 Clifford Liongue
Clifford Liongue Alister Curtis Ward
Alister Curtis Ward- 1School of Medicine, Deakin University, Geelong, VIC, Australia
- 2Strategic Research Centre in Molecular and Medical Research, Deakin University, Geelong, VIC, Australia
Granulocyte colony-stimulating factor is a cytokine able to stimulate both myelopoiesis and hematopoietic stem cell mobilization, which has seen it used extensively in the clinic to aid hematopoietic recovery. It acts specifically via the homodimeric granulocyte colony-stimulating factor receptor (G-CSFR), which is principally expressed on the surface of myeloid and hematopoietic progenitor cells. A number of pathogenic mutations have now been identified in CSF3R, the gene encoding G-CSFR. These fall into distinct classes, each of which is associated with a particular spectrum of myeloid disorders, including malignancy. This review details the various CSF3R mutations, their mechanisms of action, and contribution to disease, as well as discussing the clinical implications of such mutations.
G-CSF and Its Receptor
Granulocyte colony-stimulating factor (G-CSF, also called CSF3) augments the production and function of neutrophilic granulocytes, which play an essential role combatting infection, especially those of a bacterial or fungal nature (1–5). G-CSF acts to mobilize hematopoietic precursor cells and stimulate the proliferation and differentiation of myeloid cells, particularly along the neutrophilic lineage, as well as activate various functions of mature neutrophils (6). These properties have seen G-CSF widely used in the treatment of neutropenic conditions, including severe congenital neutropenia (SCN) (7–9), and those associated with chemotherapy (10–12). G-CSF has also been extensively used for harvesting of HSCs from the peripheral blood, thereby overcoming the requirement for bone marrow transplantations in many instances (13, 14).
The biological actions of G-CSF are mediated via docking to a homomeric receptor found on the surface of target cells, granulocyte colony-stimulating factor receptor (G-CSFR) (also called CSF3R) (15). The G-CSFR is a member of the hematopoietin receptor superfamily, which has no intrinsic tyrosine kinase activity but upon ligand-binding undergoes conformational changes to stimulate multiple tyrosine kinases associated with its cytoplasmic domain. These include Janus kinases (JAKs), especially JAK1 and JAK2 (16–19), members of the SRC kinase family, especially LYN and HCK (20–22), as well as SYK (20) and TNK (23). Important pathways activated downstream include the signal transducer and activator of transcription (STAT) proteins, particularly STAT3 and STAT5 (17, 18, 24, 25), the phosphatidyl inositol 3-kinase (PI3-K)–AKT pathway (21, 26, 27), and the RAS–MAPK pathway (28–30). Signaling via the G-CSFR is tightly regulated, including by members of the SOCS family, especially SOCS3 and CISH (31, 32), as well as the tyrosine phosphatases SHP-1 (26, 33) and SHP-2 (34, 35).
Role of G-CSFR Mutations in Myeloid Disorders
A large number of mutations in the gene encoding the G-CSFR, designated CSF3R, have now been described. These mutations can be placed into a number of distinct classes that relate to the type of mutation and their biological and clinical consequences (Figure 1). Mostly these relate to perturbations of the myeloid lineage, including SCN, Myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and chronic neutrophilic leukemia (CNL). To avoid potential confusion over mutation nomenclature, this review provides residue numbers relative to those of the mature G-CSFR in the format suggested by the Human Genome Variation Society, but with the alternate numbering that includes the cleaved signal sequence given in parenthesis in each case, since these have also been used in the literature.
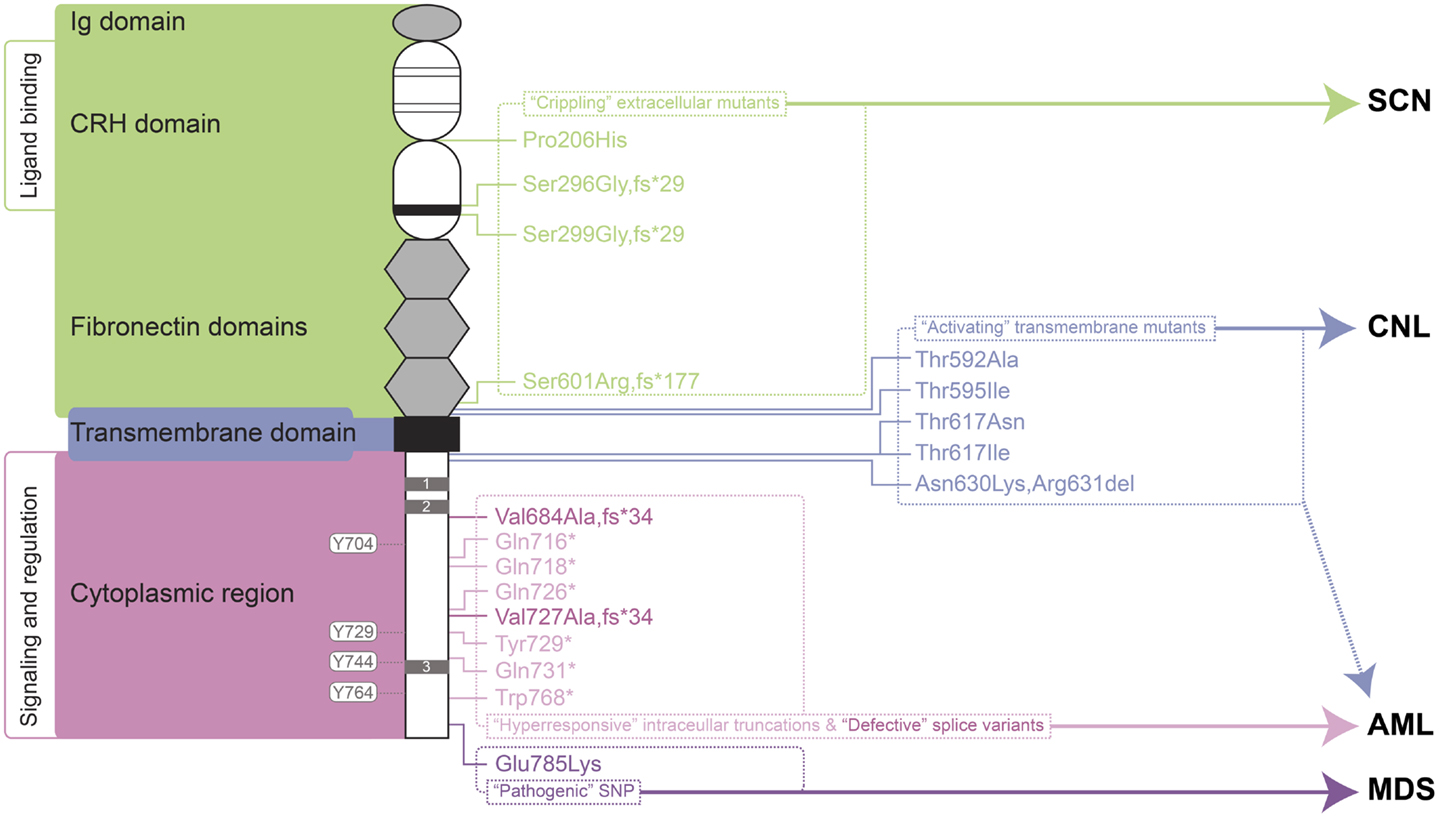
Figure 1. Granulocyte colony-stimulating factor receptor perturbations in disease. Schematic representation of the mature G-CSFR (RefSeq NP_000751.3), showing important subdomains and residues conserved among members of the hematopoietin receptor superfamily, including the N-terminal Ig domain, four conserved cysteines (thin line), and WSXWS motif (thick line) within the CRH domain, fibronectin, and transmembrane domains as well as Box 1–3 (gray rectangles) and important tyrosine residues within the cytoplasmic region. The relative positions of various classes of mutation are indicated on the right along with the respective clinical manifestations of these and other G-CSFR perturbations. Abbreviations: Ig, immunoglobulin-like; CRH, cytokine receptor homology; SCN, severe congenital neutropenia; CNL, chronic neutrophilic leukemia; MDS, myelodysplastic syndrome; AML, acute myeloid leukemia.
“Crippling” Extracellular Mutants
One class of mutations has been identified affecting the extracellular domain of the G-CSFR in patients with SCN (36–38) or chronic idiopathic neutropenia (CIN) (39). These mutations have in common the property of not only being defective themselves, but also activating in a dominant-negative manner to cripple co-expressed wild-type receptors (36–38). The first of these mutations described was a germline p.Pro206His (p.Pro229His) change that disrupted a conserved di-proline “hinge” motif located between two halves of the ligand-binding cytokine receptor homology (CRH) domain. This disrupted the normal architecture of the ligand/receptor complex, with severe consequences for G-CSF-mediated signal transduction and cellular responses (36). Two other mutants represent deletions of the CSF3R gene and concomitant alterations in reading frame that yield G-CSFR proteins consisting of extracellular regions truncated at the WSXWS motif followed by a novel sequence and a premature stop: the somatic p.Ser296Gly,fs*29 (p.Ser319Gly,fs*29) mutation (38) and the germline p.Ser299Gly,fs*29 (p.Ser322Gly,fs*29) (37). Finally, the CIN-associated p.Ser601Arg,fs*177 (p.Ser624Arg,fs*177) mutation involved a frameshift that truncates the receptor after the fibronectin domains (39). While not directly promoting malignancy, the neutropenic conditions that this class of mutation produces are likely to create susceptibility to other changes that can. Indeed, one SCN patient with this type of mutation subsequently acquired additional truncating mutations in the G-CSFR (40), while the CIN patient went on to develop acute myeloid/natural killer cell leukemia, although whether the CSF3R mutation played a role in the latter was not determined (39).
“Activating” Transmembrane Mutants
Another class of CSF3R mutations affects the transmembrane domain and adjacent residues of the encoded receptor. This class of mutations appears to act by stabilizing transmembrane helix–helix interactions in the absence of ligand, creating an active dimeric configuration that leads to constitutive (and enhanced) activation (41). These are analogous to the activating mutations found in the thrombopoietin receptor, c-MPL, which are associated with hereditary or acquired thrombocythemia (42, 43), or those in the βc chain of the heterodimeric IL-3R family identified in vitro (44, 45).
The p.Thr595Ile (p.Thr618Ile) mutation was initially described as a late somatic mutation in the development of AML in an SCN patient already bearing an alternate G-CSFR mutation (46). However, p.Thr595Ile has subsequently been identified as a common mutation in CNL (23, 47), with the adjacent p.Thr592Ala (p.Thr615Ala) mutation alternatively found in other cases of CNL (23). The p.Thr595Ile mutation is also less commonly observed in atypical chronic myelogenous leukemia (aCML) (23), chronic myelomonocytic leukemia (CMML) (48), de novo AML (23, 48, 49), as well as in cases of early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) (23). G-CSFR forms containing either the p.Thr595Ile or p.Thr592Ala mutation supported G-CSF-independent growth of Ba/F3 cells, although growth was similar to wild-type receptor at high G-CSF concentrations (48). Bone marrow transduced with the p.Thr595Ile mutant also resulted in G-CSF-independent growth (46), which could be replicated by a p.Thr595Val mutant, suggesting the change to a hydrophobic amino acid was sufficient (49). Ba/F3 cells expressing the p.Thr595Ile mutant showed constitutive activation of JAK2, SRC, TNK, STAT3, and STAT5 (23, 48), but not AKT and ERK, as well as enhanced ROS production (48). Signaling from the mutant was found to be sensitive to various JAK kinase inhibitors, including ruxolinitib and tofacitinib (23, 48), with some evidence of clinical efficacy (23), but not to dasatinib that targets a number of tyrosine kinases, including SRC and TNK (23, 48).
The p.Thr617Asn (p.Thr640Asn) mutation was first identified in a single case of AML (50). Further studies identified this – and the alternate p.Thr617Ile (p.Thr640Ile) – as rare, somatic mutations in de novo AML (49, 51). However, a germline p.Thr617Asn mutation was also identified as the cause of autosomal dominant hereditary neutrophilia, where it showed complete penetrance (52). Interestingly, one of the affected individuals progressed to a myelodysplastic syndrome type disease (52), further implicating this mutation as predisposing toward myeloid malignancy. In addition to neutrophilia, patients harboring p.Thr617Asn possessed increased numbers of CD34+ cells, which were able to proliferate and terminally differentiate in the absence of G-CSF, and induce a myeloproliferative (MPD)-like disorder in mice. Patient CD34+ cells showed constitutive phosphorylation of JAK2, STAT3, STAT5, and ERK, which were hyperactivated by G-CSF compared to wild-type cells (52). Lineage-negative bone marrow cells retrovirally transduced with the p.Thr617Asn mutant G-CSFR caused neutrophilia when transplanted into irradiated mice (52). The p.Thr617Asn mutation also supported factor-independent growth and survival in Ba/F3 cells, with weak constitutive phosphorylation of the receptor, JAK2, STAT3, and ERK, and also enabled transduced CD34+ cells to undergo myeloid differentiation in the absence of G-CSF (51).
Finally, an in-frame three nucleotide deletion has been identified in MDS that replaces two amino acids with an alternate residue, p.Asn630Lys,Arg631del (p.Asn653Lys,Arg654del). This mutation resulted in prolonged signaling following ligand stimulation (53).
“Hyperresponsive” Intracellular Truncations
By far the most studied clinical abnormalities of the CSF3R gene are a series of acquired nonsense mutations identified in a subset of SCN patients with a propensity to progress to leukemia. These somatic mutations typically affect a single allele to truncate between 82 and 98 amino acids from the carboxyl-terminus of the receptor, such as p.Gln718* (p.Gln741*) and pGln731* (p.Gln754*) (54, 55). These truncated receptors show normal affinity for G-CSF (56), but mediate heightened growth and diminished maturation in response to ligand, acting dominantly over wild-type receptors (54). Truncated G-CSFRs are not the primary cause of SCN, although they may exacerbate it to a modest extent (57–60). However, it is clear that SCN patients carrying truncating G-CSFR mutations show a particularly strong predisposition to both MDS and AML (61, 62). Indeed in SCN patients progressing to AML, the most common mutations identified are in CSF3R (82%), followed by RAS (~50%) and monosomy 7 (63), and when CSF3R mutations are present, 100% of blasts carry the mutation (54, 63). However, since mutations are not always seen in AML and can spontaneously disappear (64), progression to leukemia is not inevitable.
A mouse line carrying a truncated G-CSFR “knock-in” allele (57) or one transgenically expressing a truncated human G-CSFR (58) exhibited mild neutropenia, with an increased percentage of immature myeloid cells that were defective in maturation ex vivo (58, 65). An alternate mouse line with a targeted receptor truncation displayed normal neutrophil numbers, although the truncated form of the receptor was significantly overexpressed relative to the wild-type (59). However, all three studies revealed a hyper-responsiveness to G-CSF, with exogenous G-CSF producing elevated numbers of neutrophils compared to wild-type mice (57–59), due to increased myeloid progenitor proliferation (58, 65). Another study confirmed that G-CSFR truncations conferred a strong clonal HSC advantage that was also dependent on exogenous G-CSF (66), providing insight into how these mutants may contribute to their frequent progression to MDS/AML. Notably, expression of the truncated receptor in mice was not by itself leukemogenic, since no spontaneous leukemia has been reported in mice hetero- or homozygous for the mutation (57, 59). However, the truncated G-CSFR was found to co-operate with PML–RARa to induce AML in mice, where it decreased latency in a G-CSF-dependent manner, leading to higher blast counts and increased myelosuppression (67).
Investigation into the molecular mechanisms of G-CSFR signal transduction has helped to explain the dominant hyperproliferative function of truncated G-CSFRs. These mutant receptors exhibit higher and more sustained activation in comparison to wild-type receptors, with a heavily reduced “off-rate” (65, 68, 69). This is partly a result of impaired internalization due to the combined loss of a conserved di-leucine containing motif in Box 3 (69, 70), and a less well-defined motif spanning residues 756–769 (34). However, direct negative regulation is also blunted, due to the loss of recruitment sites on the truncated receptors, including those for the receptor-associated tyrosine phosphatases SHP-1 (at an undefined site in the C-terminus) (71) and SHP-2 (at Y724) (34), and two members of the SOCS family, CISH (at Y729 and Y744) (32) and SOCS3 (at Y729) (34), the latter exacerbated by decreased SOCS3 transcription as a result of reduced STAT3 activation by truncated receptors (34).
Cells expressing truncated G-CSFR receptors are also hypersensitive to ligand (54, 70). This is associated with an altered dose–response of STAT3:STAT5 activation, the ratio of which is drastically reduced at low concentrations of G-CSF (24). Since STAT5 contributes to G-CSF proliferative responses (72), while STAT3 is inhibitory (73–75), the reduced STAT3:STAT5 ratio may shift the balance toward proliferation, explaining the G-CSF hypersensitivity (54, 56).
Granulocyte colony-stimulating factor receptor truncation impacts on the length and magnitude of receptor activation, and particularly of STAT5 (69–71), pathways downstream of PI3-K, such as AKT (27, 76), as well as SRC (23). Dominant-negative STAT5 has been shown to inhibit the hyperproliferative function of truncated G-CSFRs in vitro (77), while the absence of STAT5 abrogated the clonal HSC advantage conferred by these receptors in vivo (66). Other pathways also contribute to proliferation and survival, including PI3-K, MAPK, and STAT3 (76–78). Interestingly, receptor truncations are sensitive to the multi-kinase inhibitor dasatinib, but not to JAK inhibitors (23), suggesting an intrinsic difference in comparison to the activating transmembrane mutants. Truncated receptors have also been shown to increase ROS production (79), potentially creating genotoxic stress to facilitate addition mutations in cells expressing these receptors.
Defective Splice Variants
A presumably somatic single base change in CSF3R adjacent to a cryptic splice-donor site has been identified in blasts of a de novo AML patient. This resulted in high expression of an alternate splice variant that generated a G-CSFR protein in which the C-terminal 130 amino acids are replaced with a different 34 amino acids from an alternate reading frame, p.Val684Ala,fs*34 (p.Val707Ala,fs*34) (80). The primary AML blast cells of this patient failed to respond to G-CSF in proliferation assays in vitro, despite responsiveness to IL-3 or GM-CSF being maintained. This variant was unable to transduce either proliferation or maturation signals in murine cell systems. By corollary, AML cells show a tendency for significantly increased levels of a normally minor CSF3R transcript, class IV (81), which encodes a similar G-CSFR protein in which the C-terminal 87 amino acids are replaced with the same alternate 34 amino acids, p.Val727Ala,fs*34 (p.Val750Ala,fs*34). The authors argue that the altered balance of class IV to normal (class I) receptors might contribute to AML, due to the ability of the class IV receptor to block maturation.
Pathogenic SNP
A CSF3R SNP that is present in ~6% of the population leads to a p.Glu785Lys (p.Glu808Lys) amino acid substitution in the intracellular region of the G-CSF, which predisposes individuals to high-risk MDS (82). Interestingly, blasts from an individual who developed AML following high-risk MDS were found to be homozygous for this allele (83), providing further evidence of the potential pathogenicity of this SNP. Although the mechanism of action remains unknown, the variant receptor appears functional, but can act in a dominant-negative manner to reduce colony formation compared to the wild-type receptor (82, 83).
Conclusion
Granulocyte colony-stimulating factor has proven to be an effective therapy in a range of life-threatening conditions or to aid in the recovery of medical treatments, such as in the treatment of neutropenia following chemotherapy. However, the evidence suggests that G-CSFR mutations contribute to several disorders, including in settings where G-CSF may be used therapeutically. It has been suggested that use of G-CSF in SCN may allow the selective expansion of clones containing truncating CSF3R mutations. However, the available data are complicated, making conclusions difficult. One study reported no significant relationship between age of MDS/AML onset and G-CSF dose or duration of therapy (63). However, another study suggested that the risk of leukemia in SCN patients increased with the degree of G-CSF therapy (84). However, higher doses may also reflect a more severe underlying disease with a higher propensity to MDS/AML. In addition, SCN patients developed AML prior to the advent of G-CSF therapy. In line with this, one SCN patient progressed to CMML in the absence of G-CSF treatment, but expressed a truncated G-CSFR (85). Thus it is possible that the mutant receptor form may have a selective advantage in the absence of treatment, perhaps due to the elevated G-CSF levels seen in SCN patients as a result of their neutropenia (63). However, G-CSF therapy is not a factor in other classes of CSF3R mutation, such as those leading to CNL. A number of pharmacologic agents are now available that target signaling pathways downstream of the G-CSFR (Figure 2), providing hope for effective treatment strategies for patients harboring G-CSFR mutations. Indeed, recent studies have begun to elucidate how these might specifically combat the aberrant signaling elicited by “activating” and “hyperresponsive” G-CSFR mutations (23, 48).
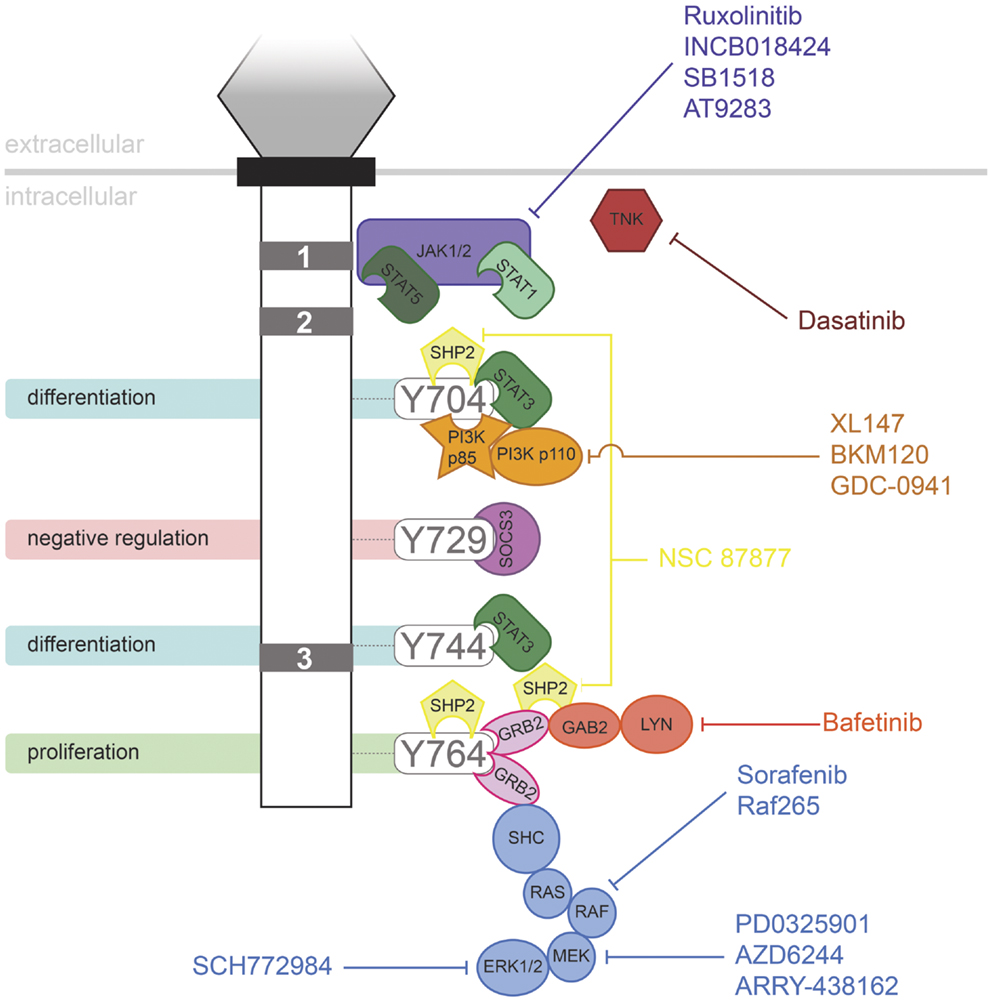
Figure 2. Inhibitors of signaling pathways downstream of G-CSFR. Schematic representation of the intracellular domain of G-CSFR, showing the important Box 1–3 sequences (gray rectangles), as well as the tyrosine residues that serve important docking sites for the downstream signaling proteins indicated. Known inhibitors of these are shown.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by funding from Deakin University via a Central Research Grant (to Alister Curtis Ward) and an Alfred Deakin Postdoctoral Research Fellowship (to Clifford Liongue).
References
1. Nicola NA. Hemopoietic cell growth factors and their receptors. Annu Rev Biochem (1989) 58:45–77. doi: 10.1146/annurev.biochem.58.1.45
2. Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor deficiency, and impaired neutrophil mobilization. Blood (1994) 84:1737–46.
3. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity (1996) 5:491–501. doi:10.1016/S1074-7613(00)80504-X
4. Santos MD, Yasuike M, Hirono I, Aoki T. The granulocyte colony-stimulating factors (CSF3s) of fish and chicken. Immunogenetics (2006) 58:422–32. doi:10.1007/s00251-006-0106-5
5. Liongue C, Hall C, O’Connell B, Crozier P, Ward AC. Zebrafish granulocyte colony-stimulating factor receptor signalling promotes myelopoiesis and myeloid cell migration. Blood (2009) 113:2535–46. doi:10.1182/blood-2008-07-171967
6. Touw IP, van de Geijn GJ. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci (2007) 12:800–15. doi:10.2741/2103
7. Bonilla M, Gillio A, Ruggeiro M, Kernan N, Brochstein J, Abboud M, et al. Effects of recombinant human granulocyte colony-stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med (1989) 320:1574–80. doi:10.1056/NEJM198906153202402
8. Dale DC, Bonilla MA, Davis MW, Nakanshi AM, Hammond WP, Kurtzberg J, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (Filgrastim) for treatment of severe chronic neutropenia. Blood (1993) 81:2496–502.
10. Hartmann LC, Tschetter LK, Habermann TM, Ebbert LP, Johnson PS, Mailliard JA, et al. Granulocyte colony-stimulating factor in severe chemotherapy-induced afebrile neutropenia. N Engl J Med (1997) 336:1776–80. doi:10.1056/NEJM199706193362502
11. Levine JE, Boxer LA. Clinical applications of hematopoietic growth factors in pediatric oncology. Curr Opin Hematol (2002) 9:222–7. doi:10.1097/00062752-200205000-00008
12. Bohlius J, Reiser M, Schwarzer G, Engert A. Impact of granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF in patients with malignant lymphoma: a systematic review. Br J Haematol (2003) 122:413–23. doi:10.1046/j.1365-2141.2003.04450.x
13. Bensinger WI, Weaver CH, Appelbaum FR, Rowley S, Demirer T, Sanders J, et al. Transplantation of allogeneic peripheral blood stem cells mobilized by recombinant human granulocyte colony-stimulating factor. Blood (1995) 85:1655–8.
14. Demirer T, Ayli M, Ozcan M, Gunel N, Haznedar R, Dagli M, et al. Mobilization of peripheral blood stem cells with chemotherapy and recombinant human granulocyte colony-stimulating factor (rhG-CSF): a randomized evaluation of different doses of rhG-CSF. Br J Haematol (2002) 116:468–74. doi:10.1046/j.1365-2141.2002.03264.x
15. Fukunaga R, Ishizaka Ikeda E, Seto Y, Nagata S. Expression cloning of a receptor for murine granulocyte colony-stimulating factor. Cell (1990) 61:341–50. doi:10.1016/0092-8674(90)90814-U
16. Nicholson SE, Oates AC, Harpur AG, Ziemiecki A, Wilks AF, Layton JE. Tyrosine kinase JAK1 is associated with the granulocyte-colony stimulating factor receptor and both become tyrosine phosphorylated after receptor activation. Proc Natl Acad Sci U S A (1994) 91:2985–8. doi:10.1073/pnas.91.8.2985
17. Nicholson SE, Novak U, Ziegler SF, Layton JE. Distinct regions of the granulocyte colony-stimulating factor receptor are required for tyrosine phosphorylation of the signaling molecules JAK2, Stat3, and p42, p44 MAPK. Blood (1995) 86:3698–704.
18. Tian S-S, Tapley P, Sincich C, Stein RB, Rosen J, Lamb P. Multiple signaling pathways induced by granulocyte colony-stimulating factor involving activation of JAKs, STAT5, and/or STAT3 are required for regulation of three distinct classes of immediate early genes. Blood (1996) 88:4435–44.
19. Shimoda K, Feng J, Murakami H, Nagata S, Watling D, Rogers NC, et al. Jak1 plays an essential role for receptor phosphorylation and Stat activation in response to granulocyte colony-stimulating factor. Blood (1997) 90:597–604.
20. Corey SJ, Burkhardt AL, Bolen JB, Geahlen RL, Tkatch LS, Tweardy DJ. Granulocyte colony-stimulating factor receptor signaling involves the formation of a three-component complex with Lyn and Syk protein-tyrosine kinases. Proc Natl Acad Sci U S A (1994) 91:4683–7. doi:10.1073/pnas.91.11.4683
21. Zhu QS, Xia L, Mills GB, Lowell CA, Touw IP, Corey SJ. G-CSF induced reactive oxygen species involves Lyn-PI 3-kinase-Akt and contributes to myeloid cell growth. Blood (2005) 107:1847–56. doi:10.1182/blood-2005-04-1612
22. Ward AC, Monkhouse JL, Csar XF, Touw IP, Bello PA. The Src-like kinase Hck is activated by granulocyte colony-stimulating factor (G-CSF), and docks to the activated G-CSF receptor. Biochem Biophys Res Commun (1998) 251:117–23. doi:10.1006/bbrc.1998.9441
23. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med (2013) 368:1781–90. doi:10.1056/NEJMoa1214514
24. Ward AC, Hermans MHA, Smith L, van Aesch YM, Schelen AM, Antonissen C, et al. Tyrosine-dependent and independent mechanisms of STAT3 activation by the human granulocyte colony-stimulating factor (G-CSF) receptor are differentially utilized depending on G-CSF concentration. Blood (1999) 93:113–24.
25. Ward AC, Smith L, de Koning JP, van Aesch Y, Touw IP. Multiple signals mediate proliferation, differentiation and survival from the granulocyte colony-stimulating factor receptor in myeloid 32D cells. J Biol Chem (1999) 274:14956–62. doi:10.1074/jbc.274.21.14956
26. Hunter MG, Avalos BR. Phosphatidylinositol 3′-kinase and SH2-containing inositol phosphatase (SHIP) are recruited by distinct positive and negative growth-regulatory domains in the granulocyte colony-stimulating factor receptor. J Immunol (1998) 160:4979–87.
27. Dong F, Larner AC. Activation of Akt kinase by granulocyte colony-stimulating factor (G-CSF): evidence for the role of a tyrosine kinase activity distinct from the Janus kinases. Blood (2000) 95:1656–62.
28. Bashey A, Healy L, Marshall CJ. Proliferative but not nonproliferative responses to granulocyte-colony stimulating factor are associated with rapid activation of the p21ras/MAP kinase signalling pathway. Blood (1994) 83:949–57.
29. de Koning JP, Schelen AM, Dong F, van Buitenen C, Burgering BM, Bos JL, et al. Specific involvement of tyrosine 764 of human granulocyte colony-stimulating factor receptor in signal transduction mediated by p145/Shc/GRB2 or p90/GRB2 complexes. Blood (1996) 87:132–40.
30. Rausch O, Marshall CJ. Cooperation of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways during granulocyte colony-stimulating factor-induced hemopoietic cell proliferation. J Biol Chem (1999) 274:4096–105. doi:10.1074/jbc.274.7.4096
31. Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S. Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J Immunol (2002) 169:1219–27.
32. Hunter MG, Jacob A, O’Donnell LC, Agler A, Druhan LJ, Coggeshall KM, et al. Loss of SHIP and CIS recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J Immunol (2004) 173:5036–45.
33. Ward AC, Oomen SPMA, Smith L, Gits J, van Leeuwen D, Soede-Bobok A, et al. The SH2-containing protein tyrosine phosphatase SHP-1 is induced by granulocyte colony-stimulating factor (G-CSF) and modulates signaling from the G-CSF receptor. Leukemia (2000) 14:1284–91. doi:10.1038/sj.leu.2401822
34. van de Geijn GJ, Gits J, Aarts LH, Heijmans-Antonissen C, Touw IP. G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood (2004) 104:667–74. doi:10.1182/blood-2003-08-2913
35. Futami M, Zhu QS, Whichard ZL, Xia L, Ke Y, Neel BG, et al. G-CSF receptor activation of the Src kinase Lyn is mediated by Gab2 recruitment of the Shp2 phosphatase. Blood (2011) 118:1077–86. doi:10.1182/blood-2009-12-261636
36. Ward AC, van Aesch YM, Gits J, Schelen AM, de Koning JP, van Leeuwen D, et al. Novel point mutation in the extracellular domain of the granulocyte colony-stimulating factor (G-CSF) receptor in a case of severe congenital neutropenia hyporesponsive to G-CSF treatment. J Exp Med (1999) 190:497–507. doi:10.1084/jem.190.4.497
37. Sinha S, Zhu QS, Romero G, Corey SJ. Deletional mutation of the external domain of the human granulocyte colony-stimulating factor receptor in a patient with severe chronic neutropenia refractory to granulocyte colony-stimulating factor. J Pediatr Hematol Oncol (2003) 25:791–6. doi:10.1097/00043426-200310000-00010
38. Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos BR. Novel mechanism of G-CSF refractoriness in patients with severe congenital neutropenia. Blood (2005) 105:584–91. doi:10.1182/blood-2004-07-2613
39. Papadaki HA, Kosteas T, Gemetzi C, Damianaki A, Anagnou NP, Eliopoulos GD. Acute myeloid/NK precursor cell leukemia with trisomy 4 and a novel point mutation in the extracellular domain of the G-CSF receptor in a patient with chronic idiopathic neutropenia. Ann Hematol (2004) 83:345–8. doi:10.1007/s00277-004-0862-y
40. Ward AC, Gits J, Majeed F, Aprikyan AA, Lewis RS, O’Sullivan LA, et al. Functional interaction between mutations in the granulocyte colony-stimulating factor receptor in severe congenital neutropenia. Br J Haematol (2008) 142:653–6. doi:10.1111/j.1365-2141.2008.07224.x
41. Touw IP, Beekman R. Severe congenital neutropenia and chronic neutrophilic leukemia: an intriguing molecular connection unveiled by oncogenic mutations in CSF3R. Haematologica (2013) 98:1490–2. doi:10.3324/haematol.2013.090571
42. Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood (2004) 103:4198–200. doi:10.1182/blood-2003-10-3471
43. Chaligne R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, et al. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia (2008) 22:1557–66. doi:10.1038/leu.2008.137
44. Jenkins BJ, D’Andrea R, Gonda TJ. Activating point mutations in the common beta subunit of the human GM-CSF, IL-3 and IL-5 receptors suggest the involvement of beta subunit dimerization and cell type-specific molecules in signalling. EMBO J (1995) 14:4276–87.
45. Jenkins BJ, Blake TJ, Gonda TJ. Saturation mutagenesis of the beta subunit of the human granulocyte-macrophage colony-stimulating factor receptor shows clustering of constitutive mutations, activation of ERK MAP kinase and STAT pathways, and differential beta subunit tyrosine phosphorylation. Blood (1998) 92:1989–2002.
46. Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood (2012) 119:5071–7. doi:10.1182/blood-2012-01-406116
47. Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia (2013) 27:1870–3. doi:10.1038/leu.2013.122
48. Mehta HM, Glaubach T, Long A, Lu H, Przychodzen B, Makishima H, et al. Granulocyte colony-stimulating factor receptor T595I (T618I) mutation confers ligand independence and enhanced signaling. Leukemia (2013) 27:2407–10. doi:10.1038/leu.2013.164
49. Beekman R, Valkhof M, van Strien P, Valk PJ, Touw IP. Prevalence of a new auto-activating colony stimulating factor 3 receptor mutation (CSF3R-T595I) in acute myeloid leukemia and severe congenital neutropenia. Haematologica (2013) 98:e62–3. doi:10.3324/haematol.2013.085050
50. Bernard T, Gale RE, Linch DC. Analysis of granulocyte colony-stimulating factor receptor isoforms, polymorphisms and mutations in normal haemopoietic and acute myeloid leukaemia blasts. Br J Haematol (1996) 93:527–33. doi:10.1046/j.1365-2141.1996.d01-1696.x
51. Forbes LV, Gale RE, Pizzey A, Pouwels K, Nathwani A, Linch DC. An activating mutation in the transmembrane domain of the granulocyte colony-stimulating factor receptor in patients with acute myeloid leukemia. Oncogene (2002) 21:5981–9. doi:10.1038/sj.onc.1205767
52. Plo I, Zhang Y, Le Couedic JP, Nakatake M, Boulet JM, Itaya M, et al. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J Exp Med (2009) 206:1701–7. doi:10.1084/jem.20090693
53. Awaya N, Uchida H, Miyakawa Y, Kinjo K, Matsushita H, Nakajima H, et al. Novel variant isoform of G-CSF receptor involved in induction of proliferation of FDCP-2 cells: relevance to the pathogenesis of myelodysplastic syndrome. J Cell Physiol (2002) 191:327–35. doi:10.1002/jcp.10102
54. Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med (1995) 333:487–93. doi:10.1056/NEJM199508243330804
55. Dong F, Dale DC, Bonilla MA, Freedman M, Fasth A, Neijens HJ, et al. Mutations in the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Leukemia (1997) 11:120–5. doi:10.1038/sj.leu.2400537
56. Dong F, van Buitenen C, Pouwels K, Hoefsloot LH, Löwenberg B, Touw IP. Distinct cytoplasmic regions of the human granulocyte colony-stimulating factor receptor involved in induction of proliferation and maturation. Mol Cell Biol (1993) 13:7774–81.
57. Hermans MHA, Ward AC, Antonissen C, Karis A, Lowenberg B, Touw IP. Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood (1998) 92:32–9.
58. Mitsui T, Watanabe S, Taniguchi Y, Hanada S, Ebihara Y, Sato T, et al. Impaired neutrophil maturation in truncated murine G-CSF receptor transgenic mice. Blood (2003) 101:2990–5. doi:10.1182/blood.V101.8.2990
59. McLemore ML, Poursine-Laurent J, Link DC. Increased granulocyte colony-stimulating factor responsiveness but normal resting granulopoiesis in mice carrying a targeted granulocyte colony-stimulating factor receptor mutation derived from a patient with severe congenital neutropenia. J Clin Invest (1998) 102:483–92. doi:10.1172/JCI3216
60. Ward AC, Dale DC. Genetic and molecular approaches to the diagnosis of severe congenital neutropenia. Curr Opin Hematol (2009) 16:9–13. doi:10.1097/MOH.0b013e32831952de
61. Germeshausen M, Ballmaier M, Welte K. Implications of mutations in hematopoietic growth factor receptor genes in congenital cytopenias. Ann N Y Acad Sci (2001) 938:305–20. doi:10.1111/j.1749-6632.2001.tb03599.x
62. Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood (2004) 103:4119–25. doi:10.1182/blood-2003-10-3518
63. Freedman MH, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol (2002) 39:128–33. doi:10.1053/shem.2002.31912
64. Bernard T, Gale RE, Evans JPM, Linch DC. Mutations of the granulocyte-colony stimulating factor receptor in patients with severe congenital neutropenia are not required for transformation to acute myeloid leukaemia and may be a bystander phenomenon. Br J Haematol (1998) 101:141–9. doi:10.1046/j.1365-2141.1998.00652.x
65. Hermans MHA, Antonissen C, Ward AC, Mayen AEM, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med (1999) 189:683–92. doi:10.1084/jem.189.4.683
66. Liu F, Kunter G, Krem MM, Eades WC, Cain JC, Tomasson MH, et al. Csf3r mutations in mice confer a strong clonal HSC advantage via activation of Stat5. J Clin Invest (2008) 118:946–55. doi:10.1172/JCI32704
67. Kunter G, Woloszynek JR, Link DC. A truncation mutant of Csf3r cooperates with PML-RARalpha to induce acute myeloid leukemia in mice. Exp Hematol (2011) 39:1136–43. doi:10.1016/j.exphem.2011.08.013
68. Dong F, Liu X, de Koning JP, Touw IP, Henninghausen L, Larner A, et al. Stimulation of Stat5 by granulocyte colony-stimulating factor (G-CSF) is modulated by two distinct cytoplasmic regions of the G-CSF receptor. J Immunol (1998) 161:6503–9.
69. Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood (1999) 93:447–58.
70. Hunter MG, Avalos BR. Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood (1999) 93:440–6.
71. Dong F, Qiu Y, Yi T, Touw IP, Larner AC. The carboxyl terminus of the granulocyte colony-stimulating factor receptor, truncated in patients with severe congenital neutropenia/acute myeloid leukemia, is required for SH2-containing phosphatase-1 suppression of Stat activation. J Immunol (2001) 167:6447–52.
72. Ilaria RL Jr, Hawley RG, Van Etten RA. Dominant negative mutants implicate STAT5 in myeloid cell proliferation and neutrophil differentiation. Blood (1999) 93:4154–66.
73. Shimozaki K, Nakajima K, Hirano T, Nagata S. Involvement of STAT3 in the granulocyte colony-stimulating factor-induced differentiation of myeloid cells. J Biol Chem (1997) 272:25184–9. doi:10.1074/jbc.272.40.25184
74. de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, et al. STAT3-mediated differentiation and survival of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27Kip1. Oncogene (2000) 19:3290–8. doi:10.1038/sj.onc.1203627
75. Lee CK, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity (2002) 17:63–72. doi:10.1016/S1074-7613(02)00336-9
76. Hunter MG, Avalos BR. Granulocyte colony-stimulating factor receptor mutations in severe congenital neutropenia transforming to acute myeloid leukemia confer resistance to apoptosis and enhance cell survival. Blood (2000) 95:2132–7.
77. Gits J, van Leeuwen D, Carroll HP, Touw IP, Ward AC. Multiple pathways contribute to the hyperproliferative responses from truncated granulocyte colony-stimulating factor receptors. Leukemia (2006) 20:2111–8. doi:10.1038/sj.leu.2404448
78. McLemore ML, Grewal S, Liu F, Archambault A, Poursine-Laurent J, Haug J, et al. STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity (2001) 14:193–204. doi:10.1016/S1074-7613(01)00101-7
79. Zhuang D, Qiu Y, Kogan SC, Dong F. Increased CCAAT enhancer binding protein e (C/EBPe) expression and premature apoptosis in myeloid cells expressing Gfi-1 N382S mutant associated with severe congenital neutropenia. J Biol Chem (2006) 281:10745–50. doi:10.1074/jbc.M510924200
80. Dong F, van Paassen M, van Buitenen C, Hoefsloot LH, Löwenberg B, Touw IP. A point mutation in the granulocyte colony-stimulating factor receptor (G-CSF-R) gene in a case of acute myeloid leukemia results in the overexpression of a novel G-CSF-R isoform. Blood (1995) 85:902–11.
81. White SM, Ball ED, Ehmann WC, Rao AS, Tweardy DJ. Increased expression of the differentiation-defective granulocyte colony-stimulating factor receptor mRNA isoform in acute myelogenous leukemia. Leukemia (1998) 12:899–906. doi:10.1038/sj.leu.2401062
82. Wolfler A, Erkeland SJ, Bodner C, Valkhof M, Renner W, Leitner C, et al. A functional single-nucleotide polymorphism of the G-CSF receptor gene predisposes individuals to high-risk myelodysplastic syndrome. Blood (2005) 105:3731–6. doi:10.1182/blood-2004-06-2094
83. Carapeti M, Soede-Bobok A, Hochhaus A, Sill H, Touw IP, Goldman JM, et al. Rarity of dominant-negative mutations of the G-CSF receptor in patients with blast crisis of chronic myeloid leukemia or de novo acute leukemia. Leukemia (1997) 11:1005–8. doi:10.1038/sj.leu.2400697
84. Donadieu J, Leblanc T, Bader Meunier B, Barkaoui M, Fenneteau O, Bertrand Y, et al. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica (2005) 90:45–53.
Keywords: G-CSF, G-CSFR, CSF3R, AML, SCN, CNL, MDS
Citation: Liongue C and Ward AC (2014) Granulocyte colony-stimulating factor receptor mutations in myeloid malignancy. Front. Oncol. 4:93. doi: 10.3389/fonc.2014.00093
Received: 31 January 2014; Accepted: 14 April 2014;
Published online: 01 May 2014.
Edited by:
Mignon Lee-Cheun Loh, University of California San Francisco, USAReviewed by:
Ken Lieuw, Uniformed Services University of the Health Sciences, USAHélène Cavé, Hôpital Robert Debré, France
Copyright: © 2014 Liongue and Ward. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alister Curtis Ward, School of Medicine, Deakin University, 75 Pigdons Road, Geelong, VIC 3216, Australia e-mail:YXdhcmRAZGVha2luLmVkdS5hdQ==