 Inga Bae-Gartz1
Inga Bae-Gartz1 Ruth Janoschek1
Ruth Janoschek1 Saida Breuer1Lisa Schmitz1Thorben Hoffmann1Nina Ferrari2Lena Branik1
Saida Breuer1Lisa Schmitz1Thorben Hoffmann1Nina Ferrari2Lena Branik1 Andre Oberthuer1
Andre Oberthuer1 Cora-Sophia Kloppe1Sarah Appel1Christina Vohlen1Jörg Dötsch1
Cora-Sophia Kloppe1Sarah Appel1Christina Vohlen1Jörg Dötsch1 Eva Hucklenbruch-Rother1*
Eva Hucklenbruch-Rother1*- 1Department of Pediatrics, University Hospital of Cologne, Cologne, Germany
- 2Heart Center, Cologne Center for Prevention in Childhood and Youth, University Hospital of Cologne, Cologne, Germany
Purpose: Maternal obesity has emerged as an important risk factor for the development of metabolic disorders in the offspring. The hypothalamus as the center of energy homeostasis regulation is known to function based on complex neuronal networks that evolve during fetal and early postnatal development and maintain their plasticity into adulthood. Development of hypothalamic feeding networks and their functional plasticity can be modulated by various metabolic cues, especially in early stages of development. Here, we aimed at determining the underlying molecular mechanisms that contribute to disturbed hypothalamic network formation in offspring of obese mouse dams.
Methods: Female mice were fed either a control diet (CO) or a high-fat diet (HFD) after weaning until mating and during pregnancy and gestation. Male offspring was sacrificed at postnatal day (P) 21. The hypothalamus was subjected to gene array analysis, quantitative PCR and western blot analysis.
Results: P21 HFD offspring displayed increased body weight, circulating insulin levels, and strongly increased activation of the hypothalamic insulin signaling cascade with a concomitant increase in ionized calcium binding adapter molecule 1 (IBA1) expression. At the same time, the global gene expression profile in CO and HFD offspring differed significantly. More specifically, manifest influences on several key pathways of hypothalamic neurogenesis, axogenesis, and regulation of synaptic transmission and plasticity were detectable. Target gene expression analysis revealed significantly decreased mRNA expression of several neurotrophic factors and co-factors and their receptors, accompanied by decreased activation of their respective intracellular signal transduction.
Conclusion: Taken together, these results suggest a potential role for disturbed neurotrophin signaling and thus impaired neurogenesis, axogenesis, and synaptic plasticity in the pathogenesis of the offspring’s hypothalamic feeding network dysfunction due to maternal obesity.
Introduction
The rising incidence of maternal obesity is a worldwide phenomenon and the relationship between maternal obesity and the offspring’s long-term predisposition for metabolic disorders, such as obesity and type-2-diabetes, is undeniable (Howie et al., 2009; Plagemann, 2011; O’Reilly and Reynolds, 2013). Human and rodent studies have led to the description of novel interactive pathways between the maternal environment and the fetus (Poston, 2012). In particular, there is a clear link between maternal adiposity and expression and function of important modulators of energy homeostasis in adipose tissue, liver and the brain of the offspring (Bouret, 2009; Catalano et al., 2009; McCurdy et al., 2009; Li et al., 2011; Maric-Bilkan et al., 2011; Rooney and Ozanne, 2011; Zhang et al., 2011).
The central control of energy homeostasis is a complex process regulated by a collection of neurons primarily located in the mediobasal hypothalamus. Specifically, the melanocortin system of the arcuate nucleus of the hypothalamus (ARC) has been shown to convey anorexigenic and orexigenic signals in response to circulating hormones and nutrients (Elias et al., 1999; Cowley et al., 2001; Aponte et al., 2011; Krashes et al., 2011). While early studies in rodents focused on analyzing the effects of hormones or nutrients on anorexigenic or orexigenic neuropeptide gene expression in the ARC, research interest shifted toward a deeper functional understanding of acute effects of substances like insulin, leptin, ghrelin, or glucose on electrophysiological properties of neuropeptide releasing ARC producing cells (Cowley et al., 2001, 2003a,b; Jobst et al., 2004; Pinto et al., 2004; van den Top et al., 2004; Dhillon et al., 2006; Konner et al., 2007). A number of studies described plasticity of synaptic input patterns as an important component of such changes in neuronal activity (Horvath and Diano, 2004; Pinto et al., 2004; Horvath and Gao, 2005; Sternson et al., 2005; Gao et al., 2007; Benani et al., 2012). For instance, leptin was shown to have a major influence on synaptic organization of orexin neurons in the lateral hypothalamus (Horvath and Gao, 2005), whereas ghrelin was found to impact synapse formation in various extra-hypothalamic brain regions (Abizaid et al., 2006; Diano et al., 2006). Furthermore, high-fat feeding in rodents was also linked to changes in the synaptic input pattern of hypothalamic feeding neurons (Benani et al., 2012), and it was found that rats prone to diet-induced obesity display a completely different pattern of synaptic inputs in the ARC compared to diet-resistant littermates before even being exposed to high-fat feeding (Horvath et al., 2010). In conclusion, there is accumulating evidence that synaptic plasticity induced by metabolic cues plays a pivotal role in determining the function of hypothalamic neuronal circuits regulating appetite.
Neurotrophins are a family of ubiquitously expressed growth factors controlling development, survival, and function of neurons (Hempstead, 2006; Reichardt, 2006) that consist of six members: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT3), neurotrophin 4/5 (NT4/5), ciliary neurotrophic factor (CNTF), and the glia cell derived neurotrophic factor (GDNF) group of ligands. Upon secretion, which is usually triggered by membrane depolarization (Blochl and Thoenen, 1996; Lessmann et al., 2003), they are capable of mediating many activity-dependent central processes, including neuronal differentiation and growth, synapse formation and plasticity of synaptic input patterns (Park and Poo, 2013). There are two distinct classes of neurotrophin receptors (NTR) that can bind to neurotrophins: p75NTR, which binds to all neurotrophins, and various subtypes of tropomyosin receptor kinase (Trk) receptors, which are each specific for different neurotrophins. Among the neurotrophins, BDNF has gained much attention in the context of energy homeostasis regulation – not for its well described positive effects on synaptogenesis and neuronal plasticity (Unger et al., 2007), but its anorexigenic effect (Rask-Andersen et al., 2011). In humans, a polymorphism in the BDNF gene has been linked to obesity (Hotta et al., 2009; Thorleifsson et al., 2009). Furthermore, BDNF-deficient mice exhibit obesity due to overfeeding (Kernie et al., 2000), a phenotype that is mirrored in mice deficient for the high-affinity BDNF receptor TrkB (Xu et al., 2003). However, to what extent BDNF’s anorexigenic effects are mediated by its action on synaptic plasticity or neuronal differentiation remains to be determined. Given the fact that an increasing number of studies indicate that synaptic plasticity in the hypothalamus plays a crucial role in the control of energy homeostasis (Pinto et al., 2004; Yang et al., 2011; Liu et al., 2012), it is very likely that BDNF and other less well characterized neurotrophins might influence hypothalamic feeding network formation and function (Vanevski and Xu, 2013).
In the context of maternal obesity and the offspring’s predisposition to obesity and diabetes in later life, the hypothalamus has emerged as a particularly vulnerable organ (Bouret, 2009). Hypothalamic dysfunction in the offspring of overfed or obese mothers has been suggested by several animal studies, predominantly showing the effect of maternal adiposity and diet on hypothalamic neuropeptide and hormone receptor expression or, more recently, reactive inflammation-associated hypothalamic neuronal dysfunction (Cottrell and Ozanne, 2007; Cottrell et al., 2009; Alfaradhi and Ozanne, 2011; Poston, 2012; Rother et al., 2012; Velloso, 2012). Furthermore, a number of studies suggested stable wiring of hypothalamic feeding networks as an important pathomechanism in hypothalamic dysfunction resulting from increased circulating hormone levels in maternal obesity (Bouret et al., 2004a, b; Pinto et al., 2004; Bouret and Simerly, 2006; Bouret, 2009). Especially, insulin and leptin have been shown to permanently change hypothalamic network formation during the critical time window of perinatal development (Bouret and Simerly, 2006; Ozanne, 2011). However, the role of neurotrophins as a potential link between a metabolically disturbed intrauterine milieu and altered formation of hypothalamic neuronal networks responsible for the regulation of feeding behavior and energy balance has not been addressed to date.
Materials and Methods
Animal Care
All animal procedures were conducted in compliance with protocols approved by the Committee on the Ethics of Animal Experiments of the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (Permit number: 37.09.292, RDA number: 84-02.04.2014.A057) and were in accordance with National Institutes of Health guidelines. Mice (C57BL/6N) were bred locally at a designated animal unit of the University Hospital of Cologne (Cologne, Germany). All animals were housed individually and were maintained at 22°C on a 12 h light, 12 h dark cycle. As bedding, spruce granulate (Lignocel FS 14; Rettenmaier & Söhne GmbH, Germany) was provided. Nestlets, mouse smart home and aspen bricks served as enrichment (Plexx B.V., The Netherlands). Animal care and use was performed by qualified individuals and supervised by a veterinarian. The manuscript complies with the Animals in Research: Reporting in vivo Experiments (ARRIVE) guidelines (Kilkenny et al., 2010).
Three weeks old female mice were fed a standard chow (#.R/M-H SSniff, Soest containing 41.2% carbohydrates, 19% protein, and 3.3% fat; 9% of calories from fat) or high-fat diet (#C1057 modified containing 26.9% carbohydrates, 21% protein, and 35.1% fat; 60% of calories from fat) for 9–10 weeks preconception and during gestation and lactation, yielding sedentary control (CO; n = 20) and high-fat diet (HFD; n = 21) group. Male breeders were standard chow-fed and had access to the HFD during mating process (48 h). Male breeders that were in contact with HFD for two times were excluded for further mating. Water and food were available ad libitum. Body weight of the dams was monitored daily from G0 to G18. Blood samples for serum analyses were collected 1 week before mating (named G0) and at G15. In order to minimize stress-induced side effects, dams and offspring were not fasted and were collected between 9 and 10o’clock in the morning. After delivery, body weight of the pups was monitored daily starting within 24 h after birth. On P2, litter size was adjusted to six for each litter. Smaller litters were excluded from the experiment (CO n = 2, HFD n = 2). On P21, male pups were sacrificed non-fasted and blood samples were collected via intracardial puncture for further analyses. Organs were excised. The hypothalamus was cut immediately caudal to the optic chiasm. The dissection was limited laterally by the hypothalamic sulci and dorsally by the mammillothalamic tract (Rother et al., 2012). The remaining brain tissue was also preserved, both epigonadal fat pads were harvested, the weight was determined and all tissues were immediately frozen at −80°C for further analysis. A maximum of two offspring per dam were analyzed to minimize litter-dependent bias. All studies were performed using male offspring. Cohorts used for this study represent a subset of animals that were also used in another study by our group, see Bae-Gartz et al. (2016).
Analytical Procedures
Serum levels of insulin and leptin were measured by ELISA using mouse standards according to the manufacturer’s guidelines (mouse insulin ELISA (EZRMI-13K and EZML-82K) with a sensitivity of 0.01 ng/ml and intra-assay variation of 1.06%; Millipore CorpBillerica, MA, United States).
Intraperitoneal Glucose Tolerance Test
Glucose tolerance tests (GTT) were performed as previously described (Bae-Gartz et al., 2016). Briefly, animals were fasted for 16 h (18:00–10:00 h). After determination of fasted blood glucose levels, each animal received an intraperitoneal (ip) injection of 20% glucose (10 ml/kg body weight = 2 g glucose/kg body weight). Blood glucose levels were measured after 15, 30, 60, and 120 min.
Quantitative PCR
Total RNA was isolated from hypothalamus of CO and HFD animals using TRI-Reagent® (Sigma-Aldrich) according to the manufacturer’s guidelines. RNA quantity and purity were determined by measuring UV absorption with a Tecan spectrophotometer (Tecan, Nano Quant infinite M200 Pro). Quantitative changes in mRNA expression for genes encoding Bdnf, Ngf, Nt3, Nt4/5, Cntf, Gdnf, TrkA, TrkB, TrkC, p75Ntr, Prr7, Arc, Egr, Syt1, Homer were assessed by quantitative real-time PCR as described previously using the 7500 Real-time PCR system (Applied Biosystem, Foster City, CA, United States) or the IQ TM SYBR-Green© Supermix and a BioRad iQ5-Cycler (Bio-Rad Laboratories, Hercules, CA, United States) (Plank et al., 2006; Rother et al., 2012). In all samples, the relative amount of specific mRNA was normalized to at least four ubiquitously expressed housekeeping genes (β-Actin, hypoxanthin-guanin-phosphoribosyltransferase (HPRT), glycerinaldehyd-3-phosphat-dehydrogenase (GAPDH), and β-glucuronidase (GUSB). Oligonucleotides were designed with Primer Express software (Perkin-Elmer, Foster City, CA, United States). Primer pairs and Taqman probes are listed in Supplementary Table 1.
Microarray Analyses of the Hypothalamus
Microarray experiments of mice hypothalamic RNA were performed as single-color experiments using 4_44K Mouse (v2) Whole Genome Arrays from Agilent Technologies (Santa Clara, CA, United States) as described before (Rother et al., 2012). First, differentially expressed genes between the CO group and HFD group, were identified using the Rank Product test (Breitling et al., 2004). Genes were called significant with a percentage false positive below 0.001. All statistical calculations were performed using R version 3.1.2 and Bioconductor. Functional Annotation Clustering was performed by DAVID Bioinformatics Resources1.
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE1358302.
Western Blot Analysis
Frozen dissected hypothalamic tissue of CO and HFD offspring was homogenized in lysis buffer as previously described (Alejandre-Alcazar et al., 2007). Protein concentration was determined with a BCA-Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States). Lysates resolved on a 10% reducing SDS-PAGE gel were transferred to a nitrocellulose membrane. Blots were probed with the following antibodies, see Supplemental Table 2. Monoclonal mouse anti-mouse-β-Actin (Cell Signaling, # 3700, 1:1000) and anti-mouse GAPDH (Cell Signaling, # 2118, 1:1000) served as a loading control. Anti-mouse IgG, horseradish peroxidase (HRP)-linked (Cell Signaling, # 7076, 1:2000), and anti-rabbit IgG, HRP-linked (Cell Signaling, # 7074, 1:2000) were used as secondary antibodies.
Statistical Analysis
Values are shown as means ± standard error of the mean (SEM). The results of realtime RT-PCR were calculated based on the ΔΔCt– method and expressed as fold induction of mRNA expression compared to the corresponding control group (1.0-fold induction). Densitometric analysis of protein bands was performed using Bio-Rad ImageLab software (Bio-Rad, Munich, Germany). Two tailed Mann-Whitney-test was used to test significance of differences between HFD and CO animals at given time points. A p-value less than 0.05 was considered significant. The calculations were performed according to a previous agreement with the Institute of Medical Statistics, Informatics and Epidemiology, University of Cologne.
Results
Metabolic Phenotype
To assess the metabolic consequences of maternal diet-induced obesity in the offspring, we first determined body weight gain and non-fasted insulin and leptin levels of the dams. Before mating, females on HFD were markedly heavier than controls (Supplemental Figure 1A). Maternal diet affected the body weight gain of the dams but at G18 the absolute body weight was similar in both groups (Supplementary Figure 1A). Serum insulin levels did not differ on a significant level between CO and HFD dams, but there was a strong insulin increase between G0 and G15 in HFD dams (Supplementary Figure 1B). Serum leptin levels revealed a marked increase in HFD dams at G0 and G15 (Supplementary Figure 1C). Litter size and sex ratio did not differ between groups (Supplementary Figures 1D,E). Portions of dams and offspring characteristics of this cohort of mice have been reported previously (Bae-Gartz et al., 2016; Schmitz et al., 2018). Offspring of obese mouse dams displayed significantly increased body weight and epigonadal fat pad weight at P21 (Figures 1A,B). Interestingly, HFD offspring had lower body weights at P1 (Figure 1A). Additionally, HFD offspring showed significantly increased non-fasted serum insulin and leptin concentrations at P21 compared to controls (Figures 1C,D). To assess the effects of maternal obesity to the offspring’s glucose metabolism, GTTs were performed. HFD offspring showed increased blood glucose levels compared with CO offspring 15 min after glucose injection in the intraperitoneal GTT (Figure 1E). At all other time points, there were no significant differences detectable.
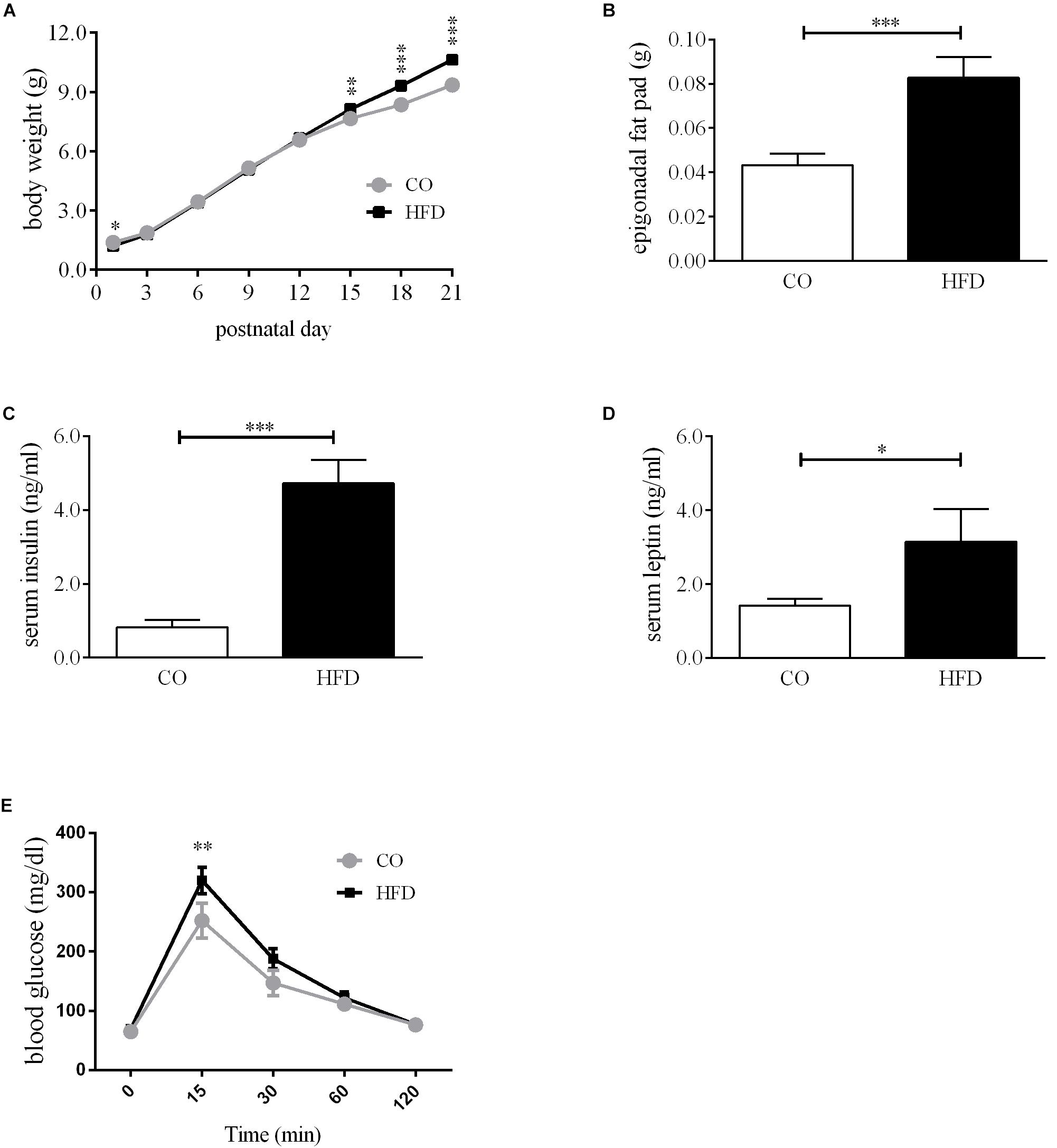
Figure 1. Offspring metabolic phenotype at P21. (A) Bodyweight gain offspring, n = 49–73. (B) Epigonadal fat pad weight, CO n = 35, HFD n = 22. (C) Serum insulin levels at P21, CO n = 8, HFD n = 6. (D) Serum leptin levels at P21, CO n = 7, HFD n = 6. (E) Intraperitoneal glucose tolerance test (GTT) at P21, CO n = 8 HFD n = 7. Data are presented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; CO, control; HFD, high fat diet; g, gram; p, postnatal day.
Hypothalamic Phenotype
To examine whether maternal obesity also affects hypothalamic insulin and leptin signal transduction, we first quantified hypothalamic insulin and leptin receptor expression. Both, insulin and leptin receptor levels were significantly increased in HFD offspring at P21 (Figures 2A,B). Furthermore, we found significantly increased levels of phosphorylated RAC-alpha serine/threonine-protein kinase (pAKT) as well as increased protein levels of glycogen synthase kinase 3ß (pGSK3ß) in HFD offspring indicating robust activation of hypothalamic PI3K signaling at P21 following maternal obesity (Figures 2C,D). Consequently, hypothalamic 5′-Adenosine monophosphate-activated protein kinase (AMPK) phosphorylation was significantly decreased in HFD offspring (Figure 2E).
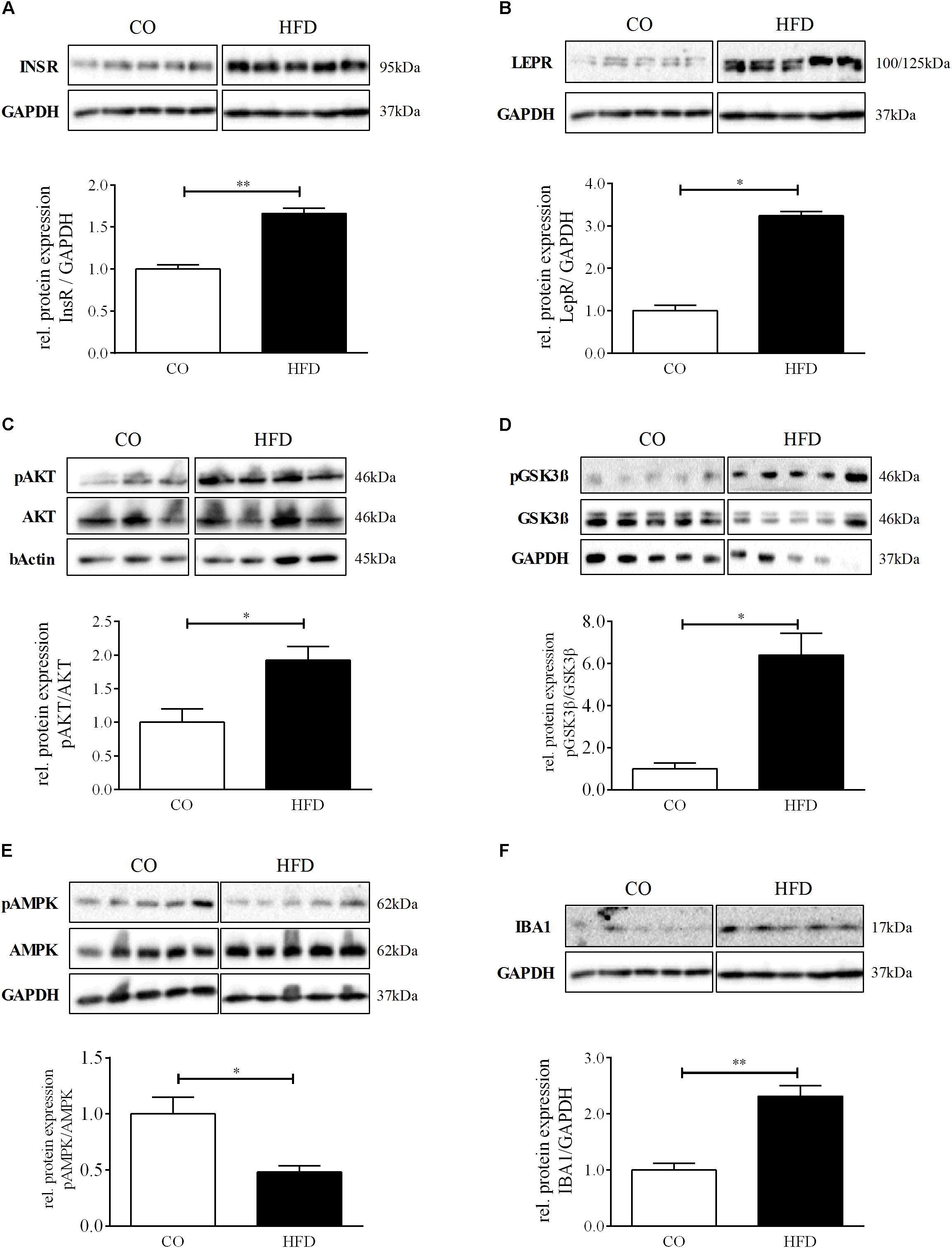
Figure 2. Hypothalamic phenotype at P21. (A–F) Representative Western Blots of INSR, LEPR, pAKT, pGSK3β, pAMPK, and IBA1 in hypothalamic tissue at P21. (A–F) Densitometric analysis of protein expression at P21: (A) INSR/GAPDH, CO n = 5, HFD n = 5. (B) LEPR/GAPDH, CO n = 5, HFD n = 5. (C) pAKT/AKT, CO n = 4, HFD n = 4. (D) pGSK3β/GSK3β, CO n = 5, HFD n = 4. (E) pAMPK/AMPK. (F) IBA1/GAPDH, CO n = 5, HFD n = 5. Data are presented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; CO, control; HFD, high fat diet.
Additionally, we assessed hypothalamic protein levels of Ionized calcium-binding adapter molecule 1 (IBA1), a marker for microglial activation and promotion of reactive gliosis, that has been associated with diet-induced obesity and hypothalamic dysfunction in mice with a just recently described role in enhancing orexigenic AgRP expression upon insulin action (Valdearcos et al., 2017; Winkler et al., 2019). Interestingly, hypothalamic IBA1 protein levels were significantly increased in HFD offspring at P21 (Figure 2F). POMC and NPY mRNA levels revealed no significant difference between CO and HFD offspring (Supplementary Figure 2).
Hypothalamic Transcriptome Analysis
To evaluate the global hypothalamic gene expression profile accompanying the observed increase in insulin and leptin signaling and microglial activation at P21, we next performed genome-wide gene expression arrays with hypothalamic tissue of CO and HFD offspring (CO n = 5, HFD n = 6). Unsupervised principal component analysis revealed that global gene expression information differed markedly between CO and HFD offspring (Figure 3). To determine the affected biological processes, we next performed Functional Annotation Clustering of the differentially expressed genes via DAVID analysis (Supplementary Tables 3, 4, gene order and probes for differentially expressed genes of hypothalamic tissue at P21 upregulated and downregulated). Interestingly, numerous GO-Terms and KEGG-Pathways related to the field of synaptic plasticity and neuron development ranged among the most prominently enriched functionally related gene groups when comparing HFD and CO offspring (Table 1).
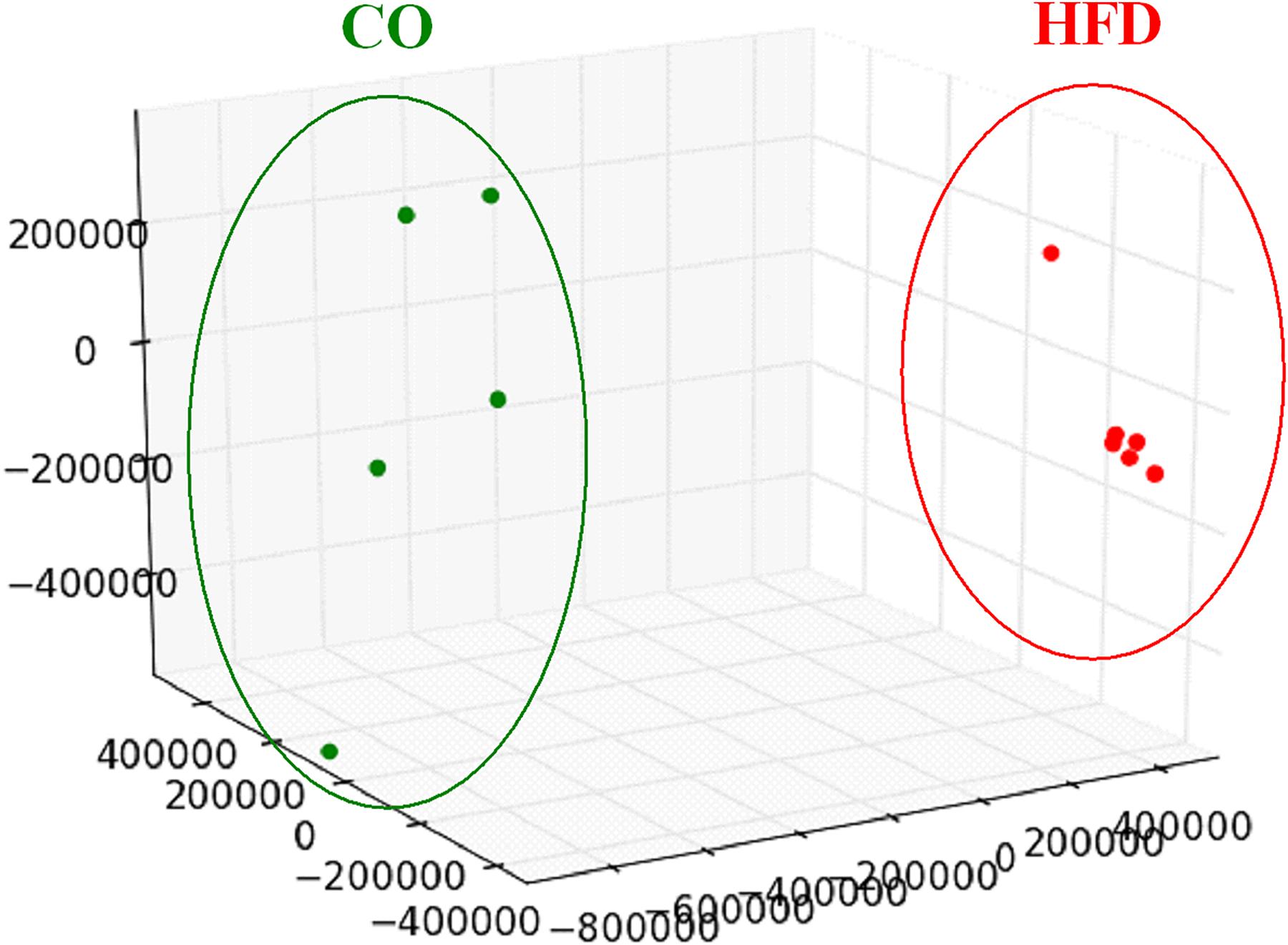
Figure 3. Hypothalamic transcriptome analysis at P21. Three-dimensional view of a principal component analysis of hypothalamic tissue performed with global gene expression information on male CO (green dots) and HFD (red dots) offspring at P21.
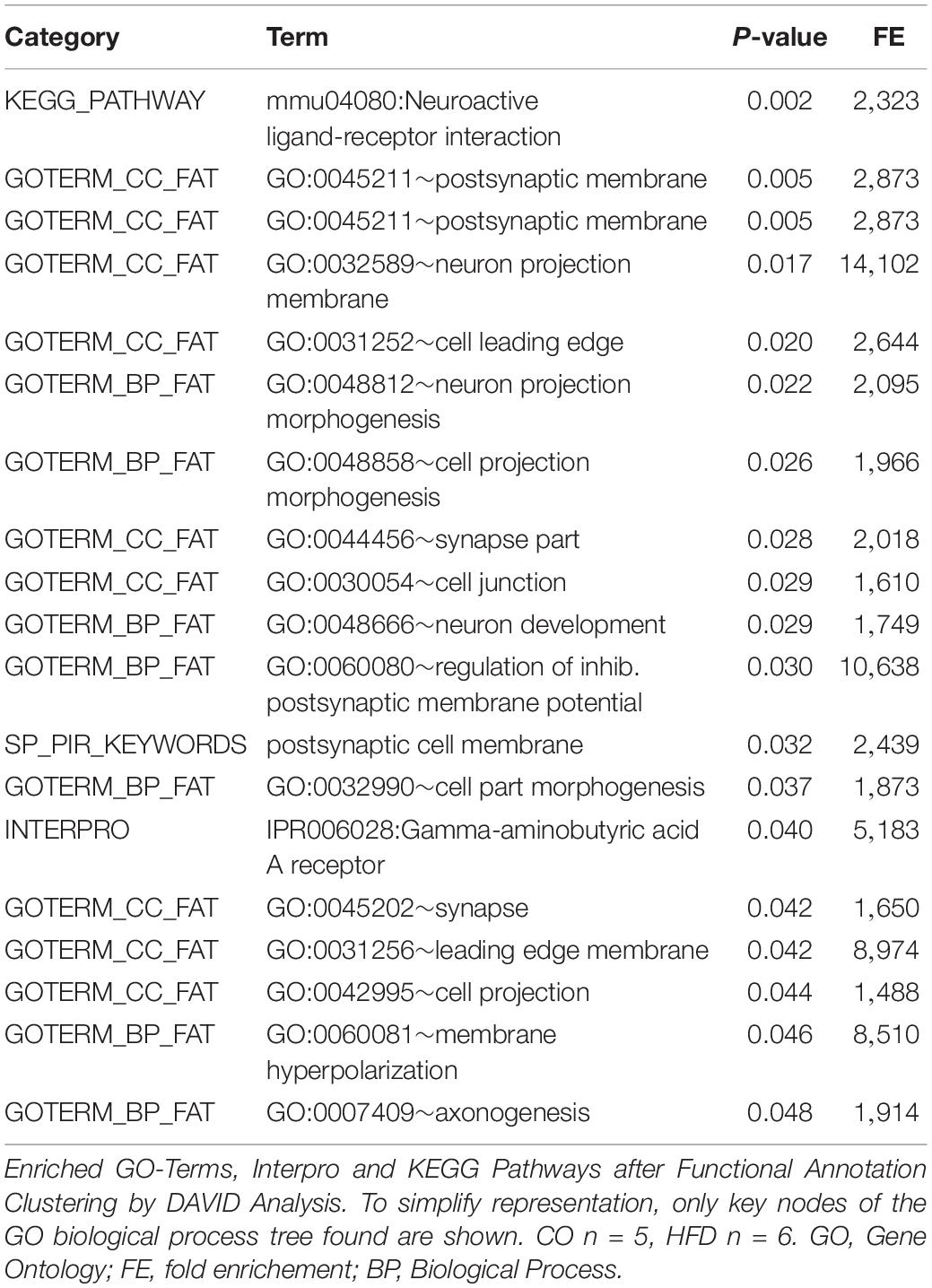
Table 1. Functional Annotation Clustering for differentially expressed genes between CO and HFD offspring.
Hypothalamic Neurotrophin Expression
With regard to the microarray results in the hypothalamus of HFD and CO offspring at P21, we first determined the mRNA expression of several neurotrophic growth factors (Bdnf, Ngf, Nt3, Nt4/5, Cntf, and Gndf) in the hypothalamus of HFD and CO offspring at P21. Interestingly, we found a significant reduction in gene expression of Bdnf (66.7% of control; p < 0.05), Ngf (56.3% of control; p < 0.01) and Nt4/5 (41.0% of control; p < 0.01) in HFD offspring compared to controls (Figure 4A). Moreover, there was also a trend toward reduced mRNA expression detectable for Nt3 and Gdnf, with 56.8 and 61.9% of control expression, respectively (p = 0.07). In contrast, for Cntf we detected a tendency toward increased gene expression as a result of maternal obesity (p = 0.10) (Figure 4A).
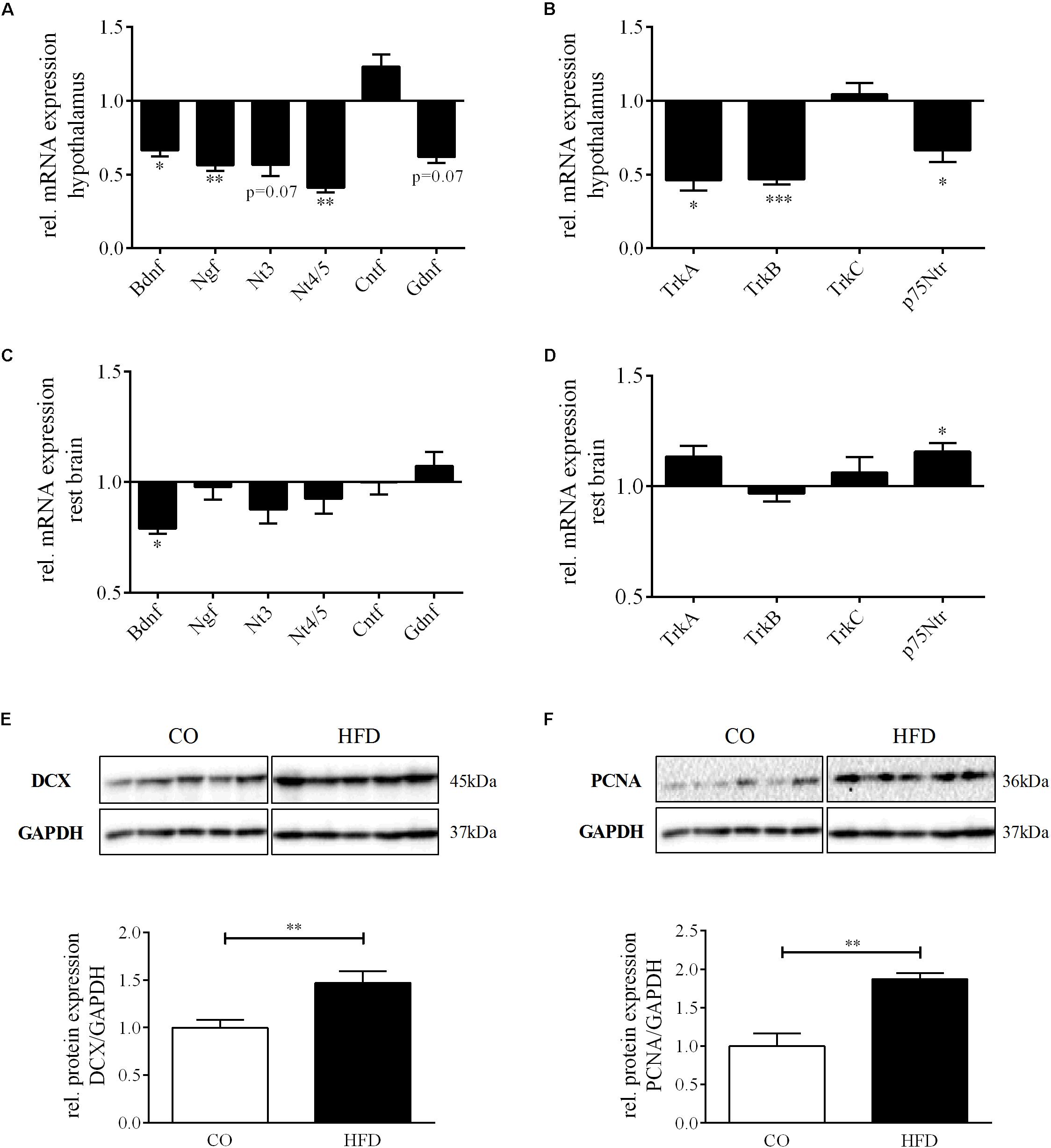
Figure 4. Neurotrophin expression and neurogenesis at P21. Relative gene expression compared to controls (= 1.0) at P21, CO n = 8, HFD n = 8. (A) Hypothalamic neurotrophins Bdnf, Ngf, Nt3, Nt4/5, Cntf, and Gdnf and (B) hypothalamic neurotrophin receptors TrkA, TrkB, TrkC, and p75Ntr. (C) Rest brain neurotrophins Bdnf, Ngf, Nt3, Nt4/5, Cntf, and Gdnf and (D) rest brain neurotrophin receptors TrkA, TrkB, TrkC, and p75Ntr. (E,F), Representative Western Blots of DCX and PCNA in hypothalamic tissue at P21. (E,F), Densitometric analysis of protein expression at P21: (E) DCX/GAPDH, CO n = 5, HFD n = 5. (F) PCNA/GAPDH, CO n = 5, HFD n = 5. Data are presented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; CO, control; HFD, high fat diet.
To determine whether altered hypothalamic gene expression as a result of maternal obesity is not only found for neurotrophic growth factors but also for their receptors, we quantified gene expression of TrkA, TrkB, TrkC, and p75Ntr in the hypothalamus of HFD and CO offspring at P21. We found no change in mRNA expression levels of TrkC, but significantly reduced gene expression for TrkA, TrkB, and p75Ntr (Figure 4B).
mRNA expression patterns of both, neurotrophins and neurotrophin receptors, in the rest of the brain did not reflect the pattern that we found in the hypothalamus (Figures 4C,D). However, mRNA expression of Bdnf was also found to be significantly reduced (p = 0.045) (Figure 4C) and p75Ntr significantly increased in the rest of the brain (0.023) (Figure 4D).
Hypothalamic Neurogenesis and Synaptic Plasticity
To next evaluate the effects of maternal obesity on hypothalamic neuronal network plasticity, we first determined protein expression of doublecortin (DCX), a specific marker for newborn neurons, in the hypothalamus of HFD and CO offspring at P21. Hypothalamic DCX protein expression was significantly increased in HFD offspring compared to controls indicating enhanced hypothalamic neurogenesis (Figure 4E). Supporting this finding, also hypothalamic protein levels of proliferating cell nuclear antigen (PCNA), a marker for DNA synthesis but also DNA repair (Frade and Ovejero-Benito, 2015), was significantly increased in HFD offspring (Figure 4F).
We additionally quantified phosphorylation of synapsin, which is known to facilitate the transport of neurotransmitter-filled vesicles to the synapse during an action potential and thereby promotes synaptic plasticity. There was no statistically significant difference between groups (Figure 5A). However, we detected significantly reduced protein levels of phosphorylated Ca2+/calmodulin-dependent protein kinase II (CAMKII) in HFD offspring at P21, indicating changes in hypothalamic synaptic plasticity and efficacy as a result of maternal obesity (Figure 5B). To next evaluate the excitatory and inhibitory synaptic tone in the hypothalamus at P21, we measured hypothalamic protein expression levels of the vesicular glutamate transporter 2 (vGLUT2), a marker for excitatory synapses, and vesicular GABA transporter (vGat), a marker for inhibitory synapses. HFD offspring displayed significantly increased levels of vGLUT2 compared to controls, while vGAT remained unaltered (Figures 5C,D), indicating an overall increased excitatory tone in the hypothalamus of HFD offspring.
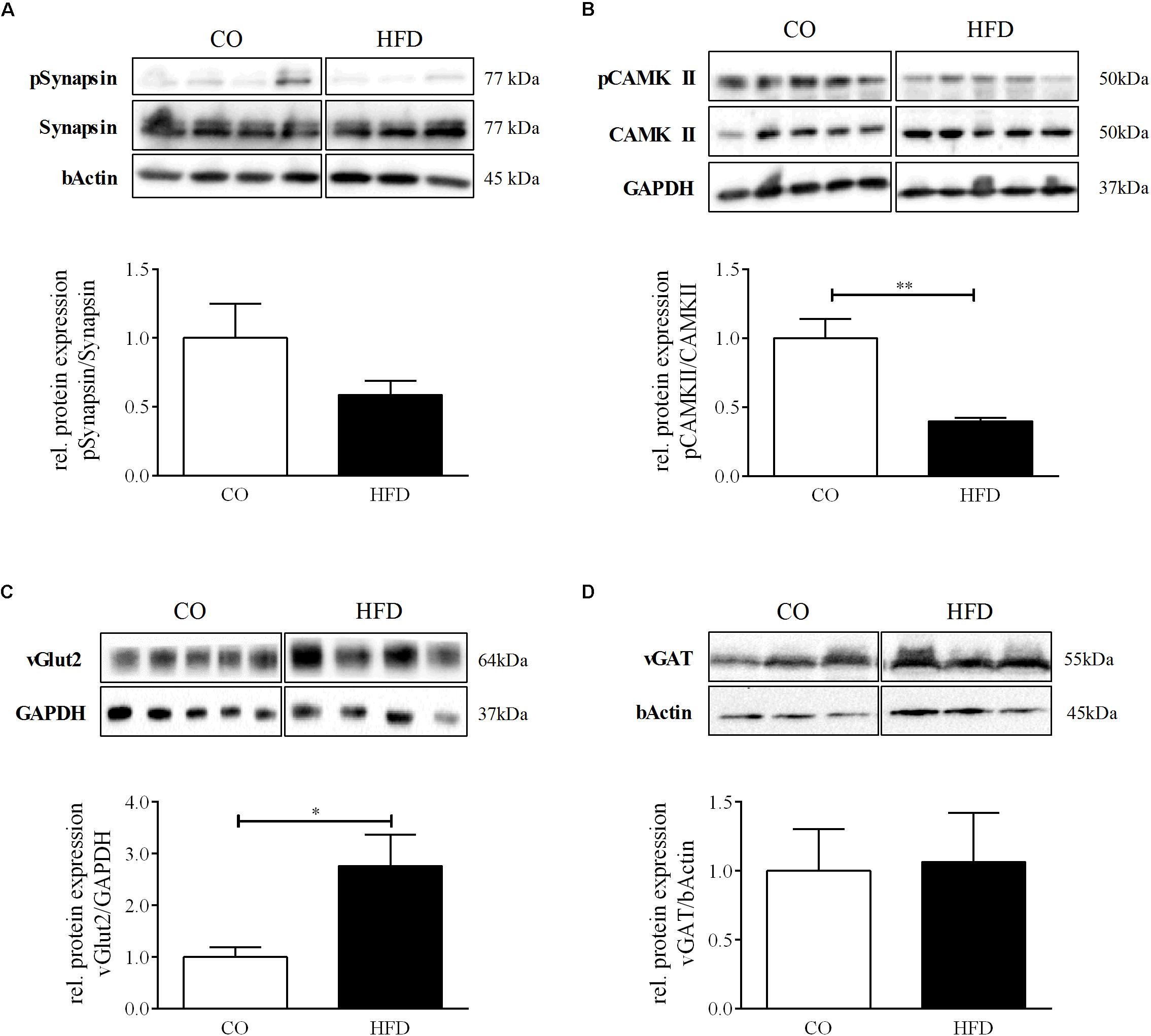
Figure 5. Hypothalamic synaptic plasticity at P21. (A–D) Representative Western Blots of pSynapsin, pCAMKII, vGlut2, and vGAT in hypothalamic tissue at P21. (A–D) Densitometric analysis of protein expression at P21: (A) pSynapsin/Synapsin, CO n = 4, HFD n = 4. (B) pCAMKII/CAMKII, CO n = 4, HFD n = 4. (C) vGLUT2/GAPDH, CO n = 5, HFD n = 4. (D) vGAT/GAPDH, CO n = 3, HFD n = 3. Data are presented as mean ± SEM. ∗p < 0.05; CO, control; HFD, high fat diet.
Hypothalamic MAPK Signaling
Neurotrophin-associated signaling has been shown to converge on the levels of mitogen activated protein kinase (MAPK) signaling (Thomas and Huganir, 2004), an intracellular cascade closely tied to synaptic plasticity in several parts of the brain (Borrie et al., 2017). To investigate whether intracellular MAPK signaling in the hypothalamus of the offspring might be affected by maternal obesity, we determined total protein amount and phosphorylation of the MAP kinases extracellular signal regulated kinases 1/2 (ERK1/2), p38, c-Jun N-terminal kinase 1 (JNK1), and JNK2/3 in the hypothalamus of offspring of obese and lean mouse dams on P21. The ratio between phosphorylated and total amount of ERK1/2 was significantly reduced to 32.9% (p = 0.041) (Figure 6A) in the hypothalamus of HFD offspring indicating a decreased hypothalamic activation of the ERK1/2 pathway following maternal obesity. Moreover, for p38 we revealed significant reduction of pp38 in relation to total p38 to 19.8% (p = 0.002), indicating reduced activation of the p38 pathway in the hypothalamus of HFD offspring (Figure 6B). There were no significant changes in phosphorylation of JNK1 and JNK2/3 in the hypothalamus at P21 (Figures 6C,D).
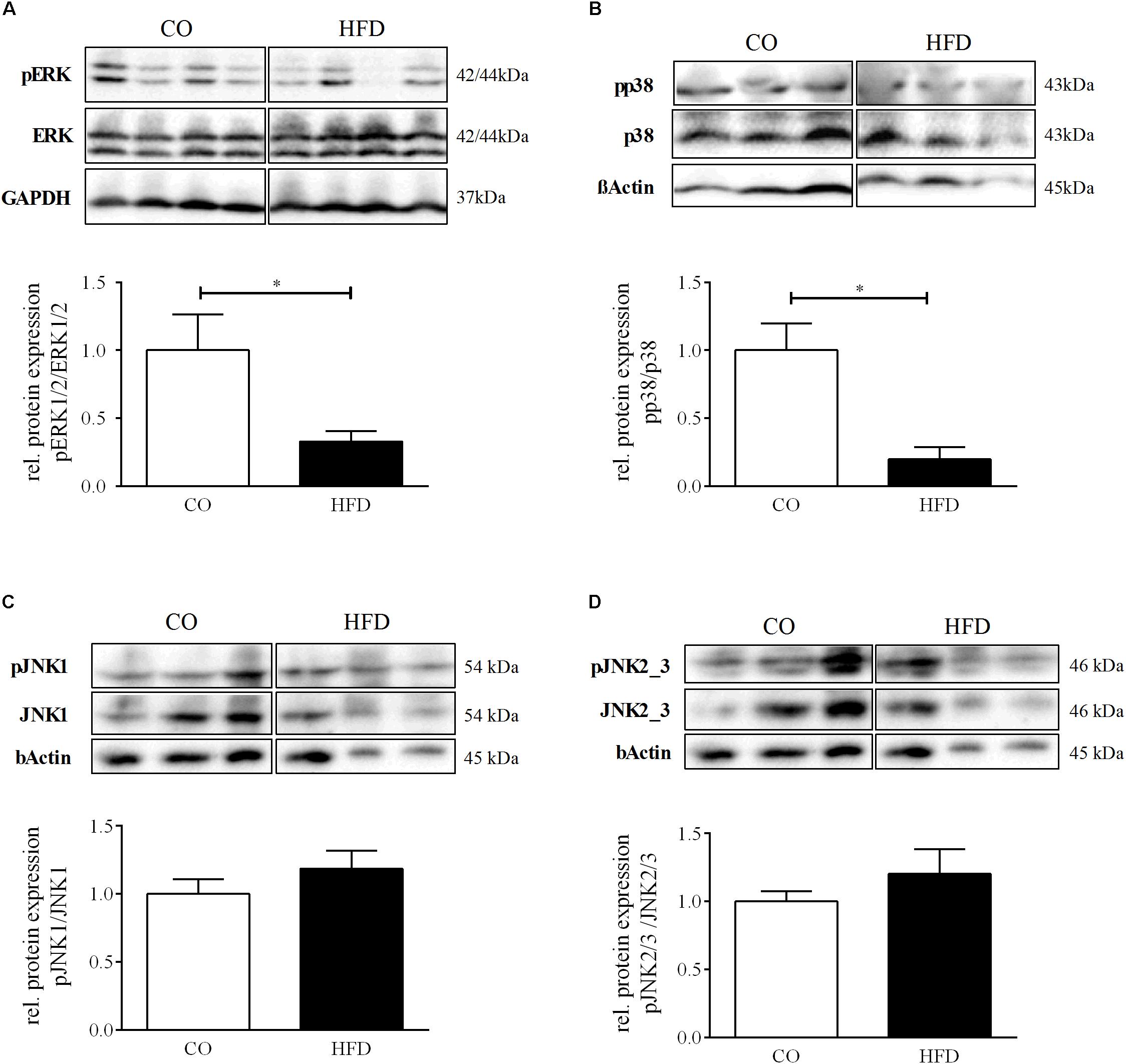
Figure 6. Hypothalamic MAP kinases signaling. (A–D) Representative Western Blots of pERK, pp38, pJNK1, pJNK2/3 in hypothalamic tissue at P21. (A–D) Densitometric analysis of protein expression at P21: (A) pERK/ERK, CO n = 4, HFD n = 4. (B) pp38/p38, CO n = 4, HFD n = 4. (C) pJNK1/JNK1, CO n = 4, HFD n = 4. (D) pJNK2/3/JNK2/3, CO n = 4, HFD n = 4. Data are presented as mean ± SEM. ∗p < 0.05; CO, control; HFD, high fat diet.
Discussion
In the present study, we determined the effects of maternal obesity on the offspring’s hypothalamic glucose sensing and insulin signaling mechanisms, assessed the global gene expression profile of the hypothalamus, and addressed hypothalamic neurotrophin expression and associated mechanisms of synaptic plasticity at P21.
We were able to describe numerous novel hypothalamic changes in HFD offspring: (a) increased IBA1 expression, (b) a global gene expression pattern indicative of major changes in neurogenesis and plasticity, and (c) reduced neurotrophin expression, changes in neurogenesis and plasticity marker expression as well as reduced MAPK signaling (ERK1/2 and p38).
In detail, we showed that maternal obesity resulted in a massive increase in hypothalamic insulin signaling at P21 and we found increased expression of hypothalamic IBA1, a marker for activated microglia. Activated microglia has been shown to promote reactive gliosis in the hypothalamus (Valdearcos et al., 2017), a mechanism directly affecting hypothalamic connectivity (Dorfman and Thaler, 2015). Just recently, acute insulin administration in mice was shown to induce both, hypothalamic AgRP expression and microglia activation (Winkler et al., 2019) suggesting a role for microglial cells in close vicinity to orexigenic neurons in facilitating the release of hunger signals to counteract hypoglycemia. In light of this hypothesis, the strong and long-lasting increase in hypothalamic insulin signaling in HFD offspring at P21 might be one reason for the observed activation of microglia in order to prevent potential hypoglycemia. This would result in increased sensation of hunger and the promotion of reactive hypothalamic gliosis with all its consequences for formation and function of hypothalamic feeding networks (Thaler et al., 2012; Valdearcos et al., 2017). This interpretation is supported by the finding of Vogt et al., who could show that blockade of insulin signaling in hypothalamic anorexigenic POMC neurons protects the offspring from maternal HFD-induced malformation of POMC neuron projections (Vogt et al., 2014). It is important to note that increased hypothalamic insulin signaling at P21 in our model of maternal obesity seems to precede insulin resistance in the brain and peripheral organs in adult offspring as shown by previous work of our group (Schmitz et al., 2018) and others (Samuelsson et al., 2008; Akyol et al., 2012; Gomes et al., 2018; George et al., 2019). Thus, it is tempting to speculate whether adult-onset hypothalamic insulin resistance might be a functional consequence of increased hypothalamic insulin signaling during early life. With regard to leptin’s effect at P21, it is worth mentioning that leptin is known to inhibit AMPK in the arcuate nucleus and reduce food intake (Hardie, 2015). Thus, it seems plausible that increased hypothalamic leptin action at P21 reflects the overfed state in HFD offspring. It would, however, be of great interest to attribute the reduction in phosphorylated hypothalamic AMPK found in our model at P21 to a specific cell type to further understand leptin’s role in hypothalamic AMPK signaling.
Furthermore, we performed microarray analysis of hypothalamic tissue of CO and HFD offspring at P21. These data revealed significantly different gene expression signatures as a result of maternal obesity. Here we present all differentially expressed genes arising from the comparison of CO and HFD offspring (Supplementary Tables 3, 4). Among the most upregulated genes in HFD offspring we found multiple zinc finger proteins. Analyzing the most prominently downregulated genes in HFD offspring, we found amongst others Map3k10 and Cntfr, a member of the MAP kinase family and a neurotrophin receptor. Furthermore, among the affected biological processes by maternal HFD, many GO-Terms referring to neurogenesis, axogenesis, and synaptic plasticity peaked out. By qPCR measurements in hypothalamic tissue, we confirmed that, indeed, HFD offspring displays significantly reduced hypothalamic mRNA expression levels for various neurotrophins and neurotrophin receptors, including Bdnf and its receptor TrkB. This finding is interesting in several ways. Besides its direct effect on synaptogenesis and neuronal plasticity (Unger et al., 2007), BDNF has been shown to have a clear anorexigenic effect (Molteni et al., 2004; Rask-Andersen et al., 2011). Heterozygous BDNF-knockout mice display hyperphagia with significantly increased body weight and elevated serum leptin and insulin levels along with decreased hypothalamic Bdnf mRNA expression (Kernie et al., 2000). Furthermore, intra-cerebroventricular application of BDNF or other TrkB ligands can reverse this effect and cause significant weight loss (Kernie et al., 2000; Burns et al., 2010; Fox et al., 2013), and BDNF injection specifically into the ventromedial nucleus of the hypothalamus leads to reduced food intake and an increase in energy expenditure (Wang et al., 2007). Moreover, also reduced TrkB receptor expression in the brain of mice leads to increased body weight and food intake (Xu et al., 2003). Keeping all that in mind, it is well conceivable that reduced hypothalamic BDNF signal transduction in offspring of obese mouse dams as indicated by reduced gene expression of both, Bdnf and TrkB, might contribute to their predisposition for obesity on P21 and permanently shape hypothalamic feeding networks to metabolic dysfunction. Future studies are needed to prove this hypothesis.
Information on the role of other members of the neurotrophin family and their receptors in energy homeostasis regulation is scarce. However, it is known that intra-cerebroventricular injection of NGF or NT4/5 also causes weight loss in mice (Kernie et al., 2000). NGF acts mainly through the TrkA receptor, whereas NT4/5 has been shown to activate TrkB receptor signaling (Glebova and Ginty, 2005). Thus, the hypothalamic gene expression pattern in the offspring of obese mouse dams in our experiment suggests an overall reduction in hypothalamic TrkA and B signaling following maternal obesity. This could contribute to the significantly reduced activation of the receptor associated MAP kinases ERK1/2 and p38 (Chaves et al., 2013; Wuhanqimuge et al., 2013). Both, ERK1/2 and p38, are sensitive to feeding status and show increased activation upon fasting in the hypothalamus of mice (Ueyama et al., 2004). At the same time, ERK1/2 signaling plays an important role in synapse formation (Huang and Reichardt, 2001; Alonso et al., 2004; Hans et al., 2004) and participates in long-term synaptic plasticity in hippocampus and sensory neurons (Martin et al., 1997). Various studies indicate that MAPKs are located in synaptic terminals influencing short- and long-term plasticity by phosphorylation of synaptic targets such as synapsin (Jovanovic et al., 2000; Sweatt, 2004; Boggio et al., 2007; Giachello et al., 2010). Thus, a reduction in Trk-mediated MAPK activation along with a reduction in synapsin phosphorylation to approximately 50% following maternal obesity (although not statistically significant) would suggest a link between maternal body weight and both, the offspring’s hypothalamic neuronal plasticity and their metabolic phenotype on P21. However, it is important to note that we did only observe a trend toward reduced synapsin phosphorylation in this study.
In addition to the above mentioned MAPK signaling pathways, neurotrophins also activate the phospholipase C gamma (PLCgamma) and the phosphatidylinositol 3-kinase (PI3K) pathways. While we did not address the role of PLCgamma signaling in this study, we determined hypothalamic AKT phosphorylation, an important downstream messenger of PI3K in HFD and CO offspring at P21. In contrast to the reduction in MAPK signaling, we found a massive increase in AKT phosphorylation in the hypothalamus of HFD offspring accompanied by increased phosphorylation of the downstream target GSK3ß. This finding cannot be explained by the observed changes in neurotrophin expression. As mentioned above, however, we found a strong increase in hypothalamic insulin signaling in HFD offspring. Thus, one could speculate that a potential effect of altered neurotrophin signaling is masked by the strong insulin action on hypothalamic cells at this time point. Being aware of the fact that reduced BDNF and NGF serum and tissue levels have been associated with hyperinsulinemic states like obesity and type-2-diabetes (Molteni et al., 2002; Geroldi et al., 2006; Suwa et al., 2006; Bullo et al., 2007; Krabbe et al., 2007; Fujinami et al., 2008; Yamanaka et al., 2008; Golden et al., 2010), even a causal relationship between overactive insulin signaling and decreased neurotrophin levels as part of a negative feedback mechanism is imaginable (Ding et al., 2006). Of course, further experiments will be needed to prove this hypothesis including deeper analysis of functional insulin sensitivity at P21.
The overlap in effects of neurotrophins and other metabolic cues on MAPKs and PI3K might also explain the effects of maternal obesity on expression of markers of synaptic plasticity in the hypothalamus of the offspring. Phosphorylation of synapsin in the hypothalamus shows a tendency toward reduction following maternal obesity along with significantly reduced pCAMKII protein levels. CAMKII is a protein involved in synaptic plasticity, memory and learning and its phosphorylated form is considered constitutively active. CAMKII activity is required for induction of long-term potentiation in the hippocampus and the associated structural plasticity of dendritic spines (Murakoshi et al., 2017). To our knowledge, CAMKII has so far not been assessed in the hypothalamus with regard to network formation of feeding circuits. However, reports on leptin modulating CAMKII activity in the hippocampus in vitro in a dose-dependent manner (Vogt et al., 2014) and altered hypothalamic CAMKII activity in diabetic rats (Ramakrishnan et al., 2005b) support our hypothesis that hormones or other metabolic factors might affect hypothalamic spine formation and synaptic activity via CAMKII-mediated processes. Interestingly, reduced hypothalamic CAMKII activity in vitro goes along with reduced p38 levels (Ramakrishnan et al., 2005a). And also, ERK1/2 has been functionally linked to synapsin expression in vitro (Chen et al., 2018). Both findings might suggest a possible direct connection between reduced MAP kinase signaling and the reduction of expression of both plasticity markers found in the hypothalamus of HFD offspring.
To investigate the overall synaptic tone in the hypothalamus, we set out to quantify vGLUT2 and vGAT protein expression representing excitatory and inhibitory synapses, respectively. While hypothalamic vGAT protein expression did not differ between groups, hypothalamic vGLUT2 protein levels were significantly increased in HFD offspring, indicating an overall increase in excitatory synapses. The ratio of excitatory and inhibitory synaptic inputs to hypothalamic feeding neurons is known to change in response to metabolic hormones, such as leptin and ghrelin (He et al., 2018). Within the hypothalamus, vGlut2 expressing neurons are most abundant in the ventromedial hypothalamic nucleus (VMH) (Tong et al., 2007). Interestingly, mice lacking vGlut2 in the VMH lack the ability to counteract hypoglycemia caused by insulin administration (Tong et al., 2007). Thus, elevated hypothalamic vGLUT2 expression in HFD offspring might directly result from sustained insulin signaling as described before. However, only recently, vGlut2 neurons in the nucleus arcuatus were found to be fast-acting anorexigenic neuronal populations that interconnect peripheral signals like CCK or leptin with other ARC neurons or directly project to the PVN (Guillebaud, 2019). Hypothalamic vGlut2 neurons were shown to be leptin sensitive (Guillebaud, 2019). Thus, increased leptin serum levels, increased hypothalamic leptin receptor protein expression and increased hypothalamic vGLUT2 expression in HFD offspring of our model confirm this recently described functional relationship. As leptin and vGlut2 are both involved in promoting satiety, upregulation of vGlut2 might be a compensatory mechanism of HFD offspring to counteract elevated body weight and fat pad weight at P21. Only future experiments that address the localization of vGlut2 within the different nuclear regions of the hypothalamus can answer the question which of these two conflicting interpretations is adequate or which effect might outweigh the other.
Interestingly, and in contrast to the impression of reduced hypothalamic plasticity marker expression in HFD offspring at P21, hypothalamic markers for proliferation and neurogenesis (DCX and PCNA) were significantly increased. This is in accordance with the only other study that addressed hypothalamic neurogenesis in the context of maternal HFD feeding so far (Chang et al., 2008). Chang et al. elegantly assessed neurogenesis in different hypothalamic nuclei at several time points during lactation and revealed that the effects on neurogenesis differ significantly between the distinct nuclei, with the most prominent increase in neurogenesis in the periventricular nucleus and the lateral hypothalamus. However, rat dams in their study received HFD only during part of the gestation and did not display an obese phenotype. In contrast, offspring exposure to gestational diabetes was shown to result in the malformation of medio-basal hypothalamic nuclei, which was suggested to be secondary to reduced neuron formation (Harder et al., 2001; Fahrenkrog et al., 2004; Franke et al., 2005; Dearden and Ozanne, 2015). Neurogenesis is generally thought to have a positive effect on network plasticity. However, recent studies reveal that in the hypothalamus, neuronal precursor cells are capable of differentiating into orexigenic or anorexigenic neurons in response to nutritional signals. Hypothalamic neurogenesis may thus act as an adaptive mechanism in order to respond to changes in food supply (Recabal et al., 2017). So, together with insulin’s postulated effect on IBA1 and vGlut2 expression in our model, increased neurogenesis (of orexigenic neurons?) in the hypothalamus of HFD offspring might thus also be interpreted as part of the physiologic response to prevent of hypoglycemia. Co-localization of DCX and orexigenic and anorexigenic cell markers would be helpful to test this hypothesis and evaluate whether, indeed, the HFD offspring’s neuronal precursor cells are more prone to differentiate into orexigenic neurons.
During pregnancy and lactation, the offspring of obese mothers is inevitably exposed to a variety of altered circulating hormonal and nutritional signals. Especially, the quality of ingested fatty acids seems to be of important matter for brain development and function of the offspring, as ingestion of increased amounts of saturated or trans fatty acids by the mother even in the absence of obesity has been shown to cause hypothalamic dysfunction in mice and rat offspring (Pimentel et al., 2011; Rother et al., 2012). We found reduced hypothalamic neurotrophin signaling following maternal obesity that was induced by ingestion of a diet rich in saturated fatty acids. In a very similar animal model, maternal obesity has been shown to cause BDNF deficiency in the hippocampus and deficits in spatial learning in the offspring (Tozuka et al., 2010). Also, omega-3-fatty acid deficiency during pregnancy and lactation has been shown to alter BDNF-TrkB-signaling in the hypothalamus and cause anxiety-like behavior in the rat (Bhatia et al., 2011). Taken together, hypothalamic neurotrophin signaling might be influenced by the quality and quantity of dietary fatty acids ingested by the dam. This hypothesis is further supported by the fact that also in adult rats, high-fat feeding causes reduced BDNF-signaling in the hippocampus, that can be restored upon reduced dietary fatty acid intake (Woo et al., 2013).
The timeliness of linking diet-induced changes in hypothalamic neurotrophin signaling to alterations in energy homeostasis is further substantiated by a report of Byerly et al. (2013) who identified neuron-derived neurotrophic factor (NENF) as a novel secreted protein in the hypothalamus regulating appetite by interacting with melanocortin signaling. Thus, neurotrophin action in the hypothalamus is gaining more and more attention as an important modulator of hypothalamic control of energy balance – either by directly modulating synaptic network formation and function or by interacting with established pathways of energy homeostasis regulation like the melanocortin system.
We aimed to shed light on the molecular mechanisms responsible for hypothalamic energy balance regulation from a completely new perspective. There are several limitations to our study – including the fact that we were not able to attribute the observed effects to specific nuclear regions of the hypothalamus and that we are lacking a direct mechanistic link between our observations and hypothalamic network formation in our animals. Yet, taken together, there is clear evidence that body weight and feeding status of the mother has an impact on the offspring’s hypothalamic neurotrophin signaling and expression of proteins involved in synaptogenesis and neurotransmitter release. Future experiments will have to show whether the observed changes are the underlying mechanism for altered wiring of hypothalamic feeding circuits and might therefore explain the predisposition for disturbed energy homeostasis as a consequence of maternal obesity. Another limitation of the study is that only male offspring were analyzed in the animal model. Some effects of developmental programming on long-term offspring health are gender-specific (Howie et al., 2009; Carter et al., 2012; de Souza et al., 2015). De Souza et al. who recently published studies examining the effects of maternal obesity on the offspring, used only male offspring. Our data is comparable with those studies (Alfaradhi et al., 2016; Frihauf et al., 2016; Liang et al., 2016; Beeson et al., 2018; Berends et al., 2018; Loche et al., 2018).
Conclusion
Taken together, we detected a global gene expression pattern in HFD offspring at P21 that was indicative of major changes in hypothalamic neurogenesis and plasticity. Targeted analysis of hypothalamic neurotrophin expression, neurogenesis and plasticity marker expression, and MAPK signaling (ERK1/2 and p38) confirmed our hypothesis that hypothalamic neurotrophin associated MAPK signaling might contribute to the pathogenesis of the offspring’s hypothalamic feeding network dysfunction in maternal obesity. Thereby, we hope to add a puzzle piece to the ongoing search for targets for the prevention and treatment of adult metabolic diseases.
Data Availability
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE1358303. The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher. Additionally, the raw data is included in the Supplementary Files.
Ethics Statement
The animal study was reviewed and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (Permit Number: 37.09.292).
Author Contributions
Each author has made an important scientific contribution to the study. IB-G, RJ, JD, and EH-R participated in the study design. IB-G and RJ carried out the animal experiments. SB, C-SK, LB, RJ, IB-G, and CV performed the molecular analyses. IB-G, RJ, and EH-R analyzed and interpreted the data. AO performed the microarray analysis. LS, NF, SB, TH, SA, and JD carefully revised the manuscript. EH-R was the guarantor of this work.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (RO 4109/2-1 to EH-R and KFO 2722 to EH-R), the University Hospital of Cologne Rotationsstellen-Pool (Grant No. 9/2015 to IB-G), and the Boll Foundation (Project No. 210-03-17 to EH-R and IB-G).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00962/full#supplementary-material
Abbreviations
(p)AKT, (phosphorylated) RAC-alpha serine/threonine-protein kinase; (p)AMPK, (phosphorylated) 5 ′ -adenosine monophosphate-activated protein kinase; (p)CAMKII, (phosphorylated) Ca2+/calmodulin-dependent protein kinase II; (p)ERK, (phosphorylated) extracellular-signal regulated kinases; (p)GSK3beta, (phosphorylated) glycogen synthase kinase 3 beta; (p)JNK1, (phosphorylated) c-JunN-terminal kinase 1; (p)JNK2, (phosphorylated) c-JunN-terminal kinase 2; (p)JNK3, (phosphorylated) c-JunN-terminal kinase 3; (p)p38, (phosphorylated) p38 mitogen-activated protein kinase; ARC, arcuate nucleus of the hypothalamus; BDNF, brain-derived neurotrophic factor; CNTF, ciliary neurotrophic factor; CO, control group; DCX, doublecortin; G, gestational day; GAPDH, glycerinaldehyd-3-phosphat-dehydrogenase; GDNF, glial cell derived neurotrophic factor; GO-terms, gene ontology terms; GTT, glucose tolerance test; HFD, high fat diet; HRP, horseradish peroxidase; IBA1, ionized calcium-binding adapter molecule 1; InsR, insulin receptor; KEGG, Kyoto Encyclopedia of Genes and Genomes; LepR, leptin receptor; MAPK, mitogen activated protein kinase; NENF, neuron-derived neurotrophic factor; NGF, nerve growth factor; NPY, neuropeptide Y; NT3, neurotrophin 3; NT4/5, neurotrophin 4/5; P, postnatal day; p75NTR, p75 neurotrophin receptor; PCLgamma, phospholipase C gamma; PCNA, proliferating cell nuclear antigen; PI3K, phosphatidylinositol 3 kinase; POMC, propriomelanocortin; PVN, paraventricular nucleus; TrkA, tropomyosin receptor kinase A; TrkB, tropomyosin receptor kinase B; TrkC, Tropomyosin receptor kinase C; vGAT, vesicular gamma-aminobutyric acid (GABA) transportervGlut2 Vesicular glutamate transporter 2.
Footnotes
- ^ https://david-d.ncifcrf.gov/
- ^ https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135830
- ^ https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135830
References
Abizaid, A., Liu, Z. W., Andrews, Z. B., Shanabrough, M., Borok, E., Elsworth, J. D., et al. (2006). Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 116, 3229–3239. doi: 10.1172/jci29867
Akyol, A., McMullen, S., and Langley-Evans, S. C. (2012). Glucose intolerance associated with early-life exposure to maternal cafeteria feeding is dependent upon post-weaning diet. Br. J. Nutr. 107, 964–978. doi: 10.1017/s0007114511003916
Alejandre-Alcazar, M. A., Kwapiszewska, G., Reiss, I., Amarie, O. V., Marsh, L. M., Sevilla-Perez, J., et al. (2007). Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L537–L549.
Alfaradhi, M. Z., Kusinski, L. C., Fernandez-Twinn, D. S., Pantaleao, L. C., Carr, S. K., Ferland-McCollough, D., et al. (2016). Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young, Lean Male Mice Offspring. Endocrinology 157, 4246–4256. doi: 10.1210/en.2016-1314
Alfaradhi, M. Z. and Ozanne, S. E. (2011). Developmental programming in response to maternal overnutrition. Front. Genet. 2:27. doi: 10.3389/fgene.2011.00027
Alonso, M., Medina, J. H., and Pozzo-Miller, L. (2004). ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn. Mem. 11, 172–178. doi: 10.1101/lm.67804
Aponte, Y., Atasoy, D., and Sternson, S. M. (2011). AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 14, 351–355. doi: 10.1038/nn.2739
Bae-Gartz, I., Janoschek, R., Kloppe, C. S., Vohlen, C., Roels, F., Oberthur, A., et al. (2016). Running exercise in obese pregnancies prevents IL-6 trans-signaling in male offspring. Med. Sci. Sports Exerc. 48, 829–838. doi: 10.1249/mss.0000000000000835
Beeson, J. H., Blackmore, H. L., Carr, S. K., Dearden, L., Duque-Guimaraes, D. E., Kusinski, L. C., et al. (2018). Maternal exercise intervention in obese pregnancy improves the cardiovascular health of the adult male offspring. Mol. Metab. 16, 35–44. doi: 10.1016/j.molmet.2018.06.009
Benani, A., Hryhorczuk, C., Gouaze, A., Fioramonti, X., Brenachot, X., Guissard, C., et al. (2012). Food intake adaptation to dietary fat involves PSA-dependent rewiring of the arcuate melanocortin system in mice. J. Neurosci. 32, 11970–11979. doi: 10.1523/jneurosci.0624-12.2012
Berends, L. M., Dearden, L., Tung, Y. C. L., Voshol, P., Fernandez-Twinn, D. S., and Ozanne, S. E. (2018). Programming of central and peripheral insulin resistance by low birthweight and postnatal catch-up growth in male mice. Diabetologia 61, 2225–2234. doi: 10.1007/s00125-018-4694-z
Bhatia, H. S., Agrawal, R., Sharma, S., Huo, Y. X., Ying, Z., and Gomez-Pinilla, F. (2011). Omega-3 fatty acid deficiency during brain maturation reduces neuronal and behavioral plasticity in adulthood. PloS One 6:e28451. doi: 10.1371/journal.pone.0028451
Blochl, A., and Thoenen, H. (1996). Localization of cellular storage compartments and sites of constitutive and activity-dependent release of nerve growth factor (NGF) in primary cultures of hippocampal neurons. Mol. Cell Neurosci. 7, 173–190. doi: 10.1006/mcne.1996.0014
Boggio, E. M., Putignano, E., Sassoe-Pognetto, M., Pizzorusso, T., and Giustetto, M. (2007). Visual stimulation activates ERK in synaptic and somatic compartments of rat cortical neurons with parallel kinetics. PloS One 2:e604. doi: 10.1371/journal.pone.0000604
Borrie, S. C., Brems, H., Legius, E., and Bagni, C. (2017). Cognitive dysfunctions in intellectual disabilities: the contributions of the ras-MAPK and PI3K-AKT-mTOR pathways. Annu. Rev. Genomics Hum. Genet. 18, 115–142. doi: 10.1146/annurev-genom-091416-035332
Bouret, S. G. (2009). Early life origins of obesity: role of hypothalamic programming. J. Pediatr. Gastroenterol. Nutr. 48(Suppl 1), S31–S38.
Bouret, S. G., Draper, S. J., and Simerly, R. B. (2004a). Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J. Neurosci. 24, 2797–2805. doi: 10.1523/jneurosci.5369-03.2004
Bouret, S. G., Draper, S. J., and Simerly, R. B. (2004b). Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304, 108–110. doi: 10.1126/science.1095004
Bouret, S. G., and Simerly, R. B. (2006). Developmental programming of hypothalamic feeding circuits. Clin. Genet. 70, 295–301. doi: 10.1111/j.1399-0004.2006.00684.x
Breitling, R., Armengaud, P., Amtmann, A., and Herzyk, P. (2004). Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 573, 83–92. doi: 10.1016/j.febslet.2004.07.055
Bullo, M., Peeraully, M. R., Trayhurn, P., Folch, J., and Salas-Salvado, J. (2007). Circulating nerve growth factor levels in relation to obesity and the metabolic syndrome in women. Eur. J. Endocrinol. 157, 303–310. doi: 10.1530/eje-06-0716
Burns, B., Schmidt, K., Williams, S. R., Kim, S., Girirajan, S., and Elsea, S. H. (2010). Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum. Mol. Genet. 19, 4026–4042. doi: 10.1093/hmg/ddq317
Byerly, M. S., Swanson, R. D., Semsarzadeh, N. N., McCulloh, P. S., Kwon, K., Aja, S., et al. (2013). Identification of hypothalamic neuron-derived neurotrophic factor as a novel factor modulating appetite. Am. J. Physiol. Regul. Integ. comp. Physiol. 304, R1085–R1095.
Carter, L. G., Lewis, K. N., Wilkerson, D. C., Tobia, C. M., Ngo Tenlep, S. Y., Shridas, P., et al. (2012). Perinatal exercise improves glucose homeostasis in adult offspring. Am. J. Physiol. Endocrinol. Metab. 303, E1061–E1068.
Catalano, P. M., Farrell, K., Thomas, A., Huston-Presley, L., Mencin, P., de Mouzon, S. H., et al. (2009). Perinatal risk factors for childhood obesity and metabolic dysregulation. Am. J. Clin. Nutr. 90, 1303–1313. doi: 10.3945/ajcn.2008.27416
Chang, G. Q., Gaysinskaya, V., Karatayev, O., and Leibowitz, S. F. (2008). Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J. Neurosci. 28, 12107–12119. doi: 10.1523/jneurosci.2642-08.2008
Chaves, R. N., Alves, A. M., Lima, L. F., Matos, H. M., Rodrigues, A. P., and Figueiredo, J. R. (2013)Role of nerve growth factor (NGF) and its receptors in folliculogenesis. Zygote 21, 187–197. doi: 10.1017/s0967199412000111
Chen, Z., Xu, Y. Y., Ge, J. F., and Chen, F. H. (2018). CRHR1 mediates the Up-regulation of synapsin I induced by nesfatin-1 through ERK 1/2 signaling in SH-SY5Y cells. Cell. Mol. Neurobiol. 38, 627–633. doi: 10.1007/s10571-017-0509-x
Cottrell, E. C., Cripps, R. L., Duncan, J. S., Barrett, P., Mercer, J. G., Herwig, A., et al. (2009). Developmental changes in hypothalamic leptin receptor: relationship with the postnatal leptin surge and energy balance neuropeptides in the postnatal rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R631–R639.
Cottrell, E. C., and Ozanne, S. E. (2007). Developmental programming of energy balance and the metabolic syndrome. Proc. Nutr. Soc. 66, 198–206. doi: 10.1017/s0029665107005447
Cowley, M. A., Cone, R., Enriori, P., Louiselle, I., Williams, S. M., and Evans, A. E. (2003a). Electrophysiological actions of peripheral hormones on melanocortin neurons. Ann. N. Y. Acad. Sci. 994, 175–186. doi: 10.1111/j.1749-6632.2003.tb03178.x
Cowley, M. A., Smith, R. G., Diano, S., Tschop, M., Pronchuk, N., Grove, K. L., et al. (2003b). The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661. doi: 10.1016/s0896-6273(03)00063-1
Cowley, M. A., Smart, J. L., Rubinstein, M., Cerdan, M. G., Diano, S., Horvath, T. L., et al. (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. doi: 10.1038/35078085
de Souza, A. P., Pedroso, A. P., Watanabe, R. L., Dornellas, A. P., Boldarine, V. T., Laure, H. J., et al. (2015). Gender-specific effects of intrauterine growth restriction on the adipose tissue of adult rats: a proteomic approach. Proteome Sci. 13:32.
Dearden, L., and Ozanne, S. E. (2015). Early life origins of metabolic disease: developmental programming of hypothalamic pathways controlling energy homeostasis. Front. Neuroendocrinol. 39, 3–16. doi: 10.1016/j.yfrne.2015.08.001
Dhillon, H., Zigman, J. M., Ye, C., Lee, C. E., McGovern, R. A., Tang, V., et al. (2006). Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203. doi: 10.1016/j.neuron.2005.12.021
Diano, S., Farr, S. A., Benoit, S. C., McNay, E. C., da Silva, I., Horvath, B., et al. (2006). Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 9, 381–388. doi: 10.1038/nn1656
Ding, Q., Vaynman, S., Akhavan, M., Ying, Z., and Gomez-Pinilla, F. (2006). Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 140, 823–833. doi: 10.1016/j.neuroscience.2006.02.084
Dorfman, M. D., and Thaler, J. P. (2015). Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 22, 325–330. doi: 10.1097/med.0000000000000182
Edgar, R., Domrachev, M., and Lash, A. E. (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. doi: 10.1093/nar/30.1.207
Elias, C. F., Aschkenasi, C., Lee, C. Kelly, J. Ahima, R. S., Bjorbaek, C., et al. (1999). Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 23, 775–786. doi: 10.1016/s0896-6273(01)80035-0
Fahrenkrog, S., Harder, T., Stolaczyk, E., Melchior, K., Franke, K., Dudenhausen, J. W., et al. (2004). Cross-fostering to diabetic rat dams affects early development of mediobasal hypothalamic nuclei regulating food intake, body weight, and metabolism. J. Nutr. 134, 648–654. doi: 10.1093/jn/134.3.648
Fox, E. A., Biddinger, J. E., Jones, K. R., McAdams, J., and Worman, A. (2013). Mechanism of hyperphagia contributing to obesity in brain-derived neurotrophic factor knockout mice. Neuroscience 229, 176–199. doi: 10.1016/j.neuroscience.2012.09.078
Frade, J. M., and Ovejero-Benito, M. C. (2015). Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 14, 712–720. doi: 10.1080/15384101.2015.1004937
Franke, K., Harder, T., Aerts, L., Melchior, K., Fahrenkrog, S., Rodekamp, E., et al. (2005). ‘Programming’ of orexigenic and anorexigenic hypothalamic neurons in offspring of treated and untreated diabetic mother rats. Brain Res. 1031, 276–283. doi: 10.1016/j.brainres.2004.11.006
Frihauf, J. B., Fekete, E. M., Nagy, T. R., Levin, B. E., and Zorrilla, E. P. (2016). Maternal Western diet increases adiposity even in male offspring of obesity-resistant rat dams: early endocrine risk markers. Am. J. Physiol. Regul. Integ. Comp. Physiol. 311, R1045–R1059.
Fujinami, A., Ohta, K., Obayashi, H., Fukui, M., Hasegawa, G., and Nakamura, N., et al. (2008). Serum brain-derived neurotrophic factor in patients with type 2 diabetes mellitus: relationship to glucose metabolism and biomarkers of insulin resistance. Clin. Biochem. 41, 812–817. doi: 10.1016/j.clinbiochem.2008.03.003
Gao, Q., Mezei, G., Nie, Y., Rao, Y., Choi, C. S., Bechmann, I., et al. (2007). Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 13, 89–94. doi: 10.1038/nm1525
George, G., Draycott, S. A. V., Muir, R., Clifford, B., Elmes, M. J., and Langley-Evans, S. C. (2019). Exposure to maternal obesity during suckling outweighs in utero exposure in programming for post-weaning adiposity and insulin resistance in rats. Sci. Rep. 9:10134.
Geroldi, D., Minoretti, P., and Emanuele, E. (2006). Brain-derived neurotrophic factor and the metabolic syndrome: more than just a hypothesis. Med. Hypotheses. 67, 195–196. doi: 10.1016/j.mehy.2006.02.001
Giachello, C. N., Fiumara, F., Giacomini, C., Corradi, A., Milanese, C., Ghirardi, M., et al. (2010). MAPK/Erk-dependent phosphorylation of synapsin mediates formation of functional synapses and short-term homosynaptic plasticity. J. Cell Sci. 123(Pt 6), 881–893. doi: 10.1242/jcs.056846
Glebova, N. O., and Ginty, D. D. (2005). Growth and survival signals controlling sympathetic nervous system development. Annu. Rev. Neurosci. 28, 191–222. doi: 10.1146/annurev.neuro.28.061604.135659
Golden, E., Emiliano, A., Maudsley, S., Windham, B. G., Carlson, O. D., Egan, J. M., et al. (2010). Circulating brain-derived neurotrophic factor and indices of metabolic and cardiovascular health: data from the baltimore longitudinal study of aging. PloS One 5:e10099. doi: 10.1371/journal.pone.0010099
Gomes, R. M., Bueno, F. G., Schamber, C. R., de Mello, J. C. P., de Oliveira, J. C., Francisco, F. A., et al. (2018). Maternal diet-induced obesity during suckling period programs offspring obese phenotype and hypothalamic leptin/insulin resistance. J. Nutr. Biochem. 61, 24–32. doi: 10.1016/j.jnutbio.2018.07.006
Guillebaud, F. (2019). VGluT2 neuronal population of the arcuate nucleus: towards a more balanced vision of food intake control. J. Physiol. 597, 2838–2837.
Hans, A., Bajramovic, J. J., Syan, S., Perret, E., Dunia, I., Brahic, M., et al. (2004). Persistent, noncytolytic infection of neurons by Borna disease virus interferes with ERK 1/2 signaling and abrogates BDNF-induced synaptogenesis. FASEB J. 18, 863–865. doi: 10.1096/fj.03-0764fje
Harder, T., Aerts, L., Franke, K., Van Bree, R., Van Assche, F. A., and Plagemann, A. (2001). Pancreatic islet transplantation in diabetic pregnant rats prevents acquired malformation of the ventromedial hypothalamic nucleus in their offspring. Neurosci. Lett. 299, 85–88. doi: 10.1016/s0304-3940(01)01495-1
Hardie, D. G. (2015). AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 33, 1–7. doi: 10.1016/j.ceb.2014.09.004
He, Z., Gao, Y., Alhadeff, A. L., Castorena, C. M., Huang, Y., Lieu, L., et al. (2018). Cellular and synaptic reorganization of arcuate NPY/AgRP and POMC neurons after exercise. Mol. Metab. 18, 107–119. doi: 10.1016/j.molmet.2018.08.011
Hempstead, B. L. (2006). Dissecting the diverse actions of pro- and mature neurotrophins. Curr. Alzheimer Res. 3, 19–24. doi: 10.2174/156720506775697061
Horvath, T. L., and Diano, S. (2004). The floating blueprint of hypothalamic feeding circuits. Nat. Rev. Neurosci. 5, 662–667. doi: 10.1038/nrn1479
Horvath, T. L., and Gao, X. B. (2005). Input organization and plasticity of hypocretin neurons: possible clues to obesity’s association with insomnia. Cell Metab. 1, 279–286. doi: 10.1016/j.cmet.2005.03.003
Horvath, T. L., Sarman, B., Garcia-Caceres, C., Enriori, P. J., Sotonyi, P., Shanabrough, M., et al. (2010). Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 14875–14880. doi: 10.1073/pnas.1004282107
Hotta, K., Nakamura, M., Nakamura, T., Matsuo, T., Nakata, Y., Kamohara, S., et al. (2009). Association between obesity and polymorphisms in SEC16B, TMEM18, GNPDA2, BDNF, FAIM2 and MC4R in a Japanese population. J. Hum. Genet. 54, 727–731. doi: 10.1038/jhg.2009.106
Howie, G. J., Sloboda, D. M., Kamal, T., and Vickers, M. H. (2009). Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J. Physiol. 587(Pt 4), 905–915. doi: 10.1113/jphysiol.2008.163477
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Jobst, E. E., Enriori, P. J., and Cowley, M. A. (2004). The electrophysiology of feeding circuits. Trends Endocrinol. Metab. 15, 488–499. doi: 10.1016/j.tem.2004.10.007
Jovanovic, J. N., Czernik, A. J., Fienberg, A. A., Greengard, P., and Sihra, T. S. (2000). Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 3, 323–329. doi: 10.1038/73888
Kernie, S. G., Liebl, D. J., and Parada, L. F. (2000). BDNF regulates eating behavior and locomotor activity in mice. Embo J. 19, 1290–1300. doi: 10.1093/emboj/19.6.1290
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M., and Altman, D. G. (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8:e1000412. doi: 10.1371/journal.pbio.1000412
Konner, A. C., Janoschek, R., Plum, L., Jordan, S. D., Rother, E., Ma, X., et al. (2007). Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metabol. 5, 438–449. doi: 10.1016/j.cmet.2007.05.004
Krabbe, K. S., Nielsen, A. R., Krogh-Madsen, R., Plomgaard, P., Rasmussen, P., Erikstrup, C., et al. (2007). Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 50, 431–438.
Krashes, M. J., Koda, S., Ye, C., Rogan, S. C., Adams, A. C., Cusher, D. S., et al. (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428. doi: 10.1172/jci46229
Lessmann, V., Gottmann, K., and Malcangio, M. (2003). Neurotrophin secretion: current facts and future prospects. Prog. Neurobiol. 69, 341–374. doi: 10.1016/s0301-0082(03)00019-4
Li, M., Sloboda, D. M., and Vickers, M. H. (2011). Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp. Diabetes Res. 2011:592408.
Liang, X., Yang, Q., Fu, X., Rogers, C. J., Wang, B., Pan, H., et al. (2016). Maternal obesity epigenetically alters visceral fat progenitor cell properties in male offspring mice. J. Physiol. 594, 4453–4466. doi: 10.1113/jp272123
Liu, T., Kong, D., Shah, B. P., Ye, C., Koda, S., Saunders, A., et al. (2012). Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 73, 511–522. doi: 10.1016/j.neuron.2011.11.027
Loche, E., Blackmore, H. L., Carpenter, A. A., Beeson, J. H., Pinnock, A., Ashmore, T. J., et al. (2018). Maternal diet-induced obesity programmes cardiac dysfunction in male mice independently of post-weaning diet. Cardiovasc. Res. 114, 1372–1384. doi: 10.1093/cvr/cvy082
Maric-Bilkan, C., Symonds, M., Ozanne, S., and Alexander, B. T. (2011). Impact of maternal obesity and diabetes on long-term health of the offspring. Exp. Diabetes Res. 2011:163438.
Martin, K. C., Michael, D., Rose, J. C., Barad, M., Casadio, A., Zhu, H., et al. (1997). MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 18, 899–912. doi: 10.1016/s0896-6273(00)80330-x
McCurdy, C. E., Bishop, J. M., Williams, S. M., Grayson, B. E., Smith, M. S., and Friedman, J. E, et al. (2009). Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 119, 323–335.
Molteni, R., Barnard, R. J., Ying, Z., Roberts, C. K., and Gomez-Pinilla, F. (2002)A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 112, 803–814. doi: 10.1016/s0306-4522(02)00123-9
Molteni, R., Wu, A., Vaynman, S., Ying, Z., Barnard, R. J., and Gomez-Pinilla, F. (2004). Exercise reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience 123, 429–440. doi: 10.1016/j.neuroscience.2003.09.020
Murakoshi, H., Shin, M. E., Parra-Bueno, P., Szatmari, E. M., Shibata, A. C. E., and Yasuda, R. (2017). Kinetics of endogenous CaMKII required for synaptic plasticity revealed by optogenetic kinase inhibitor. Neuron 94, 37–47.e5. doi: 10.1016/j.neuron.2017.02.036
O’Reilly, J. R., and Reynolds, R. M. (2013). The risk of maternal obesity to the long-term health of the offspring. Clin. Endocrinol. 78, 9–16. doi: 10.1111/cen.12055
Ozanne, S. E. (2011). Sugaring appetite development: mechanisms of neuroendocrine programming. Endocrinology 152, 4007–4009. doi: 10.1210/en.2011-1659
Park, H., and Poo, M. M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. doi: 10.1038/nrn3379
Pimentel, G. D., Lira, F. S., Rosa, J. C., Oliveira, J. L., Losinskas-Hachul, A. C., Souza, G. I., et al. (2011). Intake of trans fatty acids during gestation and lactation leads to hypothalamic inflammation via TLR4/NFkappaBp65 signaling in adult offspring. J. Nutr. Biochem. 23, 822–828.
Pinto, S., Roseberry, A. G., Liu, H., Diano, S., Shanabrough, M., Cai, X., et al. (2004). Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304, 110–115. doi: 10.1126/science.1089459
Plagemann, A. (2011). Maternal diabetes and perinatal programming. Early Hum. Dev. 87, 743–747. doi: 10.1016/j.earlhumdev.2011.08.018
Plank, C., Ostreicher, I., Hartner, A., Marek, I., Struwe, F. G., Amann, K., et al. (2006). Intrauterine growth retardation aggravates the course of acute mesangioproliferative glomerulonephritis in the rat. Kidney Int. 70, 1974–1982. doi: 10.1038/sj.ki.5001966
Poston, L. (2012). Maternal obesity, gestational weight gain and diet as determinants of offspring long term health. Best Pract. Res. Clin. Endocrinol. Metab. 26, 627–639. doi: 10.1016/j.beem.2012.03.010
Ramakrishnan, R., Kempuraj, D., Prabhakaran, K., Jayakumar, A. R., Devi, R. S., Suthanthirarajan, N., et al. (2005a). A short-term diabetes induced changes of catecholamines and p38-MAPK in discrete areas of rat brain. Life Sci. 77, 1825–1835. doi: 10.1016/j.lfs.2004.12.038
Ramakrishnan, R., Prabhakaran, K., Jayakumar, A. R., Gunasekaran, P., Sheeladevi, R., and Suthanthirarajan, N. (2005b). Involvement of Ca2+/calmodulin-dependent protein kinase II in the modulation of indolamines in diabetic and hyperglycemic rats. J. Neurosci. Res. 80, 518–528. doi: 10.1002/jnr.20499
Rask-Andersen, M., Almen, M. S., Olausen, H. R., Olszewski, P. K., Eriksson, J., Chavan, R. A., et al. (2011). Functional coupling analysis suggests link between the obesity gene FTO and the BDNF-NTRK2 signaling pathway. BMC Neurosci. 12, 1471–2202. doi: 10.1186/1471-2202-12-117
Recabal, A., Caprile, T., and Garcia-Robles, M. L. A. (2017). Hypothalamic neurogenesis as an adaptive metabolic mechanism. Front. Neurosci. 11:190. doi: 10.3389/fnins.2017.00190
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564. doi: 10.1098/rstb.2006.1894
Rooney, K., and Ozanne, S. E. (2011). Maternal over-nutrition and offspring obesity predisposition: targets for preventative interventions. Int. J. Obes. 35, 883–890. doi: 10.1038/ijo.2011.96
Rother, E., Kuschewski, R., Alcazar, M. A., Oberthuer, A., Bae-Gartz, I., Vohlen, C., et al. (2012). Hypothalamic JNK1 and IKKbeta activation and impaired early postnatal glucose metabolism after maternal perinatal high-fat feeding. Endocrinology 153, 770–781. doi: 10.1210/en.2011-1589
Samuelsson, A. M., Matthews, P. A., Argenton, M., Christie, M. R., McConnell, J. M., Jansen, E. H., et al. (2008). Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51, 383–392. doi: 10.1161/hypertensionaha.107.101477
Schmitz, L., Kuglin, R., Bae-Gartz, I., Janoschek, R., Appel, S., Mesaros, A., et al. (2018). Hippocampal insulin resistance links maternal obesity with impaired neuronal plasticity in adult offspring. Psychoneuroendocrinology 89, 46–52. doi: 10.1016/j.psyneuen.2017.12.023
Sternson, S. M., Shepherd, G. M., and Friedman, J. M. (2005). Topographic mapping of VMH –> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 8, 1356–1363. doi: 10.1038/nn1550
Suwa, M., Kishimoto, H., Nofuji, Y., Nakano, H., Sasaki, H., Radak, Z., et al. (2006). Serum brain-derived neurotrophic factor level is increased and associated with obesity in newly diagnosed female patients with type 2 diabetes mellitus. Metabolism 55, 852–857. doi: 10.1016/j.metabol.2006.02.012
Sweatt, J. D. (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 14, 311–317. doi: 10.1016/j.conb.2004.04.001
Thaler, J. P., Yi, C. X., Schur, E. A., Guyenet, S. J., Hwang, B. H., Dietrich, M. O., et al. (2012). Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 122, 153–162. doi: 10.1172/jci59660
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183. doi: 10.1038/nrn1346
Thorleifsson, G., Walters, G. B., Gudbjartsson, D. F., Steinthorsdottir, V., Sulem, P., Helgadottir, A., et al. (2009). Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41, 18–24. doi: 10.1038/ng.274
Tong, Q., Ye, C., McCrimmon, R. J., Dhillon, H., Choi, B., Kramer, M. D., et al. (2007). Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 5, 383–393. doi: 10.1016/j.cmet.2007.04.001
Tozuka, Y., Kumon, M., Wada, E., Onodera, M., Mochizuki, H., and Wada, K. (2010). Maternal obesity impairs hippocampal BDNF production and spatial learning performance in young mouse offspring. Neurochem. Int. 57, 235–247. doi: 10.1016/j.neuint.2010.05.015
Ueyama, E., Morikawa, Y., Yasuda, T., and Senba, E. (2004). Attenuation of fasting-induced phosphorylation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus in response to refeeding. Neurosci. Lett. 371, 40–44. doi: 10.1016/j.neulet.2004.08.035
Unger, T. J., Calderon, G. A., Bradley, L. C., Sena-Esteves, M., and Rios, M. (2007). Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J. Neurosci. 27, 14265–14274. doi: 10.1523/jneurosci.3308-07.2007
Valdearcos, M., Douglass, J. D., Robblee, M. M., Dorfman, M. D., Stifler, D. R., Bennett, M. L., et al. (2017). Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metabol. 26, 185–197.e3. doi: 10.1016/j.cmet.2017.05.015
van den Top, M., Lee, K., Whyment, A. D., Blanks, A. M., and Spanswick, D. (2004). Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat. Neurosci. 7, 493–494. doi: 10.1038/nn1226
Vanevski, F., and Xu, B. (2013). Molecular and neural bases underlying roles of BDNF in the control of body weight. Front. Neurosci. 7:37. doi: 10.3389/fnins.2013.00037
Velloso, L. A. (2012). Maternal consumption of high-fat diet disturbs hypothalamic neuronal function in the offspring: implications for the genesis of obesity. Endocrinology 153, 543–545. doi: 10.1210/en.2011-2077
Vogt, M. C., Paeger, L., Hess, S., Steculorum, S. M., Awazawa, M., Hampel, en-names>M., Hampel, B., et al. (2014). Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 156, 495–509. doi: 10.1016/j.cell.2014.01.008
Wang, C., Bomberg, E., Levine, A., Billington, C., and Kotz, C. M. (2007). Brain-derived neurotrophic factor in the ventromedial nucleus of the hypothalamus reduces energy intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1037–R1045.
Winkler, Z., Kuti, D., Polyak, A., Juhasz, B., Gulyas, K., Lenart, N., et al. (2019). Hypoglycemia-activated hypothalamic microglia impairs glucose counterregulatory responses. Sci. Rep. 9:6224.
Woo, J., Shin, K. O., Park, S. Y., Jang, K. S., and Kang, S. (2013). Effects of exercise and diet change on cognition function and synaptic plasticity in high fat diet induced obese rats. Lipids Health Dis. 12:144. doi: 10.1186/1476-511x-12-144
Wuhanqimuge, Itakura, A., Matsuki, Y., Tanaka, M., and Arioka, M. (2013). Lysophosphatidylcholine enhances NGF-induced MAPK and Akt signals through the extracellular domain of TrkA in PC12 cells. FEBS Open. Bio. 3, 243–251. doi: 10.1016/j.fob.2013.05.003
Xu, B., Goulding, E. H., Zang, K., Cepoi, D., Cone, R. D., Jones, K. R., et al. (2003). Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742. doi: 10.1038/nn1073
Yamanaka, M., Itakura, Y., Tsuchida, A., Nakagawa, T., and Taiji, M. (2008). Brain-derived neurotrophic factor (BDNF) prevents the development of diabetes in prediabetic mice. Biomed. Res. 29, 147–153. doi: 10.2220/biomedres.29.147
Yang, Y., Atasoy, D., Su, H. H., and Sternson, S. M. (2011). Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146, 992–1003. doi: 10.1016/j.cell.2011.07.039
Keywords: synaptic plasticity, BDNF, HFD, microarray, neurogenesis, CAMKII phosphorylation, IBA1, vGLUT2
Citation: Bae-Gartz I, Janoschek R, Breuer S, Schmitz L, Hoffmann T, Ferrari N, Branik L, Oberthuer A, Kloppe C-S, Appel S, Vohlen C, Dötsch J and Hucklenbruch-Rother E (2019) Maternal Obesity Alters Neurotrophin-Associated MAPK Signaling in the Hypothalamus of Male Mouse Offspring. Front. Neurosci. 13:962. doi: 10.3389/fnins.2019.00962
Received: 29 May 2019; Accepted: 28 August 2019;
Published: 13 September 2019.
Edited by:
Kathleen C. Page, Bucknell University, United StatesReviewed by:
Marcio Alberto Torsoni, Campinas State University, BrazilAnvita Kale, Bharati Vidyapeeth Deemed University, India
Copyright © 2019 Bae-Gartz, Janoschek, Breuer, Schmitz, Hoffmann, Ferrari, Branik, Oberthuer, Kloppe, Appel, Vohlen, Dötsch and Hucklenbruch-Rother. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Hucklenbruch-Rother, ZXZhLnJvdGhlckB1bmkta29lbG4uZGU=