
- Instituto de Biomedicina de Sevilla, Hospital Universitario Virgen del Rocío, Consejo Superior de Investigaciones Cientificas, Universidad de Sevilla, Sevilla, Spain
MAP17 is a small 17 kDa non-glycosylated membrane protein previously identified as being overexpressed in carcinomas. Breast tumor cells that overexpress MAP17 show an increased tumoral phenotype with enhanced proliferative capabilities both in the presence or the absence of contact inhibition, decreased apoptotic sensitivity, and increased migration. MAP17-expressing clones also grow better in nude mice. The increased malignant cell behavior induced by MAP17 is associated with an increase in reactive oxygen species (ROS) production, and the treatment of MAP17-expressing cells with antioxidants results in a reduction in the tumorigenic properties of these cells. The MAP17-dependent increase in ROS and tumorigenesis relies on its PDZ-binding domain because disruption of this sequence by point mutations abolishes the ability of MAP17 to enhance ROS production and tumorigenesis. MAP17 is overexpressed in a great variety of human carcinomas, including breast tumors. Immunohistochemical analysis of MAP17 during cancer progression demonstrates that overexpression of the protein strongly correlates with tumoral progression. Generalized MAP17 overexpression in human carcinomas indicates that MAP17 can be a good marker for tumorigenesis and, especially, for malignant progression.
The complex physiology of vertebrates requires the continuous renewal of most tissues, which may become damaged either by external agents or by the toxic byproducts of their own metabolism, to maintain homeostasis. Cancer arises as a consequence of genetic changes that deregulate the mechanisms that control the renewal process, either by activation of the pathways that promote survival and proliferation, or through inactivation of growth suppression pathways. In order for cancer cells to grow and metastasize, they must overcome additional barriers to their expansion by promoting angiogenesis, acquiring characteristics that allow them to survive in organs different from their origin or by evading immune surveillance mechanisms (Hanahan and Weinberg, 2000, 2011).
Tumorigenesis occurs when the mechanisms involved in the control of tissue homeostasis are disrupted and cells stop responding to physiological signals. Therefore, genes capable of desensitizing tumoral cells to physiological signals may provide a selective advantage within the tumoral mass and influence the outcome of the disease. We undertook a large-scale genetic screen to identify genes capable of altering the cellular response to physiological signals that resulted in a selective advantage during tumorigenesis (Hannon et al., 1999; Carnero et al., 2000; Vergel and Carnero, 2010). A genome-wide retroviral cDNA screen to search for genes that confer a selective advantage to cancer cells during tumorigenesis allowed us to identify MAP17 (Guijarro et al., 2007a). MAP17 is a small non-glycosylated membrane-associated 17 kDa protein that localizes to the plasma membrane and the Golgi apparatus (Blasco et al., 2003). The MAP17 protein sequence contains two transmembrane regions and a hydrophobic amino-terminus encoding a PDZ-binding domain (Jaeger et al., 2000; Figure 1). MAP17 overexpression in carcinomas was first described by using the technique of differential display (Kocher et al., 1995). MAP17 binds several PDZ domain-containing proteins, including NHeRF proteins, NaPi-IIa, and NHe3. Overexpression of MAP17 in opossum kidney cells participates in NaPi-IIa internalization to the trans-Golgi network (Lanaspa et al., 2007). In a transgenic mouse model, MAP17 hepatic overexpression resulted in PDZK1 (NHeRF3) liver deficiency, suggesting that MAP17 is an endogenous regulator of PDZK1 turnover (Silver et al., 2003). MAP17 acts as an atypical anchoring site for PDZK1 and interacts with the NaPi-IIa/PDZK1 protein complex in renal proximal tubular cells (Pribanic et al., 2003). The physiological role of MAP17 in proximal tubules is not known, but it does stimulate specific Na-dependent transport of mannose and glucose in Xenopus oocytes (Blasco et al., 2003) and some human cells (Guijarro et al., 2007a). The MAP17 gene does share regulatory elements with the stem cell leukemic gene (SCL, TAL-1), which encodes a basic Helix-Loop-Helix protein essential in the formation of the hematopoietic lineages (Gottgens et al., 2002; Delabesse et al., 2005). However, both genes show independent regulation (Guijarro et al., 2007c).
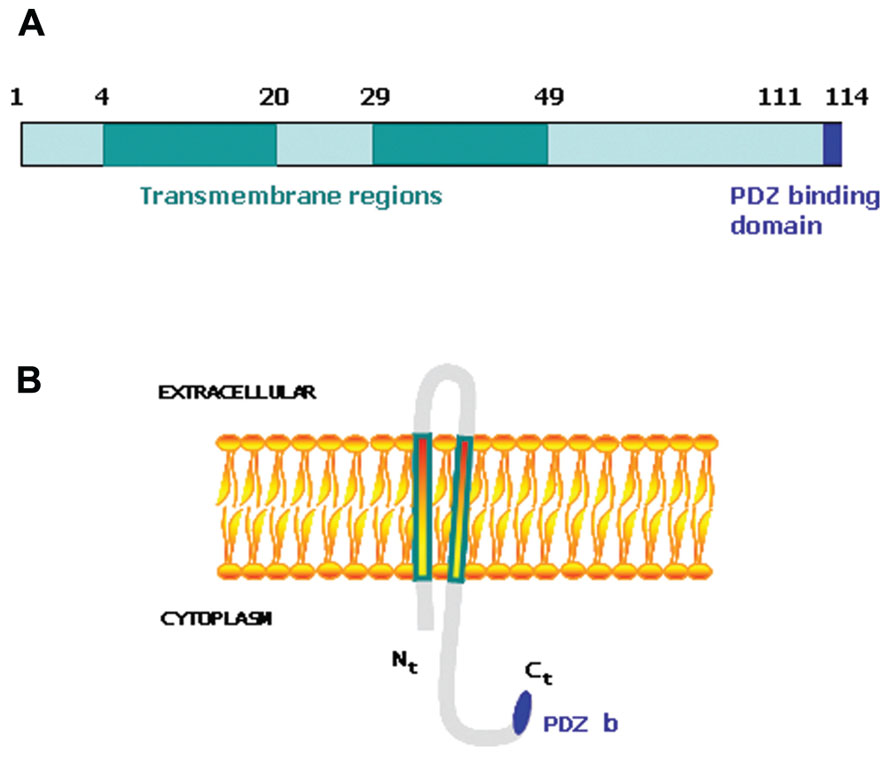
FIGURE 1. (A) Schematic representation of MAP17 protein domains. (B) Schematic representation of MAP17 disposition in the membrane.
Multiple oncogenes that activate signaling pathways directly involved in cell survival or proliferation have been discovered in previous decades. Other genes may provide an advantage to the tumoral cells, making them insensitive to physiological signals or altering their normal physiology. Although activated macrophages destroy cancer cells more effectively than normal cells, the ability to escape activated macrophages is a characteristic of tumor cells. One of the mechanisms responsible for the specific killing of tumor cells by macrophages is the production of the cytokine tumor necrosis factor-alpha (TNF-α). Therefore, resistance to TNF may provide cancer cells with a selective advantage against host elimination. Ectopic expression of MAP17 in tumor cells prevents TNF-induced G1 arrest by impairing p21waf1 induction. However, expression of MAP17 does not inhibit TNF-induced apoptosis in Me180-sensitive tumor cells. The inhibition of TNF is specific because MAP17 does not alter the response to other cytokines such as IFN-α. As described in the Xenopus oocyte system, MAP17 increases the uptake of glucose in some cells, but this effect is not responsible for TNF bypass.
MAP17 in Human Tumors
MAP17 overexpression in carcinomas occurs mostly through mRNA amplification, but promoter activation has also been observed by some oncogenes (Kocher et al., 1995; Guijarro et al., 2007c). Immunohistochemical analysis of MAP17 during cancer progression shows that overexpression of the protein strongly correlates with tumoral progression. Generalized MAP17 overexpression in human carcinomas indicates that MAP17 can be a good marker for tumorigenesis and especially for malignant progression.
MAP17 is highly expressed in renal proximal tubular cells and has been previously described to be associated with carcinomas (Kocher et al., 1995, 1996). We have performed an in-depth analysis of MAP17 overexpression in carcinomas by immunohistochemistry and mRNA expression (Figure 2). We have found that the MAP17 protein is overexpressed in a large percentage of the tumors analyzed and is significantly correlated with the tumor grade in ovarian, breast, and prostate carcinomas (Guijarro et al., 2007c, 2012). The analysis of mRNA levels by Q-PCR or by hybridization comparing tumoral vs. non-tumoral tissues of the same patient, demonstrate an even higher percentage of tumor samples with MAP17 overexpression. In tumors such as ovary, colon, stomach, cervix, and thyroid gland, the percentage of overexpression in tumor samples is higher than 70%, while in lung, uterus, and rectum it is approximately 50%. Although more samples need to be analyzed to confirm these high rates, the data suggest that MAP17 overexpression is the most common marker of tumorigenesis in carcinomas. The relevance of MAP17 as a general marker for the malignant stages of human tumors still needs to be confirmed in additional tumor types and larger cohorts. However, all tissues explored thus far have shown similar patterns of MAP17 expression. Furthermore, MAP17 expression seems to correlate with AKT phosphorylation at Ser473 (Figure 2). These expression patterns provide a mechanistic insight and a possible target for future therapies (AKT inhibition).
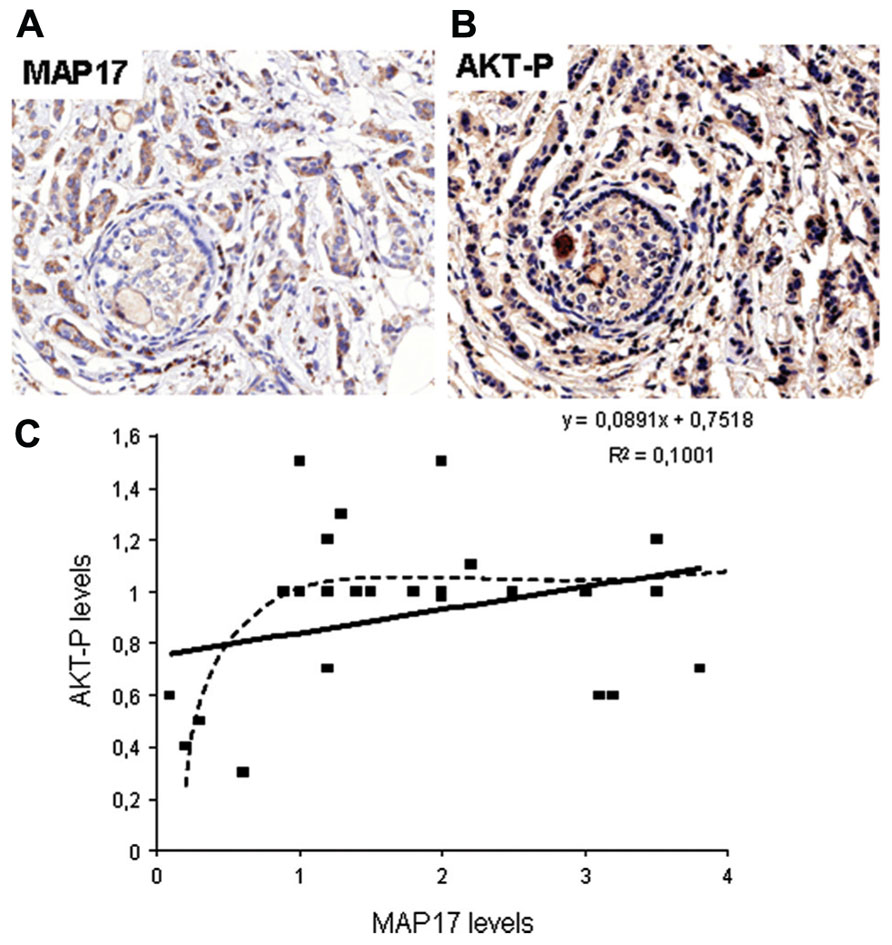
FIGURE 2. (A) Representative picture of human breast tumors overexpressing MAP17. (B) Same tumor sample showing activated AKT (phosphorylated at S473). (C) Correlation between MAP17 expression and AKT activation in breast tumor samples analyzed.
Oncogenic Activity of MAP17
Tumor cells that overexpress MAP17 show an increased tumoral phenotype with enhanced proliferative capabilities both in the presence or absence of contact inhibition, decreased apoptotic sensitivity, and increased migration. MAP17-expressing clones also grow better in nude mice. The increased malignant cell behavior induced by MAP17 is associated with an increase in reactive oxygen species (ROS) production, and the treatment of MAP17-expressing cells with antioxidants results in a reduction in the tumorigenic properties of these cells. Treatment of breast cells with inhibitors of Na+-coupled co-transporters leads to an inhibition of a ROS increase and a decrease in the malignant cell behavior in MAP17-expressing clones (Guijarro et al., 2012). Finally, MAP17-dependent increase in ROS and tumorigenesis are dependent on its PDZ-binding domain because disruption of this sequence by point mutations abolishes the ability of MAP17 to enhance ROS production and tumorigenesis (Guijarro et al., 2007b). Furthermore, expression of a MAP17 specific shRNA in protein-expressing tumor cells reduced their tumorigenic capabilities (Guijarro et al., 2012), which suggests that this effect is dependent upon MAP17 protein expression.
MAP17 significantly decreases the c-Myc induced caspase-3-like activity in Rat1 fibroblasts under low serum conditions. This decrease is in keeping with the concept of MAP17-induced PI3K/AKT signaling, in which MAP17 is able to interfere with Bax translocation to the mitochondria (Guijarro et al., 2007d). At the molecular level we have found that MAP17 protects Rat1a fibroblasts from Myc-induced apoptosis through, ROS-mediated activation of the PI3K/AKT signaling pathway (Guijarro et al., 2007d). A fraction of PTEN protein undergoes oxidation in MAP17-overexpressing cells. Furthermore, activation of AKT by MAP17 as measured by Thr308 phosphorylation was independent of PI3K activity (Figure 3). Importantly, modulation of ROS by antioxidant treatment prevented activation of AKT, thus, restoring the level of apoptosis in serum starved Rat1/c-Myc fibroblasts (Guijarro et al., 2007d). MAP17-mediated survival was associated with an absence of Bax translocation to the mitochondria and reduced caspase-3 activation. Finally, overexpression of a dominant negative mutant of AKT in MAP17-expressing clones makes them sensitive to serum depletion (Guijarro et al., 2007d). The data indicates that MAP17 protein activates AKT through ROS, and this activation is a determinant in conferring resistance to Myc-induced apoptosis in the absence of serum. These results might provide the mechanistic insight to explain the correlation between MAP17 levels and AKT phosphorylation found in tumor samples. Like ways, AKT activation has been described as responsible for TNF resistance in some tumor cell lines (Sudheerkumar et al., 2008; Xu et al., 2012).
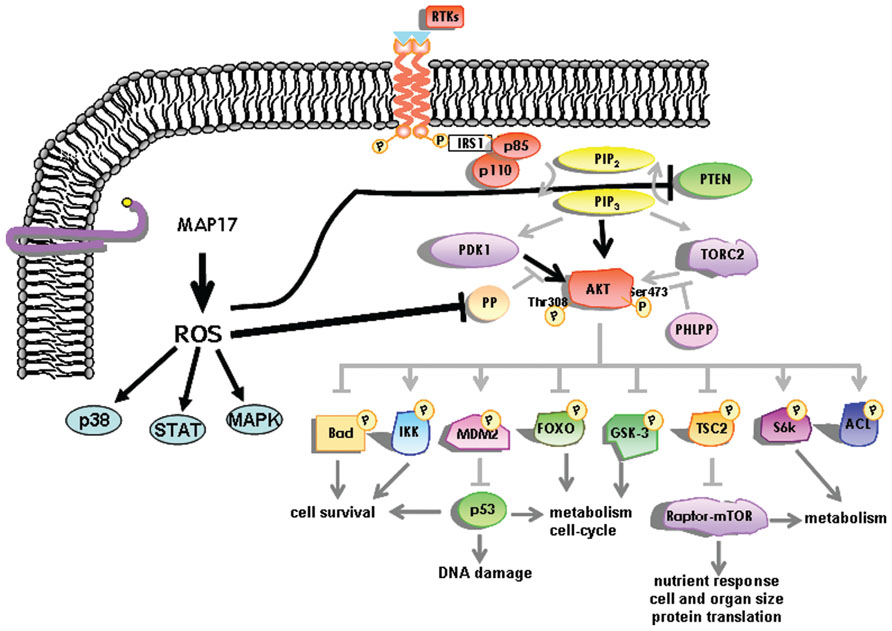
FIGURE 3. Schematic representation of the intracellular pathways activated by MAP17 through ROS. The AKT pathway is represented in more detail.
MAP17 is a Ros-Dependent Oncogene
The increased tumorigenic properties induced by MAP17 are associated with an increase in ROS because MAP17 increases endogenous ROS and the antioxidant treatment of MAP17-expressing cells entails a reduction in the tumorigenic properties of these cells. Two explanations can be offered for the mechanism by which ROS induce the transformed phenotype. First, reactive oxygen generated in the presence of MAP17 may be mutagenic, causing the transformed phenotype through the induction of mutations in oncogenes or tumor suppressor genes. Alternatively, ROS generated in a MAP17-dependent manner might function as an intracellular signal, inducing a growth-related genetic program. We have found that ROS removal by antioxidant treatments decrease the malignant cell behavior induced by MAP17; thus, the second hypothesis is favored. Accumulating evidence implicates ROS in signaling cascades related to cell proliferation and transformation (Sundaresan et al., 1995; Burdon, 1996; Irani et al., 1997). Ras-transformed fibroblasts overproduce ROS, and this overproduction is correlated with the activation of mitogenic signaling pathways (Irani et al., 1997). Loss of superoxide dismutase (SOD; which should elevate ROS levels) has also been correlated with a tumoral phenotype, and overexpression of SOD leads to the reversion of the transformed phenotype (Fernandez-Pol et al., 1982; Church et al., 1993; Yan et al., 1996). On the other hand, H2O2 is generated in response to the growth factors EGF and PDGF and is linked to growth-related signaling (Sundaresan et al., 1995; Bae et al., 1999). When overexpressed in NIH3T3 mouse fibroblasts, Nox1, a NADPH oxidase catalytic subunit, induces excessive production of ROS and a transformed phenotype with increased mitotic rates and aggressive tumor formation in athymic mice (Arnold et al., 2001). The phenotype of Nox1-transfected cells can be reversed by ROS reduction through stable expression of catalase, thereby implicating ROS as a signaling molecule (Arnold et al., 2001).
The cellular targets responsible for growth and transformation affected by ROS signaling are not fully known. DNA microarray experiments (Arnold et al., 2001) indicate that up to 2% of the genes are regulated by ROS. Furthermore, we have found that a ROS increase activates the PI3K pathway, which may be by direct oxidation and inactivation of PTEN and other AKT phosphatases, thus maintaining AKT activation even in the absence of a PI3K signal (Guijarro et al., 2007d). AKT pathway activation induced by MAP17 expression might explain some of the properties described here. However, we hypothesize that other pathways must coexist that are induced by MAP17 at the transcriptional level, as described in other systems (Klaunig et al., 1998; Droge, 2002). The p42/p44 mitogen-activated protein kinase (MAPK), p38 MAPK, p70S6k, AKT, and STAT, signaling pathways are all activated by ROS (Natarajan et al., 1993; Finkel, 1998; Bae et al., 1999; Allen and Tresini, 2000; Ray et al., 2012; Vurusaner et al., 2012). A variety of other targets can also be affected by ROS, including transcription factors such as NF-kB (Schmidt et al., 1995), AP1 (Wenk et al., 1999), and p53 (Hainaut and Milner, 1993). In most cases the activation is indirect (Min et al., 1998; Abe et al., 2000). However, a direct effect has been shown on protein tyrosine phosphatase-1B (PTP-1B), which is inhibited by oxidation of a thiol in the active site (Lee et al., 1998; Barrett et al., 1999), leading to increased phosphotyrosines on many cell proteins. ROS can directly modify signaling proteins through different modifications such as nitrosylation, carbonylation, disulfide bond formation, and glutathionylation (England and Cotter, 2005). Whatever the proximal target(s), ROS reprogram the expression of enzymes and other proteins in the cell (Klaunig et al., 1998; Droge, 2002).
However, the increased tumoral properties of carcinoma cells were not paralleled in immortal non-tumoral cells (Guijarro et al., 2012), indicating that MAP17 provides a selective advantage once tumorigenesis has begun. ROS act as a second messenger that enhances tumoral properties, but only in those cells where the senescence/apoptotic signal provided by ROS is uncoupled. In primary cells, MAP17 triggers a ROS-dependent, senescence-like response that is abolished in the absence of p38a activation. Furthermore, in human breast tumors, MAP17 activation is correlated with a lack of phosphorylation of p38a. Therefore, MAP17 is overexpressed in late-stage breast tumors, in which oncogenic activity relies on p38 insensitivity to induced intracellular ROS (Guijarro et al., 2012).
MAP17 and NHeRFs
MAP17 has been found to bind NHeRF1 and NHeRF3 (PDZK1) through its PDZ-binding motif (Pribanic et al., 2003; Silver et al., 2003; Lanaspa et al., 2007). NHeRFs are scaffolding protein defined by the presence of globular PDZ domains that assemble several proteins into functional complexes (Shenolikar et al., 2004; Cunningham et al., 2010; Claperon et al., 2011). The NHeRF proteins regulate cell surface expression and functional activity of transporters (Shenolikar et al., 2004; Lee et al., 2007). Most transporters identified as binding partners belong to the ABC family (Weinman et al., 2010). In addition to transporters, other proteins have been shown to interact with NHeRF proteins, including signaling proteins, hormone receptors, and cytoskeleton structural elements (Theisen et al., 2007). Many proteins related to the G-protein signaling pathways were found to interact with PDZK1, and they were likely to be functionally associated with transporters (Cardone et al., 2007; Theisen et al., 2007; Carnero, 2012). Furthermore, it has been shown (Dai et al., 2004) that NHeRF1 binds to the breast tumor suppressor SYK and MERLIN, the product of the tumor suppressor NF2. NHeRF1 present also mutations at the PDZ domains in breast tumors which abolishes binding to these suppressor proteins. Primary breast tumors with LoH at the NHeRF1 locus show higher aggressiveness. However, the relation of these mutations with MAP17 or other physiological alterations such as ROS of glucose uptake is at present unknown.
MAP17 form complexes with PDZK1 and NHe3 contributing to basal and calcium inhibition of NH3 activity (Cinar et al., 2007). Recently, it has been shown that PDZK1 regulates PLC β3 (Kim et al., 2012). PDZK1 also regulates the solute carriers PEPT1 (oligopeptide transporter) and OCTN2 (carnitine/organic cation transporter; Sugiura et al., 2008), the cystic fibrosis transmembrane conductance regulator (CFTR; Gentzsch et al., 2003), canalicular multispecific organic anion transport (CMOAT; Inoue et al., 2004), and the anion exchangers of the SLC26A family (Hillesheim et al., 2007). PDZK1 has also been shown to interact with AKAP10, FARP2, sodium–hydrogen antiporter 3 regulator 1, SLC22A12, SLK, SLC22A4, and SLC34A3 (Counillon et al., 2000; Gisler et al., 2003; Shenolikar et al., 2004; Ganapathy et al., 2008). As has been suggested, it is possible that the role of MAP17 is to enhance the endogenous uphill transport system (Blasco et al., 2003; Cardone et al., 2005; Chiche et al., 2010; Parks et al., 2011; Carnero, 2012).
On the other hand, MAP17 has been shown also to increase glucose uptake (Blasco et al., 2003; Guijarro et al., 2007a) thus enhancing glycolysis, contributing to Warburg’s effect and increasing intracellular oxidative stress (Bar-Even et al., 2012; Carnero, 2012). Therefore, MAP17 increase in tumor cells could be a mechanistic advantage that will permit tumor cells increase the glucose intake and in parallel decrease the intracellular pH and lactic acid by the increase of membrane bound transports (Carnero, 2012; Figure 4).
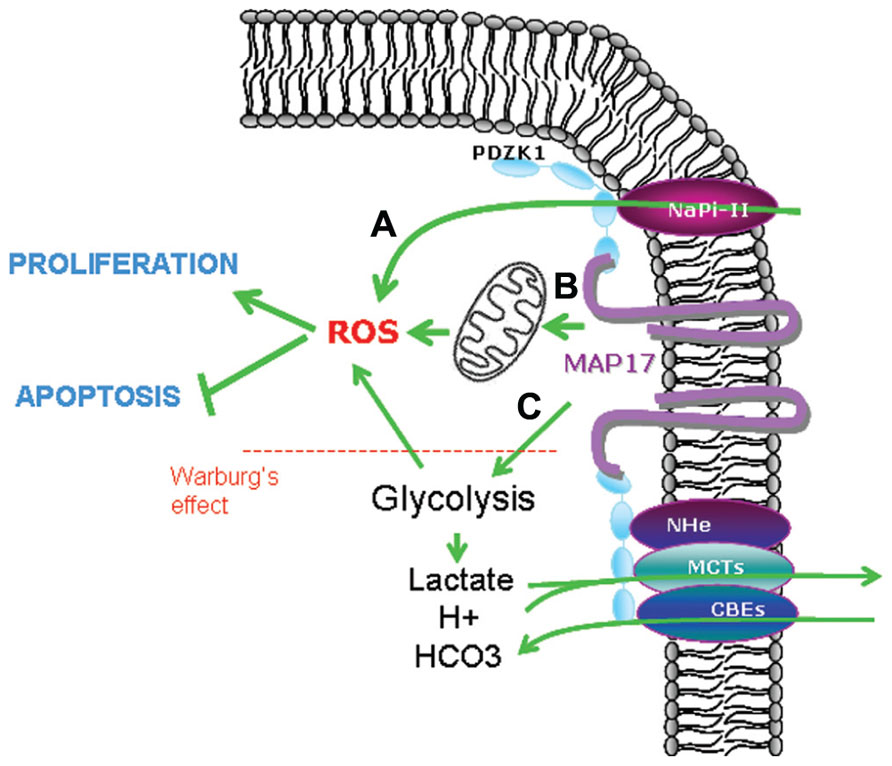
FIGURE 4. Possible mechanisms involved in MAP17-dependent increase of ROS. (A) Direct pH alteration by membrane transports, (B) increase in glucose metabolism through mitochondrial respiration, (C) increase in aerobic glycolysis (Warburg’s effect) which is allowed by acidic detoxification carried out by membrane transports bund to MAP17–NHeRFs complexes.
Concluding Remarks
In summary, MAP17 overexpression in human breast carcinomas indicates that MAP17 can be a good marker for tumorigenesis and for malignant progression. Our results indicate that this protein is likely to play an important role in carcinogenesis.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Spanish Ministry of Science and Innovation and Feder Funds (SAF2009-08605), Consejeria de Innovacion y Ciencia (CTS-6844) and Consejeria de Salud (PI-0142), Junta de Andalucia. Amancio Carnero’s Lab is also funded by a fellowship from Fundacion Oncologica FERO supported by Fundació Josep Botet. The funding source has not had a role in the study design, analysis, or interpretation of data, nor in the writing and decision to submit the manuscript.
References
Abe, J., Okuda, M., Huang, Q., Yoshizumi, M., and Berk, B. C. (2000). Reactive oxygen species activate p90 ribosomal S6 kinase via Fyn and Ras. J. Biol. Chem. 275, 1739–1748.
Allen, R. G., and Tresini, M. (2000). Oxidative stress and gene regulation. Free Radic. Biol. Med. 28, 463–499.
Arnold, R. S., Shi, J., Murad, E., Whalen, A. M., Sun, C. Q., Polavarapu, R., Parthasarathy, S., Petros, J. A., and Lambeth, J. D. (2001). Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. U.S.A. 98, 5550–5555.
Bae, G. U., Seo, D. W., Kwon, H. K., Lee, H. Y., Hong, S., Lee, Z. W., Ha, K. S., Lee, H. W., and Han, J. W. (1999). Hydrogen peroxide activates p70(S6k) signaling pathway. J. Biol. Chem. 274, 32596–32602.
Bar-Even, A., Flamholz, A., Noor, E., and Milo, R. (2012). Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat. Chem. Biol. 8, 509–517.
Barrett, W. C., DeGnore, J. P., Konig, S., Fales, H. M., Keng, Y. F., Zhang, Z. Y., Yim, M. B., and Chock, P. B. (1999). Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 38, 6699–6705.
Blasco, T., Aramayona, J., Alcalde, A., Catalan, J., Sarasa, M., and Sorribas, V. (2003). Rat kidney MAP17 induces cotransport of Na-mannose and Na-glucose in Xenopus laevis oocytes. Am. J. Physiol. Renal Physiol. 285, F799–F810.
Burdon, R. H. (1996). Control of cell proliferation by reactive oxygen species. Biochem. Soc. Trans. 24, 1028–1032.
Cardone, R. A., Bellizzi, A., Busco, G., Weinman, E. J., Dell’Aquila, M. E., Casavola, V., Azzariti, A., Mangia, A., Paradiso, A., and Reshkin, S. J. (2007). The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol. Biol. Cell 18, 1768–1780.
Cardone, R. A., Casavola, V., and Reshkin, S. J. (2005). The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 5, 786–795.
Carnero, A., Hudson, J. D., Hannon, G. J., and Beach, D. H. (2000). Loss-of-function genetics in mammalian cells: the p53 tumor suppressor model. Nucleic Acids Res. 28, 2234–2241.
Chiche, J., Brahimi-Horn, M. C., and Pouyssegur, J. (2010). Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J. Cell. Mol. Med. 14, 771–794.
Church, S. L., Grant, J. W., Ridnour, L. A., Oberley, L. W., Swanson, P. E., Meltzer, P. S., and Trent, J. M. (1993). Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc. Natl. Acad. Sci. U.S.A. 90, 3113–3117.
Cinar, A., Chen, M., Riederer, B., Bachmann, O., Wiemann, M., Manns, M., Kocher, O., and Seidler, U. (2007). NHE3 inhibition by cAMP and Ca2+ is abolished in PDZ-domain protein PDZK1-deficient murine enterocytes. J. Physiol. 581, 1235–1246.
Claperon, A., Mergey, M., and Fouassier, L. (2011). Roles of the scaffolding proteins NHERF in liver biology. Clin. Res. Hepatol. Gastroenterol. 35, 176–181.
Counillon, L., Touret, N., Bidet, M., Peterson-Yantorno, K., Coca-Prados, M., Stuart-Tilley, A., Wilhelm, S., Alper, S. L., and Civan, M. M. (2000). Na+/H+ and CI-/HCO3-antiporters of bovine pigmented ciliary epithelial cells. Pflugers Arch. 440, 667–678.
Cunningham, R., Biswas, R., Steplock, D., Shenolikar, S., and Weinman, E. (2010). Role of NHERF and scaffolding proteins in proximal tubule transport. Urol. Res. 38, 257–262.
Dai, J. L., Wang, L., Sahin, A. A., Broemeling, L. D., Schutte, M., and Pan, Y. (2004). NHERF (Na+/H+ exchanger regulatory factor) gene mutations in human breast cancer. Oncogene 23, 8681–8687.
Delabesse, E., Ogilvy, S., Chapman, M. A., Piltz, S. G., Gottgens, B., and Green, A. R. (2005). Transcriptional regulation of the SCL locus: identification of an enhancer that targets the primitive erythroid lineage in vivo. Mol. Cell. Biol. 25, 5215–5225.
Droge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95.
England, K., and Cotter, T. G. (2005). Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 10, 237–245.
Fernandez-Pol, J. A., Hamilton, P. D., and Klos, D. J. (1982). Correlation between the loss of the transformed phenotype and an increase in superoxide dismutase activity in a revertant subclone of sarcoma virus-infected mammalian cells. Cancer Res. 42, 609–617.
Ganapathy, V., Thangaraju, M., Gopal, E., Martin, P. M., Itagaki, S., Miyauchi, S., and Prasad, P. D. (2008). Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 10, 193–199.
Gentzsch, M., Cui, L., Mengos, A., Chang, X. B., Chen, J. H., and Riordan, J. R. (2003). The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J. Biol. Chem. 278, 6440–6449.
Gisler, S. M., Pribanic, S., Bacic, D., Forrer, P., Gantenbein, A., Sabourin, L. A., Tsuji, A., Zhao, Z. S., Manser, E., Biber, J., and Murer, H. (2003). PDZK1: I. A major scaffolder in brush borders of proximal tubular cells. Kidney Int. 64, 1733–1745.
Gottgens, B., Barton, L. M., Chapman, M. A., Sinclair, A. M., Knudsen, B., Grafham, D., Gilbert, J. G., Rogers, J., Bentley, D. R., and Green, A. R. (2002). Transcriptional regulation of the stem cell leukemia gene (SCL) – comparative analysis of five vertebrate SCL loci. Genome Res. 12, 749–759.
Guijarro, M. V., Castro, M. E., Romero, L., Moneo, V., and Carnero, A. (2007a). Large scale genetic screen identifies MAP17 as protein bypassing TNF-induced growth arrest. J. Cell. Biochem. 101, 112–121.
Guijarro, M. V., Leal, J. F., Blanco-Aparicio, C., Alonso, S., Fominaya, J., Lleonart, M., Castellvi, J., Ramon y Cajal, S., and Carnero, A. (2007b). MAP17 enhances the malignant behavior of tumor cells through ROS increase. Carcinogenesis 28, 2096–2104.
Guijarro, M. V., Leal, J. F., Fominaya, J., Blanco-Aparicio, C., Alonso, S., Lleonart, M., Castellvi, J., Ruiz, L., Ramon Y Cajal, S., and Carnero, A. (2007c). MAP17 overexpression is a common characteristic of carcinomas. Carcinogenesis 28, 1646–1652.
Guijarro, M. V., Link, W., Rosado, A., Leal, J. F., and Carnero, A. (2007d). MAP17 inhibits Myc-induced apoptosis through PI3K/AKT pathway activation. Carcinogenesis 28, 2443–2450.
Guijarro, M. V., Vergel, M., Marin, J. J., Munoz-Galvan, S., Ferrer, I., Cajal, S. R., Roncador, G., Blanco-Aparicio, C., and Carnero, A. (2012). p38α limits the contribution of MAP17 to cancer progression in breast tumors. Oncogene. doi: 10.1038/onc.2011.619 [Epub ahead of print].
Hainaut, P., and Milner, J. (1993). Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res. 53, 4469–4473.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Hannon, G. J., Sun, P., Carnero, A., Xie, L. Y., Maestro, R., Conklin, D. S., and Beach, D. (1999). MaRX: an approach to genetics in mammalian cells. Science 283, 1129–1130.
Hillesheim, J., Riederer, B., Tuo, B., Chen, M., Manns, M., Biber, J., Yun, C., Kocher, O., and Seidler, U. (2007). Down regulation of small intestinal ion transport in PDZK1- (CAP70/NHERF3) deficient mice. Pflugers Arch. 454, 575–586.
Inoue, J., Otsuki, T., Hirasawa, A., Imoto, I., Matsuo, Y., Shimizu, S., Taniwaki, M., and Inazawa, J. (2004). Overexpression of PDZK1 within the 1q12-q22 amplicon is likely to be associated with drug-resistance phenotype in multiple myeloma. Am. J. Pathol. 165, 71–81.
Irani, K., Xia, Y., Zweier, J. L., Sollott, S. J., Der, C. J., Fearon, E. R., Sundaresan, M., Finkel, T., and Goldschmidt-Clermont, P. J. (1997). Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275, 1649–1652.
Jaeger, C., Schaefer, B., Wallich, R., and Kramer, M. (2000). The membrane-associated protein pKe#192/MAP17 in human keratinocytes. J. Invest. Dermatol. 115, 375–380.
Kim, J. K., Kwon, O., Kim, J., Kim, E. K., Park, H. K., Lee, J. E., Kim, K. L., Choi, J. W., Lim, S., Seok, H., Lee-Kwon, W., Choi, J. H., Kang, B. H., Kim, S., Ryu, S. H., and Suh, P. G. (2012). PDZ domain-containing 1 (PDZK1) protein regulates phospholipase C-beta3 (PLC-beta3)-specific activation of somatostatin by forming a ternary complex with PLC-beta3 and somatostatin receptors. J. Biol. Chem. 287, 21012–21024.
Klaunig, J. E., Xu, Y., Isenberg, J. S., Bachowski, S., Kolaja, K. L., Jiang, J., Stevenson, D. E., and Walborg, E. F. Jr. (1998). The role of oxidative stress in chemical carcinogenesis. Environ. Health Perspect. 106(Suppl. 1), 289–295.
Kocher, O., Cheresh, P., Brown, L. F., and Lee, S. W. (1995). Identification of a novel gene, selectively up-regulated in human carcinomas, using the differential display technique. Clin. Cancer Res. 1, 1209–1215.
Kocher, O., Cheresh, P., and Lee, S. W. (1996). Identification and partial characterization of a novel mem-brane-associated protein (MAP17) up-regulated in human carcinomas and modulating cell replication and tumor growth. Am. J. Pathol. 149, 493–500.
Lanaspa, M. A., Giral, H., Breusegem, S. Y., Halaihel, N., Baile, G., Catalan, J., Carrodeguas, J. A., Barry, N. P., Levi, M., and Sorribas, V. (2007). Interaction of MAP17 with NHERF3/4 induces translocation of the renal Na/Pi IIa transporter to the trans-Golgi. Am. J. Physiol. Renal Physiol. 292, F230–F242.
Lee, S. R., Kwon, K. S., Kim, S. R., and Rhee, S. G. (1998). Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372.
Lee, Y. J., Lee, Y. J., and Han, H. J. (2007). Regulatory mechanisms of Na+/glucose cotransporters in renal proximal tubule cells. Kidney Int. Suppl. 72, S27–S35.
Min, D. S., Kim, E. G., and Exton, J. H. (1998). Involvement of tyrosine phosphorylation and protein kinase C in the activation of phospholipase D by H2O2 in Swiss 3T3 fibroblasts. J. Biol. Chem. 273, 29986–29994.
Natarajan, V., Taher, M. M., Roehm, B., Parinandi, N. L., Schmid, H. H., Kiss, Z., and Garcia, J. G. (1993). Activation of endothelial cell phospholipase D by hydrogen peroxide and fatty acid hydroperoxide. J. Biol. Chem. 268, 930–937.
Parks, S. K., Chiche, J., and Pouyssegur, J. (2011). pH control mechanisms of tumor survival and growth. J. Cell. Physiol. 226, 299–308.
Pribanic, S., Gisler, S., Bacic, D., Madjdpour, C., Hernando, N., Sorribas, V., Gantenbein, A., Biber, J., and Murer, H. (2003). Interactions of MAP17 with the NaPi-IIa/PDZK1 protein complex in renal proximal tubular cells. Am. J. Physiol. Renal Physiol. 285, F784–F791.
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990.
Schmidt, K. N., Amstad, P., Cerutti, P., and Baeuerle, P. A. (1995). The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-kappa B. Chem. Biol. 2, 13–22.
Shenolikar, S., Voltz, J. W., Cunningham, R., and Weinman, E. J. (2004). Regulation of ion transport by the NHERF family of PDZ proteins. Physiology (Bethesda) 19, 362–369.
Silver, D. L., Wang, N., and Vogel, S. (2003). Identification of small PDZK1-associated protein, DD96/MAP17, as a regulator of PDZK1 and plasma high density lipoprotein levels. J. Biol. Chem. 278, 28528–28532.
Sudheerkumar, P., Shiras, A., Das, G., Jagtap, J. C., Prasad, V., and Shastry, P. (2008). Independent activation of Akt and NF-kappaB pathways and their role in resistance to TNF-alpha mediated cytotoxicity in gliomas. Mol. Carcinog. 47, 126–136.
Sugiura, T., Kato, Y., Wakayama, T., Silver, D. L., Kubo, Y., Iseki, S., and Tsuji, A. (2008). PDZK1 regulates two intestinal solute carriers (Slc15a1 and Slc22a5) in mice. Drug Metab. Dispos. 36, 1181–1188.
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., and Finkel, T. (1995). Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270, 296–299.
Theisen, C. S., Wahl, J. K. III, Johnson, K. R., and Wheelock, M. J. (2007). NHERF links the N-cadherin/catenin complex to the platelet-derived growth factor receptor to modulate the actin cytoskeleton and regulate cell motility. Mol. Biol. Cell 18, 1220–1232.
Vergel, M., and Carnero, A. (2010). Bypassing cellular senescence by genetic screening tools. Clin. Transl. Oncol. 12, 410–417.
Vurusaner, B., Poli, G., and Basaga, H. (2012). Tumor suppressor genes and ROS: complex networks of interactions. Free Radic. Biol. Med. 52, 7–18.
Weinman, E. J., Biswas, R., Steplock, D., Douglass, T. S., Cunningham, R., and Shenolikar, S. (2010). Sodium-hydrogen exchanger regulatory factor 1 (NHERF-1) transduces signals that mediate dopamine inhibition of sodium-phosphate co-transport in mouse kidney. J. Biol. Chem. 285, 13454–13460.
Wenk, J., Brenneisen, P., Wlaschek, M., Poswig, A., Briviba, K., Oberley, T. D., and Scharffetter-Kochanek, K. (1999). Stable overexpression of manganese superoxide dismutase in mitochondria identifies hydrogen peroxide as a major oxidant in the AP-1-mediated induction of matrix-degrading metalloprotease-1. J. Biol. Chem. 274, 25869–25876.
Xu, X., Tu, L., Jiang, W., Feng, W., Zhao, C. X., and Wang, D. W. (2012). Bradykinin prevents the apoptosis of NIT-1 cells induced by TNF-alpha via the PI3K/Akt and MAPK signaling pathways. Int. J. Mol. Med. 29, 891–898.
Keywords: MAP17, cancer, oncogene, reactive oxygen species, tumorigenesis
Citation: Carnero A (2012) MAP17, a ROS-dependent oncogene. Front. Oncol. 2:112. doi: 10.3389/fonc.2012.00112
Received: 24 May 2012; Accepted: 16 August 2012;
Published online: 06 September 2012.
Edited by:
Carola A. Neumann, Medical University of South Carolina, USAReviewed by:
Olivier Micheau, Institut National de la Santé et de la Recherche Médicale, FranceVilma A. Sardao, Center for Neuroscience and Cell Biology, Portugal
Copyright: © 2012 Carnero. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Amancio Carnero, Instituto de Biomedicina de Sevilla, Hospital Universitario Virgen del Rocío, Consejo Superior de Investigaciones Cientificas, Universidad de Sevilla, Campus Hospital Universitario Virgen del Rocío, Edificio IBIS, Avda. Manuel Siurot s/n, 41013 Sevilla, Spain. e-mail:YWNhcm5lcm8taWJpc0B1cy5lcw==