 Simone Krebs
Simone Krebs Tania G. Rodríguez-Cruz
Tania G. Rodríguez-Cruz Christopher DeRenzo1,2,3
Christopher DeRenzo1,2,3 Stephen Gottschalk
Stephen Gottschalk- 1Center for Cell and Gene Therapy, Texas Children’s Hospital, Houston Methodist Hospital, Baylor College of Medicine, Houston, TX, USA
- 2Texas Children’s Cancer Center, Texas Children’s Hospital, Baylor College of Medicine, Houston, TX, USA
- 3Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
- 4Department of Pathology and Immunology, Baylor College of Medicine, Houston, TX, USA
Despite advances in surgical procedures, radiation, and chemotherapy the outcome for patients with glioblastoma (GBM) remains poor. While GBM cells express antigens that are potentially recognized by T cells, GBMs prevent the induction of GBM-specific immune responses by creating an immunosuppressive microenvironment. The advent of gene transfer has allowed the rapid generation of antigen-specific T cells as well as T cells with enhanced effector function. Here we review recent advances in the field of cell therapy with genetically modified T cells and how these advances might improve outcomes for patients with GBM in the future.
Introduction
Glioblastoma (GBM) is the second most common, but most aggressive primary brain tumor (1). Despite aggressive, multimodal therapy consisting of surgery, radiation, and chemotherapy, the outcome of patients with GBM remains poor with 5-year survival rates of <10% (1–3). Therapeutic resistance to conventional therapies is most likely caused by several factors. First, GBMs are protected by the blood-brain barrier, resulting in low concentrations of therapeutic agents at tumor sites. Second, GBMs harbor multiple mutations in key oncogenic signaling pathways including RTK/RAS/PI3K, p53, and rb pathways (4). Third, glioma-initiating cells, which are critical for the malignant phenotype of GBMs are chemo- and radiation-resistant (5, 6). Lastly, GBMs create a hostile immunosuppressive tumor microenvironment, which prevents the induction of anti-GBM-specific immune responses (7).
Immunotherapy has the potential to improve outcomes for patients with GBM since it does not rely on the cytotoxic pathways of the aforementioned conventional therapies. The most widely pursued immunotherapy for GBM is vaccines (8–10). While vaccines are safe and have prolonged survival in comparison to historical controls, few complete remissions have been observed. Nevertheless, several vaccines are currently in randomized Phase III clinical trials including one vaccine that targets an EGFR splice variant (EGFRvIII) and another that consists of tumor lysate pulsed dendritic cells (DCs) (10, 11). Conceptually, the adoptive transfer of T cells has several advantages over vaccines. T cells can be expanded ex vivo outside the immunosuppressive tumor microenvironment, and T cells can be genetically manipulated to confer specificity and enhance their effector function (12, 13). While clinical experience with genetically modified T cells for GBM is limited, recent successes in patients with melanoma, neuroblastoma, and hematological malignancies have highlighted their potent antitumor activity (14–20). Here we will review gene transfer into T cells (Table 1) and how this technology is being adapted for the immunotherapy of GBM.
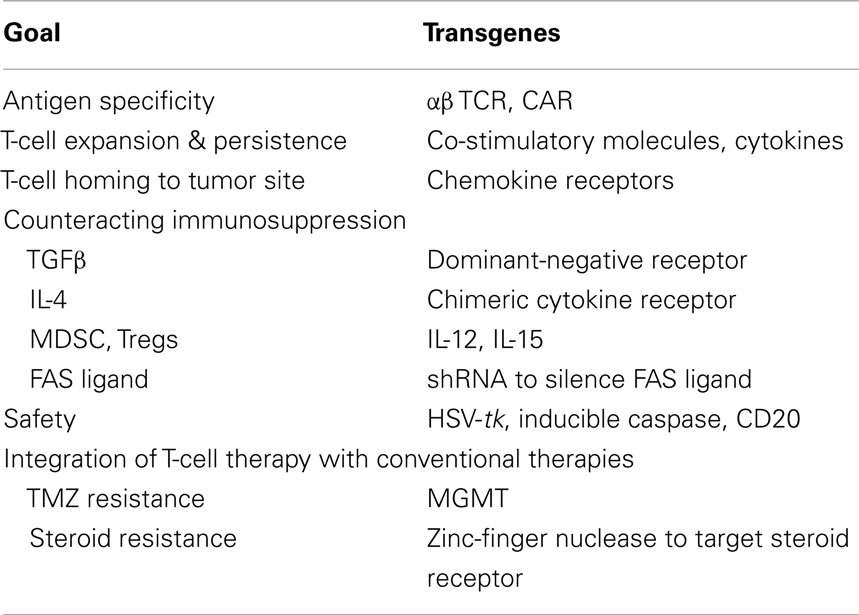
Table 1. Genetic modification of T cells.
Tumor-Associated Antigens Expressed in GBM
Glioblastomas express tumor-associated antigens (TAA) that are potential targets for immunotherapy including T-cell therapy (21, 22). TAA expressed in GBM can be classified into four categories based on their expression pattern: (i) antigens resulting from mutations, translocations, or splice variants (e.g., EGFRvIII) (23), (ii) antigens encoded by cancer-germ line genes [e.g., melanoma-associated antigen (MAGE), sarcoma antigen (SAGE), and synovial sarcoma X (SSX) families] (21, 22), (iii) antigens encoded by genes that are over expressed in GBMs [e.g., human epidermal growth factor receptor 2 (HER2), interleukin (IL)-13 receptor α2 (IL-13Rα2), erythropoietin-producing hepatocellular receptor A2 (EphA2)] (21, 24, 25), and (iv) viral antigens [e.g., pp65 and IE1 antigen of cytomegalovirus (CMV)] (26–28). Besides TAA expressed in malignant GBM cells, antigens expressed by vascular endothelial cells [e.g., vasculature endothelial growth factor receptor 2 (VEGFR2)] of the tumor vasculature or by other stromal cells are potential targets for T-cell therapy.
Genetic Modifications to Render T Cells Specific for GBM
Two genetic strategies are widely used to generate tumor-specific T cells. One approach relies on modifying T cells with T-cell receptor (TCR) genes and the other on introducing genes encoding chimeric antigen receptors (CARs) into T cells.
α/β TCR Gene Transfer
Conventional TCRs consist of two chains (α and β) that form heterodimers. TCRs recognize peptides, which are derived from proteins, in the context of major histocompatibility complex (MHC) molecules expressed on the cell surface. Isolating TCRs for adoptive T-cell therapy requires the generation of T-cell clones and subsequent isolation and cloning of the specific TCR α and β chains (29). Following isolation, α and β chain genes are cloned into viral vectors to introduce them into T cells (13). Initial studies highlighted that misspairing between endogenous α/β and transgenic α/β TCR chains is a common problem; however several approaches have been developed to overcome this limitation. For example, the introduction of disulfide bonds or use of murine sequences in the transgenic TCR genes results in preferential pairing of the introduced α/β TCR chains (30, 31). Silencing the expression of endogenous α/β TCR by shRNAs or zinc-finger nucleases are other attractive options (32, 33) that result in preferential pairing of the transgenic TCR.
α/β TCRs have been isolated for several TAA including CEA, GP100, MAGE-A3, MART1, and NY-ESO-1 (14, 34–37). While not tested in patients with GBM, some of these antigens are expressed in GBMs. The safety and efficacy of α/β TCR T-cell therapy has been evaluated in patients with melanoma, sarcoma, colon cancer, and multiple myeloma. One of the first studies in humans with α/β TCR T cells demonstrated that the infusion of autologous polyclonal T cells expressing a MART1-specific α/β TCR was safe and induced objective tumor responses in 2 out of 15 lymphodepleted patients with melanoma (34). To increase response rates, the same group infused T cells expressing high affinity MART1- and gp100-specific α/β TCRs. While response rates increased, several patients developed toxicities, including skin rash, uveitis, and/or hearing loss, that were not associated with antitumor responses (14). NY-ESO-1-specific α/β TCR T cells have also been evaluated in patients with synovial sarcoma, melanoma, and myeloma (37, 38), and encouraging antitumor responses have been observed without off-target side effects. In contrast, recognition of low levels of antigens on normal tissues by CEA-specific α/β TCR T cells has been observed in humans (36). Additionally, two adverse events have been reported in humans with affinity-matured TCRs, which recognized similar antigens (35, 39). Specifically, infusion of MAGE-A3-specific α/β TCR T cells caused both fatal neurotoxicity due to recognition of MAGE-A12, and fatal cardiac toxicities due to recognition of titin.
In conclusion, clinical studies with α/β TCR-modified T cells have not only demonstrated the potency of adoptively transferred T cells, but also some of their clinical limitations. Nevertheless, α/β TCR-modified T-cell therapy should be explored for patients with GBM, especially, since local delivery of such cells should reduce the risk of “non-CNS off target” effects.
Chimeric Antigen Receptor Gene Transfer
Chimeric antigen receptor design
Chimeric antigen receptors combine the antigen-binding property of monoclonal antibodies (MAbs) with the lytic capacity and self-renewal of T cells, and have several advantages over conventional T cells (Figure 1) (40, 41). CAR-expressing T cells recognize and kill tumor cells in an MHC unrestricted fashion, so that target cell recognition by CAR T cells is unaffected by some of the major mechanisms by which tumors avoid MHC-restricted T-cell recognition, such as downregulation of HLA class I molecules and defective antigen processing.
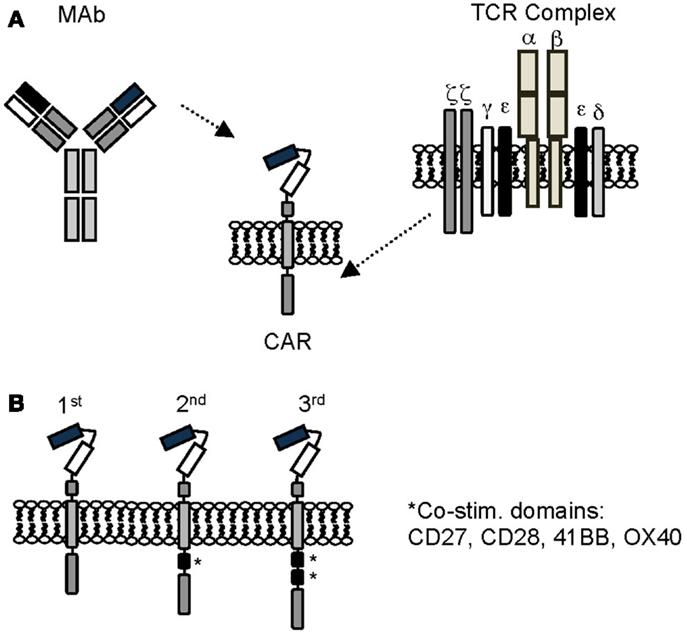
Figure 1. Chimeric antigen receptors. (A) Scheme of prototypic CAR. (B) First, second, and third generation CARs. See text for details.
Chimeric antigen receptors consist of an ectodomain, commonly derived from a single chain variable fragment (scFv), a hinge, a transmembrane domain, and an endodomain with one (first generation), two (second generation), or three (third generation) signaling domains derived from CD3ζ and/or co-stimulatory molecules (41, 42). Besides scFvs, natural ligands of receptors or peptides have also been used as antigen recognition domains (43–46). While the potency of CARs correlates with the presence of co-stimulatory domains, the structural components of the CAR, like the hinge region or the transmembrane domain, and the level of CAR expression, contribute to overall CAR function (47, 48).
While the majority of CARs only recognize target antigens expressed on the cell surface, CARs can recognize carbohydrate and glycolipid antigens, increasing the pool of potential targets. Potential CAR targets expressed on the cell surface of GBMs include IL-13Rα2, HER2, EphA2, EGFRvIII, chondroitin sulfate proteoglycan 4 (CSPG4), and NKG2D ligands (Table 2) (21, 24, 25, 49, 50). Of these only EGFRvIII is solely expressed in GBM cells, raising concerns of “on target, off organ” side effects when EphA2, IL-13Rα2, or HER2 are targeted. Targeting multiple antigens might solve this conundrum. For example, investigators have engineered T cells expressing two CARs with complementary signaling domains to allow full T-cell activation only at tumor sites where both antigens are expressed (51–53), or have engineered a single CAR with two antigen-binding domains (54). Targeting multiple antigens is also advisable to offset immune escape, which has been observed for cancer vaccine as well as T-cell therapy studies in humans (10, 55, 56).

Table 2. CAR T-cell therapy targets for GBM.
Preclinical studies with CAR T cells
Several investigators have evaluated the anti-GBM activity of CAR T cells in preclinical models (43, 57–61, 63–65). T cells expressing CARs specific for EphA2, EGFRvIII, IL-13Rα2, or HER2 recognized GBM cell lines or primary GBM samples in an antigen dependent manner as judged by cytokine production and cytolytic activity. CAR T cells also recognized and killed CD133-positive glioma-initiating cells, prevented neurosphere formation, and were able to destroy preformed neurospheres, demonstrating that radiation/chemotherapy-resistant glioma-initiating cells are sensitive to this immune-mediated killing mechanism (59, 60, 63, 64). In vivo, CAR T cells had potent antitumor activity in U87 and U373 xenograft models after local T-cell injection (43, 58, 59, 61, 65). In addition, investigators have demonstrated antitumor activity of CAR T cells using autologous GBM cells (60). Lastly, targeting two GBM antigens resulted in improved antitumor effects in one animal model (65). In summary, these studies demonstrate the potent anti-GBM activity of CAR T cells in preclinical models, warranting further active exploration. To date, CAR T-cell therapy for GBM has not been evaluated in immune-competent murine models. Immune-competent models will be critical to evaluate future combinatorial therapies in which adoptive T-cell transfer is combined with other agents to overcome the immunosuppressive tumor microenvironment.
While not tested in a GBM model, targeting the tumor vasculature with CARs is an attractive strategy to enhance the antitumor activity of CAR T cells (66, 67). Targeting the tumor vasculature with VEGFR2-specific CAR T cells in addition to tumor cells synergized in inducing tumor regression in several syngeneic, preclinical solid tumor models (68). In addition, transgenic expression of VEGFR2-specific CARs and IL-12 in T cells was sufficient to eradicate tumors, indicating that overcoming the inhibitory tumor microenvironment might potentiate effects of CAR T-cell therapies (see Section Engineering T-Cell Resistance to Immune Evasion Strategies Employed by GBMs) (69).
Clinical trials with T cells expressing CARs
So far clinical experience with CART-cell therapy for patients with GBM is limited. The safety and efficacy of intratumoral injection of T cells expressing a first generation IL-13Rα2-specific CAR (IL13-Rα2-CAR T cells) has been evaluated in one clinical study (62, 70). Infusion of IL13-Rα2-CAR T cells was well tolerated and associated with clinical benefit in several patients. In addition, two Phase I/II studies are currently in progress. In the first study the safety and efficacy of CMV-specific T cells expressing a second generation HER2-specific CAR is being evaluated in patients with recurrent GBM (NCT01109095). In the second study patients with recurrent GBM receive escalating doses of T cells expressing a third generation EGFRvIII-specific CAR after lymphodepleting chemotherapy (NCT01454596). Both studies are in progress, and clinical results should be available in the near future.
Engineering T-Cell Resistance to Immune Evasion Strategies Employed by GBMs
Similar to other malignancies, GBMs create a hostile, immunosuppressive microenvironment (7, 71–73). They: (1) secrete immunosuppressive cytokines such as transforming growth factor β (TGF-β) or IL10, (2) attract immunosuppressive cells such regulatory T cells (Tregs) or myeloid derived suppressor cells (MDSCs), (3) inhibit DC maturation, (4) express molecules on the cell surface that suppress immune cells including FAS ligand (FAS-L) and PD-L1, and (5) create a metabolic environment (e.g., high lactate, low tryptophan) that is immunosuppressive.
Different approaches are being explored to overcome tumor induced immunosuppression. Such strategies include: (1) enhancing CAR T-cell expansion and persistence by providing co-stimulation and/or lymphodepletion, (2) transgenic expression of cytokines, (3) silencing negative regulators, and (4) expression of chimeric cytokine/chemokine receptors or signaling molecules.
Increasing CAR T-Cell Expansion and Persistence
Since T-cell expansion post antigen recognition requires the provision of co-stimulation, investigators have included signaling domains in CAR endodomains derived from co-stimulatory molecules including CD27, CD28, 4-1BB, and OX40. Several preclinical studies have highlighted the benefit of added co-stimulation (74–77); however only one study so far has done a “head to head comparison” of CD19-specific CARs with a ζ− or CD28. ζ-Domain in individual patients (78). While CD28 co-stimulation resulted in enhanced expansion of adoptively transferred T cells, the effect was limited.
Thus genetic modification alone might not be sufficient to allow for T-cell expansion in vivo. Dramatic T-cell expansion and long-term persistence post infusion of adoptively transferred T cells occurs in lymphodepleted patients post hematopoietic stem cell transplantation (79, 80). Investigators have therefore lymphodepleted patients outside the transplant setting prior to T-cell transfer. The extent of lymphodepletion correlated with antitumor effects (81), and therefore many investigators currently prefer to give lymphodepleting chemotherapy before adoptive transfer of conventional or CAR T cells. Another option to boost expansion of T cells in vivo is vaccination. For example expressing CARs in T cells that are specific for viruses allows for vaccination (e.g., influenza) (82) or stimulation by latently infected cells in humans (e.g., Epstein–Barr virus) (15). Besides co-stimulation, and the status of the lymphoid compartment, it is also apparent that subsets of T cells differ in their behavior in vivo. For example, expressing CARs in effector memory T cells can enhance T-cell persistence in vivo (83, 84). In addition, the presence of CD4-positive CAR T cells in the T-cell product has correlated with long-term T-cell persistence (16).
Transgenic Expression of Cytokines
Chimeric antigen receptor T cells can be engineered to produce immunostimulatory cytokines. For example transgenic expression of IL-12 in CAR T cells reverses the immunosuppressive tumor environment by triggering apoptosis of inhibitory tumor-infiltrating macrophages, DCs, and MDSCs through a FAS-dependent pathway (85). While effective, there are safety concerns in regards to constitutive IL-12 expression. This obstacle can be overcome by using inducible promoters that are linked to the activation status of T cells, restricting IL-12 expression to tumor sites at which T cells are activated (86). Another attractive cytokine is IL-15. Transgenic expression of IL-15 (87, 88) renders T cells resistant to the inhibitory effects of Tregs through activation of the phosphoinositide 3-kinase (PI3K) (89), and improves CAR T-cell expansion and persistence in vivo.
Silencing Negative Regulators
Silencing genes that render T cells susceptible to inhibitory signals in the tumor microenvironment has the potential to improve T-cell function. For example many tumor cells express FAS-L, and silencing FAS in T cells prevents FAS-induced apoptosis (90). Other options include silencing genes that encode inhibitory molecules expressed on the T-cell surface such as CTLA-4 or PD-1 (91).
Expression of Chimeric Cytokine/Chemokine Receptors or Signaling Molecules
Transforming growth factor β is widely used by tumors as an immune evasion strategy (92), since it promotes tumor growth, limits effector T-cell function, and activates Tregs. These detrimental effects of TGF-β can be negated by modifying T cells to express a dominant-negative TGF-β receptor type II (DNR), which lacks most of the cytoplasmic kinase domain (93, 94). DNR expression interferes with TGF-β-signaling and restores T-cell effector function in the presence of TGF-β. The safety and efficacy of DNR-modified EBV-specific T cells is currently being evaluated in a Phase I/II clinical trial for patients with lymphoma (95), and if successful could be readily adapted to T-cell therapy for GBM.
T cells can also be engineered to convert inhibitory signals into stimulatory signals (96–98). For example, linking the extracellular domain of the TGF-β RII to the endodomain of toll-like receptor (TLR) four results in a chimeric receptor that not only renders T cells resistant to TGF-β, but also induces T-cell activation and expansion (98). Chimeric IL-4 receptors are another example of these “signal converters.” Many tumors secrete IL-4 to create a TH2-polarized environment, and two groups of investigators have shown that expression of chimeric IL-4 receptors, consisting of the ectodomain of the IL-4 receptor and the endodomain of the IL-7Rα or the IL-2Rβ chain, enable T cells to proliferate in the presence of IL-4 and retain their effector function including TH1-polarization (96, 97).
Another strategy to render T cells resistant to the inhibitory GBM environment is to express constitutively active signaling molecules. For example, expression of a constitutively active form of serine/threonine AKT (caAKT), which is a major component of the PI3K pathway, in T cells results in higher levels of NF-κB and elevated levels of anti-apoptotic genes such as Bcl2 conferring resistance to Tregs and TGFβ (99).
Genetic Modification of T Cells to Improve Homing to Tumor Sites
While the intravenous administration of EBV-specific T cells resulted in the regression of CNS lymphoma (100), and the adoptive transfer of tumor-infiltrating lymphocytes (TILs) resulted in the regression of brain metastases (101), the homing of T cells to GBM sites might be suboptimal, similar to clinical experience with T-cell therapy for solid tumors. For example, in one clinical study with first generation folate receptor (FR)-α specific CAR T cells for patients with ovarian cancer, infused cells did not specifically home to tumor sites as judged by 111Indium scintigraphy, and no antitumor activity was observed (102). Since then, several investigators have shown in preclinical models that the expression of chemokine receptors in CAR T cells that recognize chemokines secreted by solid tumors can enhance T-cell homing. For example, transgenic expression of chemokine receptors CCR2b or CXCR2 in T cells enhances trafficking to CCL2- or CXCL1-secreting solid tumors including melanoma and neuroblastoma (103, 104). Thus, expressing chemokine receptors in T cells or adhesion molecules that potentially facilitate the infiltration of T cells into GBM tumors has the potential to enhance the antitumor efficacy of adoptively transferred T cells.
Genetic Modification to Improve Safety of T-Cell Therapy
Potential toxicities can be divided into five categories: (1) toxicities due to genetic modification, (2) “on target organ” toxicities, (3) “on target, off organ” toxicities, (4) “off target, off organ” toxicities, and (5) systemic inflammatory syndromes. Toxicities due to genetic modification of T cells have not been observed in humans so far (105). An example of “on target organ” toxicity is the depletion of normal B cells after the infusion of CD19-speciifc CAR T cells for the treatment of B-cell malignancies (17). “On target, off organ” toxicity is exemplified by the liver toxicity observed after the infusion of carbonic anhydrase IX-specific CAR T cells to treat renal cell carcinoma (106). “Off target, off organ” toxicity is demonstrated by the recognition of MAGE-A12 in the brain or titin in the heart after the infusion of MAGE-A3-specific α/β TCR T cells for the treatment of melanoma, esophageal cancer, or myeloma (35, 39). Systemic inflammatory syndromes have been observed after CD19-specific CAR T-cell infusions for the immunotherapy of B-cell malignancies (17, 18, 107).
Genetic safety switches have been developed to selectively destroy genetically modified T cells once adverse events occur. The most widely used safety switch for T-cell therapy is the herpes simplex virus thymidine kinase (HSV-tk). HSV-tk phosphorylates acyclovir, valacyclovir, and ganciclovir to toxic nucleosides (108), and T cells transduced with the HSV-tk gene are killed in the presence of these medications. While clinical studies have demonstrated the effectiveness of this strategy, drawbacks to utilizing HSV-tk as a safety switch include the immunogenicity of HSV-tk, and that some patients require acyclovir, valacyclovir, or ganciclovir to treat herpetic diseases. Therefore, genetic safety switches using non-immunogenic human components have been developed, such as inducible caspase 9 (109, 110). With this strategy, once exposed to the dimerizer, genetically modified T cells rapidly undergo apoptosis. Another approach includes the transgenic expression of CD20, rendering T cells sensitive to the clinically approved CD20 MAb rituximab (111). While suicide gene switches can selectively kill infused cells, systemic inflammatory syndromes might be difficult to control since resident immune cells, activated by the infused T cells, most likely contribute. IL6 plays a critical role, and the infusion of the IL6 receptor MAb (tocilizumab) alone or in combinations with TNFα MAbs (infliximab) and steroids has proved to be effective (17, 18, 107).
While suicide switches are one strategy to prevent “on target, off organ” toxicities, T cells can also be engineered to only be fully activated if they encounter a unique “antigen address” at tumor sites. For example T cells expressing two CARs with different specificity of which one provides ζ-signaling and the other co-stimulation, will only be activated at tumor sites that express both antigens (51–53).
Genetic Modification of T Cells to Facilitate Integration of Cell Therapy with Current Therapies
T cells are inherently sensitive to agents that are currently used for the treatment of GBMs including steroids and temozolomide (TMZ). Gene transfer can now be used to render T cells resistant to these agents. For example, disruption of the glucocorticoid receptor gene in T cells with zinc-finger nucleases results in T cells that function in the presence of steroids (112), and this strategy is currently being evaluated in a Phase I clinical trial (NCT01082926). TMZ resistance can be conferred by expressing O(6)-methylguanine-DNA-methyltransferase (MGMT) in T cells (113), potentially allowing the infusion of T cells while GBM patients receive TMZ.
Conclusion
Preclinical studies and early clinical studies indicate that the genetic modification of T cells is a potent strategy to generate tumor-specific T cells with enhanced effector function. Not surprisingly, the greatest clinical success so far has been achieved for hematological malignancies targeting CD19. In order to develop effective CAR T-cell therapies for GBM several questions have to be addressed: which GBM antigen can be targeted without causing “on target/off cancer” side effects? How many antigens do we need to target to prevent immune escape? Do we have to target antigens expressed on non-malignant cells within the GBM microenvironment? Do we have to engineer T cells to enhance their homing to GBM sites and render them resistant to the immunosuppressive GBM environment? Do patients with GBM need to be lymphodepleted prior to T-cell transfer? Despite these unresolved issues, we believe that the results obtained so far with genetically modified T cells to target GBM are encouraging, warranting further active exploration. While genetically modified T cells are likely not the long awaited “GBM panacea,” integrating these cells into our current treatment armamentarium or combining them with other emerging targeted GBM therapies has the potential to improve outcomes for patients afflicted with this devastating cancer.
Conflict of Interest Statement
The Center for Cell and Gene Therapy has a research collaboration with Celgene and Bluebird Bio. Simone Krebs, Christopher DeRenzo, and Stephen Gottschalk have patent applications in the field of T-cell and gene-modified T-cell therapy for cancer.
Acknowledgments
The authors are supported by NIH grants 1R01CA148748-01A1, 1R01CA173750-01, P01CA094237, CPRIT RP110553, the Alex’s Lemonade Stand Foundation, the Dana Foundation, and the James S. McDonnell Foundation.
References
1. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA (2013) 310:1842–50. doi: 10.1001/jama.2013.280319
3. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi:10.1016/S1470-2045(09)70025-7
4. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol (2013) 10:14–26. doi:10.1038/nrclinonc.2012.204
5. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444:756–60. doi:10.1038/nature05236
6. Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res (2009) 7:989–99. doi:10.1158/1541-7786.MCR-09-0030
7. Albesiano E, Han JE, Lim M. Mechanisms of local immunoresistance in glioma. Neurosurg Clin N Am (2010) 21:17–29. doi:10.1016/j.nec.2009.08.008
8. Johnson LA, Sampson JH. Immunotherapy approaches for malignant glioma from 2007 to 2009. Curr Neurol Neurosci Rep (2010) 10:259–66. doi:10.1007/s11910-010-0111-9
9. Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol (2011) 29:330–6. doi:10.1200/JCO.2010.30.7744
10. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28:4722–9. doi:10.1200/JCO.2010.28.6963
11. Wheeler CJ, Black KL. DCVax-brain and DC vaccines in the treatment of GBM. Expert Opin Investig Drugs (2009) 18:509–19. doi:10.1517/13543780902841951
12. Brenner MK, Heslop HE. Adoptive T cell therapy of cancer. Curr Opin Immunol (2010) 22:251–7. doi:10.1016/j.coi.2010.01.020
13. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12:269–81. doi:10.1038/nri3191
14. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114:535–46. doi:10.1182/blood-2009-03-211714
15. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14:1264–70. doi:10.1038/nm.1882
16. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi:10.1182/blood-2011-05-354449
17. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3:95ra73. doi:10.1126/scitranslmed.3002842
18. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368:1509–18. doi:10.1056/NEJMoa1215134
19. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119:2709–20. doi:10.1182/blood-2011-10-384388
20. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5:177ra38. doi:10.1126/scitranslmed.3005930
21. Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S, et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res (2007) 13:566–75. doi:10.1158/1078-0432.CCR-06-1576
22. Zhang JG, Kruse CA, Driggers L, Hoa N, Wisoff J, Allen JC, et al. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol (2008) 88:65–76. doi:10.1007/s11060-008-9534-4
23. Humphrey PA, Wong AJ, Vogelstein B, Zalutsky MR, Fuller GN, Archer GE, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci U S A (1990) 87:4207–11. doi:10.1073/pnas.87.11.4207
24. Mintz A, Gibo DM, Slagle-Webb B, Christensen ND, Debinski W. IL-13Ralpha2 is a glioma-restricted receptor for interleukin-13. Neoplasia (2002) 4:388–99. doi:10.1038/sj.neo.7900234
25. Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res (2005) 3:541–51. doi:10.1158/1541-7786.MCR-05-0056
26. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res (2002) 62:3347–50.
27. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol (2008) 10:10–8. doi:10.1215/15228517-2007-035
28. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol (2008) 116:79–86. doi:10.1007/s00401-008-0359-1
29. Uttenthal BJ, Chua I, Morris EC, Stauss HJ. Challenges in T cell receptor gene therapy. J Gene Med (2012) 14:386–99. doi:10.1002/jgm.2637
30. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res (2007) 67:3898–903. doi:10.1158/0008-5472.CAN-06-3986
31. Goff SL, Johnson LA, Black MA, Xu H, Zheng Z, Cohen CJ, et al. Enhanced receptor expression and in vitro effector function of a murine-human hybrid MART-1-reactive T cell receptor following a rapid expansion. Cancer Immunol Immunother (2010) 59:1551–60. doi:10.1007/s00262-010-0882-5
32. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood (2012) 119:5697–705. doi:10.1182/blood-2012-01-405365
33. Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res (2009) 69:9003–11. doi:10.1158/0008-5472.CAN-09-1450
34. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:126–9. doi:10.1126/science.1129003
35. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi:10.1097/CJI.0b013e3182829903
36. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther (2011) 19:620–6. doi:10.1038/mt.2010.272
37. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi:10.1200/JCO.2010.32.2537
38. Levine BL, Rapoport AP, Stadtmauer EA, Vogl DT, Weiss B, Binder-Scholl GK, et al. Safety and correlates of clinical response in an early phase clinical trial in multiple myeloma patients post auto-SCT and adoptive immunotherapy with engineered T cells expressing an HLA-A2 restricted affinity-enhanced TCR for LAGE-1 and NY-ESO-1. Mol Ther (2013) 21:S114.
39. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi:10.1182/blood-2013-03-490565
40. Maher J. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol (2012) 2012:278093. doi:10.5402/2012/278093
41. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov (2013) 3:388–98. doi:10.1158/2159-8290.CD-12-0548
42. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A (1993) 90:720–4. doi:10.1073/pnas.90.2.720
43. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res (2004) 64:9160–6. doi:10.1158/0008-5472.CAN-04-0454
44. Shaffer DR, Savoldo B, Yi Z, Chow KK, Kakarla S, Spencer DM, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood (2011) 117:4304–14. doi:10.1182/blood-2010-04-278218
45. Davies DM, Foster J, Van Der Stegen SJ, Parente-Pereira AC, Chiapero-Stanke L, Delinassios GJ, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med (2012) 18:565–76. doi:10.2119/molmed.2011.00493
46. Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood (2005) 106:1544–51. doi:10.1182/blood-2004-11-4365
47. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T-cells. Clin Cancer Res (2013) 19(12):3153–64. doi:10.1158/1078-0432.CCR-13-0330
48. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol (2010) 184:6938–49. doi:10.4049/jimmunol.0901766
49. Svendsen A, Verhoeff JJ, Immervoll H, Brøgger JC, Kmiecik J, Poli A, et al. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol (2011) 122:495–510. doi:10.1007/s00401-011-0867-2
50. Friese MA, Platten M, Lutz SZ, Naumann U, Aulwurm S, Bischof F, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res (2003) 63:8996–9006.
51. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32:1059–70. doi:10.1007/s10875-012-9689-9
52. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol (2013) 31:71–5. doi:10.1038/nbt.2459
53. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res (2013). doi:10.1158/2326-6066.CIR-13-0008
54. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids (2013) 2:e105. doi:10.1038/mtna.2013.32
55. Gottschalk S, Ng CYC, Perez M, Smith CA, Sample C, Brenner MK, et al. An Epstein-Barr virus deletion mutant that causes fatal lymphoproliferative disease unresponsive to virus-specific T cell therapy. Blood (2001) 97:835–43. doi:10.1182/blood.V97.4.835
56. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A (2002) 99:16168–73. doi:10.1073/pnas.242600099
57. Bullain SS, Sahin A, Szentirmai O, Sanchez C, Lin N, Baratta E, et al. Genetically engineered T cells to target EGFRvIII expressing glioblastoma. J Neurooncol (2009) 94(3):373–82. doi:10.1007/s11060-009-9889-1
58. Choi BD, Suryadevara CM, Gedeon PC, Herndon IiJE, Sanchez-Perez L, Bigner DD, et al. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci (2013) 21(1):189–90. doi:10.1016/j.jocn.2013.03.012
59. Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther (2013) 21:629–37. doi:10.1038/mt.2012.210
60. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res (2010) 16:474–85. doi:10.1158/1078-0432.CCR-09-1322
61. Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res (2012) 18:5949–60. doi:10.1158/1078-0432.CCR-12-0319
62. Brown CE, Starr R, Naranjo A, Wright C, Bading J, Ressler JA, et al. Adoptive transfer of autologous IL13-zetakine+ engineered T cell clones for the treatment of recurrent glioblastoma: lessons from the clinic. Mol Ther (2011) 19:S136–7.
63. Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like tumor initiating cells isolated from IL13Ralpha2-expressing gliomas are targeted and killed by IL13-zetakine redirected T cells. Clin Cancer Res (2012) 18(8):2199–209. doi:10.1158/1078-0432.CCR-11-1669
64. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23:1043–53. doi:10.1089/hum.2012.041
65. Hegde M, Corder A, Chow KKh, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther (2013) 21:2087–101. doi:10.1038/mt.2013.185
66. Niederman TM, Ghogawala Z, Carter BS, Tompkins HS, Russell MM, Mulligan RC. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci U S A (2002) 99:7009–14. doi:10.1073/pnas.092562399
67. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest (2010) 120:3953–68. doi:10.1172/JCI43490
68. Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res (2013) 73:3371–80. doi:10.1158/0008-5472.CAN-12-3913
69. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res (2012) 18:1672–83. doi:10.1158/1078-0432.CCR-11-3050
70. Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol (2009) 6:53–8. doi:10.1038/ncponc1278
71. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res (2010) 16:461–73. doi:10.1158/1078-0432.CCR-09-1983
72. Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev (2006) 213:131–45. doi:10.1111/j.1600-065X.2006.00442.x
73. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi:10.1146/annurev.immunol.25.022106.141609
74. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A (2009) 106:3360–5. doi:10.1073/pnas.0813101106
75. Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med (2003) 9:279–86. doi:10.1038/nm827
76. Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med (2007) 13:1440–9. doi:10.1038/nm1676
77. Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther (2005) 12:933–41. doi:10.1016/j.ymthe.2005.04.016
78. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest (2011) 121:1822–6. doi:10.1172/JCI46110
79. Rooney CM, Smith CA, Ng CY, Loftin SK, Sixbey JW, Gan Y, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood (1998) 92:1549–55.
80. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood (2010) 115:925–35. doi:10.1182/blood-2009-08-239186
81. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol (2008) 26:5233–9. doi:10.1200/JCO.2008.16.5449
82. Cooper LJ, Al Kadhimi Z, Serrano LM, Pfeiffer T, Olivares S, Castro A, et al. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood (2005) 105:1622–31. doi:10.1182/blood-2004-03-1208
83. Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood (2012) 119:72–82. doi:10.1182/blood-2011-07-366419
84. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest (2008) 118:294–305. doi:10.1172/JCI32103
85. Kerkar SP, Leonardi AJ, van Panhuys N, Zhang L, Yu Z, Crompton JG, et al. Collapse of the tumor stroma is triggered by IL-12 induction of Fas. Mol Ther (2013) 21:1369–77. doi:10.1038/mt.2013.58
86. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther (2011) 19:751–9. doi:10.1038/mt.2010.313
87. Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood (2007) 110:2793–802. doi:10.1182/blood-2007-02-072843
88. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24:1160–70. doi:10.1038/leu.2010.75
89. Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, et al. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res (2013) 19:106–17. doi:10.1158/1078-0432.CCR-12-2143
90. Dotti G, Savoldo B, Pule M, Straathof KC, Biagi E, Yvon E, et al. Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood (2005) 105:4677–84. doi:10.1182/blood-2004-08-3337
91. Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol (2009) 157:9–19. doi:10.1111/j.1365-2249.2009.03912.x
92. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol (2010) 31:220–7. doi:10.1016/j.it.2010.04.002
93. Bollard CM, Rössig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood (2002) 99:3179–87. doi:10.1182/blood.V99.9.3179
94. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother (2008) 31:500–5. doi:10.1097/CJI.0b013e318177092b
95. Bollard CM, Dotti G, Gottschalk S, Mims M, Liu H, Gee AP, et al. Administration of TGF-beta resistant tumor-specific CTL to patients with EBV-associated HL and NHL. Mol Ther (2012) 20:S22.
96. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem (2010) 285:25538–44. doi:10.1074/jbc.M110.127951
97. Leen AM, Katari U, Kerinan J, Rooney CM, Brenner MK, Vera JF Improved expansion and anti-tumor activity of tumor-specific CTLs using a transgenic chimeric cytokine receptor. Mol Ther (2011) 19:S194.
98. Watanabe N, Anurathapan U, Brenner MK, Heslop HE, Leen AM, Rooney CM, et al. Transgenic expression of a novel immunosuppressive signal converter on T cells. Mol Ther (2013) 22:S153.
99. Sun J, Dotti G, Huye LE, Foster AE, Savoldo B, Gramatges MM, et al. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther (2010) 18:2006–17. doi:10.1038/mt.2010.185
100. Pakakasama S, Eames GM, Morriss MC, Huls MH, Rooney CM, Heslop HE, et al. Treatment of Epstein-Barr virus lymphoproliferative disease after hematopoietic stem-cell transplantation with hydroxyurea and cytotoxic T-cell lymphocytes. Transplantation (2004) 78:755–7. doi:10.1097/01.TP.0000129813.54517.25
101. Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res (2010) 16:4892–8. doi:10.1158/1078-0432.CCR-10-1507
102. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12:6106–15. doi:10.1158/1078-0432.CCR-06-1183
103. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (2010) 33(8):780–8. doi:10.1097/CJI.0b013e3181ee6675
104. Kershaw MH, Wang G, Bear A, Pule M, Brenner MK, Rooney CM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther (2002) 13:1971–80. doi:10.1089/10430340260355374
105. Bonini C, Brenner MK, Heslop HE, Morgan RA. Genetic modification of T cells. Biol Blood Marrow Transplant (2011) 17:S15–20. doi:10.1016/j.bbmt.2010.09.019
106. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24:e20–2. doi:10.1200/JCO.2006.05.9964
107. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi:10.1056/NEJMoa1103849
108. Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol (2009) 10:489–500. doi:10.1016/S1470-2045(09)70074-9
109. Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood (2005) 105:4247–54. doi:10.1182/blood-2004-11-4564
110. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365:1673–83. doi:10.1056/NEJMoa1106152
111. Introna M, Barbui AM, Bambacioni F, Casati C, Gaipa G, Borleri G, et al. Genetic modification of human T cells with CD20: a strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum Gene Ther (2000) 11:611–20. doi:10.1089/10430340050015798
112. Gardner RA, Reik A, Berger C, Terakura S, Gregory PD, Jensen CM et al. Disruption of glucocorticoid regulation in therapeutic human T cells with zinc finger nucleases. Mol Ther (2010) 18:S240.
Keywords: glioblastoma, immunotherapy, T-cell therapy, genetically modified T cells
Citation: Krebs S, Rodríguez-Cruz TG, DeRenzo C and Gottschalk S (2013) Genetically modified T cells to target glioblastoma. Front. Oncol. 3:322. doi: 10.3389/fonc.2013.00322
Received: 27 November 2013; Accepted: 17 December 2013;
Published online: 31 December 2013.
Edited by:
Giuseppe Merla, IRCCS Casa Sollievo della Sofferenza, ItalyReviewed by:
Nhan Le Tran, Translational Genomics Research Institute, USAPaola Parrella, IRCCS Casa Sollievo della Sofferenza, Italy
Copyright: © 2013 Krebs, Rodríguez-Cruz, DeRenzo and Gottschalk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Gottschalk, Center for Cell and Gene Therapy, Texas Children’s Hospital, Houston Methodist Hospital, Baylor College of Medicine, 1102 Bates Street, Suite 1770, Houston, TX 77030, USA e-mail:c21nb3R0c2NAdHhjaC5vcmc=