 Thuy Vu1,2,3
Thuy Vu1,2,3 Francois X. Claret1,2,3*
Francois X. Claret1,2,3*- 1 Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
- 2 Cancer Biology Program, The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, TX, USA
- 3 Experimental Therapeutic Program, The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, TX, USA
HER2-positive breast cancer accounts for 20–30% of all breast cancers and has the second-poorest prognosis among breast cancer subtypes. The approval of trastuzumab in 1998 has significantly improved patients’ outcomes and paved the way for the beginning of advent of targeted approaches in breast cancer treatment. However, primary or acquired resistance to trastuzumab has been increasingly recognized as a major obstacle in the clinical management of this disease. In addition, in clinical practice, there are currently no conclusive biomarkers for patient response to trastuzumab. Therefore, understanding the molecular mechanism of trastuzumab and the development of resistance to this drug are of interest. Such understanding will provide the guidance critically needed for the design of better combination therapy and will allow the appropriate selection of patients who are responsive to trastuzumab-based strategies. In line with that, our review highlights the well-accepted mechanisms of action and resistance to the therapy and discusses the progress that has been made toward successfully overcoming this resistance.
Introduction
The family of epidermal growth factor receptors comprises HER1, HER2, HER3, and HER4. These tyrosine kinases play a critical role in cellular growth and are frequently up-regulated in numerous cancers, including breast carcinoma (Browne et al., 2009). The overexpression or gene amplification of HER2 has been found in about 20–30% of breast cancers, which is classified as the HER2-positive subtype (Slamon et al., 1987; Seshadri et al., 1993; Owens et al., 2004). HER2-positive breast cancer has the second-poorest prognosis among breast cancer subtypes and is correlated with lower disease-free and overall survival rates (Slamon et al., 1987; Tandon et al., 1989; Seshadri et al., 1993; Andrulis et al., 1998; Paik et al., 2000). Typically, HER2 is expressed at a low level on the surface of epithelial cells and is necessary for the normal development of many tissues, including those of the breast, ovary, lung, liver, kidney, and central nervous system (Browne et al., 2009). In contrast, in breast cancer cells, immunohistochemical analyses have revealed extremely high levels of HER2, which can reach up to two million receptors per cell (Slamon et al., 1987; Browne et al., 2009). The overexpression of HER2 is of crucial importance since its activation triggers multiple downstream pathways required for the abnormal proliferation of cancer cells (Browne et al., 2009).
HER2 is activated by the formation of homodimers or heterodimers with other EGFR proteins. This dimerization results in autophosphorylation and/or transphosphorylation of specific tyrosine residues in EGFR intracellular domains, which in turn leads to the activation of the Ras/Raf/mitogen-activated protein kinase, the phosphoinositide 3-kinase/Akt, and the phospholipase Cγ (PLCγ)/protein kinase C (PKC) pathways (Browne et al., 2009). Of note, the HER2-HER3 heterodimer is the most potent stimulator of downstream pathways, particularly the PI3K/Akt, a master regulator of cell growth and survival (Alimandi et al., 1995; Lee-Hoeflich et al., 2008). Moreover, HER2 dimerization promotes the mislocalization and rapid degradation of cell cycle inhibitor p27Kip1 protein leading to cell cycle progression. Some of the well studied pathways affected by activation of HER2 are briefly described in Figure 1.
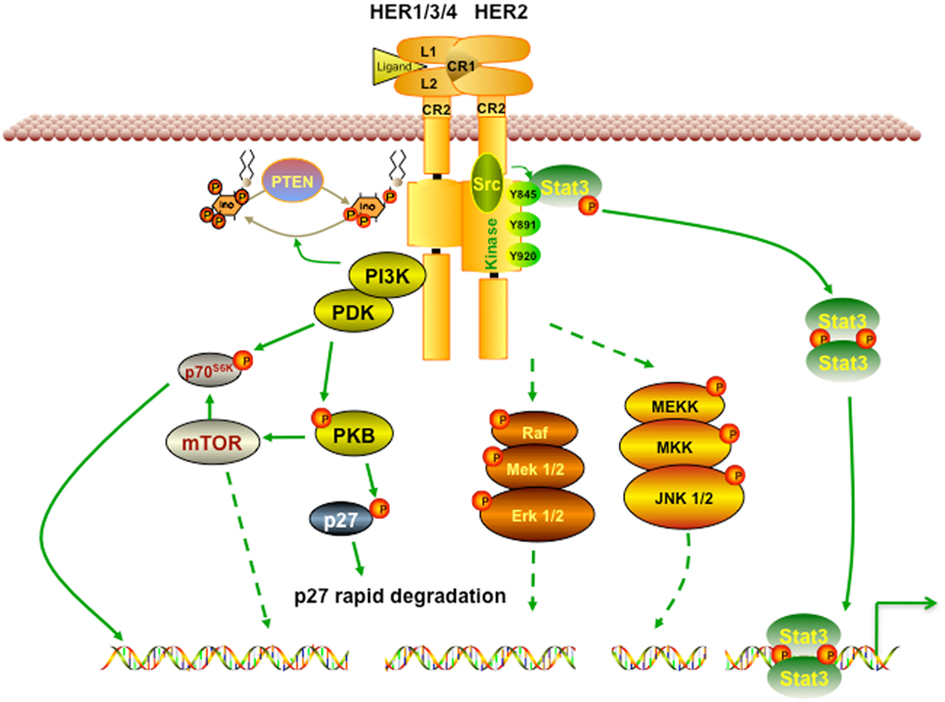
Figure 1. HER2 signaling pathway. Receptor homodimerization or heterodimerization is the prerequisite step for HER2 activation. Its activation then triggers a broad spectrum of downstream cascades to promote numerous effects, including cell growth, proliferation, and survival. PI3K/Akt is one of the most well studied pathways activated by HER2. Activated PI3K/Akt also triggers mTOR, a master positive regulator of cell metabolism. In addition, HER2 activation can activate Ras/Raf and MEK pathways, which favors cancer cells’ growth and migration.
Mechanisms of Action of Rrastuzumab
Given that HER2 is frequently up-regulated in breast cancer and is correlated with poor prognosis, targeting HER2 is an attractive therapeutic approach. The first Food and Drug Administration (FDA)-approved targeted therapy for breast cancer was trastuzumab (Gajria and Chandarlapaty, 2011). Trastuzumab (or Herceptin) was developed by Genentech Inc (San Francisco, CA, USA) as a recombinant humanized monoclonal antibody directed against the extracellular domain IV of HER2 (Carter et al., 1992), has served as a remarkable example of a successful targeted therapy in breast cancer. Clinical studies have shown that the combination of trastuzumab with standard chemotherapy produces far better response rates than does chemotherapy alone (Slamon et al., 2001; Vogel et al., 2002). Thus, the combinations that include trastuzumab have been considered as the standard of care for HER2-overexpressing breast cancer patients (Hudis, 2007). In this part, we focus on the molecular mechanisms of actions of trastuzumab, which are grouped into three major categories, summarized below.
HER2 Degradation
Trastuzumab has been proposed to trigger HER2 internalization and degradation through promoting the activity of tyrosine kinase – ubiquitin ligase c-Cbl (Klapper et al., 2000). It was observed that the binding of trastuzumab to HER2 recruits c-Cbl to its docking site, Tyr1112 where c-Cbl ubiquitinates HER2 and leads to its degradation. However, how c-Cbl is induced as trastuzumab binds to HER2 and how c-Cbl mediates HER2 degradation remain obscure.
Antibody-Dependent Cellular Cytotoxicity
As an antibody, one of the major mechanisms of trastuzumab is to attract immune cells to tumor sites that overexpress HER2, by a mechanism called antibody-dependent cellular cytotoxicity (ADCC). Xenografts study by Clynes et al. (2000) showed that natural killer cells could target HER2-overexpressing cells coated with trastuzumab via a CD16-mediated ADCC mechanism. Arnould et al. (2006) later confirmed the immune cell-modulated activity of trastuzumab by using samples from patients with locally advanced breast cancer. After treating the tumor samples with trastuzumab and docetaxel, the group observed an increase in the number of both natural killer cells and cytotoxic proteins in tumor infiltrates (Arnould et al., 2006). These findings provide strong support for ADCC as a mechanism of action of trastuzumab and help explain why cancer cells with high HER2 expression are more responsive to the drug.
MAPK and PI3K/Akt Interference
The most well known effect of trastuzumab is the inhibition of the MAPK and PI3K/Akt pathways (Figure 1), which leads to an increase in cell cycle arrest, and the suppression of cell growth and proliferation. It is widely accepted that by interfering with the dimerization of HER2, trastuzumab inhibits HER2 activation and suppresses Akt phosphorylation (Junttila et al., 2009). Other groups critically showed that trastuzumab, by binding to HER2, can block tyrosine kinase Src signaling and thus, increases PTEN level and activity. This also results in the suppression of PI3K/Akt signaling and reduction in cell growth and survival (Nagata et al., 2004; Zhang et al., 2011). Moreover, trastuzumab was demonstrated to induce cell cycle arrest by restoring p27 and suppressing CDK2 activity in BT474 and SKBR3 breast cancer cells.
In general, trastuzumab, by binding to extracellular domain of HER2, can potently suppress cancer cells growth, proliferation, and survival in both direct and indirect manners.
Mechanisms of Trastuzumab Resistance
Even though trastuzumab has significantly improved outcome in breast cancer patients and has paved the way for the era of targeted therapy in breast cancer treatment, the median duration of response is modest (Gajria and Chandarlapaty, 2011; Zhang et al., 2011). This is due to either primary or secondary mechanisms of resistance to the therapy. Less than 35% of patients with HER2-positive breast cancer initially respond to trastuzumab (Wolff et al., 2007; Narayan et al., 2009), which means these patients are “primarily” or “inherently” resistant to the drug. On the other hand, about 70% of patients who initially responded experience progression to metastatic disease within a year (Gajria and Chandarlapaty, 2011), suggesting that “secondary” or “acquired” resistance to trastuzumab frequently develops. Some tumor suppressor proteins and oncoproteins, such as PTEN and Src, have biological significance and play overlapping roles in both primary and acquired resistance (Nagata et al., 2004; Zhang et al., 2011). On the other hand, little information on the basis of primary and secondary resistance is available (Narayan et al., 2009). In this review, we classified the potential mechanisms of resistance to trastuzumab according to the inter- or intra-networks of HER2 signaling. These are summarized in three major categories, briefly described in Figure 2: (1) the steric effects, such as the structural mutation in HER2 protein, (2) the alternative elevations of other tyrosine kinase receptors, such as insulin-like growth factor receptor (IGFR), or (3) the intracellular alterations in HER2 downstream signaling, notably phosphatase and tensin homolog (PTEN) deficiency and/or PI3K/Akt constitutive active.
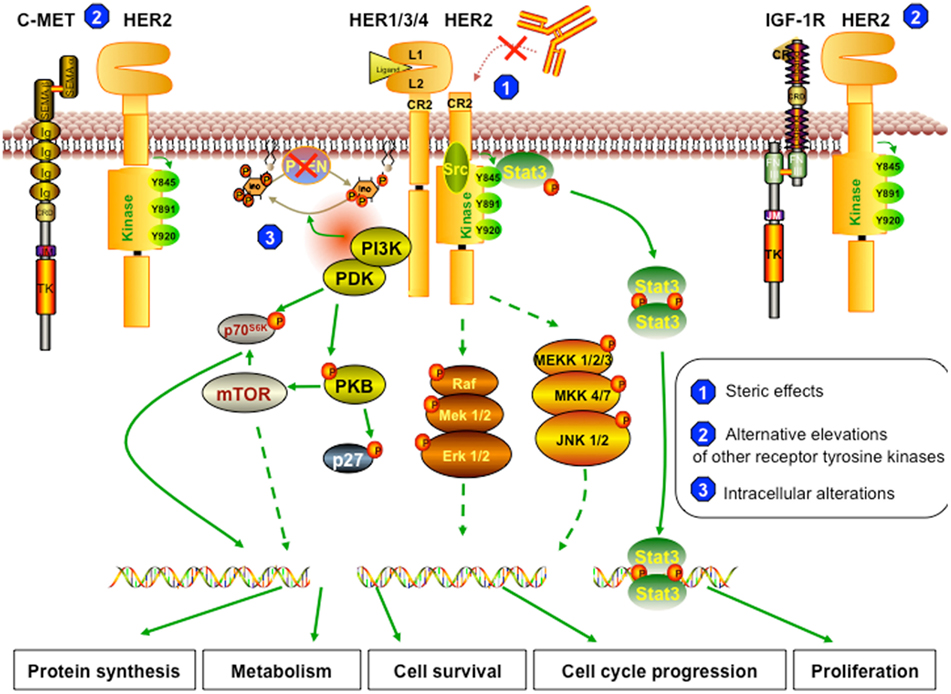
Figure 2. Resistant mechanism to trastuzumab. (1) Steric effects or masking of the trastuzumab-binding sites. The truncated form of HER2 (p95HER2) that lacks trastuzumab-binding domain can no longer be inhibited by trastuzumab. The remaining structure can still dimerize with other receptors and therefore can still trigger downstream cascades. (2) Alternative elevations of other receptor tyrosine kinases. The overexpression of other growth factor receptors, such as c-Met and IGF-1R, the two most commonly reported, can trigger similar effects on cell behavior, even though HER2 signaling is inhibited by trastuzumab. (3) Intracellular alterations. In trastuzumab resistant cells, PTEN function is lost; thus, Akt is constitutively active. Also, resistant cells can harbor activating mutations in PI3K or Akt, which can trigger other downstream signaling pathways even when HER2 is blocked by trastuzumab or when PTEN is active.
Steric Effects
HER2 can mutate to “cut off” its extracellular domain, which inhibits the binding of trastuzumab to the receptor (Scott et al., 1993; Christianson et al., 1998). This process is called proteolysis, which generates the truncated p95HER2 isoform with constitutive kinase activity (Scott et al., 1993; Christianson et al., 1998; Scaltriti et al., 2007). This mutated isoform promotes the continuous activation of oncogenic signaling and bypass trastuzumab’s effects. In a study of tumor cells from 46 patients with metastatic breast cancer, those who acquired the p95HER2 mutation were less likely to respond to trastuzumab than were those with full-length HER2 (Scaltriti et al., 2007). However, how cancer cells can induce HER2 proteolysis in response to trastuzumab has not been examined. Another mechanism that may contribute to the “masking” of the trastuzumab-binding site on HER2 is the elevated expression of mucin-4. Mucin-4, a highly O-glycosylated membrane protein, is also the putative partner of HER2, and its expression was correlated with poor prognosis in not only breast cancer but also other carcinoma types (Nagy et al., 2005). However, how Mucin-4 is overexpressed in trastuzumab resistant cells and by what mechanism Mucin-4 confers resistance to the therapy required further studies.
Overexpression of Other Tyrosine Kinase Receptors
Since trastuzumab does not prevent HER3 dimerization (Wehrman et al., 2006), the overexpression of HER3 could overcome trastuzumab-mediated inhibition of HER2 signaling. It was previously found that co-expression of HER3 and HER2 is required for the transformation of NIH-3T3 cells (Alimandi et al., 1995) and that HER2-HER3 dimerization results in increased tyrosine phosphorylation of HER3 which subsequently increases PI3K recruitment and downstream activation (Alimandi et al., 1995; Holbro et al., 2003). These findings imply that cancer cells, by utilizing HER3 signaling, can compensate for the inhibition of HER2 signaling mediated by trastuzumab.
Another promising target whose aberrant expression may confer resistance to trastuzumab is insulin-like growth factor 1 receptor (IGF-1R). Lu et al. (2001) found that ectopic expression of IGF-1R drives trastuzumab-sensitive SKBR3 cells to resistant to the therapy. Also, IGF-1R-overexpressing SKBR3 cells show significantly low levels of p27Kip1, p21Cip1 proteins, and a high level of CDK2 kinase activity (Lu et al., 2001). This indicates that IGF-1R-overexpressing cells, by overcoming cell cycle arrest mediated by trastuzumab contribute to the drug resistance. Interestingly, the resistance is reversed by suppression of IGF-1R signaling with recombinant IGF-binding protein-3 (IGFBP3) or by gradual heat-induced expression of the dominant-negative IGF-1R 486/STOP (Lu et al., 2001; Jerome et al., 2006). This further confirms that the elevation of IGF-1R significantly contributes to trastuzumab resistance.
Notably, the overexpression of tyrosine kinase c-Met has emerged as a potential contributor to trastuzumab. c-Met is a critical oncogene that encodes for a receptor tyrosine kinase composed of an α chain disulfide linked to a β subunit (Bottaro et al., 1991). c-Met, until now, is reported to have only one known ligand called hepatocyte growth factor (HGF) and is normally expressed in epithelial and endothelial cells (Bottaro et al., 1991). Although Met signaling is actively involved in the regeneration and development of various organs, c-Met and HGF have been reported to be overexpressed in human breast carcinomas and tumor stroma, respectively. Moreover, c-Met overexpression is correlated with poor prognosis in breast cancer patients (Kang et al., 2003). Furthermore, c-Met was shown to be highly elevated in HER2-positive breast cancer cell lines and in 25% of HER2-positive breast cancer patients’ tissues (Yamashita et al., 1994; Lindemann et al., 2007; Shattuck et al., 2008). More importantly, c-Met has recently been demonstrated to physically interact with HER2 (Agarwal et al., 2009). These findings suggest that c-Met synergizes with HER2 signaling to confer resistance to trastuzumab. Sweeney’s group, indeed, revealed that depletion of c-Met makes the cells more sensitive to trastuzumab and that trastuzumab rapidly up-regulates c-Met expression in vitro (Shattuck et al., 2008). These findings imply that c-Met overexpression is critical for treatment-acquired resistance to trastuzumab. However, further studies are needed to strengthen our knowledge of the contribution of c-Met to trastuzumab resistance and to understand how c-Met is elevated in resistant cells.
Moreover, Liang et al. (2010) found that recombinant human erythropoietin (rHuEPO), which has long been used to manage the anemia and fatigue caused by cancer treatment, antagonizes trastuzumab’s effects on breast cancer models both in vitro and in vivo. This group demonstrated that, in a panel of 10 primary breast cancer cell lines, 5 express the erythropoietin receptor (EpoR), and 2 have relatively high levels of both HER2 and EpoR. These observations indicate that EpoR and HER2 are overexpressed in the same cancer cells. Furthermore, using the same cell lines, they found that rHuEPO significantly reduces the cell growth inhibition induced by trastuzumab. rHuEPO also triggers the PI3K/Akt and Src pathways, leading to the suppression of PTEN and trastuzumab’s effects. This was confirmed by a mouse xenografts study in which rHuEPO introduction promotes tumor growth suppressed by trastuzumab (Liang et al., 2010). These elegant models suggested that rHuEPO administration contributes to the resistance to trastuzumab.
Intracellular Alterations
One of the most under-investigated “driving forces” involved in trastuzumab resistance is PTEN. Nagata et al. (2004) reported that the loss of PTEN was observed in 36% of HER2-positive primary breast tumor specimens from patients with stage IV disease. In addition, they also showed that patients with PTEN-deficient tumors had remarkably lower overall response rates to trastuzumab than did patients with wild-type PTEN. Later on, other group reported 25% of PI3KCA activating mutation and 20–25% of PTEN deficiency in tumors from a cohort of 55 patients resistant to trastuzumab (Berns et al., 2007; Kataoka et al., 2010). They found that trastuzumab-treated patients with activating PI3KCA mutations had significantly shorter progression-free survival than did those without mutations. The group also demonstrated that HER2-positive breast cancer cell lines with activating PIK3CA mutations (E545K and H1047R) are more resistant to trastuzumab than are HER2-positive cell lines without those mutations (Berns et al., 2007; Kataoka et al., 2010). These studies suggest that constitutive active PI3K/Akt pathway, either due to PTEN loss or activating mutations in PIK3CA, significantly contributes to both inherent and acquired resistance to trastuzumab.
PTEN deficiency that may confer resistance to the therapy was recently confirmed by Yu’s group (Zhang et al., 2011). Of interest, the group also found that Src is activated in breast cancer cell lines which is associated with both primary and acquired resistance to trastuzumab. Specifically, they revealed that patients with active Src were significantly less responsive to trastuzumab than were those without Src activation. In terms of molecular networking, they showed that Src is the central mediator of all resistant pathways. Those features enable Src a promising “global” therapeutic target for patients resistant to trastuzumab. Furthermore, Src inhibition can reverse trastuzumab resistance in vitro and in vivo, specifically through effectively blocking Akt phosphorylation and downstream signaling of EGFR, even in PTEN-deficient cells. This poses a novel promising approach of combining trastuzumab and Src inhibition for better clinical benefit in a large population of patients with poor response to trastuzumab.
Concluding Remarks
A number of critical studies have been conducted to identify the mechanisms that account for trastuzumab’s effects and the resistance to the therapy. Notably, multiple mechanisms may act simultaneously to confer resistance to the drug, due to the accumulation of intracellular alterations and the heterogeneity of breast cancer. For this reason, it is clinically unfeasible to extensively combine various therapeutic targets. Therefore, further studies are urgently needed to distinguish “driving force” from “bypass” mechanisms. This, together with the clarification of primary and acquired resistance, will ultimately help in designing more specific and effective combination therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Cancer Institute (R01-CA90853), Cancer Prevention and Research Institute of Texas (CPRIT) grant (RP12045101) to Francois X. Claret and the Vietnam Education Foundation to TTV. We thank Tamara K. Locke for editing the manuscript.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; Akt, protein kinase B; c-Cbl, Casitas B-lineage lymphoma; CDK2, cyclin-dependent kinase 2; EGFR, epidermal growth factor receptor; EpoR, erythropoietin receptor; HGF, hepatocyte growth factor; IGFBP3, IGF-binding protein-3; IGFR, insulin-like growth factor receptor; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol triphosphate kinase; PTEN, phosphatase and tensin homolog; rHuEPO, recombinant human erythropoietin.
References
Agarwal, S., Zerillo, C., Kolmakova, J., Christensen, J. G., Harris, L. N., Rimm, D. L., Digiovanna, M. P., and Stern, D. F. (2009). Association of constitutively activated hepatocyte growth factor receptor (Met) with resistance to a dual EGFR/Her2 inhibitor in non-small-cell lung cancer cells. Br. J. Cancer 100, 941–949.
Alimandi, M., Romano, A., Curia, M. C., Muraro, R., Fedi, P., Aaronson, S. A., Di Fiore, P. P., and Kraus, M. H. (1995). Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 10, 1813–1821.
Andrulis, I. L., Bull, S. B., Blackstein, M. E., Sutherland, D., Mak, C., Sidlofsky, S., Pritzker, K. P., Hartwick, R. W., Hanna, W., Lickley, L., Wilkinson, R., Qizilbash, A., Ambus, U., Lipa, M., Weizel, H., Katz, A., Baida, M., Mariz, S., Stoik, G., Dacamara, P., Strongitharm, D., Geddie, W., and McCready, D. (1998). neu/erbB-2 amplification identifies a poor-prognosis group of women with node-negative breast cancer. J. Clin. Oncol. 16, 1340–1349.
Arnould, L., Gelly, M., Penault-Llorca, F., Benoit, L., Bonnetain, F., Migeon, C., Cabaret, V., Fermeaux, V., Bertheau, P., Garnier, J., Jeannin, J. F., and Coudert, B. (2006). Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 94, 259–267.
Berns, K., Horlings, H. M., Hennessy, B. T., Madiredjo, M., Hijmans, E. M., Beelen, K., Linn, S. C., Gonzalez-Angulo, A. M., Stemke-Hale, K., Hauptmann, M., Beijersbergen, R. L., Mills, G. B., van de Vijver, M. J., and Bernards, R. (2007). A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12, 395–402.
Bottaro, D. P., Rubin, J. S., Faletto, D. L., Chan, A. M., Kmiecik, T. E., Vande Woude, G. F., and Aaronson, S. A. (1991). Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 251, 802–804.
Browne, B. C., O’Brien, N., Duffy, M. J., Crown, J., and O’Donovan, N. (2009). HER-2 signaling and inhibition in breast cancer. Curr. Cancer Drug Targets 9, 419–438.
Carter, P., Presta, L., Gorman, C. M., Ridgway, J. B., Henner, D., Wong, W. L., Rowland, A. M., Kotts, C., Carver, M. E., and Shepard, H. M. (1992). Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 89, 4285–4289.
Christianson, T. A., Doherty, J. K., Lin, Y. J., Ramsey, E. E., Holmes, R., Keenan, E. J., and Clinton, G. M. (1998). NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 58, 5123–5129.
Clynes, R. A., Towers, T. L., Presta, L. G., and Ravetch, J. V. (2000). Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446.
Gajria, D., and Chandarlapaty, S. (2011). HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 11, 263–275.
Holbro, T., Beerli, R. R., Maurer, F., Koziczak, M., Barbas, C. F. III, and Hynes, N. E. (2003). The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 100, 8933–8938.
Hudis, C. A. (2007). Trastuzumab – mechanism of action and use in clinical practice. N. Engl. J. Med. 357, 39–51.
Jerome, L., Alami, N., Belanger, S., Page, V., Yu, Q., Paterson, J., Shiry, L., Pegram, M., and Leyland-Jones, B. (2006). Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res. 66, 7245–7252.
Junttila, T. T., Akita, R. W., Parsons, K., Fields, C., Lewis Phillips, G. D., Friedman, L. S., Sampath, D., and Sliwkowski, M. X. (2009). Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440.
Kang, J. Y., Dolled-Filhart, M., Ocal, I. T., Singh, B., Lin, C. Y., Dickson, R. B., Rimm, D. L., and Camp, R. L. (2003). Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res. 63, 1101–1105.
Kataoka, Y., Mukohara, T., Shimada, H., Saijo, N., Hirai, M., and Minami, H. (2010). Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann. Oncol. 21, 255–262.
Klapper, L. N., Waterman, H., Sela, M., and Yarden, Y. (2000). Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 60, 3384–3388.
Lee-Hoeflich, S. T., Crocker, L., Yao, E., Pham, T., Munroe, X., Hoeflich, K. P., Sliwkowski, M. X., and Stern, H. M. (2008). A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 68, 5878–5887.
Liang, K., Esteva, F. J., Albarracin, C., Stemke-Hale, K., Lu, Y., Bianchini, G., Yang, C. Y., Li, Y., Li, X., Chen, C. T., Mills, G. B., Hortobagyi, G. N., Mendelsohn, J., Hung, M. C., and Fan, Z. (2010). Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell 18, 423–435.
Lindemann, K., Resau, J., Nahrig, J., Kort, E., Leeser, B., Annecke, K., Welk, A., Schafer, J., Vande Woude, G. F., Lengyel, E., and Harbeck, N. (2007). Differential expression of c-Met, its ligand HGF/SF and HER2/neu in DCIS and adjacent normal breast tissue. Histopathology 51, 54–62.
Lu, Y., Zi, X., Zhao, Y., Mascarenhas, D., and Pollak, M. (2001). Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 93, 1852–1857.
Nagata, Y., Lan, K. H., Zhou, X., Tan, M., Esteva, F. J., Sahin, A. A., Klos, K. S., Li, P., Monia, B. P., Nguyen, N. T., Hortobagyi, G. N., Hung, M. C., and Yu, D. (2004). PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127.
Nagy, P., Friedlander, E., Tanner, M., Kapanen, A. I., Carraway, K. L., Isola, J., and Jovin, T. M. (2005). Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 65, 473–482.
Narayan, M., Wilken, J. A., Harris, L. N., Baron, A. T., Kimbler, K. D., and Maihle, N. J. (2009). Trastuzumab-induced HER reprogramming in “resistant” breast carcinoma cells. Cancer Res. 69, 2191–2194.
Owens, M. A., Horten, B. C., and Da Silva, M. M. (2004). HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin. Breast Cancer 5, 63–69.
Paik, S., Bryant, J., Tan-Chiu, E., Yothers, G., Park, C., Wickerham, D. L., and Wolmark, N. (2000). HER2 and choice of adjuvant chemotherapy for invasive breast cancer: National Surgical Adjuvant Breast and Bowel Project Protocol B-15. J. Natl. Cancer Inst. 92, 1991–1998.
Scaltriti, M., Rojo, F., Ocana, A., Anido, J., Guzman, M., Cortes, J., Di Cosimo, S., Matias-Guiu, X., Ramon y Cajal, S., Arribas, J., and Baselga, J. (2007). Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 99, 628–638.
Scott, G. K., Robles, R., Park, J. W., Montgomery, P. A., Daniel, J., Holmes, W. E., Lee, J., Keller, G. A., Li, W. L., Fendly, B. M., Wood, W. I., Shepard, M. H., and Benz, C. C. (1993). A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol. Cell. Biol. 13, 2247–2257.
Seshadri, R., Firgaira, F. A., Horsfall, D. J., McCaul, K., Setlur, V., and Kitchen, P. (1993). Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J. Clin. Oncol. 11, 1936–1942.
Shattuck, D. L., Miller, J. K., Carraway, K. L. III, and Sweeney, C. (2008). Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 68, 1471–1477.
Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., and McGuire, W. L. (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182.
Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter, J., Pegram, M., Baselga, J., and Norton, L. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792.
Tandon, A. K., Clark, G. M., Chamness, G. C., Ullrich, A., and McGuire, W. L. (1989). HER-2/neu oncogene protein and prognosis in breast cancer. J. Clin. Oncol. 7, 1120–1128.
Vogel, C. L., Cobleigh, M. A., Tripathy, D., Gutheil, J. C., Harris, L. N., Fehrenbacher, L., Slamon, D. J., Murphy, M., Novotny, W. F., Burchmore, M., Shak, S., Stewart, S. J., and Press, M. (2002). Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 20, 719–726.
Wehrman, T. S., Raab, W. J., Casipit, C. L., Doyonnas, R., Pomerantz, J. H., and Blau, H. M. (2006). A system for quantifying dynamic protein interactions defines a role for Herceptin in modulating ErbB2 interactions. Proc. Natl. Acad. Sci. U.S.A. 103, 19063–19068.
Wolff, A. C., Hammond, M. E., Schwartz, J. N., Hagerty, K. L., Allred, D. C., Cote, R. J., Dowsett, M., Fitzgibbons, P. L., Hanna, W. M., Langer, A., McShane, L. M., Paik, S., Pegram, M. D., Perez, E. A., Press, M. F., Rhodes, A., Sturgeon, C., Taube, S. E., Tubbs, R., Vance, G. H., van de Vijver, M., Wheeler, T. M., and Hayes, D. F. American Society of Clinical Oncology and College of American Pathologists. (2007). American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 25, 118–145.
Yamashita, J., Ogawa, M., Yamashita, S., Nomura, K., Kuramoto, M., Saishoji, T., and Shin, S. (1994). Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res. 54, 1630–1633.
Zhang, S., Huang, W. C., Li, P., Guo, H., Poh, S. B., Brady, S. W., Xiong, Y., Tseng, L. M., Li, S. H., Ding, Z., Sahin, A. A., Esteva, F. J., Hortobagyi, G. N., and Yu, D. (2011). Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 17, 461–469.
Keywords: HER2/ERBB2, HER3, trastuzumab, herceptin, targeted therapies, breast cancer
Citation: Vu T and Claret FX (2012) Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2:62. doi: 10.3389/fonc.2012.00062
Received: 04 April 2012; Accepted: 30 May 2012;
Published online: 18 June 2012.
Edited by:
Ruggero De Maria, Istituto Superiore di Sanità, ItalyReviewed by:
George Calin, MD Anderson Cancer Center, USASukyung Woo, University of Oklahoma Health Science Center, USA
Copyright: © 2012 Vu and Claret. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Francois X. Claret, Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77054, USA. e-mail:ZnhjbGFyZXRAbWRhbmRlcnNvbi5vcmc=