Chem. Neuroanat. 37, 196–205.
Kaplan, M. S., and Hinds, J. W. (1977). Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 197, 1092–1094.
Karpowicz, P., Morshead, C., Kam, A., Jervis, E., Ramunas, J., Cheng, V., and van der Kooy, D. (2005). Support for the immortal strand hypothesis: neural stem cells partition DNA asymmetrically in vitro. J. Cell Biol. 170, 721–732.
Kempermann, G., Jessberger, S., Steiner, B., and Kronenberg, G. (2004). Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 27, 447–452.
Kim, K. K., Adelstein, R. S., and Kawamoto, S. (2009). Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J. Biol. Chem. 284, 31052–31061.
Knoth, R., Singec, I., Ditter, M., Pantazis, G., Capetian, P., Meyer, R. P., Horvat, V., Volk, B., and Kempermann, G. (2010). Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS ONE 5, e8809. doi: 10.1371/journal.pone.0008809
Koketsu, D., Furuichi, Y., Maeda, M., Matsuoka, N., Miyamoto, Y., and Hisatsune, T. (2006). Increased number of new neurons in the olfactory bulb and hippocampus of adult non-human primates after focal ischemia. Exp. Neurol. 199, 92–102.
Kornack, D. R., and Rakic, P. (1999). Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc. Natl. Acad. Sci. U.S.A. 96, 5768–5773.
Kornack, D. R., and Rakic, P. (2001). The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc. Natl. Acad. Sci. U.S.A. 98, 4752–4757.
Kukekov, V. G., Laywell, E. D., Suslov, O., Davies, K., Scheffler, B., Thomas, L. B., O’Brien, T. F., Kusakabe, M., and Steindler, D. A. (1999). Multipotent stem/progenitor cells with similar properties arise from two neurogenic regions of adult human brain. Exp. Neurol. 156, 333–344.
Kuruba, R., Hattiangady, B., and Shetty, A. K. (2009). Hippocampal neurogenesis and neural stem cells in temporal lobe epilepsy. Epilepsy Behav. 14(Suppl. 1), 65–73.
Lazarov, O., Mattson, M. P., Peterson, D. A., Pimplikar, S. W., and van Praag, H. (2010). When neurogenesis encounters aging and disease. Trends Neurosci. 33, 569–579.
Lemmens, M. A., Steinbusch, H. W., Rutten, B. P., and Schmitz, C. (2010). Advanced microscopy techniques for quantitative analysis in neuromorphology and neuropathology research: current status and requirements for the future. J. Chem. Neuroanat. 40, 199–209.
Lendahl, U., Zimmerman, L. B., and McKay, R. D. (1990). CNS stem cells express a new class of intermediate filament protein. Cell 60, 585–595.
Leonard, B. W., Mastroeni, D., Grover, A., Liu, Q., Yang, K., Gao, M., Wu, J., Pootrakul, D., van den Berge, S. A., Hol, E. M., and Rogers, J. (2009). Subventricular zone neural progenitors from rapid brain autopsies of elderly subjects with and without neurodegenerative disease. J. Comp. Neurol. 515, 269–294.
Liimatainen, T. J., Erkkila, A. T., Valonen, P., Vidgren, H., Lakso, M., Wong, G., Grohn, O. H., Yla-Herttuala, S., and Hakumaki, J. M. (2008). 1H MR spectroscopic imaging of phospholipase-mediated membrane lipid release in apoptotic rat glioma in vivo. Magn. Reson. Med. 59, 1232–1238.
Lin, T. N., Sun, S. W., Cheung, W. M., Li, F., and Chang, C. (2002). Dynamic changes in cerebral blood flow and angiogenesis after transient focal cerebral ischemia in rats. Evaluation with serial magnetic resonance imaging. Stroke 33, 2985–2991.
Lin, W., Celik, A., and Paczynski, R. P. (1999). Regional cerebral blood volume: a comparison of the dynamic imaging and the steady state methods. J. Magn. Reson. Imaging 9, 44–52.
Liu, Y. W., Curtis, M. A., Gibbons, H. M., Mee, E. W., Bergin, P. S., Teoh, H. H., Connor, B., Dragunow, M., and Faull, R. L. (2008). Doublecortin expression in the normal and epileptic adult human brain. Eur. J. Neurosci. 28, 2254–2265.
Lledo, P. M., Alonso, M., and Grubb, M. S. (2006). Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 7, 179–193.
Lois, C., and Alvarez-Buylla, A. (1993). Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. U.S.A. 90, 2074–2077.
Lois, C., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1996). Chain migration of neuronal precursors. Science 271, 978–981.
Lopez-Toledano, M. A., and Shelanski, M. L. (2007). Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind). J. Alzheimers Dis. 12, 229–240.
Lucassen, P. J., Muller, M. B., Holsboer, F., Bauer, J., Holtrop, A., Wouda, J., Hoogendijk, W. J., De Kloet, E. R., and Swaab, D. F. (2001). Hippocampal apoptosis in major depression is a minor event and absent from subareas at risk for glucocorticoid overexposure. Am. J. Pathol. 158, 453–468.
Ma, D. K., Kim, W. R., Ming, G. L., and Song, H. (2009). Activity-dependent extrinsic regulation of adult olfactory bulb and hippocampal neurogenesis. Ann. N. Y. Acad. Sci. 1170, 664–673.
Ma, L. H., Li, Y., Djuric, P. M., and Maletic-Savatic, M. (2011). “Systems biology approach to imaging of neural stem cells,” in Magnetic Resonance Neuroimaging Methods and Protocols, eds M. B. Modo and J. W. M. Bulte (New York: Springer), 421–435.
Macas, J., Nern, C., Plate, K. H., and Momma, S. (2006). Increased generation of neuronal progenitors after ischemic injury in the aged adult human forebrain. J. Neurosci. 26, 13114–13119.
Malberg, J. E., Eisch, A. J., Nestler, E. J., and Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104–9110.
Maletic-Savatic, M., Vingara, L. K., Manganas, L. N., Li, Y., Zhang, S., Sierra, A., Hazel, R., Smith, D., Wagshul, M. E., Henn, F., Krupp, L., Enikolopov, G., Benveniste, H., Djuric, P. M., and Pelczer, I. (2008). Metabolomics of neural progenitor cells: a novel approach to biomarker discovery. Cold Spring Harb. Symp. Quant. Biol. 73, 389–401.
Manganas, L. N., Zhang, X., Li, Y., Hazel, R. D., Smith, S. D., Wagshul, M. E., Henn, F., Benveniste, H., Djuric, P. M., Enikolopov, G., and Maletic-Savatic, M. (2007). Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science 318, 980–985.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Marti-Fabregas, J., Romaguera-Ros, M., Gomez-Pinedo, U., Martinez-Ramirez, S., Jimenez-Xarrie, E., Marin, R., Marti-Vilalta, J. L., and Garcia-Verdugo, J. M. (2010). Proliferation in the human ipsilateral subventricular zone after ischemic stroke. Neurology 74, 357–365.
Mattiesen, W. R., Tauber, S. C., Gerber, J., Bunkowski, S., Bruck, W., and Nau, R. (2009). Increased neurogenesis after hypoxic-ischemic encephalopathy in humans is age related. Acta Neuropathol. 117, 525–534.
McEwen, B. S. (2001). Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann. N. Y. Acad. Sci. 933, 265–277.
Mikkonen, M., Soininen, H., Kalvianen, R., Tapiola, T., Ylinen, A., Vapalahti, M., Paljarvi, L., and Pitkanen, A. (1998). Remodeling of neuronal circuitries in human temporal lobe epilepsy: increased expression of highly polysialylated neural cell adhesion molecule in the hippocampus and the entorhinal cortex. Ann. Neurol. 44, 923–934.
Miller, M. W., and Nowakowski, R. S. (1988). Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 457, 44–52.
Minturn, J. E., Geschwind, D. H., Fryer, H. J., and Hockfield, S. (1995). Early postmitotic neurons transiently express TOAD-64, a neural specific protein. J. Comp. Neurol. 355, 369–379.
Miyata, T., Maeda, T., and Lee, J. E. (1999). NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 13, 1647–1652.
Modo, M. B., and Bulte J. W. M. (eds). (2011). Magnetic Resonance Neuroimaging, Vol. 711. New York: Springer.
Moe, M. C., Varghese, M., Danilov, A. I., Westerlund, U., Ramm-Pettersen, J., Brundin, L., Svensson, M., Berg-Johnsen, J., and Langmoen, I. A. (2005). Multipotent progenitor cells from the adult human brain: neurophysiological differentiation to mature neurons. Brain 128, 2189–2199.
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765.
Mullen, R. J., Buck, C. R., and Smith, A. M. (1992). NeuN, a neuronal specific nuclear protein in vertebrates. Development 116, 201–211.
Nacher, J., Blasco-Ibanez, J. M., and McEwen, B. S. (2002). Non-granule PSA-NCAM immunoreactive neurons in the rat hippocampus. Brain Res. 930, 1–11.
Naumann, N., Alpar, A., Ueberham, U., Arendt, T., and Gartner, U. (2010). Transgenic expression of human wild-type amyloid precursor protein decreases neurogenesis in the adult hippocampus. Hippocampus 20, 971–979.
Pagano, S. F., Impagnatiello, F., Girelli, M., Cova, L., Grioni, E., Onofri, M., Cavallaro, M., Etteri, S., Vitello, F., Giombini, S., Solero, C. L., and Parati, E. A. (2000). Isolation and characterization of neural stem cells from the adult human olfactory bulb. Stem Cells 18, 295–300.
Palmer, T. D., Willhoite, A. R., and Gage, F. H. (2000). Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 425, 479–494.
Paradisi, M., Fernandez, M., Del Vecchio, G., Lizzo, G., Marucci, G., Giulioni, M., Pozzati, E., Antonelli, T., Lanzoni, G., Bagnara, G. P., Giardino, L., and Calzà, L. (2010). Ex vivo study of dentate gyrus neurogenesis in human pharmacoresistant temporal lobe epilepsy. Neuropathol. Appl. Neurobiol. 36, 535–550.
Parent, J. M. (2002). The role of seizure-induced neurogenesis in epileptogenesis and brain repair. Epilepsy Res. 50, 179–189.
Parent, J. M., Valentin, V. V., and Lowenstein, D. H. (2002). Prolonged seizures increase proliferating neuroblasts in the adult rat subventricular zone-olfactory bulb pathway. J. Neurosci. 22, 3174–3188.
Parent, J. M., Yu, T. W., Leibowitz, R. T., Geschwind, D. H., Sloviter, R. S., and Lowenstein, D. H. (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 17, 3727–3738.
Pencea, V., Bingaman, K. D., Freedman, L. J., and Luskin, M. B. (2001). Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp. Neurol. 172, 1–16.
Pereira, A. C., Huddleston, D. E., Brickman, A. M., Sosunov, A. A., Hen, R., McKhann, G. M., Sloan, R., Gage, F. H., Brown, T. R., and Small, S. A. (2007). An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 104, 5638–5643.
Pryce, C. R., and Seifritz, E. (2011). A translational research framework for enhanced validity of mouse models of psychopathological states in depression. Psychoneuroendocrinology 36, 308–329
Rakic, P. (1985). DNA synthesis and cell division in the adult primate brain. Ann. N. Y. Acad. Sci. 457, 193–211.
Rakic, P. (2002b). Neurogenesis in adult primate neocortex: an evaluation of the evidence. Nat. Rev. Neurosci. 3, 65–71.
Rakic, P., and Nowakowski, R. S. (1981). The time of origin of neurons in the hippocampal region of the rhesus monkey. J. Comp. Neurol. 196, 99–128.
Ramm, P., Couillard-Despres, S., Plotz, S., Rivera, F. J., Krampert, M., Lehner, B., Kremer, W., Bogdahn, U., Kalbitzer, H. R., and Aigner, L. (2009). A nuclear magnetic resonance biomarker for neural progenitor cells: is it all neurogenesis? Stem Cells 27, 420–423.
Reif, A., Fritzen, S., Finger, M., Strobel, A., Lauer, M., Schmitt, A., and Lesch, K. P. (2006). Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol. Psychiatry 11, 514–522.
Reynolds, B. A., and Weiss, S. (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710.
Rodriguez, J. J., Jones, V. C., Tabuchi, M., Allan, S. M., Knight, E. M., LaFerla, F. M., Oddo, S., and Verkhratsky, A. (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3, e2935. doi: 10.1371/journal.pone.0002935
Rodriguez, J. J., Jones, V. C., and Verkhratsky, A. (2009). Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. Neuroreport 20, 907–912.
Romer, B., Sartorius, A., Inta, D., Vollmayr, B., and Gass, P. (2008). Imaging new neurons in vivo: a pioneering tool to study the cellular biology of depression? Bioessays 30, 806–810.
Roy, N. S., Wang, S., Jiang, L., Kang, J., Benraiss, A., Harrison-Restelli, C., Fraser, R. A., Couldwell, W. T., Kawaguchi, A., Okano, H., Nedergaard, M., and Goldman, S. A. (2000). In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat. Med. 6, 271-277.
Sakakibara, S., and Okano, H. (1997). Expression of neural RNA-binding proteins in the postnatal CNS: implications of their roles in neuronal and glial cell development. J. Neurosci. 17, 8300–8312.
Sanai, N., Berger, M. S., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (2007). Comment on “human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension”. Science 318, 393; author reply 393.
Sanai, N., Tramontin, A. D., Quinones-Hinojosa, A., Barbaro, N. M., Gupta, N., Kunwar, S., Lawton, M. T., McDermott, M. W., Parsa, A. T., Manuel-Garcia Verdugo, J., Berger, M. S., and Alvarez-Buylla, A. (2004). Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427, 740–744.
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., and Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809.
Scharfman, H. E., Goodman, J. H., and Sollas, A. L. (2000). Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J. Neurosci. 20, 6144–6158.
Seevinck, P. R., Deddens, L. H., and Dijkhuizen, R. M. (2010). Magnetic resonance imaging of brain angiogenesis after stroke. Angiogenesis 13, 101–111.
Seress, L., Abraham, H., Tornoczky, T., and Kosztolanyi, G. (2001). Cell formation in the human hippocampal formation from mid-gestation to the late postnatal period. Neuroscience 105, 831–843.
Sgubin, D., Aztiria, E., Perin, A., Longatti, P., and Leanza, G. (2007). Activation of endogenous neural stem cells in the adult human brain following subarachnoid hemorrhage. J. Neurosci. Res. 85, 1647–1655.
Sheline, Y. I., Wang, P. W., Gado, M. H., Csernansky, J. G., and Vannier, M. W. (1996). Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. U.S.A. 93, 3908–3913.
Shen, J., Xie, L., Mao, X., Zhou, Y., Zhan, R., Greenberg, D. A., and Jin, K. (2008). Neurogenesis after primary intracerebral hemorrhage in adult human brain. J. Cereb. Blood Flow Metab. 28, 1460–1468.
Sibtain, N. A., Howe, F. A., and Saunders, D. E. (2007). The clinical value of proton magnetic resonance spectroscopy in adult brain tumours. Clin. Radiol. 62, 109–119.
Sierra, A., Encinas, J. M., Deudero, J. J., Chancey, J. H., Enikolopov, G., Overstreet-Wadiche, L. S., Tsirka, S. E., and Maletic-Savatic, M. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495.
Soares, D. P., and Law, M. (2009). Magnetic resonance spectroscopy of the brain: review of metabolites and clinical applications. Clin. Radiol. 64, 12–21.
Sohur, U. S., Emsley, J. G., Mitchell, B. D., and Macklis, J. D. (2006). Adult neurogenesis and cellular brain repair with neural progenitors, precursors and stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1477–1497.
Spalding, K. L., Bhardwaj, R. D., Buchholz, B. A., Druid, H., and Frisen, J. (2005). Retrospective birth dating of cells in humans. Cell 122, 133–143.
Spector, R., and Johanson, C. E. (2007). The origin of deoxynucleosides in brain: implications for the study of neurogenesis and stem cell therapy. Pharm. Res. 24, 859–867.
Stockmeier, C. A., Mahajan, G. J., Konick, L. C., Overholser, J. C., Jurjus, G. J., Meltzer, H. Y., Uylings, H. B., Friedman, L., and Rajkowska, G. (2004). Cellular changes in the postmortem hippocampus in major depression. Biol. Psychiatry 56, 640–650.
Stoeber, K., Tlsty, T. D., Happerfield, L., Thomas, G. A., Romanov, S., Bobrow, L., Williams, E. D., and Williams, G. H. (2001). DNA replication licensing and human cell proliferation. J. Cell. Sci. 114, 2027–2041.
Suzuki, K., Okada, K., Wakuda, T., Shinmura, C., Kameno, Y., Iwata, K., Takahashi, T., Suda, S., Matsuzaki, H., Iwata, Y., Hashimoto, K., and Mori, N. (2010). Destruction of dopaminergic neurons in the midbrain by 6-hydroxydopamine decreases hippocampal cell proliferation in rats: reversal by fluoxetine. PLoS ONE 5, e9260. doi: 10.1371/journal.pone.0009260
Takasaki, Y., Deng, J. S., and Tan, E. M. (1981). A nuclear antigen associated with cell proliferation and blast transformation. J. Exp. Med. 154, 1899–1909.
Tata, D. A., and Anderson, B. J. (2010). The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: implications for hippocampal volume reductions in depression. Physiol. Behav. 99, 186–193.
Tattersfield, A. S., Croon, R. J., Liu, Y. W., Kells, A. P., Faull, R. L., and Connor, B. (2004). Neurogenesis in the striatum of the quinolinic acid lesion model of Huntington’s disease. Neuroscience 127, 319–332.
Van der Borght, K., Kobor-Nyakas, D. E., Klauke, K., Eggen, B. J., Nyakas, C., Van der Zee, E. A., and Meerlo, P. (2009). Physical exercise leads to rapid adaptations in hippocampal vasculature: temporal dynamics and relationship to cell proliferation and neurogenesis. Hippocampus 19, 928–936.
van Praag, H., Shubert, T., Zhao, C., and Gage, F. H. (2005). Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 25, 8680–8685.
Veeraraghavalu, K., Choi, S. H., Zhang, X., and Sisodia, S. S. (2010). Presenilin 1 mutants impair the self-renewal and differentiation of adult murine subventricular zone-neuronal progenitors via cell-autonomous mechanisms involving notch signaling. J. Neurosci. 30, 6903–6915.
Verret, L., Jankowsky, J. L., Xu, G. M., Borchelt, D. R., and Rampon, C. (2007). Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J. Neurosci. 27, 6771–6780.
Verwer, R. W., Sluiter, A. A., Balesar, R. A., Baayen, J. C., Noske, D. P., Dirven, C. M., Wouda, J., van Dam, A. M., Lucassen, P. J., and Swaab, D. F. (2007). Mature astrocytes in the adult human neocortex express the early neuronal marker doublecortin. Brain 130, 3321–3335.
Vollmayr, B., Mahlstedt, M. M., and Henn, F. A. (2007). Neurogenesis and depression: what animal models tell us about the link. Eur. Arch. Psychiatry Clin. Neurosci. 257, 300–303.
von Bohlen Und Halbach, O. (2007). Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 329, 409–420.
Watts, C., McConkey, H., Anderson, L., and Caldwell, M. (2005). Anatomical perspectives on adult neural stem cells. J. Anat. 207, 197–208.
Wegiel, J., Kuchna, I., Nowicki, K., Imaki, H., Marchi, E., Ma, S. Y., Chauhan, A., Chauhan, V., Bobrowicz, T. W., de Leon, M., Louis, L. A., Cohen, I. L., London, E., Brown, W. T., and Wisniewski, T. (2010). The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 119, 755–770.
Weickert, C. S., Webster, M. J., Colvin, S. M., Herman, M. M., Hyde, T. M., Weinberger, D. R., and Kleinman, J. E. (2000). Localization of epidermal growth factor receptors and putative neuroblasts in human subependymal zone. J. Comp. Neurol. 423, 359–372.
Whitman, M. C., and Greer, C. A. (2009). Adult neurogenesis and the olfactory system. Prog. Neurobiol. 89, 162–175.
Winner, B., Geyer, M., Couillard-Despres, S., Aigner, R., Bogdahn, U., Aigner, L., Kuhn, G., and Winkler, J. (2006). Striatal deafferentation increases dopaminergic neurogenesis in the adult olfactory bulb. Exp. Neurol. 197, 113–121.
Wong, M. L., and Licinio, J. (2001). Research and treatment approaches to depression. Nat. Rev. Neurosci. 2, 343–351.
Yerushalmi, R., Woods, R., Ravdin, P. M., Hayes, M. M., and Gelmon, K. A. (2010). Ki67 in breast cancer: prognostic and predictive potential. Lancet Oncol. 11, 174–183.
Yoshimi, K., Ren, Y. R., Seki, T., Yamada, M., Ooizumi, H., Onodera, M., Saito, Y., Murayama, S., Okano, H., Mizuno, Y., and Mochizuki, H. (2005). Possibility for neurogenesis in substantia nigra of parkinsonian brain. Ann. Neurol. 58, 31–40.
Yu, T. S., Zhang, G., Liebl, D. J., and Kernie, S. G. (2008). Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J. Neurosci. 28, 12901–12912.
Yu, Y., He, J., Zhang, Y., Luo, H., Zhu, S., Yang, Y., Zhao, T., Wu, J., Huang, Y., Kong, J., Tan, Q., and Li, X. M. (2009). Increased hippocampal neurogenesis in the progressive stage of Alzheimer’s disease phenotype in an APP/PS1 double transgenic mouse model. Hippocampus 19, 1247–1253.
Zaharchuk, G. (2007). Theoretical basis of hemodynamic MR imaging techniques to measure cerebral blood volume, cerebral blood flow, and permeability. AJNR Am. J. Neuroradiol. 28, 1850–1858.
Zhang, R. L., Zhang, Z. G., Zhang, L., and Chopp, M. (2001). Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience 105, 33–41.
Zhang, Z. G., and Chopp, M. (2009). Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 8, 491–500.
Zhao, M., Momma, S., Delfani, K., Carlen, M., Cassidy, R. M., Johansson, C. B., Brismar, H., Shupliakov, O., Frisen, J., and Janson, A. M. (2003). Evidence for neurogenesis in the adult mammalian substantia nigra. Proc. Natl. Acad. Sci. U.S.A. 100, 7925–7930.
Keywords: adult, neurogenesis, neural stem cells, human, methods, BrdU, cerebral blood volume, metabolomics
Citation: Sierra A, Encinas JM and Maletic-Savatic M (2011) Adult human neurogenesis: from microscopy to magnetic resonance imaging. Front. Neurosci. 5:47. doi: 10.3389/fnins.2011.00047
Received: 20 January 2011;
Accepted: 23 March 2011;
Published online: 04 April 2011.
Edited by:
Silvia De Marchis, University of Turin, ItalyReviewed by:
Adam C. Puche, University of Maryland, USANader Sanai, Barrow Neurological Institute, USA
Copyright: © 2011 Sierra, Encinas and Maletic-Savatic. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Mirjana Maletic-Savatic and Amanda Sierra, Department of Pediatrics, Baylor College of Medicine, Neurological Research Institute, 1250 Morsund St, MS NR1250.01, Houston, TX 77030, USA. e-mail:bWFsZXRpY3NAYmNtLmVkdQ==;YW1hbmRhLnNpZXJyYXNhYXZlZHJhQGdtYWlsLmNvbQ==