 Turid Hellevik
Turid Hellevik Iñigo Martinez-Zubiaurre
Iñigo Martinez-Zubiaurre- 1Department of Oncology, University Hospital of Northern-Norway, Tromsø, Norway
- 2Translational Cancer Research Group, Department of Clinical Medicine, University of Tromsø, Tromsø, Norway
Ionizing radiation is a non-specific but highly effective way to kill malignant cells. However, tumor recurrence sustained by a minor fraction of surviving tumor cells is a commonplace phenomenon caused by the activation of both cancer cell intrinsic resistance mechanisms, and also extrinsic intermediaries of therapy resistance, represented by non-malignant cells and structural components of the tumor stroma. The improved accuracy offered by advanced radiotherapy (RT)-technology permits reduced volume of healthy tissue in the irradiated field, and has been triggering an increase in the prescription of high-dose oligo-fractionated regimens in the clinics. Given the remarkable clinical success of high-dose RT and the current therapeutic shift occurring in the field, in this review we revise the existing knowledge on the effects that different radiation regimens exert on the different compartments of the tumor microenvironment, and highlight the importance of anti-tumor immunity and other tumor cell extrinsic mechanisms influencing therapeutic responses to high-dose radiation.
Introduction
Radiotherapy (RT) is a long-standing pillar in both curative and palliative cancer treatment, and about 50% of all cancer patients are receiving RT some time during their disease (1). However, despite the proved efficacy of RT, many treated cancer patients suffer from locally recurrent disease and/or metastatic spread of tumor growth (2). In the clinics, implementation of advanced imaging, computer technology together with technical perfection in radiation therapy planning and performance, has revolutionized the field (3). Consequently, a different therapeutic strategy commonly referred to as stereotactic ablative radiotherapy (SART), stereotactic body radiotherapy (SBRT), or stereotactic radiosurgery (SRS) is now gaining terrain in the clinics for the treatment of cancers with difficult cures such as lung, liver, pancreas, or brain (4, 5).
In line with the notion that solid tumors are complex and heterotypic tissues (6, 7), it is currently recognized that cancer cell responses to therapeutic treatments are greatly influenced by their microenvironment (8–10). This includes the physical contact of tumor cells with structural elements such as ECM via integrins, associations with resident and temporary host cells such as inflammatory cells, vascular cells, fibroblasts, and circulating progenitor cells, and interactions with miscellaneous diffusible molecules of autocrine, paracrine, and endocrine origin that influence cell behavior (2, 11). In RT, the nature of both external and internal RT is unspecific in the sense that it does not exclusively target neoplastic cells but necessarily affects all tumor-associated cells. Accordingly, secondary effects inflicted by RT on benign tumor constituents markedly guide the therapeutic outcomes post-treatment. Intriguingly, the radiation-induced changes in different stromal constituents of tumors have been demonstrated to depend on physical parameters such as total radiation dose, fraction-size, fraction intervals, or number of fractions. In view of that, recent reports have underscored the existence of threshold doses (and regimens) that are able to switch on different damage programs that profoundly affect responses to therapy. For instance, it has been suggested that local high-dose irradiation exerts more potent immune-responses than standard low-dose radiation, therefore promoting the eradication of cancer cells that escape the radiation-induced death (12). Studies with experimental tumors show that irradiation with doses higher than 10 Gy in a single-fractioned or up to 60 Gy in an oligo-fractionated manner causes severe vascular damages leading to strong alterations of the tumor microenvironment and indirect death of tumor cells (13). Furthermore, exposure of tumor fibroblasts to high radiation doses but not to small doses induces permanent DNA-damage responses and irreversible cellular senescence, which in turn might influence therapeutic upshots by the altered release of cytokines, chemokines, and growth factors (14). Given the acknowledged role played by the tumor microenvironment on guiding responses to therapy and the growing interest on using hypofractionated high-dose RT on cancers with poor prognosis, here we have aimed at presenting an overview on existing knowledge on how the different cellular compartments in tumors respond to different radiation schemes, and their potential impact on treatment outcomes. Summarized key literature is presented in Table 1.
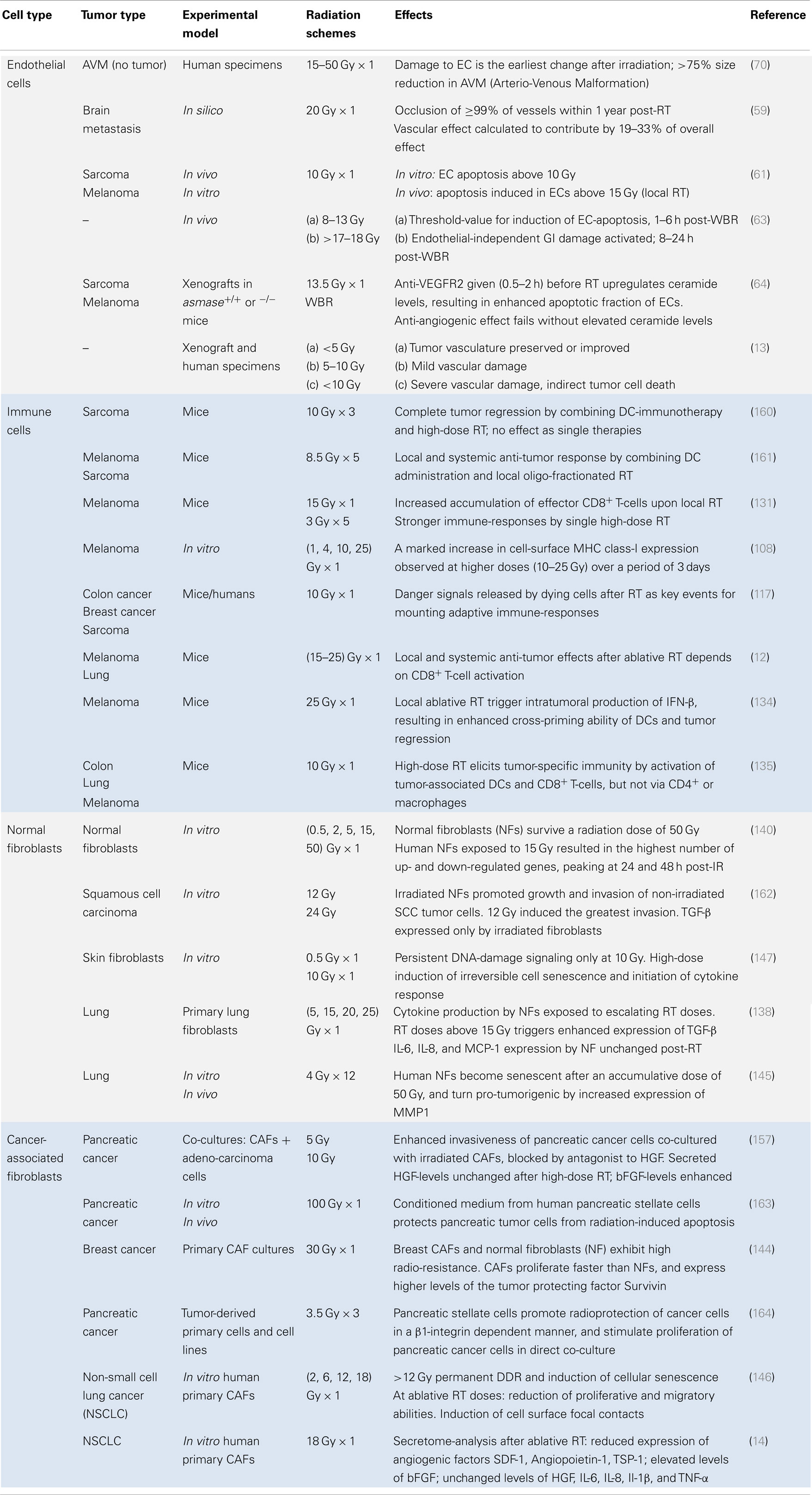
Table 1. Responses of the various cellular components of the tumor stroma to diverse radiation regimens.
The Evolution of Radiation Therapy and Treatment Regimens
Landmark observations about 80 years ago suggested that normal tissues could be spared if the radiation dose was delivered in fractions over several days (15). Since then, curative RT has typically been delivered as multi-fractionated rather than single-dose regimens, with daily doses of 2 Gy (5×/week), to an accumulative dose of 50–70 Gy. Big leaps within RT-related imaging and technology have led to enhanced accuracy, thus reducing the need of including large safety margins (of healthy normal tissue) in the treatment field. Hence, on the condition of rigorous quality assurances (16, 17), high-doses of ionizing radiation (IR) may now be delivered safely to patients.
The first attempts to deliver ablative or high-dose RT to patients in a single session derive from 1951, when neurosurgeon Leksell constructed an immobilizing stereotactic head-frame for delivery of highly focused IR with extreme precision to small volumes of non-malignant lesions of the brain (18). Later on, modified versions of this non-invasive technique, named “SRS,” have been applied to small primary tumors and single metastases of the brain (19, 20). Single fractions of 20–30 Gy are typical in the context of SRS, resulting in high rates of local tumor control, preservation of cerebral functions, and option of repeated treatment (21). Recently, oligo-fractionated SRS has proven beneficial also for brain tumors that are large (>3 cm) (22, 23) or located near critical structures (24), with a recommended radiation dose corresponding to 20 Gy × 1, 11.6 Gy × 2, or 8.5 Gy × 3 to gain local tumor control (25). The clinical success of SRS encouraged the development of a corresponding technique to treat extracranial tumors, named SBRT or SART (26). Hence, SART is defined as a method to deliver high-dose radiation by ultra-precision external-beam RT to extracranial parts of the body using few fractions (typically 1–5) (27).
In lung cancers, high-dose RT-regimens have revolutionized treatment outcome and overall survival for early stage inoperable peripheral tumors (28–30), and are currently considered also for operable lung cancers (31). In an overview by Heinzerling et al. (28), stage I NSCLC lung tumors receiving 45–60 Gy in three fractions versus 30–34 Gy in one single-fraction demonstrated 3-year post-RT tumor control rates of 92–98% and 80%, respectively. Centrally located lung tumors, being more prone to normal tissue toxicity than peripheral tumors, may gain similar numbers of tumor control (>85%) and acceptable toxicity with a less potent dose per fraction, i.e., eight fractions of 7.5 Gy (32, 33). In addition to lung cancers, SART (15–18 Gy × 3) has also generated encouraging results in primary and secondary (34, 35) hepatic tumors (36–38) and metastatic spinal lesions (39, 40).
The successful application of high-dose RT to large brain tumors and centrally located lung tumors illustrates the concept of “risk-adapted” fractionation regimens, designed to avoid high-grade toxicity upon high-dose RT for tumors located close to critical organs (41). However, clinical data have indicated that local tumor control is not only exclusively determined by dose and fractionation but also influenced by dose-prescription (41) and tumor size (42). Accordingly, recent tumor-volume-adapted dosing-approaches demonstrated excellent local control also for SART of large-volume lung tumors (43). Additionally, by re-plotting clinical data for tumor control probability as a function of biological effective dose for stage I non-small cell lung cancer, it was suggested that there is no difference in tumor control for single-fraction versus multi-fraction SART (44). Important in this context is a clear distinction between novel high-dose oligo-fractionated regimens and standard protracted regimens with 25–36 fractions of 2 Gy.
In the clinics, effects of varying dose and fractionation are usually guided by the linear–quadratic (LQ)-model (45–47). This mathematical tool was developed to estimate radiation damage on normal (and neoplastic tissue), and has proved useful for designing and comparing effects of new fractionation protocols (48). The LQ-model is suggested valid up to 8–10 Gy per fraction (49), but considered inappropriate for the high-dose regimens delivered by SRS and (lung) SART (50), presumably underestimating tumor control in the high-dose range (51, 52). Therefore, based on the recent success and widespread use of ablative RT, adapted tools for comparing dose and fractionation are being discussed (53–56).
Endothelial Cells and the Different RT-Regimens
The importance of angiogenesis and sprouting of resident endothelial cells (ECs) for continued tumor growth and metastasis has been advocated by Folkman since 1971, who also proposed the idea of killing tumor cells indirectly by targeting the ECs (57). The leaky and fragile ECs in tumors are proliferating considerably faster than ECs in normal tissues, and were early on suggested as a suitable therapeutic target (58). In the context of RT, a computer model fitted to clinical data proposed that the vascular effect contributes by 19–33% to the overall effect from single high-dose (20 Gy) radiosurgery (59). Of note, radiation effects on the tumor vasculature go beyond direct damage to the nuclear matter. Kolesnick and coworkers have long suggested that a rapid wave of ceramide-mediated apoptosis of ECs is the principal target for radiation injury and epithelial stem cell damage after high-dose RT (60–62). This group proposed a threshold dose of ~10 Gy for induction of apoptosis in tumor-associated ECs in culture (60, 63). More recently, anti-angiogenic agents (anti-VEGF or anti-VEGFR2) introduced right before high-dose RT demonstrated induction of up-regulated ceramide levels that resulted in augmented fraction of apoptotic ECs (64).
Radiation-induced changes in tumor blood vessels are known to be highly variable, depending markedly on the radiation dose and regimens [review in Ref. (13)]. Thus, during conventional (low-dose) fractionated RT, the status of the vasculature is somehow preserved, and occasionally improved or normalized (65), at least during the early part of a treatment course. These results are in line with a reported low-dose (≤5 Gy) RT stimulation of angiogenesis (66) and/or vasculogenesis (67, 68) in EC in vitro models. Also, in a study of radiation responses to the microcirculation of muscle flaps in vivo, a single dose of 8 Gy could only reduce capillary perfusion by 4.3%, implying that 8 Gy causes minimal damage to microvessels and the EC lining (69). However, at radiation doses exceeding 10 Gy/fraction, the tumor vessels become severely damaged (70) resulting in reduced blood perfusion and indirect tumor cell killing due to starvation (13). Despite such potentially, beneficial effects of high-dose RT, studies performed in animal models of intracranial glioblastoma multiforme (GBM) suggest that following local irradiation and ablation of the tumor vasculature, blood flow may be reconstituted several weeks after completed RT (71). The phenomena of such in-field recurrences are particularly common for the highly angiogenic glioblastomas, and represent a significant therapeutic challenge. Assuming that the process of angiogenesis (sprouting of vital resident ECs) is largely abrogated upon local high-dose RT (15–20 Gy), tumors rely on infiltration of circulating endothelial and other progenitor cells for revascularization and regrowth. This secondary “backup” pathway termed vasculogenesis represents a novel strategy to enhance local tumor control by RT (72, 73). Notably, even though endothelial progenitor cells (EPCs) are recognized for homing to ischemic areas where they contribute to neovascularization (74, 75), extensive research efforts has lead to the conclusion that bone-marrow-derived EPCs are contributing only marginally to (re)growth of irradiated tumors (71, 76). The source of circulating ECs (or EPCs) that are homing to irradiated tumors is therefore currently undefined (72, 77). However, it is well established that recruitment of pro-angiogenic myeloid (but not endothelial) (78) bone-marrow-derived progenitor cells (BMDCs) (71, 79) is pivotal for vascular-repair (80) and tumor regrowth, although they presumably do not incorporate into the tumor vasculature (81). So far, the role of non-bone-marrow-derived EPCs (75) in a setting of tumor relapses post-RT has been little explored (81). Intriguingly, recent studies in GBM-models point to the possibility that a fraction of tumor-associated ECs may be derived from trans-differentiation of (radioresistant) cancer stem-like cells (82–84).
Blood vessels of tumors are hyperpermeable and structurally abnormal, with a tortuous and dilated appearance and low coverage of pericytes that in total creates a microenvironment that is hypoxic and acidic, with high interstitial pressure. Hence, around 90% of all solid tumors have median oxygen levels that are lower than those appearing in normal tissues (85, 86). Tumor hypoxia is a well-known cause of radiation resistance (85), due to the fact that irradiated hypoxic cells are about 2.5–3 times less radiosensitive than normoxic cells (87). The idea that low-oxygen values contribute to radio-protect tumor cells correlates with clinical observations; severely, hypoxic tumors and high expression of hypoxia-inducible genes are generally associated with an aggressive phenotype, treatment failure, enhanced metastasis, and poor prognosis (88, 89). Considerable endeavors have been dedicated to improve radioresponses by targeting hypoxia (2, 85, 90, 91). In patients, the benefit of fractionated and protracted (low-dose) radiation regimens is partly explained by the phenomenon of tumor re-oxygenation, where surviving hypoxic cells become oxygenated between fractions (92). Conversely, a consequence of single high-dose RT is disrupted vasculature and invalidated angiogenesis, which may lead to a large increase in tumor hypoxia (71). Currently, there is an ongoing debate among scholars whether the dose-boosting offered by oligo-fractionated SART is sufficient to counterbalance the loss/reduced contribution of re-oxygenation (93) or if SART should be combined with a hypoxic radiosensitizer (42, 94). The latter alternative offers the possibility of achieving similar tumor control rates with a reduced radiation dose, and hence reduced toxicity, in patients (42, 90).
Prevention of revascularization post-radiation represents an appealing strategy to radio-sensitize tumors. Restoration of (radiation-) damaged vasculature by colonizing progenitor cells is initiated by HIF-1 (hypoxia-inducible factor-1) (95), the HIF-1-dependent and -independent (67) expression of SDF-1 (stromal cell-derived factor-1) (96) and the potent angiogenesis regulator VEGF (vascular endothelial growth factor). In a preclinical study with local (whole brain) irradiation of GBM-xenografts using a single high-dose (15 Gy), perfusion was reduced to a minimum after 2 weeks, whereas hypoxia, HIF-1 and SDF-1 was maximal at the same time point (71). SDF-1, secreted mainly by reactive tumor-associated fibroblasts (97), is retained in hypoxic tissues where it binds to the receptors CXCR-4 (96) and CXCR-7 expressed on monocytes and ECs (72), respectively. Recent studies indicate that hypoxia increases the expression of CXCR-7 in the pulmonary endothelium (98). Moreover, CXCR-7 is a scavenger receptor (99) responsible for uptake and degradation of SDF-1 (100) as well as mediating EC regeneration, repair, and proliferation (98, 101). Interestingly, inhibitors of CXCR-4, CXCR-7, and SDF-1 have all demonstrated delayed (or blocked) tumor recurrence post-RT in animal tumor growth-delay experiments (72). Regarding regulation of SDF-1 expression by RT, some opposite observations have been reported. Whereas secreted levels of SDF-1 from cultured lung (NSCLC)-CAFs are significantly reduced after 18 Gy × 1 (14), local high-dose irradiation to mice xenografts of GBM, breast, and lung tumors followed by detection in tissue-sections has indicated increased intratumoral SDF-1 quantity already 2 days post-RT (78), with maximum levels appearing after 14 days (71). In the latter system, the vasculogenesis-pathway could be blocked by interfering with the SDF-1/CXCR-4 interactions, and resulted in tumor control at radiation doses that alone were insufficient for sterilizing the tumors (71). The HIF-1 target-gene VEGF is another important factor required for homing and retention of circulating mononuclear myeloid cells (80). However, inhibition of VEGF was demonstrated to be less efficient than inhibition of the SDF-1/CXCR-4 interactions in abrogating tumor reperfusion and regrowth post-RT (71). Interestingly, in the same GBM-model, the radiation-induced influx of BMDCs was demonstrated to be dose-dependent, with doubled levels appearing after local (whole brain) irradiation with 15 versus 8 Gy (71). Nevertheless, when combining inhibition of SDF-1/CXCR-4 interactions with radiation, a fractionated low-dose regimen (2 Gy × 5) and a single high-dose (15 Gy) both resulted in complete anti-tumor responses (71).
Immune Cells and the Different RT-Regimens
Immune escape is the process in which malignant cells become unrecognized by the host immune system. The steady integration of multiple immune escape mechanisms by tumors during its progressive growth is now acknowledged as one of the hallmarks in cancer (6). During tumor development, not only malignant cells but also many other tumor-infiltrating cells undergo a process of immune editing, developing mechanisms by which the tumor tissue escapes immune recognition and elimination. Similar to angiogenesis, IR may act as a double-edged sword on inflammation and the immune system. Originally, RT was considered to be immuno-suppressive rather than immuno-stimulatory, largely due to the procedure of whole-body-radiation to minimize the immune-responses connected with bone-marrow transplantation. Also, it has been clinically documented that low-dose X-ray irradiation exerts an anti-inflammatory effect on benign diseases and chronic degenerative disorders (102). Additionally, immune-suppressive effects may come from the collateral damage exerted on tissue-resident antigen-presenting cells. On the other hand, irradiation may induce inflammation and immune reactions by the generation of tumor antigens, oxygen radicals, and alarm signals (8). Interestingly, an abscopal off-target response to RT has occasionally been reported in metastatic cancer patients, denoting the phenomenon of remote tumor regression outside the irradiated field after local RT (103, 104). The renewed interest of using RT as an immune adjuvant has already stimulated scientific efforts to decipher the mechanisms behind radiation-induced immune-responses and has generated novel concepts as “immunogenic cell death” to explain these events (105). In the following text, we present some of the important immunogenic pathways regulated by RT and the impact RT may exert on the different cellular components of the immune system.
One of the principal mechanisms for immune escape of tumor cells is the reduction of MHC-I levels on their plasma membrane. Notably, RT has been shown to upregulate MHC-I expression (106, 107). Moreover, MHC-I up-regulation is dose and time dependent, having a significant effect at doses above 4 Gy in melanoma cell-lines and at 8–20 Gy in murine colon carcinoma MC38 cells (108, 109). A potential explanation to this observation may be found in the induction of IFN-γ in the tumor microenvironment mainly by infiltrating T-lymphocytes and NK cells (110). RT is also able to modulate other surface molecules with immune modulating functions. ICAM-1 induction by RT has been observed in an array of different human and murine cell-lines, but mainly after high-dose radiation (111). On the other hand, radiation-induced up-regulation of the death-receptor CD95 is observed in various tumor types after different doses of radiation (112). The prominent role of RT on modulating phenotypic markers on neoplastic cells significant for their immunogenicity is reflected by the clear synergistic effect between irradiation and immunotherapeutic approaches using either specific blockage of immune-regulatory receptors (113), intratumoral administration of immune-stimulatory cytokines (114), or adoptive transfer of tumor-specific CTLs (115).
Damage-associated molecular patterns (DAMPs), or danger signals, are an important part for initiation of effective immune activation. DAMPs are small molecules released by either tumor cells or the miscellaneous inflammatory cells present in the tumor stroma. These molecules are generally released in response to stress situations as the ones created by RT. Some of the best characterized are high motility group protein 1 (HMGB1), calreticulin (CRT), heat shock proteins (HSPs) 70 and 90, adenosine triphosphate (ATP) and uric acid (105). Radiation can induce different cell death pathways depending on multiple factors such as fraction numbers and doses, tumor-type, tumor stage, and microenvironment. Tumor cell apoptosis is triggered most likely at low doses of radiation. Apoptotic cell death shows patterns of both tolerogenic and immunogenic cell death, viewed as CD47 up-regulation and oxidized HMGB1 release, and CRT externalization and reduced HMGB1 release, respectively. Radiation at high-doses on the other hand promotes necrotic cell death (116, 117). During necrosis, the cell membrane becomes disintegrated, which lead to an uncontrolled release of the cytoplasmic content into the extracellular space; this could include DAMPs and pro-inflammatory cytokines. Necrotic cell death is thus considered immunogenic but only if the process is accompanied by the release of stress signals. Radiation can also modulate the expression of certain chemokines and cytokines released by both tumor cells and stromal cells (8, 118). However, each single molecule can be both stimulatory and inhibitory depending on the target cells, concentrations, microenvironmental conditions, as well as tumor stage and type. Importantly, many of these factors are directly responsible for the systemic effects of local irradiation. Among others, factors such as IFN-γ, GM-CSF, TNF-α, IL-6, IL-10, or TGF-β account as powerful immune-modulating molecules potentially modified by IR with properties that can mediate systemic effects (118–121). Of note, the origin of these cytokines is not restricted to tumor cells, as also host cells from the tumor microenvironment constitute an important source (14, 122, 123).
Macrophage (Mφ) infiltration into tumors is a frequently observed phenomenon, usually accumulated at the border of malignant tissues and close to necrotic areas, with a highly variable abundance that depends on tumor-type and stage (124, 125). The role of Mφ in anti-tumor immunity is reliant on their phenotype, type-1 being considered immune-stimulating and type-2 immune-suppressive. Previous reports have suggested that local irradiation favors the Mφ-2 phenotype with tolerogenic functions, most likely due to RT-induction of immune inhibitory molecules such as COX-2/PGE2 and NO (126). This behavior seems to be associated with both single high-dose (20 Gy) and fractionated regimens (4 Gy × 15). Moreover, some groups have observed that tumor-associated Mø (TAM) in high numbers correlates with increased radio-resistance of tumors (127). Likewise, others have demonstrated that selective elimination of TAMs increases radiosensitivity in B16 murine melanomas in vivo (128). Interestingly, recent reports are indicating that low-dose irradiation redirects macrophage differentiation from the immune-suppressive state (M2) to one that enables recruitment of cytotoxic T-cells (65) (M1). CD8+ T-cell recruitment and expansion in tumors is a good prognostic factor in many tumor types, whereas intratumoral Tregs are important promoters and stabilizers of immuno-suppressive conditions, therefore associated with poor prognosis (129). Preclinical studies on radiation-mediated effects on lymphocyte levels and profiles are somewhat contradictory, with some reports showing positive effects and others showing the opposite (130, 131). Most likely, the conflicting literature available underlines the fact that the immunological profile of cancers is tumor-type specific, and that local microenvironmental parameters such as degree of hypoxia, intratumoral pH, tumor stroma composition, and cytokine milieu are indeed key factors influencing local and systemic immune-responses after RT. Dendritic cells are also key elements for proper initiation of adaptive immune reactions, but are frequently present at low numbers within the tumor tissues, and very often the infiltrating DC is plasmacytoid DC or immature DC with a clear immuno-suppressive phenotype (132). Immunotherapy aimed at reversing the immuno-suppressive status of DC contemplates the administration of cytokines that stimulate DC maturation and/or numbers, or the local injection of exogenously activated autologous DC (133). Radiation-induced DAMPs during cell death and the enhanced amounts of tumor antigens released may exert an impact on DC activation; thus, following the aforementioned explanations, different radiation doses and regimens may influence differently the activation of DC.
Apposite stimulation of the immune system by RT is probably the most illustrative example of indirect mechanisms contributing to tumor control, working in parallel to the more widely perceived idea of DNA damage in neoplastic tumor cells. Some of the first issues that need clarification for optimal induction of immune-responses are related to radiation dose and fractionation (103). Several authors claim that high-dose RT mounts a stronger immune attack against tumors than low-dose RT. This effect is supported by activation of different immunogenic mechanisms such as (a) secretion of higher levels of alarmin protein HMGB1 by dying tumor cells (10 Gy × 1) (117); (b) increased intratumoral production of type-I IFN by tumor-associated DCs (25 Gy × 1) (134); (c) increased number of CD45+ cells infiltrating tumors (15 Gy × 1), and a higher number of lymphocytes expressing IFN-γ (131); (d) increased number of tumor-specific CD8+ T-cells supported by the activation of tumor-infiltrating DCs (10 Gy × 1) (135); or (e) inducing more efficient maturation of antigen-presenting DCs (135) with enhanced cell-surface expression of MHC-I (10 Gy × 1) (108). Notably, two reports have suggested that RT-induced immune-responses depend on (oligo)fractionated rather than single-dose regimens (113, 136). Local irradiation of B16-OVA melanoma in mice revealed that a single (medium-high) dose of 7.5 Gy and above, but not 5 Gy, generated tumor immunity (136). An intriguing observation in that study was the fact that at the highest dose tested (15 Gy × 1), the authors reported intratumoral accumulation of T-regulatory cells, a reaction that could hinder the adaptive tumor immunity exerted by RT. In line with the last observation, the immuno-suppressive cytokine TGF-β (137) in growth medium of irradiated fibroblasts demonstrated base-line levels after 5 Gy, but significantly increased levels after 15 and 20 Gy (138). In the study by Dewan et al. (113), an abscopal effect was observed only when immunotherapy (anti-CTLA-4 antibodies) was combined with (3–5) fractions of medium–high-dose RT but not with single-high-dose (20 Gy × 1). The introduction of such medium–high radiation doses in the clinics has resulted in corresponding novel oligo-fractionated regimens that clearly contrasts the classical protracted 2 Gy-regimens typically utilized in curative settings for the last 80 years.
Fibroblasts and the Different RT-Regimens
Much of our acquired knowledge on fibroblast responses to IR emerge from studies aimed at understanding normal tissue reactions to RT, using normal connective tissue fibroblasts as experimental models. Genome-wide studies have tried to decode the overall changes in gene-expression on normal tissue fibroblasts exposed to IR (139–141). After functional categorization of differentially expressed genes, those reports clearly reveal that some central cellular pathways become persistently altered by radiation including DNA-damage responses, regulation of cell-cycle and proliferation, programed cell death, p53 target-genes, and some growth factor receptor signaling pathways (142). Similar responses have been described also for lung-CAFs (143). From a clinical point-of-view, parameters such as fraction-size and number of fractions seem to play an important role on fibroblast responses to IR. Hence, studies on transcriptome alterations induced by IR on fibroblasts have revealed marked differences at escalating radiation doses (140). In another study, ROS scavenging pathways and ECM remodeling pathways were more potently altered when fibroblasts were challenged with larger total dose and fractionated regimens than with single low doses (139).
A common observation made by many laboratories is that the fibroblast is a rather radioresistant cell-type, surviving single doses above 50 Gy in cell culture (140, 144–146). However, when the radiation dose exceeds 10 Gy, the cellular phenotype becomes profoundly altered, characterized by induction of irreversible DNA-damage response and concomitant development of stress-induced cellular senescence (146, 147). Senescent fibroblasts are by definition trapped in a state of permanent growth-arrest, but are still metabolically active and may therefore influence tissue responses to damage. In fact, numerous reports have highlighted the strong influence of senescent fibroblasts in the regulation of inflammation and the growth of adjacent epithelial cells (148). In a cancer setting, senescent fibroblasts are considered to play deleterious roles by contributing to sustained chronic inflammatory reactions, promoting angiogenesis and nourishing growth and invasion of neoplastic cells. These pro-malignant events are primarily driven by the release of soluble paracrine factors globally referred to as the senescence-associated secretory phenotype or SASP (149, 150). The SASP involves secretion of numerous inflammatory mediators, extracellular matrix proteins, proteases, and growth factors that collectively can render the microenvironment favorable to tumor growth. However, some authors argue in the opposite direction. Studies performed on tissue specimens from the post-irradiated breast indicate that malignant cells may be observed in irradiated tissues, while tumors often remain in a dormant state (151). A potential explanation to this observation may rely not only on the intrinsic susceptibility of cancer cells to the therapy but also on changes in the supportive stroma that makes the environment non-permissible for tumor regrowth (152).
Cancer-Associated Fibroblasts and the Different RT-Regimens
Collectively, most previous literature highlights the pro-malignant phenotype acquired by fibroblasts after radiation exposure. However, it should be noticed that the large majority of these studies have been conducted with normal tissue fibroblasts and/or fibroblast cell-lines, but not with resident and activated tumor-associated fibroblasts. To understand the contribution of CAFs to therapeutic outcomes after RT, it is utterly important to take into consideration the activated nature of tumor-resident fibroblasts (123, 153, 154). Non-irradiated reactive lung-CAFs actively produce numerous tumor-promoting molecules such as matrix metalloproteases, inflammatory cytokines, pro-angiogenic factors, and other growth factors (14). Also non-irradiated (activated) CAFs from skin, breast, and pancreatic cancer possess a pro-inflammatory signature, including up-regulated levels of IL-6 (155), SDF-1 (97), and TGF-β (156). Lung-CAFs exposed to ablative RT (18 Gy) demonstrated reduced expression of pro-angiogenic molecules such as SDF-1, angiogenin, and angiopoietin-1, and a concomitant reduction in migration-rates of ECs. In the context of high-dose RT, while human diploid fibroblasts demonstrated enhanced levels of IL-6 (147), primary normal fibroblasts (138) or lung-CAFs did not show altered expression of this cytokine. Regarding the growth factors HGF and bFGF, two separate reports have shown unchanged levels of the first and enhanced levels of the second, after high-dose RT in lung-CAFs (14) and PCC-CAFs (157). Activation of HGF-receptor is suggested to contribute to the enhanced invasiveness of PCCs grown in co-culture with CAFs (157). In xenograft models, senescent fibroblasts co-transplanted with cancer cells were found to increase tumorigenic effects on cancer cells (145, 158). However, the overall tumor regulatory properties of senescent fibroblasts remain controversial. It is still unresolved if this phenomenon is tumor-type specific or organ specific, or whether the senescent phenotype acquired after high-dose radiation exposure is comparable to other forms of senescence. In this regard, some authors have suggested that cancer promotion by senescent stromal cells may be restricted to certain organs and tissue types and claim that the importance of senescent cells needs to be validated in other tissues than subcutaneously grown tumors (159). All in all, existing literature on the contribution of irradiated fibroblasts to cancer relapse or eradication remains puzzling. Dose and fractionation regimens may play a central role on the ultimate response of CAFs to treatment. Additionally, important aspects such as the fibroblast-mediated immune-regulatory effects after irradiation still need to be explored.
Conclusions and Future Perspectives
Today it is widely acknowledged that the non-malignant components of solid neoplasms play central roles in the overall responses of cancers to therapies. Radiation therapy is a unique therapeutic modality that targets a defined tumor-volume, which implies that all cell types within the radiation field receive the prescribed radiation dose. This insult provokes the activation of cell death mechanisms and cell damage programs that greatly influence the therapeutic outcomes. Accordingly, it becomes a priority task for the scientific community to uncover all potential side signals and events occurring during and after the course of radiation in non-malignant tumor components that could influence tumor evolution and even the fate of distant metastasis.
Stereotactic ablative RT has recently emerged as a therapeutic alternative that is already proving its efficacy for treating cancers with very poor prognosis. The hype created around this therapeutic modality is fostering the interest of scientists to investigate on the different radiobiological responses elicited on different tumor constituents by different radiation regimens. In the context of RT in general, it is utterly important to consider the different scenarios represented by variables such as dose-per-fraction, number of fractions, total dose, and overall treatment time. Thus, conventional protracted regimens in the clinics comprise multiple doses (20–36) given at low doses (~2 Gy). On the other hand, SART-regimens are meant to be given in few fractions (1–5) and higher doses (5–30 Gy). However, in vivo and in vitro radiobiological studies have revealed substantially different responses triggered by different regimens. For instance, important variations are observed between single high- or oligo high-doses, both regimens within the frame of SART. Also, medium–high-doses (5–10 Gy) may exert critically different responses than high-doses (>10 Gy). All these observations should alert researchers and practitioners at the time of choosing radiation regimens that are meant to give optimal outcomes.
Not only malignant cells, but also tumor-infiltrating host cells respond differently to different radiation regimens. ECs and the tumor vasculature seem to be preserved at the low doses used in conventional RT-regimens. On the contrary, damages to tumor blood vessels are mild at doses ranging 5–10 Gy, and severe at doses above 10 Gy. On the consequences brought by damages inflicted to the vasculature by RT, still two confronted arguments remain: does the preservation and/or normalization of tumor blood vessels in conventional protracted regimens permit a better oxygenation of tumor cells and thus provide higher radiosensitivity, or will severe ablation of tumor vasculature by SART aid to a more efficient tumor cell killing by starvation? The rate of EC damage and the extent of hypoxia are two important and interconnected factors that add certain degree of complexity to predict beneficial or detrimental effects elicited by SART. With the recent enthusiasm and extended clinical application of high-dose RT in curative settings, exemplified by SART for lung cancers, these previous results are gaining clinical relevance.
Tumor-associated fibroblasts are able to repair DNA damages induced by low radiation doses; however, at doses above 10–12 Gy, the DNA-damage response becomes permanent and the cells enter in a permanent stage of senescence. Senescent fibroblasts are metabolically active and may secrete soluble signals and enzymes that in turn could regulate the growth and behavior of neighboring cancer cells. It remains to be elucidated if irradiated fibroblasts or CAFs certainly contribute to tumor evolution post-RT, and if so which molecules and pathways are implicated in this regulation. Also, it remains uncertain if normal tissue fibroblasts and CAFs respond in the same way to RT, or if medium–high-doses (8–10 Gy) exert different effects than higher doses (15–20 Gy) in these cells.
The notion of using RT for boosting anti-tumor immunity is not new; however, at present we know that conventional RT is unable to mount efficient anti-tumor immune-responses as a single treatment modality. In the new era of SART, the interest on using radiation as an efficacious immune-stimulant is being renewed. Evidences in this field are demonstrating that high-dose regimens are more efficient to trigger both innate and adaptive anti-tumor immunity than low-dose regimens. Expression of alarm signals, induced intratumoral levels of interferons, efficient maturation of DCs, and controlled immune-regulatory mechanisms count as some of the important mechanisms behind the effect. Importantly, some reports argue that oligo-fractionated regimens work better than single-high-dose regimens, and also that medium–high (7–10 Gy) doses may work better than very high doses (>15 Gy). It is now most widely assumed that radiation probably can only amplify or augment the pro-immunogenic status in tumors but will hardly be able to change by itself a well-settled immuno-suppressive environment into an immune-stimulating one. Therefore, current research is now focused on the successful combination of immune modulating therapies and the optimal RT protocols.
Different cellular components are found in tumors, and as such, different responses are taking place at the same time incited by RT. Only if we globally consider all events, in a system biology way of thinking, could we predict more precisely the ultimate consequences of the therapy. Dissecting the responses undergone by the different constituents of the tumor stroma under different RT-regimens is aiding us to more efficiently choose the adequate treatment schedules in RT. Finally, concepts such as stereotactic radiation or high-dose radiation are getting too broad and may lead to undesired misinterpretations. Given the striking different responses of cells to small variations in the schedules, a more precise terminology would be needed among professionals to define radiation regimens and treatment schedules.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The project has been supported by grants from The Norwegian Cancer Society and The Northern Norway Regional Health Authorities (Helse-Nord).
References
1. Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements. Nat Rev Cancer (2004) 4:737–47. doi:10.1038/nrc1451
2. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer (2011) 11:239–53. doi:10.1038/nrc3007
3. De Ruysscher D, Belderbos J, Reymen B, van Elmpt W, van Baardwijk A, Wanders R, et al. State of the art radiation therapy for lung cancer 2012: a glimpse of the future. Clin Lung Cancer (2013) 14:89–95. doi:10.1016/j.cllc.2012.06.006
4. Lo SS, Fakiris AJ, Chang EL, Mayr NA, Wang JZ, Papiez L, et al. Stereotactic body radiation therapy: a novel treatment modality. Nat Rev Clin Oncol (2010) 7:44–54. doi:10.1038/nrclinonc.2009.188
5. Salama JK, Kirkpatrick JP, Yin FF. Stereotactic body radiotherapy treatment of extracranial metastases. Nat Rev Clin Oncol (2012) 9:654–65. doi:10.1038/nrclinonc.2012.166
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
7. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi:10.1016/j.ccr.2012.02.022
8. Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment – tumorigenesis and therapy. Nat Rev Cancer (2005) 5:867–75. doi:10.1038/nrc1735
9. Chargari C, Clemenson C, Martins I, Perfettini JL, Deutsch E. Understanding the functions of tumor stroma in resistance to ionizing radiation: emerging targets for pharmacological modulation. Drug Resist Updat (2013) 16:10–21. doi:10.1016/j.drup.2013.01.001
10. Good JS, Harrington KJ. The hallmarks of cancer and the radiation oncologist: updating the 5Rs of radiobiology. Clin Oncol (R Coll Radiol) (2013) 25:569–77. doi:10.1016/j.clon.2013.06.009
11. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer (2001) 1:46–54. doi:10.1038/35094059
12. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood (2009) 114:589–95. doi:10.1182/blood-2009-02-206870
13. Park HJ, Griffin RJ, Hui S, Levitt SH, Song CW. Radiation-induced vascular damage in tumors: implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS). Radiat Res (2012) 177:311–27. doi:10.1667/RR2773.1
14. Hellevik T, Pettersen I, Berg V, Bruun J, Bartnes K, Busund LT, et al. Changes in the secretory profile of NSCLC-associated fibroblasts after ablative radiotherapy: potential impact on angiogenesis and tumor growth. Transl Oncol (2013) 6:66–74. doi:10.1593/tlo.13640
15. Coutard H. Roentgentherapy of epitheliomas of the tonsillar region; hypopharynx and larynx from 1920 to 1926. Am J Roentgenol (1932) 28:313–31 and 343–348.
16. De Ruysscher D, Faivre-Finn C, Nestle U, Hurkmans CW, Le Péchoux C, Price A, et al. European Organisation for Research and Treatment of Cancer recommendations for planning and delivery of high-dose, high-precision radiotherapy for lung cancer. J Clin Oncol (2010) 28:5301–10. doi:10.1200/JCO.2010.30.3271
17. Potters L, Steinberg M, Rose C, Timmerman R, Ryu S, Hevezi JM, et al. American Society for Therapeutic Radiology and Oncology and American College of Radiology practice guideline for the performance of stereotactic body radiation therapy. Int J Radiat Oncol Biol Phys (2004) 60:1026–32. doi:10.1016/j.ijrobp.2004.07.701
18. Leksell L. The stereotaxic method and radiosurgery of the brain. Acta Chir Scand (1951) 102:316–9.
19. Sturm V, Kober B, Höver KH, Schlegel W, Boesecke R, Pastyr O, et al. Stereotactic percutaneous single dose irradiation of brain metastases with a linear accelerator. Int J Radiat Oncol Biol Phys (1987) 13:279–82. doi:10.1016/0360-3016(87)90140-4
20. Kelly PJ, Lin YB, Yu AY, Alexander BM, Hacker F, Marcus KJ, et al. Stereotactic irradiation of the postoperative resection cavity for brain metastasis: a frameless linear accelerator-based case series and review of the technique. Int J Radiat Oncol Biol Phys (2012) 82:95–101. doi:10.1016/j.ijrobp.2010.10.043
21. Maranzano E, Trippa F, Casale M, Costantini S, Anselmo P, Carletti S, et al. Reirradiation of brain metastases with radiosurgery. Radiother Oncol (2012) 102:192–7. doi:10.1016/j.radonc.2011.07.018
22. Wegner RE, Leeman JE, Kabolizadeh P, Rwigema JC, Mintz AH, Burton SA, et al. Fractionated stereotactic radiosurgery for large brain metastases. Am J Clin Oncol (2013). doi:10.1097/COC.0b013e31828aadac
23. Minniti G, Esposito V, Clarke E, Scaringi C, Lanzetta G, Salvati M, et al. Multidose stereotactic radiosurgery (9 Gy × 3) of the postoperative resection cavity for treatment of large brain metastases. Int J Radiat Oncol Biol Phys (2013) 86:623–9. doi:10.1016/j.ijrobp.2013.03.037
24. Kim YJ, Cho KH, Kim JY, Lim YK, Min HS, Lee SH, et al. Single-dose versus fractionated stereotactic radiotherapy for brain metastases. Int J Radiat Oncol Biol Phys (2011) 81:483–9. doi:10.1016/j.ijrobp.2010.05.033
25. Wiggenraad R, Verbeek-de Kanter A, Kal HB, Taphoorn M, Vissers T, Struikmans H. Dose-effect relation in stereotactic radiotherapy for brain metastases. A systematic review. Radiother Oncol (2011) 98:292–7. doi:10.1016/j.radonc.2011.01.011
26. Blomgren H, Lax I, Naslund I, Svanstrom R. Stereotactic high dose fraction radiation therapy of extracranial tumors using an accelerator. Clinical experience of the first thirty-one patients. Acta Oncol (1995) 34:861–70. doi:10.3109/02841869509127197
27. Potters L, Gaspar LE, Kavanagh B, Galvin JM, Hartford AC, Hevezi JM, et al. American Society for Therapeutic Radiology and Oncology (ASTRO) and American College of Radiology (ACR) practice guidelines for image-guided radiation therapy (IGRT). Int J Radiat Oncol Biol Phys (2010) 76:319–25. doi:10.1016/j.ijrobp.2009.09.041
28. Heinzerling JH, Kavanagh B, Timmerman RD. Stereotactic ablative radiation therapy for primary lung tumors. Cancer J (2011) 17:28–32. doi:10.1097/PPO.0b013e31820a7f80
29. Palma D, Senan S. Stereotactic radiation therapy: changing treatment paradigms for stage I nonsmall cell lung cancer. Curr Opin Oncol (2011) 23:133–9. doi:10.1097/CCO.0b013e328341ee11
30. Kavanagh BD, Miften M, Rabinovitch RA. Advances in treatment techniques: stereotactic body radiation therapy and the spread of hypofractionation. Cancer J (2011) 17:177–81. doi:10.1097/PPO.0b013e31821f7dbd
31. Timmerman RD, Fernando HC. A radiation oncologist’s and thoracic surgeon’s view on the role of stereotactic ablative radiotherapy for operable lung cancer. Semin Thorac Cardiovasc Surg (2013) 25:8–13. doi:10.1053/j.semtcvs.2013.01.006
32. Senthi S, Haasbeek CJ, Slotman BJ, Senan S. Outcomes of stereotactic ablative radiotherapy for central lung tumours: a systematic review. Radiother Oncol (2013) 106:276–82. doi:10.1016/j.radonc.2013.01.004
33. Nuyttens JJ, van derVoort van Zyp NC, Praag J, Aluwini S, van Klaveren RJ, Verhoef C, et al. Outcome of four-dimensional stereotactic radiotherapy for centrally located lung tumors. Radiother Oncol (2012) 102:383–7. doi:10.1016/j.radonc.2011.12.023
34. Chang DT, Swaminath A, Kozak M, Weintraub J, Koong AC, Kim J, et al. Stereotactic body radiotherapy for colorectal liver metastases: a pooled analysis. Cancer (2011) 117:4060–9. doi:10.1002/cncr.25997
35. Schefter TE, Kavanagh BD. Radiation therapy for liver metastases. Semin Radiat Oncol (2011) 21:264–70. doi:10.1016/j.semradonc.2011.05.006
36. Liu E, Stenmark MH, Schipper MJ, Balter JM, Kessler ML, Caoili EM, et al. Stereotactic body radiation therapy for primary and metastatic liver tumors. Transl Oncol (2013) 6:442–6. doi:10.1593/tlo.12448
37. Dewas S, Bibault JE, Mirabel X, Fumagalli I, Kramar A, Jarraya H, et al. Prognostic factors affecting local control of hepatic tumors treated by Stereotactic Body Radiation Therapy. Radiat Oncol (2012) 7:166. doi:10.1186/1748-717X-7-166
38. Hiniker SM, Chen DS, Reddy S, Chang DT, Jones JC, Mollick JA, et al. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol (2012) 5:404–7. doi:10.1593/tlo.12280
39. Yamada Y, Bilsky MH, Lovelock DM, Venkatraman ES, Toner S, Johnson J, et al. High-dose, single-fraction image-guided intensity-modulated radiotherapy for metastatic spinal lesions. Int J Radiat Oncol Biol Phys (2008) 71:484–90. doi:10.1016/j.ijrobp.2007.11.046
40. Wang XS, Rhines LD, Shiu AS, Yang JN, Selek U, Gning I, et al. Stereotactic body radiation therapy for management of spinal metastases in patients without spinal cord compression: a phase 1–2 trial. Lancet Oncol (2012) 13:395–402. doi:10.1016/S1470-2045(11)70384-9
41. Senan S, Palma DA, Lagerwaard FJ. Stereotactic ablative radiotherapy for stage I NSCLC: recent advances and controversies. J Thorac Dis (2011) 3:189–96. doi:10.3978/j.issn.2072-1439.2011.05.03
42. Brown JM, Diehn M, Loo BW Jr. Stereotactic ablative radiotherapy should be combined with a hypoxic cell radiosensitizer. Int J Radiat Oncol Biol Phys (2010) 78:323–7. doi:10.1016/j.ijrobp.2010.04.070
43. Trakul N, Chang CN, Harris J, Chapman C, Rao A, Shen J, et al. Tumor volume-adapted dosing in stereotactic ablative radiotherapy of lung tumors. Int J Radiat Oncol Biol Phys (2012) 84:231–7. doi:10.1016/j.ijrobp.2011.10.071
44. Brown JM, Brenner DJ, Carlson DJ. Dose escalation, not “new biology,” can account for the efficacy of stereotactic body radiation therapy with non-small cell lung cancer. Int J Radiat Oncol Biol Phys (2013) 85:1159–60. doi:10.1016/j.ijrobp.2012.11.003
45. Thames HD Jr, Withers HR, Peters LJ, Fletcher GH. Changes in early and late radiation responses with altered dose fractionation: implications for dose-survival relationships. Int J Radiat Oncol Biol Phys (1982) 8:219–26. doi:10.1016/0360-3016(82)90517-X
46. Barendsen GW. Dose fractionation, dose rate and iso-effect relationships for normal tissue responses. Int J Radiat Oncol Biol Phys (1982) 8:1981–97. doi:10.1016/0360-3016(82)90459-X
47. Travis EL, Tucker SL. Isoeffect models and fractionated radiation therapy. Int J Radiat Oncol Biol Phys (1987) 13:283–7. doi:10.1016/0360-3016(87)90141-6
48. Brenner DJ, Sachs RK, Peters LJ, Withers HR, Hall EJ. We forget at our peril the lessons built into the alpha/beta model. Int J Radiat Oncol Biol Phys (2012) 82:1312–4. doi:10.1016/j.ijrobp.2011.12.045
49. Brenner DJ. The linear–quadratic model is an appropriate methodology for determining isoeffective doses at large doses per fraction. Semin Radiat Oncol (2008) 18:234–9. doi:10.1016/j.semradonc.2008.04.004
50. Kirkpatrick JP, Meyer JJ, Marks LB. The linear–quadratic model is inappropriate to model high dose per fraction effects in radiosurgery. Semin Radiat Oncol (2008) 18:240–3. doi:10.1016/j.semradonc.2008.04.005
51. Otsuka S, Shibamoto Y, Iwata H, Murata R, Sugie C, Ito M, et al. Compatibility of the linear–quadratic formalism and biologically effective dose concept to high-dose-per-fraction irradiation in a murine tumor. Int J Radiat Oncol Biol Phys (2011) 81:1538–43. doi:10.1016/j.ijrobp.2011.05.034
52. Stuschke M, Pottgen C. Altered fractionation schemes in radiotherapy. Front Radiat Ther Oncol (2010) 42:150–6. doi:10.1159/000262470
53. Park C, Papiez L, Zhang S, Story M, Timmerman RD. Universal survival curve and single fraction equivalent dose: useful tools in understanding potency of ablative radiotherapy. Int J Radiat Oncol Biol Phys (2008) 70:847–52. doi:10.1016/j.ijrobp.2007.10.059
54. Guerrero M, Li XA. Extending the linear–quadratic model for large fraction doses pertinent to stereotactic radiotherapy. Phys Med Biol (2004) 49:4825–35. doi:10.1088/0031-9155/49/20/012
55. Brenner DJ, Hlatky LR, Hahnfeldt PJ, Hall EJ, Sachs RK. A convenient extension of the linear–quadratic model to include redistribution and reoxygenation. Int J Radiat Oncol Biol Phys (1995) 32:379–90. doi:10.1016/0360-3016(95)00544-9
56. Strigari L, Benassi M, Sarnelli A, Polico R, D’Andrea M. A modified hypoxia-based TCP model to investigate the clinical outcome of stereotactic hypofractionated regimes for early stage non-small-cell lung cancer (NSCLC). Med Phys (2012) 39:4502–14. doi:10.1118/1.4730292
57. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med (1971) 285:1182–6. doi:10.1056/NEJM197111182852108
58. Denekamp J. Vascular endothelium as the vulnerable element in tumours. Acta Radiol Oncol (1984) 23:217–25. doi:10.3109/02841868409136015
59. Kocher M, Treuer H, Voges J, Hoevels M, Sturm V, Müller RP. Computer simulation of cytotoxic and vascular effects of radiosurgery in solid and necrotic brain metastases. Radiother Oncol (2000) 54:149–56. doi:10.1016/S0167-8140(99)00168-1
60. Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science (2001) 293:293–7. doi:10.1126/science.1060191
61. Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science (2003) 300:1155–9. doi:10.1126/science.1082504
62. Fuks Z, Alfieri A, Haimovitz-Friedman A, Seddon A, Cordon-Cardo C. Intravenous basic fibroblast growth factor protects the lung but not mediastinal organs against radiation-induced apoptosis in vivo. Cancer J Sci Am (1995) 1:62–72.
63. Ch’ang HJ, Maj JG, Paris F, Xing HR, Zhang J, Truman JP, et al. ATM regulates target switching to escalating doses of radiation in the intestines. Nat Med (2005) 11:484–90. doi:10.1038/nm1237
64. Truman JP, García-Barros M, Kaag M, Hambardzumyan D, Stancevic B, Chan M, et al. Endothelial membrane remodeling is obligate for anti-angiogenic radiosensitization during tumor radiosurgery. PLoS One (2010) 5:e12310. doi:10.1371/journal.pone.0012310
65. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24:589–602. doi:10.1016/j.ccr.2013.09.014
66. Sofia Vala I, Martins LR, Imaizumi N, Nunes RJ, Rino J, Kuonen F, et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS One (2010) 5:e11222. doi:10.1371/journal.pone.0011222
67. Lerman OZ, Greives MR, Singh SP, Thanik VD, Chang CC, Seiser N, et al. Low-dose radiation augments vasculogenesis signaling through HIF-1-dependent and -independent SDF-1 induction. Blood (2010) 116:3669–76. doi:10.1182/blood-2009-03-213629
68. Thanik VD, Chang CC, Lerman OZ, Greives MR, Le H, Warren SM, et al. Cutaneous low-dose radiation increases tissue vascularity through upregulation of angiogenic and vasculogenic pathways. J Vasc Res (2010) 47:472–80. doi:10.1159/000313875
69. Siemionow M, Mee J, Porvasnik S, Krapohl BD, Ozer K, Piza P, et al. Effects of 8-Gy radiation on the microcirculation of muscle flaps in the rat. Plast Reconstr Surg (1999) 104:1372–8. doi:10.1097/00006534-199910000-00020
70. Schneider BF, Eberhard DA, Steiner LE. Histopathology of arteriovenous malformations after gamma knife radiosurgery. J Neurosurg (1997) 87:352–7. doi:10.3171/jns.1997.87.3.0352
71. Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest (2010) 120:694–705. doi:10.1172/JCI40283
72. Martin BJ. Inhibiting vasculogenesis after radiation: a new paradigm to improve local control by radiotherapy. Semin Radiat Oncol (2013) 23:281–7. doi:10.1016/j.semradonc.2013.05.002
73. Chen FH, Chiang CS, Wang CC, Fu SY, Tsai CS, Jung SM, et al. Vasculatures in tumors growing from preirradiated tissues: formed by vasculogenesis and resistant to radiation and antiangiogenic therapy. Int J Radiat Oncol Biol Phys (2011) 80:1512–21. doi:10.1016/j.ijrobp.2011.02.055
74. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science (1997) 275:964–7. doi:10.1126/science.275.5302.964
75. Aicher A, Rentsch M, Sasaki K, Ellwart JW, Fändrich F, Siebert R, et al. Nonbone marrow-derived circulating progenitor cells contribute to postnatal neovascularization following tissue ischemia. Circ Res (2007) 100:581–9. doi:10.1161/01.RES.0000259562.63718.35
76. Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell (2008) 13:193–205. doi:10.1016/j.ccr.2007.11.032
77. Kozin SV, Duda DG, Munn LL, Jain RK. Neovascularization after irradiation: what is the source of newly formed vessels in recurring tumors? J Natl Cancer Inst (2012) 104:899–905. doi:10.1093/jnci/djs239
78. Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res (2010) 70:5679–85. doi:10.1158/0008-5472.CAN-09-4446
79. Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol (2013) 4:157. doi:10.3389/fphys.2013.00157
80. Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell (2006) 124:175–89. doi:10.1016/j.cell.2005.10.036
81. Kozin SV, Duda DG, Munn LL, Jain RK. Is vasculogenesis crucial for the regrowth of irradiated tumours? Nat Rev Cancer (2011) 11:532. doi:10.1038/nrc2007-c1
82. Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature (2010) 468:824–8. doi:10.1038/nature09557
83. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature (2010) 468:829–33. doi:10.1038/nature09624
84. Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A (2011) 108:4274–80. doi:10.1073/pnas.1016030108
85. Brown JM. The hypoxic cell: a target for selective cancer therapy – eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res (1999) 59:5863–70.
86. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer (2008) 8:967–75. doi:10.1038/nrc2540
87. Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol (1953) 26:638–48. doi:10.1259/0007-1285-26-312-638
88. Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res (1996) 56:4509–15.
89. Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol (2005) 77:18–24. doi:10.1016/j.radonc.2005.06.038
90. Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol (2007) 25:4066–74. doi:10.1200/JCO.2007.12.7878
91. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol (2012) 9:674–87. doi:10.1038/nrclinonc.2012.171
92. Kallman RF. The phenomenon of reoxygenation and its implications for fractionated radiotherapy. Radiology (1972) 105:135–42.
93. Ruggieri R, Naccarato S, Nahum AE. Severe hypofractionation: non-homogeneous tumour dose delivery can counteract tumour hypoxia. Acta Oncol (2010) 49:1304–14. doi:10.3109/0284186X.2010.486796
94. Carlson DJ, Keall PJ, Loo BW Jr, Chen ZJ, Brown JM. Hypofractionation results in reduced tumor cell kill compared to conventional fractionation for tumors with regions of hypoxia. Int J Radiat Oncol Biol Phys (2011) 79:1188–95. doi:10.1016/j.ijrobp.2010.10.007
95. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell (2008) 13:206–20. doi:10.1016/j.ccr.2008.01.034
96. Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med (2006) 12:557–67. doi:10.1038/nm0806-978
97. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121:335–48. doi:10.1016/j.cell.2005.02.034
98. Costello CM, McCullagh B, Howell K, Sands M, Belperio JA, Keane MP, et al. A role for the CXCL12 receptor, CXCR7, in the pathogenesis of human pulmonary vascular disease. Eur Respir J (2012) 39:1415–24. doi:10.1183/09031936.00044911
99. Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes HG, et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One (2010) 5:e9175. doi:10.1371/journal.pone.0009175
100. Hoffmann F, Müller W, Schütz D, Penfold ME, Wong YH, Schulz S, et al. Rapid uptake and degradation of CXCL12 depend on CXCR7 carboxyl-terminal serine/threonine residues. J Biol Chem (2012) 287:28362–77. doi:10.1074/jbc.M111.335679
101. Zhang XY, Su C, Cao Z, Xu SY, Xia WH, Xie WL, et al. CXCR7 upregulation is required for early endothelial progenitor cell-mediated endothelial repair in patients with hypertension. Hypertension (2014) 63:383–9. doi:10.1161/HYPERTENSIONAHA.113.02273
102. Rödel F, Frey B, Manda K, Hildebrandt G, Hehlgans S, Keilholz L, et al. Immunomodulatory properties and molecular effects in inflammatory diseases of low-dose x-irradiation. Front Oncol (2012) 2:120. doi:10.3389/fonc.2012.00120
103. Kroemer G, Zitvogel L. Abscopal but desirable: the contribution of immune responses to the efficacy of radiotherapy. Oncoimmunology (2012) 1:407–8. doi:10.4161/onci.20074
104. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med (2012) 366:925–31. doi:10.1056/NEJMoa1112824
105. Golden EB, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol (2012) 2:88. doi:10.3389/fonc.2012.00088
106. Santin AD, Hermonat PL, Hiserodt JC, Chiriva-Internati M, Woodliff J, Theus JW, et al. Effects of irradiation on the expression of major histocompatibility complex class I antigen and adhesion costimulation molecules ICAM-1 in human cervical cancer. Int J Radiat Oncol Biol Phys (1997) 39:737–42. doi:10.1016/S0360-3016(97)00372-6
107. Hauser SH, Calorini L, Wazer DE, Gattoni-Celli S. Radiation-enhanced expression of major histocompatibility complex class I antigen H-2Db in B16 melanoma cells. Cancer Res (1993) 53:1952–5.
108. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med (2006) 203:1259–71. doi:10.1084/jem.20052494
109. Santin AD, Rose GS, Hiserodt JC, Fruehauf J, Eck LM, Garcia RI, et al. Effects of cytokines combined with high-dose gamma irradiation on the expression of major histocompatibility complex molecules and intercellular adhesion molecule-1 in human ovarian cancers. Int J Cancer (1996) 65:688–94. doi:10.1002/(SICI)1097-0215(19960301)65:5<688::AID-IJC21>3.3.CO;2-E
110. Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol (2008) 180:3132–9.
111. Hareyama M, Imai K, Oouchi A, Takahashi H, Hinoda Y, Tsujisaki M, et al. The effect of radiation on the expression of intercellular adhesion molecule-1 of human adenocarcinoma cells. Int J Radiat Oncol Biol Phys (1998) 40:691–6. doi:10.1016/S0360-3016(97)00860-2
112. Sheard MA, Uldrijan S, Vojtesek B. Role of p53 in regulating constitutive and X-radiation-inducible CD95 expression and function in carcinoma cells. Cancer Res (2003) 63:7176–84.
113. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res (2009) 15:5379–88. doi:10.1158/1078-0432.CCR-09-0265
114. Yasuda K, Nirei T, Tsuno NH, Nagawa H, Kitayama J. Intratumoral injection of interleukin-2 augments the local and abscopal effects of radiotherapy in murine rectal cancer. Cancer Sci (2011) 102:1257–63. doi:10.1111/j.1349-7006.2011.01940.x
115. Wei S, Egenti MU, Teitz-Tennenbaum S, Zou W, Chang AE. Effects of tumor irradiation on host T-regulatory cells and systemic immunity in the context of adoptive T-cell therapy in mice. J Immunother (2013) 36:124–32. doi:10.1097/CJI.0b013e31828298e6
116. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ (2008) 15:3–12. doi:10.1038/sj.cdd.4402269
117. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi:10.1038/nm1622
118. Du S, Barcellos-Hoff MH. Tumors as organs: biologically augmenting radiation therapy by inhibiting transforming growth factor beta activity in carcinomas. Semin Radiat Oncol (2013) 23:242–51. doi:10.1016/j.semradonc.2013.05.001
119. Santin AD, Hiserodt JC, DiSaia PJ, Pecorelli S, Granger GA. Differential effects of high-dose gamma irradiation on the production of transforming growth factor-beta in fresh and established human ovarian cancer. Gynecol Oncol (1996) 61:403–8. doi:10.1006/gyno.1996.0164
120. Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA. Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest (1994) 93:892–9. doi:10.1172/JCI117045
121. Huang M, Wang J, Lee P, Sharma S, Mao JT, Meissner H, et al. Human non-small cell lung cancer cells express a type 2 cytokine pattern. Cancer Res (1995) 55:3847–53.
122. Mishra P, NBanerjee D, Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J Leukoc Biol (2011) 89: doi:10.1189/jlb.0310182
123. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer (2006) 6:392–401. doi:10.1038/nrc1877
124. Hashimoto I, Kodama J, Seki N, Hongo A, Miyagi Y, Yoshinouchi M, et al. Macrophage infiltration and angiogenesis in endometrial cancer. Anticancer Res (2000) 20:4853–6.
125. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 66:605–12. doi:10.1158/0008-5472.CAN-05-4005
126. Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys (2007) 68:499–507. doi:10.1016/j.ijrobp.2007.01.041
127. Milas L. Tumor bed effect in murine tumors: relationship to tumor take and tumor macrophage content. Radiat Res (1990) 123:232–6. doi:10.2307/3577551
128. Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, et al. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res (2010) 70:1534–43. doi:10.1158/0008-5472.CAN-09-2995
129. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi:10.1038/nm1093
130. Qinfeng S, Depu W, Xiaofeng Y, Shah W, Hongwei C, Yili W. In situ observation of the effects of local irradiation on cytotoxic and regulatory T lymphocytes in cervical cancer tissue. Radiat Res (2013) 179:584–9. doi:10.1667/RR3155.1
131. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol (2005) 174:7516–23.
132. Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res (2009) 69:3086–94. doi:10.1158/0008-5472.CAN-08-2826
133. Nishisaka N, Maini A, Kinoshita Y, Yasumoto R, Kishimoto T, Jones RF, et al. Immunotherapy for lung metastases of murine renal cell carcinoma: synergy between radiation and cytokine-producing tumor vaccines. J Immunother (1999) 22:308–14. doi:10.1097/00002371-199907000-00004
134. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res (2011) 71:2488–96. doi:10.1158/0008-5472.CAN-10-2820
135. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol (2012) 189:558–66. doi:10.4049/jimmunol.1200563
136. Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys (2012) 83:1306–10. doi:10.1016/j.ijrobp.2011.09.049
137. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10:554–67. doi:10.1038/nri2808
138. Kuhlmann UC, Chwieralski CE, Reinhold D, Welte T, Buhling F. Radiation-induced matrix production of lung fibroblasts is regulated by interleukin-8. Int J Radiat Biol (2009) 85:138–43. doi:10.1080/09553000802641136
139. Rodningen OK, Overgaard J, Alsner J, Hastie T, Borresen-Dale AL. Microarray analysis of the transcriptional response to single or multiple doses of ionizing radiation in human subcutaneous fibroblasts. Radiother Oncol (2005) 77:231–40. doi:10.1016/j.radonc.2005.09.020
140. Tachiiri S, Katagiri T, Tsunoda T, Oya N, Hiraoka M, Nakamura Y. Analysis of gene-expression profiles after gamma irradiation of normal human fibroblasts. Int J Radiat Oncol Biol Phys (2006) 64:272–9. doi:10.1016/j.ijrobp.2005.08.030
141. Kis E, Szatmári T, Keszei M, Farkas R, Esik O, Lumniczky K, et al. Microarray analysis of radiation response genes in primary human fibroblasts. Int J Radiat Oncol Biol Phys (2006) 66:1506–14. doi:10.1016/j.ijrobp.2006.08.004
142. Andreassen CN, Alsner J. Genetic variants and normal tissue toxicity after radiotherapy: a systematic review. Radiother Oncol (2009) 92:299–309. doi:10.1016/j.radonc.2009.06.015
143. Martinez-Zubiaurre I, Fenton C, Taman H, Pettersen I, Hellevik T, Paulssen R. Tumorigenic responses of cancer-associated stromal fibroblasts after ablative radiotherapy: a transcriptome-profiling study. J Cancer Ther (2013) 4:208–50. doi:10.4236/jct.2013.41031
144. Hawsawi NM, Ghebeh H, Hendrayani SF, Tulbah A, Al-Eid M, Al-Tweigeri T, et al. Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res (2008) 68:2717–25. doi:10.1158/0008-5472.CAN-08-0192
145. Papadopoulou A, Kletsas D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int J Oncol (2011) 39:989–99. doi:10.3892/ijo.2011.1132
146. Hellevik T, Pettersen I, Berg V, Winberg JO, Moe BT, Bartnes K, et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat Oncol (2012) 7:59. doi:10.1186/1748-717X-7-59
147. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol (2009) 11:973–9. doi:10.1038/ncb1909
148. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol (2011) 192:547–56. doi:10.1083/jcb.201009094
149. Velarde MC, Demaria M, Campisi J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip Top Gerontol (2013) 38:17–27. doi:10.1159/000343572
150. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol (2013) 75:685–705. doi:10.1146/annurev-physiol-030212-183653
151. Friedman N. The effects of irradiation on breast cancer and the breast. CA Cancer J Clin (1988) 38:368–71. doi:10.3322/canjclin.38.6.368
152. Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med (2011) 17:320–9. doi:10.1038/nm.2328
153. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432:332–7. doi:10.1038/nature03096
154. Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle (2006) 5:1597–601. doi:10.4161/cc.5.15.3112
155. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell (2010) 17:135–47. doi:10.1016/j.ccr.2009.12.041
156. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A (2010) 107:20009–14. doi:10.1073/pnas.1013805107
157. Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res (2004) 64:3215–22. doi:10.1158/0008-5472.CAN-03-2464
158. Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res (2007) 67:3117–26. doi:10.1158/0008-5472.CAN-06-3452
159. Liu D, Hornsby PJ. Fibroblast stimulation of blood vessel development and cancer cell invasion in a subrenal capsule xenograft model: stress-induced premature senescence does not increase effect. Neoplasia (2007) 9:418–26. doi:10.1593/neo.07205
160. Nikitina EY, Gabrilovich DI. Combination of gamma-irradiation and dendritic cell administration induces a potent antitumor response in tumor-bearing mice: approach to treatment of advanced stage cancer. Int J Cancer (2001) 94:825–33. doi:10.1002/1097-0215(20011215)94:6<825::AID-IJC1545>3.0.CO;2-5
161. Teitz-Tennenbaum S, Li Q, Rynkiewicz S, Ito F, Davis MA, McGinn CJ, et al. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res (2003) 63:8466–75.
162. Kamochi N, Nakashima M, Aoki S, Uchihashi K, Sugihara H, Toda S, et al. Irradiated fibroblast-induced bystander effects on invasive growth of squamous cell carcinoma under cancer-stromal cell interaction. Cancer Sci (2008) 99:2417–27. doi:10.1111/j.1349-7006.2008.00978.x
163. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res (2008) 68:918–26. doi:10.1158/0008-5472.CAN-07-5714
Keywords: stereotactic ablative radiotherapy, tumor microenvironment, hypoxia, angiogenesis, cancer-immunity, cancer-associated fibroblasts
Citation: Hellevik T and Martinez-Zubiaurre I (2014) Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front. Oncol. 4:1. doi: 10.3389/fonc.2014.00001
Received: 31 October 2013; Accepted: 03 January 2014;
Published online: 21 January 2014.
Edited by:
Juan José Lasarte, Centro de Investigación Médica Aplicada, SpainReviewed by:
Agnès Noël, University of Liege, BelgiumMichael Gough, Providence Portland Medical Center, USA
Copyright: © 2014 Hellevik and Martinez-Zubiaurre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iñigo Martinez-Zubiaurre, Translational Cancer Research Group, Department of Clinical Medicine, University of Tromsø, N-9037 Tromsø, Norway e-mail:aW5pZ28ubWFydGluZXpAdWl0Lm5v