 Panagiotis A. Konstantinopoulos
Panagiotis A. Konstantinopoulos Ursula A. Matulonis
Ursula A. Matulonis- Medical Gynecologic Oncology Program, Department of Medical Oncology, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
Epithelial ovarian cancer (EOC) is the most lethal gynecologic malignancy and the fifth most common cause of female cancer death in the United States. Although important advances in surgical and chemotherapeutic strategies over the last three decades have significantly improved the median survival of EOC patients, the plateau of the survival curve has not changed appreciably. Given that EOC is a genetically and biologically heterogeneous disease, identification of specific molecular abnormalities that can be targeted in each individual ovarian cancer on the basis of predictive biomarkers promises to be an effective strategy to improve outcome in this disease. However, for this promise to materialize, appropriate preclinical experimental platforms that recapitulate the complexity of these neoplasms and reliably predict antitumor activity in the clinic are critically important. In this review, we will present the current status and evolution of preclinical models of EOC, including cell lines, immortalized normal cells, xenograft models, patient-derived xenografts, and animal models, and will discuss their potential for oncology drug development.
Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynecologic malignancy and the fifth most common cause of female cancer death in the United States (1). Advanced stage at diagnosis for most women with this cancer and emergence of resistance to conventional chemotherapy are primarily responsible for this dire outcome. Although important advances in surgical and chemotherapeutic strategies over the last three decades have significantly improved the quality of life and median survival of EOC patients, the overall cure rate has not improved appreciably (2–5). EOC is a genetically and biologically heterogeneous disease and is traditionally divided into two types (types I and II) with distinct genotypic and phenotypic characteristics which are summarized in Table 1 (6–8). Type I tumors frequently harbor somatic mutations in KRAS, BRAF, PIK3CA, PTEN, CTNNB1, and ARID1A genes, and exhibit low genomic instability without genome-wide copy number changes (9) while type II tumors are characterized by high degree of genomic instability with high frequency of DNA copy number changes and p53 mutations (6, 7, 10).
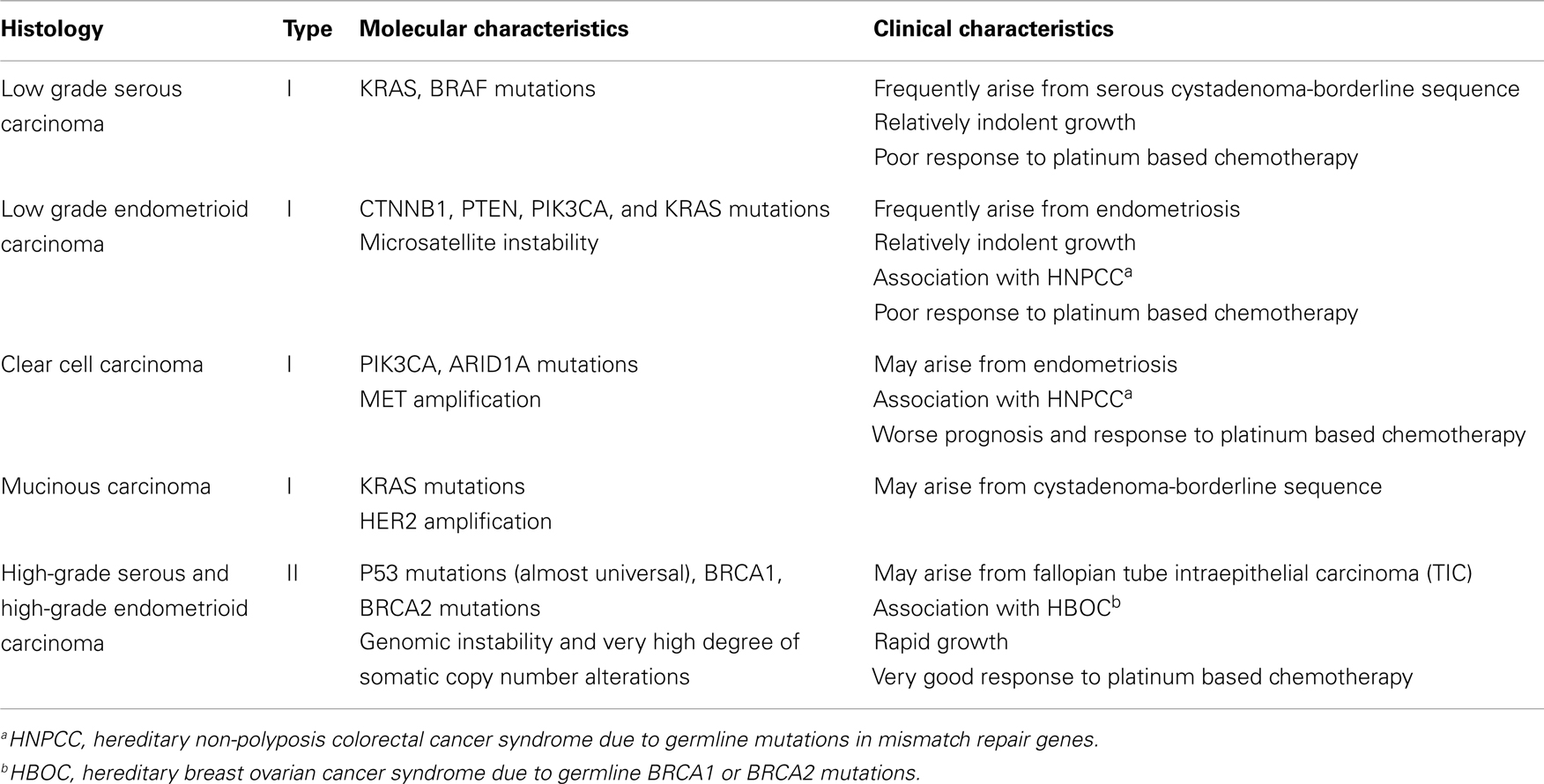
Table 1. Molecular and clinical characteristics of EOC subtypes.
High-grade serous carcinomas (HGSCs) represent the most common type II histologic subtype and account for approximately 70% of all EOCs. These tumors exhibit histological features that are identical to those of primary peritoneal and fallopian tube serous cancers and are treated similarly to these neoplasms. A number of molecular studies and most recently The Cancer Genome Atlas (TCGA) project have shown that HGSCs are characterized by frequent genetic and epigenetic alterations in gene members of the homologous recombination (HR) DNA repair pathway, including the BRCA1 and BRCA2 genes (10). Furthermore, the NOTCH, FOXM1, RB, and PI3K/RAS signaling pathways have also been implicated in the pathogenesis of HGSCs (10). These important advances in our understanding of the molecular pathogenesis and heterogeneity of EOC hold promise for the development of novel therapies against these tumors. However, for this promise to materialize, appropriate preclinical experimental platforms that recapitulate the complexity of these neoplasms and reliably predict antitumor activity in the clinic are critically important. In this review, we will discuss the current status and evolution of preclinical models of EOC focusing on their potential for oncology drug development.
Cell Lines
Historically, ovarian cancer cell lines have been the most frequently used tumor models to prescreen experimental anticancer agents in vitro and to select specific histologic subtypes of EOC for further exploration of these agents. These cell lines have undergone a high degree of evolutionary selection pressure in vitro as they have been in passage for several years (or even decades in some cases). As a result, their genomic profiles have been irreversibly altered and rarely recapitulate the genetic and pathologic characteristics of the parental cells (11–13). Furthermore, cancer cell lines lack the molecular heterogeneity of the parental tumor and are molecularly skewed toward affinity to grow in monolayers.
In a recently published study, Domcke and colleagues used available molecular profiles (copy number changes, mutations, and mRNA expression profiles) of cell lines from the Cancer Cell Line Encyclopedia (CCLE) and of tumor samples from the TCGA to evaluate the suitability of 47 EOC cell lines as in vitro models of HGSCs (14). The investigators showed significant differences in the molecular profiles between commonly used EOC cell lines and HGSC samples and reported that the presumed histologic subtype for several of these cell lines did not correspond to their molecular profiles. Of note, the two most frequently used cell lines, SKOV3, and A2780 were deemed unsuitable as HGSC models, while other rarely used cell lines such as KURAMOCHI, OVSAHO, and SNU119 closely resembled the molecular profiles of HGSC samples. Interestingly, the suitability of these cell lines as HGSC models did not correlate with time of their derivation suggesting that number of passages may not correlate with model suitability. Among the cell lines deemed most suitable to use as HGSC models, three cell lines harbored BRCA mutations i.e., KURAMACHI (BRCA2), COV362 (BRCA1), and JHOS2 (BRCA1) and therefore may be useful as in vitro models for BRCA-associated EOC.
This study may provide molecular explanation for the challenges of translating preclinical observations from ovarian cancer cell lines into the clinic, a problem that is not unique to ovarian cancer but transcends multiple tumor types (14, 15). However, this study also highlights that certain EOC lines may still hold value as HGSCs models and underscores the importance of evaluating and screening them to confirm their origin and molecular resemblance with HGSC. This is now feasible given the increasing availability of large scale genomic data from studies such as the TCGA, the CCLE, and the Sanger Cancer Cell Line project (10, 16). Cell line models whose molecular identity has been confirmed using targeted sequencing and copy number profiling may be extremely valuable as preclinical models, particularly those with well defined molecular alterations such as BRCA1/2 or PI3K mutations in order to assess the potential of experimental drugs in patient populations with specific molecular alterations. In this regard, the promise of PARP inhibitors in the management of BRCA-deficient EOC was first realized in BRCA1/2 deficient cell lines (17, 18). In the era of advanced molecular profiling, using cell lines with molecular similarities with patient samples may increase the possibility that in vitro observations will be eventually translatable to the clinic.
Immortalized Normal Cells and Stem Cells
Several investigators have reported isolation, in vitro propagation and immortalization of human ovarian surface epithelial (OSE) and fallopian tube epithelial (FTE) cells which are considered the cells of origin of ovarian carcinomas. Retroviral transduction of either the human papilloma virus E6/E7 oncogenes or the simian virus 40 T-Antigen (SV40-TAg) in human OSE cells leads to increased and sustained proliferation even after multiple passages but does not induce transformation (19, 20). For immortalization to occur, additional retroviral constructs targeting TP53, hTERT, or RB are required (21, 22). Besides retroviral transduction, RNA interference technology has been successful in immortalizing human OSE cells as exemplified by the work of Yang and colleagues who successfully immortalized OSE cells via siRNA knockdown of p53 and Rb (23, 24). As with human OSE cells, Karst and colleagues immortalized normal human FTE cells via retroviral transduction of hTERT and either of SV40-TAg or an shRNA targeting p53 and mutant CDK4R24C, while transformation occurred via further ectopic expression of either MYC or HRAS oncogenes (25). When injected in immunocompromised mice, these cells developed tumors resembling HGSCs both histologically and clinically. Shan and colleagues used a similar approach of hTERT and SV40-TAg overexpression for immortalization and of additional ectopic HRAS expression for transformation of human FTE cells while similar results have been reported by Jazaeri and colleagues (26, 27).
Although presence of ovarian cancer stem cells has been reported, definite characterization of these cells is still lacking (28). Furthermore, the stem cell niche of the OSE which regenerates after each ovulation has not been determined. There have been several reports of ovarian cancer stem cells isolation which have been based on markers and protocols used to define stem cells in other tumors including leukemia, colon, and breast cancers (29–31). In a seminal study, Flesken-Nikitin and colleagues proposed that the hilum region of the mouse ovary is a stem cell niche of the OSE (32). Specifically, the investigators showed that hilum cells express stem cell markers ALDH1, LGR5, LEF1, CD133, and CK6B, display long-term stem cell properties ex vivo and in vivo and exhibit increased transformation potential after inactivation of TP53 and RB1.
Xenografts
Xenograft models have been extensively used in ovarian cancer research and are still very important experimental platforms for preclinical drug development (33–36). These models require use of immunodeficient mice strains, i.e., athymic nude mice lacking T lymphocytes, severe combined immunodeficient (SCID) mice which lack functional B and T lymphocytes, or the NOD/SCID/IL2Rγnull mice which also exhibit inactive innate immunity due to abrogation of maturation of natural killer (NK) T cells (37). The requirement of immunodeficiency has often been cited as one of the main reasons why xenografts have shown limited predictive value in the clinic (38, 39). Specifically, tumor xenografts in immunocompromised mice cannot recapitulate either the contributions of immune factors on tumor development and progression or the extensive interactions of the human host tumor microenvironment (stroma, extracellular matrix, and vasculature) with the tumor cells.
Traditionally, xenograft models rely on implantation of established EOC cell lines subcutaneously, intraperitoneally, or orthotopically. Subcutaneous implantation offers the advantage of easy quantification of tumor volume which is ideal for assessing antitumor efficacy of experimental agents, but rarely results in ascites formation or intraperitoneal (IP) seeding of the tumor, and thereby fails to reflect the clinical course of human EOC. Conversely, IP and orthotopic implantation (OI) frequently result in peritoneal carcinomatosis and development of malignant ascites. The most commonly used xenograft model in ovarian cancer was developed by IP injection of a subpopulation of the drug resistant cell line NIH:OVCAR-3 (40) (isolated by serial in vitro and in vivo selection of cells) into athymic mice which resulted in development of ascites and peritoneal carcinomatosis (33). The NIH:OVCAR-3 cell line has been molecularly ranked as possibly of HGSC origin on a rank of likely, possibly and unlikely, and this xenograft model is still widely used today (14). The OVCAR-3 and other xenograft models have been used in the preclinical evaluation of antiangiogenic agents (41, 42). Specifically, these models demonstrated the ability of a monoclonal antibody (mAb) to human vascular endothelial growth factor (VEGF) to prevent ascites formation and that combination therapy with inhibitors of VEGF plus paclitaxel exhibits synergistic reduction of tumor growth and ascites in ovarian cancer. These observations were subsequently confirmed in clinical trials of bevacizumab as single agent and in combination with paclitaxel in EOC (43–45).
Orthotopic implantation involves injecting EOC cells into their natural position adjacent to the ovaries which in mice corresponds to the ovarian bursa, a thin membrane that encapsulates the ovaries (46). OI is usually accomplished by direct injection within the ovarian bursa via the infundibulum (47, 48). OI recapitulates initiation of EOC growth in the ovaries, does not require selection of EOC cell lines, and preserves tumor histology and the potential for peritoneal dissemination and ascites formation. Furthermore, several studies have indicated increased tumor take rates with OI thereby reflecting a more favorable microenvironment for tumor growth and metastatic dissemination (48, 49). Unlike subcutaneous xenografts, orthotopic and IP xenografts pose a challenge for accurately quantifying tumor volume and monitoring disease progression thus making them less appealing as models for preclinical drug development. However, this challenge may be overcome by advances in non-invasive imaging of tumors in mice [magnetic resonance imaging (MRI), ultrasound (US), positron emission tomography (PET), computed tomography (CT), and single photon emission computed tomography (SPECT)] and/or use of fluorescent or bioluminescent reporters with optical imaging [fluorescent imaging (FLI) or bioluminescent imaging (BLI)] and/or use of serum tumor biomarkers such as CA125 (50).
Patient-Derived Xenografts
Patient-derived xenografts (PDXs) represent an evolution of the cell line xenograft model whereby fresh tumor tissue, obtained directly from patients, is implanted subcutaneously or orthotopically into immunodeficient mice (51, 52). After a variable period of time, PDXs enter a logarithmic growth phase which allows for harvesting and reimplantation in successive mice generations with reported tumor engraftment rates higher than 75% (53–55). The time to engraftment depends on the individual tumor, the site of implantation and the type of immunodeficient mice used (NOD/SCID/IL2Rγnull mice are associated with superior engraftment efficiency) and is generally between 2 and 4 months. PDXs have been successfully established from primary or metastatic tumors (56, 57), from untreated or heavily pretreated tumors (58, 59) thereby potentially capturing chemotherapy-refractory tumor populations and permitting the study of molecular changes that occur at the time of development of resistance.
A growing body of literature suggests that PDXs hold significant promise as models for preclinical drug development because they closely resemble and recapitulate tumor growth in humans (Table 2). In a seminal study by Hidalgo and colleagues, the investigators treated PDXs from 14 patients with various advanced solid tumors with 63 drugs in 232 treatment regimens, and showed that there was an excellent correlation between response in the PDX models and patient response to these regimens (60). Of note, in some cases, the treatment administered to patients based on the PDX response was not the first choice of the oncologist treating these patients. This study highlights the potential of PDXs as experimental platforms for preclinical drug development. PDXs represent significant improvement over the standard cell line xenografts because they maintain the principal characteristics of the original patients’ tumors including histology, mutational status, DNA copy number changes, gene-expression patterns and clinical behavior while they remain biologically stable when passaged in mice. Specifically, genome-wide expression analysis in non-small cell lung cancer has demonstrated that PDXs exhibit similar gene-expression profiles and maintain the key gene and pathway activity of the primary tumors (61). Furthermore, mutational and expression analysis in pancreatic PDXs has shown that there is excellent concordance between primary tumors and PDX models (62). Several studies have also shown that PDXs maintain their molecular similarity (histology, protein expression, tumor biomarkers, genomic, and genetic status) with the primary tumors during sequential passage (63–65). This molecular similarity is even higher when PDX models are generated using patient tumors that are immediately implanted into immunocompromised mice without an intermediate in vitro culture step (66, 67). Another key feature of PDXs is the maintenance of the original tumor architecture and histopathological characteristics, including a component of human stroma as well as tumor microvasculature although there is a controversy over how long this is maintained. Specifically, in one study of pancreatic PDXs, vessels with human endothelial cells were maintained or even increased over time while in a similar study with renal cell cancer PDXs, a decrease in human-derived tumor microvasculature was observed (68, 69). Of note, maintenance of human tumor-associated leukocytes including memory T cells for up to 9 weeks after implantation has been reported in lung cancer PDXs implanted into NOD/SCID/IL2Rγnull mice. Furthermore, preservation of pluripotent CD133+ stem cells in PDXs following repeated orthotopic subtransplantations has been reported and in these studies the CD133+ cells continued to exhibit multi-lineage differentiation capacity in vitro (70–73). PDXs (particularly early passage PDXs) may therefore be excellent preclinical platforms to study stromal-tumor interactions and cancer stem cell biology as well as to assess novel anticancer agents or drug combinations.
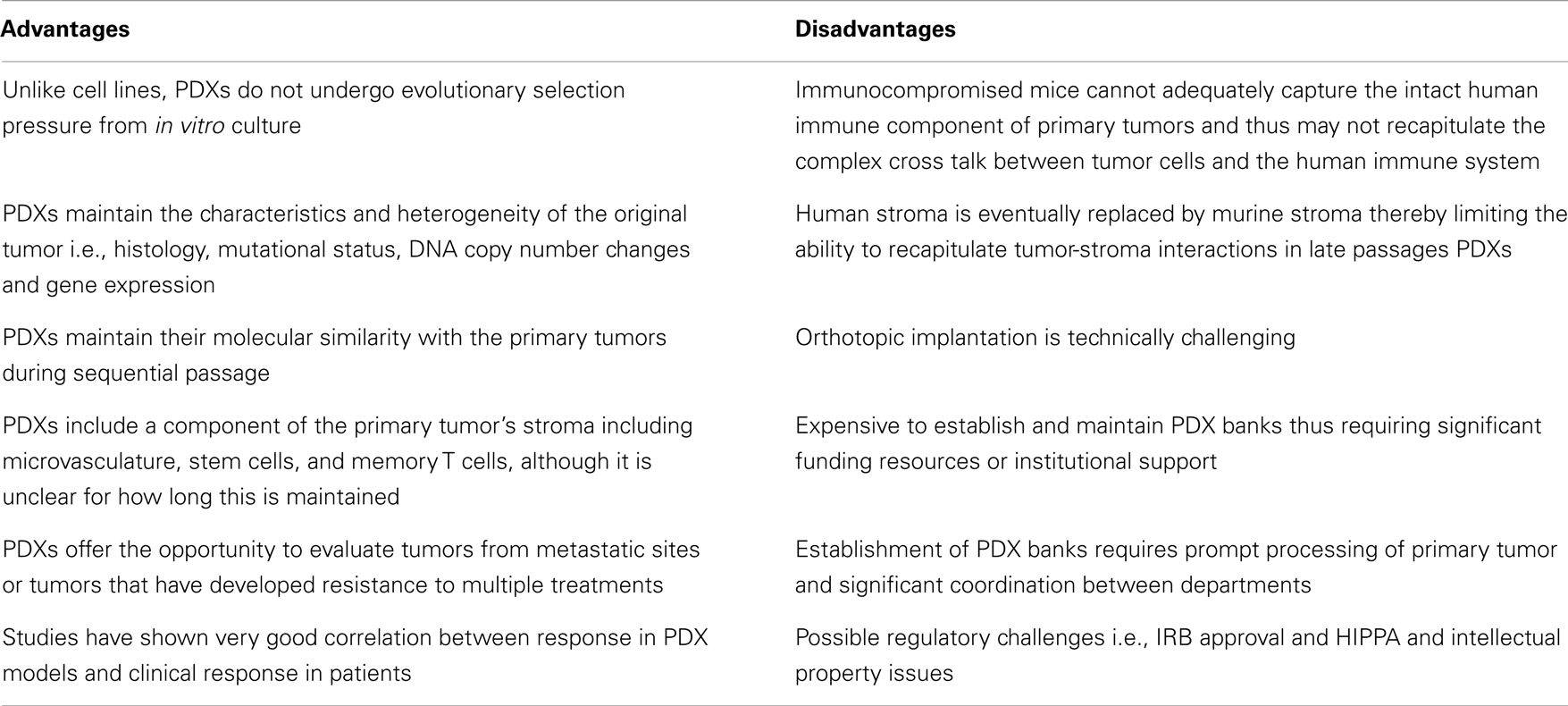
Table 2. Advantages and disadvantages of PDX models.
Several limitations of PDXs exist (Table 2). A major limitation of PDXs is the requirement to use immunodeficient mice which limits the number of drugs that can be evaluated (i.e., alternative models are necessary for immune-modulating agents) (74, 75). Furthermore, severely immunocompromised mice cannot adequately capture the intact human immune component of the primary tumors and thus may not recapitulate the complex cross talk between tumor cells, stroma, and the human immune system. One approach to circumvent this problem may be transplantation of human CD34+ cord blood cells enriched for human hematopoietic stem cells that may reconstitute a human innate and adaptive immune system in mice (76). However, development of PDX models in mice with a reconstituted human immune system is technically challenging and would require that the xenografted tumors and the human immune cell component are HLA matched. Furthermore, the eventual replacement of human stroma by murine stroma is an important disadvantage of PDX models given the importance of tumor-stroma interactions in mediating drug response and resistance. Therefore drugs that target the tumor-stroma or microvasculature such as antiangiogenic agents may also require alternative models for evaluation. Murine models are also known to be imperfect models of drug metabolism and distribution in humans. For example, an overestimation of response may occur when drugs are tolerated at higher doses in mice while an underestimation may occur when mice are less tolerant to drugs compared to humans. There also several logistic challenges including financial and personnel resources that are necessary to establish and maintain PDX banks and the ability to freeze and reestablish tumors after months of storage. Compared to the inexpensive cell line experiments, the cost burden of PDX tumor models is substantial and will likely require significant institutional and national funding to support widespread use of PDXs as experimental models.
In EOC, Kolfschoten and colleagues have reported development of a panel of 15 human ovarian cancer xenografts (12 from fresh tumor derived from patients and 3 from EOC cell lines) grown subcutaneously in the flank of athymic nude mice (77). They assessed the sensitivity of these xenografts to six commonly used anticancer agents and showed that their panel reflected the response rates known for similar drugs in ovarian cancer patients. This study, together with several analogous studies in other tumor types, suggests that PDXs may be used for drug screening in EOC. In our institution, in collaboration with the Belfer Institute of Applied Cancer Research we have embarked on building a platform of ovarian cancer PDXs. The goal of this project is to provide a resource for evaluating efficacy of experimental agents and to identify novel predictive and pharmacodynamic biomarkers. Ovarian cancer cells taken from consented patients are implanted intraperitoneally into immunodeficient mice and these tumors grow and disseminate in the peritoneal cavity similar to human EOC (manuscript in progress, personal communication, Joyce Liu). In order to accurately quantify tumor growth and assess response to experimental therapies, ovarian cancer cells derived from the initial passages are tagged with luciferase and reimplanted into mice for non-invasive BLI. In addition, surrogate biomarkers such as CA125 are evaluated in each of the models to monitor response to therapy.
In the era of personalized medicine, patient-centric PDX models for tumor growth and assessment of drug efficacy may be a valuable resource for the preclinical development of experimental anticancer agents. However, as in the case of cell lines, periodic molecular assessment of these models examining the fidelity to the patients’ original tumors in terms of genetics and histology, two factors that are major determinants of their eventual predictive ability.
Animal Models
Spontaneous EOC models including the aging hen, the cynomolgus macaque, and the rhesus macaque are rarely used in preclinical drug development due to their low incidence rates and long interval until cancer development (78–80). However, because of its anatomic resemblance to humans, the cynomolgus macaque has been occasionally used to evaluate novel agents such as chimeric antibodies or antibody-cytotoxic conjugates (81, 82). Similar to spontaneous EOC models, chemically or hormonally induced models of EOC are rarely used because their histopathological features are not always predictable and their individual molecular alterations are not well defined (83). Conversely, genetically engineered animal models may be promising platforms for preclinical drug development and will be reviewed below (48, 84).
Virus-Mediated Gene Delivery
The first successful mouse model of EOC using a retroviral gene delivery system was reported in 2002 by Orsulic and colleagues (85) who isolated OSE cells from transgenic mice which carried the avian tumor virus receptor A (TVA) under the transcriptional control of the b-actin or keratin 5. Using this TVA retroviral delivery system, they infected OSE cells from TVA; p53−/− mice with any combination of two or three of the c-MYC, KRAS, and AKT oncogenes, and reimplanted them in the TVA; p53−/− mice resulting in rapid formation of tumors 8 weeks later. The resulting tumors exhibited poorly differentiated histology with areas of papillary structures resembling HGSCs. This model was subsequently used to assess sensitivity to molecular pathway inhibitors; for example tumors with AKT and c-MYC oncogenes or AKT and KRAS were sensitive to mTOR inhibitor rapamycin while tumors with all three oncogenes (KRAS, c-MYC, and AKT) were resistant to rapamycin but sensitive to a combination of mTOR inhibitor and MEK inhibitor (i.e., rapamycin and PD98059). These experiments highlight how such models may be used to test the efficacy of molecular targeted agents in EOC. A similar experimental strategy was also employed for development of a BRCA1-associated EOC model whereby expression of c-MYC resulted in transformation of BRCA1 and p53 deficient murine OSEs (86). When implanted intraperitoneally in mice, these cells developed tumors with several characteristic of BRCA1-associated HGSCs, i.e., papillary architecture, peritoneal carcinomatosis, development of malignant ascites, and enhanced sensitivity to cisplatin.
Transgenic Models
A transgenic EOC model was developed by Connolly and colleagues (87) by expressing the early region of SV40-TAg under the transcriptional control of Mullerian Inhibitory Substance Receptor II (MISRII). Fifty percent of the transgenic founder mice developed very aggressive tumors (poorly differentiated carcinomas with rapid development of peritoneal carcinomatosis and ascites) but none of them were fertile. In a subsequent report (88), the same group reported a stable transgenic line from a male transgenic founder (TgMISRII-Tag-DR26) whereby all female offsprings developed bilateral EOCs resembling HGSCs. This is the first transgenic model of HGSC and it has been used for evaluation of experimental agents in clinical trials (89).
Conditional Models
Genetically engineered mouse models using conditional expression of tumor suppressor genes via Cre-recombinase-mediated excision of LoxP flanked sequences have been reported extensively in ovarian cancer literature. Given that there are currently no transgenic mice that express Cre-recombinase only in ovarian epithelial cells, localized delivery of recombinant adenovirus expressing Cre-recombinase in the ovarian bursa of mice is required to achieve Cre-LoxP-mediated gene inactivation solely in the ovarian epithelium. Flesken-Nikitin and colleagues (90) first reported intrabursal administration of Ad-Cre for conditional inactivation of p53 and Rb in p53LoxP/LoxP; RbLoxP/LoxP mice which resulted in ovarian tumor formation in 97% of them (39% low grade serous, 45% poorly differentiated, and 15% undifferentiated carcinomas). Peritoneal carcinomatosis and ascites were present in 27 and 24% of the cases respectively. Dinulescu and colleagues (91) developed the first model of endometrioid EOC by conditional expression of an activating KRAS mutation and inactivation of PTEN via intrabursal administration of Ad-Cre in LoxP-Stop-LoxP-KRASG12D/+; PTENLoxP/LoxP mice. Endometrioid EOCs developed in all mice as early as 7 weeks after injection and were associated with ascites, peritoneal carcinomatosis, and lymph node involvement. Endometrioid EOCs also developed in PTENLoxP/LoxP; APCLoxP/LoxP mice after conditional inactivation of PTEN and APC using intrabursal injection with Ad-Cre (92). These tumors had short latency, 100% penetrance and were associated with peritoneal carcinomatosis and ascites in 21 and 76% of the cases. Importantly, the gene-expression profiles of these tumors closely resembled those of human endometrioid EOCs, particularly those with mutations in the Wnt/b-catenin and PI3K/PTEN pathways suggesting that these models may be promising preclinical experimental platforms for evaluation of novel anticancer agents for these tumors. Another conditional model was reported by Kinross and colleagues (93) whereby intrabursal administration of Ad-Cre for conditional activation of the PI3KCA-H1047R mutation and inactivation of PTEN resulted in ovarian serous adenocarcinomas and granulosa cell tumors.
Finally, a HGSC model was reported by Kim and colleagues (94) by conditionally deleting DICER, a key gene for microRNA synthesis, and PTEN using anti-Mullerian hormone receptor type 2-directed Cre (Amhr2-Cre). HGSCs developed from the fallopian tube in DICERLoxP/LoxP; PTENLoxP/LoxP; Amhr2cre/+ mice and spread to encapsulate the ovaries and then metastasize throughout the abdominal cavity killing all mice by 13 months. These fallopian tube HGSCs exhibited molecular similarity with human high-grade serous ovarian cancers suggesting that they may be used as preclinical models for drug development. Interestingly, removal of fallopian tubes but not of the ovaries prevented cancer formation confirming the fallopian tube origin of these cancers and providing further support to the hypothesis that the fallopian tube is the primary origin of high-grade serous ovarian cancer (95).
Limitations of Animal Models for Preclinical Evaluation of Experimental Agents
Although certain genetically engineered mouse models of EOC mimic the origin, histopathology, clinical behavior (peritoneal carcinomatosis, ascites formation, lymph node involvement, and sensitivity to platinum), and molecular fingerprints (gene-expression profiling and mutational events) of EOC, there are several limitations of these models particularly relevant to their use for preclinical evaluation of novel anticancer agents (84). The most significant challenge is the species-specific differences between humans and mice. Telomerase is active in most mouse cells (unlike human cells where it is inactive) and therefore mice tumors require fewer genetic alterations for malignant transformation compared to human tumors. Mouse telomerase activity prevents adequate modeling of the genomic instability of human tumors, particularly of HGSCs which are characterized by high degree of genomic instability. Furthermore, fundamental differences in drug metabolism (protein binding, metabolic rate, and pathways of metabolism) between mice and humans represent a major challenge when mouse models are used for preclinical testing.
Another issue is that mouse models rely on specific oncogenes and tumor suppressor genes while ignoring other aspects of tumor development such as the host immune system and the tumor microenvironment. Due to the limited number of genetic alterations that induce the development of mouse tumors, mouse models are relatively homogeneous and thus may not adequately recapitulate the significant molecular heterogeneity of human tumors which is an essential element of a good preclinical model. Finally, logistical issues including cost, technical challenges in generating GEM models especially GEMs with multiple genetic alterations, long interval until development of tumors and variable penetrance are important limitations of GEM models for preclinical evaluation of novel anticancer drugs.
Conclusion
Despite significant advances in surgical and medical management, EOC remains a highly lethal malignancy for which new therapeutic strategies are urgently needed. Appropriate experimental platforms that recapitulate the complexity of these tumors are critically important for evaluation of novel therapeutics. Table 3 presents the cell/animal models used for preclinical evaluation of selected experimental agents in EOC and shows the outcome of clinical phase II/III evaluation of these agents. In the first two cases (antiangiogenic agents and PARP inhibitors), cell lines and xenograft models successfully predicted the activity of these agents in phase II/III clinical trials, while in the case of anti-CA125 antibodies and anti-HER-2 agents, preclinical evaluation did not correlate with their phase II/III evaluation. These examples highlight the challenges of preclinical evaluation of novel agents in EOC and underscore the need for appropriate preclinical platforms for a wide variety of experimental agents, i.e., immunotherapies, targeted agents, etc.
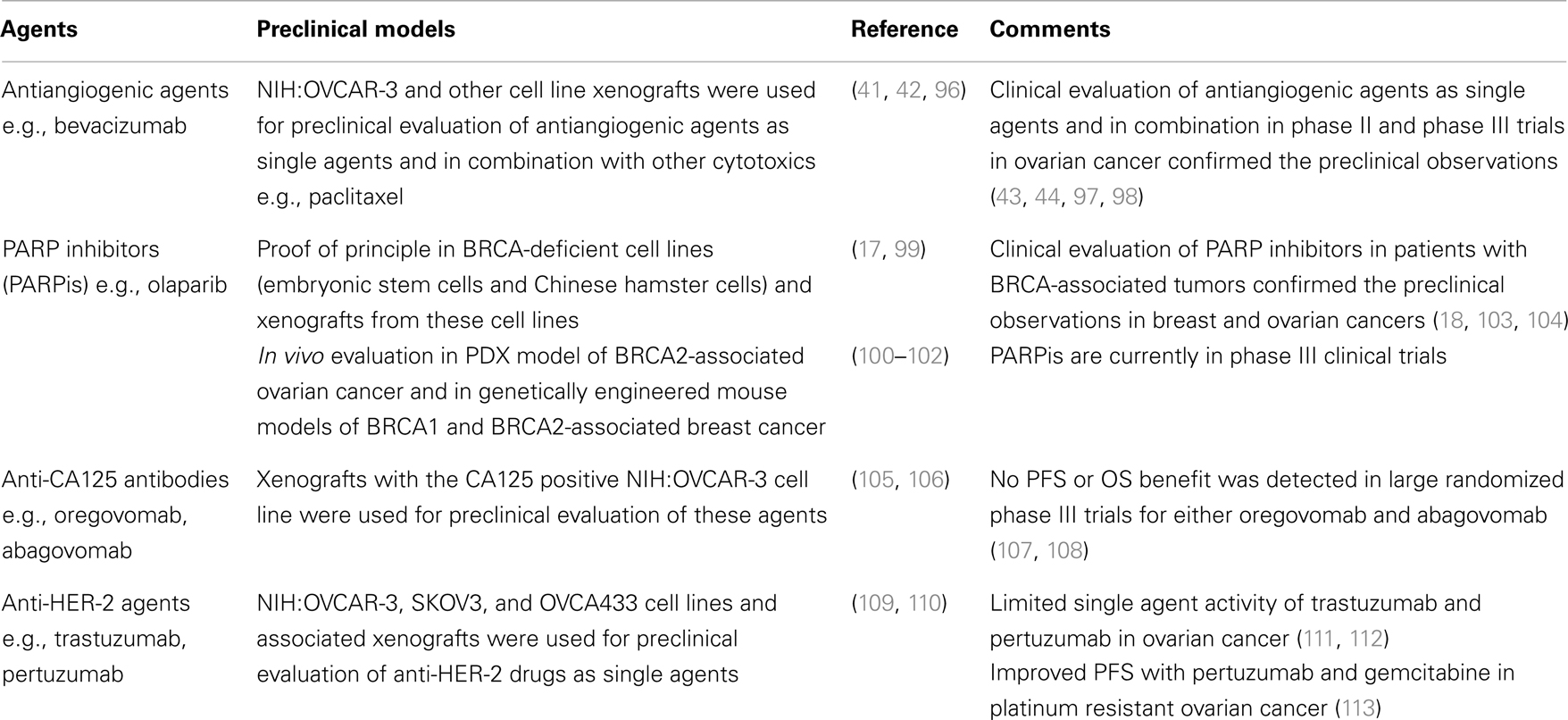
Table 3. Preclinical evaluation of selected experimental agents used against EOC.
In conclusion, cell lines with confirmed molecular identity using targeted sequencing and copy number profiling may be extremely valuable as in vitro models, particularly those with well defined molecular alterations such as BRCA1/2 or PI3K mutations. Xenograft models of established EOC cell lines are still commonly used in preclinical drug development, but are increasingly giving place to PDXs which offer the important advantage of closely resembling original patients’ tumors and adequately capturing the molecular and intratumoral heterogeneity of the original tumors. Finally, genetically engineered mouse models hold promise as they may mimic all major elements of human EOCs including stromal-tumor interactions without the requirement of an immunodeficient background. Clearly, there is no one best preclinical EOC model. Rather, preclinical evaluation of experimental anticancer agents should include multiple model systems in order to increase the possibility of correctly predicting their clinical activity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63:11–30. doi:10.3322/caac.21166
2. Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med (2006) 354:34–43. doi:10.1056/NEJMoa052985
3. Konstantinopoulos PA, Awtrey CS. Management of ovarian cancer: a 75-year-old woman who has completed treatment. JAMA (2012) 307:1420–9. doi:10.1001/jama.2012.269
4. McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med (1996) 334:1–6. doi:10.1056/NEJM199601043340101
5. Winter WE III, Maxwell GL, Tian C, Carlson JW, Ozols RF, Rose PG, et al. Prognostic factors for stage III epithelial ovarian cancer: a gynecologic oncology group study. J Clin Oncol (2007) 25:3621–7. doi:10.1200/JCO.2006.10.2517
6. Cho KR, Shih IeM. Ovarian cancer. Annu Rev Pathol (2009) 4:287–313. doi:10.1146/annurev.pathol.4.110807.092246
7. Meinhold-Heerlein I, Bauerschlag D, Hilpert F, Dimitrov P, Sapinoso LM, Orlowska-Volk M, et al. Molecular and prognostic distinction between serous ovarian carcinomas of varying grade and malignant potential. Oncogene (2005) 24:1053–65. doi:10.1038/sj.onc.1208298
8. Shih IeM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol (2004) 164:1511–8. doi:10.1016/S0002-9440(10)63708-X
9. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med (2010) 363:1532–43. doi:10.1056/NEJMoa1008433
10. The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474:609–15.
11. Stein WD, Litman T, Fojo T, Bates SE. A serial analysis of gene expression (sage) database analysis of chemosensitivity: comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res (2004) 64:2805–16. doi:10.1158/0008-5472.CAN-03-3383
12. Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci U S A (2011) 108:18708–13. doi:10.1073/pnas.1111840108
13. Sandberg R, Ernberg I. Assessment of tumor characteristic gene expression in cell lines using a tissue similarity index (TSI). Proc Natl Acad Sci U S A (2005) 102:2052–7. doi:10.1073/pnas.0408105102
14. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun (2013) 4:2126. doi:10.1038/ncomms3126
15. Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res (2011) 17:6083–96. doi:10.1158/1078-0432.CCR-11-0945
16. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature (2012) 483:603–7. doi:10.1038/nature11003
17. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434:917–21. doi:10.1038/nature03445
18. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361:123–34. doi:10.1056/NEJMoa0900212
19. Tsao SW, Mok SC, Fey EG, Fletcher JA, Wan TS, Chew EC, et al. Characterization of human ovarian surface epithelial cells immortalized by human papilloma viral oncogenes (HPV-E6E7 ORFS). Exp Cell Res (1995) 218:499–507. doi:10.1006/excr.1995.1184
20. Maines-Bandiera SL, Kruk PA, Auersperg N. Simian virus 40-transformed human ovarian surface epithelial cells escape normal growth controls but retain morphogenetic responses to extracellular matrix. Am J Obstet Gynecol (1992) 167:729–35. doi:10.1016/S0002-9378(11)91579-8
21. Li NF, Kocher HM, Salako MA, Obermueller E, Sandle J, Balkwill F. A novel function of colony-stimulating factor 1 receptor in hTERT immortalization of human epithelial cells. Oncogene (2009) 28:773–80. doi:10.1038/onc.2008.412
22. Davies BR, Steele IA, Edmondson RJ, Zwolinski SA, Saretzki G, von Zglinicki T, et al. Immortalisation of human ovarian surface epithelium with telomerase and temperature-sensitive SV40 large T antigen. Exp Cell Res (2003) 288:390–402. doi:10.1016/S0014-4827(03)00218-0
23. Yang G, Rosen DG, Colacino JA, Mercado-Uribe I, Liu J. Disruption of the retinoblastoma pathway by small interfering RNA and ectopic expression of the catalytic subunit of telomerase lead to immortalization of human ovarian surface epithelial cells. Oncogene (2007) 26:1492–8. doi:10.1038/sj.onc.1209905
24. Yang G, Rosen DG, Mercado-Uribe I, Colacino JA, Mills GB, Bast RC Jr., et al. Knockdown of p53 combined with expression of the catalytic subunit of telomerase is sufficient to immortalize primary human ovarian surface epithelial cells. Carcinogenesis (2007) 28:174–82. doi:10.1093/carcin/bgl115
25. Karst AM, Levanon K, Drapkin R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc Natl Acad Sci U S A (2011) 108:7547–52. doi:10.1073/pnas.1017300108
26. Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, et al. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia (2011) 13:899–911.
27. Shan W, Mercado-Uribe I, Zhang J, Rosen D, Zhang S, Wei J, et al. Mucinous adenocarcinoma developed from human fallopian tube epithelial cells through defined genetic modifications. Cell Cycle (2012) 11:2107–13. doi:10.4161/cc.20544
28. Curley MD, Garrett LA, Schorge JO, Foster R, Rueda BR. Evidence for cancer stem cells contributing to the pathogenesis of ovarian cancer. Front Biosci (Landmark Ed) (2011) 16:368–92. doi:10.2741/3693
29. Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res (2008) 68:4311–20. doi:10.1158/0008-5472.CAN-08-0364
30. Alvero AB, Fu HH, Holmberg J, Visintin I, Mor L, Marquina CC, et al. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells (2009) 27:2405–13. doi:10.1002/stem.191
31. Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. Cd24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene (2010) 29:2672–80. doi:10.1038/onc.2010.35
32. Flesken-Nikitin A, Hwang CI, Cheng CY, Michurina TV, Enikolopov G, Nikitin AY. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature (2013) 495:241–5. doi:10.1038/nature11979
33. Hamilton TC, Young RC, Louie KG, Behrens BC, McKoy WM, Grotzinger KR, et al. Characterization of a xenograft model of human ovarian carcinoma which produces ascites and intraabdominal carcinomatosis in mice. Cancer Res (1984) 44:5286–90.
34. Kelland LR, Jones M, Abel G, Valenti M, Gwynne J, Harrap KR. Human ovarian-carcinoma cell lines and companion xenografts: a disease-oriented approach to new platinum anticancer drug discovery. Cancer Chemother Pharmacol (1992) 30:43–50. doi:10.1007/BF00686484
35. Massazza G, Tomasoni A, Lucchini V, Allavena P, Erba E, Colombo N, et al. Intraperitoneal and subcutaneous xenografts of human ovarian carcinoma in nude mice and their potential in experimental therapy. Int J Cancer (1989) 44:494–500. doi:10.1002/ijc.2910440320
36. Ward BG, Wallace K. Localization of the monoclonal antibody hmfg2 after intravenous and intraperitoneal injection into nude mice bearing subcutaneous and intraperitoneal human ovarian cancer xenografts. Cancer Res (1987) 47:4714–8.
37. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol (2005) 174:6477–89.
38. Molthoff CF, Calame JJ, Pinedo HM, Boven E. Human ovarian cancer xenografts in nude mice: characterization and analysis of antigen expression. Int J Cancer (1991) 47:72–9. doi:10.1002/ijc.2910470114
39. Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther (2004) 10:1032–42. doi:10.1016/j.ymthe.2004.08.013
40. Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, et al. Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res (1983) 43:5379–89.
41. Hu L, Hofmann J, Zaloudek C, Ferrara N, Hamilton T, Jaffe RB. Vascular endothelial growth factor immunoneutralization plus paclitaxel markedly reduces tumor burden and ascites in athymic mouse model of ovarian cancer. Am J Pathol (2002) 161:1917–24. doi:10.1016/S0002-9440(10)64467-7
42. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature (1993) 362:841–4. doi:10.1038/362841a0
43. Burger RA, Sill MW, Monk BJ, Greer BE, Sorosky JI. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a gynecologic oncology group study. J Clin Oncol (2007) 25:5165–71. doi:10.1200/JCO.2007.11.5345
44. Cannistra SA, Matulonis UA, Penson RT, Hambleton J, Dupont J, Mackey H, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol (2007) 25:5180–6. doi:10.1200/JCO.2007.12.0782
45. Shaw D, Clamp A, Jayson GC. Angiogenesis as a target for the treatment of ovarian cancer. Curr Opin Oncol (2013) 25:558–65. doi:10.1097/CCO.0b013e328363e0da
46. Fu X, Hoffman RM. Human ovarian carcinoma metastatic models constructed in nude mice by orthotopic transplantation of histologically-intact patient specimens. Anticancer Res (1993) 13:283–6.
47. Bao R, Connolly DC, Murphy M, Green J, Weinstein JK, Pisarcik DA, et al. Activation of cancer-specific gene expression by the survivin promoter. J Natl Cancer Inst (2002) 94:522–8. doi:10.1093/jnci/94.7.522
48. Connolly DC. Animal models of ovarian cancer. Cancer Treat Res (2009) 149:353–91. doi:10.1007/978-0-387-98094-2_17
49. Vanderhyden BC, Shaw TJ, Ethier JF. Animal models of ovarian cancer. Reprod Biol Endocrinol (2003) 1:67. doi:10.1186/1477-7827-1-67
50. Olive KP, Tuveson DA. The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res (2006) 12:5277–87. doi:10.1158/1078-0432.CCR-06-0436
51. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol (2012) 9:338–50. doi:10.1038/nrclinonc.2012.61
52. Jin K, Teng L, Shen Y, He K, Xu Z, Li G. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol (2010) 12:473–80. doi:10.1007/s12094-010-0540-6
53. Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc (2007) 2:247–50. doi:10.1038/nprot.2007.25
54. Rubio-Viqueira B, Hidalgo M. Direct in vivo xenograft tumor model for predicting chemotherapeutic drug response in cancer patients. Clin Pharmacol Ther (2009) 85:217–21. doi:10.1038/clpt.2008.200
55. Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res (2006) 66:3351–4. discussion 3354, doi:10.1158/0008-5472.CAN-05-3627
56. Huang S, Pan W, Zhang J, Zhao B, Wang H, Liu F, et al. Heterogeneity-related anticancer therapy response differences in metastatic colon carcinoma: new hints to tumor-site-based personalized cancer therapy. Hepatogastroenterology (2013). doi:10.5754/hge13457
57. Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat (2012) 135:415–32. doi:10.1007/s10549-012-2164-8
58. Yang M, Shan B, Li Q, Song X, Cai J, Deng J, et al. Overcoming erlotinib resistance with tailored treatment regimen in patient-derived xenografts from naive Asian NSCLC patients. Int J Cancer (2013) 132:E74–84. doi:10.1002/ijc.27813
59. Kim MP, Truty MJ, Choi W, Kang Y, Chopin-Lally X, Gallick GE, et al. Molecular profiling of direct xenograft tumors established from human pancreatic adenocarcinoma after neoadjuvant therapy. Ann Surg Oncol (2012) 19(Suppl 3):S395–403. doi:10.1245/s10434-011-1839-4
60. Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther (2011) 10:1311–6. doi:10.1158/1535-7163.MCT-11-0233
61. Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res (2008) 14:6456–68. doi:10.1158/1078-0432.CCR-08-0138
62. Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res (2006) 12:4652–61. doi:10.1158/1078-0432.CCR-06-0113
63. Verschraegen CF, Hu W, Du Y, Mendoza J, Early J, Deavers M, et al. Establishment and characterization of cancer cell cultures and xenografts derived from primary or metastatic mullerian cancers. Clin Cancer Res (2003) 9:845–52.
64. Schmidt KF, Ziu M, Schmidt NO, Vaghasia P, Cargioli TG, Doshi S, et al. Volume reconstruction techniques improve the correlation between histological and in vivo tumor volume measurements in mouse models of human gliomas. J Neurooncol (2004) 68:207–15. doi:10.1023/B:NEON.0000033364.43142.bf
65. Verstijnen CP, Arends JW, Moerkerk P, Schutte B, van der Linden E, Kuypers-Engelen B, et al. Culturing and xenografting of primary colorectal carcinoma cells: comparison of in vitro, and in vivo model and primary tumor. Anticancer Res (1988) 8:1193–200.
66. Reyal F, Guyader C, Decraene C, Lucchesi C, Auger N, Assayag F, et al. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Res (2012) 14:R11. doi:10.1186/bcr3095
67. Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res (2009) 69:3364–73. doi:10.1158/0008-5472.CAN-08-4210
68. Sanz L, Cuesta AM, Salas C, Corbacho C, Bellas C, Alvarez-Vallina L. Differential transplantability of human endothelial cells in colorectal cancer and renal cell carcinoma primary xenografts. Lab Invest (2009) 89:91–7. doi:10.1038/labinvest.2008.108
69. Gray DR, Huss WJ, Yau JM, Durham LE, Werdin ES, Funkhouser WK Jr., et al. Short-term human prostate primary xenografts: an in vivo model of human prostate cancer vasculature and angiogenesis. Cancer Res (2004) 64:1712–21. doi:10.1158/0008-5472.CAN-03-2700
70. Grisanzio C, Seeley A, Chang M, Collins M, Di Napoli A, Cheng SC, et al. Orthotopic xenografts of RCC retain histological, immunophenotypic and genetic features of tumours in patients. J Pathol (2011) 225:212–21. doi:10.1002/path.2929
71. Shu Q, Wong KK, Su JM, Adesina AM, Yu LT, Tsang YT, et al. Direct orthotopic transplantation of fresh surgical specimen preserves cd133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells (2008) 26:1414–24. doi:10.1634/stemcells.2007-1009
72. Yu L, Baxter PA, Voicu H, Gurusiddappa S, Zhao Y, Adesina A, et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol (2010) 12:580–94. doi:10.1093/neuonc/nop056
73. James CD. Tumor-initiating cells: an influential paradigm for xenograft research. Neuro Oncol (2010) 12:519. doi:10.1093/neuonc/noq053
74. Williams SA, Anderson WC, Santaguida MT, Dylla SJ. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest (2013) 93:970–82. doi:10.1038/labinvest.2013.92
75. Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin Cancer Res (2012) 18:5160–2. doi:10.1158/1078-0432.CCR-12-2408
76. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in nod/scid/il2 receptor {gamma} chain(null) mice. Blood (2005) 106:1565–73. doi:10.1182/blood-2005-02-0516
77. Kolfschoten GM, Pinedo HM, Scheffer PG, Schluper HM, Erkelens CA, Boven E. Development of a panel of 15 human ovarian cancer xenografts for drug screening and determination of the role of the glutathione detoxification system. Gynecol Oncol (2000) 76:362–8. doi:10.1006/gyno.1999.5689
78. Fredrickson TN. Ovarian tumors of the hen. Environ Health Perspect (1987) 73:35–51. doi:10.1289/ehp.877335
79. Rodriguez GC, Walmer DK, Cline M, Krigman H, Lessey BA, Whitaker RS, et al. Effect of progestin on the ovarian epithelium of macaques: cancer prevention through apoptosis? J Soc Gynecol Investig (1998) 5:271–6. doi:10.1016/S1071-5576(98)00017-3
80. Brewer M, Baze W, Hill L, Utzinger U, Wharton JT, Follen M, et al. Rhesus macaque model for ovarian cancer chemoprevention. Comp Med (2001) 51:424–9.
81. Hsieh FY, Tengstrand E, Lee JW, Li LY, Silverman L, Riordan B, et al. Drug safety evaluation through biomarker analysis – a toxicity study in the cynomolgus monkey using an antibody-cytotoxic conjugate against ovarian cancer. Toxicol Appl Pharmacol (2007) 224:12–8. doi:10.1016/j.taap.2007.06.009
82. Hassan R, Ebel W, Routhier EL, Patel R, Kline JB, Zhang J, et al. Preclinical evaluation of MORAB-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun (2007) 7:20.
83. Stewart SL, Querec TD, Ochman AR, Gruver BN, Bao R, Babb JS, et al. Characterization of a carcinogenesis rat model of ovarian preneoplasia and neoplasia. Cancer Res (2004) 64:8177–83. doi:10.1158/0008-5472.CAN-04-1702
84. Cheon DJ, Orsulic S. Mouse models of cancer. Annu Rev Pathol (2011) 6:95–119. doi:10.1146/annurev.pathol.3.121806.154244
85. Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell (2002) 1:53–62. doi:10.1016/S1535-6108(01)00002-2
86. Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res (2006) 66:8949–53. doi:10.1158/0008-5472.CAN-06-1495
87. Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res (2003) 63:1389–97.
88. Hensley H, Quinn BA, Wolf RL, Litwin SL, Mabuchi S, Williams SJ, et al. Magnetic resonance imaging for detection and determination of tumor volume in a genetically engineered mouse model of ovarian cancer. Cancer Biol Ther (2007) 6:1717–25. doi:10.4161/cbt.6.11.4830
89. Mabuchi S, Altomare DA, Connolly DC, Klein-Szanto A, Litwin S, Hoelzle MK, et al. Rad001 (everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res (2007) 67:2408–13. doi:10.1158/0008-5472.CAN-06-4490
90. Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and rb1 in the mouse ovarian surface epithelium. Cancer Res (2003) 63:3459–63.
91. Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med (2005) 11:63–70. doi:10.1038/nm1173
92. Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell (2007) 11:321–33. doi:10.1016/j.ccr.2007.02.016
93. Kinross KM, Montgomery KG, Kleinschmidt M, Waring P, Ivetac I, Tikoo A, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest (2012) 122:553–7. doi:10.1172/JCI59309
94. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A (2012) 109:3921–6. doi:10.1073/pnas.1117135109
95. Crum CP, Drapkin R, Miron A, Ince TA, Muto M, Kindelberger DW, et al. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol (2007) 19:3–9. doi:10.1097/GCO.0b013e328011a21f
96. Mesiano S, Ferrara N, Jaffe RB. Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am J Pathol (1998) 153:1249–56. doi:10.1016/S0002-9440(10)65669-6
97. Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med (2011) 365:2473–83. doi:10.1056/NEJMoa1104390
98. Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med (2011) 365:2484–96. doi:10.1056/NEJMoa1103799
99. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434:913–7. doi:10.1038/nature03443
100. Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res (2009) 69:3850–5. doi:10.1158/0008-5472.CAN-08-2388
101. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of brca1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A (2008) 105:17079–84. doi:10.1073/pnas.0806092105
102. Kortmann U, McAlpine JN, Xue H, Guan J, Ha G, Tully S, et al. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clin Cancer Res (2010) 17:783–91. doi:10.1158/1078-0432.CCR-10-1382
103. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet (2010) 376:245–51. doi:10.1016/S0140-6736(10)60893-8
104. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol (2010) 28:2512–9. doi:10.1200/JCO.2009.26.9589
105. Schultes BC, Zhang C, Xue LY, Noujaim AA, Madiyalakan R. Immunotherapy of human ovarian carcinoma with OvaRex MAb-B43.13 in a human-PBL-SCID/BG mouse model. Hybridoma (1999) 18:47–55. doi:10.1089/hyb.1999.18.47
106. Pfisterer J, Harter P, Simonelli C, Peters M, Berek J, Sabbatini P, et al. Abagovomab for ovarian cancer. Expert Opin Biol Ther (2011) 11:395–403. doi:10.1517/14712598.2011.553598
107. Sabbatini P, Harter P, Scambia G, Sehouli J, Meier W, Wimberger P, et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: a phase III trial of the AGO OVAR, COGI, GINECO, and GEICO – the MIMOSA study. J Clin Oncol (2013) 31:1554–61. doi:10.1200/JCO.2012.46.4057
108. Berek J, Taylor P, McGuire W, Smith LM, Schultes B, Nicodemus CF. Oregovomab maintenance monoimmunotherapy does not improve outcomes in advanced ovarian cancer. J Clin Oncol (2009) 27:418–25. doi:10.1200/JCO.2008.17.8400
109. Delord JP, Allal C, Canal M, Mery E, Rochaix P, Hennebelle I, et al. Selective inhibition of HER2 inhibits AKT signal transduction and prolongs disease-free survival in a micrometastasis model of ovarian carcinoma. Ann Oncol (2005) 16:1889–97. doi:10.1093/annonc/mdi405
110. Takai N, Jain A, Kawamata N, Popoviciu LM, Said JW, Whittaker S, et al. 2C4, a monoclonal antibody against HER2, disrupts the HER kinase signaling pathway and inhibits ovarian carcinoma cell growth. Cancer (2005) 104:2701–8. doi:10.1002/cncr.21533
111. Bookman MA, Darcy KM, Clarke-Pearson D, Boothby RA, Horowitz IR. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the gynecologic oncology group. J Clin Oncol (2003) 21:283–90. doi:10.1200/JCO.2003.10.104
112. Gordon MS, Matei D, Aghajanian C, Matulonis UA, Brewer M, Fleming GF, et al. Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: potential predictive relationship with tumor HER2 activation status. J Clin Oncol (2006) 24:4324–32. doi:10.1200/JCO.2005.05.4221
Keywords: epithelial ovarian cancer, high-grade serous, preclinical models, personalized therapy, cell lines, xenografts, mouse models, patient-derived xenografts
Citation: Konstantinopoulos PA and Matulonis UA (2013) Current status and evolution of preclinical drug development models of epithelial ovarian cancer. Front. Oncol. 3:296. doi: 10.3389/fonc.2013.00296
Received: 29 September 2013; Accepted: 23 November 2013;
Published online: 11 December 2013.
Edited by:
Elise Kohn, National Cancer Institute, USAReviewed by:
Elise Kohn, National Cancer Institute, USAIvan Garcia-Bassets, University of California San Diego, USA
Copyright: © 2013 Konstantinopoulos and Matulonis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ursula A. Matulonis, Medical Gynecologic Oncology Program, Department of Medical Oncology, Dana Farber Cancer Institute, Harvard Medical School, 450 Brookline Avenue, Boston, MA 02215, USA e-mail:dXJzdWxhX21hdHVsb25pc0BkZmNpLmhhcnZhcmQuZWR1