 Panagiotis Christidis1
Panagiotis Christidis1 Abhya Vij2
Abhya Vij2 Stamatios Petousis3Javid Ghaemmaghami4Bhairav V. Shah5
Stamatios Petousis3Javid Ghaemmaghami4Bhairav V. Shah5 Ioannis Koutroulis6
Ioannis Koutroulis6 Panagiotis Kratimenos4,7*
Panagiotis Kratimenos4,7*- 1Laboratory of Physiology, Faculty of Health Sciences, School of Medicine, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 2Department of Pediatrics, Boston Children's Hospital and Harvard Medical School, Boston, MA, United States
- 32nd Department of Obstetrics and Gynecology, “Hippokrateion” General Hospital of Thessaloniki, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 4Center for Neuroscience Research, Children's National Research Institute, Washington, DC, United States
- 5Division of Pediatric Surgery, Department of Pediatrics, School of Medicine, Prisma Health Children's Hospital-Midlands, University of South Carolina, Columbia, SC, United States
- 6Department of Pediatrics, Division of Emergency Medicine, Children's National Hospital, George Washington University School of Medicine and Health Sciences, Washington, DC, United States
- 7Division of Neonatology, Department of Pediatrics, Children's National Hospital, George Washington University School of Medicine and Health Sciences, Washington, DC, United States
Background: Hypoxic-ischemic encephalopathy (HIE) is a major cause of neonatal morbidity and mortality worldwide. While the application of therapeutic hypothermia has improved neurodevelopmental outcomes for some survivors of HIE, this lone treatment option is only available to a subset of affected neonates. Src kinase, an enzyme central to the apoptotic cascade, is a potential pharmacologic target to preserve typical brain development after HIE. Here, we present evidence of the neuroprotective effects of targeting Src kinase in preclinical models of HIE.
Methods: We performed a comprehensive literature search using the National Library of Medicine's MEDLINE database to compile studies examining the impact of Src kinase regulation on neurodevelopment in animal models. Each eligible study was assessed for bias.
Results: Twenty studies met the inclusion criteria, and most studies had an intermediate risk for bias. Together, these studies showed that targeting Src kinase resulted in a neuroprotective effect as assessed by neuropathology, enzymatic activity, and neurobehavioral outcomes.
Conclusion: Src kinase is an effective neuroprotective target in the setting of acute hypoxic injury. Src kinase inhibition triggers multiple signaling pathways of the sub-membranous focal adhesions and the nucleus, resulting in modulation of calcium signaling and prevention of cell death. Despite the significant heterogeneity of the research studies that we examined, the available evidence can serve as proof-of-concept for further studies on this promising therapeutic strategy.
Introduction
One in four perinatal deaths is attributed to hypoxic-ischemic (HI) brain injury, a term that describes acute interruptions in oxygenated blood flow to the brain (Lawn et al., 2005; Pauliah et al., 2013). Following the perinatal asphyxiation insult, HI involves a cascade of biochemical events that cause cerebral edema, inflammation, neuronal cell injury, and ultimately, neuronal cell death over a period of hours to days (Gunn and Thoresen, 2006; Cilio and Ferriero, 2010; Juul and Ferriero, 2014; Hagberg et al., 2015; Van Bel and Groenendaal, 2016; Delivoria-Papadopoulos et al., 2018). While there have been improvements in survival rates after neonatal HI, many of these patients suffer ongoing neurological impairments that both lessen quality of life and incur burdensome healthcare costs (Blencowe et al., 2013; Eunson, 2015).
The only evidence-based treatment currently available for neonatal HI is therapeutic hypothermia (TH). This approach is targeted at modulating the deleterious cytotoxic and inflammatory processes that occur during HI by tightly regulating temperature (Wyatt et al., 2007), blood pressure, ventilation, and glucose metabolism; for some patients, application of TH has resulted in improved neurological outcomes (Filan et al., 2006; Tam et al., 2012; Wong et al., 2013).
While conceptually promising, the application of TH has several limitations. First, it is mainly available in high-resource countries. Moreover, the protocol requires that total body cooling be initiated within 6 h of birth, leaving clinicians with a narrow window to establish the diagnosis, assess the severity of HI and implement treatment with TH. This time is further compressed for centers that do not have the necessary advanced equipment, staffing, monitoring and experience to provide neonatal TH, as attempts are made to transfer the patient to a suitable facility (Olsen et al., 2013). Finally, despite the use of TH, both the overall mortality rates and the disability rates following application of TH after neonatal HI in published trials remain high (Shankaran et al., 2012).
Most strikingly, even when TH is timely applied in an advanced neonatal intensive care unit, only a subset of neonates with HI have been shown to benefit (Gunn and Thoresen, 2006). Thus, there is a critical and urgent need to develop additional therapeutic strategies that address both morbidity and mortality following neonatal HI. To this end, multiple pre-clinical in vivo studies have focused primarily on (a) elucidating the molecular biology underlying HI, (b) identifying potential molecular targets in pathways integral to cerebral injury, (c) optimizing cooling strategies, and (d) recognizing adjuvant therapies that could augment the neuroprotective effects of TH (Jacobs et al., 2013). These studies implicate potential targets of the apoptotic cascade that may, when modulated by pharmacological intervention, offer additional or alternative therapies for HI, critical in cases where TH is not available, particularly in low-resource countries (Robertson et al., 2008; Pauliah et al., 2013; Montaldo et al., 2015), and in treatment of patients who do not respond or respond insufficiently to TH.
In particular, these studies have shown that Src kinase is involved in numerous activated intracellular pathways during HI (Paul et al., 2001; Mishra et al., 2009; Haass and Mandelkow, 2010; Ittner et al., 2010; Delivoria-Papadopoulos et al., 2011; Liu and Sharp, 2011; Hossain et al., 2012; Angelis and Delivoria-Papadopoulos, 2017a,b; Kratimenos et al., 2017). However, there is conflicting evidence regarding its regulatory role, which may differ depending upon brain maturation. Hossain et al. demonstrated that Src kinase activation improves neuronal survival in primary cortical cell cultures (Paul et al., 2001; Haass and Mandelkow, 2010; Ittner et al., 2010; Liu and Sharp, 2011; Hossain et al., 2012), whereas several other studies have shown that Src kinase phosphorylation causes neuronal damage in ischemic stroke, intracerebral hemorrhage, and Alzheimer's disease (Haass and Mandelkow, 2010; Ittner et al., 2010; Liu and Sharp, 2011; Hossain et al., 2012). Porcine experimental models have been used to examine the deleterious effects of Src kinase in neonatal HI and have shown that it can induce the production of free radicals, causing secondary inflammation and excitotoxicity (Kratimenos et al., 2017, 2018, 2022).
Selective Src inhibitors (Src-i) exhibit effectiveness against neuronal cell injury in neonatal and developing animal models and offered neuroprotection as demonstrated by histologic, biochemical, and neurobehavioral assessments (Mishra et al., 2009; Delivoria-Papadopoulos et al., 2011; Angelis and Delivoria-Papadopoulos, 2017a,b; Kratimenos et al., 2017). Contrary to those results, experiments in adult mice showed that Src kinase inhibition worsens cerebral injury (Wang et al., 2004; Guo et al., 2006; Wu et al., 2008; Hu et al., 2009; Tian et al., 2009). In addition, several studies have highlighted the role of Src kinase in neuronal survival after ischemia/reperfusion (I/R) through interactions with the extracellular signal-regulated kinase (ERK) (Wang et al., 2004; Guo et al., 2006; Wu et al., 2008; Hu et al., 2009; Tian et al., 2009).
This systematic review aims to examine the current knowledge regarding the role of Src kinase in neonatal HI and the potential neuroprotective effects of selective Src manipulation.
Materials and methods
Protocol
A review of relevant preclinical studies was performed to summarize the current knowledge regarding the role of Src kinase inhibition and its potential benefits on the neonatal hypoxic-ischemic brain. We utilized the CAMARADES (Collaborative Approach to Meta-Analysis and Review of Animal Data from Experimental Studies) guidelines in the methodology (De Vries et al., 2015).
Literature search
A literature search for the Medline electronic database for all studies up to July 01, 2022. Below, the search strategy for the database is presented: (((“hypoxia”[MeSH Terms] OR “hypoxia”[All Fields]) OR (“ischaemia”[All Fields] OR “ischemia”[MeSH Terms] OR “ischemia”[All Fields])) OR ((“cerebrum”[MeSH Terms] OR “cerebrum”[All Fields] OR “cerebral”[All Fields] OR “brain”[MeSH Terms] OR “brain”[All Fields]) OR (“brain”[MeSH Terms] OR “brain”[All Fields]))) AND Src kinase[tiab]. The reference lists of the retrieved articles were subsequently manually reviewed to identify any additional studies that would be considered for inclusion.
Inclusion criteria
Preclinical studies were included. For inclusion, experimental study protocols were required to involve animals treated with any kind of Src kinase inhibitor before or after the induction of HI. The outcome measures of the studies analyzed were required to include “neuroprotection” defined by histologic, biochemical, and/or neurobehavioral findings.
The current review defines HI as an acute interruption of blood flow and oxygen to the brain. In preclinical animal models, HI is typically induced via ligation or occlusion of the common carotid artery or by decreasing oxygen concentration in mechanically ventilated animals. Given the bilaterality of neonatal HI, we focused on experimental protocols that induced global brain hypoxia rather than unilateral hypoxia. Global transient hypoxia could be induced by four-vessel occlusion (4VO) of both vertebral arteries and common carotid arteries, by bilateral occlusion of common carotid arteries (2VO), or by titration of the FiO2 below 0.21 (drop of FiO2 to 0.05–0.006 within 5 min, maintained for the 60 min period and titrated to achieve a 40 % reduction in systolic BP from baseline) in a controlled environment for a period of time (Traystman, 2003).
Exclusion criteria
We excluded all studies that were based on cell lines and in vitro experiments. Articles written in languages other than English were also excluded. We also excluded studies that treated animals with unilateral ligation of one of the carotid arteries or with occlusion of fewer than four vessels since these techniques are commonly used in stroke models. Studies that did not use selective Src kinase inhibitors or studies in which the intervention did not directly result in Src kinase modulation were not eligible for inclusion.
Risk of bias assessment
Assessment of risk for bias was based on the Systematic Review Center for Laboratory Animal Experimentation (SYRCLE) Risk of Bias (RoB) tool (Hooijmans et al., 2014), which was derived by the Cochrane Risk of Bias tool. SYRCLE RoB tool consists of nine questions adjusted for the specific characteristics of bias contributing to the results of interventional preclinical studies. Each question was marked as “Yes,” “No,” or “Unclear.”
Data extraction
From each study, the following data were extracted: authors' names, year of publication, sample size, type of animal model and age, type of Src kinase inhibitor and timing of administration, method for HI induction, Src kinase inhibition outcomes, role of Src kinases (protective/damaging on neuron's survival) and whether reperfusion took place.
Results
Eligible studies
Results from the Medline database were combined by a citation manager software (Mendeley Desktop, Version 1.19.8), and duplicate entries were removed. Our search yielded 443 studies that were then screened by title and abstract to meet our criteria. Eighty-nine studies were found relevant to this review and required further analysis. Access to full-text articles, despite repeated efforts, was not possible for six studies, thus these were excluded. The remaining eighty-three studies were screened using our pre-defined inclusion and exclusion criteria. Sixty-three studies did not meet our inclusion criteria. Most studies (twenty-one) were excluded due to the use of cell cultures. Other common reasons for exclusion were the lack of an Src kinase inhibitor in the protocol (16 studies) or of a hypoxic event (nine studies). Six studies described unilateral brain hypoxia (mimicking stroke), seven studies were literature reviews and, finally, four articles were published in a non-English language. Table 1 summarizes the reasoning used for exclusion of the sixty-three studies. Ultimately, twenty studies were considered eligible for this analysis. Due to the variability in experimental methods and outcomes, it was impossible to perform a meta-analysis on the effect of Src kinase inhibition on the neonatal brain after hypoxia, so a qualitative approach was taken instead. An illustration of our methodology and approach is demonstrated in Figure 1.

Table 1. Studies not eligible based upon the exclusion criteria.
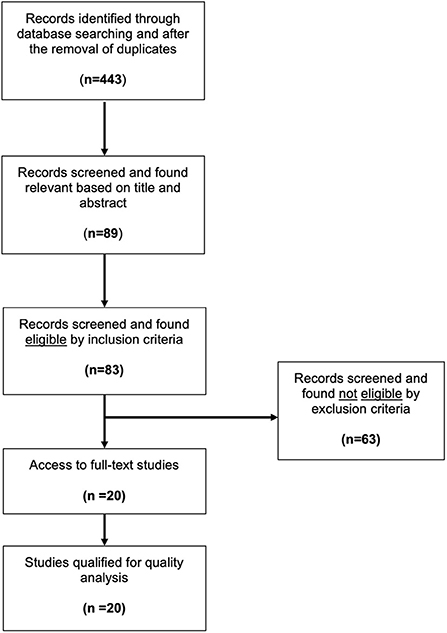
Figure 1. Flow diagram of the literature search. Modified from the preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) and modified to accurately depict the literature research (Moher et al., 2009).
Risk for bias assessment
As previously described, the evaluation of risk for bias was based on the SYRCLE RoB tool (Hooijmans et al., 2014). The twenty studies that were included in this review were evaluated for selection, detection, performance, and attrition bias. All but six studies (14/20, 70%) were prone to selection bias because the authors did not adequately describe the methodology used for assigning the animals to the experimental groups. In all twenty studies (20/20, 100%), the investigators were not blinded to the intervention (performance bias), the animals were not housed randomly (performance bias), or the animals were not randomized for outcome assessment (attrition bias). Only two studies (2/20, 10%) had two independent assessors review their results (neuropathology scores) and only two studies (2/20, 10%) specifically described the allocation concealment process. All the studies had animals that were comparable at baseline prior to group assignment, did not selectively present their outcomes, and appropriately addressed incomplete data. The results from the risk for bias analysis are shown in Table 2.
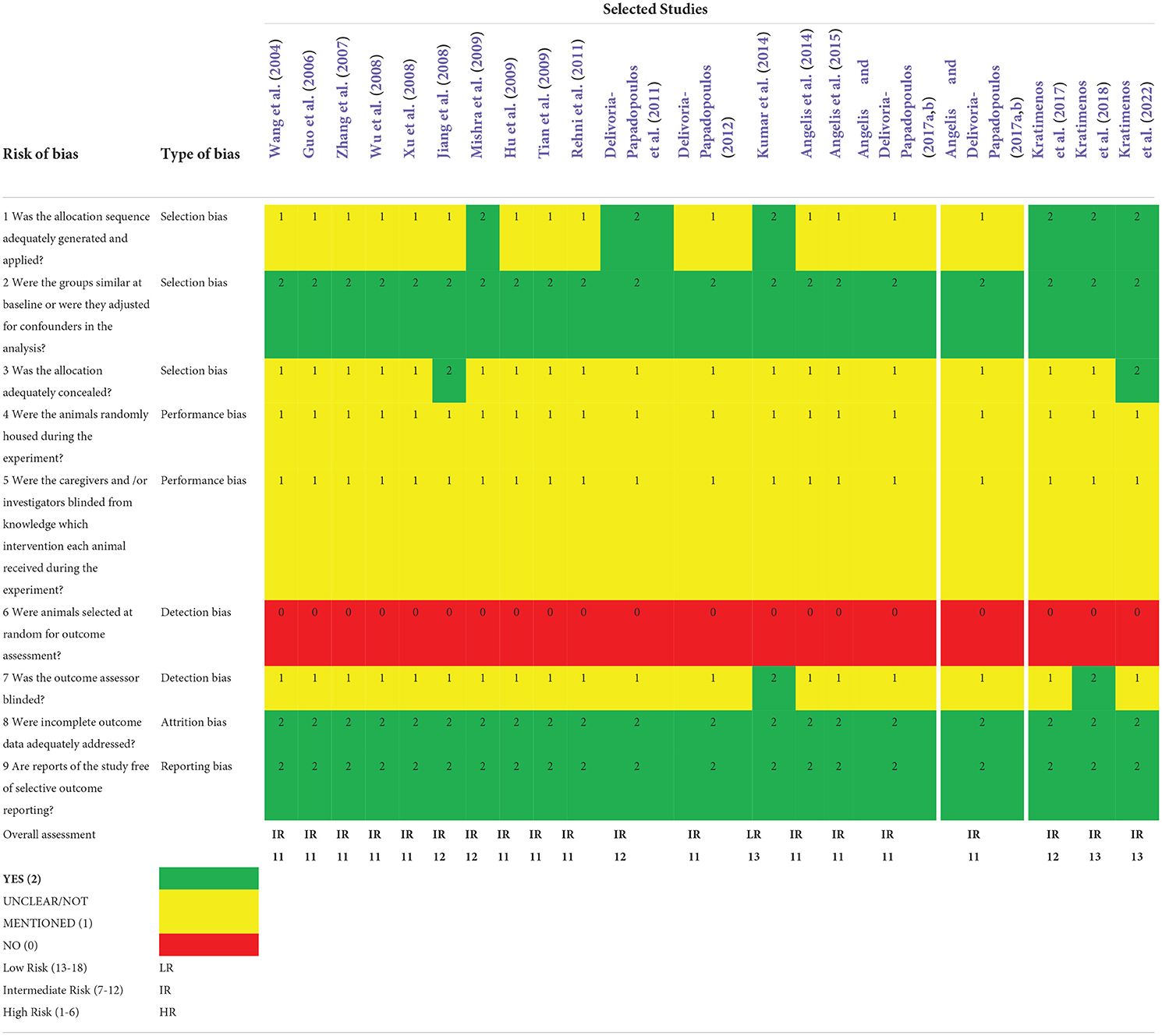
Table 2. Assessment of risk of bias based upon SYRCLE RoB tool.
Study characteristics
Four different species of animals were used in the included studies: Sprangue-Dawley rats (nstudies = 7, 35%), Yorkshire newborn piglets (nstudies = 10, 50%), Swiss albino mice (nstudies = 2, 10%), and mice CD1 strain (nstudies = 1, 5%). Twelve studies (nstudies = 12, 60%) specified the total number of animals used; the remaining studies (nstudies = 8, 40%) did not document the number of animals and the size of the assigned groups.
In all studies, a single dose of Src inhibitor was administered. In eighteen studies (nstudies = 18, 90%) the investigators administered a selective Src kinase inhibitor (PP1, PP2, PP3, or SU6656). The remaining two studies (nstudies = 2, 10%) included two different non-selective inhibitors of Src kinase, the neuronal nitric oxide synthase inhibitor (nNOSi) and (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA), which also modulated the activity of the Src kinase after the hypoxic insult. In addition, one study (nstudies = , 5%) combined therapeutic hypothermia with Src-i. The Src-i and their characteristics are presented in Supplementary Table 1.
Different models of HI were used in the included studies, primarily depending on the animal species involved. Eleven studies (nstudies = 11, 55%) used FiO2 titration to establish transient cerebral hypoxia; most of these studies (10/11, 91%) utilized piglets. In two studies (nstudies = 2, 10%), investigators performed bilateral carotid occlusion (2VO) in Swiss albino mice and in seven of them (nstudies = 7, 35%) they used the method of 4VO in Sprangue-Dawley rats. According to the data that were extracted, reperfusion ensued after HI in every study. Analyzed studies, the species and number of animals, method of HI and type of Src-i used are summarized in Table 3.

Table 3. The included studies and their descriptive statistics.
Concerning the timing of intervention, the experimental compound was administered after the onset of the HI event in only three of the twenty studies (nstudies = 3, 15%), mimicking the actual sequence of events in clinical practice. In the remaining seventeen studies (nstudies = 17, 85%), the compound was given prior to the induction of the hypoxic insult. Two studies (nstudies = 2, 10%) described conditioning which entails several brief repetitive cycles of ischemia with intermittent reperfusion prior to or subsequently to prolonged ischemia (Rehni et al., 2011; Kumar et al., 2014). Both studies demonstrated the neuroprotective properties of the Src kinase under HI conditions. It is worth mentioning that studies (nstudies = 12, 60%) that focused exclusively on biochemical analyses did not include a sham or control group for direct comparisons (treated vs. non-treated) of the effectiveness of the therapy with Src-i.
The inclusion of eligible studies led to the assessment of histologic, biochemical, and neurobehavioral outcomes. Histologic analyses included cortical and striatal lesions. Moreover, biochemical parameters, such as the enzymatic expression or activity related to neurological damage, and the cerebral energy status as quantified by ATP and phosphocreatine (PCr) concentrations, were evaluated. In ten out of twenty studies (nstudies = 10, 50%) sufficient cerebral hypoxia was induced for both the treatment and control groups as confirmed by energy production levels. Neurobehavioral indicators were examined with neurobehavioral tests designed to measure cognitive function (memory) and motor coordination. Two studies (nstudies = 2, 10%) included both histologic and neurobehavioral outcomes, eleven studies (nstudies = 11, 55%) only biochemical outcomes and six (nstudies = 6, 30%) reported both biochemical and histologic outcomes. It is worth mentioning that one study (nstudies = 1, 5%) utilized experimental data to create and validate a computational model of the critical intracellular signaling components of HI in neonatal brain (Kratimenos et al., 2022).
Of the studies included in our analysis, six (nstudies = 6, 30%) demonstrated that Src kinase phosphorylation was neuroprotective, whereas 14 studies (nstudies = 14, 70%) provided evidence that the effects of Src kinase can be deleterious while its inhibition provides neuroprotection. Six out of twenty studies (nstudies = 6, 30%), where Src kinase activity had a beneficial impact, were conducted on adult animal models. The studies in which Src kinase activity led to worse outcomes were conducted on either newborn animals (nstudies = 9, 45%) or adult animals (nstudies = 5, 25%). The time of intervention, type of outcome measures, and the role of Src kinase in selected studies are presented in Table 4.
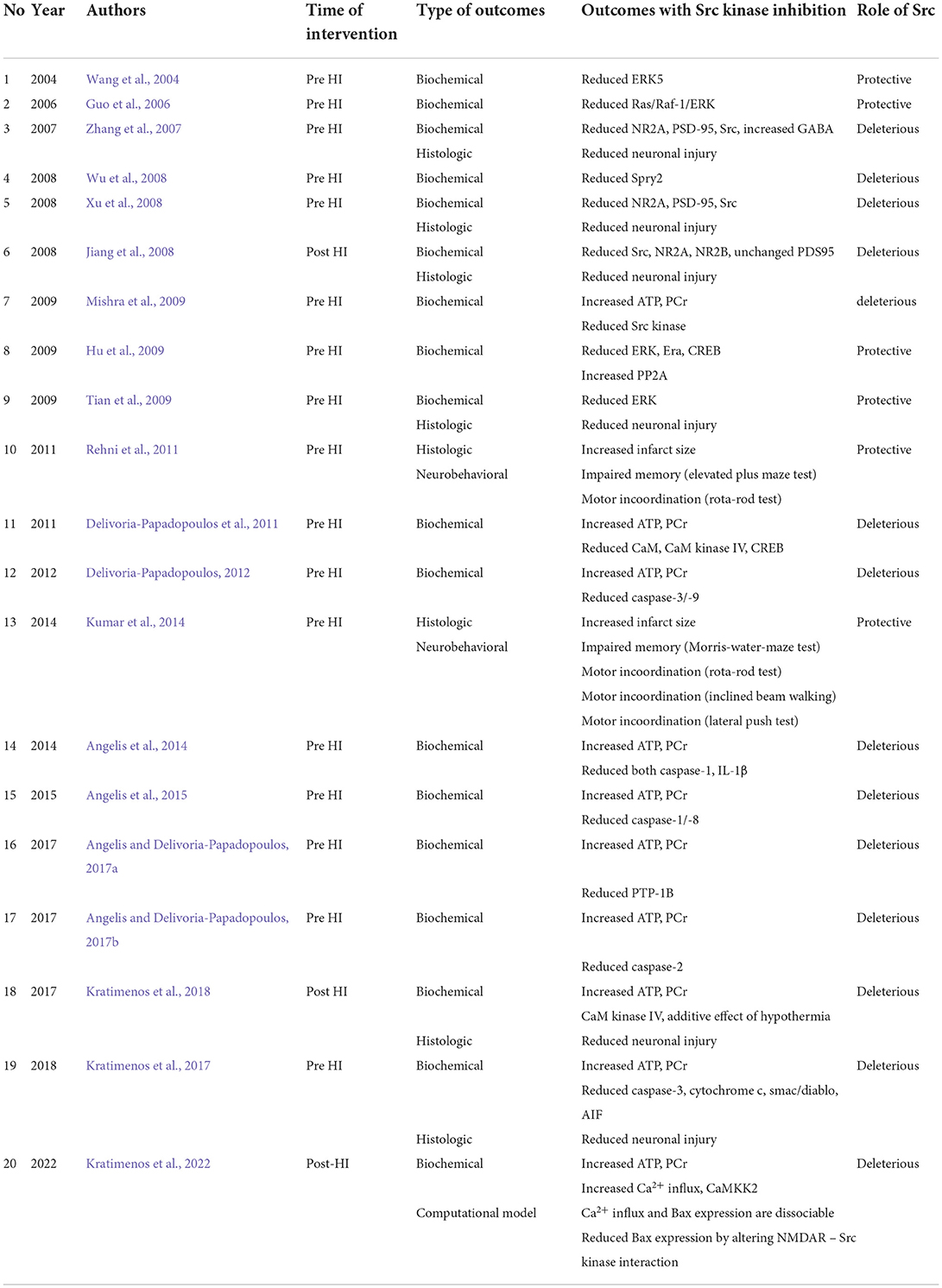
Table 4. The included studies and their descriptive statistics.
Discussion
This paper highlights the current evidence on neuroprotective effects of Src kinase modulation as demonstrated by histologic, biochemical, and neurobehavioral outcomes in twenty eligible studies. Notably, all the included studies were based on a single dose regimen, but only 15% of the studies administered Src-i post-HI. This treatment timing is a key consideration, as in actual clinical practice, treatment with Src-i would likewise occur post-HI.
Although various animal models were utilized in the studies included, all inhibitors used were selective for Src kinase. Each preclinical study that examined models of the neonatal age group reported neuroprotective effects of treatment with Src-i, whereas studies in adult rats showed the opposite effect. This discrepancy may be attributed to the pathophysiologic differences between neonatal and adult brains as it pertains to susceptibility to injury, plasticity and cell death pathway activation (Sands et al., 1979; Clancy et al., 2007; Pressler and Auvin, 2013).
Methodological quality assessment using the SYRCLE's RoB tool yielded an intermediate risk for bias scores for the evaluated studies in this review. Due to the variability of experimental methods and outcomes used, it was not possible to perform a meta-analysis on the effect of Src kinase inhibition on the neonatal brain after hypoxia.
Pathophysiology of HI in neonatal brain
There are three major mechanisms of neuronal cell death during global ischemia: generation of free radicals, excitotoxicity, and inflammation. Each mechanism is mediated through inflammatory cascades that require phosphorylation of enzymatic regulatory sites by Src kinases (Mishra et al., 2009; Delivoria-Papadopoulos et al., 2011; Delivoria-Papadopoulos, 2012; Angelis et al., 2014, 2015; Angelis and Delivoria-Papadopoulos, 2017a,b; Kratimenos et al., 2017, 2018). Aligned with these findings, experiments on newborn piglets demonstrated that inhibition of Src kinase phosphorylation after HI by a selective antagonist (Src-i) is a novel mechanism of neuroprotection (Mishra et al., 2009; Delivoria-Papadopoulos et al., 2011; Delivoria-Papadopoulos, 2012; Angelis et al., 2014, 2015; Angelis and Delivoria-Papadopoulos, 2017a; Kratimenos et al., 2017, 2018). The proposed mechanism of the apoptosis-induced cell death in a developing neuron is illustrated in Figure 2.
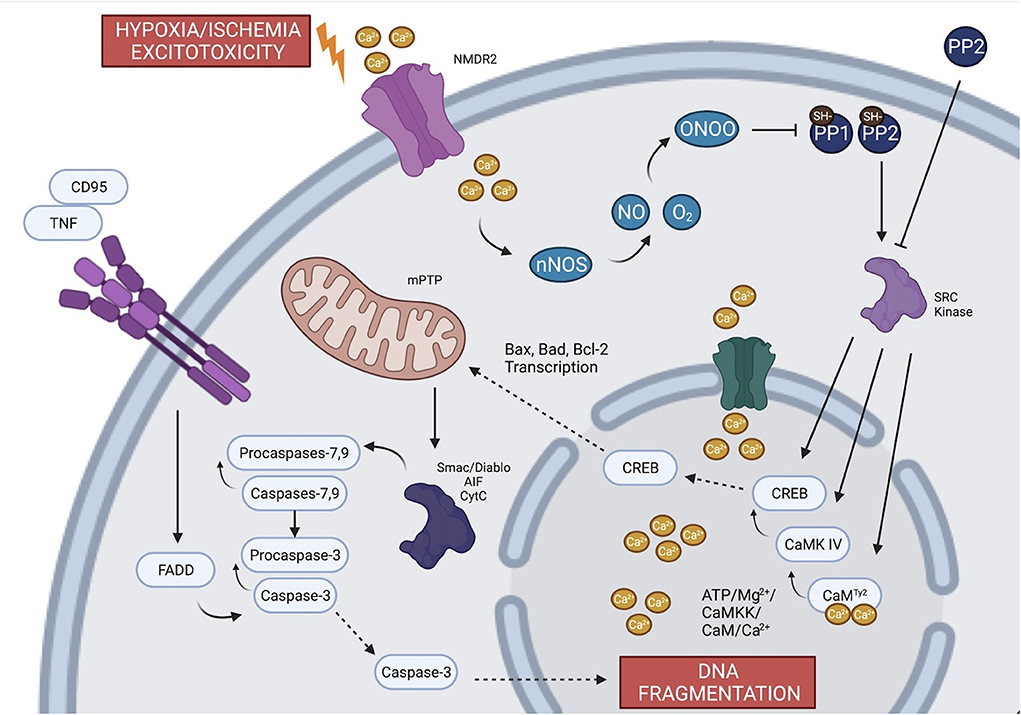
Figure 2. The proposed mechanism of the apoptotic pathway in a developing neuron. During global ischemia, evidence suggests that the three major mechanisms of neuronal cell death (excitoxicity, generation of free radicals and inflammation) are mediated through the phosphorylation of regulatory sites by Src kinases. Failure of the oxidative phosphorylation gives rise to intracellular Ca2+ which triggers ecotoxicity and potentiates calcium-dependent apoptotic pathways. Calcium contributes to the generation of free radicals through NO and nNOS and leads to transcription of factors that induces leakage of apoptotic proteins from mitochondria. The inflammation and the extrinsic apoptotic pathway are mediated through TNF and different types of caspases. Created with BioRender.com.
The activation of Src kinases in HI is dependent, in part, upon formation of free radicals, which are known to develop during HI in the setting of decreased oxidative phosphorylation given decreased O2. Nitric oxide (NO) free radicals, which are produced by neuronal nitric oxide synthase (nNOS), react with superoxide to form peroxynitrate. Peroxynitrate then inactivates protein tyrosine phosphatases, SH-PTP-1 and SH-PTP-2 via reduction mechanisms on cysteine residues (Lee et al., 1998; Barrett et al., 1999; Takakura et al., 1999), facilitating the activation of Src kinases (Mishra et al., 2009).
In addition to the generation of free radicals as oxidative phosphorylation fails, this metabolic alteration also induces excitotoxicity through the depolarization and activation of voltage-gated Ca2+-channels with a subsequent rise in intracellular Ca2+ (White et al., 2000). Depolarization of those channels results in the release of excitatory neurotransmitters such as glutamate into the synaptic cleft (Fujimoto et al., 2004) and inability of the glutamate reuptake mechanisms to clear glutamate from the cleft. This cascade of events leads to the upregulation of gated NMDARs, further increasing intracellular Ca2+ and activating calcium-dependent apoptotic pathways (excitotoxicity) (Arundine and Tymianski, 2004; Delivoria-Papadopoulos et al., 2011). Additionally, excitotoxicity is augmented by the concurrent phosphorylation of certain subunits of NMDAR by Src kinase (Chen et al., 2003; Salter and Kalia, 2004).
Calcium itself plays a critical role in HI-mediated neuronal cell injury (Delivoria-Papadopoulos et al., 2007). During hypoxia, nuclear Ca2+ forms a complex with calmodulin (CaM), which activates an apoptotic cascade (Delivoria-Papadopoulos et al., 2011). This cascade involves Ca2+-dependent kinases such as the CaM kinase-dependent kinase (CaMKK) and CaM kinase IV (CaMKIV), primarily located in the neuronal cell nucleus. CaMKK directly activates CaMKIV via phosphorylation of threonine 200 (Thr200) or threonine 196 (Thr196) in a process that requires Ca2+/CaM and ATP/Mg2+. Activated CaMKIV mediates DNA transcription by phosphorylating cyclic AMP response binding protein (CREB) (Mishra et al., 2006; Delivoria-Papadopoulos et al., 2007, 2011; Hornick et al., 2007). During HI, Src kinase phosphorylates CaM at tyrosine 99 (Tyr99), CaMKIV at Thr200 or Thr196 and CREB protein at Ser133, promoting the expression of pro-apoptotic proteins (Delivoria-Papadopoulos et al., 2011). As discussed, inhibition of Src kinase by a potent selective inhibitor has been shown to ameliorate the impact of HI in the cerebral cortex of newborn piglets (Delivoria-Papadopoulos et al., 2011).
Programmed cell death occurs through two pathways: intrinsic and extrinsic. The intrinsic pathway is mediated by the generation of free radicals (oxidative stress) and caspases, whereas the extrinsic pathway is mediated by inflammation and tumor necrosis factor (TNF) (Bleicken et al., 2013; Lukyanova and Kirova, 2015). The Bcl-2 family protein includes several anti-apoptotic (Bcl-2, Bcl-xL, and Bcl-w) and pro-apoptotic (Bax, BAD, Bak, or Bok) proteins, whose transcription is induced by HI (Bleicken et al., 2013; Lukyanova and Kirova, 2015). Activation of the CREB protein leads to apoptosis by the transcription of Bax and suppression of Bcl-2 (Kratimenos et al., 2017). Porcine animal models have shown that activated Bax forms pores in the mitochondrial membrane, allowing the leakage of apoptosis-inducing factor (AIF), Smac/Diablo and cytochrome c in the cytosol (Kratimenos et al., 2017). The release of these molecules is further facilitated by the action of Src kinases that expand the opening of the mitochondrial permeability transition pore (mPTP) (Kratimenos et al., 2017). The translocation of apoptotic factors activates caspases −3, −7, and −9, which results in DNA fragmentation and neuronal cell death (Kratimenos et al., 2017). These caspases mediate the activation of the intrinsic apoptotic pathway, whereas the extrinsic pathway is activated by TNF through the binding of Fas ligand (CD95L) to the CD95 receptor. This process results in the formation of the complex FAS-associated death domain (FADD) which then activates caspase-8 (Boatright et al., 2003). Caspase-8 induces apoptosis via caspase-3 activity. Additionally, in vivo studies have demonstrated the inhibitory effect of Protein Phosphatase 2 (PP2) on caspase-8, which would in this case ameliorate the observed apoptosis (Angelis et al., 2015).
Research indicates that activity of both caspase-8 and caspase-1 acutely increases in the newborn piglet brain following hypoxia (Angelis et al., 2015). Capsase-1 activation is directly related to neuro-inflammation and contributes to the production of IL-1β through the formation of inflammasomes (Angelis et al., 2015). Furthermore, Src kinase inhibition protects cortical neurons from the deleterious sequelae of inflammation caused by HI (Angelis et al., 2015). Additionally, there is evidence that Src kinase is involved in apoptotic cascade activation and pro-neuroinflammatory pathways, ultimately leading to neuronal cell death (Angelis et al., 2015).
N-methyl-D-aspartate receptors (NMDARs) in HI
The balance between neuronal inhibition and excitation is an essential homeostatic mechanism for typical brain function and development. HI disrupts this homeostasis by upregulating excitation, resulting in apoptosis and neuronal cell death (Seeburg, 1993; Hollmann and Heinemann, 1994). The excitatory component of this mechanism is mediated by glutamate and the NMDARs (Kumari and Ticku, 2000). NMDARs and their associated signaling pathways are located at the electron-dense matrix beneath the postsynaptic membrane of excitatory synapses, called postsynaptic density (PSD) (Kennedy, 1997; Martone et al., 1999). HI-induced activation of Src kinases upregulates NMDARs, increasing excitation by phosphorylating tyrosine residues on the NR2A and NR2B subunits. In addition, transient ischemia causes changes in the structure and protein composition of the PSD, enhancing their association with certain proteins (Kennedy, 1997; Martone et al., 1999).
The interaction between NR2A, Src kinase and post-synaptic density protein 95 (PSD-95) has been implicated in HI-induced neuronal injury. Liu et al. suggest that this mechanism also includes the activation proline-rich kinase 2 (Pyk2). After ischemia-reperfusion (I/R), activated Pyk2 binds to Src kinase, promoting phosphorylation of NR2A and calcium overload via PSD-95 (Liu et al., 2005). PP2 can block the NR2A-PSD95-Src signaling pathway and alleviate neuronal cell injury (Zhang et al., 2007; Jiang et al., 2008). Other than PP2, MK-801, a selective antagonist of NMDARs, can reverse Src kinase's activation and its effect on NR2A during HI. A novel approach using a computational model also predicted that Src-i can modulate the interaction between the NMDARs and Src and can significantly reduce Bax expression (39).
In the setting of HI-induced excitotoxicity, targeting gamma-aminobutyric acid (GABA) signaling is a potentially effective therapeutic strategy. GABA is the primary inhibitory neurotransmitter in the CNS that balances the excitatory effects of glutamate (Oja et al., 1990; Rosenbaum et al., 1990; Johansen and Diemer, 1991; Sivilotti and Nistri, 1991). Several researchers have proposed that enhancing GABAergic activity could potentially alleviate the excitotoxic effects of ischemic brain injury (Oja et al., 1990; Rosenbaum et al., 1990; Johansen and Diemer, 1991; Sivilotti and Nistri, 1991). The effects of GABA on ischemia are mediated by the activation of GABAA, which increases Cl− permeability and hyperpolarizes cells. Hyperpolarized cells demonstrate reduced excitability due to decreased glutamate concentrations and calcium influx (Oja et al., 1990; Rosenbaum et al., 1990; Johansen and Diemer, 1991; Sivilotti and Nistri, 1991). Furthermore, Zhang et al. demonstrated that muscimol and baclofen, both GABA receptor agonists, prevent hippocampal CA1 neurons' death during cerebral I/R via suppression of the phosphorylation of excitatory NMDA receptor subunit NR2A (Zhang et al., 2007). Interestingly, muscimol's and baclofen's neuroprotective properties are linked to the downregulation of the phosphorylation of Src kinase and NMDARs. In addition, administration of (RS)-alpha-amino-3-hydroxy-5-tert-butyl-4-isoxazolepropionic acid (ATPA), an agonist of GluR5 (glutamate receptor 5)-containing kainate receptor, also demonstrated neuroprotective effects (Zhang et al., 2007). Xu et al. hypothesized that this was secondary to increased GABA release and inhibition of the NR2A-PSD95-Src signaling pathway (Xu et al., 2008). However, some researchers have also reported conflicting results, indicating that increased GABA signaling after HI may accelerate neuronal cell loss (Rosenbaum et al., 1990; Stokes et al., 2001).
The dual role of Src kinase following ischemia-reperfusion (I/R)
Several studies have highlighted Src kinase's role in neuronal survival after I/R through interactions with the extracellular signal-regulated kinase (ERK) (Wang et al., 2004; Guo et al., 2006; Wen et al., 2008; Wu et al., 2008; Hu et al., 2009; Tian et al., 2009). Following I/R, Src kinase and NMDARs upregulate ERK, increasing neuronal survival (Wang et al., 2004; Guo et al., 2006). HI-induced activation of Src kinase leads to the phosphorylation of Raf at the Tyr340/341 position. Raf-1, an upstream molecule of the ERK pathway, subsequently induces the phosphorylation of estrogen receptor a (ERa) and CREB at Ser133 position, promoting neuronal cell survival (Wang et al., 2004; Guo et al., 2006; Wu et al., 2008; Hu et al., 2009). Despite some reports of Src kinase-related increases in neuronal survival, several studies showed that Src kinase inhibition by PP2A was neuroprotective following I/R. Wu et al. demonstrated that Src kinase's induction of neuronal apoptosis following I/R, is mediated by the phosphorylation of Spry2, a down-regulator of Raf/ERK pathway (Wu et al., 2008). Administration of PP2 or SU6656 shows an attenuation of Src kinase's negative effect on cellular death in rat hippocampi (Wang et al., 2004; Guo et al., 2006; Hu et al., 2009; Tian et al., 2009). The results of the aforementioned studies cannot be translated to clinical neonatology practice because of the use of adult mice and their differences in pathophysiology, mainly on the mechanism of injury and recovery when compared to neonatal mice (Sands et al., 1979; Clancy et al., 2007; Pressler and Auvin, 2013). Moreover, intact Src kinase is correlated with improved neuronal survival, whereas in conditions of excitotoxicity, calpain cleavage of Src kinase generates a neurotoxic truncated Src fragment (Hossain et al., 2015).
Pre-/post-conditioning
As mentioned previously, Src kinase plays a pivotal role in neuronal health among multiple disease models, including conditioning. Conditioning involves the intermittent reperfusion that precedes or follows prolonged ischemia. These are termed ischemic preconditioning (IPrCo) and ischemic post-conditioning (IPoCo), respectively (Kumar et al., 2014). Studies have shown that both IPrCo and IPoCo prevent cerebral infarct formation by ischemia-reperfusion and prevent neurobehavioral impairment in Swiss albino mice (Rehni et al., 2008a, 2011; Kumar et al., 2014). IPrCo's neuroprotective effect on the brain is likely attributable to amelioration of the ischemia and reperfusion sequela through the activation of the Akt/p38-mitogen/ERK pathway (Bochelen et al., 1999; Rehni et al., 2008b, 2009; Kumar et al., 2014). However, Rehni et al. demonstrated that the effect of IPrCo is exerted through phosphorylation of Src kinase, even though the exact activation transduction pathway is not yet well understood (Rehni et al., 2008a, 2011). Neuronal cell injury was also ameliorated by IPoCo through the activation of Src kinase (Kumar et al., 2014). Although the exact mechanism remains unknown, Kumar et al. suggested that it also involves the activation of Akt/p38-mitogen/ERK (Kumar et al., 2014). In a clinical setting, IPrCo is not feasible due to the inability to predict the onset of ischemia. IPoCo, on the other hand, is clinically relevant.
Why are findings for Src-i not yet translatable?
Although Src kinase inhibition exhibits a neuroprotective effect on neonatal animal models, this has not yet been confirmed in clinical trials. The variability of experimental results and animal models used has prevented the introduction of Src kinase inhibition as a therapeutic approach in clinical trials. Neurobehavioral experiments could provide additional data to support the use of Src-i, however they require a longer follow-up period with highly trained personnel, as well as validated scoring systems for accuracy and consistency. In the present analysis, only two studies provided a complete set of outcomes that were evaluated with multiple different approaches (Rehni et al., 2011; Kumar et al., 2014). The use of histologic and biochemical markers has been proven to be a cost-effective alternative approach. Further investigations are required to elucidate the precise mechanism by which Src kinase affect cortical neurons. Many Src kinase inhibitors like dasatinib are currently being tested as adjuvant therapies in cancer. Pharmacokinetic data of such inhibitors including PP2, a more selective Src kinase inhibitor used in thirteen out of the 20 included studies, are still lacking. Finally, the potential addition of therapeutic hypothermia to Src-i has not been sufficiently explored.
Future directions
To date, Src-i have only been examined in clinical trials for cancer and neurodegenerative conditions such as Alzheimer's and Parkinson's disease (ClinicalTrials.gov Identifier: NCT00779389, NCT02167256, and NCT03661125). We anticipate that further research will involve large animals and primates. Large animal studies, although expensive, offer the greatest potential of translation to humans, due to the similarities in brain size, gray/white matter ratio, developmental ages and morphology, as well as the localization of injury after HI (Odden et al., 1989; Thoresen et al., 1996; Haaland et al., 1997; Björkman et al., 2006). Additionally, larger sample sizes can help to decrease bias and improve study validity. Standard reporting methods for preclinical studies focused on Src-i are also necessary to minimize reporting bias. Future work needs to focus on HI pathophysiologic mechanisms and Src-i dosage, timing, route of administration, and potential adverse events. Moreover, as therapeutic hypothermia is considered standard of care for HI in neonates, the additive effects of a combined hypothermia/Src kinase inhibition protocol should be further investigated (Kratimenos et al., 2018). Recently, our team validated a computational model with experimental measurements of critical intracellular signaling components and captured key molecular trends in this pathway (Kratimenos et al., 2022). Our computational model indicated that Src-i disassociates Ca2+ influx from Bax expression and modulates the interaction between the NMDAR and Src reducing Bax expression (Kratimenos et al., 2022). This model could provide a translational platform to design and screen drugs in neonatal hypoxic brain (Kratimenos et al., 2022).
Strengths and limitations of the study
To our knowledge, this is the first attempt to systematically evaluate the current literature on preclinical evidence supporting the use of Src kinase inhibitors in models of HI. Notably, we included ten studies in which large animals, including Yorkshire newborn piglets, were treated with Src-i following HI. Yorkshire newborn piglets' brains share many characteristics with the human brain (Odden et al., 1989; Thoresen et al., 1996; Haaland et al., 1997; Björkman et al., 2006). Human neonates suffer from somatosensory cortical and basal ganglia damage after perinatal asphyxia, which has many similarities to findings in term piglets aged 1–5 days after similar insults (Thoresen et al., 1996).
Although every effort was made for a thorough literature search, it is possible that some relevant studies were missed. It was not feasible to perform a quantitative analysis (meta-analysis) of studies with a focus on Src-i in neonatal HI as there was a significant variability in experimental animals used, sample size and reported outcomes (histologic, biochemical, neurobehavioral). Most of those studies were using inhibitors of the Src kinase to investigate mechanistic questions rather than examining its role as a therapeutic target. The lack of neurobehavioral assessments did not allow for the study of HI-induced visual, motor, and cognitive impairments. Moreover, we are unable to examine the clinical safety of Src-i due to the lack of long term follow-up. Most of the studies were characterized as intermediate when assessed for risk for bias, which can be attributed to the insufficient description of experimental methods, protocols and interventions as evaluated by the SYRCLE's RoB tool. Following the ARRIVE (Animal Research: Reporting of in vivo Experiments) guidelines could have improved the reporting of results by minimizing publication bias (Hooijmans et al., 2014). However, these guidelines were not published prior to June 2010, and many studies that were included in our review were conducted before that time. Not all studies explicitly added control groups to compare effectiveness against the treatment group again likely because the studies were not designed to examine therapeutic effects. Despite the moderate quality assigned to the examined studies by the SYRCLE RoB tool, the evidence presented still indicates the potential benefits of Src kinase inhibition in neonates suffering perinatal asphyxia.
Conclusions
This systematic review demonstrates that inhibition of Src during hypoxia-ischemia results in neuroprotection. However, these protective properties were assessed based on varying animal models, study designs, and intervention characteristics. Further preclinical studies on large animals and specific experimental models are required to examine the pharmacokinetics of Src-i and its exact role in programmed neuronal death. While heterogeneity and risk for bias were limiting factors, the overall results indicate that Src-i neuroprotective properties could be a promising therapeutic strategy to neonates after hypoxic events.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
PK and PC conceptualized the manuscript. PC wrote the manuscript with contribution from IK, AV, JG, and SP. PK, BS, and IK edited the manuscript. All authors have read and approved the final manuscript.
Funding
This work was funded by K12HD001399 (NIH/NICHD) Child Health Research Career Development Award (CHRCDA) (PI: PK), Children's National Board of Visitors Grant (PI: PK), and K12HD001399-20 (NIH/NICHD) Child Health Research Career Development Award (CHRCDA) (PI: IK).
Acknowledgments
We dedicate this manuscript to our beloved mentor and friend Dr. Maria Delivoria-Papadopoulos, whose laboratory studied the role of Src kinase in neonatal hypoxic brain injury.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2022.1049655/full#supplementary-material
References
Angelis, D., and Delivoria-Papadopoulos, M. (2017a). Effects of Src kinase inhibition on expression of pro-caspase-2 after brain hypoxia in a piglet animal model. NeuroReport. 28, 770–773. doi: 10.1097/WNR.0000000000000835
Angelis, D., and Delivoria-Papadopoulos, M. (2017b). Effects of Src kinase inhibition on expression of protein tyrosine phosphatase 1b after brain hypoxia in a piglet animal model. Mediat. Inflam. 2017, 1–7. doi: 10.1155/2017/2810295
Angelis, D., Fontánez Nieves, T. D., and Delivoria-Papadopoulos, M. (2015). Temporal changes in caspase-1 and caspase-8 activities following brain hypoxia with and without src kinase inhibition in a piglet animal model. Neurochem. Res. 40, 2270–2279. doi: 10.1007/s11064-015-1717-8
Angelis, D., Fontánez-Nieves, T. D., and Delivoria-Papadopoulos, M. (2014). The role of src kinase in the caspase-1 pathway after hypoxia in the brain of newborn piglets. Neurochem. Res. 39, 2118–2126. doi: 10.1007/s11064-014-1404-1
Arundine, M., and Tymianski, M. (2004). Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Molec. Life Sci. 61, 657–668. doi: 10.1007/s00018-003-3319-x
Barrett, W. C., DeGnore, J. P., Keng, Y. F., Zhang, Z. Y., Yim, M. B., and Chock, P. B. (1999). Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J. Biol. Chem. 274, 34543–6. doi: 10.1074/jbc.274.49.34543
Björkman, S. T., Foster, K. A., O'driscoll, S. M., Healy, G. N., Lingwood, B. E., Burke, C., et al. (2006). Hypoxic/Ischemic models in newborn piglet: Comparison of constant FiO2 versus variable FiO2 delivery. Brain Res. 1100, 110–117. doi: 10.1016/j.brainres.2006.04.119
Bleicken, S., Landeta, O., Landajuela, A., Basañez, G., and García-Sáez, A. J. (2013). Proapoptotic Bax and Bak proteins form stable protein-permeable pores of tunable size. J. Biol. Chem. 288, 33241–33252. doi: 10.1074/jbc.M113.512087
Blencowe, H., Vos, T., Lee, A. C., Philips, R., Lozano, R., Alvarado, M. R., et al. (2013). Estimates of neonatal morbidities and disabilities at regional and global levels for 2010: introduction, methods overview and relevant findings from the Global Burden of Disease study. Pediatr. Res. 74, 4–16. doi: 10.1038/pr.2013.203
Boatright, K. M., Renatus, M., Scott, F. L., Sperandio, S., Shin, H., Pedersen, I. M., et al. (2003). A unified model for apical caspase activation. Molec. Cell, 11, 529–41. doi: 10.1016/S1097-2765(03)00051-0
Bochelen, D., Rudin, M., and Sauter, A. (1999). Calcineurin inhibitors FK506 and SDZ ASM 981 alleviate the outcome of focal cerebral ischemic/reperfusion injury. J. Pharmacol. Exper. Therap. 288, 653–9.
Chen, M., Hou, X., and Zhang, G. (2003). Tyrosine kinase and tyrosine phosphatase participate in regulation of interactions of NMDA receptor subunit 2A with Src and Fyn mediated by PSD-95 after transient brain ischemia. Neurosci. Lett. 339, 29–32. doi: 10.1016/S0304-3940(02)01439-8
Cilio, M. R., and Ferriero, D. M. (2010). Synergistic neuroprotective therapies with hypothermia. Semin. Fetal. Neonatal. Med. 15, 293–298. doi: 10.1016/j.siny.2010.02.002
Clancy, B., Finlay, B. L., Darlington, R. B., and Anand, K. J. (2007). Extrapolating brain development from experimental species to humans. NeuroToxicol. 28, 931–937. doi: 10.1016/j.neuro.2007.01.014
De Vries, R. B., Hooijmans, C. R., Langendam, M. W., van Luijk, J., Leenaars, M., Ritskes-Hoitinga, M., et al. (2015). A protocol format for the preparation, registration and publication of systematic reviews of animal intervention studies. Evidence-based Preclin. Med. 2, 1–9. doi: 10.1002/ebm2.7
Delivoria-Papadopoulos, M. (2012). Mechanism of caspase-9 activation during hypoxia in the cerebral cortex of newborn piglets: The role of Src kinase. Neurosci. Lett. 523, 19–23. doi: 10.1016/j.neulet.2012.06.029
Delivoria-Papadopoulos, M., Ashraf, Q. M., and Mishra, O. P. (2007). Differential expression of apoptotic proteins following hypoxia-induced CREB phosphorylation in the cerebral cortex of newborn piglets. Neurochem. Res. 32, 1256–1263. doi: 10.1007/s11064-007-9301-5
Delivoria-Papadopoulos, M., Ashraf, Q. M., and Mishra, O. P. (2011). Mechanism of CaM kinase IV activation during hypoxia in neuronal nuclei of the cerebral cortex of newborn piglets: The role of Src kinase. Neurochem. Res. 36, 1512–1519. doi: 10.1007/s11064-011-0477-3
Delivoria-Papadopoulos, M., Kratimenos, P., and Anday, E. K. (2018). Biochemical Basis of Hypoxic-Ischemic Encephalopathy. Neonatology. 11, 2143–2164. doi: 10.1007/978-3-319-29489-6_272
Eunson, P. (2015). The long-term health, social, and financial burden of hypoxic-ischaemic encephalopathy. Develop. Med. Child Neurol. 57, 48–50. doi: 10.1111/dmcn.12727
Filan, P. M., Inder, T. E., Cameron, F. J., Kean, M. J., and Hunt, R. W. (2006). Neonatal hypoglycemia and occipital cerebral injury. J. Pediatrics 148, 552–555. doi: 10.1016/j.jpeds.2005.11.015
Fujimoto, S., Katsuki, H., Kume, T., Kaneko, S., and Akaike, A. (2004). Mechanisms of oxygen glucose deprivation-induced glutamate release from cerebrocortical slice cultures. Neurosci. Res. 50, 179–187. doi: 10.1016/j.neures.2004.06.013
Gunn, A. J., and Thoresen, M. (2006). Hypothermic neuroprotection. NeuroRX 3, 154–169. doi: 10.1016/j.nurx.2006.01.007
Guo, J., Wu, H. W., Hu, G., Han, X., De, W, and Sun, Y. J. (2006). Sustained activation of Src-family tyrosine kinases by ischemia: A potential mechanism mediating extracellular signal-regulated kinase cascades in hippocampal dentate gyrus. Neuroscience 143, 827–836. doi: 10.1016/j.neuroscience.2006.08.031
Haaland, K., Løberg, E. M., Steen, P. A., and Thoresen, M. (1997). Posthypoxic Hypothermia in Newborn Piglets. Pediatric Res. 41, 505–512. doi: 10.1203/00006450-199704000-00009
Haass, C., and Mandelkow, E. (2010). Fyn-tau-amyloid: a toxic triad. Cell 142, 356–358. doi: 10.1016/j.cell.2010.07.032
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Ann. Rev. Neurosci. 17, 31–108. doi: 10.1146/annurev.ne.17.030194.000335
Hooijmans, C. R., Rovers, M. M., de Vries, R., Leenaars, M., Ritskes-Hoitinga, M., and Langendam, M. W. (2014). SYRCLEs risk of bias tool for animal studies. BMC Med. Res. Methodol. 14, 43. doi: 10.1186/1471-2288-14-43
Hornick, K., Chang, E., Zubrow, A. B., Mishra, O. P., and Delivoria-Papadopoulos, M. (2007). Mechanism of Ca(2+)/calmodulin-dependent protein kinase IV activation and of cyclic AMP response element binding protein phosphorylation during hypoxia in the cerebral cortex of newborn piglets. Brain Res. 1150, 40–45. doi: 10.1016/j.brainres.2007.02.079
Hossain, M. I., Hoque, A., Lessene, G., Kamaruddin, M. A., Chu, P. W., Ng, I. H., et al. (2015). Dual role of Src kinase in governing neuronal survival. Brain Res. 1594, 1–14. doi: 10.1016/j.brainres.2014.10.040
Hossain, M. I., Kamaruddin, M. A., and Cheng, H.-C. (2012). Aberrant regulation and function of Src family tyrosine kinases: Their potential contributions to glutamate-induced neurotoxicity. Clin. Exper. Pharmacol. Physiol. 39, 684–691. doi: 10.1111/j.1440-1681.2011.05621.x
Hu, X., Wu, X., Xu, J., Zhou, J., Han, X., and Guo, J. (2009). Src kinase up-regulates the ERK cascade through inactivation of protein phosphatase 2A following cerebral ischemia. BMC Neurosci. 10, 74. doi: 10.1186/1471-2202-10-74
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in alzheimers disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jacobs, S. E., Berg, M., Hunt, R., Tarnow-Mordi, W. O., Inder, T. E., and Davis, P. G. (2013). Cooling for newborns with hypoxic ischaemic encephalopathy. Cochr. Datab. System. Rev. 1, CD003311. doi: 10.1002/14651858.CD003311.pub3
Jiang, X., Mu, D., Biran, D., Faustino, J., Chang, S., Rinćon, C. M., et al. (2008). Activated Src kinases interact with the N -methyl-D-aspartate receptor after neonatal brain ischemia. Ann. Neurol. 63, 632–641. doi: 10.1002/ana.21365
Johansen, F. F., and Diemer, N. H. (1991). Enhancement of GABA neurotransmission after cerebral ischemia in the rat reduces loss of hippocampal CA1 pyramidal cells. Acta Neurol. Scand. 84, 1–6. doi: 10.1111/j.1600-0404.1991.tb04893.x
Juul, S. E., and Ferriero, D. M. (2014). Pharmacologic neuroprotective strategies in neonatal brain injury. Clin. Perinatol. 41, 119–131. doi: 10.1016/j.clp.2013.09.004
Kennedy, M. B. (1997). The postsynaptic density at glutamatergic synapses. Trends Neurosci. 20, 264–8. doi: 10.1016/S0166-2236(96)01033-8
Kratimenos, P., Koutroulis, I., Agarwal, B., Theocharis, S., and Delivoria-Papadopoulos, M. (2017). Effect of Src Kinase inhibition on Cytochrome c, Smac/DIABLO and apoptosis inducing factor (AIF) following cerebral hypoxia-ischemia in newborn piglets. Sci. Rep. 7, 16664. doi: 10.1038/s41598-017-16983-1
Kratimenos, P., Koutroulis, I., Jain, A., Malaeb, S., and Delivoria-Papadopoulos, M. (2018). Effect of concurrent src kinase inhibition with short-duration hypothermia on Ca2+/calmodulin kinase IV activity and neuropathology after hypoxia-ischemia in the newborn swine brain. Neonatology 113, 37–43. doi: 10.1159/000480067
Kratimenos, P., Vij, A., Vidva, R., Koutroulis, I., Delivoria-Papadopoulos, M., Gallo, V., et al. (2022). Computational analysis of cortical neuronal excitotoxicity in a large animal model of neonatal brain injury. J. Neurodevelop. Diso. 14, 1–16. doi: 10.1186/s11689-022-09431-3
Kumar, A., Jaggi, A. S., and Singh, N. (2014). Pharmacological investigations on possible role of Src kinases in neuroprotective mechanism of ischemic postconditioning in mice. Int. J. Neurosci. 124, 777–786. doi: 10.3109/00207454.2013.879869
Kumari, M., and Ticku, M. K. (2000). Regulation of NMDA receptors by ethanol. Progr. Drug Res. 54, 152–89. doi: 10.1007/978-3-0348-8391-7_5
Lawn, J. E., Cousens, S., Zupan, J., and Lancet Neonatal Survival Steering Team. (2005). 4 million neonatal deaths: when? Where? Why?. Lancet 365, 891–900. doi: 10.1016/S0140-6736(05)71048-5
Lee, S. R., Kwon, K. S., Kim, S. R., and Rhee, S. G. (1998). Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–72. doi: 10.1074/jbc.273.25.15366
Liu, D.-Z., and Sharp, F. R. (2011). The dual role of SRC kinases in intracerebral hemorrhage. Acta Neurochirurgica. 111, 77–81. doi: 10.1007/978-3-7091-0693-8_13
Liu, Y., Zhang, G. Y., Yan, J. Z., and Xu, T. L. (2005). Suppression of Pyk2 attenuated the increased tyrosine phosphorylation of NMDA receptor subunit 2A after brain ischemia in rat hippocampus. Neurosci. Lett. 379, 55–58. doi: 10.1016/j.neulet.2004.12.054
Lukyanova, L. D., and Kirova, Y. I. (2015). Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 9, 320. doi: 10.3389/fnins.2015.00320
Martone, M. E., Jones, Y. Z., Young, S. J., Ellisman, M. H., Zivin, J. A., and Hu, B. R. (1999). Modification of postsynaptic densities after transient cerebral ischemia: a quantitative and three-dimensional ultrastructural study. J. Neurosci. 19, 1988–97. doi: 10.1523/JNEUROSCI.19-06-01988.1999
Mishra, O. P., Ashraf, Q. M., and Delivoria-Papadopoulos, M. (2009). NO-mediated activation of Src kinase during hypoxia in the cerebral cortex of newborn piglets. Neurosci. Lett. 460, 61–65. doi: 10.1016/j.neulet.2009.05.041
Mishra, O. P., Zubrow, A. B., Ashraf, Q. M., and Delivoria-Papadopoulos, M. (2006). Nuclear Ca++-influx, Ca++/calmodulin-dependent protein kinase IV activity and CREB protein phosphorylation during post-hypoxic reoxygenation in neuronal nuclei of newborn piglets: The role of nitric oxide. Neurochem. Res. 31, 1463–1471. doi: 10.1007/s11064-006-9204-x
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G., and PRISMA Group. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6, e1000097. doi: 10.1371/journal.pmed.1000097
Montaldo, P., Pauliah, S. S., Lally, P. J., Olson, L., and Thayyil, S. (2015). Cooling in a low-resource environment: Lost in translation. Semin. Fetal Neonatal Med. 20, 72–79. doi: 10.1016/j.siny.2014.10.004
Odden, J. P., Stiris, T., Hansen, T. W., and Bratlid, D. (1989). Cerebral blood flow during experimental hypoxaemia and ischaemia in the newborn piglet. Acta Paediatr. Scand. 360, 13–9. doi: 10.1111/j.1651-2227.1989.tb11276.x
Oja, S. S., Korpi, E. R., and Saransaari, P. (1990). Modification of chloride flux across brain membranes by inhibitory amino acids in developing and adult mice. Neurochem. Res. 15, 797–804. doi: 10.1007/BF00968557
Olsen, S. L., DeJonge, M., Kline, A., Liptsen, E., Song, D., Anderson, B., et al. (2013). Optimizing therapeutic hypothermia for neonatal encephalopathy. Pediatrics. 131, e591–e603. doi: 10.1542/peds.2012-0891
Paul, R., Zhang, Z. G., Eliceiri, B. P., Jiang, Q., Boccia, A. D., Zhang, R. L., et al. (2001). Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 7, 222–227. doi: 10.1038/84675
Pauliah, S. S., Shankaran, S., Wade, A., Cady, E. B., and Thayyil, S. (2013). Therapeutic hypothermia for neonatal encephalopathy in low- and middle-income countries: a systematic review and meta-analysis. PLoS ONE. 8, e58834. doi: 10.1371/journal.pone.0058834
Pressler, R., and Auvin, S. (2013). Comparison of brain maturation among species: an example in translational research suggesting the possible use of bumetanide in newborn. Front. Neurol. 4, 36. doi: 10.3389/fneur.2013.00036
Rehni, A. K., Bhateja, P., and Singh, N. (2009). Diethyl dithiocarbamic acid, a possible nuclear factor kappa B inhibitor, attenuates ischemic postconditioning-induced attenuation of cerebral ischemia–reperfusion injury in mice. Canad. J. Physiol. Pharmacol. 87, 63–68. doi: 10.1139/Y08-100
Rehni, A. K., Bhateja, P., Singh, N., and Jaggi, A. S. (2008a). Implication of mast cell degranulation in ischemic preconditioning-induced prevention of cerebral injury. Fundam. Clin. Pharmacol. 22, 179–188. doi: 10.1111/j.1472-8206.2008.00567.x
Rehni, A. K., Singh, T. G., Jaggi, A. S., and Singh, N. (2008b). Pharmacological preconditioning of the brain: a possible interplay between opioid and calcitonin gene related peptide transduction systems. Pharmacol. Rep. 60, 904–13.
Rehni, A. K., Singh, T. G., Kakkar, T., and Arora, S. (2011). Involvement of src-kinase activation in ischemic preconditioning induced protection of mouse brain. Life Sci. 88, 825–829. doi: 10.1016/j.lfs.2011.02.024
Robertson, N. J., Nakakeeto, M., Hagmann, C., Cowan, F. M., Acolet, D., Iwata, O., et al. (2008). Therapeutic hypothermia for birth asphyxia in low-resource settings: a pilot randomised controlled trial. Lancet 372, 801–803. doi: 10.1016/S0140-6736(08)61329-X
Rosenbaum, D. M., Grotta, J. C., Pettigrew, L. C., Ostrow, P., Strong, R., Rhoades, H., et al. (1990). Baclofen does not protect against cerebral ischemia in rats. Stroke, 21, 138–40. doi: 10.1161/01.STR.21.1.138
Salter, M. W., and Kalia, L. V. (2004). Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328. doi: 10.1038/nrn1368
Sands, J., Dobbing, J., and Gratrix, C. A. (1979). Cell number and cell size: organ growth and development and the control of catch-up growth in rats. Lancet. 2, 503–5. doi: 10.1016/S0140-6736(79)91556-3
Seeburg, P. H. (1993). The TINS/TiPS Lecture. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 16, 359–65. doi: 10.1016/0166-2236(93)90093-2
Shankaran, S., Pappas, A., McDonald, S. A., Vohr, B. R., Hintz, S. R., Yolton, K., et al. (2012). Childhood outcomes after hypothermia for neonatal encephalopathy. New Engl. J. Med. 366, 2085–2092. doi: 10.1056/NEJMoa1112066
Sivilotti, L., and Nistri, A. (1991). GABA receptor mechanisms in the central nervous system. Progr. Neurobiol. 36, 35–92. doi: 10.1016/0301-0082(91)90036-Z
Stokes, A. H., Bernard, L. P., Nicklas, W. J., and Zeevalk, G. D. (2001). Attenuation of malonate toxicity in primary mesencephalic cultures using the GABA transport blocker, NO-711. J. Neurosci. Res. 64, 43–52. doi: 10.1002/jnr.1052
Takakura, K., Beckman, J. S., MacMillan-Crow, L. A., and Crow, J. P. (1999). Rapid and Irreversible Inactivation of Protein Tyrosine Phosphatases PTP1B, CD45, and LAR by Peroxynitrite. Arch. Biochem. Biophys. 369, 197–207. doi: 10.1006/abbi.1999.1374
Tam, E. W., Haeusslein, L. A., Bonifacio, S. L., Glass, H. C., Rogers, E. E., Jeremy, R. J., et al. (2012). Hypoglycemia is associated with increased risk for brain injury and adverse neurodevelopmental outcome in neonates at risk for encephalopathy. J. Pediatr. 161, 88–93. doi: 10.1016/j.jpeds.2011.12.047
Thoresen, M., Haaland, K., Løberg, E. M., Whitelaw, A., Apricena, F., Hankø, E., et al. (1996). A piglet survival model of posthypoxic encephalopathy. Pediatr. Res. 40, 738–748. doi: 10.1203/00006450-199611000-00014
Tian, H. P., Huang, B. S., Zhao, J., Hu, X. H., Guo, J., and Li, L. X. (2009). Non-receptor tyrosine kinase Src is required for ischemia-stimulated neuronal cell proliferation via Raf/ERK/CREB activation in the dentate gyrus. BMC Neurosci. 10, 139. doi: 10.1186/1471-2202-10-139
Traystman, R. J. (2003). Animal models of focal and global cerebral ischemia. ILAR J. 44, 85–95. doi: 10.1093/ilar.44.2.85
Van Bel, F., and Groenendaal, F. (2016). Drugs for neuroprotection after birth asphyxia: Pharmacologic adjuncts to hypothermia. Semin. Perinatol. 40, 152–159. doi: 10.1053/j.semperi.2015.12.003
Wang, R.-M., Zhang, Q.-G., and Zhang, G.-Y. (2004). Activation of ERK5 is mediated by N-methyl-D-aspartate receptor and L-type voltage-gated calcium channel via Src involving oxidative stress after cerebral ischemia in rat hippocampus. Neurosci. Lett. 357, 13–16. doi: 10.1016/j.neulet.2003.11.061
Wen, X. R., Li, C., Zong, Y. Y., Yu, C. Z., Xu, J., Han, D., et al. (2008). Dual inhibitory roles of geldanamycin on the c-Jun NH2-terminal kinase 3 signal pathway through suppressing the expression of mixed-lineage kinase 3 and attenuating the activation of apoptosis signal-regulating kinase 1 via facilitating the activation of. Neuroscience. 156, 483–497. doi: 10.1016/j.neuroscience.2008.08.006
White, B. C., Sullivan, J. M., DeGracia, D. J., O'Neil, B. J., Neumar, R. W., Grossman, L. I., et al. (2000). Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J. Neurol. Sci. 179, 1–33. doi: 10.1016/S0022-510X(00)00386-5
Wong, D. S., Poskitt, K. J., Chau, V., Miller, S. P., Roland, E., Hill, A., et al. (2013). Brain injury patterns in hypoglycemia in neonatal encephalopathy. Am. J. Neuroradiol. 34, 1456–1461. doi: 10.3174/ajnr.A3423
Wu, H., Li, H., and Guo, J. (2008). Spry2-mediated inhibition of the Ras/ERK pathway through interaction with Src kinase following cerebral ischemia. Brain Injury 22, 275–281. doi: 10.1080/02699050801911295
Wyatt, J. S., Gluckman, P. D., Liu, P. Y., Azzopardi, D., Ballard, R., Edwards, A. D., et al. (2007). Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics 119, 912–921. doi: 10.1542/peds.2006-2839
Xu, J., Liu, Y., and Zhang, G.-Y. (2008). Neuroprotection of GluR5-containing kainate receptor activation against ischemic brain injury through decreasing tyrosine phosphorylation of N-methyl-D-aspartate receptors mediated by Src kinase. J. Biol. Chem. 283, 29355–29366. doi: 10.1074/jbc.M800393200
Zhang, F., Li, C., Wang, R., Han, D., Zhang, Q., Zhou, G., et al. (2007). Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neuroscience. 150, 938–949. doi: 10.1016/j.neuroscience.2007.09.070
Keywords: Src, hypoxic-ischemic encephalopathy (HIE), hypoxia, neonatal brain, neuroprotection
Citation: Christidis P, Vij A, Petousis S, Ghaemmaghami J, Shah BV, Koutroulis I and Kratimenos P (2022) Neuroprotective effect of Src kinase in hypoxia-ischemia: A systematic review. Front. Neurosci. 16:1049655. doi: 10.3389/fnins.2022.1049655
Received: 20 September 2022; Accepted: 02 November 2022;
Published: 24 November 2022.
Edited by:
Apostolos Zarros, Pharmacological Research Observatory, United KingdomReviewed by:
Xue Wang, Wenzhou Medical University, ChinaTong Li, Qingdao University Medical College, China
Copyright © 2022 Christidis, Vij, Petousis, Ghaemmaghami, Shah, Koutroulis and Kratimenos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis Kratimenos, cGtyYXRpbWVuMkBjaGlsZHJlbnNuYXRpb25hbC5vcmc=