

- Department of Cell Biology, Oncology, Nerviano Medical Sciences, Milan, Italy
In 2007, the ALK tyrosine kinase was described as a potential therapeutic target for a subset of non-small-cell lung cancer patients. Clinical proof of concept, culminating in the recent approval by the Food and Drug Administration of the Pfizer drug crizotinib followed in record time. The drug was approved together with a companion diagnostic for detection of patients eligible for therapy. This remarkable example of the coming of age of personalized medicine in cancer therapy is hopefully only an auspice of things to come in a rapidly developing field. Perhaps unsurprisingly, however, the appearance of clinical acquired resistance to crizotinib was observed early on in clinical testing, with the identification of several ALK secondary point mutations which diminish drug efficacy and which open the way for development of second-generation inhibitors. It is also emerging that acquired resistance to crizotinib may additionally occur through ALK-independent mechanisms, which still need to be elucidated in detail. Here we discuss the factors that led to such a rapid approval of a targeted agent, and we describe the second-generation compounds currently in development.
Introduction
The ALK (anaplastic lymphoma kinase) gene encodes a tyrosine kinase belonging to the insulin receptor superfamily. ALK is abundantly expressed in neural tissue during embryogenesis, but levels fall during early development, so that in adults it is expressed only in rare scattered neural cells (Iwahara et al., 1997; Morris et al., 1997; Webb et al., 2009; Ardini et al., 2010). ALK was originally identified in anaplastic large cell lymphoma (ALCL) cells as the product of a recurring chromosomal translocation, t(2;5; p23;q35), between the ALK gene on chromosome 2 and the nucleophosmin (NPM) gene on chromosome 5, which gives rise to expression of the NPM–ALK fusion protein (Morris et al., 1994). The oncogenic potential of NPM–ALK, which contains a constitutively activated ALK kinase domain, was subsequently demonstrated in several different preclinical models, confirming its role in the pathogenesis of ALCL (Shiota et al., 1994; Drexler et al., 2000). In addition to ALCL, ALK gene translocations or activating point mutations were identified in other rare tumor types, including inflammatory myofibroblastic tumors (IMT) and neuroblastoma (Webb et al., 2009; Ardini et al., 2010). IMT is a rare tumor of mesenchymal origin that affects young individuals, with approximately 50% of cases bearing a chromosomal translocation involving the ALK gene, fused to many different N-terminal partners (Griffin et al., 1999), while neuroblastoma is a rare pediatric solid tumor and originates from neural cell-derived tissue, giving tumor masses localized mainly at the level of the adrenal glands (Maris et al., 2007). In neuroblastoma, ALK gene amplification and point mutations are found as recurring events, rather than gene translocation.
Notwithstanding the substantial evidence linking activated ALK kinase to tumorigenesis in these rare tumors, it is fair to say that the considerable current enthusiasm for ALK as target for cancer therapy is largely driven by the relatively recent finding of a recurring ALK gene translocation in a significant subset (ca. 5%) of non-small-cell lung cancer (NSCLC, Rikova et al., 2007; Soda et al., 2007). In ALK-positive NSCLC, ALK gene rearrangement most often involves an inversion within the short arm of chromosome 2 (between loci 2p21 and 2p23), leading to expression of echinoderm microtubule associated protein like 4 (EML4)–ALK, an oncogenic fusion protein composed of the N-terminal portion of EML4 and the entire intracellular portion of ALK. As with NPM–ALK, there is much convincing preclinical evidence in support of the oncogenic nature of EML4–ALK, the requirement for ALK kinase activity in maintenance of EML4–ALK-dependent tumor cell growth and of the capacity of selective small molecule kinase inhibitors of ALK to induce cell death in such tumors (Christensen et al., 2007; Galkin et al., 2007; Choi et al., 2008; Koivunen et al., 2008; McDermott et al., 2008; Soda et al., 2008). Subsequent studies of tissue samples from NSCLC patients aimed at further characterizing ALK-positive NSCLC have led to the identification of a relatively well defined potential patient population, characterized by specific clinical–pathological features. It appears that ALK-positive patients tend to be younger than the median age for lung cancer patients and are, in general, never-smokers, or former light smokers, while at the histological level, ALK-positive tumors are almost exclusively adenocarcinomas, with a clear component of the signet-ring cell type (Inamura et al., 2008; Shaw et al., 2009; Solomon et al., 2009; Kwak et al., 2010). The presence of EML4–ALK rearrangement appears to be mutually exclusive with KRAS and EGFR mutations, further supporting a role for ALK as a unique driver of malignancy in these patients, though interestingly, an exception is possibly represented by the recent description of a small fraction of crizotinib-naïve patients reported to possess both EML4–ALK rearrangement and EGFR mutations (Sasaki et al., 2011), as will be further commented below.
Clinical Validation of ALK as a Therapeutic Target in ALK-Positive NSCLC
Crizotinib is an orally available drug that was originally discovered and optimized as an inhibitor of c-Met kinase (Christensen et al., 2007; Zou et al., 2007). Prior to designation of the International Non-proprietary Name of “crizotinib” the drug was known as PF-02341066 (the Pfizer company’s chemical collection number for the compound) and it is now also known as Xalkori®, a Pfizer brand name, but we will subsequently only refer to it in this text as crizotinib. Studies with c-Met kinase revealed that crizotinib has a classical ATP-competitive mechanism of action and as is often the case for such inhibitors, it was subsequently found to cross-react with a few “off-target” kinases. In particular, potent activity of the drug on ALK was revealed through selectivity profiling in biochemical assay and ALK-driven cellular models (Christensen et al., 2007). A multi-indication Phase I clinical trial of crizotinib in solid tumors and lymphomas had already been initiated, with the drug described as a “c-Met/Hepatocyte Growth Factor tyrosine kinase inhibitor” (ClinicalTrials.gov Identifier: NCT00585195), when identification of the genetic rearrangement involving ALK in NSCLC was first reported (Soda et al., 2007). In 2008, while preclinical data supporting a therapeutic rationale for targeting ALK in NSCLC was still emerging, ALK-positive patients started to be enrolled in this already ongoing Phase I trial. ALK cross-reactivity of crizotinib, apparently initially seen as a possible path for registration of the compound in niche indications such as chemotherapy resistant ALCL, now became a major opportunity. Thus, patient screening and enrollment of ALK-positive subjects into the trial was initiated, using a methodology based on the break apart probe FISH (fluorescent in situ hybridization) technique, with a kit specifically developed for detecting ALK translocation in patient tumor samples (Perner et al., 2008). Within a few months, impressive preliminary data on clinical response in these patients became available. A dedicated Phase I/II clinical trial focused on ALK-positive NSCLC patients was completed in 2010 (Kwak et al., 2010), barely 3 years after the first description of this genetic lesion. After the standard dose escalation Phase I that defined the recommended dose of 250 mg twice a day per 28-day cycle, an expanded cohort of ALK-positive NSCLC was selected for treatment. Approximately 1500 NSCLC patients were screened by FISH, identifying 82 patients considered eligible and then enrolled in the expanded cohort study. Most of these patients had received previous therapy and almost half were heavily pre-treated. The overall objective response rate in this study was 57% (47 out of 82 patients, with 46 confirmed partial response and 1 complete response), with a further 33% of patients (27 out of 82) in stable disease. The estimated probability of 6-month progression-free survival was 72%. To date, the median overall survival time from initiation of crizotinib has not been determined, but 1-year overall survival was 74% and 2-year overall survival was 54% (Kwak et al., 2010; Shaw et al., 2011).
The spectacular efficacy observed for crizotinib in this challenging setting was associated with relatively mild side effects. The most frequently reported were gastrointestinal toxicities, with grade 1 nausea and diarrhea and visual disturbances, but with no abnormalities detected in ophthalmological examination. Increased levels of hepatic transaminases were also observed, but only reaching grade 3 in a limited number of patients (5 and 6% for ALT and AST, respectively). Two randomized Phase III clinical trials in ALK-positive NSCLC are currently underway to compare the activity of crizotinib to standard of care. Nevertheless, based on the impressive responses observed in Phase I/II trial, the Food and Drug Administration (FDA) approved crizotinib for treatment of ALK rearranged NSCLC, under its accelerated approval program, on August 26, 2011. The National Comprehensive Cancer network guidelines recommend the use of crizotinib as first line therapy for ALK-positive selected NSCLC patients (www.nccn.org).
Other patients affected by rare malignancies for which a clear involvement of ALK had been demonstrated in preclinical studies, were also enrolled in the trial with crizotinib. For at least two patients with ALK-positive ALCL treated at the recommended Phase II dose, signs of clinical benefit were seen within a remarkably short treatment period, with a PR and a CR achieved (Gambacorti-Passerini and Messa, 2011). Two patients with IMT were enrolled already in the dose escalation phase: for one of these, a rapid and sustained partial response was seen. The other patient had no response to crizotinib, but retrospective genetic analysis showed that this IMT tumor lacked ALK rearrangement (Butrynski et al., 2010).
Current Treatment of ALK-Positive Tumors: Successes and Challenges
Current publicly available data indicate that crizotinib therapy of ALK-positive NSCLC patients is associated with a median progression-free survival time of circa 10 months. However, soon after publication of efficacy results of Phase I/II trials, early data on relapse to crizotinib due to newly acquired secondary mutations in the ALK kinase domain were also reported (Choi et al., 2010; Sasaki et al., 2010). This observation poignantly reflects previous clinical experience with other inhibitors that selectively target kinases to which “oncogene addiction” appears to be a driving force for tumor growth. A wealth of clinical data has been accumulated, for example, with the EGFR inhibitors gefitinib and erlotinib in NSCLC patients bearing EGFR mutations, with imatinib and sunitinib in c-Kit dependent GIST tumors and with imatinib in Bcr–Abl positive CML patients. It has been amply demonstrated that relapse to these agents is often linked to acquired resistance to the inhibitor due to secondary mutations in the target kinase domain which compromise drug inhibitory activity (Shah et al., 2002; Tamborini et al., 2004; Carter et al., 2005; Kobayashi et al., 2005). In fact, that crizotinib might also be susceptible to such a resistance mechanism had been suggested by preclinical studies with kinase domain point mutants of ALK corresponding to those found in neuroblastoma. Several different single amino acid mutations of ALK are known in this disease, all mapping to the cytoplasmic portion of the receptor and most of which induce constitutive kinase activity of the full length receptor (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). Intriguingly, biochemical and cellular studies revealed that not all neuroblastoma mutants are equally susceptible to inhibition by ATP-competitive kinase inhibitors, including crizotinib. For example, crizotinib maintains activity against the R1275Q mutant, but drastically loses activity against F1174L, another frequently occurring mutant (Wood et al., 2009; Sakamoto et al., 2011; Schönherr et al., 2011). These findings indicate that the ALK kinase domain can naturally undergo single point mutations which result in loss of sensitivity to crizotinib compared with the wild-type domain.
Perhaps unsurprisingly, therefore, DNA sequence analyses performed in three relapsed NSCLC patients and in the IMT case which, after successful treatment with crizotinib for a few months, had acquired resistance to therapy, have identified four different de novo secondary mutations that are compellingly linked to acquired drug resistance. The L1196M gatekeeper mutation and the C1156Y and L1152R mutants were identified in the relapsed NSCLC cases, and the F1174L mutation in the relapsed IMT (Choi et al., 2010; Sasaki et al., 2010, 2011). The mechanisms underlying decreased activity of crizotinib on these secondary ALK mutants were investigated by structural and biochemical analyses, together with cellular data generated in engineered in vitro models. For the L1196M, C1156Y, and L1152R mutants, it appears that binding of the inhibitor to ALK may be negatively affected by steric hindrance or conformational changes in the enzyme. F1174L, which recapitulates the primary mutation found in neuroblastoma previously shown to render the enzyme insensitive to crizotinib, appears instead to induce a conformational change in the protein which results in increased affinity for ATP itself. This latter type of resistance mechanism is highly reminiscent of that previously described for resistance of EGFR to gefitinib (Iressa) and erlotinib (Tarceva) in NSCLC patients as a result of the T790M secondary mutation in EGFR (Yun et al., 2008) and by analogy, all ALK inhibitors with an ATP-competitive binding mechanism may be destined to display decreased inhibitory activity when the F1174L mutation appears. Consequently, effective targeting of this mutant may require extremely high affinity or irreversible inhibitors.
Thus, after the initial wave of enthusiasm to crizotinib in the scientific and NSCLC patient communities, the need for second-generation ALK inhibitors became quickly evident.
However, another key finding emerging from the clinical data available to date is that not all cases of acquired resistance to crizotinib are necessarily due to secondary mutations in ALK itself, since in some relapsed NSCLC lesions, no secondary ALK mutation is detectable (Sasaki et al., 2010). Mechanisms underlying ALK-independent resistance have not yet been extensively elucidated, but it is likely that in some patients relapse is due to activation of alternative signal transduction pathways, so that the tumor is no longer solely critically dependent upon ALK signaling. The only firm data available to date in this regard are derived from a single NSCLC case in which increased levels of EGFR signaling were detected after acquired resistance to crizotinib and a limited genetic study in treatment-naïve ALK rearranged NSCLC cases suggests that EGFR mutations may be present in circa 5% of such cases (Sasaki et al., 2011). In this context, it is interesting to note that in addition to secondary mutation of EGFR, c-Met amplification has been associated with acquired resistance to the EGFR inhibitors gefitinib and erlotinib in NSCLC patients bearing activating mutations of EGFR (Engelman et al., 2007). A similar scenario is seen in B-raf mutated melanoma patients in relapse from therapy with the B-raf inhibitor vemurafenib, in which progression does not appear, at least to date, to be associated with acquisition of secondary mutations in B-raf V600E, but rather with induction of alternative pathways for MAPK activation, including Cot, PDGFR, and MEK1 dependent signaling (Johannessen et al., 2010; Nazarian et al., 2010).
We can thus envisage that in future, specimens from relapsed patients will be subjected to detailed molecular analyses aimed at verifying firstly whether secondary ALK mutation is present and then whether other resistance phenomena such as EGFR mutation or c-Met amplification have by-passed ALK signaling (see Figure 1 for a schematic view of crizotinib mode of action in ALK-positive NSCLC and the two currently postulated mechanisms for acquired resistance).
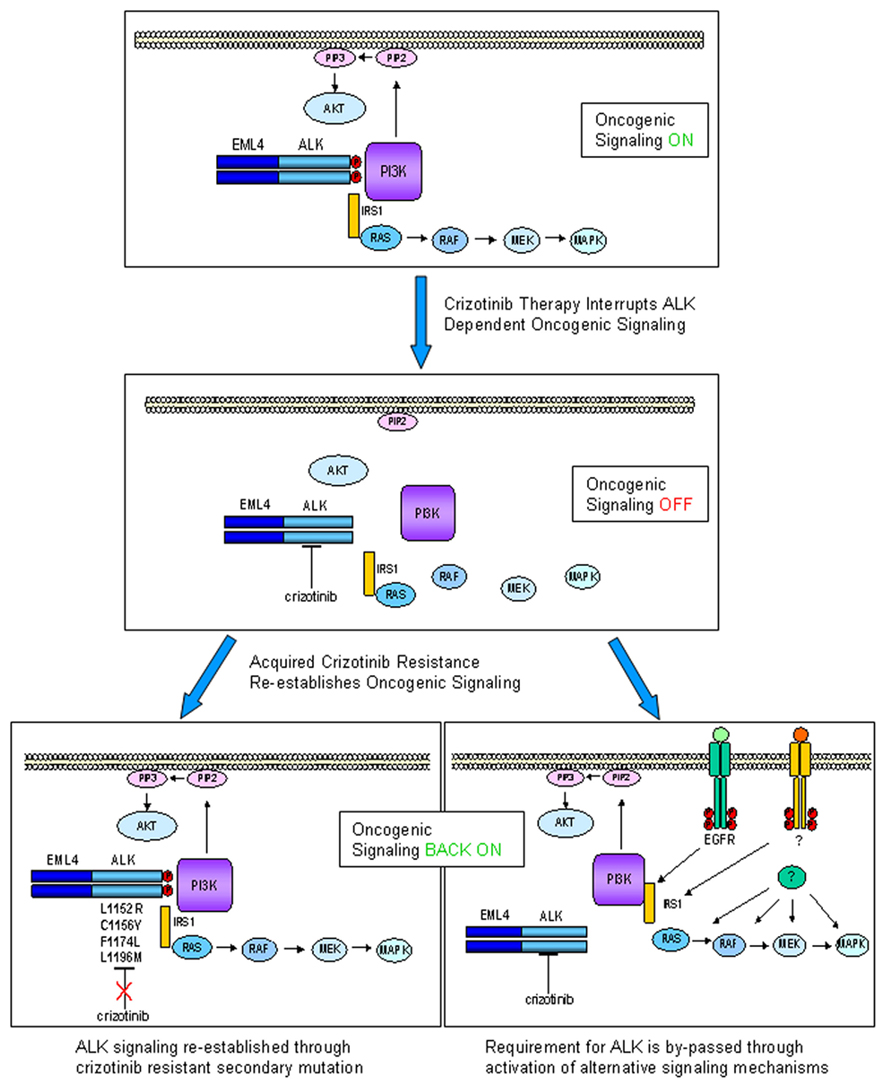
Figure 1. Echinoderm microtubule associated protein like 4–ALK is a driver oncogene in ca. 5% of NSCLC, engaging multiple downstream signaling pathways, including the MAPK and AKT pathways (upper panel). Crizotinib inactivates EML4–ALK kinase activity, disengaging oncogenic signaling pathways, thus inducing growth arrest and cell death (middle panel). Upon sustained selective pressure following ALK inhibition by crizotinib, tumors can acquire resistance through two types of mechanism: ALK-dependent (lower left panel) or ALK-independent (lower right panel). In ALK-dependent acquired resistance, ALK can undergo secondary mutations which abrogate ability of crizotinib to inhibit its kinase activity (some known clinically relevant mutations are indicated). For this type of resistance second-generation ALK inhibitors might be particularly effective. In ALK-independent acquired resistance, activation of alternative signaling pathways (e.g., EGFR, c-MET, or downstream signaling components) occurs collateral to ALK activation, removing the absolute requirement for ALK as a driver oncogene. In this case, it can be hypothesized that ALK is still present and inhibited by drug, so that combination therapy with an ALK inhibitor would be a therapeutic option.
This would in theory open a perspective of combination of crizotinib with other targeted therapies for treatment of a subset of ALK-positive patients with acquired resistance. For example, in the case of acquired ALK inhibitor resistance due to collateral EGF receptor activation, such an approach would be immediately feasible by combination of crizotinib with already approved agents such as gefitinib and erlotinib. In the case of c-Met amplification as a potential crizotinib resistance mechanism in NSCLC, as has been amply described for the EGFR inhibitors, it will be extremely interesting to see whether or not this occurs, given that crizotinib cross-reacts strongly with c-Met and that emerging clinical evidence indicates that the drug has activity in a c-Met-amplified context (Lennerz et al., 2011).
New Drugs Targeting ALK
To date, crizotinib remains the only drug that has been evaluated in clinical trials beyond Phase I. However, several new ALK kinase inhibitors have been described, with some already in early clinical development (Table 1). Clinical development strategies for the most advanced molecules appear to be based on two approaches: a first “all-comer” approach including both crizotinib-naïve patients and patients who developed acquired crizotinib resistance after initial response and a second focusing solely on patients with acquired resistance (Hallberg and Palmer, 2011; Sasaki and Jänne, 2011).
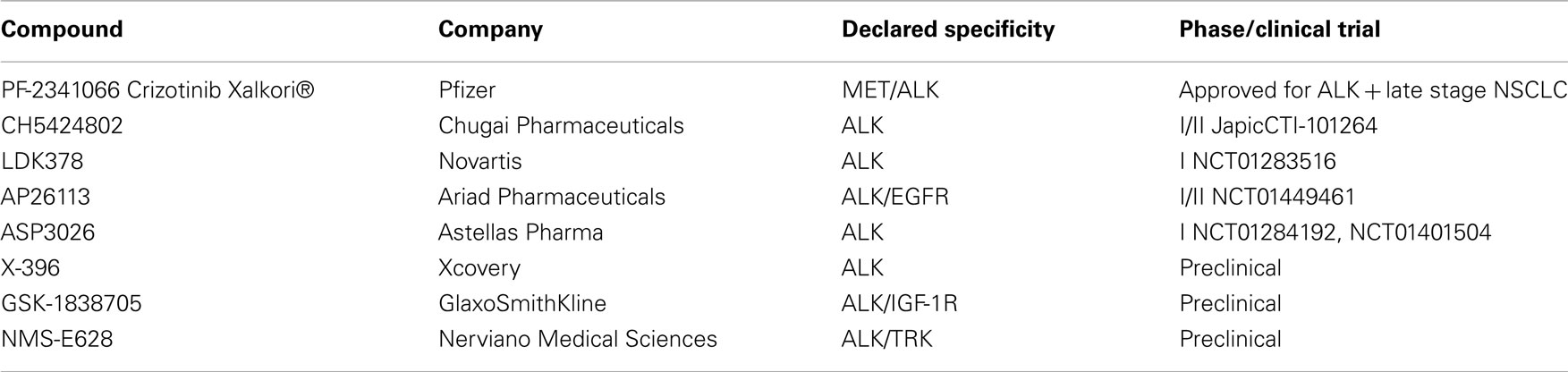
Table 1. ALK small molecule inhibitors.
CH5424802 (Chugai Pharmaceutical, a subsidiary of Roche) is a potent, selective, and orally available kinase inhibitor of ALK. It is an ATP-competitive inhibitor and displays strong anti-proliferative activity in different ALK–driven tumor models in vitro, as well as in vivo, with impressive anti-tumor activity in ALK-positive NSCLC, ALCL, and neuroblastoma xenografts (Sakamoto et al., 2011). Preclinical characterization of the drug included evaluation of the potency of CH5424802 on ALK mutants using both biochemical enzyme assays and engineered cellular models. Good biochemical potency was reported on L1196M, C1156Y, and F1174L mutated proteins, with low nanomolar IC50 or Ki values, comparable to that found on wild-type ALK. In vitro studies performed on Ba/F3 cells expressing mutated ALK kinase forms supported the biochemical data, confirming potent inhibition of L1196M and C1156Y mutants in a cellular setting. In vivo efficacy was described only for the L1196M gatekeeper mutation, confirming a higher potency with respect to crizotinib in inhibiting the in vivo growth of ALK–L1196M driven Ba/F3 cells. For the F1174L mutant, activity in Ba/F3 cells was not described, but the compound was able to effectively inhibit proliferation of a neuroblastoma cell line naturally bearing the mutation.
CH5424802 is currently under clinical evaluation in an open-labeled Phase I/II trial in NSCLC patients in Japan (ClinicalTrials.gov Identifier: Japic CTI-101264). The trial is scheduled to be completed in March 2014.
LDK378 (Novartis) is an orally available ALK inhibitor that is being evaluated in an open-label dose escalation Phase I trial in ALK rearranged tumors (ClinicalTrials.gov Identifier: NCT01283516). Three different arms are foreseen, including ALK-positive crizotinib-naïve NSCLC patients, ALK-positive NSCLC patients previously treated with other ALK inhibitors and all ALK-positive tumors other than NSCLC, respectively. Limited information on preclinical evaluation are publicly available for this drug. LDK378 appears very efficacious in vivo, inducing complete and durable tumor regression in an ALK-positive NSCLC dependent model and was also described to be active in tumors bearing the C1156Y mutation that confers crizotinib resistance (Li et al., 2011).
AP26113 (Ariad Pharmaceuticals) is a potent and orally available inhibitor of ALK whose chemical structure has not been disclosed. Biochemical characterization shows that in addition to ALK, the compound cross-reacts with a number of other kinases (>45 inhibited with IC50 < 200 nM), among which EGFR is inhibited with an IC50 of 129 nM. Considering that EGFR is a well validated target per se in NSCLC and that in at least one case, resistance to crizotinib was associated with EGFR activation, this cross-reactivity was considered an opportunity by the company and the compound is in clinical testing as a dual ALK/EGFR inhibitor. Additionally, AP26113 was evaluated on the crizotinib resistant gatekeeper mutant L1196M both in vitro and in vivo and appeared to be able to overcome resistance to crizotinib. Ki determination demonstrated a very similar biochemical potency on wild-type ALK and the L1196M ALK mutant (0.09 and 0.08 nM respectively), with both cellular and in vivo data (using engineered Ba/F3 cells) indicating that growth of ALK–L1196M mutant driven-cells is inhibited at similar, albeit slightly higher, doses which inhibit cells harboring wild-type ALK (Katayama et al., 2011). AP26113 was also described to be active on a series of in vitro induced crizotinib resistant mutations, which however have not been observed to date in clinical cases of acquired crizotinib resistance (Zhang et al., 2010).
Clinical development of this drug has initiated recently (September 2011), with a Two-Stage development strategy (ClinicalTrials.gov Identifier: NCT01449461). The initial dose escalation will be conducted in patients with advanced cancers, particularly NSCLC. The expanded cohort of patients treated at the RP2D will include four genetically defined patient populations: including: patients with ALK-positive NSCLC who have not previously received an ALK inhibitor, patients with ALK-positive NSCLC who are resistant to at least one ALK inhibitor, patients with EGFR positive NSCLC who are resistant to at least one prior EGFR inhibitor and patients with other cancers expressing ALK.
ASP3026 (Astellas) is an orally available ALK inhibitor, for which no preclinical data are publicly available. The compound is being evaluated in a phase I, non-randomized, open-label, study in patients with solid tumors (ClinicalTrials.gov Identifier: NCT01401504). The trial initiated in December 2010 and is scheduled to be completed in April 2013.
X-296/X-396 (Xcovery) are aminopyridazine-based ALK kinase inhibitors which display good anti-tumor activity in vitro and in vivo on different ALK-dependent tumor models. X-396 was also evaluated on L1196M and C1156Y mutations and data suggest that it can potentially overcome at least these crizotinib resistance mutations. Pharmacokinetic properties and toxicity profiles are described as favorable for X-396 and suggest that this might be a future candidate for clinical testing (Lovly et al., 2011). Additionally, data regarding the distribution of X-396 in brain tissue suggest that this drug might also possess activity against ALK-positive brain metastases.
GSK1838705A, a compound originally identified as potent, ATP-competitive inhibitor of IGF-1R and insulin receptor, has been described to be highly active against ALK kinase (Sabbatini et al., 2009). In vivo, tumor growth inhibition in ALK-positive xenograft models was observed, with minimal and transient effects on glucose homeostasis, suggesting that, despite potential diabetogenic effects, an acceptable therapeutic window might be achieved by schedule modulation. No information are available for this compound regarding activity against crizotinib resistant ALK mutants.
NMS-E628 (Nerviano Medical Sciences), from the authors’ own group, is an orally available small molecule inhibitor of ALK kinase activity for which preclinical characterization has been completed, with the compound approaching clinical development.
NMS-E628 selectively inhibits ALK-dependent cell proliferation at 100 nM or below and is able to induce tumor regression in several preclinical ALK-dependent models, with some animals remaining tumor-free for extended periods of time after the end of the treatment. Preclinical pharmacokinetic studies show that NMS-E628 is able to efficiently cross the blood–brain barrier. Efficacy following oral administration in an intracranial growth model of the H2228 NSCLC xenograft confirmed that efficacious exposures can be reached in the brain and support the possible use of NMS-E628 in patients bearing brain metastases (Ardini et al., 2009). When tested in Ba/F3 cells driven by ALK mutants identified in crizotinib-relapsed patients, NMS-E628 is circa fivefold more potent than crizotinib in inhibiting the proliferation of L1196M ALK and C1156Y ALK-driven-cells in vitro and in vivo (Ardini et al., 2011). Thus, on the basis of increased potency and ability to cross the blood–brain barrier, NMS-E628 might represent a valid therapeutic opportunity for crizotinib-relapsed patients that experience acquired resistance to specific ALK mutations.
Future Perspectives
Crizotinib recently received accelerated approval from the FDA, coming hot on the heels of the B-raf inhibitor vemurafenib (Zelboraf®, approved August 17, 2011 for melanoma patients with the B-raf V600E mutation). Significantly, both agents were approved not for a broad indication, but for a molecularly defined subset of patients and both were approved with a companion diagnostic test. Unlike vemurafenib, crizotinib is an “off-the shelf” inhibitor in the sense that it was already in clinical development when the molecular setting for which it was eventually approved was discovered. This certainly gave the compound a strong competitive advantage over those originating within ALK-focused programs, but what contributed vitally to its successful registration was the efficiency with which ALK-positive NSCLC patients, which represent only circa 5% of the indication, were detected and selected for treatment within the expansion arm of the first Phase I/II studies. Such logistic efficiency, organization, and vision on the part of Pfizer and collaborators is praiseworthy and indeed registration and marketing of the drug were made possible by the parallel availability of a companion diagnostic test, the Vysis ALK Break Apart FISH Probe Kit (Abbott Molecular, Inc.) which was approved alongside crizotinib for detection of patients eligible for treatment with the drug (FDA News, August 26, 2011).
Data available to date and comparison with other kinase inhibitors approved for NSCLC, such as gefitinib and erlotinib, indicate that in most cases, treatment of ALK-driven tumors with crizotinib will not be curative, but that relapse will occur with at least two types of mechanism, differing on the basis of whether or not tumors retain ALK-dependency. In the case of ALK-dependent relapse, current evidence indicates that acquired resistance to crizotinib will certainly occur through secondary ALK mutations, resulting in variants which are intrinsically less sensitive to the drug, but it has also been suggested that crizotinib may possess other weaknesses, such as inability of the drug to act effectively in pharmacological “sanctuary” sites, for example beyond the blood–brain barrier (Camidge, 2011). This is an important consideration for a disease in which circa 40–50% of cases experience brain metastases (Eichler et al., 2011). For ALK-dependent progressive disease, several second-generation compounds, originating from ALK-focused programs, are currently undergoing, or will soon enter clinical testing and it is likely that efficacious new agents will emerge amongst these within the next couple of years.
With regards to ALK-independent acquired resistance to crizotinib, it is not yet clear how frequently this will occur and which signaling pathways will be involved. However, we expect that approaches such as “deep” DNA sequencing of relapsed lesions (e.g., Ross and Cronin, 2010) and genome-wide functional genetic studies (e.g., Ashworth and Bernards, 2010) will define major resistance mechanisms, some of which, such as EGF receptor activation, may be suitable for targeting in combination with ALK inhibition.
From a Pharmaceutical point of view, it is clear that ALK was relatively neglected as a target for drug discovery until the emergence of its role in NSCLC. Despite the great interest this finding has since generated, ALK targeting nonetheless remains a relatively niche area for drug discovery, given that only ca. 5% of NSCLC patients harbor the rearrangement and that the other ALK-driven malignancies known to date are very rare tumor types. Several factors therefore came into play in the relatively rapid clinical development of crizotinib and appearance on the scene of second-generation ALK inhibitors. Firstly, the great deal of emphasis that both large pharmas and small biotechs have placed on developing kinase programs in the last two decades means that kinases are amongst the best characterized enzyme classes from the pharmacological point of view, with inhibitors now available, on the benchtop at least, for hundreds of kinases (e.g., Posy et al., 2011). The ever-increasing understanding of “chemical space” for targeting this class of enzyme means that today, identification of kinase inhibitors is a relatively rapid and low-cost process compared to other classes of drug target.
Another important factor that was key for the successful clinical development of crizotinib is the rapid definition of the molecular characteristics of patients likely to benefit from therapy and the application of a reliable diagnostic method for upfront identification of these patients during clinical trials. The Phase I/II responses observed in patients with rearranged ALK allowed crizotinib to be reviewed under the FDA’s accelerated approval program, which permits conditional approval of a drug for a serious disease based on reasonable likelihood of clinical benefit. In the field of cancer, in which patients with the specific genetic context required for drug activity may be relatively rare within a given tumor type, this type of provision considerably facilitates clinical development of targeted agents in the very well defined molecular contexts for which they were designed. It is probable that development of next generation compounds targeting secondary mutations of ALK will follow a similar strategy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ardini, E., Magnaghi, P., Orsini, P., Galvani, A., and Menichincheri, M. (2010). Anaplastic lymphoma kinase: role in specific tumors and development of small molecule inhibitors for cancer therapy. Cancer Lett. 299, 81–94.
Ardini, E., Menichincheri, M., Banfi, P., Saccardo, M. B., Rusconi, L., Avanzi, N., Amboldi, N., Casero, D., Cribioli, S., Isacchi, A., Donati, D., Pesenti, E., and Galvani, A. (2011). “In vitro and in vivo activity of NMS-E628 against ALK mutations resitant to Xalkori,” AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, San Francisco, CA, abstr. # A232.
Ardini, E., Menichincheri, M., De Ponti, C., Amboldi, N., Saccardo, M. B., Texido, G., Russo, M., Orsini, P., Bandiera, T., Lombardi Borgia, A., Isacchi, A., Pesenti, E., Colotta, F., Magnaghi, P., and Galvani, A. (2009). “Characterization of NMS-E628, a small molecule inhibitor of anaplastic lymphoma kinase with antitumor efficacy in ALK-dependent lymphoma and non-small cell lung cancer models,” AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Boston, MA, abstr. # A244.
Ashworth, A., and Bernards, R. (2010). Using functional genetics to understand breast cancer biology. Cold Spring Harb. Perspect. Biol. 2, a003327.
Butrynski, J. E., D’Adamo, D. R., Hornick, J. L., Dal Cin, P., Antonescu, C. R., Jhanwar, S. C., Ladanyi, M., Capelletti, M., Rodig, S. J., Ramaiya, N., Kwak, E. L., Clark, J. W., Wilner, K. D., Christensen, J. G., Jänne, P. A., Maki, R. G., Demetri, G. D., and Shapiro, G. I. (2010). Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 363, 1727–1733.
Camidge, D. (2011). ALK inhibitors in lung cancer. European Society for Medical Oncology (ESMO) Meeting, Stockholm, abstr. #186.
Carter, T. A., Wodicka, L. M., Shah, N. P., Velasco, A. M., Fabian, M. A., Treiber, D. K., Milanov, Z. V., Atteridge, C. E., Biggs, W. H., Edeen, P. T., Floyd, M., Fored, J. M., Grotzfeld, R. M., Herrgard, S., Insko, D. E., Metha, S. A., Patel, H. K., Pao, W., Sawyers, C. L., Varmus, H., Zarrinkar, P. P., and Lockhart, D. J. (2005). Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc. Natl. Acad. Sci. U.S.A. 102, 1011–1016.
Chen, Y., Takita, J., Choi, Y. L., Kato, M., Ohira, M., Sanada, M., Wang, L., Soda, M., Kikuchi, A., Igarashi, T., Nakagawara, A., Hayashi, Y., Mano, H., and Ogawa, S. (2008). Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974.
Choi, Y. L., Soda, M., Yamashita, Y., Ueno, T., Takashima, J., Nakajima, T., Yatabe, Y., Takeuchi, K., Hamada, T., Haruta, H., Ishikawa, Y., Kimura, H., Mitsudomi, T., Tanio, Y., and Mano, H. (2010). EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 363, 1734–1739.
Choi, Y. L., Takeuchi, K., Soda, M., Inamura, K., Togashi, Y., Hatano, S., Enomoto, M., Hamada, T., Haruta, H., Watanabe, H., Kurashina, K., Hatanaka, H., Ueno, T., Takada, S., Yamashita, Y., Sugiyama, Y., Ishikawa, Y., and Mano, H. (2008). Identification of novel isoforms of the EML4–ALK transforming gene in non-small cell lung cancer. Cancer Res. 68, 4971–4976.
Christensen, J. G., Zou, H. Y., Arango, M. E., Li, Q., Lee, J. H., McDonnell, S. R., Yamazaki, S., Alton, G. R., Mroczkowski, B., and Los, G. (2007). Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 6, 3314–3322.
Drexler, H. G., Gignac, S. M., von Wasielewski, R., Werner, M., and Dirks, W. G. (2000). Pathobiology of NPM-ALK and variant fusion genes in anaplastic large cell lymphoma and other lymphomas. Leukemia 14, 1533–1559.
Eichler, A. F., Euiheon, C., Kodack, D. P., Loeffler, J. S., Fukumura, D., and Jain, R. K. (2011). The biology of brain metastases-translation to new therapies. Nat. Rev. Clin. Oncol. 8, 344–356.
Engelman, J. A., Zejnullahu, K., Mitsudomi, T., Song, Y., Hyland, C., Park, J. O., Lindeman, N., Gale, C. M., Zhao, X., Christensen, J., Kosaka, T., Holmes, A. J., Rogers, A. M., Cappuzzo, F., Mok, T., Lee, C., Johnson, B. E., Cantley, L. C., and Jänne, P. A. (2007). MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043.
Galkin, A. V., Melnick, J. S., Kim, S., Hood, T. L., Li, N., Li, L., Xia, G., Steensma, R., Chopiuk, G., Jiang, J., Wan, Y., Ding, P., Liu, Y., Sun, F., Schultz, P. G., Gray, N. S., and Warmuth, M. (2007). Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc. Natl. Acad. Sci. U.S.A. 104, 270–275.
Gambacorti-Passerini, C., and Messa, C. (2011). Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 364, 775–776.
George, R. E., Sanda, T., Hanna, M., Fröhling, S., Luther, W. II, Zhang, J., Ahn, Y., Zhou, W., London, W. B., McGrady, P., Xue, L., Zozulya, S., Gregor, V. E., Webb, T. R., Gray, N. S., Gilliland, D. G., Diller, L., Greulich, H., Morris, S. W., Meyerson, M., and Look, A. T. (2008). Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978.
Griffin, C. A., Hawkins, A. L., Dvorak, C., Henkle, C., Ellingham, T., and Perlman, E. J. (1999). Recurrent involvement of 2p23 in inflammatory myofoibroblastic tumors. Cancer Res. 59, 2776–2780.
Hallberg, B., and Palmer, R. H. (2011). ALK and NSCLC: targeted therapy with ALK inhibitors. F1000 Med. Rep. 3, 21.
Inamura, K., Takeuchi, K., Togashi, Y., Nomura, K., Ninomiya, H., Okui, M., Satoh, Y., Okumura, S., Nakagawa, K., Soda, M., Lim Choi, Y., Niki, T., Mano, H., and Ishikawa, Y. (2008). EML4–ALK fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol. 3, 13–17.
Iwahara, T., Fujimoto, J., Wen, D., Cupples, R., Bucay, N., Arakawa, T., Mori, S., Ratzkin, B., and Yamamoto, T. (1997). Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 14, 439–449.
Janoueix-Lerosey, I., Lequin, D., Brugières, L., Ribeiro, A., de Pontual, L., Combaret, V., Raynal, V., Puisieux, A., Schleiermacher, G., Pierron, G., Valteau-Couanet, D., Frebourg, T., Michon, J., Lyonnet, S., Amiel, J., and Delattre, O. (2008). Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967–970.
Johannessen, C. M., Boehm, J. S., Kim, S. Y., Thomas, S. R., Wardwell, L., Johnson, L. A., Emery, C. M., Stransky, N., Cogdill, A. P., Barretina, J., Caponigro, G., Hieronymus, H., Murray, R. R., Salehi-Ashtiani, K., Hill, D. E., Vidal, M., Zhao, J. J., Yang, X., Alkan, O., Kim, S., Harris, J. L., Wilson, C. J., Myer, V. E., Finan, P. M., Root, D. E., Roberts, T. M., Golub, T., Flaherty, K. T., Dummer, R., Weber, B. L., Sellers, W. R., Schlegel, R., Wargo, J. A., Hahn, W. C., and Garraway, L. A. (2010). COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972.
Katayama, R., Khan, T. M., Benes, C., Lifshits, E., Ebi, H., Rivera, V. M., Shakespeare, W. C., Iafrate, A. J., Engelman, J. A., and Shaw, A. T. (2011). Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. U.S.A. 108, 7535–7540.
Kobayashi, S., Boggon, T. J., Dayaram, T., Jänne, P. A., Kocher, O., Meyerson, M., Johnson, B. E., Eck, J., Tenen, D. G., and Halmos, B. N. (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792.
Koivunen, J. P., Mermel, C., Zejnullahu, K., Murphy, C., Lifshits, E., Holmes, A. J., Choi, H. G., Kim, J., Chiang, D., Thomas, R., Lee, J., Richards, W. G., Sugarbaker, D. J., Ducko, C., Lindeman, N., Marcoux, J. P., Engelman, J. A., Gray, N. S., Lee, C., Meyerson, M., and Jänne, P. A. (2008). EML4–ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 14, 4275–4283.
Kwak, E. L., Bang, Y. J., Camidge, D. R., Shaw, A. T., Solomon, B., Maki, R. G., Ou, S. H., Dezube, B. J., Jänne, P. A., Costa, D. B., Varella-Garcia, M., Kim, W. H., Lynch, T. J., Fidias, P., Stubbs, H., Engelman, J. A., Sequist, L. V., Tan, W., Gandhi, L., Mino-Kenudson, M., Wei, G. C., Shreeve, S. M., Ratain, M. J., Settleman, J., Christensen, J. G., Haber, D. A., Wilner, K., Salgia, R., Shapiro, G. I., Clark, J. W., and Iafrate, A. J. (2010). Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N. Engl. J. Med. 363, 1693–1703.
Lennerz, J. K., Kwak, E. L., Ackerman, A., Michael, M., Fox, S. B., Bergethon, K., Lauwers, G. Y., Christensen, J. G., Wilner, K. D., Haber, D. A., Salgia, R., Bang, Y. J., Clark, J. W., Solomon, B. J., and Iafrate, A. J. (2011). MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 29, 4803–4810.
Li, N., Michellys, P.-Y., Sungjon, K., Culazzo Pferdekamper, A., Li, J., Kasibhatla, S., Tompkins, C. S., Steffy, A., Li, A., Sun, F., Sun, X., Hua, S., Tiedt, R., Sarkisova, Y., Marsilje, T. H., McNamara, P., and Harris, J. (2011). “Activity of a potent and selective phase I ALK inhibitor LDK378 in naïve and crizotinib-resistant preclinical tumor models,” AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, San Francisco, CA, abstr. # B232.
Lovly, C. M., Heuckmann, J. M., de Stanchina, E., Chen, H., Thomas, R. K., Liang, C., and Pao, W. (2011). Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 71, 4920–4931.
Maris, J. M., Hogarty, M. D., Bagatell, R., and Cohn, S. L. (2007). Neuroblastoma. Lancet 369, 2106–2120.
McDermott, U., Iafrate, A. J., Gray, N. S., Shioda, T., Classon, M., Maheswaran, S., Zhou, W., Choi, H. G., Smith, S. L., Dowell, L., Ulkus, L. E., Kuhlmann, G., Greninger, P., Christensen, J. G., Haber, D. A., and Settleman, J. (2008). Genomic alterations of anaplastic lymphoma kinase may sensitize tumours to anaplastic lymphoma kinase inhibitors. Cancer Res. 68, 3389–3395.
Morris, S. W., Kirstein, M. N., Valentine, M. B., Dittmer, K. G., Shapiro, D. N., Saltman, D. L., and Look, A. T. (1994). Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263, 1281–1284.
Morris, S. W., Naeve, C., Mathew, P., James, P. L., Kirstein, M. N., Cui, X., and Witte, D. P. (1997). ALK, the chromosome 2 gene locus altered by the t(2,5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 14, 2175–2188.
Mossé, Y. P., Laudenslager, M., Longo, L., Cole, K. A., Wood, A., Attiyeh, E. F., Laquaglia, M. J., Sennett, R., Lynch, J. E., Perri, P., Laureys, G., Speleman, F., Kim, C., Hou, C., Hakonarson, H., Torkamani, A., Schork, N. J., Brodeur, G. M., Tonini, G. P., Rappaport, E., Devoto, M., and Maris, J. M. (2008). Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935.
Nazarian, R., Shi, H., Wang, Q., Kong, X., Koya, R. C., Lee, H., Chen, Z., Lee, M. K., Attar, N., Sazegar, H., Chodon, T., Nelson, S. F., McArthur, G., Sosman, J. A., Ribas, A., and Lo, R. S. (2010). Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAF upregulation. Nature 468, 973–977.
Perner, S., Wagnaer, P. L., Demichelis, F., Mehra, R., LaFargue, C. J., Moss, B. J., Arbogast, S., Soltermann, A., Weder, W., Giordano, T. J., Beer, D. G., Rickman, D. S., Chinnaiyan, A. M., Moch, H., and Rubin, M. A. (2008). EML4-ALK fusion lung cancer: a rare acquired event. Neoplasia 10, 298–302.
Posy, S. L., Hermsmeier, M. A., Vaccaro, W., Ott, K. H., Todderud, G., Lippy, J. S., Trainor, G. L., Loughney, D. A., and Johnson, S. R. (2011). Trends in kinase selectivity: insights for target class-focused library screening. J. Med. Chem. 54, 54–66.
Rikova, K., Guo, A., Zeng, O., Possemato, A., Yu, J., Haack, H., Nardone, J., Lee, K., Reeves, C., Li, Y., Hu, Y., Tan, Z., Stokes, M., Sullivan, L., Mitchell, J., Wetzel, R., MacNeill, J., Ren, J. M., Yuan, J., and Bakalarski, C. E. (2007). Global survey of phosphotyrosine signalling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203.
Ross, J. S., and Cronin, M. (2010). Whole cancer genome sequencing by next-generation methods. Am. J. Clin. Pathol. 136, 527–539.
Sabbatini, P., Korenchuk, S., Rowand, J. L., Groi, A., Liu, Q., Leperi, D., Aktins, C., Dumble, M., Yang, J., Anderson, K., Kruger, R. G., Gontarek, R. R., Maksimchuk, K. R., Suravajjala, S., Lapierre, R. R., Shotwell, J. B., Wilson, J. W., Chamberlain, S. D., Rabindran, S. K., and Kumar, R. (2009). GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancer. Mol. Cancer Ther. 8, 2811–2820.
Sakamoto, H., Tsukaguchi, T., Hiroshima, S., Kodama, T., Kobayashi, T., Fukami, T. A., Oikawa, N., Tsukuda, T., Ishii, N., and Aoki, Y. (2011). CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 19, 679–690.
Sasaki, T., and Jänne, P. A. (2011). New strategies for treatment of ALK rearranged non-small cell lung cancers. Clin. Cancer Res. 17, 7213–7218.
Sasaki, T., Koivunen, J., Ogino, A., Yanagita, M., Nikiforow, S., Zheng, W., Lathan, C., Marcoux, J. P., Du, J., Okuda, K., Capelletti, M., Shimamura, T., Ercan, D., Stumpfova, M., Xiao, Y., Weremowicz, S., Butaney, M., Heon, S., Wilner, K., Christensen, J. G., Eck, M. J., Wong, K. K., Lindeman, N., Gray, N. S., Rodig, S. J., and Jänne, P. A. (2011). A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 71, 6051–6060.
Sasaki, T., Okuda, K., Zheng, W., Butrynski, J., Capelletti, M., Wang, L., Gray, N. S., Wilner, K., Christensen, J. G., Demetri, G., Shapiro, G. I., Rodig, S. J., Eck, M. J., and Jänne, P. A. (2010). The neuroblastoma-associated F1174L ALK mutation cause resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 70, 10038–10043.
Schönherr, C., Ruuth, K., Yamazaki, Y., Eriksson, T., Christensen, J., Palmer, R. H., and Hallberg, B. (2011). Activating ALK mutations found in neuroblastoma are inhibited by crizotinib and NVP-TAE684. Biochem. J. 440, 405–413.
Shah, N. P., Nicoll, J. M., Nagar, B., Gorre, M. E., Paquette, R. L., Kuriyan, J., and Sawyers, C. L. (2002). Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125.
Shaw, A. T., Yeap, B. Y., Mino-Kenudson, M., Digumarthy, S. R., Costa, D. B., Heist, R. S., Solomon, B., Stubbs, H., Admane, S., McDermott, U., Settleman, J., Kobayashi, S., Mark, E. J., Rodig, S. J., Chirieac, L. R., Kwak, E. L., Lynch, T. J., and Iafrate, A. J. (2009). Clinical features and outcome of patients with non-small-cell lung cancer who harbour EML4-ALK. J. Clin. Oncol. 27, 4247–4253.
Shaw, A. T., Yeap, B. Y., Solomon, B. J., Riely, G. J., Gainor, J., Engelman, J. A., Shapiro, G. I., Costa, D. B., Ou, S. H., Butaney, M., Salgia, R., Maki, R. G., Varella-Garcia, M., Doebele, R. C., Bang, Y. J., Kulig, K., Selaru, P., Tang, Y., Wilner, K. D., Kwak, E. L., Clark, J. W., Iafrate, A. J., and Camidge, D. R. (2011). Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 11, 1004–1012.
Shiota, M., Fujimoto, J., Semba, T., Satoh, H., Yamamoto, T., and Mori, S. (1994). Hyperphosphorylation of a novel 80 kDa protein tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 9, 1567–1574.
Soda, M., Choi, Y. L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., Fujiwara, S.-I., Watanabe, H., Kurashina, K., Hatanaka, H., Bando, M., Ohno, S., Ishikawa, Y., Aburatani, H., Niki, T., Sohara, Y., Sugiyama, Y., and Mano, H. (2007). Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566.
Soda, M., Takada, S., Takeuchi, K., Choi, Y. L., Enomoto, M., Ueno, T., Haruta, H., Hamada, T., Yamashita, Y., Ishikawa, Y., Sugiyama, Y., and Mano, H. (2008). A mouse model for EML4–ALK-positive lung cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 19893–19897.
Solomon, B., Varella-Garcia, M., and Camidge, D. R. (2009). ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J. Thorac. Oncol. 4, 1450–1454.
Tamborini, E., Bonadiman, L., Greco, A., Albertini, V., Negri, T., Gronchi, A., Bertulli, R., Colecchia, M., Casali, P. G., Pierotti, M. A., and Pilotti, S. (2004). A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 127, 294–299.
Webb, T. R., Slavish, J., George, R. E., Look, A. T., Xue, L., Jiang, Q., Cui, X., Rentrop, W. B., and Morris, S. W. (2009). Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev. Anticancer Ther. 9, 331–356.
Wood, A. C., Laudenslager, M., Haglund, E. A., Attiyeh, E. F., Pawel, B., Courtright, J., Plegaria, J., Christensen, J. G., Maris, J. M., and Mossé, Y. P. (2009). Inhibition of ALK mutated neuroblastomas by the selective inhibitor PF-02341066. J. Clin. Oncol. 27, 15S.
Yun, C.-H., Mengwasser, K. E., Toms, A. V., Woo, M. S., Greulich, H., Wong, K.-K., Meyerson, M., and Eck, M. J. (2008). The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 105, 2070–2075.
Zhang, S., Wang, F., Keats, J., Ning, Y., Wardwell, S. D., Moran, L., Mohemmad, Q. K., Ye, E., Anjum, R., Wang, Y., Zhu, X., Miret, J. J., Dalgarno, D., Narasimhan, N. I., Clackson, T., Shakespeare, W. C., and Rivera, V. M. (2010). AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066. 101st AACR Annual Meeting, Washington, DC.
Zou, H. Y., Li, Q., Lee, J. H., Arango, M. E., McDonnell, S. R., Yamazaki, S., Koudriakova, T. B., Alton, G., Cui, J. J., Kung, P.-P., Nambu, M. D., Los, G., Bender, S. L., Mroczkowski, B., and Christensen, J. G. (2007). An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 67, 4408–4417.
Keywords: ALK, inhibitor, crizotinib, NSCLC, IMT, ALCL, neuroblastoma, resistance
Citation: Ardini E and Galvani A (2012) ALK inhibitors, a pharmaceutical perspective. Front. Oncol. 2:17. doi: 10.3389/fonc.2012.00017
Received: 10 November 2011; Paper pending published: 13 December 2011;
Accepted: 06 February 2012; Published online: 22 February 2012.
Edited by:
Giuseppe Giaccone, National Institutes of Health, USAReviewed by:
Michelle A. Rudek, Johns Hopkins University, USAJacalyn Rosenblatt, Beth Israel Deaconess Medical Center, USA
Copyright: © 2012 Ardini and Galvani. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Elena Ardini, Department of Cell Biology, Oncology, Nerviano Medical Sciences, Viale Pasteur 10, 20014 Nerviano, Milan, Italy. e-mail:ZWxlbmEuYXJkaW5pQG5lcnZpYW5vbXMuY29t