 Alba Di Pardo
Alba Di Pardo Vittorio Maglione
Vittorio Maglione- IRCCS Neuromed, Pozzilli, Italy
Neurodegenerative diseases represent a class of fatal brain disorders for which the number of effective therapeutic options remains limited with only symptomatic treatment accessible. Multiple studies show that defects in sphingolipid pathways are shared among different brain disorders including neurodegenerative diseases and may contribute to their complex pathogenesis. In this mini review, we discuss the hypothesis that modulation of sphingolipid metabolism and their related signaling pathways may represent a potential therapeutic approach for those devastating conditions. The plausible “druggability” of sphingolipid pathways is greatly promising and represent a relevant feature that brings real advantage to the development of new therapeutic options for these conditions. Indeed, several molecules that selectively target sphingolipds are already available and many of them currently in clinical trial for human diseases. A deeper understanding of the “sphingolipid scenario” in neurodegenerative disorders would certainly enhance therapeutic perspectives for these conditions, by taking advantage from the already available molecules and by promoting the development of new ones.
Introduction
Sphingolipids have long been viewed as merely ubiquitous components of the cell membrane, and exert a critical role in regulating vital cell functions and formation of membrane microdomain “lipid rafts” for integrating cell signaling (Gault et al., 2010; Olsen and Faergeman, 2017). Sphingolipids synthesis can occur via de novo biosynthetic pathway or the hydrolysis of sphingomyelin, or can also derive by the “salvage pathway” which determines the recovery of sphingosine, by the recycling of complex sphingolipids (gangliosides) through a coordinated action of several enzymes (see Figure 1) (Gault et al., 2010).
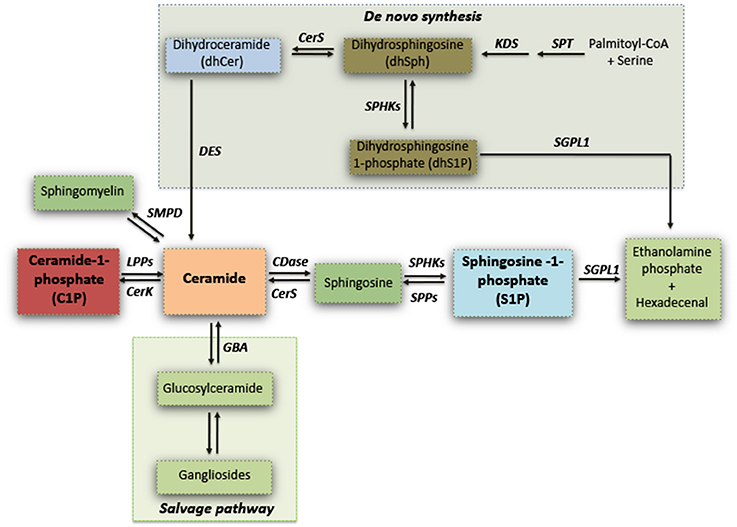
Figure 1. Simplified schematic representation of sphingolipid biosynthesis. Serine palmitoyltransferase (SPT) catalyzes the initial reaction of the de novo biosynthesis of sphingolipids. Dihydrosphingosine (dhSPH) is generated after an intermediate step by the action of 3-keto-dihydrosphingosine reductase (KDS). Successively, dhSph can be either phosphorylated, with the generation of dhSphingosine-1-phosphate by sphingosine kinases (SPHKs), or acetylated by ceramide synthase (CERS) and desaturated by ceramide desaturase (DES) to form ceramide. Ceramide may also derive from the Salvage pathway through either the hydrolysis of sphingomyelin or by the recycling of gangliosides by Sphingomyelin Phosphodiesterase (SMPD) and Glucosylceramidase (GBA) respectively. Ceramide can be phosphorylated by Ceramide kinase (CerK) with the generation of ceramide-1-phosphate (C1P) which in turn can be re-converted in ceramide by Lipid Phosphate Phosphatases (LPPs). Ceramide can be subsequently metabolized by Ceramidase (CDase) to generate sphingosine which, in turn, produces sphingosine-1-phosphate (S1P) through phosphorylation by SPHKs. S1P can be either dephosphorylated and re-converted to sphingosine by S1P Phosphatases (SPPs), or irreversible catabolized into hexadecenal + phospho-ethanolamine by S1P Lyase (SGPL1).
Enzymatic pathways result in the formation of several different and tightly regulated lipid mediators such as ceramide, ceramide-1-phosphate (C1P), sphingosine, and sphingosine-1-phosphate (S1P) (Gault et al., 2010; Goñi et al., 2014).
Ceramide is pivotal in the synthesis of sphingolipids (Mullen et al., 2012). It benefits early growth and development of neuronal cells and, at low levels it promotes cell survival and division (Schwarz and Futerman, 1997; Brann et al., 1999). Defects in its metabolism may have deleterious effects, often resulting in its abnormal accumulation that causes an increase of the programmed cell death in different cell types including neurons (Tohyama et al., 1999; Jana et al., 2009; Mullen and Obeid, 2012; Czubowicz and Strosznajder, 2014).
Direct phosphorylation of ceramide by ceramide kinase (CerK) is the major identified mechanism for generation of C1P in cells (Bajjalieh et al., 1989). The role of C1P has been mainly studied in blood cells in which it regulates proliferation, migration, and survival (Hoeferlin et al., 2013). The function of C1P in the brain is still poorly investigated. However, the presence of CerK in synaptic vesicles (Bajjalieh et al., 1989), along with the evidence that C1P regulates photoreceptor homeostasis (Miranda et al., 2011) as well as P-glycoprotein transport at the blood-brain barrier (Mesev et al., 2017), strongly suggests that C1P may be involved also in the homeostasis of the central nervous system (CNS).
Similarly, S1P is a potent signaling molecule that, beside governing essential physiological processes like vascular, bone formation (Hla et al., 2008; Xiong and Hla, 2014; Holmes, 2015) and inflammatory response (Huang et al., 2013; Aoki et al., 2016), regulates many of the molecular events crucial for brain development and neuronal survival (Mendelson et al., 2014; van Echten-Deckert et al., 2014). S1P acts either in the intracellular or in the extracellular compartments (Strub et al., 2010; Mendelson et al., 2014). Intracellularly, S1P may play different roles depending on its subcellular localization and normally regulates mitochondria function (Strub et al., 2011; Shen et al., 2014), gene expression by inhibiting histone deacetylases (HDACs) (Hait et al., 2009; Riccio, 2010) and ER stress (Lépine et al., 2011; Park et al., 2016).
Outside the cell, S1P acts as a high affinity agonist at five known G protein-coupled receptors, S1PR1 -S1PR5, which in the brain are expressed by many CNS cell types (Blaho and Hla, 2014; Martin and Sospedra, 2014) and have been shown to influence cell proliferation and migration, cell differentiation and survival as well as neurite outgrowth and neurogenesis (Toman et al., 2004; Anderson and Maes, 2014; Blaho and Hla, 2014; Martin and Sospedra, 2014; Ye et al., 2016).
Several studies showed that these sphingolipid mediators and their enzymes are likely to have an integral role in different cell processes including proliferation, inflammation, apoptosis, and migration (Zheng et al., 2006; Maceyka et al., 2012).
S1P metabolism involves a number of different highly specialized enzymes. S1P is normally synthesized by sphingosine kinase-1 and−2 (SPHK1 and 2) and degraded by sphingosine-1-phosphate phosphatase (SGPP) or lyase (SGPL1) (see Figure 1) (Le Stunff et al., 2002; Morozov et al., 2013). SPHK1 activity is mainly associated with cell survival (Le Stunff et al., 2002; Morozov et al., 2013; Di Pardo et al., 2017a), while SPHK2 is widely described as being a dual-function protein whose activity may either guarantee the proper occurrence of physiological events like mitochondrial function and homeostasis as well as regulation of gene expression through inhibition of Class I HDACs or result detrimental mainly suppressing cell growth and promoting apoptosis (Maceyka et al., 2005; Hait et al., 2009; Riccio, 2010; Gomez et al., 2011; Strub et al., 2011).
On the other hand, the degradative enzymes SGPP and SGPL1 represent key regulators for the maintenance of balanced S1P levels and other sphingolipid intermediates that may control cell growth, proliferation and death (Serra and Saba, 2010). Uncontrolled up-regulation of SGPL1 typically results in a reduction of S1P availability and accumulation of hexadecenal, a biochemical condition previously reported to be cytotoxic (Kumar et al., 2011; Maceyka et al., 2012) that may likely contribute to neurodegenerative processes.
This mini-review will shortly highlight the current knowledge of research that now supports the idea that sphingolipids are intimately involved in disease progression and, together with altered expression/activity of metabolizing enzymes and associated receptors, can provide effective drug targets for the treatment of pathological states.
Alteration of Sphingolipid Metabolism in Neurodegenerative Disorders
A fine balance between synthesis of sphingolipids and their degradation is normally required for many biological processes (Gault et al., 2010; Mullen et al., 2012), thus changes in their metabolism may profoundly affect brain homeostasis and function. Over the past few years, perturbed metabolism of the interconvertible bioactive sphingolipids, ceramide and S1P is increasingly becoming recognized as potential pathogenic factor in different neurodegenerative disorders (Table 1). Indeed, a plethora of new information identifying the importance of sphingolipids and their signaling pathways in these conditions has accumulated.
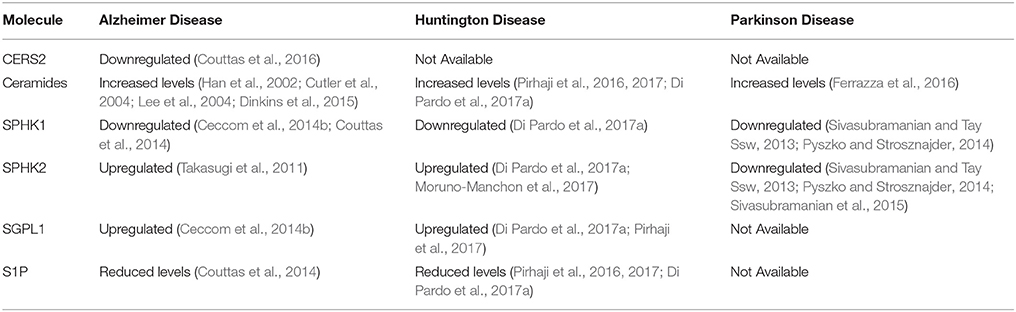
Table 1. Molecular alterations of sphingolipid metabolism detected in neurodegenerative disorders.
Alzheimer's Disease
Alzheimer's disease (AD) is the most common neurodegenerative disease and the leading cause of dementia in elderly people (Kumar et al., 2015). Progressive neurodegeneration in brain regions involved in learning and memory results in cognitive decline, loss of memory and changes of social and emotional behavior (Robins Wahlin and Byrne, 2011; Sona et al., 2013; Levenson et al., 2014). AD is characterized by extracellular accumulation of amyloid β-peptide (Aβ) toxic aggregates and intracellular deposits of abnormally phosphorylated tau protein (Blennow et al., 2006; Gouras et al., 2010).
A number of evidence demonstrates a key role of aberrant sphingolipid metabolism in the pathogenesis of the disease (Chakrabarti et al., 2016). Activity of ceramide synthetic enzyme 2 (CERS2), normally involved in the formation of long chain ceramide species (Levy and Futerman, 2010), has been recently found to be reduced in multiple brain regions of subjects at the preclinical stage of the disease (Couttas et al., 2016). Conversely, expression of genes involved in the de novo synthesis of sphingolipids is upregulated early in the disease progression (Katsel et al., 2007). Consistently, accumulation of ceramide has been reported in brain tissues from AD patients even at early stages of disease (Han et al., 2002; Cutler et al., 2004; He et al., 2010; Filippov et al., 2012) and may contribute to neurotoxic action of Aβ (Lee et al., 2004; Dinkins et al., 2015).
A number of evidence also indicates that, along with ceramide abnormalities, metabolism of other sphingolipids is affected in AD (Zheng et al., 2006; Mielke and Lyketsos, 2010). Also, alteration in the expression and/or in the activity of S1P-metabolizing enzymes as well as reduced levels of S1P has been widely reported in AD human brains (Katsel et al., 2007; Ceccom et al., 2014a,b; Couttas et al., 2014).
In particular, loss of SPHK1 and reduced bioavailability of S1P have been found early in the pathogenesis of the disease even before clinical diagnosis (Couttas et al., 2014). Conversely, upregulation of SPHK2 has been described to modulate Aβ release (Takasugi et al., 2011). Further indication of global derangement of sphingolipid metabolism in AD comes from the evidence of concomitant reduction in the levels of SPHK1 and S1P receptor 1 with enhanced expression of SGPL1 in post-mortem brains from human patients (Ceccom et al., 2014b).
Huntington's Disease
Huntington's disease (HD) is the most common dominantly inherited brain disorder, characterized by progressive striatal and cortical neurodegeneration which results in motor, cognitive and behavioral disturbance (Mccolgan and Tabrizi, 2018). The disease derives from the expansion of a polyglutamine stretch (polyQ) (> 36 repeats) in the N-terminal region of the protein huntingtin (Htt) (Jimenez-Sanchez et al., 2017). Although the function of this protein is not completely known, expansion of the polyQ stretch endows mutant Htt (mHtt) with toxic properties, resulting in the development of a number of deleterious effects in both neuronal and non-neuronal cells (Maglione et al., 2005, 2006a,b; Carroll et al., 2015; Jimenez-Sanchez et al., 2017).
Among all cellular dysfunctions and biochemical defects, classically associated with the disease, defective metabolism of sphingolipids seems to play a key role in its pathogenesis (Di Pardo et al., 2017a).
Expression of S1P-metabolizing enzymes was reported to be aberrant in multiple HD settings (Di Pardo et al., 2017a,b; Pirhaji et al., 2017). Levels of SPHK1 was found reduced in brain tissues form two fully manifest HD mouse models (R6/2 and YAC128 mice), and most importantly, in brain tissues from HD patients (Di Pardo et al., 2017a). Conversely, levels of SGPL1 were increased in the brain of multiple disease animal models and in early manifest R6/2 mice (Di Pardo et al., 2017a), indicating that defect in sphingolipid metabolism occurs early in the disease stage likely contributing to its pathogenic process. First signs of early defective sphingolipid metabolism in HD have been reported also (Di Pardo et al., 2017a; Pirhaji et al., 2017) and such further evidence corroborates the hypothesis that similar alterations may conceivably contribute to the pathogenesis of the disease.
The imbalance in S1P-metabolizing enzymes results in decreased bioavailability of S1P and increased levels of ceramide species as reported in HD models (Pirhaji et al., 2016, 2017; Di Pardo et al., 2017a).
Ultimately, synthesis of de novo sphingolipids is also affected in HD animals, even at early stage of the disease (Di Pardo et al., 2017b). This alteration determines a robust reduction of certain dihydroceramide species along with dihydrosphingosine and dihydroS1P (Di Pardo et al., 2017b).
Parkinson's Disease
Parkinson's disease (PD) is a neurodegenerative movement disorder with a prevalence of approximately 1 to 2% of the population over 65 years which increases up to 5% in people over 85 years old (Fahn, 2003; Obeso et al., 2010).
The pathological hallmarks of PD include the loss of dopaminergic neurons in the substantia nigra pars compacta and the formation of Lewy bodies mainly composed of aggregated alpha-synuclein (a-syn) protein and other components, including lipids (Gai et al., 2000; Halliday et al., 2005; Dickson et al., 2009).
Several studies demonstrated that defective ceramide metabolism may contribute to the pathogenesis of PD (Bras et al., 2008; Haughey, 2010; Fabelo et al., 2011). Mutations in the gene encoding for Glucocerebrosidase (GBA), a lysosomal enzyme converting glucosyl-ceramides into ceramide (see also Figure 1), increase the risk of developing sporadic PD (O'Regan et al., 2017).
Alterations in the expression of ceramide synthase genes as well as in the levels of certain ceramide species have been reported in post-mortem brain tissues from sporadic PD patients (Abbott et al., 2014). Consistently, mice knock out for Leucine-rich repeat kinase 2 (LRRK2), whose gene mutations cause inherited PD (Li et al., 2014), show a significant increase in the content of brain ceramide (Ferrazza et al., 2016).
Although very little, defects in S1P metabolism have been also reported in PD. Levels of SPHK1 and 2 have been described aberrant in both in-vitro and in-vivo models highlighting a potential contribution of S1P metabolism to the pathogenesis of the disease (Sivasubramanian and Tay Ssw, 2013; Sivasubramanian et al., 2015). Consistently, further studies demonstrate altered expression of S1P metabolizing enzymes with reduced activity for both SPHK1 and 2 in an in-vitro PD model (neuronal dopaminergic SH-SY5Y cells) induced by 1-methyl-4-phenylpyridinium (MPP+) (Pyszko and Strosznajder, 2014).
Sphingolipid Pathways as Target for the Development of Potential Therapeutic Interventions
The study of bioactive sphingolipids as novel therapeutic targets for the treatment of brain disorders is still relatively young, however efforts are being made to question how therapeutics may be designed to take advantage of sphingolipid signaling pathways. Over the past few years it has been established that modulation of sphingolipid metabolism can lead to improved efficacy in many brain disorders ranging from neurodevelopmental to neurodegenerative settings (Deogracias et al., 2012; Asle-Rousta et al., 2013a; Di Pardo et al., 2014; Miguez et al., 2015; Moruno Manchon et al., 2015; Pirhaji et al., 2017; Zhao et al., 2017).
Cell survival and proper functioning of neuronal circuits is critical to the effectiveness of therapies in brain disorders. Sphingolipids are key signaling molecules regulating many of these cellular processes, thus it is very likely that manipulation of sphingolipid metabolism may represent a great way to develop more effective and targeted therapeutic strategies.
Alzheimer's Disease
Very little is still known about the therapeutic effect that the modulation of sphingolipid metabolism may have in AD, however there is evidence demonstrating that stimulation of S1P receptors is beneficial in pre-clinical models of the diseases (Asle-Rousta et al., 2013a,b, 2014; Hemmati et al., 2013; Takasugi et al., 2013). Infusion of FTY720 (fingolimod), a non-selective S1P receptor modulator and FDA- approved drug for the treatment of Multiple Sclerosis, prevents spatial learning and memory impairments as well as Aβ induced-changes in the expression of some pro-apoptotic and inflammatory markers in AD animal models (Asle-Rousta et al., 2013a, 2014; Hemmati et al., 2013). Consistently, treatment with fingolimod leads to reduction of Aβ species in both in-vitro and in-vivo disease models (Takasugi et al., 2013).
Activation of specific S1P receptors also exerts beneficial effect in animal models of the disease. Indeed, administration of SEW2871, a S1PR1 selective agonist, ameliorates spatial memory impairments and attenuates the Aβ1-induced hippocampal neuronal loss in an AD rat model (Asle-Rousta et al., 2013b).
Huntington's Disease
The effective therapeutic potential of the modulation of both levels and activity of S1P-metabolizing enzyme in HD is increasingly becoming evident in the last few years (Moruno Manchon et al., 2015; Di Pardo et al., 2017a; Pirhaji et al., 2017).
Recent evidence demonstrates that SPHK1 co-localizes with autophagosomes and its over expression favors autophagy-mediated clearance of mutant Htt exon-1 construct in vitro (Moruno Manchon et al., 2015). Coherently, stimulation of SPHK1, by the selective pharmacological activator K6PC-5 (Ji et al., 2015), significantly reduces apoptosis in mouse striatal derived HD cell lines and leads to the activation of pro-survival signaling pathways (Di Pardo et al., 2017a). The potential therapeutic validity of this pharmacological approach in HD is further supported by the beneficial effects that modulation of the kinase has in human HD iPSC-derived neurons (Di Pardo et al., 2017a).
Inhibition of SPHK2 or SGPL1 also represents a possible pharmacological strategy to increase survival in HD cell models (Di Pardo et al., 2017a; Moruno-Manchon et al., 2017; Pirhaji et al., 2017). Stimulation of S1P receptors is an additional therapeutically effective approach in this context. It stimulates the production of neurotrophins, activates neuronal pro-survival pathways and ultimately delays disease progression in HD mouse models (Di Pardo et al., 2014; Miguez et al., 2015).
Parkinson's Disease
Modulation of S1P pathways has been recently explored in experimental models of PD. Pharmacological inhibition of SPHK1 reduces cell survival and increases oxygen reactive species in MPP+ human dopaminergic neuronal cells (Pyszko and Strosznajder, 2014). Conversely, treatment with S1P significantly reduced apoptosis in the same experimental model by regulating the expression of S1PR1 (Pyszko and Strosznajder, 2014).
Coherently, inhibition of SPHK2 leads to downregulation of mitochondrial-related genes such as proliferator-activated receptor γ coactivator-1α (PGC-1α) and its downstream targets nuclear respiratory factor 1 (NRF-1) and mitochondrial transcription factor A (TFAM) in multiple PD experimental models (Sivasubramanian et al., 2015). Furthermore, treatment with S1P enhance the expression of PGC-1α and NRF-1 in a mouse model of the disease and exert neuroprotective effect of dopaminergic neurons via S1PR1 (Sivasubramanian et al., 2015).
The potential role of S1PR1 stimulation in PD has been further explored. Treatment with FTY720 attenuates motor deficit and prevent dopaminergic neuronal loss in two chemical-induced PD models (Zhao et al., 2017). The neuroprotective effect of the drug is associated with the activation of pro-survival kinase ERK in both in-vitro and in-vivo PD models (Zhao et al., 2017). The benefit of FTY720 was abolished by the co-treatment with W146, a S1PR1 selective antagonist, indicating that action of fingolimod was mediated, at least in part, by its binding at S1PR1.
Concluding Remarks and Future Perspectives
In the last few years, the recognition of aberrant sphingolipid metabolism is becoming more evident in neurodegenerative disorders and, its deeper investigation may either strongly contribute to a better understanding of the disease pathogenesis or support the development of novel and more targeted therapeutic approaches.
Defects in this metabolism may profoundly affect the CNS and may interfere with selective biological pathways, whose dysregulation can explain some of the molecular and cellular alterations underlying neurodegeneration.
The hypothesis of candidating sphingolipid pathways as attractive therapeutic targets is strongly supported by evidence that demonstrates that modulation of such pathways has beneficial effects in different neurodegenerative conditions. To date, what makes sphingolipid metabolism a promising target with a real potential to be successfully translated into clinical practice of brain disorders is the evidence that some of the drugs targeting S1P and its receptors are already marketed or in advanced phases of clinical development for the treatment of human disorders (Hatoum et al., 2017; O'Sullivan and Dev, 2017). This could certainly allow to take advantage from the already available molecules and eventually to promote the development of new ones.
Takin into account the diverse functions that some sphingolipids may have, reducing ceramide accumulation and/or targeting its conversion into other sphingolipid species may represent an alternative approach for brain disorders (Brodowicz et al., 2018). In this context, the feasibility of lowering ceramide levels and its effectiveness is supported by a number of pre-clinical studies and by the evidence that some drugs that target ceramide production are currently approved for clinical use (Kornhuber et al., 2010). On the other hand, boosting ceramide metabolism toward the synthesis of C1P and/or S1P, through the use of specific kinase activators (Kwon et al., 2007; Tada et al., 2010), may also represent a fascinating approach.
Author Contributions
AD and VM co-wrote the manuscript.
Funding
This research was supported by Fondazione Neuromed and funded by Italian Ministry of Health Ricerca Corrente funding program granted to VM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, S. K., Li, H., Muñoz, S. S., Knoch, B., Batterham, M., Murphy, K. E., et al. (2014). Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson's disease. Mov. Disord. 29, 518–526. doi: 10.1002/mds.25729
Anderson, G., and Maes, M. (2014). Reconceptualizing adult neurogenesis: role for sphingosine-1-phosphate and fibroblast growth factor-1 in co-ordinating astrocyte-neuronal precursor interactions. CNS Neurol. Disord. Drug Targets 13, 126–136. doi: 10.2174/18715273113126660132
Aoki, M., Aoki, H., Ramanathan, R., Hait, N. C., and Takabe, K. (2016). Sphingosine-1-phosphate signaling in immune cells and inflammation: roles and therapeutic potential. Mediators Inflamm. 2016:8606878. doi: 10.1155/2016/8606878
Asle-Rousta, M., Kolahdooz, Z., Dargahi, L., Ahmadiani, A., and Nasoohi, S. (2014). Prominence of central sphingosine-1-phosphate receptor-1 in attenuating abeta-induced injury by fingolimod. J. Mol. Neurosci. 54, 698–703. doi: 10.1007/s12031-014-0423-3
Asle-Rousta, M., Kolahdooz, Z., Oryan, S., Ahmadiani, A., and Dargahi, L. (2013a). FTY720 (fingolimod) attenuates beta-amyloid peptide (Abeta42)-induced impairment of spatial learning and memory in rats. J. Mol. Neurosci. 50, 524–532. doi: 10.1007/s12031-013-9979-6
Asle-Rousta, M., Oryan, S., Ahmadiani, A., and Rahnema, M. (2013b). Activation of sphingosine 1-phosphate receptor-1 by SEW2871 improves cognitive function in Alzheimer's disease model rats. EXCLI J. 12, 449–461.
Bajjalieh, S. M., Martin, T. F., and Floor, E. (1989). Synaptic vesicle ceramide kinase. A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. J. Biol. Chem. 264, 14354–14360.
Blaho, V. A., and Hla, T. (2014). An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 55, 1596–1608. doi: 10.1194/jlr.R046300
Blennow, K., De Leon, M. J., and Zetterberg, H. (2006). Alzheimer's disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Brann, A. B., Scott, R., Neuberger, Y., Abulafia, D., Boldin, S., Fainzilber, M., et al. (1999). Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J. Neurosci. 19, 8199–8206.
Bras, J., Singleton, A., Cookson, M. R., and Hardy, J. (2008). Emerging pathways in genetic Parkinson's disease: potential role of ceramide metabolism in Lewy body disease. FEBS J. 275, 5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x
Brodowicz, J., Przegalinski, E., Müller, C. P., and Filip, M. (2018). Ceramide and its related neurochemical networks as targets for some brain disorder therapies. Neurotox. Res. 33, 474–484. doi: 10.1007/s12640-017-9798-6
Carroll, J. B., Bates, G. P., Steffan, J., Saft, C., and Tabrizi, S. J. (2015). Treating the whole body in Huntington's disease. Lancet Neurol. 14, 1135–1142. doi: 10.1016/S1474-4422(15)00177-5
Ceccom, J., Delisle, M. B., and Cuvillier, O. (2014a). [Sphingosine 1-phosphate as a biomarker for Alzheimer's disease? Med. Sci. 30, 493–495. doi: 10.1051/medsci/20143005006
Ceccom, J., Loukh, N., Lauwers-Cances, V., Touriol, C., Nicaise, Y., Gentil, C., et al. (2014b). Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer's disease. Acta Neuropathol. Commun. 2:12. doi: 10.1186/2051-5960-2-12
Chakrabarti, S. S., Bir, A., Poddar, J., Sinha, M., Ganguly, A., and Chakrabarti, S. (2016). Ceramide and sphingosine-1-phosphate in cell death pathways: relevance to the pathogenesis of Alzheimer's disease. Curr. Alzheimer Res. 13, 1232–1248. doi: 10.2174/1567205013666160603004239
Couttas, T. A., Kain, N., Daniels, B., Lim, X. Y., Shepherd, C., Kril, J., et al. (2014). Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer's disease pathogenesis. Acta Neuropathol. Commun. 2:9. doi: 10.1186/2051-5960-2-9
Couttas, T. A., Kain, N., Suchowerska, A. K., Quek, L. E., Turner, N., Fath, T., et al. (2016). Loss of ceramide synthase 2 activity, necessary for myelin biosynthesis, precedes tau pathology in the cortical pathogenesis of Alzheimer's disease. Neurobiol. Aging 43, 89–100. doi: 10.1016/j.neurobiolaging.2016.03.027
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 2070–2075. doi: 10.1073/pnas.0305799101
Czubowicz, K., and Strosznajder, R. (2014). Ceramide in the molecular mechanisms of neuronal cell death. The role of sphingosine-1-phosphate. Mol. Neurobiol. 50, 26–37. doi: 10.1007/s12035-013-8606-4
Deogracias, R., Yazdani, M., Dekkers, M. P., Guy, J., Ionescu, M. C., Vogt, K. E., et al. (2012). Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 109, 14230–14235. doi: 10.1073/pnas.1206093109
Dickson, D. W., Fujishiro, H., Orr, C., Delledonne, A., Josephs, K. A., Frigerio, R., et al. (2009). Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat. Disord. 15(Suppl. 3), S1–S5. doi: 10.1016/S1353-8020(09)70769-2
Dinkins, M. B., Dasgupta, S., Wang, G., Zhu, G., He, Q., Kong, J. N., et al. (2015). The 5XFAD mouse model of Alzheimer's disease exhibits an age-dependent increase in anti-ceramide IgG and exogenous administration of ceramide further increases anti-ceramide titers and amyloid plaque burden. J. Alzheimers. Dis. 46, 55–61. doi: 10.3233/JAD-150088
Di Pardo, A., Amico, E., Basit, A., Armirotti, A., Joshi, P., Neely, D. M., et al. (2017a). Defective sphingosine-1-phosphate metabolism is a druggable target in Huntington's disease. Sci. Rep. 7:5280. doi: 10.1038/s41598-017-05709-y
Di Pardo, A., Amico, E., Favellato, M., Castrataro, R., Fucile, S., Squitieri, F., et al. (2014). FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum. Mol. Genet. 23, 2251–2265. doi: 10.1093/hmg/ddt615
Di Pardo, A., Basit, A., Armirotti, A., Amico, E., Castaldo, S., Pepe, G., et al. (2017b). De novo synthesis of sphingolipids is defective in experimental models of Huntington's Disease. Front. Neurosci. 11:698. doi: 10.3389/fnins.2017.00698
Fabelo, N., Martín, V., Santpere, G., Marin, R., Torrent, L., Ferrer, I., et al. (2011). Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson's disease and incidental Parkinson's disease. Mol. Med. 17, 1107–1118. doi: 10.2119/molmed.2011.00119
Fahn, S. (2003). Description of Parkinson's disease as a clinical syndrome. Ann. N.Y. Acad. Sci. 991, 1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x
Ferrazza, R., Cogo, S., Melrose, H., Bubacco, L., Greggio, E., Guella, G., et al. (2016). LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 478, 1141–1146. doi: 10.1016/j.bbrc.2016.08.082
Filippov, V., Song, M. A., Zhang, K., Vinters, H. V., Tung, S., Kirsch, W. M., et al. (2012). Increased ceramide in brains with Alzheimer's and other neurodegenerative diseases. J. Alzheimers. Dis. 29, 537–547. doi: 10.3233/JAD-2011-111202
Gai, W. P., Yuan, H. X., Li, X. Q., Power, J. T., Blumbergs, P. C., and Jensen, P. H. (2000). In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in lewy bodies. Exp. Neurol. 166, 324–333. doi: 10.1006/exnr.2000.7527
Gault, C. R., Obeid, L. M., and Hannun, Y. A. (2010). An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 688, 1–23. doi: 10.1007/978-1-4419-6741-1_1
Gomez, L., Paillard, M., Price, M., Chen, Q., Teixeira, G., Spiegel, S., et al. (2011). A novel role for mitochondrial sphingosine-1-phosphate produced by sphingosine kinase-2 in PTP-mediated cell survival during cardioprotection. Basic Res. Cardiol. 106, 1341–1353. doi: 10.1007/s00395-011-0223-7
Goñi, F. M., Sot, J., and Alonso, A. (2014). Biophysical properties of sphingosine, ceramides and other simple sphingolipids. Biochem. Soc. Trans. 42, 1401–1408. doi: 10.1042/BST20140159
Gouras, G. K., Tampellini, D., Takahashi, R. H., and Capetillo-Zarate, E. (2010). Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 119, 523–541. doi: 10.1007/s00401-010-0679-9
Hait, N. C., Allegood, J., Maceyka, M., Strub, G. M., Harikumar, K. B., Singh, S. K., et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. doi: 10.1126/science.1176709
Halliday, G. M., Ophof, A., Broe, M., Jensen, P. H., Kettle, E., Fedorow, H., et al. (2005). Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson's disease. Brain 128, 2654–2664. doi: 10.1093/brain/awh584
Han, X., D, M. H., Mckeel, D. W. Jr., Kelley, J., and Morris, J. C. (2002). Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J. Neurochem. 82, 809–818. doi: 10.1046/j.1471-4159.2002.00997.x
Hatoum, D., Haddadi, N., Lin, Y., Nassif, N. T., and McGowan, E. M. (2017). Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: challenges for SphK as an oncotarget. Oncotarget 8, 36898–36929. doi: 10.18632/oncotarget.16370
Haughey, N. J. (2010). Sphingolipids in neurodegeneration. Neuromole. Med. 12, 301–305. doi: 10.1007/s12017-010-8135-5
He, X., Huang, Y., Li, B., Gong, C. X., and Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol. Aging 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
Hemmati, F., Dargahi, L., Nasoohi, S., Omidbakhsh, R., Mohamed, Z., Chik, Z., et al. (2013). Neurorestorative effect of FTY720 in a rat model of Alzheimer's disease: comparison with memantine. Behav. Brain Res. 252, 415–421. doi: 10.1016/j.bbr.2013.06.016
Hla, T., Venkataraman, K., and Michaud, J. (2008). The vascular S1P gradient-cellular sources and biological significance. Biochim. Biophys. Acta 1781, 477–482. doi: 10.1016/j.bbalip.2008.07.003
Hoeferlin, L. A., Wijesinghe, D. S., and Chalfant, C. E. (2013). The role of ceramide-1-phosphate in biological functions. Handb. Exp. Pharmacol. 215, 153–166. doi: 10.1007/978-3-7091-1368-4_8
Holmes, D. (2015). Bone: key role for S1P in bone remodelling. Nat. Rev. Endocrinol. 11:3. doi: 10.1038/nrendo.2014.190
Huang, W. C., Nagahashi, M., Terracina, K. P., and Takabe, K. (2013). Emerging role of Sphingosine-1-phosphate in inflammation, cancer, and lymphangiogenesis. Biomolecules 3, 408–434. doi: 10.3390/biom3030408
Jana, A., Hogan, E. L., and Pahan, K. (2009). Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J. Neurol. Sci. 278, 5–15. doi: 10.1016/j.jns.2008.12.010
Ji, F., Mao, L., Liu, Y., Cao, X., Xie, Y., Wang, S., et al. (2015). K6PC-5, a novel sphingosine kinase 1 (SphK1) activator, alleviates dexamethasone-induced damages to osteoblasts through activating SphK1-Akt signaling. Biochem. Biophys. Res. Commun. 458, 568–575. doi: 10.1016/j.bbrc.2015.02.007
Jimenez-Sanchez, M., Licitra, F., Underwood, B. R., and Rubinsztein, D. C. (2017). Huntington's disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 7:a024240. doi: 10.1101/cshperspect.a024240
Katsel, P., Li, C., and Haroutunian, V. (2007). Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer's disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer's disease? Neurochem. Res. 32, 845–856. doi: 10.1007/s11064-007-9297-x
Kornhuber, J., Tripal, P., Reichel, M., Mühle, C., Rhein, C., Muehlbacher, M., et al. (2010). Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 26, 9–20. doi: 10.1159/000315101
Kumar, A., Byun, H. S., Bittman, R., and Saba, J. D. (2011). The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell. Signal. 23, 1144–1152. doi: 10.1016/j.cellsig.2011.02.009
Kumar, A., Singh, A., and Ekavali (2015). A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol. Rep. 67, 195–203. doi: 10.1016/j.pharep.2014.09.004
Kwon, Y. B., Kim, C. D., Youm, J. K., Gwak, H. S., Park, B. D., Lee, S. H., et al. (2007). Novel synthetic ceramide derivatives increase intracellular calcium levels and promote epidermal keratinocyte differentiation. J. Lipid Res. 48, 1936–1943. doi: 10.1194/jlr.M700185-JLR200
Lee, J. T., Xu, J., Lee, J. M., Ku, G., Han, X., Yang, D. I., et al. (2004). Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 164, 123–131. doi: 10.1083/jcb.200307017
Lépine, S., Allegood, J. C., Park, M., Dent, P., Milstien, S., and Spiegel, S. (2011). Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 18, 350–361. doi: 10.1038/cdd.2010.104
Le Stunff, H., Peterson, C., Liu, H., Milstien, S., and Spiegel, S. (2002). Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim. Biophys. Acta 1582, 8–17. doi: 10.1016/S1388-1981(02)00132-4
Levenson, R. W., Sturm, V. E., and Haase, C. M. (2014). Emotional and behavioral symptoms in neurodegenerative disease: a model for studying the neural bases of psychopathology. Annu. Rev. Clin. Psychol. 10, 581–606. doi: 10.1146/annurev-clinpsy-032813-153653
Levy, M., and Futerman, A. H. (2010). Mammalian ceramide synthases. IUBMB Life 62, 347–356. doi: 10.1002/iub.319
Li, J. Q., Tan, L., and Yu, J. T. (2014). The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 9:47. doi: 10.1186/1750-1326-9-47
Maceyka, M., Harikumar, K. B., Milstien, S., and Spiegel, S. (2012). Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 22, 50–60. doi: 10.1016/j.tcb.2011.09.003
Maceyka, M., Sankala, H., Hait, N. C., Le Stunff, H., Liu, H., Toman, R., et al. (2005). SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 280, 37118–37129. doi: 10.1074/jbc.M502207200
Maglione, V., Cannella, M., Gradini, R., Cislaghi, G., and Squitieri, F. (2006a). Huntingtin fragmentation and increased caspase 3, 8 and 9 activities in lymphoblasts with heterozygous and homozygous Huntington's disease mutation. Mech. Ageing Dev. 127, 213–216. doi: 10.1016/j.mad.2005.09.011
Maglione, V., Cannella, M., Martino, T., De Blasi, A., Frati, L., and Squitieri, F. (2006b). The platelet maximum number of A2A-receptor binding sites (Bmax) linearly correlates with age at onset and CAG repeat expansion in Huntington's disease patients with predominant chorea. Neurosci. Lett. 393, 27–30. doi: 10.1016/j.neulet.2005.09.037
Maglione, V., Giallonardo, P., Cannella, M., Martino, T., Frati, L., and Squitieri, F. (2005). Adenosine A2A receptor dysfunction correlates with age at onset anticipation in blood platelets of subjects with Huntington's disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 139B, 101–105. doi: 10.1002/ajmg.b.30223
Martin, R., and Sospedra, M. (2014). Sphingosine-1 phosphate and central nervous system. Curr. Top. Microbiol. Immunol. 378, 149–170. doi: 10.1007/978-3-319-05879-5_7
Mccolgan, P., and Tabrizi, S. J. (2018). Huntington's disease: a clinical review. Eur. J. Neurol. 25, 24–34. doi: 10.1111/ene.13413
Mendelson, K., Evans, T., and Hla, T. (2014). Sphingosine 1-phosphate signalling. Development 141, 5–9. doi: 10.1242/dev.094805
Mesev, E. V., Miller, D. S., and Cannon, R. E. (2017). Ceramide 1-phosphate increases P-Glycoprotein transport activity at the blood-brain barrier via prostaglandin E2 signaling. Mol. Pharmacol. 91, 373–382. doi: 10.1124/mol.116.107169
Mielke, M. M., and Lyketsos, C. G. (2010). Alterations of the sphingolipid pathway in Alzheimer's disease: new biomarkers and treatment targets? Neuromolecular Med. 12, 331–340. doi: 10.1007/s12017-010-8121-y
Miguez, A., García-Díaz, G., Brito, V., Straccia, M., Giralt, A., Ginés, S., et al. (2015). Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington's disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 24, 4958–4970. doi: 10.1093/hmg/ddv218
Miranda, G. E., Abrahan, C. E., Agnolazza, D. L., Politi, L. E., and Rotstein, N. P. (2011). Ceramide-1-phosphate, a new mediator of development and survival in retina photoreceptors. Invest. Ophthalmol. Vis. Sci. 52, 6580–6588. doi: 10.1167/iovs.10-7065
Morozov, V. I., Sakuta, G. A., and Kalinski, M. I. (2013). Sphingosine-1-phosphate: distribution, metabolism and role in the regulation of cellular functions. Ukr. Biokhim. Zh. 85, 5–21. doi: 10.15407/ubj85.01.005
Moruno-Manchon, J. F., Uzor, N. E., Blasco-Conesa, M. P., Mannuru, S., Putluri, N., Furr-Stimming, E. E., et al. (2017). Inhibiting sphingosine kinase 2 mitigates mutant Huntingtin-induced neurodegeneration in neuron models of Huntington disease. Hum. Mol. Genet. 26, 1305–1317. doi: 10.1093/hmg/ddx046
Moruno Manchon, J. F., Uzor, N. E., Dabaghian, Y., Furr-Stimming, E. E., Finkbeiner, S., and Tsvetkov, A. S. (2015). Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci. Rep. 5:15213. doi: 10.1038/srep15213
Mullen, T. D., Hannun, Y. A., and Obeid, L. M. (2012). Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 441, 789–802. doi: 10.1042/BJ20111626
Mullen, T. D., and Obeid, L. M. (2012). Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer. Agents Med. Chem. 12, 340–363. doi: 10.2174/187152012800228661
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson's disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
Olsen, A. S. B., and Færgeman, N. J. (2017). Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol. 7:170069. doi: 10.1098/rsob.170069
O'Regan, G., Desouza, R. M., Balestrino, R., and Schapira, A. H. (2017). Glucocerebrosidase mutations in Parkinson disease. J. Parkinsons. Dis. 7, 411–422. doi: 10.3233/JPD-171092
O'Sullivan, S., and Dev, K. K. (2017). Sphingosine-1-phosphate receptor therapies: advances in clinical trials for CNS-related diseases. Neuropharmacology 113, 597–607. doi: 10.1016/j.neuropharm.2016.11.006
Park, K., Ikushiro, H., Seo, H. S., Shin, K. O., Kim, Y. I., Kim, J. Y., et al. (2016). ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc. Natl. Acad. Sci. U.S.A. 113, E1334–E1342. doi: 10.1073/pnas.1504555113
Pirhaji, L., Milani, P., Dalin, S., Wassie, B. T., Dunn, D. E., Fenster, R. J., et al. (2017). Identifying therapeutic targets by combining transcriptional data with ordinal clinical measurements. Nat. Commun. 8:623. doi: 10.1038/s41467-017-00353-6
Pirhaji, L., Milani, P., Leidl, M., Curran, T., Avila-Pacheco, J., Clish, C. B., et al. (2016). Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 13, 770–776. doi: 10.1038/nmeth.3940
Pyszko, J. A., and Strosznajder, J. B. (2014). The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson's disease. Folia Neuropathol. 52, 260–269. doi: 10.5114/fn.2014.45567
Riccio, A. (2010). New endogenous regulators of class I histone deacetylases. Sci. Signal. 3:pe1. doi: 10.1126/scisignal.3103pe1
Robins Wahlin, T. B., and Byrne, G. J. (2011). Personality changes in Alzheimer's disease: a systematic review. Int. J. Geriatr. Psychiatry 26, 1019–1029. doi: 10.1002/gps.2655
Schwarz, A., and Futerman, A. H. (1997). Distinct roles for ceramide and glucosylceramide at different stages of neuronal growth. J. Neurosci. 17, 2929–2938. doi: 10.1523/JNEUROSCI.17-09-02929.1997
Serra, M., and Saba, J. D. (2010). Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzyme Regul. 50, 349–362. doi: 10.1016/j.advenzreg.2009.10.024
Shen, Z., Liu, C., Liu, P., Zhao, J., and Xu, W. (2014). Sphingosine 1-phosphate (S1P) promotes mitochondrial biogenesis in Hep G2 cells by activating Peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha). Cell Stress Chaperones 19, 541–548. doi: 10.1007/s12192-013-0480-5
Sivasubramanian, M., and Tay Ssw, D. S. (2013). Alteration in the sphingolipid metabolism leads to activation of the apoptotic cascade in the MPTP induced mouse model of Parkinson's disease FASEB J. 27, 533–538.
Sivasubramanian, M., Kanagaraj, N., Dheen, S. T., and Tay, S. S. (2015). Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson's disease and in MPP+ -treated MN9D cells in vitro. Neuroscience 290, 636–648. doi: 10.1016/j.neuroscience.2015.01.032
Sona, A., Ellis, K. A., and Ames, D. (2013). Rapid cognitive decline in Alzheimer's disease: a literature review. Int. Rev. Psychiatry 25, 650–658. doi: 10.3109/09540261.2013.859128
Strub, G. M., Maceyka, M., Hait, N. C., Milstien, S., and Spiegel, S. (2010). Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 688, 141–155. doi: 10.1007/978-1-4419-6741-1_10
Strub, G. M., Paillard, M., Liang, J., Gomez, L., Allegood, J. C., Hait, N. C., et al. (2011). Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612. doi: 10.1096/fj.10-167502
Tada, E., Toyomura, K., Nakamura, H., Sasaki, H., Saito, T., Kaneko, M., et al. (2010). Activation of ceramidase and ceramide kinase by vanadate via a tyrosine kinase-mediated pathway. J. Pharmacol. Sci. 114, 420–432. doi: 10.1254/jphs.10181FP
Takasugi, N., Sasaki, T., Ebinuma, I., Osawa, S., Isshiki, H., Takeo, K., et al. (2013). FTY720/fingolimod, a sphingosine analogue, reduces amyloid-beta production in neurons. PLoS ONE 8:e64050. doi: 10.1371/journal.pone.0064050
Takasugi, N., Sasaki, T., Suzuki, K., Osawa, S., Isshiki, H., Hori, Y., et al. (2011). BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 31, 6850–6857. doi: 10.1523/JNEUROSCI.6467-10.2011
Tohyama, J., Oya, Y., Ezoe, T., Vanier, M. T., Nakayasu, H., Fujita, N., et al. (1999). Ceramide accumulation is associated with increased apoptotic cell death in cultured fibroblasts of sphingolipid activator protein-deficient mouse but not in fibroblasts of patients with Farber disease. J. Inherit. Metab. Dis. 22, 649–662. doi: 10.1023/A:1005590316064
Toman, R. E., Payne, S. G., Watterson, K. R., Maceyka, M., Lee, N. H., Milstien, S., et al. (2004). Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J. Cell Biol. 166, 381–392. doi: 10.1083/jcb.200402016
van Echten-Deckert, G., Hagen-Euteneuer, N., Karaca, I., and Walter, J. (2014). Sphingosine-1-phosphate: boon and bane for the brain. Cell. Physiol. Biochem. 34, 148–157. doi: 10.1159/000362991
Xiong, Y., and Hla, T. (2014). S1P control of endothelial integrity. Curr. Top. Microbiol. Immunol. 378, 85–105. doi: 10.1007/978-3-319-05879-5_4
Ye, Y., Zhao, Z., Xu, H., Zhang, X., Su, X., Yang, Y., et al. (2016). Activation of sphingosine 1-phosphate receptor 1 enhances hippocampus neurogenesis in a rat model of Traumatic brain injury: an involvement of MEK/Erk signaling pathway. Neural Plast. 2016:8072156. doi: 10.1155/2016/8072156
Zhao, P., Yang, X., Yang, L., Li, M., Wood, K., Liu, Q., et al. (2017). Neuroprotective effects of fingolimod in mouse models of Parkinson's disease. FASEB J. 31, 172–179. doi: 10.1096/fj.201600751R
Zheng, W., Kollmeyer, J., Symolon, H., Momin, A., Munter, E., Wang, E., et al. (2006). Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 1758, 1864–1884. doi: 10.1016/j.bbamem.2006.08.009
Keywords: neurodegenerative diseases, sphingolipid metabolism, S1P, ceramide, FTY720, S1PRs
Citation: Di Pardo A and Maglione V (2018) Sphingolipid Metabolism: A New Therapeutic Opportunity for Brain Degenerative Disorders. Front. Neurosci. 12:249. doi: 10.3389/fnins.2018.00249
Received: 29 October 2017; Accepted: 29 March 2018;
Published: 17 April 2018.
Edited by:
Gian Carlo Bellenchi, Institute of Genetics and Biophysics (CNR), ItalyReviewed by:
Nataliya G. Kolosova, Institute of Cytology and Genetics (RAS), RussiaRiccardo Ghidoni, Università degli Studi di Milano, Italy
Copyright © 2018 Di Pardo and Maglione. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vittorio Maglione, dml0dG9yaW8ubWFnbGlvbmVAbmV1cm9tZWQuaXQ=