 Antonino B. D’Assoro
Antonino B. D’Assoro Tufia Haddad
Tufia Haddad Evanthia Galanis
Evanthia Galanis- 1Department of Medical Oncology, Mayo Clinic College of Medicine, Rochester, MN, USA
- 2Department of Biochemistry and Molecular Biology, Mayo Clinic College of Medicine, Rochester, MN, USA
- 3Department of Molecular Medicine, Mayo Clinic College of Medicine, Rochester, MN, USA
Mammalian Aurora family of serine/threonine kinases are master regulators of mitotic progression and are frequently overexpressed in human cancers. Among the three members of the Aurora kinase family (Aurora-A, -B, and -C), Aurora-A and Aurora-B are expressed at detectable levels in somatic cells undergoing mitotic cell division. Aberrant Aurora-A kinase activity has been implicated in oncogenic transformation through the development of chromosomal instability and tumor cell heterogeneity. Recent studies also reveal a novel non-mitotic role of Aurora-A activity in promoting tumor progression through activation of epithelial–mesenchymal transition reprograming resulting in the genesis of tumor-initiating cells. Therefore, Aurora-A kinase represents an attractive target for cancer therapeutics, and the development of small molecule inhibitors of Aurora-A oncogenic activity may improve the clinical outcomes of cancer patients. In the present review, we will discuss mitotic and non-mitotic functions of Aurora-A activity in oncogenic transformation and tumor progression. We will also review the current clinical studies, evaluating small molecule inhibitors of Aurora-A activity and their efficacy in the management of cancer patients.
Introduction
Cell division in normal cells is a tightly regulated process by which replicated DNA is equally distributed into two daughter cells (1). Key players that orchestrate cell division are the centrosomes and mitotic spindles that ensure correct chromosome alignment on the metaphase plate and equal chromosome segregation, resulting in the maintenance of a genomic stable diploid karyotype (2). Due to the complexity of the mitotic machinery, several checkpoint surveillance mechanisms have evolved to safeguard accurate temporal and spatial coordination of cell cycle events (3). Abrogation of cell cycle checkpoints impairs the fidelity of correct chromosome segregation and induces chromosomal instability (CIN), a driving force of oncogenic transformation and tumor progression (4, 5). Aurora serine/threonine kinases are key mitotic regulators required for the maintenance of chromosomal stability (6). In mammalian cells, Aurora kinases consist of three members termed Aurora-A, -B, and -C that are expressed in a cell cycle-dependent fashion. These mitotic kinases are highly conserved through evolution and guarantee the precise coordination of cytoskeletal and chromosomal events through modulation of centrosome duplication, maturation, and separation, as well as proper mitotic spindle assembly resulting in equal chromosome distribution into daughter cells (7). While all three Aurora kinases are expressed in human cancer cells, Aurora-A and Aurora-B are best characterized because they are expressed at high levels in aneuploid tumors (8, 9). Aurora-A and Aurora-B share about 70% homology in the carboxyl terminus catalytic domain and three conserved Aurora box motifs in their varying amino terminal domain (10). However, they control cell cycle progression and mitosis by interacting with different proteins. Aurora-A is localized primarily on centrosomes, spindle poles, and transiently along the spindle microtubules as cells progress through mitosis (Figure 1) (11, 12). By contrast, Aurora-B interacts with the chromosomal passenger complex (CPC) that localizes to the inner centromere during prophase through metaphase and then moves to the spindle midzone and the midbody during late mitosis and cytokinesis (13). While some studies have shown that Aurora-B kinase is overexpressed in cancer cells (14, 15), it is not clear whether Aurora-B overexpression is merely associated with the high proliferative activity of cancer cells or if it plays a causative role in tumorigenesis. Due to the lack of definitive evidence that Aurora-B strictly functions as an oncogene, Aurora-A kinase represents a better candidate target for cancer therapeutics. In the last decade, several small molecule inhibitors of Aurora kinases have been developed, though only a few are selective for Aurora-A; they represent promising drugs to impair the progression of aggressive tumors (16).
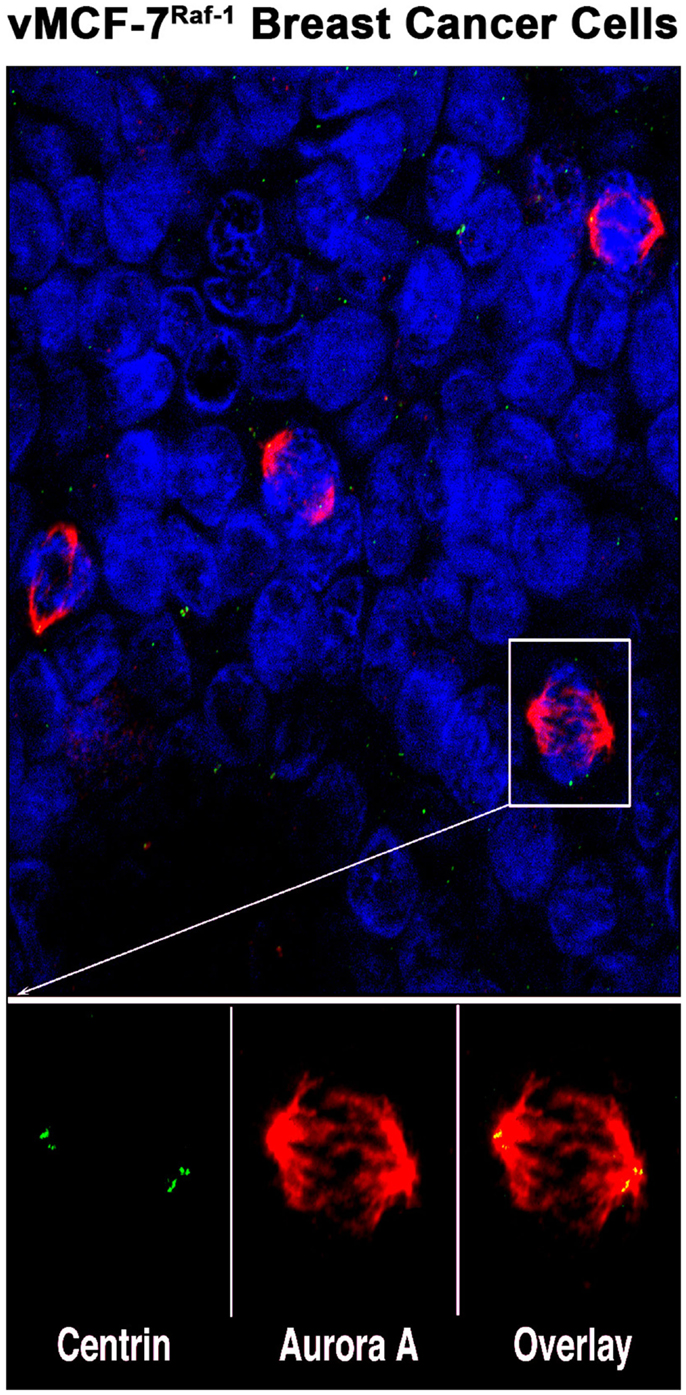
Figure 1. Aurora-A localization in human breast cancer cells: representative image of mitotic figures from MCF-7 breast cancer cell line engineered to express the Raf-1 oncoprotein (vMCF-7Raf-1). Centrosomes are labeled in green with 20H5 centrin mouse monoclonal antibody (Mayo Clinic), mitotic spindles are labeled in red with Aurora-A rabbit polyclonal antibody (Abcam, Cambridge, MA, USA) and nuclei are labeled in blue with DAPI (Thermo Fischer Scientific, Rockford, IL, USA). Centrosomal co-localization of Aurora-A is observed in the overlay image (yellow).
Aurora-A Expression in Cancer Cells
The mammalian Aurora-A protein contains 403 amino acids and has a molecular weight of 46 kDa. Aurora-A was first isolated as the product of gene BTAK (breast tumor amplified kinase, also named STK15) on chromosome 20q13, a region that is frequently amplified in breast, colorectal, and bladder tumors as well as ovarian, prostate, neuroblastoma, and cervical cancer cell lines (17–21). Although gene amplification represents a well-established mechanism to induce Aurora-A overexpression in cancer cells, transcriptional and post-translational mechanisms also play an important role to enhance Aurora-A expression in the absence of BTAK gene amplification. In normal cells, the abundance of Aurora-A is down-regulated through APC/C–Cdh1-dependent, proteasome-mediated proteolysis, leading to the organization of the anaphase spindle at the end of mitosis. APC/C–Cdh1-dependent degradation of human Aurora-A requires a destruction box (D-box) in the C-terminal region and a motif in the N-terminus (A-box) (22). Importantly, the phosphorylation state of a serine residue (Ser51) in the A-box inhibits degradation of Aurora-A, as mutants mimicking constitutive phosphorylation of this site cannot be degraded by the APC/C–Cdh1 (23). Furthermore, we have showed that HER-2 oncogenic signaling induces Aurora-A phosphorylation, thereby increasing Aurora-A stability and expression in breast cancer cells (24). These findings indicate a functional link between deregulation of Aurora-A stability and tumorigenesis. Conversely, tumor suppressors involved in the control of cell cycle progression promote Aurora-A degradation. The mitotic checkpoint protein Chfr physically interacts with Aurora-A and ubiquitinates Aurora-A both in vitro and in vivo, ensuring the proper control of mitotic events and maintenance of chromosomal stability (25). Loss of Chfr expression in cancer cells induces aberrant Aurora-A kinase activity, CIN, and promotes tumorigenesis (26). The tumor suppressor p53 modulates Aurora-A expression via both transcriptional and post-translational regulation. Specifically, p53 knockdown in cancer cells promotes the activation of E2F3 transcriptional factor that in turn induces Aurora-A gene expression. p53 deficiency also increases Aurora-A expression through the downregulation of Fbw7α, a key component of e3 ligase of Aurora-A involved in its degradation (27). A separate study demonstrated that highly invasive primary tumors harboring mutant p53 also exhibited Aurora-A overexpression (28). Taken together, these findings strongly demonstrate that Chfr and p53 are key negative regulators of Aurora-A kinase signaling, and their loss of function promotes a growth advantage for cancer cells through increased expression of Aurora-A.
Aurora-A Promotes Centrosome Amplification, Aneuploidy, and CIN
Aurora-A is overexpressed in a variety of solid tumors, indicative of the critical role that aberrant Aurora-A kinase activity plays in tumorigenesis. Several studies demonstrate the causative function of Aurora-A overexpression in promoting cell transformation in vitro and tumor growth in vivo employing NIH 3T3 cells and Rat1 fibroblasts (17, 29). The majority of research aims to identify the mechanisms responsible for Aurora-A-induced tumorigenesis has focused on the role of Aurora-A kinase in the control of centrosome duplication and mitosis. Accurate centrosome duplication plays a central role in the maintenance of a normal diploid karyotype. In order to give rise to a bipolar mitotic spindle responsible for the equal segregation of chromosomes to dividing cells, the centrosome must be duplicated once, and only once during each cell cycle (30). Cell cycle checkpoints are essential surveillance mechanisms that guarantee the coordination between centrosome duplication, DNA replication, and mitosis during cell cycle progression (31). Abrogation of cell cycle checkpoints in cancer cells induces centrosome amplification, a pathological condition characterized by the presence of more than two centrosomes within a cell. Centrosome amplification may result from inactivation of the G1/S checkpoint leading to centrosome overduplication or from abrogation of the G2/M checkpoint leading to cytokinesis failure, endoreduplication, and consequent centrosome accumulation (2). Centrosome amplification due to cytokinesis failure is exacerbated in cancer cells lacking the “G1 phase post-mitotic checkpoint” that is dependent on the integrity of p53/Rb axis (32–34). One of the major consequences of centrosome amplification is the formation of multipolar or pseudo-bipolar mitotic spindles that will result in unequal chromosome segregation and aneuploidy (35–37). Aneuploidy is characterized by gains and/or losses of whole chromosomes during cell division and occurs in early stages of tumor development, playing a critical role in both tumorigenesis and tumor progression (38). Significantly, while aneuploidy represents the state of an aberrant karyotype, the continuous generation of chromosome variations in cancer cells is defined as CIN that will ultimately drive genetic heterogeneity, tumor recurrence, and poor outcome (39). Several lines of evidence have established that centrosome amplification drives CIN and genetic heterogeneity in aneuploid tumors (40–42). Elegant studies have demonstrated that deregulated expression of Aurora-A is functionally linked to centrosome amplification and CIN (43–45). The major mechanism by which aberrant Aurora-A kinase activity induces centrosome amplification and CIN is through cytokinesis failure and consequent multinucleation leading to centrosome accumulation (46). Aurora-A induces cytokinesis failure and centrosome amplification mainly through its interaction with key tumor suppressor gene products that control cell cycle checkpoints, centrosome duplication, and chromosomal stability. Aurora-A phosphorylates the tumor suppressor p53 on Ser215 residue, abrogating the DNA-binding and transactivation activity of p53 that results in the inhibition of the downstream target gene p21 involved in the control of centrosome duplication (47). Moreover, Aurora-A-mediated phosphorylation of p53 on Ser315 residue will increase the affinity of p53 with Mdm2 that in turn will promote p53 degradation (48). The tumor suppressors BRCA1 and BRCA2 play a central role in the maintenance of chromosomal stability and germline mutations in BRCA1 and BRCA2 genes have been detected in approximately 90% of hereditary breast/ovarian cancers (49). Specifically, BRCA1 monitors the physical integrity of DNA following genotoxic stress and coordinates DNA replication with centrosome duplication cycle (50). It has been demonstrated that Aurora-A directly binds to BRCA1 and phosphorylates it on Ser308 residue. Deregulated Aurora-A-mediated BRCA1 phosphorylation on Ser308 residue induces abrogation of the G2/M checkpoint leading to centrosome amplification and CIN (51). Moreover, Aurora-A is required to activate polo-like kinase 1 (PLK1) that plays a key role in promoting centrosome duplication and mitotic entry (52, 53). These findings indicate that Aurora-A overexpression induces aberrant Plk1 activity that will drive centrosome amplification, improper segregation of chromosomes, CIN, and tumorigenesis. Leontovich et al. uncovered a novel mechanism by which Cyclin-A/Cdk2 oncogenic signaling favors Aurora-A centrosomal localization that in turn induces centrosome overduplication in breast cancer cells (54). Taken together, these studies strongly demonstrate that deregulated Aurora-A kinase activity induces centrosome amplification in cancer cells through different mechanisms and results in the development of CIN, a driving force for genetic heterogeneity and tumor progression.
Non-Mitotic Function of Aurora-A in Tumorigenesis
Although Aurora-A-mediated centrosome amplification and CIN represents a well-recognized mechanism that promotes oncogenic transformation, the kinase activity of Aurora-A is essential to acquire a transformed phenotype regardless of the induction of centrosome amplification (55). These findings led to the discovery that Aurora-A kinase also phosphorylates proteins unrelated to centrosome function that play a central role in tumorigenesis. Taga et al. showed in U2OS human osteosarcoma cells that Aurora-A induces phosphorylation of Akt and mTOR oncoproteins that is required to increase U2OS tumorigenicity (56). In agreement with these results, aberrant Aurora-A kinase activity promotes resistance to cisplatin, etoposide, and paclitaxel-induced apoptosis by phosphorylating Akt in wild-type p53 ovarian cancer cells (57). Other studies have revealed the direct role of Aurora-A kinase activity in mediating cancer cell motility and distant metastases. Aurora-A promotes breast cancer metastases by dephosphorylation of cofilin and activation of cofilin–F-actin pathway, which accelerates actin reorganization and polymerization (58). Furthermore, inhibition of phosphatidylinositol 3-kinase (PI3K) oncogenic signaling blocked Aurora-A-mediated cofilin dephosphorylation, actin reorganization, and cell migration. These results uncover a novel crosstalk between PI3K signaling and Aurora-A in tumor progression. In esophageal squamous cell carcinoma cells, Aurora-A overexpression induces cell migration and invasion as well as secretion and expression of matrix metalloproteinase-2 (MMP-2). This mechanism is mediated by Aurora-A-induced phosphorylation of p38 MAPK and Akt protein kinases (59). Aberrant Aurora-A kinase activity also induces activation of Rap1, a member of the Ras family of small GTPases, leading to the development of distant metastases originating from oral cavity squamous cell carcinomas (60). Du and Hannon demonstrated that Aurora-A kinase activity inhibits the function of Nm23-H1 protein that is involved in the suppression of distant metastases, facilitating tumor progression (61).
Moreover, recent studies revealed a novel function of Aurora-A in the progression of solid tumors through activation of epithelial–mesenchymal transition (EMT) and stemness reprograming. Cammareri et al. demonstrated that Aurora-A overexpression is restricted in colorectal cancer stem cells (CR-CSC), and Aurora-A inhibition restored chemosensitivity and compromised the tumor initiating ability of CR-CSC to form tumor xenografts in immunocompromised mice (62). The causative role of Aurora-A overexpression in promoting EMT and tumor progression through stabilization of Snail transcription factor has been shown in head and neck cancer cells (63). Significantly, we have defined for the first time the essential role of Aurora-A in promoting breast cancer progression through activation of EMT and the genesis of breast cancer stem cells responsible for the onset of distant metastases (24). Moreover, Aurora-A-induced EMT and onset of distant metastases was functionally linked to SMAD5 and SOX2 expression, two master transcription factors involved in the development of EMT, tumor self-renewal, and an invasive, basal-like phenotype. In the same study, we have uncovered the causative role of Aurora-A overexpression in inducing expansion of cancer stem cells through impairment of asymmetric divisions. These results are in agreement with a previous study showing that a phosphorylation cascade triggered by the activation of Aurora-A kinase is responsible for the asymmetric localization of Numb during mitosis (64). Taken together, these studies highlight an essential role of Aurora-A kinase in driving tumor progression by modulating the activity of key oncogenic pathways involved in cell migration, chemoresistance, tumor initiating ability, and onset of distant metastases.
Aurora-A as a Novel Biomarker Prognostic of Poor Clinical Outcome
Several studies have shown that Aurora-A kinase is overexpressed in a variety of tumors, suggesting that Aurora-A may represent a promising prognostic biomarker. Reiter et al. reported that increased expression of Aurora-A in head and neck squamous cell carcinomas was significantly associated with shorter disease-free and overall survival of patients (65). Likewise, Aurora-A overexpression is associated with centrosome amplification and shorter survival in an extensive proportion of ovarian tumors (66, 67). Gastrointestinal tumors also display deregulation of Aurora-A expression that is linked to high risk of recurrence and tumor progression. Employing tissue microarrays from a retrospective cohort of 343 patients with colorectal cancer liver metastases, Goos et al. showed that Aurora-A levels were increased in liver metastatic lesions compared to corresponding primary tumors and was associated with poor clinical outcome (68). Wang et al. showed that Aurora-A overexpression was an independent prognostic marker of poor survival in gastric cancer patients without lymph node metastases (69). Samaras et al. performed a comparative immunohistochemical analysis of Aurora-A and Aurora-B expression in 40 patients with primary glioblastomas to identify possible correlations with Ki-67 proliferation index and clinical outcomes (70). While Aurora-A was overexpressed in glioblastomas with high Ki-67 expression and was associated with poor survival, Aurora-B expression was not correlated with Ki-67 expression and patient survival. Aurora-A overexpression has also been established as a valuable biomarker prognostic of poor clinical outcome in breast carcinomas. Nadler et al. demonstrated in a tissue microarray containing primary breast tumor tissue from 638 patients with 15-year follow-up that aberrant expression of Aurora-A, but not Aurora-B, was an independent prognostic marker strongly correlated with decreased survival (71). High Aurora-A expression was also associated with high nuclear grade and elevated HER-2/neu and progesterone receptor expression. In 48 cases of operable triple-negative breast tumors, Yamamoto et al. established that basal-like subtype was significantly associated with high levels of Aurora-A and shorter disease-free and overall survival compare to non-basal-like breast tumors (72). Using microarray-based gene expression data from three independent cohorts of 766 node-negative breast cancer patients, Siggelkow et al. demonstrated that patients harboring high Aurora-A expression had a shorter metastasis-free survival in the molecular subtype estrogen receptor-positive (ER+)/HER2− carcinomas, but not in ER−/HER2− or HER2+ carcinomas (73). A recent study reported, in a cohort of 426 patients with primary breast cancer, that elevated expression of Aurora-A and SURVIVIN, together with BTAK gene amplification, is correlated with increased CIN and shorter survival (74). Taken together, these studies highlight Aurora-A as a novel, independent prognostic biomarker of poor clinical outcome that could identify patients at high risk of tumor recurrence or progression.
Pharmacologic Targeting of Aurora-A Kinase Activity in Cancer Therapy
In the last decade, at least 13 different inhibitors of the Aurora kinases have been evaluated in phase I clinical trials in patients with various hematologic and solid tumor malignancies. Nearly all of the initial agents studied were pan-inhibitors of Aurora-A, -B, and -C, and several of them furthermore inhibited other kinases, such as bcr–abl (T135I), Flt3, VEGFR2, and JAK 2/3. Some of these trials were suspended and not completed or published. Some inhibitors have not continued beyond phase I evaluation due to significant toxicities at clinically effective doses or limited clinical antitumor activity. Only a limited number of these pan-Aurora and multi-kinase inhibitors have been pursued in phase II clinical trials (AT-9283, MK-0457, ENMD-2076, PHA-739358). Three of the Aurora kinase inhibitors developed were selective for Aurora-A (MLN 8054, MLN 8237, TAS-119). Of all the inhibitors, only MLN 8237 (alisertib) has proceeded to phase III evaluation.
The first of the selective Aurora-A kinase inhibitors to enter into human studies was MLN 8054. In Phase I dose escalation studies in patients with advanced solid cancers, the observed dose limiting toxicity (DLT) was reversible somnolence, attributed to GABAA α-1 benzodiazepine off-target binding (75, 76). With the aim of improving the therapeutic window, the chemical structure of the molecule was modified, and of potential new agents, MLN 8237 (alisertib) was selected for further development based on preclinical evidence demonstrating its increased potency in Aurora-A enzymatic inhibition, reduced degree of brain partitioning, and while GABAA binding potency was comparable to MLN 8054, alisertib displayed a greater selectivity ratio of Aurora-A inhibition to GABAA α-1 benzodiazepine site binding affinity (77).
In 2007, the first clinical trial opened to evaluate alisertib, an orally administered, small molecule inhibitor that is selective for Aurora-A kinase. To date, well over 1000 patients with hematological or solid tumor malignancies have participated in clinical trials with the agent as monotherapy or in combination with chemotherapy or other targeted agents (78, 79). In the original phase I trials, different formulations of the drug, doses, and schedules were evaluated (80, 81). Stomatitis and neutropenia were the most common DLTs consistent with its antiproliferative effect. Somnolence was evident in patients receiving once daily dosing of alisertib at the highest dose levels; however, the frequency and severity of these episodes were reduced with twice daily dosing of alisertib at lower individual doses, which reduced peak plasma levels while maintaining overall systemic exposures. Other common low-grade toxicities included alopecia, nausea, diarrhea, anemia, and fatigue. The recommended phase II dose was 50 mg twice daily on days 1–7 of a 21-day cycle, and the preferred formulation was the enteric-coated tablet; both were confirmed in the industry-sponsored study of alisertib as monotherapy in patients with advanced solid tumor malignancies (82). Encouraging clinical activity was demonstrated in this trial. In the cohort of heavily pre-treated women with hormone receptor-positive metastatic breast cancer (n = 26), 23% had an objective response (complete or partial response) and 31% achieved stable disease for at least 6 months, resulting in a clinical benefit rate of 54%. Median PFS was 7.9 months. In the chemotherapy-refractory, relapsed small cell lung cancer (SCLC) cohort (n = 12), a response rate of 25% was observed with a median duration of response of 4.3 months. A phase II trial of alisertib alone or combined with paclitaxel for second-line therapy of SCLC is currently active (NCT02038647). Based on promising activity observed in relapsed/refractory peripheral T-cell lymphoma (83, 84), a phase III clinical trial of alisertib versus treatment of investigator’s choice (NCT01482962) was pursued but subsequently terminated enrollment at a pre-specified interim analysis due to projections that the study was unlikely to meet the primary endpoint of superior PFS.
An alternative 28-day regimen with alisertib given days 1–3, 8–10, and 15–17 was studied in combination with paclitaxel in breast and ovarian cancer models, and it is associated with equivalent drug levels, decreased incidence of dose limiting neutropenia with negligible compromise to efficacy (85). The safety and tolerability of this schedule in combination with fulvestrant is currently being explored in an ongoing phase I trial in patients with hormone receptor-positive, advanced breast cancer (NCT 02219789).
TAS-119 is the only other selective Aurora-A kinase inhibitor to enter into clinical evaluation. It is being studied as monotherapy and in combination with taxane-based chemotherapy in two separate, active phase I clinical trials (NCT02134067, NCT02448589).
Conclusion
One of the major hallmarks of cancer is aneuploidy and the development of CIN characterized by the relentless generation of chromosome variations that will ultimately drive genetic heterogeneity and tumor progression. In normal cells, cell division is monitored by checkpoints that are safeguard mechanisms to guarantee the accurate temporal and spatial coordination of cell cycle events. Abrogation of cell cycle checkpoints induces centrosome amplification that impairs the fidelity of correct chromosome segregation, promoting aneuploidy and CIN. Members of the Aurora serine/threonine kinase family are key mitotic regulators required for the maintenance of chromosomal stability. Overexpression and aberrant activation of Aurora-A kinase has been functionally linked to oncogenic transformation mainly through development of centrosome amplification and CIN. Significantly, recent studies have also demonstrated that Aurora-A kinase mediates MAPK-induced distant metastases through activation of EMT and stemness reprograming (Figure 2). Taken together, these findings demonstrate that Aurora-A kinase represents a critical “druggable target” in cancer, controlling key oncogenic pathways associated with drug resistance and poor patient outcome. For this reason, several small molecule inhibitors of Aurora-A kinase activity have been developed and their efficacy is being tested in clinical trials (Table 1). Significantly, recent studies have highlighted the incremental therapeutic efficacy when combining Aurora-A inhibitors with conventional anti-cancer drugs to restore chemosensitivity and inhibit tumor progression, a strategy expected to further build on the clinical benefit potential of Aurora-A inhibition.
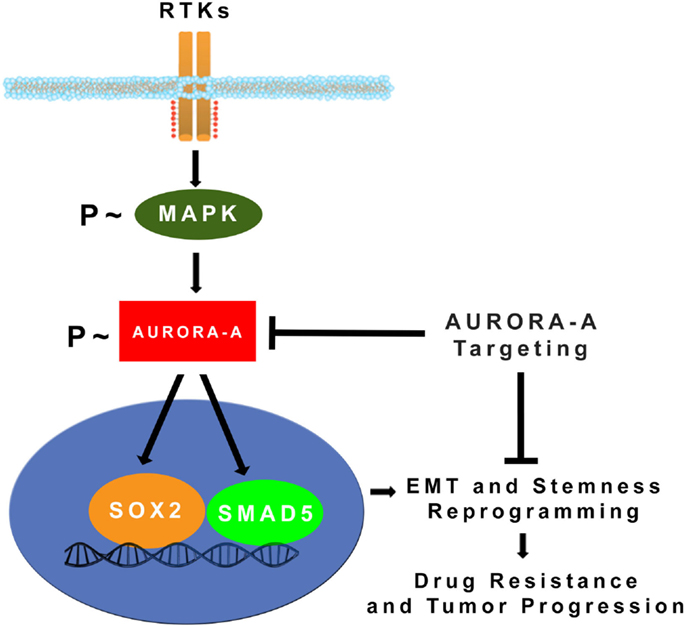
Figure 2. MAPK-induced activation of Aurora-A kinase promotes EMT, stemness, and tumor progression: constitutive activation of MAPK oncogenic signaling during tumor growth leads to stabilization and accumulation of Aurora-A kinase. Aberrant Aurora-A kinase activity induces activation of SMAD5 and SOX2 transcription factors that in turn will orchestrate EMT and stemness reprograming leading to drug resistance and tumor progression (24). Pharmacologic targeting of Aurora-A kinase activity can be effective for eliminating highly invasive cancer stem cells and defeat tumor progression.
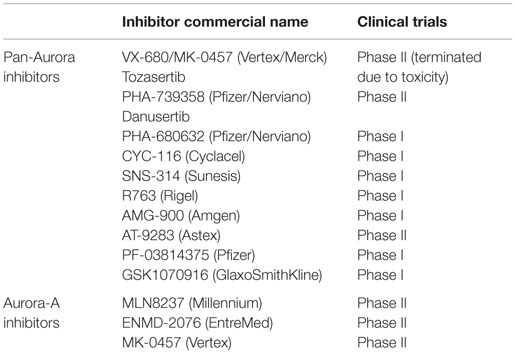
Table 1. Aurora kinase inhibitors in clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Nan Sawyer fund (to AD), the Mayo Clinic K12 Training grant (5K12CA090628 to TH), an Atwater Foundation grant (to EG), and the Mayo Clinic Breast Cancer SPORE (P50 CA 116201).
References
1. Saka Y, Giuraniuc CV, Ohkura H. Accurate chromosome segregation by probabilistic self-organisation. BMC Biol (2015) 13:65. doi:10.1186/s12915-015-0172-y
2. D’Assoro AB, Lingle WL, Salisbury JL. Centrosome amplification and the development of cancer. Oncogene (2002) 21(40):6146–53. doi:10.1038/sj.onc.1205772
3. Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science (1989) 246(4930):629–34. doi:10.1126/science.2683079
4. D’Assoro AB, Busby R, Suino K, Delva E, Almodovar-Mercado GJ, Johnson H, et al. Genotoxic stress leads to centrosome amplification in breast cancer cell lines that have an inactive G1/S cell cycle checkpoint. Oncogene (2004) 23(23):4068–75. doi:10.1038/sj.onc.1207568
5. Knauf JA, Ouyang B, Knudsen ES, Fukasawa K, Babcock G, Fagin JA. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J Biol Chem (2006) 281(7):3800–9. doi:10.1074/jbc.M511690200
6. Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev (2003) 22(4):451–64. doi:10.1023/A:1023789416385
7. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol (2003) 4(11):842–54. doi:10.1038/nrm1245
8. Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer (2001) 84(6):824–31. doi:10.1054/bjoc.2000.1684
9. González-Loyola A, Fernández-Miranda G, Trakala M, Partida D, Samejima K, Ogawa H, et al. Aurora B overexpression causes aneuploidy and p21Cip1 repression during tumor development. Mol Cell Biol (2015) 35(20):3566–78. doi:10.1128/MCB.01286-14
10. Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol (2009) 21(6):796–805. doi:10.1016/j.ceb.2009.09.008
11. Stenoien DL, Sen S, Mancini MA, Brinkley BR. Dynamic association of a tumor amplified kinase, Aurora-A, with the centrosome and mitotic spindle. Cell Motil Cytoskeleton (2003) 55(2):134–46. doi:10.1002/cm.10120
12. Shim SY, Perez de Castro I, Neumayer G, Wang J, Park SK, Sanada K, et al. Phosphorylation of targeting protein for Xenopus kinesin-like protein 2 (TPX2) at threonine 72 in spindle assembly. J Biol Chem (2015) 290(14):9122–34. doi:10.1074/jbc.M114.591545
13. Gohard FH, St-Cyr DJ, Tyers M, Earnshaw WC. Targeting the INCENP IN-box-Aurora B interaction to inhibit CPC activity in vivo. Open Biol (2014) 4(11):140163. doi:10.1098/rsob.140163
14. Lin ZZ, Jeng YM, Hu FC, Pan HW, Tsao HW, Lai PL, et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B overexpression in HCC. BMC Cancer (2010) 10:461. doi:10.1186/1471-2407-10-461
15. Hegyi K, Egervári K, Sándor Z, Méhes G. Aurora kinase B expression in breast carcinoma: cell kinetic and genetic aspects. Pathobiology (2012) 79(6):314–22. doi:10.1159/000338082
16. Malumbres M, Pérez de Castro I. Aurora kinase A inhibitors: promising agents in antitumoral therapy. Expert Opin Ther Targets (2014) 18(12):1377–93. doi:10.1517/14728222
17. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet (1998) 20(2):189–93. doi:10.1038/2496
18. Hu W, Kavanagh JJ, Deaver M, Johnston DA, Freedman RS, Verschraegen CF, et al. Frequent overexpression of STK15/Aurora-A/BTAK and chromosomal instability in tumorigenic cell cultures derived from human ovarian cancer. Oncol Res (2005) 15(1):49–57.
19. Sen S, Zhou H, Zhang RD, Yoon DS, Vakar-Lopez F, Ito S, et al. Amplification/overexpression of a mitotic kinase gene in human bladder cancer. J Natl Cancer Inst (2002) 94(17):1320–9. doi:10.1093/jnci/94.17.1320
20. Baba Y, Nosho K, Shima K, Irahara N, Kure S, Toyoda S, et al. Aurora-A expression is independently associated with chromosomal instability in colorectal cancer. Neoplasia (2009) 11(5):418–25. doi:10.1593/neo.09154
21. Twu NF, Yuan CC, Yen MS, Lai CR, Chao KC, Wang PH, et al. Expression of Aurora kinase A and B in normal and malignant cervical tissue: high Aurora A kinase expression in squamous cervical cancer. Eur J Obstet Gynecol Reprod Biol (2009) 142(1):57–63. doi:10.1016/j.ejogrb.2008.09.012
22. Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1 dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev (2002) 16(17):2274–85. doi:10.1101/gad.1007302
23. Kitajima S, Kudo Y, Ogawa I, Tatsuka M, Kawai H, Pagano M, et al. Constitutive phosphorylation of Aurora-A on ser51 induces its stabilization and consequent overexpression in cancer. PLoS One (2007) 2(9):e944. doi:10.1371/journal.pone.0000944
24. D’Assoro AB, Liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, et al. The mitotic kinase Aurora – a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERα(+) breast cancer cells. Oncogene (2014) 33(5):599–610. doi:10.1038/onc.2012.628
25. Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, et al. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet (2005) 37(4):401–6. doi:10.1038/ng1538
26. Privette LM, Petty EM. CHFR: a novel mitotic checkpoint protein and regulator of tumorigenesis. Transl Oncol (2008) 1(2):57–64. doi:10.1593/tlo.08109
27. Wu CC, Yang TY, Yu CT, Phan L, Ivan C, Sood AK, et al. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle (2012) 11(18):3433–42. doi:10.4161/cc.21732
28. Takahashi T, Futamura M, Yoshimi N, Sano J, Katada M, Takagi Y, et al. Centrosomal kinases, HsAIRK1 and HsAIRK3, are overexpressed in primary colorectal cancers. Jpn J Cancer Res (2000) 91(10):1007–14. doi:10.1111/j.1349-7006.2000.tb00878.x
29. Fukuda T, Mishina Y, Walker MP, DiAugustine RP. Conditional transgenic system for mouse Aurora A kinase: degradation by the ubiquitin proteasome pathway controls the level of the transgenic protein. Mol Cell Biol (2005) 25(12):5270–81. doi:10.1128/MCB.25.12.5270-5281.2005
30. Hinchcliffe EH. Centrosomes and the art of mitotic spindle maintenance. Int Rev Cell Mol Biol (2014) 313:179–217. doi:10.1016/B978-0-12-800177-6.00006-2
31. Hook SS, Lin JJ, Dutta A. Mechanisms to control rereplication and implications for cancer. Curr Opin Cell Biol (2007) 19(6):663–71. doi:10.1016/j.ceb.2007.10.007
32. Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol (1998) 18(2):1055–64. doi:10.1128/MCB.18.2.1055
33. Borel F, Lohez OD, Lacroix FB, Margolis RL. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc Natl Acad Sci U S A (2002) 99(15):9819–24. doi:10.1073/pnas.152205299
34. Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell (2011) 19(6):701–14. doi:10.1016/j.ccr.2011.04.017
35. D’Assoro AB, Barrett SL, Folk C, Negron VC, Boeneman K, Busby R, et al. Amplified centrosomes in breast cancer: a potential indicator of tumor aggressiveness. Breast Cancer Res Treat (2002) 75(1):25–34. doi:10.1023/A:1016550619925
36. D’Assoro AB, Busby R, Acu ID, Quatraro C, Reinholz MM, Farrugia DJ, et al. Impaired p53 function leads to centrosome amplification, acquired ERalpha phenotypic heterogeneity and distant metastases in breast cancer MCF-7 xenografts. Oncogene (2008) 27(28):3901–11. doi:10.1038/onc.2008.18
37. D’Assoro AB, Leontovich A, Amato A, Ayers-Ringler JR, Quatraro C, Hafner K, et al. Abrogation of p53 function leads to metastatic transcriptome networks that typify tumor progression in human breast cancer xenografts. Int J Oncol (2010) 37(5):1167–76.
38. Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol (2015) 16(8):473–85. doi:10.1038/nrm4025
39. Lingle WL, Barrett SL, Negron VC, D’Assoro AB, Boeneman K, Liu W, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A (2002) 99(4):1978–83. doi:10.1073/pnas.032479999
40. Das TK, Dana D, Paroly SS, Perumal SK, Singh S, Jhun H, et al. Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis (2013) 2:e69. doi:10.1038/oncsis.2013.34
41. Telentschak S, Soliwoda M, Nohroudi K, Addicks K, Klinz FJ. Cytokinesis failure and successful multipolar mitoses drive aneuploidy in glioblastoma cells. Oncol Rep (2015) 33(4):2001–8. doi:10.3892/or.2015.3751
42. McBride M, Rida PC, Aneja R. Turning the headlights on novel cancer biomarkers: inspection of mechanics underlying intratumor heterogeneity. Mol Aspects Med (2015) 45:3–13. doi:10.1016/j.mam.2015.05.001
43. Lentini L, Amato A, Schillaci T, Di Leonardo A. Simultaneous Aurora-A/STK15 overexpression and centrosome amplification induce chromosomal instability in tumour cells with a MIN phenotype. BMC Cancer (2007) 7:212. doi:10.1186/1471-2407-7-212
44. Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci U S A (2004) 101(52):18123–8. doi:10.1073/pnas.0408273101
45. Miyoshi Y, Iwao K, Egawa C, Noguchi S. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer (2001) 92(3):370–3. doi:10.1002/ijc.1200
46. Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta (2008) 1786(1):60–72. doi:10.1016/j.bbcan.2008.07.003
47. Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem (2004) 279(50):52175–82. doi:10.1074/jbc.M406802200
48. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet (2004) 36(1):55–62. doi:10.1038/ng1279
49. Carter RF. BRCA1, BRCA2 and breast cancer: a concise clinical review. Clin Invest Med (2001) 24(3):147–57.
50. Shimada M, Kobayashi J, Hirayama R, Komatsu K. Differential role of repair proteins, BRCA1/NBS1 and Ku70/DNA-PKcs, in radiation-induced centrosome overduplication. Cancer Sci (2010) 101(12):2531–7. doi:10.1111/j.1349-7006.2010.01702.x
51. Ouchi M, Fujiuchi N, Sasai K, Katayama H, Minamishima YA, Ongusaha PP, et al. BRCA phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem (2004) 279(19):19643–8. doi:10.1074/jbc.M311780200
52. Liu X, Erikson RL. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc Natl Acad Sci U S A (2002) 99(13):8672–6. doi:10.1073/pnas.132269599
53. Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by Aurora A to promote checkpoint recovery. Nature (2008) 455(7209):119–23. doi:10.1038/nature07185
54. Leontovich AA, Salisbury JL, Veroux M, Tallarita T, Billadeau D, McCubrey J, et al. Inhibition of Cdk2 activity decreases Aurora-A kinase centrosomal localization and prevents centrosome amplification in breast cancer cells. Oncol Rep (2013) 29(5):1785–8. doi:10.3892/or.2013.2313
55. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of Drosophila Aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J (1998) 17(11):3052–65. doi:10.1093/emboj/17.11.3052
56. Taga M, Hirooka E, Ouchi T. Essential roles of mTOR/Akt pathway in Aurora-A cell transformation. Int J Biol Sci (2009) 5(5):444–50. doi:10.7150/ijbs.5.444
57. Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer (2006) 119(10):2304–12. doi:10.1002/ijc.22154
58. Wang LH, Xiang J, Yan M, Zhang Y, Zhao Y, Yue CF, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res (2010) 70(22):9118–28. doi:10.1158/0008-5472.CAN-10-1246
59. Wang X, Lu N, Niu B, Chen X, Xie J, Cheng N. Overexpression of Aurora-A enhances invasion and matrix metalloproteinase-2 expression in esophageal squamous cell carcinoma cells. Mol Cancer Res (2012) 10(5):588–96. doi:10.1158/1541-7786.MCR-11-0416
60. Chen CH, Chuang HC, Huang CC, Fang FM, Huang HY, Tsai HT, et al. Overexpression of Rap-1A indicates a poor prognosis for oral cavity squamous cell carcinoma and promotes tumor cell invasion via Aurora-A modulation. Am J Pathol (2013) 182(2):516–28. doi:10.1016/j.ajpath.2012.10.023
61. Du J, Hannon GJ. The centrosomal kinase Aurora-A/STK15 interacts with a putative tumor suppressor NM23-H1. Nucleic Acids Res (2002) 30(24):5465–75. doi:10.1093/nar/gkf678
62. Cammareri P, Scopelliti A, Todaro M, Eterno V, Francescangeli F, Moyer MP, et al. Aurora-A is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells. Cancer Res (2010) 70(11):4655–65. doi:10.1158/0008-5472.CAN-09-3953
63. Chou CH, Yang NK, Liu TY, Tai SK, Hsu DS, Chen YW, et al. Chromosome instability modulated by BMI1-AURKA signaling drives progression in head and neck cancer. Cancer Res (2013) 73(2):953–66. doi:10.1158/0008-5472.CAN-12-2397
64. Wirtz-Peitz F, Nishimura T, Knoblich JA. Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell (2008) 135(1):161–73. doi:10.1016/j.cell.2008.07.049
65. Reiter R, Gais P, Jütting U, Steuer-Vogt MK, Pickhard A, Bink K, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res (2006) 12(17):5136–41. doi:10.1158/1078-0432.CCR-05-1650
66. Landen CN Jr, Lin YG, Immaneni A, Deavers MT, Merritt WM, Spannuth WA, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res (2007) 13(14):4098–104. doi:10.1158/1078-0432.CCR-07-0431
67. Lassmann S, Shen Y, Jütting U, Wiehle P, Walch A, Gitsch G, et al. Predictive value of Aurora-A/STK15 expression for late stage epithelial ovarian cancer patients treated by adjuvant chemotherapy. Clin Cancer Res (2007) 13(14):4083–91. doi:10.1158/1078-0432.CCR-06-2775
68. Goos JA, Coupe VM, Diosdado B, Delis-Van Diemen PM, Karga C, Beliën JA, et al. Aurora kinase A (AURKA) expression in colorectal cancer liver metastasis is associated with poor prognosis. Br J Cancer (2013) 109(9):2445–52. doi:10.1038/bjc.2013.608
69. Wang J, Yang S, Zhang H, Song Y, Zhang X, Qian H, et al. Aurora-A as an independent molecular prognostic marker in gastric cancer. Oncol Rep (2011) 26(1):23–32. doi:10.3892/or.2011.1250
70. Samaras V, Stamatelli A, Samaras E, Arnaoutoglou C, Arnaoutoglou M, Stergiou I, et al. Comparative immunohistochemical analysis of Aurora-A and Aurora-B expression in human glioblastomas. Associations with proliferative activity and clinicopathological features. Pathol Res Pract (2009) 205(11):765–73. doi:10.1016/j.prp.2009.06.011
71. Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res (2008) 14(14):4455–62. doi:10.1158/1078-0432.CCR-07-5268
72. Yamamoto Y, Ibusuki M, Nakano M, Kawasoe T, Hiki R, Iwase H. Clinical significance of basal-like subtype in triple-negative breast cancer. Breast Cancer (2009) 16(4):260–7. doi:10.1007/s12282-009-0150-8
73. Siggelkow W, Boehm D, Gebhard S, Battista M, Sicking I, Lebrecht A, et al. Expression of aurora kinase A is associated with metastasis-free survival in node-negative breast cancer patients. BMC Cancer (2012) 12:562. doi:10.1186/1471-2407-12-562
74. Roylance R, Endesfelder D, Jamal-Hanjani M, Burrell RA, Gorman P, Sander J, et al. Expression of regulators of mitotic fidelity are associated with intercellular heterogeneity and chromosomal instability in primary breast cancer. Breast Cancer Res Treat (2014) 148(1):221–9. doi:10.1007/s10549-014-3153-x
75. Dees EC, Infante JR, Cohen RB, O’Neil BH, Jones S, von Mehren M, et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol (2011) 67:945–54. doi:10.1007/s00280-010-1377-y
76. Macarulla T, Cervantes A, Elez E, Rodriguez-Braun E, Baselga J, Rosello S, et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol Cancer Ther (2010) 9:2844–52. doi:10.1158/1535-7163.MCT-10-0299
77. Sells TB, Chau R, Ecsedy JA, Gershman RE, Hoar K, Huck J, et al. MLN8054 and Alisertib (MLN8237): discovery of selective oral Aurora A inhibitors. ACS Med Chem Lett (2015) 6(6):630–4. doi:10.1021/ml500409n
78. Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol (2012) 127(1):63–9. doi:10.1016/j.ygyno.2012.06.040
79. Goldberg SL, Fenaux P, Craig MD, Gyan E, Lister J, Kassis J, et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk Res Rep (2014) 3(2):58–61. doi:10.1016/j.lrr.2014.06.003
80. Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective Aurora A kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res (2012) 18(17):4764–74. doi:10.1158/1078-0432.CCR-12-0571
81. Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, et al. Phase I study of Aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res (2012) 18:4775–84. doi:10.1158/1078-0432.CCR-12-0589
82. Melichar FB, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol (2015) 16:395–405. doi:10.1016/S1470-2045(15)70051-3
83. Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol (2014) 32:44–50. doi:10.1200/JCO.2012.46.8793
84. Barr PM, Li H, Spier C, Mahadevan D, LeBlanc M, Ul Haq M, et al. Phase II intergroup trial of alisertib in relapsed and refractory peripheral T-cell lymphoma and transformed mycosis fungoides: SWOG 1108. J Clin Oncol (2015) 33(21):2399–404. doi:10.1200/JCO.2014.60.6327
85. Huck JJ, Zhang M, Mettetal J, Chakravarty A, Venkatakrishnan K, Zhou X, et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational Aurora A kinase inhibitor MLN8237 (alisertib). Mol Cancer Ther (2014) 13:2170–83. doi:10.1158/1535-7163.MCT-14-0027
Keywords: mitotic kinase, cell cycle, cancer, tumor progression, targeted therapy
Citation: D’Assoro AB, Haddad T and Galanis E (2016) Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front. Oncol. 5:295. doi: 10.3389/fonc.2015.00295
Received: 22 September 2015; Accepted: 11 December 2015;
Published: 06 January 2016
Edited by:
Claude Prigent, CNRS, FranceReviewed by:
Valerio Donato, New York University Medical Center, USAAdriana De Siervi, Instituto de Biologia y Medicina Experimental, Argentina
Sourisseau Tony, Gustave Roussy Cancer Campus, France
Copyright: © 2016 D’Assoro, Haddad and Galanis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evanthia Galanis, Z2FsYW5pcy5ldmFudGhpYUBtYXlvLmVkdQ==