
 Nirmala Pandeya1
Nirmala Pandeya1
- 1 School of Population Health, The University of Queensland, Brisbane, QLD, Australia
- 2 Population Health Department, Queensland Institute of Medical Research, Brisbane, QLD, Australia
Esophageal adenocarcinoma is the most common type of esophageal cancer in most Western countries and is an important contributor to overall cancer mortality. Most cases of esophageal adenocarcinoma are believed to arise from Barrett’s esophagus. Esophageal adenocarcinoma occurs more frequently in white men over 50 years old, as well as in people with frequent symptoms of gastroesophageal reflux, in smokers and in people who are obese. Higher consumption of fruit and vegetables, use of non-steroidal anti-inflammatory drugs, and infection with Helicobacter pylori have all been shown to reduce the risk of esophageal adenocarcinoma. Here, we review the epidemiological evidence for the major risk factors of esophageal adenocarcinoma and also discuss perspectives for future research.
Introduction
Worldwide, esophageal cancer is the eighth most common malignancy and the sixth leading cause of cancer mortality. Recent estimates suggest 482,000 new cases of esophageal cancer arise worldwide each year, resulting in 407,000 deaths (Ferlay et al., 2010). The lifetime risk of esophageal cancer in the United States is about 1 in 125 men and about 1 in 400 women (American Cancer Society, 2011). Two predominant histopathologic types of esophageal cancer are recognized: squamous cell carcinoma (ESCC) and adenocarcinoma (EAC). In Western countries, the rate of increase in EAC incidence during the past four decades has been among the highest for any cancer (Pohl and Welch, 2005). Prognosis for patients with EAC is strongly related to stage at diagnosis, however most patients are diagnosed with late-stage disease and less than 20% survive for 5 years (Stavrou et al., 2009).
The last two decades have seen increasing research attention given to the epidemiology of EAC. Numerous population-based epidemiological studies have examined EAC risk factors; however the small size of these individual studies has limited the precision of resulting risk estimates and reconciling inconsistent findings have proven difficult. Valuable insights have been gained from the output from the Barrett’s and Esophageal Adenocarcinoma Consortium (BEACON), a collaborative project which has pooled the data from 12 of these studies (http://bea.tlvcloud.org/), thereby conferring greater statistical precision. The analyses combine 10 case–control studies and 2 cohort studies from the United States, Canada, the United Kingdom, Ireland, Australia, and Sweden, involving more than 2,100 EAC patients and almost 14,000 population controls (Cook et al., 2010). Much of the discussion and conclusions regarding risk factors in this review is based on these pooled analyses; where appropriate other articles have also been cited.
Most cases of EAC are believed to arise from underlying Barrett’s esophagus (BE), a premalignant condition in which the normal stratified squamous epithelium of the distal esophagus is replaced by specialized columnar epithelium (Falk, 2002). Here, we will provide an overview of the descriptive epidemiology of EAC and BE, and provide a summary of the risk factors for these conditions. This review is restricted to the effects of host characteristics and environmental exposures, and does not address risks associated with constitutional genotypes, since several large-scale genome-wide association studies are being conducted currently and will publish their findings shortly.
Descriptive Epidemiology
Incidence rates for EAC have increased sharply during the past four decades in developed countries, with a reported seven-fold (0.36–2.56 per 100,000) increase in the United States between 1973 and 2006 (Pohl et al., 2010). Increases in incidence of similar magnitude have been reported among populations residing in Australia, Denmark, Finland, Norway, Sweden, Switzerland, and the United Kingdom (Botterweck et al., 2000; Vizcaino et al., 2002; Bosetti et al., 2008; Cook et al., 2009; Stavrou et al., 2009). As a result of these increases, EAC became the most common form of esophageal cancer in the United States and most other Western countries in the late 1990s (Curado et al., 2007; Holmes and Vaughan, 2007). In all populations, the greatest increase in incidence has been observed among older white men. However, incidence rates have also increased significantly among other ethnic groups, in women, and in people less than 65 years old (Holmes and Vaughan, 2007; Brown et al., 2008).
Figure 1 presents the age-adjusted annual incidence rates for EAC from a broad geographical range of cancer registries and highlights the considerable international variation in EAC incidence (Curado et al., 2007). Notably, rates also vary among different ethnic groups within a particular country. For example, in the United States, compared with non-Hispanic whites, the incidence of EAC is significantly lower among Hispanic whites, Blacks, Asians, and Pacific Islanders (Cook et al., 2009). Migration studies have shown that EAC rates tend to approach those of the country of adoption rather than the country of origin, suggesting variation within countries is unlikely to be fully explained by racial or genetic differences. Another remarkable feature is the sex ratio, with most populations reporting more than five-fold higher incidence in men than women (Curado et al., 2007). EAC is rare among young persons (80% of EAC cases are aged 55–70 years) and incidence increases with age (mean age at diagnosis is 60 years; Yang and Davis, 1988; Lagergren, 2005).
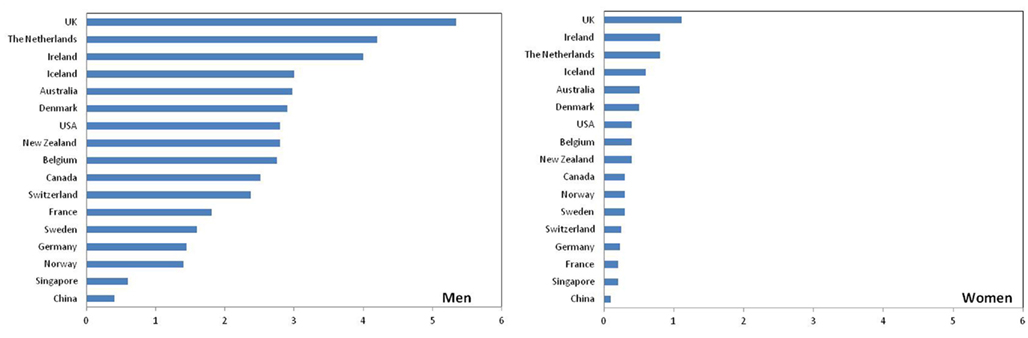
Figure 1. Age-adjusted annual incidence of esophageal adenocarcinoma (per 100,000; Data source: Curado et al., 2007).
Barrett’s Esophagus
As the precursor to EAC, BE is frequently asymptomatic, prevalence is largely unknown and it is thought that a large percentage of the general population may have undiagnosed BE (Cameron et al., 1990). Population prevalence estimates for BE have been gained from autopsy and endoscopy series. In an autopsy study in Olmsted County, Minnesota, the prevalence of BE was reported to be as low as 0.4% (Cameron et al., 1990). Data from endoscopic surveys completed in Sweden (Ronkainen et al., 2005) and Italy (Zagari et al., 2008) suggest that 1.3–1.6% of the population have BE. However, according to data from the United States, the upper bound of population prevalence of BE may be as high as 6% (Rex et al., 2003; Hayeck et al., 2010).
Analyses of temporal trends for BE patients have failed to determine whether BE incidence has truly increased. While several studies have investigated the recent increases in diagnosis of BE, it remains unclear whether the observed increase in incidence of BE is real or whether increased awareness and widespread use of endoscopy have resulted in more diagnoses (Prach et al., 1997; van Soest et al., 2005; Kendall and Whiteman, 2006; Musana et al., 2008).
Overall, the associations between sociodemographic characteristics and the risk of BE are similar to those for EAC. Epidemiological data suggest a 2:1 male predominance for BE (Cook et al., 2005). There is consensus in the literature that BE develops more frequently in older people, with most BE patients diagnosed between 50 and 60 years of age (Corley et al., 2009). Finally, comparable with EAC, BE is more commonly diagnosed in whites than in other ethnic groups (including Blacks, Asians, and Hispanics; Ford et al., 2005; Corley et al., 2009).
Cancer Risk in Barrett’s Esophagus
Adenocarcinoma is thought to arise through a progressive sequence, whereby the abnormal columnar epithelium that characterizes BE progresses to low-grade dysplasia, then to high-grade dysplasia and finally to carcinoma (Hamilton and Smith, 1987; Hameeteman et al., 1989; Miros et al., 1991). The absolute risk of EAC in patients with BE is unclear however, with earlier studies reporting higher risks than more recent studies (Shaheen et al., 2000; Sikkema et al., 2010). Although previously assumed to be between 5 and 10 per 1,000 person-years, a recent large, population-based study in Denmark reported a much lower absolute risk of progression to EAC of 1.2 per 1,000 person-years (Hvid-Jensen et al., 2011). The risk is substantially higher however in BE patients with dysplasia (5.1 vs. 1.0 cases per 1,000 person-years among patients with low-grade dysplasia compared with patients without dysplasia; Hvid-Jensen et al., 2011). It is unclear why adenocarcinoma develops in only a small subset of those with BE, as the pathways leading to EAC remain ill-defined. The progressively lower estimates of annual risk of EAC in BE patients raise questions about the feasibility of endoscopic surveillance in patients who have BE. Future research will need to develop better tools for stratifying patients based on risk of progression. Numerous approaches are being investigated currently, including clinical decision algorithms, serological markers, and tissue-based prognostic markers (Rabinovitch et al., 2001; Risques et al., 2007; Sato et al., 2008; Merlo et al., 2010).
Environmental Exposures
Esophageal adenocarcinoma is a multifactorial disease. The temporal trends in epidemiology and the effect of migration on incidence suggest that environmental factors play an important role in the etiology of EAC (Table 1).
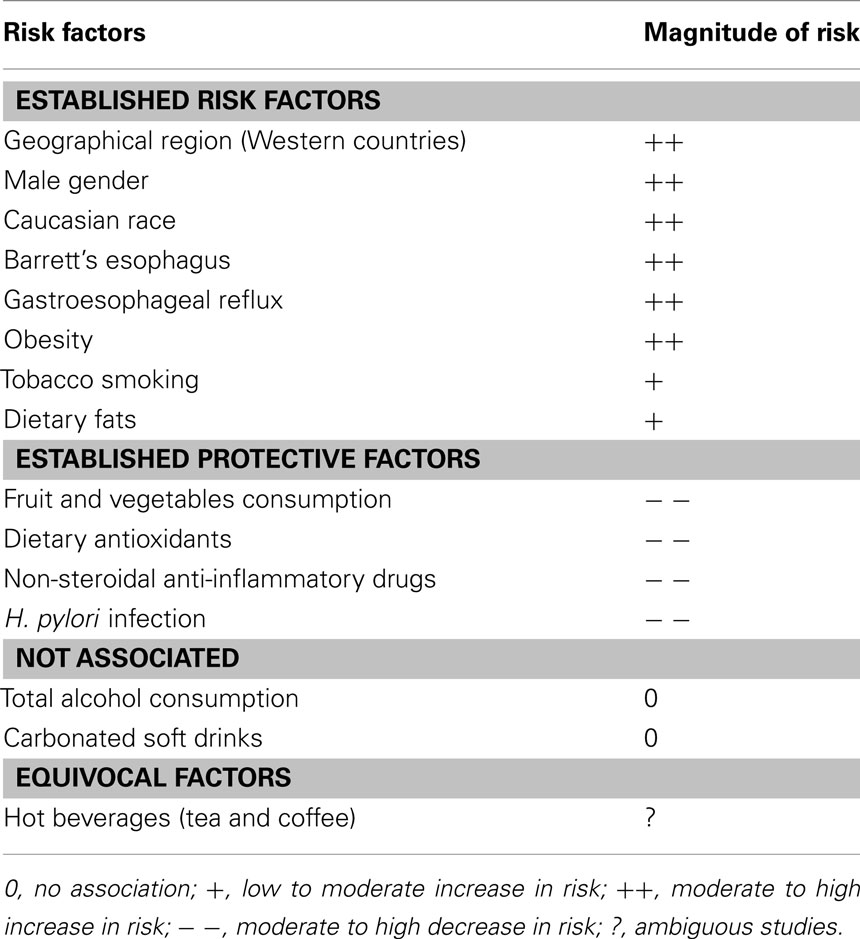
Table 1. Summary of risk factors for esophageal adenocarcinoma.
Gastroesophageal Reflux
Gastroesophageal reflux (GER), a condition that occurs when the lower esophageal sphincter allows stomach acid to flow back into the esophagus, is becoming increasingly common in Western populations (Ness-Jensen et al., 2011). Long-standing GER is a major risk factor predisposing to the development of BE and EAC, and is thought to play a role in progression from BE to cancer. A recent meta analysis of five large population-based case–control studies reported that the relative risks of EAC associated with at least weekly GER symptoms and daily symptoms are around five and seven, respectively (Rubenstein and Taylor, 2010). There is also general agreement that chronic GER is the main cause of BE (Falk, 2002), with frequent GER symptoms associated with 10-fold greater risk of developing BE (Anderson et al., 2007; Kubo et al., 2009a; Smith et al., 2009). The evidence regarding the effect of longer duration of GER symptoms and risks of EAC and BE is inconclusive (Anderson et al., 2007; Rubenstein and Taylor, 2010).
Due to the role of GER in the development of EAC, various acid suppression therapies have been used to reduce esophageal acid exposure in BE patients. Findings to date suggest that acid-suppressant medications may reduce the risk of progression from BE to cancer (El-Serag et al., 2004; Hillman et al., 2004, 2008; Cooper et al., 2006; Nguyen et al., 2009), but definite evidence from clinical trials is lacking. Furthermore, it is not known whether these medications may prevent the development of BE in the first place.
The biological mechanisms whereby GER causes BE and influences the development of EAC is still unclear. Two main hypotheses have arisen in response to experimental and epidemiological data. The prevailing hypothesis is that chronic reflux of acid or bile injures the esophageal epithelium, inducing a cascade of cytokine responses that result in inflammation and cell proliferation, thereby initiating the metaplasia–dysplasia–neoplasia sequence (Yoshida, 2007). The second hypothesis is that GER may cause the production of nitrous oxide from ingested nitrites, leading to elevated levels of DNA damage and enhanced risk of disease through increasing the likelihood of genetic change (Clemons et al., 2007). These hypotheses are not mutually exclusive however, and other mechanisms are also possible. Research to fill these gaps in our knowledge is ongoing.
Obesity
Obesity is one of the strongest risk factors for EAC, but less so for BE. Population-based studies have consistently reported that a body mass index (BMI) greater than 30.0 kg/m2 is associated with two- to three-fold increased risk of EAC (Corley et al., 2008b; Whiteman et al., 2008). A recent meta analysis found some evidence of an association between BMI and BE (Kamat et al., 2009), however more recent attention has focused on whether body fat distribution (in particular, more centralized visceral fat) may be a better marker of risk than BMI. Increased central adiposity (waist circumference, waist-to-thigh ratio, and waist-to-hip ratio) has been reported to strongly and significantly increase the risk of BE, independent of BMI (Corley et al., 2007; Edelstein et al., 2007). Since fat is more likely to be distributed around the abdomen in men, and since visceral fat produces high levels of inflammation-related cytokines (such as leptin; Williams et al., 2000; Considine, 2001), this may explain some of the gender-related difference in the incidence of BE. However, while leptin is known to be up-regulated in obesity and in vitro studies have shown that leptin promotes proliferation in EAC cells, the evidence regarding the effect of leptin on BE risk is inconclusive. Although high leptin levels have been reported to increase the risk of BE in two studies, one study observed a stronger effect in men (Kendall et al., 2008), and the only other study observed a stronger effect in women (Thompson et al., 2010). Resolution of the relationship between leptin and risk of BE requires more data from larger studies.
Despite the strong associations between BMI and EAC risk, there is some doubt as to whether the rise in obesity explains all of the increase in EAC incidence and whether BMI increases risk independently of obesity-related inflammation. A recent study has suggested that the increase in EAC incidence preceded the rise in obesity prevalence by a decade (Abrams et al., 2011), and data from a simulation model indicated that increasing obesity may only explain a small percentage (6.5%) of the rise in EAC incidence (Kong et al., 2011). More work is needed to help disassociate the role of obesity from chronic inflammation in EAC risk and to understand the drivers of the shift in EAC incidence.
Smoking
Studies have consistently found a strong association between tobacco smoking and EAC and, to a lesser extent, BE. Pooled analyses estimate an approximate two-fold increased risk of EAC associated with ever smoking, and a strong dose–response association with cumulative exposure (Cook et al., 2010). For BE, the association is unclear, with some studies (Johansson et al., 2007; Smith et al., 2009), but not all (Anderson et al., 2007; Kubo et al., 2009a), reporting an approximately two-fold increased risk among ever smokers but no consistent trend of increasing risk with increasing quantity of cigarettes consumed. Recent research suggests that tobacco smoking is also strongly associated with progression from BE to cancer, with a two-fold increased risk associated with ever smoking (Coleman et al., 2012). At a mechanistic level, relatively little is understood about how smoking might cause metaplasia and neoplasia of the distal esophagus.
Diet
Dietary factors may partially explain aspects of risk variation by sex, ethnicity and nationality, and may also account for some of the changes in EAC incidence observed among migrants. While many aspects of the role of diet on EAC and BE etiology remain unclear, observational studies have reported that high levels of consumption of saturated fat and processed meat, low fruit and vegetable consumption, low dietary antioxidant intake, and low intakes of certain minerals are all associated with increased risks of both EAC and BE (Mayne et al., 2001; Anderson et al., 2007; Kubo and Corley, 2007; Wu et al., 2007; Kubo et al., 2008a,b, 2009; Mulholland et al., 2009; Thompson et al., 2009; Murphy et al., 2010). The effects of meat and fish consumption are unclear. Evidence to date suggests that neither hot beverages nor carbonated soft drinks are consistently related to EAC risk (Lagergren et al., 2006; Mayne et al., 2006; Ibiebele et al., 2008, 2010; Islami et al., 2009; Ren et al., 2010). There is also no evidence that alcohol consumption increases the risk of EAC or BE; indeed, recent studies have suggested a possible inverse association with wine consumption (Anderson et al., 2009; Kubo et al., 2009b; Freedman et al., 2011; Thrift et al., 2011c).
Medication Use
A prominent hypothesis posits that medications inducing relaxation of the lower esophageal sphincter (including calcium channel blockers, benzodiazepines, and asthma medications, among others) may promote GER, thereby indirectly increasing the risks of BE and EAC. Epidemiological studies have examined their association with EAC, with inconsistent findings (Chow et al., 1995; Vaughan et al., 1998; Farrow et al., 2000; Lagergren et al., 2000; Ranka et al., 2006; Fortuny et al., 2007). Few studies have investigated associations between use of these medications and BE risk (Corley et al., 2006; Ladanchuk et al., 2010). While results suggest that regular users of asthma medications may have an increased risk of EAC, residual confounding may explain these findings since users of asthma medications report higher frequency of GER symptoms, and reflux is strongly associated with EAC, BE, and reflux-associated asthma symptoms.
Non-Steroidal Anti-Inflammatory Drugs
There is consistent evidence from observational studies that frequent users of non-steroidal anti-inflammatory drugs (NSAIDs) have reduced risks of EAC (Corley et al., 2003; Liao et al., 2012), and that NSAID use among BE patients may also reduce their risk of progression to cancer by up to 70% (Vaughan et al., 2005). However, to date, findings have been inconsistent for BE, with one study reporting a halving of risk of BE among NSAID users (Anderson et al., 2006), while the only other study reported no association (Thrift et al., 2011b). In this respect, the discordant findings for EAC and BE may be analogous to those observed for colon cancer and polyps, whereby aspirin and other NSAID use may stop progression from pre-cancer to cancer, but not the initial development of pre-cancer. Definitive evidence to resolve this issue awaits the findings from randomized trials (Jankowski and Moayyedi, 2004).
Helicobacter pylori Infection
Helicobacter pylori is a Gram-negative bacterium that persistently colonizes the human stomach (Suerbaum and Michetti, 2002; Blaser and Atherton, 2004; Atherton and Blaser, 2009), and is a major cause of gastric cancer (Huang et al., 1998; Atherton and Blaser, 2009). In contrast, epidemiological studies have reported that persons infected with H. pylori have half the risks of EAC and BE than those uninfected (Rokkas et al., 2007; Anderson et al., 2008; Corley et al., 2008a; Islami and Kamangar, 2008; Whiteman et al., 2010; Thrift et al., 2011a). It is postulated that the decline in infection rates for H. pylori in Western populations may have contributed to the changing epidemiology of EAC. A possible explanation for the inverse association is reduced acid secretion in those infected, with ongoing research focusing on the mechanisms through which H. pylori mediates its effects on esophageal epithelium.
Conclusion
Since the 1970s, the incidence of EAC has increased sharply in most Western countries. Explanations commonly offered to explain this change in epidemiology of EAC include the rising prevalences of central obesity and reflux among white men, although quantifying the overall contribution of these factors remains the subject of investigation. Given the rising prevalence of obesity worldwide, especially in developing countries, and the links between obesity, inflammation, and cancer, it is clear that a better understanding of these relationships is required to yield more targeted strategies for cancer prevention. It is anticipated that research currently being conducted will provide a better understanding of the interplay between inherited susceptibility and environmental factors. In the future, it is possible that such knowledge will be applied to develop tools which will enable patients and clinicians to quantify an individual’s absolute risk of developing esophageal cancer and thereby make appropriate decisions to improve outcomes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BE, Barrett’s esophagus; EAC, esophageal adenocarcinoma; ESCC, esophageal squamous cell carcinoma; GER, gastroesophageal reflux; NSAIDs, non-steroidal anti-inflammatory drugs.
References
Abrams, J. A., Sharaiha, R. Z., Gonsalves, L., Lightdale, C. J., and Neugut, A. I. (2011). Dating the rise of esophageal adenocarcinoma: analysis of Connecticut Tumor Registry data, 1940–2007. Cancer Epidemiol. Biomarkers Prev. 20, 183–186.
American Cancer Society. (2011). Cancer Facts and Figures 2011. Atlanta, GA: American Cancer Society.
Anderson, L. A., Cantwell, M. M., Watson, R. G. P., Johnston, B. T., Murphy, S. J., Ferguson, H. R., McGuigan, J., Comber, H., Reynolds, J. V., and Murray, L. J. (2009). The association between alcohol and reflux esophagitis, Barrett’s esophagus, and esophageal adenocarcinoma. Gastroenterology 136, 799–805.
Anderson, L. A., Johnston, B. T., Watson, R. G. P., Murphy, S. J., Ferguson, H. R., Comber, H., McGuigan, J., Reynolds, J. V., and Murray, L. J. (2006). Nonsteroidal anti-inflammatory drugs and the esophageal inflammation-metaplasia-adenocarcinoma sequence. Cancer Res. 66, 4975–4982.
Anderson, L. A., Murphy, S. J., Johnston, B. T., Watson, R. G. P., Ferguson, H. R., Bamford, K. B., Ghazy, A., McCarron, P., McGuigan, J., Reynolds, J. V., Comber, H., and Murray, L. J. (2008). Relationship between Helicobacter pylori infection and gastric atrophy and the stages of the oesophageal inflammation, metaplasia, adenocarcinoma sequence: results from the FINBAR case-control study. Gut 57, 734–739.
Anderson, L. A., Watson, R. G., Murphy, S. J., Johnston, B. T., Comber, H., Mc Guigan, J., Reynolds, J. V., and Murray, L. J. (2007). Risk factors for Barrett’s oesophagus and oesophageal adenocarcinoma: results from the FINBAR study. World J. Gastroenterol. 13, 1585–1594.
Atherton, J. C., and Blaser, M. J. (2009). Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest. 119, 2475–2487.
Blaser, M. J., and Atherton, J. C. (2004). Helicobacter pylori persistence: biology and disease. J. Clin. Invest. 113, 321–333.
Bosetti, C., Levi, F., Ferlay, J., Garavello, W., Lucchini, F., Bertuccio, P., Negri, E., and La Vecchia, C. (2008). Trends in oesophageal cancer incidence and mortality in Europe. Int. J. Cancer 122, 1118–1129.
Botterweck, A. A., Schouten, L. J., Volovics, A., Dorant, E., and van den Brandt, P. A. (2000). Trends in incidence of adenocarcinoma of the oesophagus and gastric cardia in ten European countries. Int. J. Epidemiol. 29, 645–654.
Brown, L. M., Devesa, S. S., and Chow, W. H. (2008). Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J. Natl. Cancer Inst. 100, 1184–1187.
Cameron, A. J., Zinsmeister, A. R., Ballard, D. J., and Carney, J. A. (1990). Prevalence of columnar-lined (Barrett’s) esophagus. Comparison of population-based clinical and autopsy findings. Gastroenterology 99, 918–922.
Chow, W. H., Finkle, W. D., McLaughlin, J. K., Frankl, H., Ziel, H. K., and Fraumeni, J. F. (1995). The relation of gastroesophageal reflux disease and its treatment to adenocarcinomas of the esophagus and gastric cardia. JAMA 274, 474–477.
Clemons, N. J., McColl, K. E., and Fitzgerald, R. C. (2007). Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology 133, 1198–1209.
Coleman, H. G., Bhat, S., Johnston, B. T., McManus, D., Gavin, A. T., and Murray, L. J. (2012). Tobacco smoking increases the risk of high-grade dysplasia and cancer among patients with Barrett’s esophagus. Gastroenterology 42, 233–240.
Cook, M. B., Chow, W. H., and Devesa, S. S. (2009). Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977–2005. Br. J. Cancer 101, 855–859.
Cook, M. B., Kamangar, F., Whiteman, D. C., Freedman, N. D., Gammon, M. D., Bernstein, L., Brown, L. M., Risch, H. A., Ye, W., Sharp, L., Pandeya, N., Webb, P. M., Wu, A. H., Ward, M. H., Giffen, C., Casson, A. G., Abnet, C. C., Murray, L. J., Corley, D. A., Nyren, O., Vaughan, T. L., and Chow, W. H. (2010). Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J. Natl. Cancer Inst. 102, 1344–1353.
Cook, M. B., Wild, C. P., and Forman, D. (2005). A systematic review and meta-analysis of the sex ratio for Barrett’s esophagus, erosive reflux disease, and nonerosive reflux disease. Am. J. Epidemiol. 162, 1050–1061.
Cooper, B. T., Chapman, W., Neumann, C. S., and Gearty, J. C. (2006). Continuous treatment of Barrett’s oesophagus patients with proton pump inhibitors up to 13 years: observations on regression and cancer incidence. Aliment. Pharmacol. Ther. 23, 727–733.
Corley, D. A., Kerlikowske, K., Verma, R., and Buffler, P. (2003). Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology 124, 47–56.
Corley, D. A., Kubo, A., Levin, T. R., Block, G., Habel, L., Rumore, G., Quesenberry, C., and Buffler, P. (2009). Race, ethnicity, sex and temporal differences in Barrett’s oesophagus diagnosis: a large community-based study, 1994–2006. Gut 58, 182–188.
Corley, D. A., Kubo, A., Levin, T. R., Block, G., Habel, L., Zhao, W., Leighton, P., Quesenberry, C., Rumore, G. J., and Buffler, P. A. (2007). Abdominal obesity and body mass index as risk factors for Barrett’s esophagus. Gastroenterology 133, 34–41.
Corley, D. A., Kubo, A., Levin, T. R., Block, G., Habel, L., Zhao, W., Leighton, P., Rumore, G., Quesenberry, C., Buffler, P., and Parsonnet, J. (2008a). Helicobacter pylori infection and the risk of Barrett’s oesophagus: a community-based study. Gut 57, 727–733.
Corley, D. A., Kubo, A., and Zhao, W. (2008b). Abdominal obesity and the risk of esophageal and gastric cardia carcinomas. Cancer Epidemiol. Biomarkers Prev. 17, 352–358.
Corley, D. A., Levin, T. R., Habel, L. A., and Buffler, P. A. (2006). Barrett’s esophagus and medications that relax the lower esophageal sphincter. Am. J. Gastroenterol. 101, 937–944.
Curado, M. P., Edwards, B., Shin, H. R., Storm, H., Ferlay, J., Heanue, M., and Boyle, P. (eds). (2007). Cancer Incidence in Five Continents, Vol. IX: IARC Scientific Publications No. 160 (Lyon: IARC).
Edelstein, Z. R., Farrow, D. C., Bronner, M. P., Rosen, S. N., and Vaughan, T. L. (2007). Central adiposity and risk of Barrett’s esophagus. Gastroenterology 133, 403–411.
El-Serag, H. B., Aguirre, T. V., Davis, S., Kuebeler, M., Bhattacharyya, A., and Sampliner, R. E. (2004). Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am. J. Gastroenterol. 99, 1877–1883.
Farrow, D. C., Vaughan, T. L., Sweeney, C., Gammon, M. D., Chow, W. H., Risch, H. A., Stanford, J. L., Hansten, P. D., Mayne, S. T., Schoenberg, J. B., Rotterdam, H., Ahsan, H., West, A. B., Dubrow, R., Fraumeni, J. F., and Blot, W. J. (2000). Gastroesophageal reflux disease, use of H-2 receptor antagonists, and risk of esophageal and gastric cancer. Cancer Causes Control 11, 231–238.
Ferlay, J., Shin, H. R., Bray, F., Forman, D., Mathers, C., and Parkin, D. M. (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917.
Ford, A. C., Forman, D., Reynolds, P. D., Cooper, B. T., and Moayyedi, P. (2005). Ethnicity, gender, and socioeconomic status as risk factors for esophagitis and Barrett’s esophagus. Am. J. Epidemiol. 162, 454–460.
Fortuny, J., Johnson, C. C., Bohlke, K., Chow, W. H., Hart, G., Kucera, G., Mujumdar, U., Ownby, D., Wells, K., Yood, M. U., and Engel, L. S. (2007). Use of anti-inflammatory drugs and lower esophageal sphincter-relaxing drugs and risk of esophageal and gastric cancers. Clin. Gastroenterol. Hepatol. 5, 1154–1159.
Freedman, N. D., Murray, L. J., Kamangar, F., Abnet, C. C., Cook, M. B., Nyrén, O., Ye, W., Wu, A. H., Bernstein, L., Brown, L. M., Ward, M. H., Pandeya, N., Green, A. C., Casson, A. G., Giffen, C., Risch, H. A., Gammon, M. D., Chow, W. H., Vaughan, T. L., Corley, D. A., and Whiteman, D. C. (2011). Alcohol intake and risk of esophageal adenocarcinoma: a pooled analysis from the BEACON Consortium. Gut 60, 1029–1037.
Hameeteman, W., Tytgat, G. N., Houthoff, H. J., and Vandentweel, J. G. (1989). Barrett’s esophagus: development of dysplasia and adenocarcinoma. Gastroenterology 96, 1249–1256.
Hamilton, S. R., and Smith, R. R. (1987). The relationship between columnar epithelial dysplasia and invasive adenocarcinoma arising in Barrett’s esophagus. Am. J. Clin. Pathol. 87, 301–312.
Hayeck, T. J., Kong, C. Y., Spechler, S. J., Gazelle, G. S., and Hur, C. (2010). The prevalence of Barrett’s esophagus in the US: estimates from a simulation model confirmed by SEER data. Dis. Esophagus 23, 451–457.
Hillman, L. C., Chiragakis, L., Shadbolt, B., Kaye, G. L., and Clarke, A. C. (2004). Proton-pump inhibitor therapy and the development of dysplasia in patients with Barrett’s oesophagus. Med. J. Aust. 180, 387–391.
Hillman, L. C., Chiragakis, L., Shadbolt, B., Kaye, G. L., and Clarke, A. C. (2008). Effect of proton pump inhibitors on markers of risk for high-grade dysplasia and oesophageal cancer in Barrett’s oesophagus. Aliment. Pharmacol. Ther. 27, 321–326.
Holmes, R. S., and Vaughan, T. L. (2007). Epidemiology and pathogenesis of esophageal cancer. Semin. Radiat. Oncol. 17, 2–9.
Huang, J. Q., Sridhar, S., Chen, Y., and Hunt, R. H. (1998). Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 114, 1169–1179.
Hvid-Jensen, F., Pedersen, L., Drewes, A. A., Sørensen, H. T., and Funch-Jensen, P. (2011). Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 365, 1375–1383.
Ibiebele, T. I., Hughes, M. C., O’Rourke, P., Webb, P. M., Whiteman, D. C., and Australian Cancer Study. (2008). Cancers of the esophagus and carbonated beverage consumption: a population-based case-control study. Cancer Causes Control 19, 577–584.
Ibiebele, T. I., Taylor, A. R., Whiteman, D. C., and van der Pols, J. C. (2010). Eating habits and risk of esophageal cancers: a population-based case-control study. Cancer Causes Control 21, 1475–1484.
Islami, F., Boffetta, P., Ren, J. S., Pedoeim, L., Khatib, D., and Kamangar, F. (2009). High-temperature beverages and foods and esophageal cancer risk: a systematic review. Int. J. Cancer 125, 491–524.
Islami, F., and Kamangar, F. (2008). Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev. Res. (Phila.) 1, 329–338.
Jankowski, J., and Moayyedi, P. (2004). Re: cost-effectiveness of aspirin chemoprevention for Barrett’s esophagus. J. Natl. Cancer Inst. 96, 885–887.
Johansson, J., Hakansson, H. O., Mellblom, L., Kempas, A., Johansson, K. E., Granath, F., and Nyren, O. (2007). Risk factors for Barrett’s oesophagus: a population-based approach. Scand. J. Gastroenterol. 42, 148–156.
Kamat, P., Wen, S. J., Morris, J., and Anandasabapathy, S. (2009). Exploring the association between elevated body mass index and Barrett’s esophagus: a systematic review and meta-analysis. Ann. Thorac. Surg. 87, 655–662.
Kendall, B. J., Macdonald, G. A., Hayward, N. K., Prins, J. B., Brown, I., Walker, N., Pandeya, N., Green, A. C., Webb, P. M., Whiteman, D. C., and Study of Digestive Health. (2008). Leptin and the risk of Barrett’s oesophagus. Gut 57, 448–454.
Kendall, B. J., and Whiteman, D. C. (2006). Temporal changes in the endoscopic frequency of new cases of Barrett’s esophagus in an Australian health region. Am. J. Gastroenterol. 101, 1178–1182.
Kong, C. Y., Nattinger, K. J., Hayeck, T. J., Omer, Z. B., Wang, Y. C., Spechler, S. J., McMahon, P. M., Gazelle, G. S., and Hur, C. (2011). The impact of obesity on the rise in esophageal adenocarcinoma incidence: estimates from a disease simulation model. Cancer Epidemiol. Biomarkers Prev. 20, 2450–2456.
Kubo, A., Block, G., Quesenberry, C. P., Buffler, P., and Corley, D. A. (2009). Effects of dietary fiber, fats, and meat intakes on the risk of Barrett’s esophagus. Nutr. Cancer 61, 607–616.
Kubo, A., and Corley, D. A. (2007). Meta-analysis of antioxidant intake and the risk of esophageal and gastric cardia adenocarcinoma. Am. J. Gastroenterol. 102, 2323–2330.
Kubo, A., Levin, T. R., Block, G., Rumore, G., Quesenberry, C. P., Buffler, P., and Corley, D. A. (2009a). Cigarette smoking and the risk of Barrett’s esophagus. Cancer Causes Control 20, 303–311.
Kubo, A., Levin, T. R., Block, G., Rumore, G. J., Quesenberry, C. P., Buffler, P., and Corley, D. A. (2009b). Alcohol types and sociodemographic characteristics as risk factors for Barrett’s esophagus. Gastroenterology 136, 806–815.
Kubo, A., Levin, T. R., Block, G., Rumore, G. J., Quesenberry, C. P., Buffler, P., and Corley, D. A. (2008a). Dietary antioxidants, fruits, and vegetables and the risk of Barrett’s esophagus. Am. J. Gastroenterol. 103, 1614–1623.
Kubo, A., Levin, T. R., Block, G., Rumore, G. J., Quesenberry, C. P., Buffler, P., and Corley, D. A. (2008b). Dietary patterns and the risk of Barrett’s esophagus. Am. J. Epidemiol. 167, 839–846.
Ladanchuk, T. C., Johnston, B. T., Murray, L. J., Anderson, L. A., and FINBAR Study Group. (2010). Risk of Barrett’s oesophagus, oesophageal adenocarcinoma and reflux oesophagitis and the use of nitrates and asthma medications. Scand. J. Gastroenterol. 45, 1397–1403.
Lagergren, J. (2005). Adenocarcinoma of oesophagus: what exactly is the size of the problem and who is at risk? Gut 54, i1–i5.
Lagergren, J., Bergstrom, R., Adami, H. O., and Nyren, O. (2000). Association between medications that relax the lower esophageal sphincter and risk for esophageal adenocarcinoma. Ann. Intern. Med. 133, 165–175.
Lagergren, J., Viklund, P., and Jansson, C. (2006). Carbonated soft drinks and risk of esophageal adenocarcinoma: a population-based case-control study. J. Natl. Cancer Inst. 98, 1158–1161.
Liao, L. M., Vaughan, T. L., Corley, D. A., Cook, M. B., Casson, A. G., Kamangar, F., Abnet, C. C., Risch, H. A., Giffen, C., Freedman, N. D., Chow, W. H., Sadeghi, S., Pandeya, N., Whiteman, D. C., Murray, L. J., Bernstein, L., Gammon, M. D., and Wu, A. H. (2012). Non-steroidal anti-inflammatory drug use reduces risk for adenocarcinomas of the esophagus and esophagogastric junction in a pooled analysis. Gastroenterology. doi: 10.1053/j.gastro.2011.11.019. [Epub ahead of print].
Mayne, S. T., Risch, H. A., Dubrow, R., Chow, W. H., Gammon, M. D., Vaughan, T. L., Borchardt, L., Schoenberg, J. B., Stanford, J. L., West, A. B., Rotterdam, H., Blot, W. J., and Fraumeni, J. F. (2006). Carbonated soft drink consumption and risk of esophageal adenocarcinoma. J. Natl. Cancer Inst. 98, 72–75.
Mayne, S. T., Risch, H. A., Dubrow, R., Chow, W. H., Gammon, M. D., Vaughan, T. L., Farrow, D. C., Schoenberg, J. B., Stanford, J. L., Ahsan, H., West, A. B., Rotterdam, H., Blot, W. J., and Fraumeni, J. F. (2001). Nutrient intake and risk of subtypes of esophageal and gastric cancer. Cancer Epidemiol. Biomarkers Prev. 10, 1055–1062.
Merlo, L. M., Shah, N. A., Li, X., Blount, P. L., Vaughan, T. L., Reid, B. J., and Maley, C. C. (2010). A comprehensive survey of clonal diversity measures in Barrett’s esophagus as biomarkers of progression to esophageal adenocarcinoma. Cancer Prev. Res. (Phila.) 3, 1388–1397.
Miros, M., Kerlin, P., and Walker, N. (1991). Only patients with dysplasia progress to adenocarcinoma in Barrett’s oesophagus. Gut 32, 1441–1446.
Mulholland, H. G., Cantwell, M. M., Anderson, L. A., Johnston, B. T., Watson, R. G., Murphy, S. J., Ferguson, H. R., McGuigan, J., Reynolds, J. V., Comber, H., and Murray, L. J. (2009). Glycemic index, carbohydrate and fiber intakes and risk of reflux esophagitis, Barrett’s esophagus, and esophageal adenocarcinoma. Cancer Causes Control 20, 279–288.
Murphy, S. J., Anderson, L. A., Ferguson, H. R., Johnston, B. T., Watson, P. R., McGuigan, J., Comber, H., Reynolds, J. V., Murray, L. J., and Cantwell, M. M. (2010). Dietary antioxidant and mineral intake in humans is associated with reduced risk of esophageal adenocarcinoma but not reflux esophagitis or Barrett’s esophagus. J. Nutr. 140, 1757–1763.
Musana, A. K., Resnick, J. M., Torbey, C. F., Mukesh, B. N., and Greenlee, R. T. (2008). Barrett’s esophagus: incidence and prevalence estimates in a rural mid-western population. Am. J. Gastroenterol. 103, 516–524.
Ness-Jensen, E., Lindam, A. P., Lagergren, J., and Hveem, K. (2011). Changes in prevalence, incidence and spontaneous loss of gastro-oesophageal reflux symptoms: a prospective population-based cohort study, the HUNT study. Gut. doi: 10.1136/guyjnl-2011-300715. [Epub ahead of print].
Nguyen, D. M., El-Serag, H. B., Henderson, L., Stein, D., Bhattacharyya, A., and Sampliner, R. E. (2009). Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin. Gastroenterol. Hepatol. 7, 1299–1304.
Pohl, H., Sirovich, B., and Welch, H. G. (2010). Esophageal adenocarcinoma incidence: are we reaching the peak? Cancer Epidemiol. Biomarkers Prev. 19, 1468–1470.
Pohl, H., and Welch, H. G. (2005). The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J. Natl. Cancer Inst. 97, 142–146.
Prach, A. T., MacDonald, T. A., Hopwood, D. A., and Johnston, D. A. (1997). Increasing incidence of Barrett’s oesophagus: education, enthusiasm, or epidemiology? Lancet 350, 933.
Rabinovitch, P. S., Longton, G., Blount, P. L., Levine, D. S., and Reid, B. J. (2001). Predictors of progression in Barrett’s esophagus III: baseline flow cytometric variables. Am. J. Gastroenterol. 96, 3071–3083.
Ranka, S., Gee, J. M., Johnson, I. T., Skinner, J., Hart, A. R., and Rhodes, M. (2006). Non-steroidal anti-inflammatory drugs, lower oesophageal sphincter-relaxing drugs and oesophageal cancer: a case-control study. Digestion 74, 109–115.
Ren, J. S., Freedman, N. D., Kamangar, F., Dawsey, S. M., Hollenbeck, A. R., Schatzkin, A., and Abnet, C. C. (2010). Tea, coffee, carbonated soft drinks and upper gastrointestinal tract cancer risk in a large United States prospective cohort study. Eur. J. Cancer 46, 1873–1881.
Rex, D. K., Cummings, O. W., Shaw, M., Cumings, M. D., Wong, R. K., Vasudeva, R. S., Dunne, D., Rahmani, E. Y., and Helper, D. J. (2003). Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology 125, 1670–1677.
Risques, R. A., Vaughan, T. L., Li, X., Odze, R. D., Blount, P. L., Ayub, K., Gallaher, J. L., Reid, B. J., and Rabinovitch, P. S. (2007). Leukocyte telomere length predicts cancer risk in Barrett’s esophagus. Cancer Epidemiol. Biomarkers Prev. 16, 2649–2655.
Rokkas, T., Pistiolas, D., Sechopoulos, P., Robotis, I., and Margantinis, G. (2007). Relationship between Helicobacter pylori infection and esophageal neoplasia: a meta-analysis. Clin. Gastroenterol. Hepatol. 5, 1413–1417.
Ronkainen, J., Aro, P., Storskrubb, T., Johansson, S. E., Lind, T., Bolling-Sternevald, E., Vieth, M., Stolte, M., Talley, N. J., and Agreus, L. (2005). Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology 129, 1825–1831.
Rubenstein, J. H., and Taylor, J. B. (2010). Meta-analysis: the association of oesophageal adenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment. Pharmacol. Ther. 32, 1222–1227.
Sato, F., Jin, Z., Schulmann, K., Wang, J., Greenwald, B. D., Ito, T., Kan, T., Hamilton, J. P., Yang, J., Paun, B., David, S., Olaru, A., Cheng, Y., Mori, Y., Abraham, J. M., Yfantis, H. G., Wu, T. T., Fredericksen, M. B., Wang, K. K., Canto, M., Romero, Y., Feng, Z., and Meltzer, S. J. (2008). Three-tiered risk stratification model to predict progression in Barrett’s esophagus using epigenetic and clinical features. PLoS ONE 3, e1890. doi:10.1371/journal.pone.0001890
Shaheen, N. J., Crosby, M. A., Bozymski, E. M., and Sandler, R. S. (2000). Is there publication bias in the reporting of cancer risk in Barrett’s esophagus? Gastroenterology 119, 333–338.
Sikkema, M., De Jonge, P. J., Steyerberg, E. W., and Kuipers, E. J. (2010). Risk of esophageal adenocarcinoma and mortality in patients with Barrett’s esophagus: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 8, 235–244.
Smith, K. J., O’Brien, S. M., Green, A. C., Webb, P. M., Whiteman, D. C., and Study of Digestive Health. (2009). Current and past smoking significantly increase risk for Barrett’s esophagus. Clin. Gastroenterol. Hepatol. 7, 840–848.
Stavrou, E. P., McElroy, H. J., Baker, D. F., Smith, G., and Bishop, J. F. (2009). Adenocarcinoma of the oesophagus: incidence and survival rates in New South Wales, 1972–2005. Med. J. Aust. 191, 310–314.
Suerbaum, S., and Michetti, P. (2002). Medical progress: Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186.
Thompson, O. M., Beresford, S. A., Kirk, E. A., Bronner, M. P., and Vaughan, T. L. (2010). Serum leptin and adiponectin levels and risk of Barrett’s esophagus and intestinal metaplasia of the gastroesophageal junction. Obesity (Silver Spring) 18, 2204–2211.
Thompson, O. M., Beresford, S. A., Kirk, E. A., and Vaughan, T. L. (2009). Vegetable and fruit intakes and risk of Barrett’s esophagus in men and women. Am. J. Clin. Nutr. 89, 890–896.
Thrift, A. P., Pandeya, N., Smith, K. J., Green, A. C., Hayward, N. K., Webb, P. M., and Whiteman, D. C. (2011a). Helicobacter pylori infection and the risks of Barrett’s oesophagus: a population-based case-control study. Int. J. Cancer. doi: 10.1002/ijc.26242. [Epub ahead of print]..
Thrift, A. P., Pandeya, N., Smith, K. J., Green, A. C., Webb, P. M., and Whiteman, D. C. (2011b). The use of nonsteroidal anti-inflammatory drugs and the risk of Barrett’s oesophagus. Aliment. Pharmacol. Ther. 34, 1235–1244.
Thrift, A. P., Pandeya, N., Smith, K. J., Mallitt, K. A., Green, A. C., Webb, P. M., and Whiteman, D. C. (2011c). Lifetime alcohol consumption and risk of Barrett’s esophagus. Am. J. Gastroenterol. 106, 1220–1230.
van Soest, E. M., Dieleman, J. P., Siersema, P. D., Sturkenboom, M., and Kuipers, E. J. (2005). Increasing incidence of Barrett’s oesophagus in the general population. Gut 54, 1062–1066.
Vaughan, T. L., Dong, L. M., Blount, P. L., Ayub, K., Odze, R. D., Sanchez, C. A., Rabinovitch, P. S., and Reid, B. J. (2005). Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett’s oesophagus: a prospective study. Lancet Oncol. 6, 945–952.
Vaughan, T. L., Farrow, D. C., Hansten, P. D., Chow, W. H., Gammon, M. D., Risch, H. A., Stanford, J. L., Schoenberg, J. B., Mayne, S. T., Rotterdam, H., Dubrow, R., Ahsan, H., West, A. B., Blot, W. J., and Fraumeni, J. F. (1998). Risk of esophageal and gastric adenocarcinomas in relation to use of calcium channel blockers, asthma drugs, and other medications that promote gastroesophageal reflux. Cancer Epidemiol. Biomarkers Prev. 7, 749–756.
Vizcaino, A. P., Moreno, V., Lambert, R., and Parkin, D. M. (2002). Time trends incidence of both major histologic types of esophageal carcinomas in selected countries, 1973–1995. Int. J. Cancer 99, 860–868.
Whiteman, D. C., Parmar, P., Fahey, P., Moore, S. P., Stark, M., Zhao, Z. Z., Montgomery, G. W., Green, A. C., Hayward, N. K., Webb, P. M., and Australian Cancer Study. (2010). Association of Helicobacter pylori infection with reduced risk for esophageal cancer is independent of environmental and genetic modifiers. Gastroenterology 139, 73–83.
Whiteman, D. C., Sadeghi, S., Pandeya, N., Smithers, B. M., Gotley, D. C., Bain, C. J., Webb, P. M., Green, A. C., and Australian Cancer Study. (2008). Combined effects of obesity, acid reflux and smoking on the risk of adenocarcinomas of the oesophagus. Gut 57, 173–180.
Williams, L. B., Fawcett, R. L., Waechter, A. S., Zhang, P. L., Kogon, B. E., Jones, R., Inman, M., Huse, J., and Considine, R. V. (2000). Leptin production in adipocytes from morbidly obese subjects: stimulation by dexamethasone, inhibition with troglitazone, and influence of gender. J. Clin. Endocrinol. Metab. 85, 2678–2684.
Wu, A. H., Tseng, C. C., Hankin, J., and Bernstein, L. (2007). Fiber intake and risk of adenocarcinomas of the esophagus and stomach. Cancer Causes Control 18, 713–722.
Yang, P. C., and Davis, S. (1988). Incidence of cancer of the esophagus in the United States by histologic type. Cancer 61, 612–617.
Yoshida, N. (2007). Inflammation and oxidative stress in gastroesophageal reflux disease. J. Clin. Biochem. Nutr. 40, 13–23.
Zagari, R. M., Fuccio, L., Wallander, M. A., Johansson, S., Fiocca, R., Casanova, S., Farahmand, B. Y., Winchester, C. C., Roda, E., and Bazzoli, F. (2008). Gastro-oesophageal reflux symptoms, oesophagitis and Barrett’s oesophagus in the general population: the Loiano-Monghidoro study. Gut 57, 1354–1359.
Keywords: esophageal adenocarcinoma, Barrett’s esophagus, epidemiology, risk factors
Citation: Thrift AP, Pandeya N and Whiteman DC (2012) Current status and future perspectives on the etiology of esophageal adenocarcinoma. Front. Oncol. 2:11. doi: 10.3389/fonc.2012.00011
Received: 30 November 2011;
Paper pending published: 20 December 2011;
Accepted: 17 January 2012;
Published online: 13 February 2012.
Edited by:
Mohandas K. Mallath, Tata Memorial Centre, IndiaReviewed by:
Farhad Islami, Mount Sinai School of Medicine, USAFarin Kamangar, Morgan State University, USA
Copyright: © 2012 Thrift, Pandeya and Whiteman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David C. Whiteman, Cancer Control Laboratory, Queensland Institute of Medical Research, Locked Bag 2000, Royal Brisbane Hospital, QLD 4029, Australia. e-mail:ZGF2aWQud2hpdGVtYW5AcWltci5lZHUuYXU=