



- 1 Medical Clinic 1, University of Luebeck, Luebeck, Germany
- 2 Computational Mathematics, Technical University of Braunschweig, Braunschweig, Germany
The brain occupies a special hierarchical position in human energy metabolism. If cerebral homeostasis is threatened, the brain behaves in a “selfish” manner by competing for energy resources with the body. Here we present a logistic approach, which is based on the principles of supply and demand known from economics. In this “cerebral supply chain” model, the brain constitutes the final consumer. In order to illustrate the operating mode of the cerebral supply chain, we take experimental data which allow assessing the supply, demand and need of the brain under conditions of psychosocial stress. The experimental results show that the brain under conditions of psychosocial stress actively demands energy from the body, in order to cover its increased energy needs. The data demonstrate that the stressed brain uses a mechanism referred to as “cerebral insulin suppression” to limit glucose fluxes into peripheral tissue (muscle, fat) and to enhance cerebral glucose supply. Furthermore psychosocial stress elicits a marked increase in eating behavior in the post-stress phase. Subjects ingested more carbohydrates without any preference for sweet ingredients. These experimentally observed changes of cerebral demand, supply and need are integrated into a logistic framework describing the supply chain of the selfish brain.
Stress and Eating Behavior
How stress, the stress response, and the adaption to the stress-response influence our eating behavior is a central question in brain research and medicine. The causal relationship between stress and food intake however cannot easily be untangled. An obvious paradox has been observed in first-year students at the University College of London (Oliver and Wardle, 1999; Serlachius et al., 2007). A majority of students reported an effect of stress on eating behavior: in particular, 42% reported decreased food intake, whereas 38% reported increased food intake. Later investigations of this university student population revealed reported weight gain in 55%, weight loss in 12% of the students, and stable weight over the 1 year period in 33%. Paradoxically, stress was associated with both the risk of weight gain and weight loss.
How can it happen that stress is linked with both weight gain and weight loss? Many of the British first-year students experienced repeated stressful events or chronic stress at least in the mild form. According to the concept of “allostatic regulation,” which has been formulated by Sterling (2004) and further developed by McEwen (2007), a considerable number of students has been exposed to an allostatic load. The concept of allostatic load has served as a framework for a large body of research on the integrative health psychology, epidemiology, and demography of aging. It is based on the hypothesis that there is a cumulative physiological risk associated with exposure to psychosocial stressors over the life-course. On this background, changes in body weight might be considered as an allostatic regulation, which would have allowed the study participants to adapt to the challenging conditions in their first student year. McEwen (1998) distinguishes two types of subjects and how they react with their stress systems to an allostatic load: type A displays a lack of adaptation and keeps the stress responses high on repeated occasions; in contrast, type B does show adaptation, i.e., habituation of the stress responsiveness by time. Of course, the “distinct” types represent extreme patterns of adaptation, which belong to a continuous spectrum ranging from weak to strong adaptive responses. It has been shown recently, that it is the endocannabinoid system, which plays an essential role in determining the degree of stress-response adaptation (Hill et al., 2010). As stress-responses in general are closely linked to the cerebral energetic state, the interesting question arises here, whether different patterns of stress responses and different patterns of body weight changes constitute allostatic regulations, which all serve to maintain cerebral energy homeostasis.
According to the “Selfish Brain” theory, cerebral energy homeostasis has highest priority in human energy metabolism (Peters et al., 2004). The pathologist Marie Krieger was the first to provide evidence that the brain behaves in a “selfish” manner. She showed that the human brain mass is preserved during inanition, while all the organs of the body such as heart, liver, spleen, kidneys, and pancreas lose about 40% of their mass (Krieger, 1921). Her finding has been confirmed both in humans and animals in adult and fetal life by using modern state-of-the-art techniques (Goodman et al., 1984; Gong et al., 1998; Miller et al., 2002; Kind et al., 2005; Muhlau et al., 2007; Peters et al., 2011). These observations provide clear evidence that the brain must actively demand energy from the body in order to maintain cerebral energy homeostasis.
Does the brain under psychosocial stress conditions also prioritize its own energy procurement? Using the invasive Kety–Schmidt technique to assess brain energy metabolism, it could be shown that during a mild laboratory mental stress (e.g., Wisconsin card sorting test) the energy supply of the human brain increases by 12% (Madsen et al., 1995). Such an instantaneous augmentation of brain supply also suggests that underlying cerebral demand mechanisms do exist. Similarly, brain glucose consumption is enhanced during focal physiological neural activity in humans (Fox et al., 1988). Both during mental stress and increased focal neural activity, a concomitant rise in brain oxygen consumption is far less marked. These findings suggest that the extra glucose taken up is not immediately oxidized. Animal experiments studying brain metabolism during cerebral activation imply that intermediary metabolic products accumulate in the brain which are oxidized subsequently (Madsen et al., 1999). On the basis of these findings it remains unclear whether the energetic need of the brain actually increases during stress.
The Supply Chain of the Selfish Brain
Need, supply, and demand are terms used in the field of economics and logistics. In a previous paper, we made use of supply chain principles to describe the central and peripheral energy metabolism (Peters and Langemann, 2009). In this focused review, we make use of the principles of logistic supply chains (Slack et al., 2004), in order to address the question, why stress may evoke apparently “paradoxical” changes in eating behavior and body weight. The supply chain of the brain – with the central nervous system as the final consumer – describes the energy fluxes from the remote environment to the near environment through the body toward the brain (Figure 1). The supply chain is branched, i.e., it is possible to store energy in side buffers such as muscle and fat tissue. It is a general principle in economic supply chains that the flux can be determined by both the supplier (push-component) and the receiver (pull-component). In other words, the fluxes are regulated by offer and demand. In this connection, the “brain-pull” functions to demand energy from the body. In the previous publication on neuroenergetics (Peters and Langemann, 2009) we presented the concept that a healthy organism is maintained by a competent brain-pull – which serves systemic homeostasis – and that the underlying cause of body weight gain is an incompetent brain-pull (e.g., that the brain is unable to properly demand and receive glucose from the body).
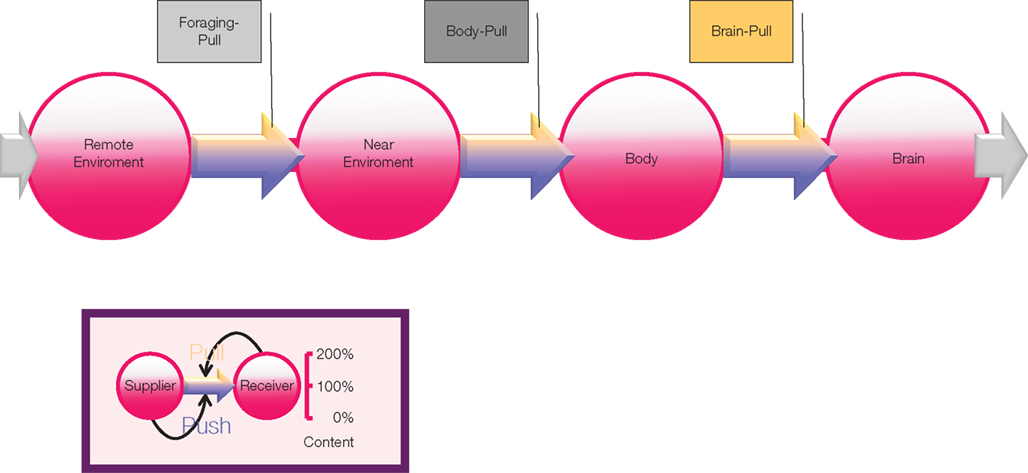
Figure 1. Supply chain of the human brain. Energy from the remote environment is brought to the immediate environment, it is then taken up by the body, and from there a large part of it enters the brain. In a general supply chain, the flux of energy is determined principally by the supplier (previous step) and the receiver (proximate step). Insert: the share of the flux which is determined by the supplier is called the “push component” (blue part of the arrows); the share which is determined by the receiver is called the “pull-component” (yellow part of the arrows). When the brain needs energy, it activates brain-pull mechanisms in order to demand energy from the body. Upon energy need in the body, body-pull mechanisms are activated to demand for energy from the environment, i.e., they initiate eating. Foraging-Pull mechanisms initiate processes to explore and get energy from the remote environment.
Brain-Pull Mechanisms
When focusing on the proximal, final part of the supply chain, we find strong experimental evidence identifying two brain-pull mechanisms, a direct and an allocative one. Of note, these two components act synergistically to supply the brain. Direct brain-pull mechanisms have been found which enable astrocytes to actively take up glucose depending on energetic conditions: firstly their need and secondly their adjacent neuron’s energy needs. Astrocytes are equipped with mechanisms which enable them to monitor changes in their own ATP concentrations. The glucose transporter 1 (GLUT1) has been found to mediate glucose transport in a manner that depends on cytoplasmic ATP (Blodgett et al., 2007). GLUT1 are abundantly located on the luminal and abluminal side of endothelial cells forming the blood–brain barrier (Cornford and Hyman, 2005) and on the end feet of the adjacent astrocytes which make contact to the endothelial cells (Kacem et al., 1998). It has been shown that astrocytes enhance their GLUT1 mediated glucose uptake once they consume ATP (Pellerin and Magistretti, 1997). Thus, astrocytes feature mechanisms that fulfill pull-functions serving to control their energy balance.
Pellerin and Magistretti (1994) discovered that astrocytes particularly respond to activity of their neighboring neurons. The researchers found that astrocytes actively pull glucose from their vicinity, when their neighboring neurons are at work, and termed this process “energy on demand” (Magistretti et al., 1999). The conception of such a “pull-mechanism” has come to be known as the astrocyte-neuron lactate shuttle hypothesis (ANLSH; Pellerin et al., 2007). Upon neuronal excitation, glutamate released in the synaptic cleft is taken up by astrocytes to be recycled via the specific glutamate transporters. In this way, neurons send glutamergic signals to their astrocytes conveying that energy is needed in these neurons at this very moment. Then astrocytes take up glucose from the blood, convert glucose to lactate, and pass on lactate molecules to the working neurons, which use oxidative phosphorylation to cover their large needs.
Furthermore, mechanisms, given in more detail beneath, which exert “allocative brain-pull mechanism” have been experimentally located (Figure 2). Such mechanisms are capable of limiting peripheral energy storage in favor of the brain. Those experiments have sustained the “Selfish Brain” theory, whose foundations have been laid from 1998 to 2004 (Peters et al., 2004), postulating the existence of such allocative mechanisms and implementing them as functional elements serving to maintain the brain’s high energy content at the expense of the body. According to that concept, the brain simultaneously represents the highest regulatory authority and the consumer with the highest priority – it looks after itself first. In this respect, a competition among all organs drives energy allocation. In the following, we take a closer look at the mechanisms that can fulfill the function of allocative brain-pull.
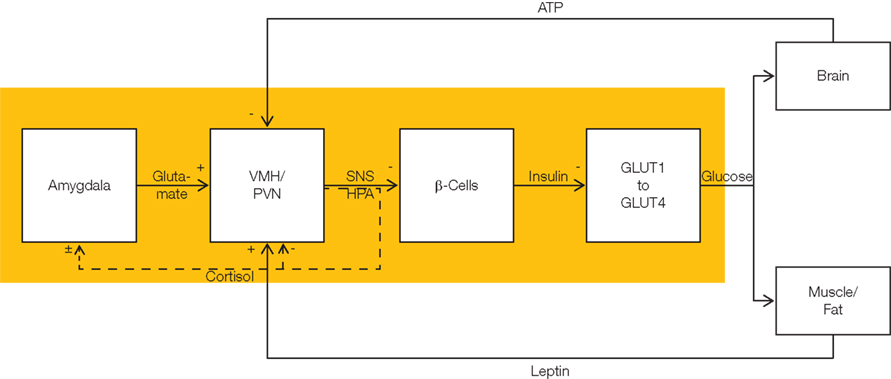
Figure 2. Flow chart showing allocative brain-pull mechanisms. The stress system fulfills brain-pull function (yellow area). It allows the brain to actively demand energy from the body. The stress system is hierarchically organized. At the level of the cerebral hemispheres, amygdala neurons convey an energy-on-demand signal via glutamatergic input into the VMH and the paraventricular nucleus (PVN). With activation of these nuclei at the hypothalamic level, the sympathetic nervous system (SNS) and the hypothalamus pituitary adrenal (HPA) systems are stimulated. These hypothalamic nuclei act via autonomic efferences and via cortisol upon the level of the autonomic viscero- and endocrine secretomotoneurons, i.e., they suppress beta cell insulin secretion. Cerebral insulin suppression (CIS) results in an increased ratio of GLUT1- to GLUT4-mediated glucose uptake. While insulin stimulates (insulin-dependent) GLUT4-mediated glucose uptake into muscle and fat tissue, it does not affect (insulin-independent) GLUT1-mediated glucose uptake across the blood–brain barrier. In this way, brain-pull mechanisms favor glucose fluxes directed toward the brain. There are three feedback loops tuning the brain-pull activity. First, intraneuronal ATP exerts negative feedback at the level of the VMH in order to limit further energy demand. Second, leptin which indicates the energy fill content in muscle and fat exerts positive feedback on VMH-neurons. Third, the stress system is a self-organizing system which optimizes its brain-pull function under the feedback influence of cortisol released from the adrenals. Cortisol exerts feedback inhibition at the hypothalamic level (PVN), and also modulates the processes of long-term potentiation and long-term depression at synapses which convey cerebral input signal directed to the amygdala neurons. In all, the brain-pull system is a hierarchically self-organized feedback-control system which matches the brain’s energy supply with the cerebral energy needs.
In the ventromedial hypothalamus (VMH), neurons have been found which sensitively respond to changes in cerebral intracellular ATP (Miki et al., 2001). In this brain area, ATP-sensitive potassium (KATP) channels have been shown to monitor ATP (Spanswick et al., 1997). When cerebral intracellular ATP levels fall, presynaptic KATP channels alter the release of GABA within the VMH, hereupon the postsynaptic VMH-neurons release glutamate, and in so doing act via the sympatho-adrenal system in order to inhibit insulin secretion and action (Ahren, 2000; Mulder et al., 2005; Chan et al., 2007; Tong et al., 2007). Because insulin is required for glucose uptake in the peripheral fat and muscle tissue via glucose transporter 4 (GLUT4), these postsynaptic VMH-neurons are able to limit glucose outflow from the blood into the peripheral energy buffers. In this way, these allocative mechanisms are able to preserve blood glucose as a source for the insulin-independent glucose uptake via GLUT1 into the brain. These VMH-triggered mechanisms can be regarded as pull-mechanisms because they safeguard blood glucose availability, a necessity for adequate brain supply. It has been shown with the help of experimental data that in the cerebral supply chain, a competent brain-pull is indispensable to maintain systemic energy homeostasis (Peters and Langemann, 2009). This means, that even small changes in the efficiency of the brain-pull cause considerable changes in the systemic energy homeostasis and thus in body weight and blood glucose concentrations.
Body-Pull Mechanisms
When looking up at the more distal part of the supply chain, another pull-mechanism can be identified: body-pull, which controls eating behavior. Body-pull is initiated when blood glucose concentrations decline (Figure 1). Recent experimental evidence suggests that orexigenic neurons in the lateral hypothalamus (LH) fulfill such a pull-function. Glucose from the blood circulation crosses the endothelial cells of the blood–brain-barrier via glucose transporters 1 and enters the brain interstitial space. Glucose acts at an extracellular site on the orexin neurons. These LH neurons respond to changes in extracellular glucose concentrations by modulating their tandem-pore K+ channels (Burdakov et al., 2006; Gonzalez et al., 2008). Upon a fall in extracellular glucose, these neurons depolarize. With activation of these neurons, appetite increases and ingestive behavior is initiated. The LH neurons can fulfill the functions of body-pull mechanisms that aim to replenish energy in blood and peripheral buffers.
How the Brain Organizes Its Supply and Demand during Acute Psychosocial Stress
In our recent publication in Frontiers in Neuroenergetics changes in the cerebral supply chain had been investigated under the conditions of acute psychosocial stress (Hitze et al., 2010). Young healthy men were exposed to a Trier Social Stress Test. They were offered a rich buffet immediately after the stress intervention. Carbohydrate intake from the rich buffet was increased by 34 g (when compared with the non-stress condition). The subjects did not prefer specific food when stressed, particularly subjects did not display preferences for sweet or non-sweet carbohydrates. These results demonstrate that a social stress intervention increases the carbohydrate intake from a rich buffet without special emphasis on sweet food. These findings confirm a recent report on enhanced carbohydrate intake after an acute stress test (Rutters et al., 2009).
Cerebral Insulin Suppression (CIS)
Do these stress extra carbohydrates alter blood glucose concentrations? Blood glucose concentrations increased in the post-stress replenishment phase after rich buffet ingestion (Hitze et al., 2010). As expected the meal induced hyperglycemia was more pronounced in the stress compared to the non-stress intervention. Higher hyperglycemia was obviously due to the 34 g extra carbohydrates ingested. Interestingly postprandial insulin concentrations were not different between stress- and the non-stress intervention (Figure 3). Noteworthy, these serum insulin concentrations were equal despite different blood glucose concentrations. Thus, the extra carbohydrates ingested after stress failed to elicit a correspondent rise in insulin.
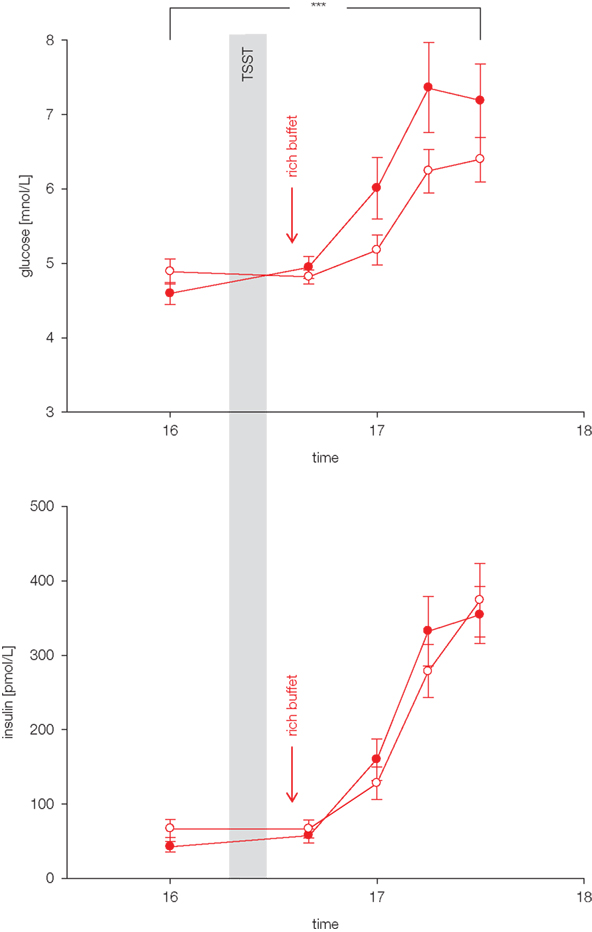
Figure 3. Glucose and insulin concentrations during stress and non-stress intervention in 10 men offered a rich buffet. Values are means ± SEM; closed symbols, stress intervention and open symbols, non-stress intervention; glucose concentrations are significantly different from non-stress intervention, interaction time × stress intervention: F = 3.8, ***p < 0.001, by ANOVA for repeated measures; insulin concentrations are equal between stress and non-stress intervention, interaction time × stress intervention: F = 2.5, p > 0.05, by ANOVA for repeated measures.
Furthermore, social stress elicited a robust sympatho-adrenal stress response. Social stress markedly increased concentrations of epinephrine, norepinephrine, ACTH, cortisol, and autonomic symptoms. Cortisol was found to be associated with the pattern of insulin suppression. A higher increase in cortisol after stress was associated with a steeper increase in glucose concentrations after rich buffet ingestion, whereas cortisol concentrations were not associated with an increase in insulin concentrations after rich buffet ingestion. This pattern was independent of carbohydrate intake. These findings support the notion that CIS was operative in these study subjects.
Cerebral Energy Needs and Mood
Since subjects ate 34 g extra carbohydrates when stressed, and insulin was suppressed in the postprandial replenishment phase, it is likely that the main energy flux was directed to the brain. A neuroglycopenic state developed at normal blood glucose concentrations after stress (Figure 4). Subjects showed more neuroglycopenic symptoms (assessed by a standard questionnaire usually applied in hypoglycemia research) immediately after social stress intervention. Of note, neuroglycopenic symptoms which typically occur only during hypoglycemia occurred in the presence of normal blood glucose concentrations. When subjects were provided high energy under stress conditions, post-stress neuroglycopenic symptoms were resolved. However, when subjects were offered a meager salad only the post-stress neuroglycopenic state persisted for at least 90 min.
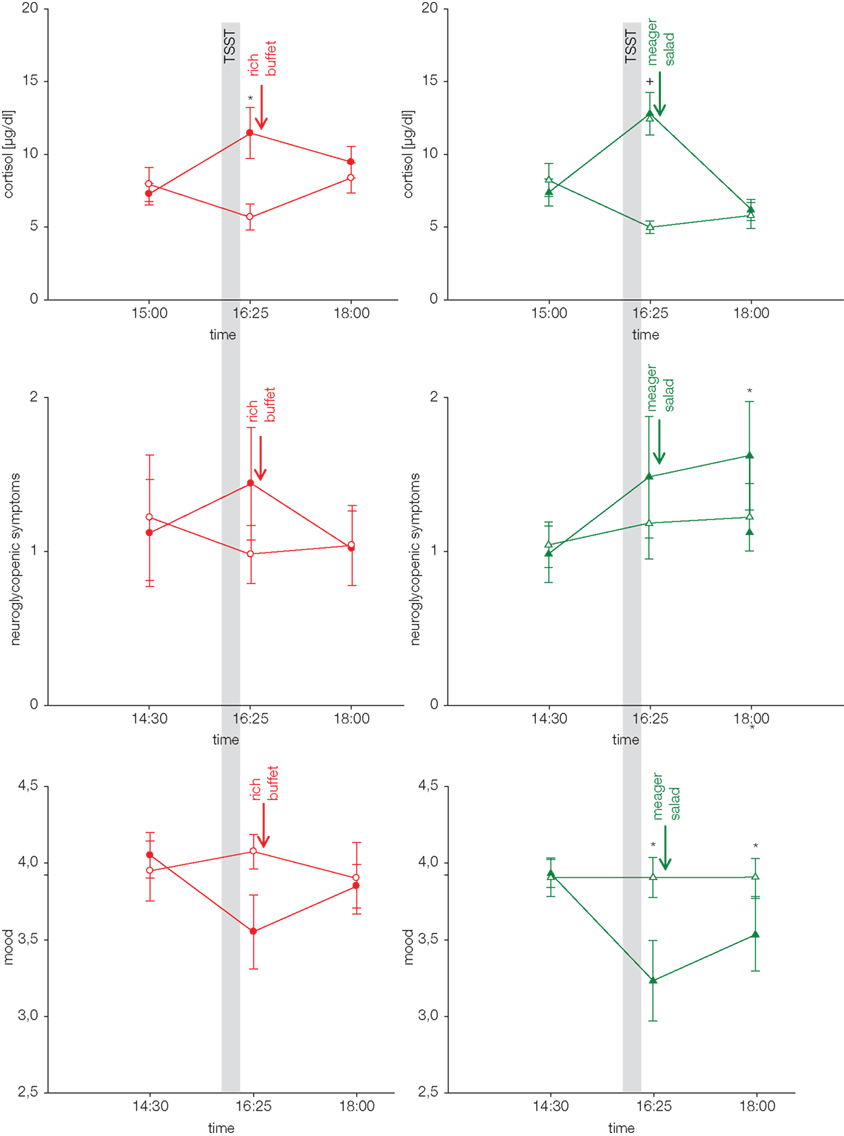
Figure 4. Cortisol response, neuroglycopenic symptoms, and mood during stress and non-stress intervention in subjects offered a rich buffet or a meager salad. Values are means ± SEM; closed symbols, stress intervention and open symbols, non-stress intervention; *p < 0.05, +p < 0.01, significantly different from non-stress intervention, by paired t-test. For complete statistical analysis (including ANOVA for repeated measures) see original paper Hitze et al. (2010).
Mood (assessed by a short form of the multidimensional mood state questionnaire, MDBF; Steyer et al., 1997) deteriorated during psychosocial stress, but was normalized subsequently, when subjects were provided with high energy (Figure 4). However, when they were offered a meager salad only, the mood did not recover. Thus, recovery of the SNS/HPA-response is not sufficient to let the mood return to normal. The reestablishment of cerebral energy homeostasis is also necessary to regain a balanced emotional state.
These findings support the notion that the extra carbohydrates ingested after stress were mainly allocated to the brain in order to reestablish brain energy homeostasis. The experiment presented in that study shows how the brain can demand for energy from the body: by making use of CIS. In this way, CIS can be interpreted as a brain-pull mechanism. Moreover, psychosocial stress did increase the brain’s need. After psychosocial stress, energy intake was found markedly increased to supply the brain and the body with energy for replenishment. The concept of CIS has already been proposed by Steve Woods and Daniel Porte in the 1970’s (Woods and Porte Jr., 1974). Here we confirm the role of CIS in humans undergoing acute psychosocial stress. The described stress experiments show the interplay between brain-pull and body-pull in the supply chain of the human brain.
The “Energy on Demand” Principle
Is CIS also evident in other pathological states? The presence of a CIS-brain-pull mechanism has been found in two animal experiments studying the effects of cerebral ischemia. First, when mice underwent cerebral artery occlusions (MCAO) and received a glucose load one day later, they failed to increase their insulin secretion, while control rats (sham MCAO) exhibited a marked insulin rise (Harada et al., 2009). These findings are compatible with the presence of a CIS-mechanism, suggesting that the brain activates demand mechanisms to compensate for the cerebral energy depletion. In a second experiment, adult rats displayed the features of CIS after MCAO, and CIS was particularly pronounced in those rats, which had additionally undergone postnatal stress and which consequently displayed elevated serum cortisol in their later adult life (McPherson et al., 2009). As reported earlier, CIS is also known to occur in various kinds of stress states threatening cerebral energy homeostasis including myocardial infarction, hemorrhagic shock, hypoxia (Woods and Porte Jr., 1974), and caloric restriction (Peters et al., 2011). Thus, cerebral energy demand mechanisms like CIS are not only present during psychosocial stress, but also during other kinds of cerebral metabolic crisis.
In summary, these experiments on acute psychosocial stress show that the brain is able to actively demand energy from the body by using mechanisms referred to as CIS.
The Adaption to Chronic Stress and the Change of Body Weight
Lack of Stress-Response Adaptation – Preserved Brain-Pull Competence
What changes will occur in the cerebral supply chain, if the conditions of psychosocial stress persist chronically? We used the mathematical model of the cerebral supply chain that predicts the effect of a long-term increase of the cerebral energy need on the body’s energy stores (muscle, fat, tissue; Figure 5A). Under conditions of increased cerebral need the supply chain model predicts the increase of brain-pull activity, that the energy flux from the body to the brain is augmented, that the energetic equilibrium in blood and muscle fat compartment is burdened, and that the peripheral energy stores will decline. These observations within the supply chain are similar to those that occurred in the 42% of first-year students who participated in the two British studies and reported decreased food intake during stress or weight loss (type A). Thus, it is conceivable that weight loss in an individual is driven by a persistent SNS/HPA stress response or in other words by a persistent competent and overactive brain-pull.
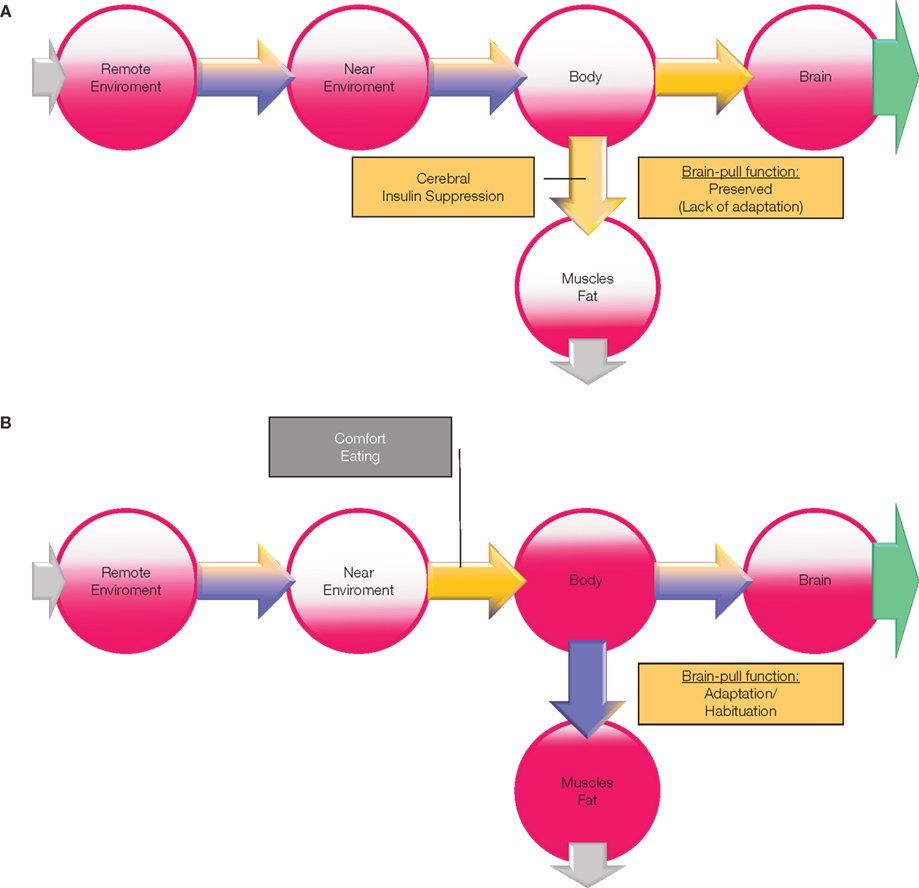
Figure 5. (A) The effect of stress on energy metabolism. Changes are based on a simulation study using the “supply chain” model as published in Peters and Langemann (2009); the results are depicted in a semiquantitative manner. In this case (type A), brain-pull function (stress-response) lacks adaptation to a chronic allostatic load, i.e., brain-pull function is preserved. If the cerebral energy consumption increases (green arrow), the cerebral energy content slightly decreases and prompts a strong activation of the brain-pull. As a consequence of overactive brain-bull (CIS) energy content in the body compartment declines, particularly in muscle and fat. “Push component” (blue part of the arrows); “pull-component” (yellow part of the arrows). (B) The effect of the adaptation of the stress response on energy metabolism. In this case (type B), brain-pull function (stress-response) shows adaptation (habituation) to a chronic allostatic load, i.e., brain-pull function becomes incompetent. While the brain decreases the brain-pull on the one hand, it compensatorily increases the ingestive body-pull on the other hand. In this way, cerebral energy homeostasis is maintained. However, the change of energy fluxes leads to a build-up in the supply chain, resulting in obesity and type 2 diabetes. “Push component” (blue part of the arrows); “pull-component” (yellow part of the arrows).
Habituation of the Stress-Response – Brain-Pull Incompetence
Which changes will occur under the same conditions, if the brain-pull is incompetent, i.e., that the ability of the brain to actively demand energy from the body is compromised?
We repeated the investigation of the cerebral supply chain under conditions of increased cerebral need, but this time tested its changes, when the brain-pull was incompetent. The following peripheral metabolic abnormalities were evident (Figure 5B): the brain-pull activity is not increased, but instead food intake is increased compensatory; as a result the brain energy content is found to be maintained. As an adverse event of that “comfort eating” brain-supply strategy, energy is found to accumulate in blood and in peripheral tissues as a build-up in the cerebral supply chain. Under these conditions the brain is supplied by the increased push-component from the blood, and thus the overactive brain-pull can be unburdened. It could be proven analytically that a general property is inherent in the presented cerebral supply chain: the fat compartment increases with decreasing brain-pull efficiency (Peters and Langemann, 2009). Hence the development of obesity is predicted to occur with an inadequate sympathoadrenal stress response. These observations of occurrences in the supply chain as described here are similar to those abnormalities that have been found in the subjects who participated in a large epidemiological study in Norway (Flaa et al., 2008). The intriguing paper on this large cohort by Flaa et al. (2008) demonstrated that low sympathoadrenal activity predicts body weight gain during an 18 year follow-up study. These researchers performed a mental stress test in 99 healthy men of normal weight. In the 18 year follow-up investigation, they found that a body weight gain could be predicted by a low sympathoadrenal response to the stress test at baseline. This study provides decisive support of the view that an inadequate sympathoadrenal responsiveness to a mental stressor is a crucial cause for the development and progression of obesity. On this background, the changes predicted by the supply chain model are also in agreement with those changes that have been observed in the other 38% of first-year students in the British studies, who ate more or gained weight (type B). Thus, the cerebral supply chain model provides a systemic understanding of both first-year student groups.
What are the causal factors that may reduce the responsiveness of the SNS/HPA system in people who develop obesity? Chronic stress in adult life may play a major role in adaption of the stress response. The topical paper by Block et al. (2009) provides new insights into the association between stress and long-term weight gain in a large representative cohort. The researchers used a longitudinal cohort of 1355 subjects and showed that several domains of psychosocial stress were associated with weight gain over a period of 9 years in both, women and men with higher body mass index. Among the people with high baseline body mass index weight gain was associated with increasing levels of psychosocial stress related to “job related demands” and “difficulty paying bills.” In men, additional factors such as “lack of skill discretion” (variety of work and opportunity for use of skills) and “lack of decision authority” were important; in women, “perceived constraints in life” and “strain in the relation with family” were important. Thus adaption to chronic stressors could play a role in reducing brain-pull competence and in this way promote the development of obesity.
Conclusions
The experiments presented here show how the brain under conditions of acute psychosocial stress organizes its supply and demand. To cover its increased need during stress the brain actively demands for energy by using the brain-pull mechanism referred to as CIS. In this way the brain prevents storage of energy in peripheral tissues and in so doing enhances its own glucose supply.
Referring to the above mentioned concept of allostatic load, the British first-year students may represent two distinct types with respect to their adaption to chronic stress. According to the predictions of the supply chain model, those students (who eat less and lose weight) may belong to the group of people (type A) who lack an adaptation and keep their stress response high on repeated stressful occasions. The other group of students (who eat more and gain weight) may belong to those (type B) who show adaption of their stress response, i.e., that their SNS/HPA responsiveness decreases by time. The British study, the concept of allostasis with the different types to responses to chronic stress, and the predictions of the supply chain model support the following view, which may be regarded as a solution of the apparent “stress-eating paradox”: Chronic stress leads to weight loss in those people who keep their stress response high, but to weight gain in those who habituate with their stress response to chronic stress.
Future Directions
There are still open questions with respect to the cerebral energy supply and demand in obese people: first, are they also able to exert CIS during acute psychosocial stress? And if not, are they also capable – as healthy subjects are – to enhance their need during a psychosocial challenge?
The recent research presented in this focused review supports the view that an adaption of the SNS/HPA stress response to a chronic persistent stressor involves a metabolic strategy of the brain that on the one hand relieves the overloaded brain-pull and improves mood, but on the other hand makes it necessary to increase eating behavior. Thus evidence accumulates that the stressed mind can choose a metabolic coping strategy by switching its supply mode from brain-pull to body-pull, i.e., to “comfort eating.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the German Research Foundation for the large financial support of the SELFISH BRAIN research group. We thank Sabine Wittnebel for expert assistance in the preparation of the manuscript. This work was supported by a grant from the German Research Foundation (Clinical Research Group KFO-126).
Key Concepts
The concept of “allostatic load,” which has been formulated bySterling (2004) and further developed by McEwen (2007), is based on the hypothesis that there is a cumulative physiological risk associated with exposure to psychosocial stressors over the life-course. Adaptation in the face of stressful situations and stimuli causes allostatic load and this, over time, leads to diseases.
The selfish brain theory describes the characteristic of the brain to cover its own energy requirements with the highest priority when regulating energy fluxes in the body. The brain behaves selfishly in this respect. The selfish brain theory has been founded and formulated mathematically between 1998 and 2004. Meanwhile, the interdisciplinary Selfish Brain research group, supported by the German Research Foundation, has provided experimental evidence for the validity of the theory’s axioms.
The brain regulates energy homeostasis via “brain-pull mechanisms,” which function by demanding energy from the body. Two brain-pull mechanisms have been detected so far: first, allocative brain-pull mechanisms, which activate the stress system to favor glucose allocation to the brain, and, second, direct astrocytic brain-pull mechanisms, which enhance glucose transport through the blood–brain barrier.
The body-pull functions to demand energy from the near environment by initiating ingestive behavior to replenish body stores and blood glucose concentrations. In this connection, mechanisms driven by extracellular cerebral glucose, which is closely related to blood glucose, fulfill the function of exerting ingestive body-pull.
The supply chain of the brain – with the central nervous system as the final consumer – describes the energy fluxes from the remote environment to the near environment, through the body and finally toward the brain. The brain regulates energy homeostasis via brain-pull mechanisms.
Cerebral insulin suppression (CIS)
With cerebral activation of the stress systems, energy – particularly glucose – is allocated to the brain. With activation of the sympathetic nervous system, insulin secretion from the beta cells is suppressed and the insulin-dependent glucose uptake into body periphery becomes limited. As a consequence of CIS, glucose is available via insulin-independent transport across the blood–brain barrier. In this way, CIS can be interpreted as a brain-pull mechanism.
References
Ahren, B. (2000). Autonomic regulation of islet hormone secretion – implications for health and disease. Diabetologia 43, 393–410.
Block, J. P., He, Y., Zaslavsky, A. M., Ding, L., and Ayanian, J. Z. (2009). Psychosocial stress and change in weight among US adults. Am. J. Epidemiol. 170, 181–192.
Blodgett, D. M., De Zutter, J. K., Levine, K. B., Karim, P., and Carruthers, A. (2007). Structural basis of GLUT1 inhibition by cytoplasmic ATP. J. Gen. Physiol. 130, 157–168.
Burdakov, D., Jensen, L. T., Alexopoulos, H., Williams, R. H., Fearon, I. M., O’Kelly, I., Gerasimenko, O., Fugger, L., and Verkhratsky, A. (2006). Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron 50, 711–722.
Chan, O., Lawson, M., Zhu, W., Beverly, J. L., and Sherwin, R. S. (2007). ATP-sensitive K(+) channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes 56, 1120–1126.
Cornford, E. M., and Hyman, S. (2005). Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. NeuroRx 2, 27–43.
Flaa, A., Sandvik, L., Kjeldsen, S. E., Eide, I. K., and Rostrup, M. (2008). Does sympathoadrenal activity predict changes in body fat? An 18-y follow-up study. Am. J. Clin. Nutr. 87, 1596–1601.
Fox, P. T., Raichle, M. E., Mintun, M. A., and Dence, C. (1988). Nonoxidative glucose consumption during focal physiologic neural activity. Science 241, 462–464.
Gong, Q. Y., Roberts, N., Garden, A. S., and Whitehouse, G. H. (1998). Fetal and fetal brain volume estimation in the third trimester of human pregnancy using gradient echo MR imaging. Magn. Reson. Imaging 16, 235–240.
Gonzalez, J. A., Jensen, L. T., Fugger, L., and Burdakov, D. (2008). Metabolism-independent sugar sensing in central orexin neurons. Diabetes 57, 2569–2576.
Goodman, M. N., Lowell, B., Belur, E., and Ruderman, N. B. (1984). Sites of protein conservation and loss during starvation: influence of adiposity. Am. J. Physiol. 246, E383–E390.
Harada, S., Fujita, W. H., Shichi, K., and Tokuyama, S. (2009). The development of glucose intolerance after focal cerebral ischemia participates in subsequent neuronal damage. Brain Res. 1279, 174–181.
Hill, M. N., McLaughlin, R. J., Bingham, B., Shrestha, L., Lee, T. T., Gray, J. M., Hillard, C. J., Gorzalka, B. B., and Viau, V. (2010). Endogenous cannabinoid signaling is essential for stress adaptation. Proc. Natl. Acad. Sci. U.S.A. 107, 9406–9411.
Hitze, B., Hubold, C., van Dyken, R., Schlichting, K., Lehnert, H., Entringer, S., and Peters, A. (2010). How the selfish brain organizes its ‘supply and demand’. Front. Neuroenergetics 2, 7. doi: 10.3389/fnene.2010.00007
Kacem, K., Lacombe, P., Seylaz, J., and Bonvento, G. (1998). Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia 23, 1–10.
Kind, K. L., Roberts, C. T., Sohlstrom, A. I., Katsman, A., Clifton, P. M., Robinson, J. S., and Owens, J. A. (2005). Chronic maternal feed restriction impairs growth but increases adiposity of the fetal guinea pig. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R119–R126.
Krieger, M. (1921). Über die Atrophie der menschlichen Organe bei Inanition [On the atrophy of human organs in inanition]. Z. Angew. Anat. Konstitutionsl. 7, 87–134.
Madsen, P. L., Cruz, N. F., Sokoloff, L., and Dienel, G. A. (1999). Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J. Cereb. Blood Flow Metab. 19, 393–400.
Madsen, P. L., Hasselbalch, S. G., Hagemann, L. P., Olsen, K. S., Bulow, J., Holm, S., Wildschiodtz, G., Paulson, O. B., and Lassen, N. A. (1995). Persistent resetting of the cerebral oxygen/glucose uptake ratio by brain activation: evidence obtained with the Kety-Schmidt technique. J. Cereb. Blood Flow Metab. 15, 485–491.
Magistretti, P. J., Pellerin, L., Rothman, D. L., and Shulman, R. G. (1999). Energy on demand. Science 283, 496–497.
McEwen, B. S. (1998). Protective and damaging effects of stress mediators. N. Engl. J. Med. 338, 171–179.
McEwen, B. S. (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 87, 873–904.
McPherson, R. J., Mascher-Denen, M., and Juul, S. E. (2009). Postnatal stress produces hyperglycemia in adult rats exposed to hypoxia-ischemia. Pediatr. Res. 66, 278–282.
Miki, T., Liss, B., Minami, K., Shiuchi, T., Saraya, A., Kashima, Y., Horiuchi, M., Ashcroft, F., Minokoshi, Y., Roeper, J., and Seino, S. (2001). ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat. Neurosci. 4, 507–512.
Miller, S. L., Green, L. R., Peebles, D. M., Hanson, M. A., and Blanco, C. E. (2002). Effects of chronic hypoxia and protein malnutrition on growth in the developing chick. Am. J. Obstet. Gynecol. 186, 261–267.
Muhlau, M., Gaser, C., Ilg, R., Conrad, B., Leibl, C., Cebulla, M. H., Backmund, H., Gerlinghoff, M., Lommer, P., Schnebel, A., Wohlschlager, A. M., Zimmer, C., and Nunnemann, S. (2007). Gray matter decrease of the anterior cingulate cortex in anorexia nervosa. Am. J. Psychiatry 164, 1850–1857.
Mulder, A. H., Tack, C. J., Olthaar, A. J., Smits, P., Sweep, F. C., and Bosch, R. R. (2005). Adrenergic receptor stimulation attenuates insulin-stimulated glucose uptake in 3T3-L1 adipocytes by inhibiting GLUT4 translocation. Am. J. Physiol. Endocrinol. Metab. 289, E627–E633.
Oliver, G., and Wardle, J. (1999). Perceived effects of stress on food choice. Physiol. Behav. 66, 511–515.
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R., and Magistretti, P. J. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262.
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A 91, 10625–10629.
Pellerin, L., and Magistretti, P. J. (1997). Glutamate uptake stimulates Na+,K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J. Neurochem. 69, 2132–2137.
Peters, A., Bosy-Westphal, A., Kubera, B., Langemann, D., Goele, K., Later, W., Heller, M., Hubold, C., and MÜller, M. J. (2011). Why doesn’t the brain lose weight, when obese people diet? Obes. Facts 4, 151–157.
Peters, A., and Langemann, D. (2009). Build-ups in the supply chain of the brain: on the neuroenergetic cause of obesity and type 2 diabetes mellitus. Front. Neuroenergetics 1, 2. doi: 10.3389/neuro.14.002.2009
Peters, A., Schweiger, U., Pellerin, L., Hubold, C., Oltmanns, K. M., Conrad, M., Schultes, B., Born, J., and Fehm, H. L. (2004). The selfish brain: competition for energy resources. Neurosci. Biobehav. Rev. 28, 143–180.
Rutters, F., Nieuwenhuizen, A. G., Lemmens, S. G., Born, J. M., and Westerterp-Plantenga, M. S. (2009). Acute stress-related changes in eating in the absence of hunger. Obesity (Silver Spring) 17, 72–77.
Serlachius, A., Hamer, M., and Wardle, J. (2007). Stress and weight change in university students in the United Kingdom. Physiol. Behav. 92, 548–553.
Spanswick, D., Smith, M. A., Groppi, V. E., Logan, S. D., and Ashford, M. L. (1997). Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390, 521–525.
Sterling, P. (2004). “Principle of allostasis: optimal design, predictive regulation, pathophysiology and rational therapeutics,” in Allostasis, Homeostasis, and the Costs of Adaptation, ed. J. Schulkin (Cambridge: Cambridge University Press), 17–64.
Steyer, R., Schwenkmezger, O., Notz, P., and Eid, M. (1997). Der Mehrdimensionale Befindlichkeitsfragebogen (MDBF). Göttingen: Verlag fÜr Psychologie Dr C J Hogrefe.
Tong, Q., Ye, C., McCrimmon, R. J., Dhillon, H., Choi, B., Kramer, M. D., Yu, J., Yang, Z., Christiansen, L. M., Lee, C. E., Choi, C. S., Zigman, J. M., Shulman, G. I., Sherwin, R. S., Elmquist, J. K., and Lowell, B. B. (2007). Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 5, 383–393.
Keywords: stress, eating behavior, selfish brain theory, brain-pull, cerebral insulin suppression
Citation: Peters A, Kubera B, Hubold C and Langemann D (2011) The selfish brain: stress and eating behavior. Front. Neurosci. 5:74. doi: 10.3389/fnins.2011.00074
Received: 31 January 2011;
Accepted: 09 May 2011;
Published online: 30 May 2011.
Edited by:
Ranulfo Romo, Universidad Nacional Autónoma de México, MexicoReviewed by:
Federico Bermudez-Rattoni, Universidad Nacional Autónoma de México, MexicoJose Bargas, Universidad Nacional Autónoma de México, Mexico
Copyright: © 2011 Peters, Kubera, Hubold and Langemann. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Achim Peters, University of Luebeck, Medical Clinic 1, Luebeck, Germany,YWNoaW0ucGV0ZXJzQHVrLXNoLmRl