 Stephanie Shishido
Stephanie Shishido Halvard Bönig
Halvard Bönig Yong-Mi Kim
Yong-Mi Kim- 1Division of Hematology and Oncology, Department of Pediatrics, Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, Los Angeles, CA, USA
- 2Institute for Transfusion Medicine and Immunohematology, German Red Cross Blood Service Baden-Wuerttemberg-Hessen, Goethe University, Frankfurt, Germany
Chemotherapeutic drug resistance in acute lymphoblastic leukemia (ALL) is a significant problem, resulting in poor responsiveness to first-line treatment or relapse after transient remission. Classical anti-leukemic drugs are non-specific cell cycle poisons; some more modern drugs target oncogenic pathways in leukemia cells, although in ALL these do not play a very significant role. By contrast, the molecular interactions between microenvironment and leukemia cells are often neglected in the design of novel therapies against drug resistant leukemia. It was shown however, that chemotherapy resistance is promoted in part through cell–cell contact of leukemia cells with bone marrow (BM) stromal cells, also called cell adhesion-mediated drug resistance (CAM-DR). Incomplete response to chemotherapy results in persistence of resistant clones with or without detectable minimal residual disease (MRD). Approaches for how to address CAM-DR and MRD remain elusive. Specifically, studies using anti-functional antibodies and genetic models have identified integrin alpha4 as a critical molecule regulating BM homing and active retention of normal and leukemic cells. Pre-clinical evidence has been provided that interference with alpha4-mediated adhesion of ALL cells can sensitize them to chemotherapy and thus facilitate eradication of ALL cells in an MRD setting. To this end, Andreeff and colleagues recently provided evidence of stroma-induced and alpha4-mediated nuclear factor-κB signaling in leukemia cells, disruption of which depletes leukemia cells of strong survival signals. We here review the available evidence supporting the targeting of alpha4 as a novel strategy for treatment of drug resistant leukemia.
Introduction
Relapse of leukemia due to incomplete eradication of leukemia stem cells by conventional chemotherapy remains a problem in adult and childhood leukemia patients. The elimination of chemotherapy-refractory relapse-initiating acute lymphoblastic leukemia (ALL) cells thus remains a significant challenge and novel approaches for targeting residual leukemia cells are warranted. Even though chemotherapy kills the bulk of ALL cells, some can evade the toxicity of cytoreductive chemotherapy. Minimal residual disease (MRD) refers to the presence of these surviving cells that can be detected by flow cytometry or by PCR for informative genetic markers (1–3). The challenge is to eliminate the relapse-initiating ALL cells, and novel approaches specifically targeting residual leukemia cells are warranted (4–6).
Hematopoietic stem cells (HSCs) are located in the osteoblastic and the perivascular niches of the bone marrow (BM) (7, 8). Adhesion of leukemia cells to the BM has been found to contribute to chemoresistance of residual leukemia cells (6, 9–11). Integrins, a family of glycoprotein cell surface receptors composed of two subunits, alpha and beta (12), are responsible for cell adhesion to the extracellular matrix (ECM). The beta1 integrins are also known as very-late-activation antigens (VLAs). The integrin alpha4-chain (alpha4, also known as CD49d) non-covalently associates with the beta1 integrin chain, CD29, to form very-late-antigen-4 (VLA-4). VLA-4 is also referred as alpha4/beta1 or CD49d/CD29 and although there is an alternative beta-partner for alpha4, beta7, to form MadCAM, CD49d/CD29 is referred to throughout this manuscript as “the alpha4 integrin.” The alpha4 integrin binds to its counter receptors, including vascular cell adhesion molecule-1 (VCAM-1), fibronectin, or osteopontin (OPN) (13), and regulates retention and mobilization as well as to some degree cell cycle activity of immature progenitors in the BM (14). Alpha4 is expressed, among many other blood cells, on pre-B ALL cells (15, 16) and was recently quantified by real-time polymerase chain reaction in leukemia cells from 56 patients with relapsed ALL enrolled in the ALL-REZ BFM 2002 trial of the Berlin–Frankfurt–Münster study group (17). High alpha4 integrin mRNA expression was identified as an adverse risk factor in childhood ALL at first relapse. Therefore, we review here integrin alpha4 as a therapeutic target in drug resistance of leukemia.
Integrin Structure
Integrins are a family of 24 heterodimeric cell surface proteins sharing significant structural and functional commonalities (13, 18, 19). They consist of an alpha- and a beta-chain, which are non-covalently bound. The alpha-chain predominantly defines the ligand specificity of the integrin. Conformational changes induced by approximation of the transmembrane regions of the integrin heterodimer open the ligand binding pocket. Ligand binding further affects integrin conformation, stretching the heterodimer from a bent to an extended conformation, shown in Figure 1 (20). The high-affinity form of integrins is stabilized by insertion of certain proteins from a multi-protein complex associated with the intracellular domain of integrins, e.g., talin (21). In the active conformation, integrins elicit intracellular (outside-in) signals through a variety of cell-type-dependent pathways, which include survival/apoptosis, cell cycle, metabolism, among others (12, 22–29). Integrins thus mediate physical adhesion to suitable matrices/ligands while at the same time providing outside-in signals regulating cell fate.
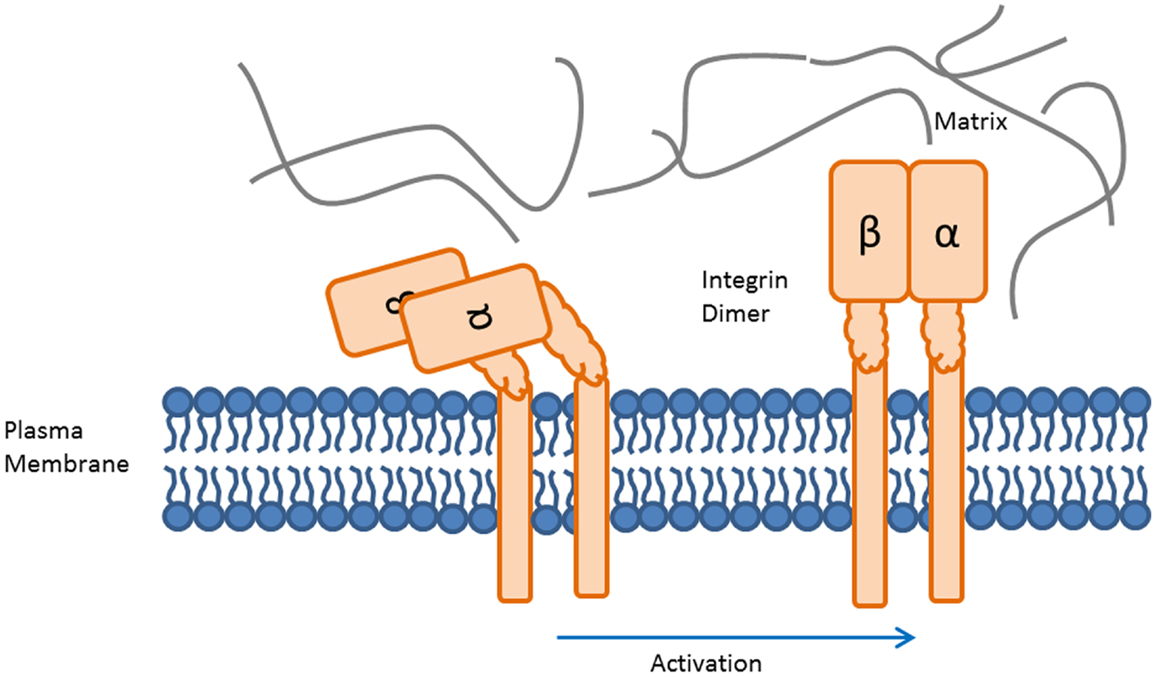
Figure 1. Activation of the integrin heterodimer induces a conformational change. The conformational states of the integrin heterodimer determine whether it functions for cellular adhesion or migration. The bent, inactive form of the integrin heterodimer prevents binding of ligands to the recognition region (left). Activation induced conformational change results in availability of the ligand binding region (right).
Integrin Ligands
Integrin alpha4 binds to VCAM-1, a cell surface protein expressed on activated vascular endothelium and a host of other cells, as well as several ECM proteins, including OPN and the highly abundant fibronectin. VCAM-1 is a type I membrane protein expressed mainly on the surface of vascular endothelial cells throughout the vascular tree, but also on hematopoietic cells, although its function on these cells remains elusive. VCAM-1 expression on blood vessels is induced after cytokine stimulation, which is regulated either by increasing VCAM1 mRNA expression or mRNA stabilization (30). An alternative receptor for VCAM-1 to alpha4 is MadCAM, the alpha4/beta7 integrin heterodimer (31).
Fibronectin, a large glycoprotein dimer, binds primarily other matrix proteins, such as collagens and heparan sulfates, but contains moieties for integrin alpha4/beta1 binding (within the V-domain) as well as for alpha5 and the platelet integrin alphaV/beta3 (the RGD domain) (32). Experiments with molecules antagonizing fibronectin versus VCAM-1 binding, however, did not detect apparent effects of fibronectin-blockade on the trafficking of immature hematopoietic cells, whereas VCAM-1 inhibition interfered quantitatively with the interaction between hematopoietic stem/progenitor cells and BM (33). Matsunaga et al. have reported a fibronectin peptide based blockade of acute myeloid leukemia (AML) (34). As to the relevant ligand for alpha4 integrin in the stroma niche, contradictory data have been published. Specifically, Cradock and colleagues identified VCAM1 as the relevant ligand for normal hematopoietic cells, while the CS1 motif was apparently redundant (33). By contrast, Matsunaga et al. proposed adhesion to fibronectin as the molecular mechanism driving alpha4 integrin-mediated chemoresistance (35). If this was a general rule, then targeting the VLA-4-fibronectin interface would represent an even more leukemia cell specific target than VLA-4 proper. However, our own data show contributory roles of both ligands for chemoresistance of leukemia cells (and most strongly for VCAM1) at least in vitro (36), so that the situation currently remains unresolved.
Osteopontin is a negatively charged ECM glycoprotein, and has been described as a ligand for alpha4 integrin (37, 38), but alternative receptors include the alpha9 integrins (39). A role for OPN as a negative regulator of HSC proliferation and a mediator of HSC localization within BM has been proposed (40, 41). Which one of these alpha4 ligands is most critical for leukemia cell attachment-mediated drug resistance remains elusive.
Integrin Intracellular Signaling
Integrins can elicit intracellular signaling both directly and indirectly through other receptors (42). These are complex signaling mechanisms, which are briefly summarized here: Indirect intracellular signaling involves integrins forming complexes with receptor tyrosine kinase (RTK), which then interferes with activation of RTK by its normal ligand (43). A main structural and signaling protein involved in direct integrin signaling is integrin-linked kinase (ILK), which binds integrins (Figure 2). ILK forms multi-protein complexes with several key components involved with the cytoskeletal dynamics and intracellular signaling cascades. ILK kinase activity is dependent on PI3K and requires binding of PtdIns(3,4,5)P3 (PIP3) (44–46). Key players in cellular signaling that bind ILK specifically at the kinase domain include: PDK1, Akt, Rictor, Src. Rictor directly interacts with ILK, leading to the phosphorylation of Akt at serine 473 (26). This regulates cellular survival via caspase activation and nuclear factor-κB (NF-κB) stimulation (44, 47). ILK phosphorylates glycogen synthase kinase-3β (GSK3β) through phosphorylation on serine 9, resulting in the activation of activator protein 1 (AP-1), which then stimulates cyclin D1 and matrix metalloprotease 9 (MMP9) (44, 45, 48). Tabe et al. showed that ILK/Akt is a signaling pathway critical for survival of leukemic cells (49). Specifically, they demonstrated in a co-culture system of leukemic NB4 cells with BM-derived stromal mesenchymal stem cells (MSC), activation of ILK/Akt, extracellular signal-regulated kinase 1/2 (ERK1/2), signal transducers and activators of transcription 3 (STAT3), as well as Notch1/Hes. Blockade of PI3K or ILK signaling with pharmacologic inhibitors, LY294002 or QLT0267, resulted in induction of apoptosis in both leukemic cell lines and in primary AML samples. Muranyi et al. showed that targeting ILK and FMS-like tyrosine kinase-3 (FLT3) with an inhibitor of ILK and FLT3, OLT0267, is cytotoxic to AML stem cells using a long-term suspension culture system and a NOD/SCID mouse leukemia-initiating assay (50).
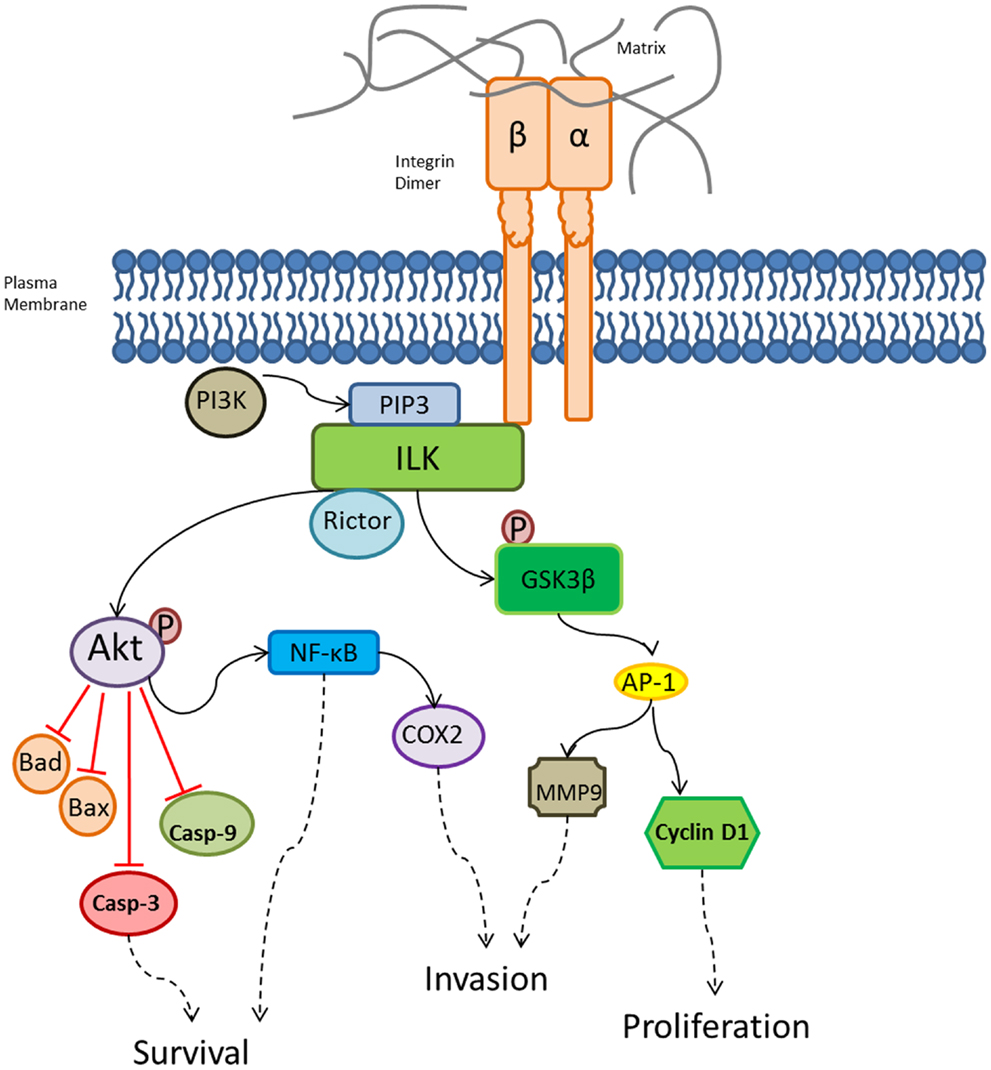
Figure 2. Integrin intracellular signaling pathways regulated by ILK. A variety of biological processes are regulated by ILK, which is a central player in multiple signaling cascades crucial for tissue homeostasis. ILK activation results in downstream effects responsible for survival, invasion, and proliferation. AP-1, activator protein 1; casp, caspase; GSK β, glycogen synthase kinase-3β; MMP9, matrix metalloprotease 9; NF-κB, nuclear factor-κB; P, phosphate; PI3K, phosphatidylinositol 3-kinase; PIP3, PtdIns(3,4,5)P3. Solid black arrows indicate activation, dashed black arrows indicate downstream effects, and the red lines indicate inhibitory effects.
Direct intracellular signaling involves direct activation of tyrosine kinases by integrins. It has been described that integrin clustering activates tyrosine phosphorylation via focal adhesion kinase (FAK) (51). Integrin intracellular signaling involved the recruitment and activation of Src-family kinases (SFKs), which recruit FAK through the beta subunit (Figure 3). FAK can activate signaling from phosphatidylinositol 3-kinase (PI3K) to AKT/protein kinase B (PKB) through phosphatidylinositol-3,4,5-trisphosphate [PtdIns(3,4,5)P3], as well as recruiting Src to focal adhesions. Src can then phosphorylate CAS and paxillin, which further recruits the Crk–DOCK180 complex that results in the activation of Rac (52). The activation of Rac further activates p21-activated kinase (PAK), Jun amino-terminal kinase (JNK), and NF-κB (52–54).
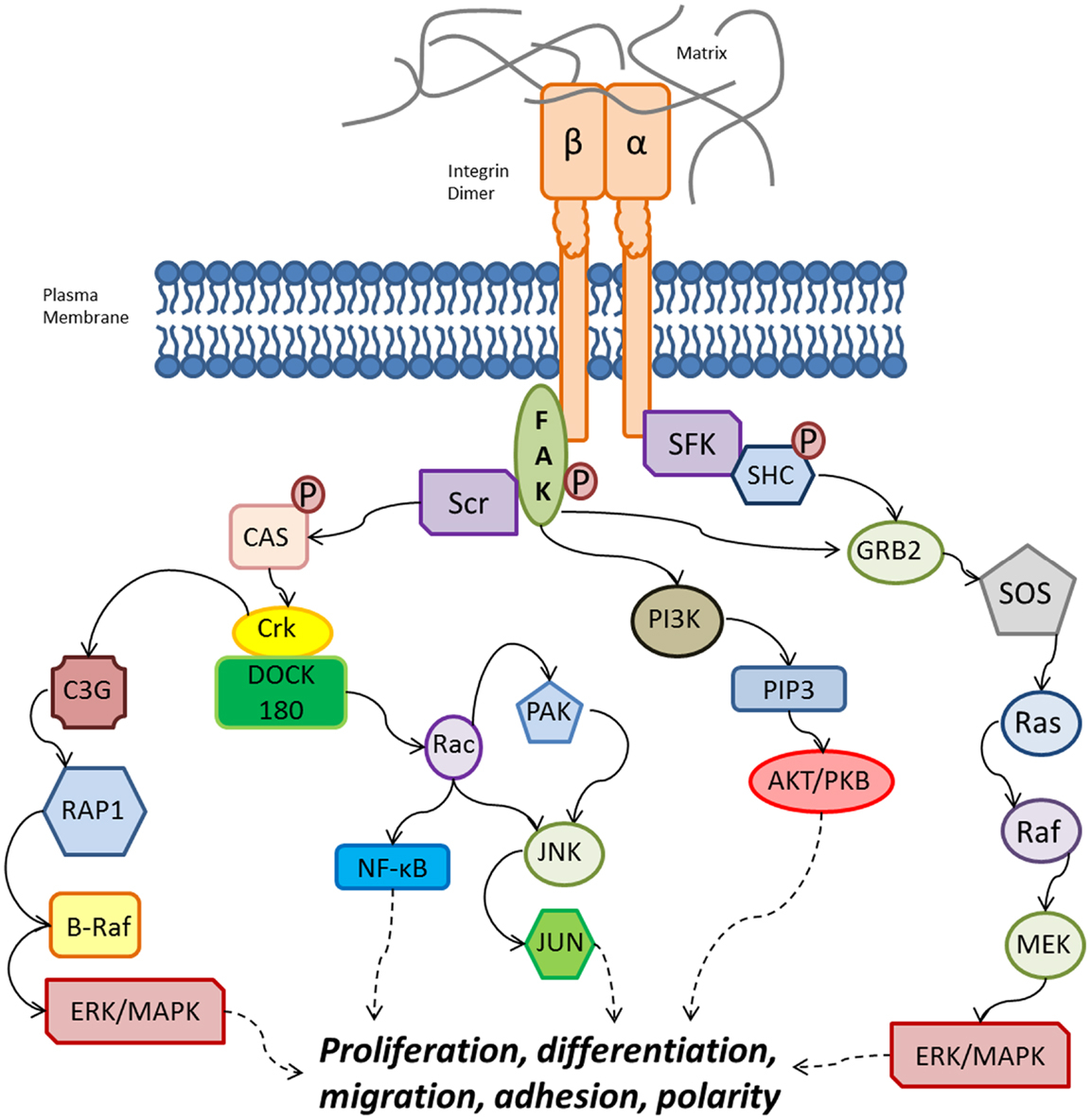
Figure 3. Overview of integrin intracellular signaling cascades from both the alpha and beta subunits, leading to the activation of various cellular functions. Binding of an alpha/beta integrin to the extracellular matrix ligands leads to activation of FAK. Note that other signaling pathways are stimulated by integrin heterodimers, but are not included for clarity and conciseness. FAK: focal adhesion kinase; GRB2, growth-factor-receptor-bound-2; P, phosphate group; PAK, p21-activated kinase; PI3K, phosphatidylinositol 3-kinase; PIP3, PtdIns(3,4,5)P3; PKB, protein kinase B; SFKs, Src-family kinases; SOS, son-of-sevenless. Solid black arrows indicate activation, dashed black arrows indicate downstream effects, and the red lines indicate inhibitory effects.
Focal adhesion kinase can activate extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) via two pathways. First FAK can recruit C3G and RAP1 via Crk (55), which induces B-Raf activity and ERK/MAPK activation (12). The second pathway involves the growth-factor-receptor-bound-2 (GRB2) and son-of-sevenless (SOS) complex, which activates Ras–ERK/MAPK. The alpha subunit is also able to activate ERK/MAPK via SFK coupling, which phosphorylates the SHC, activating the GRB2–SOS complex and ERK/MAPK signaling downstream of Ras (56, 57).
The extracellular domain of alpha4 integrins mediates cell adhesion, while the cytoplasmic domain couples signaling and linkage with the cytoskeleton (13). The cytoplasmic tail of alpha4 integrin has been reported to bind to the signaling adaptor paxillin (58). The tight association of paxillin with the alpha4 tail leads to distinct biochemical and biological responses to integrin-mediated cell adhesion (58).
Lim et al. have reported that localized cAMP-dependent protein kinase (PKA) activation in pseudopodia of migrating cells phosphorylates alpha4 integrins to provide spatial cues governing cell motility. Specifically, they have shown that the alpha4 cytoplasmic domain is a Type I PKA-specific a-kinase anchoring proteins (AKAP) (59).
Rivera Rosado et al. have shown that cytoplasmic alpha4 tail associates with non-muscle myosin IIA (MIIA) independent of paxillin binding indicating a new mechanism for linking integrins to the actomyosin cytoskeleton and for regulating cell migration by integrins (60).
The interactions outlined above may regulate alpha4-specific adhesion in leukocytes, and it is conceivable, although it remains to be seen, that they may impact MRD.
In addition, Liu et al. have shown that chemoprotection of Jurkat T-ALL cells is integrin alpha4- or alpha5-mediated (61). The cytoplasmic domains of alpha-integrins have few sequences in common with the exception of the highly conserved membrane-proximal KXGFFKR motif (62). Interesting, Liu et al. have shown that reconstituted expression of alpha4δ, a truncated alpha4 integrin with KxGFFKR as cytoplasmic motif, in alpha4-deficient cells promoted chemoresistance to doxorubicin independent of alpha4-mediated adhesion of T-ALL cells (61).
Taken together, these observations indicate that the chemoprotective effects of leukemia cells associated with alpha4 integrin are at least in part mediated through integrin signaling and not through alpha4-mediated adhesion alone, although contributory roles of adhesion cannot be ruled out.
Integrin-Bone Marrow Stromal Interactions Mediate Survival and Resistance of Leukemia Cells to Chemotherapy
As the BM is the most frequent relapse site for ALL (63), the BM has been considered a protective niche for leukemia cells (64). Previous studies have shown that ALL cell adhesion is mediated by alpha4 (65) and also that AML cell adhesion is specifically mediated by beta1 integrin, which leads to cell adhesion-mediated drug resistance (CAM-DR) (66). Mudry et al. conducted an in vitro investigation of one T-ALL and three pre-B ALL cell lines either in co-culture with stroma cells or on proteins isolated from the stromal matrix (11). When ALL cells were able to make direct contact with the stromal cells, the co-culture system maintained leukemia cell proliferation despite the presence of the chemotherapeutic agents, cytarabine or etoposide. This pro-survival effect was mediated through VCAM-1. Chemotherapeutic protection was not observed when leukemia cells were cultured in suspension above the stroma or on fibronectin alone. An interesting function of integrins beyond simple physical adherence is the activation of “outside-in” signals upon binding to their extracellular ligands, which apparently contribute to the integrin-mediated chemoprotective effects. Some of these chemoprotective signaling changes have been described below, as well as in Figures 2 and 3.
Astier and colleagues observed the inhibition of capase-3 and -7 activation by stimulation of β1 integrin in pre-B ALL cells. In the co-culture of BM stroma with pre-B ALL cells during chemotherapeutic treatment, the expression of the pro-apoptotic 23 kDa Bcl-2 protein was reduced (67). Wang et al. reported reduced levels of both PARP and cleaved Bcl-2 in ALL cells post-chemotherapy treatment, which was shown to be Akt-mediated (68). Previous studies by Fortney et al. showed that caspase 3 activity can be induced in ALL cells after treatment with the chemotherapeutic compounds, cytarabine or etoposide (69). Interestingly, this apoptotic effect is prevented in ALL cells co-cultured with human BM stroma cells.
The PI3K/Akt pathway (Figure 3) has been implicated in stroma cell-mediated chemoprotection and survival of leukemia cells (68). Additionally, gene expression analysis of leukemia cells with high versus low alpha4 expression identified 27 differentially expressed genes involved in ephrin, Rho GTPase, and PI3K/Akt pathways (17). More recently, it has been implicated that B-ALL cells interact with the BM-derived mesenchymal stromal cells through Notch-3 and -4 signaling (70). In a study by Bertrand et al., the inhibition of MEK with either mTOR or PI3K in the B-ALL cell line BLIN-2 resulted in rapid apoptosis of the leukemic cells (71).
Pillozzi et al. reported similar data in a co-culture system of ALL cells and BM mesenchymal stroma cells where hERG1 (human ether-à-go-go-related gene 1) channels have a role in the survival of leukemia cells via a signaling complex also containing beta1 integrin and CXCR4 (72). In addition, the PI3K/Akt pathway has been linked to regulation of drug transporting pumps, which may contribute to drug resistance (73): Ma showed high expression level of ST6GAL (β-galactoside α2, 6-sialyltransferase)-1 gene in leukemia cells. ST6GAL1 modulated the activity of (PI3K)/Akt signaling and regulated the expression of P-glycoprotein (P-gp) and multidrug resistance related protein 1 (MRP1) in leukemia cell lines. These data indicate that changes in P-gp based drug efflux mechanisms downstream of integrin signaling may contribute to chemoresistance. However, others have reported that MRP1, but not P-gp expression is under the control of the PI3K/Akt axis in AML blasts (74), so that definitive conclusions cannot be drawn at this point.
Recently, Andreeff and colleagues provided evidence of stroma-induced and alpha4-mediated NF-κB signaling in leukemia cells (75): Jacamo et al. have shown that VCAM1/VLA-4 activates NF-κB activation in leukemia and BM stromal cells and this crosstalk contributes to chemoresistance of leukemia cells (75).
Taken together, these studies demonstrate that integrin-BM stromal interactions trigger intracellular signaling changes, which then mediate chemoprotection. Interruption of this chemoprotective interaction-induced signaling would potentially overcome drug resistance. These findings indicate also that the effect of targeting integrin alpha4 goes beyond physical dislodgement of leukemia cells from the BM niche.
Targeting of Alpha4 Integrin
Pre-clinical and clinical testing of new therapies against various integrins involved in different diseases is a rapidly emerging field (76–81). Over 260 anti-integrin drugs are currently in clinical evaluation, but only a few have been approved for clinical use. One of these is the anti-functional antibody, natalizumab (82), which targets both alpha4/beta1 and alpha4/beta7 (Table 1). Natalizumab, which is almost fully humanized, has been available in the clinic for treatment of relapsing–remitting multiple sclerosis as well as of complicated refractory inflammatory bowel disease with very high efficaciousness and for the most part good safety and tolerability. Long-term treatment has been associated in some patients with induction of neutralizing antibodies against the few remaining murine amino acid residues of the antibody, a scenario presenting as mild infusion-associated serum sickness and loss of efficacy (83). Infrequently, JC-virus-associated progressive multifocal leukoencephalopathy (PML) has been described after prolonged use, a complication, which is associated with a poor prognosis despite aggressive plasmapheresis (84). Long-term effects on hematopoiesis have been studied and did not provide any evidence for hematopoietic exhaustion in agreement with specific evidence in mice deficient for alpha4 integrin on hematopoietic cells (85, 86). We have evaluated the use of natalizumab in a xenograft model of primary leukemia and observed prolonged survival of mice treated with the combination of vincristine, dexamethasone, and l-asparaginase plus natalizumab (36). These data indicate the promise of this clinically approved antibody for ALL treatment. Another humanized antibody which is shown to inhibit alpha4beta7-mediated cellular adhesion is MLN-02 (87), which is a selective antagonist of alpha4/beta7 currently in clinical evaluation against Crohn’s disease and Ulcerative Colitis (88–90).
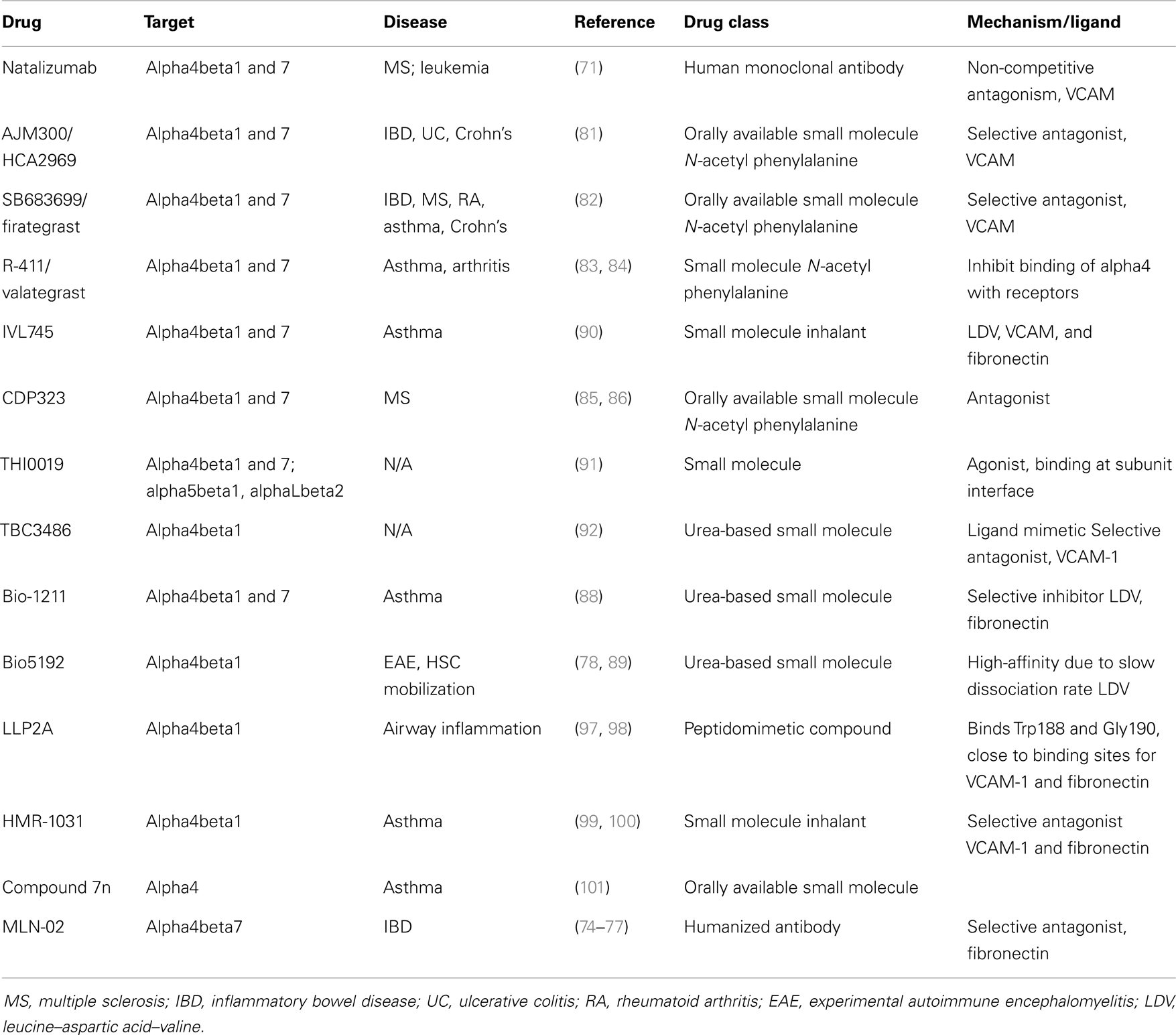
Table 1. Summary of integrin alpha4 targeting drugs.
Alternative molecules targeting alpha4 are under evaluation for a variety of diseases and have been summarized in more detail (78, 79, 91–93) (Table 1), although none are approved so far for clinical use in leukemia. There are two main classes of alpha4 integrin antagonists: urea-based and phenylalanine-based antagonists. The N-acetyl phenylalanine group consists of the small molecules AJM300 (94), SB683699 (Firategrast) (95), R-411 (Valategrast) (96, 97), and CDP323 (98, 99). These phenylalanine derivatives are novel compounds with improved potency against alpha4 integrins, through the formation of a cyclic peptide (100).
The urea-based antagonists are Bio-1211 (101), Bio5192 (92, 102), IVL745 (103), and TBC3486 (104), which is the parent molecule to THI0019 (105). These small molecule antagonists can further be categorized by the presence of a leucine–aspartic acid–valine (LDV) motif. The LDV motif contains a crucial aspartate recognized by integrin alpha4 (106–108). Alpha4 integrin has three LDV motifs in the extracellular sequence. The active-site motifs of integrin ligands can be reproduced synthetically as small molecules to provide both information regarding receptor–ligand binding and the development of therapeutic agents. For example, R-411 (valategrast), is metabolized to its active form RO0270608, which immediately reversed the binding of leukocytes to VCAM-1 (96). In early clinical trials, this molecule has shown encouraging results regarding pharmacokinetics and toxicity in patients with asthma (96, 97, 109). Of the LDV antagonists, Bio5192 is the only selective antagonists specifically for alpha4/beta1, while the others are dual antagonists. Another antagonist discussed here is a novel peptidomimetic compound that is specific for the biding site on alpha4/beta1 integrin heterodimer called LLP2A (110). LLP2A is a high-affinity ligand due to the replacement of the LDV motif with unnatural, modified amino acids (111). HMR 1031 is a selective VLA-4 receptor antagonist that blocks the binding of VCAM1 and fibronectin (112), but has shown poor efficacy in subjects with clinically persistent asthma (113). Compound 7n is a zwitterionic compound specifically modified from the VLA-4 antagonist compound 3, which is a 2-(phenylamino)benzoxazole derivative to improve oral bioavailability, which showed favorable efficacy in a mouse model of asthma (114). It remains to be seen if these novel alpha4 integrin inhibitors will be approved for clinical use.
Conclusion
Integrins have been implicated in adhesion-mediated drug resistance of leukemic cells. As integrin alpha4 has been described in particular to be highly expressed in ALL (58, 102), this review focused on summarizing recent studies of integrin alpha4-associated cell survival and how to interrupt this chemoprotective binding. We have shown that integrin alpha4 is a promising target in the therapy against ALL (102). Interference with alpha4 not only deadheres leukemia cells physically from the chemoprotective BM niche, but may interrupt intracellular signaling changes critical for survival and resistance of leukemia cells. Several ways to target integrin alpha4 are under pre-clinical evaluation. Whether targeting of alpha4 integrin should be a strategy to prevent relapse, or whether it should be a treatment of relapsed ALL remains to be determined. Currently, the only clinically available anti-integrin alpha4 antagonist is natalizumab. Pre-clinical evidence has been provided that interference with alpha4-mediated adhesion of ALL cells can sensitize them to chemotherapy and thus facilitate eradication of ALL cells in an MRD setting (36). Finally, it remains to be determined whether toxicity of standard-risk ALL treatment can be decreased by combining administration of anti-alpha4 inhibitions with a reduced-dose chemotherapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Halvard Bönig acknowledges funding by the German Federal Ministry for Education and Research (BMBF, Ci3), the Federal State of Hesse (LOEWE CGT), Deutsche Krebshilfe and DFG (BO3553/1-1). Yong-Mi Kim acknowledges funding by NIH R01CA172896, Hyundai Hope on Wheels, St. Baldrick’s Foundation and V-Foundation.
References
1. Campana D. Role of minimal residual disease monitoring in adult and pediatric acute lymphoblastic leukemia. Hematol Oncol Clin North Am (2009) 23(5):1083–98. doi:10.1016/j.hoc.2009.07.010
2. Coustan-Smith E, Song G, Clark C, Key L, Liu P, Mehrpooya M, et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood (2011) 117(23):6267–76. doi:10.1182/blood-2010-12-324004
3. Cavé H, van der Werfften Bosch J, Suciu S, Guidal C, Waterkeyn C, Otten J, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer – Childhood Leukemia Cooperative Group. N Engl J Med (1998) 339(9):591–8. doi:10.1056/NEJM199808273390904
4. Nair RR, Tolentino J, Hazlehurst LA. The bone marrow microenvironment as a sanctuary for minimal residual disease in CML. Biochem Pharmacol (2010) 80(5):602–12. doi:10.1016/j.bcp.2010.04.003
5. Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat (2009) 12(4–5):103–13. doi:10.1016/j.drup.2009.06.001
6. Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol (2011) 29(5):591–9. doi:10.1200/JCO.2010.31.0904
7. Kiel MJ, Acar M, Radice GL, Morrison SJ. Hematopoietic stem cells do not depend on N-cadherin to regulate their maintenance. Cell Stem Cell (2009) 4(2):170–9. doi:10.1016/j.stem.2008.10.005
8. Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol (2008) 8(4):290–301. doi:10.1038/nri2279
9. Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood (1992) 79(9):2370–7.
10. Kumagai M, Manabe A, Pui CH, Behm FG, Raimondi SC, Hancock ML, et al. Stroma-supported culture in childhood B-lineage acute lymphoblastic leukemia cells predicts treatment outcome. J Clin Invest (1996) 97(3):755–60. doi:10.1172/JCI118474
11. Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood (2000) 96(5):1926–32.
12. Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol (2004) 5(10):816–26. doi:10.1038/nrm1490
13. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110(6):673–87. doi:10.1016/S0092-8674(02)00971-6
14. Priestley GV, Scott LM, Ulyanova T, Papayannopoulou T. Lack of alpha4 integrin expression in stem cells restricts competitive function and self-renewal activity. Blood (2006) 107(7):2959–67. doi:10.1182/blood-2005-07-2670
15. Hara J, Matsuda Y, Fujisaki H, Tokimasa S, Ohta H, Osagi Y, et al. Expression of adhesion molecules in childhood B-lineage-cell neoplasms. Int J Hematol (2000) 72(1):69–73.
16. Liesveld JL, Winslow JM, Frediani KE, Ryan DH, Abboud CN. Expression of integrins and examination of their adhesive function in normal and leukemic hematopoietic cells. Blood (1993) 81(1):112–21.
17. Shalapour S, Hof J, Kirschner-Schwabe R, Bastian L, Eckert C, Prada J, et al. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica (2011) 96(11):1627–35. doi:10.3324/haematol.2011.047993
18. Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol (2010) 2(12):a005066. doi:10.1101/cshperspect.a005066
19. Juliano RL. Signal transduction by cell adhesion receptors and the cytoskeleton: functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu Rev Pharmacol Toxicol (2002) 42:283–323. doi:10.1146/annurev.pharmtox.42.090401.151133
20. Chigaev A, Zwartz GJ, Buranda T, Edwards BS, Prossnitz ER, Sklar LA. Conformational regulation of alpha 4 beta 1-integrin affinity by reducing agents. “Inside-out” signaling is independent of and additive to reduction-regulated integrin activation. J Biol Chem (2004) 279(31):32435–43. doi:10.1074/jbc.M404387200
21. Ye F, Snider AK, Ginsberg MH. Talin and kindlin: the one-two punch in integrin activation. Front Med (2014) 8(1):6–16. doi:10.1007/s11684-014-0317-3
22. Boudreau NJ, Jones PL. Extracellular matrix and integrin signalling: the shape of things to come. Biochem J (1999) 339(Pt 3):481–8. doi:10.1042/0264-6021:3390481
23. Parsons JT. Integrin-mediated signalling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol (1996) 8(2):146–52. doi:10.1016/S0955-0674(96)80059-7
24. Herter J, Zarbock A. Integrin regulation during leukocyte recruitment. J Immunol (2013) 190(9):4451–7. doi:10.4049/jimmunol.1203179
25. Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood (2013) 121(12):2301–10. doi:10.1182/blood-2012-09-457085
26. McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase – essential roles in physiology and cancer biology. J Cell Sci (2008) 121(Pt 19):3121–32. doi:10.1242/jcs.017996
27. Liu W, Draheim KM, Zhang R, Calderwood DA, Boggon TJ. Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol Cell (2013) 49(4):719–29. doi:10.1016/j.molcel.2012.12.005
28. Aoudjit F, Vuori K. Integrin signaling in cancer cell survival and chemoresistance. Chemother Res Pract (2012) 2012:283181. doi:10.1155/2012/283181
29. Margadant C, Monsuur HN, Norman JC, Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol (2011) 23(5):607–14. doi:10.1016/j.ceb.2011.08.005
30. Iademarco MF, Barks JL, Dean DC. Regulation of vascular cell adhesion molecule-1 expression by IL-4 and TNF-alpha in cultured endothelial cells. J Clin Invest (1995) 95(1):264–71. doi:10.1172/JCI117650
31. Katayama Y, Hidalgo A, Peired A, Frenette PS. Integrin alpha4beta7 and its counterreceptor MAdCAM-1 contribute to hematopoietic progenitor recruitment into bone marrow following transplantation. Blood (2004) 104(7):2020–6. doi:10.1182/blood-2003-12-4157
32. Garcia-Pardo A, Wayner EA, Carter WG, Ferreira OC Jr. Human B lymphocytes define an alternative mechanism of adhesion to fibronectin. The interaction of the alpha 4 beta 1 integrin with the LHGPEILDVPST sequence of the type III connecting segment is sufficient to promote cell attachment. J Immunol (1990) 144(9):3361–6.
33. Craddock CF, Nakamoto B, Elices M, Papayannopoulou T. The role of CS1 moiety of fibronectin in VLA mediated haemopoietic progenitor trafficking. Br J Haematol (1997) 97(1):15–21. doi:10.1046/j.1365-2141.1997.d01-2120.x
34. Matsunaga T, Fukai F, Miura S, Nakane Y, Owaki T, Kodama H, et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia (2008) 22(2):353–60. doi:10.1038/sj.leu.2405017
35. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med (2003) 9(9):1158–65. doi:10.1038/nm909
36. Hsieh YT, Gang EJ, Geng H, Park E, Huantes S, Chudziak D, et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood (2013) 121(10):1814–8. doi:10.1182/blood-2012-01-406272
37. Bayless KJ, Meininger GA, Scholtz JM, Davis GE. Osteopontin is a ligand for the alpha4beta1 integrin. J Cell Sci (1998) 111(Pt 9):1165–74.
38. Bogetto L, Gabriele E, Cariati R, Dolcetti R, Spessotto P, Doglioni C, et al. Bidirectional induction of the cognate receptor-ligand alpha4/VCAM-1 pair defines a novel mechanism of tumor intravasation. Blood (2000) 95(7):2397–406.
39. Yokosaki Y, Palmer EL, Prieto AL, Crossin KL, Bourdon MA, Pytela R, et al. The integrin alpha 9 beta 1 mediates cell attachment to a non-RGD site in the third fibronectin type III repeat of tenascin. J Biol Chem (1994) 269(43):26691–6.
40. Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood (2005) 106(4):1232–9. doi:10.1182/blood-2004-11-4422
41. Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med (2005) 201(11):1781–91. doi:10.1084/jem.20041992
42. Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem J (2009) 418(3):491–506. doi:10.1042/BJ20081948
43. Cabodi S, Moro L, Bergatto E, Boeri EE, Di SP, Turco E, et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem Soc Trans (2004) 32(Pt 3):438–42. doi:10.1042/BST0320438
44. Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature (1996) 379(6560):91–6. doi:10.1038/379091a0
45. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A (1998) 95(19):11211–6. doi:10.1073/pnas.95.19.11211
46. Pasquali C, Bertschy-Meier D, Chabert C, Curchod ML, Arod C, Booth R, et al. A chemical proteomics approach to phosphatidylinositol 3-kinase signaling in macrophages. Mol Cell Proteomics (2007) 6(11):1829–41. doi:10.1074/mcp.T600066-MCP200
47. Legate KR, Montanez E, Kudlacek O, Fassler RILK. PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol (2006) 7(1):20–31. doi:10.1038/nrm1789
48. Guo W, Jiang H, Gray V, Dedhar S, Rao Y. Role of the integrin-linked kinase (ILK) in determining neuronal polarity. Dev Biol (2007) 306(2):457–68. doi:10.1016/j.ydbio.2007.03.019
49. Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W, et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res (2007) 67(2):684–94. doi:10.1158/0008-5472.CAN-06-3166
50. Muranyi AL, Dedhar S, Hogge DE. Targeting integrin linked kinase and FMS-like tyrosine kinase-3 is cytotoxic to acute myeloid leukemia stem cells but spares normal progenitors. Leuk Res (2010) 34(10):1358–65. doi:10.1016/j.leukres.2010.01.006
51. Kornberg LJ, Earp HS, Turner CE, Prockop C, Juliano RL. Signal transduction by integrins: increased protein tyrosine phosphorylation caused by clustering of beta 1 integrins. Proc Natl Acad Sci U S A (1991) 88(19):8392–6. doi:10.1073/pnas.88.19.8392
52. Parsons JT, Parsons SJ. Src family protein tyrosine kinases: cooperating with growth factor and adhesion signaling pathways. Curr Opin Cell Biol (1997) 9(2):187–92. doi:10.1016/S0955-0674(97)80062-2
53. Cary LA, Han DC, Guan JL. Integrin-mediated signal transduction pathways. Histol Histopathol (1999) 14(3):1001–9.
54. Schlaepfer DD, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs? Trends Cell Biol (1998) 8(4):151–7. doi:10.1016/S0962-8924(97)01172-0
55. Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol (2009) 9(9):630–44. doi:10.1038/nri2606
56. Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell (1996) 87(4):733–43. doi:10.1016/S0092-8674(00)81392-6
57. Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell (1998) 94(5):625–34. doi:10.1016/S0092-8674(00)81604-9
58. Liu S, Thomas SM, Woodside DG, Rose DM, Kiosses WB, Pfaff M, et al. Binding of paxillin to alpha4 integrins modifies integrin-dependent biological responses. Nature (1999) 402(6762):676–81. doi:10.1038/45264
59. Lim CJ, Han J, Yousefi N, Ma Y, Amieux PS, McKnight GS, et al. Alpha4 integrins are type I cAMP-dependent protein kinase-anchoring proteins. Nat Cell Biol (2007) 9(4):415–21. doi:10.1038/ncb1561
60. Rivera Rosado LA, Horn TA, McGrath SC, Cotter RJ, Yang JT. Association between alpha4 integrin cytoplasmic tail and non-muscle myosin IIA regulates cell migration. J Cell Sci (2011) 124(Pt 3):483–92. doi:10.1242/jcs.074211
61. Liu CC, Leclair P, Yap SQ, Lim CJ. The membrane-proximal KXGFFKR motif of alpha-integrin mediates chemoresistance. Mol Cell Biol (2013) 33(21):4334–45. doi:10.1128/MCB.00580-13
62. Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol (2009) 27:339–62. doi:10.1146/annurev.immunol.021908.132554
63. Gaynon PS, Qu RP, Chappell RJ, Willoughby ML, Tubergen DG, Steinherz PG, et al. Survival after relapse in childhood acute lymphoblastic leukemia: impact of site and time to first relapse – the Children’s Cancer Group Experience. Cancer (1998) 82(7):1387–95. doi:10.1002/(SICI)1097-0142(19980401)82:7<1387::AID-CNCR24>3.0.CO;2-1
64. Bradstock KF, Gottlieb DJ. Interaction of acute leukemia cells with the bone marrow microenvironment: implications for control of minimal residual disease. Leuk Lymphoma (1995) 18(1–2):1–16. doi:10.3109/10428199509064917
65. Filshie R, Gottlieb D, Bradstock K. VLA-4 is involved in the engraftment of the human pre-B acute lymphoblastic leukaemia cell line NALM-6 in SCID mice. Br J Haematol (1998) 102(5):1292–300. doi:10.1046/j.1365-2141.1998.00899.x
66. Hazlehurst LA, Argilagos RF, Dalton WS. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br J Haematol (2007) 136(2):269–75. doi:10.1111/j.1365-2141.2006.06435.x
67. Fortney JE, Hall BM, Bartrug L, Gibson LF. Chemotherapy induces bcl-2 cleavage in lymphoid leukemic cell lines. Leuk Lymphoma (2002) 43(11):2171–8. doi:10.1080/1042819021000033024
68. Wang L, Fortney JE, Gibson LF. Stromal cell protection of B-lineage acute lymphoblastic leukemic cells during chemotherapy requires active Akt. Leuk Res (2004) 28(7):733–42. doi:10.1016/j.leukres.2003.10.033
69. Fortney JE, Zhao W, Wenger SL, Gibson LF. Bone marrow stromal cells regulate caspase 3 activity in leukemic cells during chemotherapy. Leuk Res (2001) 25(10):901–7. doi:10.1016/S0145-2126(01)00051-0
70. Nwabo Kamdje AH, Krampera M. Notch signaling in acute lymphoblastic leukemia: any role for stromal microenvironment? Blood (2011) 118(25):6506–14. doi:10.1182/blood-2011-08-376061
71. Bertrand FE, Spengemen JD, Shelton JG, McCubrey JA. Inhibition of PI3K, mTOR and MEK signaling pathways promotes rapid apoptosis in B-lineage ALL in the presence of stromal cell support. Leukemia (2005) 19(1):98–102. doi:10.1038/sj.leu.2403560
72. Pillozzi S, Masselli M, De LE, Accordi B, Cilia E, Crociani O, et al. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood (2011) 117(3):902–14. doi:10.1182/blood-2010-01-262691
73. Ma H, Cheng L, Hao K, Li Y, Song X, Zhou H, et al. Reversal effect of ST6GAL 1 on multidrug resistance in human leukemia by regulating the PI3K/Akt pathway and the expression of P-gp and MRP1. PLoS One (2014) 9(1):e85113. doi:10.1371/journal.pone.0085113
74. Tazzari PL, Cappellini A, Ricci F, Evangelisti C, Papa V, Grafone T, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia (2007) 21(3):427–38. doi:10.1038/sj.leu.2404523
75. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood (2014) 123(17):2691–702. doi:10.1182/blood-2013-06-511527
76. Elices MJ. The integrin alpha 4 beta 1 (VLA-4) as a therapeutic target. Ciba Found Symp (1995) 189:79–85.
77. Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov (2010) 9(10):804–20. doi:10.1038/nrd3266
78. Millard M, Odde S, Neamati N. Integrin targeted therapeutics. Theranostics (2011) 1:154–88. doi:10.7150/thno/v01p0154
79. Goodman SL, Picard M. Integrins as therapeutic targets. Trends Pharmacol Sci (2012) 33(7):405–12. doi:10.1016/j.tips.2012.04.002
80. Chigaev A, Wu Y, Williams DB, Smagley Y, Sklar LA. Discovery of very late antigen-4 (VLA-4, alpha4beta1 integrin) allosteric antagonists. J Biol Chem (2011) 286(7):5455–63. doi:10.1074/jbc.M110.162636
81. Jackson DY. Alpha 4 integrin antagonists. Curr Pharm Des (2002) 8(14):1229–53. doi:10.2174/1381612023394737
82. Yu Y, Schurpf T, Springer TA. How natalizumab binds and antagonizes alpha4 integrins. J Biol Chem (2013) 288(45):32314–25. doi:10.1074/jbc.M113.501668
83. Weber MS, Menge T, Lehmann-Horn K, Kronsbein HC, Zettl U, Sellner J, et al. Current treatment strategies for multiple sclerosis – efficacy versus neurological adverse effects. Curr Pharm Des (2012) 18(2):209–19. doi:10.2174/138161212799040501
84. Clifford DB, De LA, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol (2010) 9(4):438–46. doi:10.1016/S1474-4422(10)70028-4
85. Neumann F, Zohren F, Haas R. The role of natalizumab in hematopoietic stem cell mobilization. Expert Opin Biol Ther (2009) 9(8):1099–106. doi:10.1517/14712590903055011
86. Bonig H, Wundes A, Chang KH, Lucas S, Papayannopoulou T. Increased numbers of circulating hematopoietic stem/progenitor cells are chronically maintained in patients treated with the CD49d blocking antibody natalizumab. Blood (2008) 111(7):3439–41. doi:10.1182/blood-2007-09-112052
87. Tilg H, Kaser A. Vedolizumab, a humanized mAb against the alpha4beta7 integrin for the potential treatment of ulcerative colitis and Crohn’s disease. Curr Opin Investig Drugs (2010) 11(11):1295–304.
88. Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn’s disease and ulcerative colitis. Inflammopharmacology (2012) 20(1):1–18. doi:10.1007/s10787-011-0104-6
89. Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, et al. Treatment of active Crohn’s disease with MLN0002, a humanized antibody to the alpha4beta7 integrin. Clin Gastroenterol Hepatol (2008) 6(12):1370–7. doi:10.1016/j.cgh.2008.06.007
90. Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med (2005) 352(24):2499–507. doi:10.1056/NEJMoa042982
91. Rettig MP, Ansstas G, DiPersio JF. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia (2012) 26(1):34–53. doi:10.1038/leu.2011.197
92. Ramirez P, Rettig MP, Uy GL, Deych E, Holt MS, Ritchey JK, et al. Bio5192, a small molecule inhibitor of VLA-4, mobilizes hematopoietic stem and progenitor cells. Blood (2009) 114(7):1340–3. doi:10.1182/blood-2008-10-184721
93. Becker PS, Appelbaum FR, Chien S, Zhao X, Bonig H, Mukasa R, et al. Oral small molecule inhibitor of VLA-4 overcomes adhesion mediated chemotherapy resistance of acute myeloid leukemia (AML) blasts in vitro, without impairment of normal blood cell recovery when combined with chemotherapy in vivo. Blood (2008) 112:858.
94. Sugiura T, Kageyama S, Andou A, Miyazawa T, Ejima C, Nakayama A, et al. Oral treatment with a novel small molecule alpha 4 integrin antagonist, AJM300, prevents the development of experimental colitis in mice. J Crohns Colitis (2013) 7(11):e533–42. doi:10.1016/j.crohns.2013.03.014
95. Miller DH, Weber T, Grove R, Wardell C, Horrigan J, Graff O, et al. Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol (2012) 11(2):131–9. doi:10.1016/S1474-4422(11)70299-X
96. Hijazi Y, Welker H, Dorr AE, Tang JP, Blain R, Renzetti LM, et al. Pharmacokinetics, safety, and tolerability of R411, a dual alpha4beta1-alpha4beta7 integrin antagonist after oral administration at single and multiple once-daily ascending doses in healthy volunteers. J Clin Pharmacol (2004) 44(12):1368–78. doi:10.1177/0091270004270147
97. Hijazi Y, Welker H, Dorr AE, Frank K, Renzetti LM, Patel I. Evaluation of the effect of multiple-dose administration of R411, a dual alpha4beta1-alpha4beta7 integrin antagonist on the major CYP isoform activities in healthy volunteers. Eur J Clin Pharmacol (2006) 62(1):83–5. doi:10.1007/s00228-005-0067-x
98. Wolf C, Sidhu J, Otoul C, Morris DL, Cnops J, Taubel J, et al. Pharmacodynamic consequences of administration of VLA-4 antagonist CDP323 to multiple sclerosis subjects: a randomized, double-blind phase 1/2 study. PLoS One (2013) 8(3):e58438. doi:10.1371/journal.pone.0058438
99. Chanteux H, Rosa M, Delatour C, Prakash C, Smith S, Nicolas JM. In vitro hydrolysis and transesterification of CDP323, an alpha4beta1/alpha4beta7 integrin antagonist ester prodrug. Drug Metab Dispos (2014) 42(1):153–61. doi:10.1124/dmd.113.054049
100. Chen L, Tilley J, Trilles RV, Yun W, Fry D, Cook C, et al. N-acyl-l-phenylalanine derivatives as potent VLA-4 antagonists that mimic a cyclic peptide conformation. Bioorg Med Chem Lett (2002) 12(2):137–40. doi:10.1016/S0960-894X(01)00711-9
101. Lin K, Ateeq HS, Hsiung SH, Chong LT, Zimmerman CN, Castro A, et al. Selective, tight-binding inhibitors of integrin alpha4beta1 that inhibit allergic airway responses. J Med Chem (1999) 42(5):920–34. doi:10.1021/jm980673g
102. Leone DR, Giza K, Gill A, Dolinski BM, Yang W, Perper S, et al. An assessment of the mechanistic differences between two integrin alpha 4 beta 1 inhibitors, the monoclonal antibody TA-2 and the small molecule Bio5192, in rat experimental autoimmune encephalomyelitis. J Pharmacol Exp Ther (2003) 305(3):1150–62. doi:10.1124/jpet.102.047332
103. Norris V, Choong L, Tran D, Corden Z, Boyce M, Arshad H, et al. Effect of IVL745, a VLA-4 antagonist, on allergen-induced bronchoconstriction in patients with asthma. J Allergy Clin Immunol (2005) 116(4):761–7. doi:10.1016/j.jaci.2005.04.045
104. Vanderslice P, Woodside DG, Caivano AR, Decker ER, Munsch CL, Sherwood SJ, et al. Potent in vivo suppression of inflammation by selectively targeting the high affinity conformation of integrin alpha4beta1. Biochem Biophys Res Commun (2010) 400(4):619–24. doi:10.1016/j.bbrc.2010.08.114
105. Vanderslice P, Biediger RJ, Woodside DG, Brown WS, Khounlo S, Warier ND, et al. Small molecule agonist of very late antigen-4 (VLA-4) integrin induces progenitor cell adhesion. J Biol Chem (2013) 288(27):19414–28. doi:10.1074/jbc.M113.479634
106. Wayner EA, Garcia-Pardo A, Humphries MJ, McDonald JA, Carter WG. Identification and characterization of the T lymphocyte adhesion receptor for an alternative cell attachment domain (CS-1) in plasma fibronectin. J Cell Biol (1989) 109(3):1321–30. doi:10.1083/jcb.109.3.1321
107. Mould AP, Wheldon LA, Komoriya A, Wayner EA, Yamada KM, Humphries MJ. Affinity chromatographic isolation of the melanoma adhesion receptor for the IIICS region of fibronectin and its identification as the integrin alpha 4 beta 1. J Biol Chem (1990) 265(7):4020–4.
108. Komoriya A, Green LJ, Mervic M, Yamada SS, Yamada KM, Humphries MJ. The minimal essential sequence for a major cell type-specific adhesion site (CS1) within the alternatively spliced type III connecting segment domain of fibronectin is leucine-aspartic acid-valine. J Biol Chem (1991) 266(23):15075–9.
109. Woodside DG, Vanderslice P. Cell adhesion antagonists: therapeutic potential in asthma and chronic obstructive pulmonary disease. BioDrugs (2008) 22(2):85–100. doi:10.2165/00063030-200822020-00002
110. Kenyon NJ, Liu R, O’Roark EM, Huang W, Peng L, Lam KS. An alpha4beta1 integrin antagonist decreases airway inflammation in ovalbumin-exposed mice. Eur J Pharmacol (2009) 603(1–3):138–46. doi:10.1016/j.ejphar.2008.11.063
111. Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS. Combinatorial chemistry identifies high-affinity peptidomimetics against alpha4beta1 integrin for in vivo tumor imaging. Nat Chem Biol (2006) 2(7):381–9. doi:10.1038/nchembio798
112. Shah B, Jensen BK, Zhang J, Hunt T, Rohatagi S. Effect of food on pharmacokinetics of an inhaled drug: a case study with a VLA-4 antagonist, HMR1031. J Clin Pharmacol (2003) 43(12):1341–9. doi:10.1177/0091270003258172
113. Diamant Z, Kuperus J, Baan R, Nietzmann K, Millet S, Mendes P, et al. Effect of a very late antigen-4 receptor antagonist on allergen-induced airway responses and inflammation in asthma. Clin Exp Allergy (2005) 35(8):1080–7. doi:10.1111/j.1365-2222.2005.02296.x
114. Setoguchi M, Iimura S, Sugimoto Y, Yoneda Y, Chiba J, Watanabe T, et al. A novel, potent, and orally active VLA-4 antagonist with good aqueous solubility: trans-4-[1-[[2-(5-Fluoro-2-methylphenylamino)-7-fluoro-6-benzoxazolyl]acetyl]-(5S)-[methoxy(methyl)amino]methyl-(2S)-pyrrolidinylmethoxy]cyclohexanecarboxylic acid. Bioorg Med Chem (2013) 21(1):42–61. doi:10.1016/j.bmc.2012.11.003
Keywords: integrin alpha4, CD49d, adhesion, drug resistance, acute lymphoblastic leukemia
Citation: Shishido S, Bönig H and Kim Y-M (2014) Role of integrin alpha4 in drug resistance of leukemia. Front. Oncol. 4:99. doi: 10.3389/fonc.2014.00099
Received: 10 January 2014; Accepted: 22 April 2014;
Published online: 23 May 2014.
Edited by:
Mignon Lee-Cheun Loh, University of California San Francisco, USAReviewed by:
Hema Dave, Children’s National Medical Center, USAChinten James Lim, University of British Columbia, Canada
Copyright: © 2014 Shishido, Bönig and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Mi Kim, Division of Hematology and Oncology, Department of Pediatrics, Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, 4650 Sunset Boulevard, MS #57, Los Angeles, CA 90027, USA e-mail:eW1raW1AY2hsYS51c2MuZWR1