 Peder Rustøen Braadland
Peder Rustøen Braadland Håkon Ramberg
Håkon Ramberg Helene Hartvedt Grytli1
Helene Hartvedt Grytli1 Kristin Austlid Taskén
Kristin Austlid Taskén- 1Department of Tumor Biology, Institute of Cancer Research, Division of Cancer Medicine, Transplantation and Surgery, Oslo University Hospital, Oslo, Norway
- 2Institute of Clinical Medicine, University of Oslo, Oslo, Norway
Enhanced sympathetic signaling, often associated with obesity and chronic stress, is increasingly acknowledged as a contributor to cancer aggressiveness. In prostate cancer, intact sympathetic nerves are critical for tumor formation, and sympathectomy induces apoptosis and blocks tumor growth. Perineural invasion, involving enrichment of intra-prostatic nerves, is frequently observed in prostate cancer and is associated with poor prognosis. β2-adrenergic receptor (ADRB2), the most abundant receptor for sympathetic signals in prostate luminal cells, has been shown to regulate trans-differentiation of cancer cells to neuroendocrine-like cells and to affect apoptosis, angiogenesis, epithelial–mesenchymal transition, migration, and metastasis. Epidemiologic studies have shown that use of β-blockers, inhibiting β-adrenergic receptor activity, is associated with reduced prostate cancer-specific mortality. In this review, we aim to present an overview on how β-adrenergic receptor and its downstream signaling cascade influence the development of aggressive prostate cancer, primarily through regulating neuroendocrine differentiation.
Introduction
Most men die with and not from prostate cancer. Despite this, prostate cancer was the primary cause of death in more than 300,000 men worldwide in 2012, with an estimated 630,000 deaths to be expected in 2035 (1). Neuroendocrine prostate cancer, a poorly defined clinical phenotype of aggressive disease, is predicted to cause approximately 10–25% of the prostate cancer-specific deaths (2–4). Drugs targeting androgen receptor activity promote development of a neuroendocrine prostate cancer phenotype (5) and increases the prevalence of neuroendocrine cells (6), and as more drugs in this category reach the clinic the occurrence is expected to rise. Neuroendocrine-like cancer cells are differentiated to a varying extent and may express luminal, mesenchymal, and/or stem cell markers in addition to neuroendocrine markers (7–11). This reflects the high plasticity of these cells. Although the molecular mechanisms underlying neuroendocrine differentiation in vivo are poorly understood, inflammation (12), androgen deprivation (13), ionizing radiation therapy (14), and activation of the β-adrenergic receptor (ADRB) have been shown to induce trans-differentiation of prostate cancer cell lines to neuroendocrine-like cells in vitro.
Over the last decade, epidemiologic studies have indicated that use of β-blockers may have beneficial effects on cancer progression, metastasis, and mortality (15–24). β-blockers form a group of commonly prescribed drugs used as treatment for hypertension, cardiac heart failure, and arrhythmias, as well as for migraine prophylaxis. In two Norwegian cohorts of patients with aggressive prostate cancer, it was reported that use of β-blocker was associated with reduced prostate cancer-specific mortality (21, 22). In contrast, a nested case-control study of prostate cancer patients in the UK Clinical Practice Research Datalink cohort did not observe an effect of β-blocker usage after diagnosis on prostate cancer-specific deaths (25). However, use of β-blocker has been reported to be inversely associated with progression of breast, ovarian, and non-small cell lung cancer (23). Moreover, β-blocker use has been associated with longer relapse-free survival (15) and lower risk of tumor recurrence (17), distant metastasis (17), and cancer-specific mortality (16, 17) in breast cancer patients. Indeed, pre-clinical and epidemiological evidence have led to the initiation of clinical phase II studies evaluating the effect of administering the β-blocker propranolol to ovarian, cervix, colorectal, and breast cancer patients (ClinicalTrials.gov identifiers: NCT01504126, NCT01308944, NCT01902966, NCT00888797, and NCT01847001). Together this indicates that more studies on prostate cancer cohorts are needed.
In this review, we will focus on how β-adrenergic activity, primarily via the β2-adrenergic receptor (ADRB2) and the subsequent cyclic AMP (cAMP) signaling pathway, affects development of aggressive prostate cancer by regulating neuroendocrine differentiation, metastasis, angiogenesis, and apoptosis-resistance.
Adrenergic Receptor’s Functional Role in the Prostate
The β-adrenergic receptors (ADRBs) are part of the sympathetic nervous system, the general role of which is to ensure that the body responds fast and targeted upon danger, as well as to regulate the whole body energy expenditure. The receptors are activated by catecholamines; norepinephrine released by adrenergic nerves, innervating most major organs, and epinephrine produced by chromaffin cells (26). Chromaffin cells are most highly abundant in the adrenal medulla, but paraganglia has also been observed in proximity to sympathetic nerves within the prostate (27, 28). Macrophages, aside from exerting an immunosuppressive activity following catecholamine stimulation (29), also have the capacity to produce catecholamines themselves to a minor extent (30). Interestingly, infiltration of macrophages has been reported to be associated with prostate cancer aggressiveness (31, 32).
The prostate is highly innervated (33), and the nerves are required for formation of the prostate during embryogenesis, maturation during puberty, and maintenance of the adult phenotype (34). Thus, like androgen stimulation, sympathetic stimuli contribute to prostatic differentiation in vivo (35). Interestingly, most prostate cancers originate from the peripheral zone, which is part of the posterior region where the majority of nerves are located (36). Whereas, parasympathetic nerves are uniformly spread from the base to the apex and innervate the epithelium, sympathetic nerves are slightly enriched toward the base and are in close contact with the smooth muscle cells (36–39). The adrenergic nerves fire during ejaculation, promoting contraction of smooth muscle cells expressing α-adrenergic receptors (40). In addition, adrenergic stimulation facilitates secretion from the luminal cells predominately expressing β-adrenergic receptors (41).
The interplay between nerves and cancer cells is an emerging field in prostate cancer research. Intact sympathetic nerves were recently shown to be essential for tumor formation as sympathectomy induced apoptosis and blocked prostatic intraepithelial neoplasia formation and tumor growth in a mouse model (42). Furthermore, perineural invasion is a phenomenon whereby cancer cells are frequently observed to surround or track the nerve fiber (43). An increasing number of studies conclude that perineural invasion is a prognostic marker in prostate cancer (44, 45). The nerve density is enriched in cancer areas and higher in prostatic tissue from high-risk compared to low-risk prostate cancer patients (42, 46), indicating that neurogenesis may occur during cancer development (47).
Prostate cancer cells proximal to areas of perineural invasion have been shown to exhibit reduced apoptosis and increased proliferation compared to distant cancer cells (47). In a prostate cancer case study, increased frequency of neuroendocrine-like cells was observed in the proximity to perineural invasion (48). Interestingly, in a pancreatic cancer study, catecholamine exposure from co-cultured dorsal root ganglia was shown to promote perineural invasion both in vitro and in animal experiments (49). A possible mechanism explaining this observation is that norepinephrine secreted by the sympathetic nerves acts as a chemoattractant, promoting cancer cell migration toward innervated areas (49), with subsequent metastasis through the perineural space. Studies are wanted to unravel whether this mechanism is involved in stress-induced metastatic prostate cancer.
ADRB2 Regulation and Downstream Signaling in Prostate Cancer
The prostate is highly enriched in β-adrenergic receptors with ADRB2 being the dominating isoform in luminal cells [ADRB1: (50, 51); ADRB2: (41, 51–53); and ADRB3: (54)]. More than 95% of the β-adrenergic receptor binding activity in PC-3 cells is mediated through ADRB2 (51), and the main ADRB isoform in LNCaP cells is the β2 subtype (52). β2- and β3-adrenergic receptors have been observed in stromal cells (55, 56), although immunohistochemical staining using ADRB2 antibodies showed predominantly epithelial localization in both benign and malignant prostate tissue (57, 58). In the first immunohistochemical staining report of β2-adrenergic receptor in human prostate, ADRB2 was only observed in malignant tissue (59). Most gene expression profiles show up-regulation of ADRB2 mRNA in prostatic adenocarcinomas (57, 60), and the general consensus in the literature is that the protein expression level of ADRB2 is increased in prostate cancer cells compared to benign prostate cells (57, 58). Following castration in mice and during androgen deprivation therapy of prostate cancer patients, low β-adrenergic activity, and down-regulation of ADRB2 mRNA, respectively, has been reported (57, 61). Although ADRB2 is up-regulated in malignant cells, the expression level seems to decrease during progression as ADRB2 is inversely correlated with PSA recurrence-free survival (58). In metastatic prostate cancer, the situation is more complex as both high and low levels of ADRB2 have been observed (57, 58). ADRB2 is assumed to be up-regulated in castration-resistant prostate cancer to support sensitization of the androgen receptor, but it is down-regulated in the androgen independent sub-line LNCaP-abl at the mRNA-level (62), and at the protein level in LNCaP-Rf (57), both compared to the parental LNCaP cell line. Amplification of ADRB2 has, however, been reported in 3 out of 28 cases in a cohort of castrated metastatic prostate cancer patients (60). More data are needed to test whether ADRB2 is involved in development of castration-resistant prostate cancer.
Besides being regulated by thyroid hormones in LNCaP cells (57), ADRB2 has been shown to be an androgen receptor target gene (35, 63–65). Interestingly, ADRB2 is also a target gene of two important markers in prostate cancer that are involved in transcriptional regulation; v-ets avian erythroblastosis virus E26 oncogene homolog (ERG) (66) and Enhancer of zeste homolog 2 (EZH2) (58). Both ERG and EZH2 exert repressive action on ADRB2 transcription in vitro, through direct binding and epigenetic silencing, respectively (66). Furthermore, ERG up-regulates the expression of EZH2 (66). This suggests that ERG and EZH2 antagonize the stimulatory effect of androgen on ADRB2 expression. The overall effect, however, based on analysis of data from cBioPortal is that ADRB2 as well as ERG and EZH2 are either up-regulated or unaltered at the mRNA level in malignant compared to benign prostate tissue (60). This does not rule out the possibility that ERG and/or EZH2 exert a more dominating effect on ADRB2 expression, as suggested by Yu et al. (58, 66), in advanced diseases. ERG was recently shown to inhibit luminal and neuroendocrine differentiation in a transgenic prostate cancer mouse model (67), suggesting that ERG can be linked to de-differentiation of cancer cells. This would fit into the hypothesis that ADRB2 is positively and ERG negatively correlated with a differentiated phenotype. Although the prognostic value of TMPRSS2-ERG is controversial, this hypothesis would also support a role of ERG as prognostic marker (67–69).
An overview of known ADRB2 downstream signaling pathways in prostate cancer cell lines is summarized in Figure 1. ADRB2 is a seven-trans membrane G-protein coupled receptor primarily acting through the cAMP-signaling pathway. Ligand binding to ADRB2 stimulates adenylyl cyclase activity and cAMP production via Gαs. Induction of cAMP in response to adrenergic stimulation has been shown in a number of prostate cancer cell lines (70–73). Most effects of cAMP are mediated through the cAMP-dependent protein kinase (PKA), and among other proteins regulated by cAMP are exchange proteins activated by cAMP (EPAC) and cyclic nucleotide-gated ion channels. It is not known whether these are activated in response to adrenergic stimulation of prostate cancer cells, but activation of EPAC, using an EPAC-specific cAMP analog, affects the MAP kinase, RhoA (74), and AKT-p70S6K signaling pathways in prostatic epithelial cells (75, 76). These pathways are also regulated by adrenergic activation in prostate epithelial cells as described below. Noteworthy, treatment of LNCaP cells with an EPAC analog indicated that PKA is the dominating mediator of neuroendocrine differentiation in these cells (77).
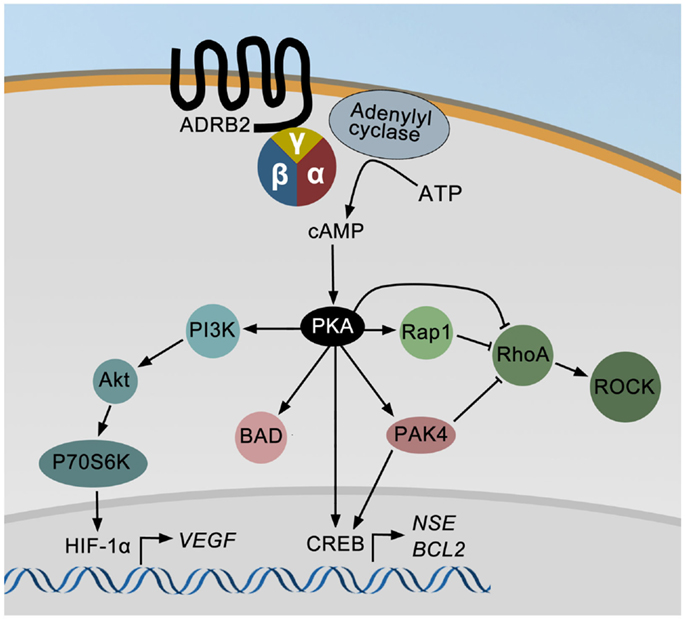
Figure 1. The ADRB2 signaling pathways in prostate cancer. Ligand binding to ADRB2 increases the intracellular level of cAMP, which activates cAMP-dependent protein kinase (PKA). PKA may either directly or through PAK4 stimulate CREB activity and thereby induce the expression of ENO2 and BCL2. PKA can also directly or indirectly via PAK4 or Rap1 inhibit RhoA and ROCK activities and thereby induce neurite outgrowth. Finally, VEGF expression is up-regulated by adrenergic stimulation via PI3K/AKT/p70S6K mediated activation of HIF-1α.
Cyclic AMP produced in response to adrenergic stimulation binds the regulatory subunit of PKA and the activated catalytic subunit is released. The catalytic subunit may translocate to the nucleus and phosphorylate cAMP responsive element binding protein (CREB), which induces the expression of e.g., neuron specific enolase/enolase 2 (ENO2, a neuroendocrine marker), and B-cell CLL/lymphoma 2 (BCL2, encoding an anti-apoptotic protein) (78). PKA-induced phosphorylation of CREB may either be direct or indirect through regulation of p21-activated protein kinase 4 (PAK4) and/or ERK activity. Stress may also promote apoptosis-resistance through PKA-dependent phosphorylation of BCL2-associated agonist of cell death (BAD), as shown in Figure 1 (79). Furthermore, PKA may inhibit the ras homolog family member A (RhoA) – Rho-associated PKA (ROCK) pathway leading to neurite outgrowth either directly or mediated through either Rap1, a member of the RAS oncogene family, or PAK4 (80). Rap1 is also possibly involved in PKA-induced regulation of ERK activity (not shown in Figure 1). Finally, PKA-mediated effects of adrenergic stimuli up-regulate vascular endothelial growth factor (VEGF) levels and HUVEC capillary tube formation via the PI3K/AKT/p70S6K/HIF-1α pathway (81).
Besides regulating the transcription factor activity of CREB and HIF-1α, the ADRB2/cAMP/PKA signaling pathway has been shown to stimulate the androgen receptor responsive gene transcription (57, 72). The putative molecular mechanisms involved in ADRB2/PKA-mediated regulation of androgen receptor activity have been thoroughly described in a review by Merkle and Hoffmann (82).
Much is still to be learned about the ADRB2 signaling pathway in prostatic luminal cells. β-arrestin is instrumental in the desensitization and internalization/sequestration of β-adrenergic receptors (83). One study reported increased formation of a β-arrestin-SRC complex following ADRB2 stimulation in LNCaP cells over-expressing β-arrestin2 (73). How this affects the functional effects of adrenergic signaling is unknown.
Adrenergic Regulation of Neuroendocrine Differentiation
β-adrenergic stimulation is a well-known inducer of neuroendocrine differentiation of prostatic adenocarcinoma cell lines (70, 84–86). Data linking sympathetic stimuli to neuroendocrine differentiation in vivo, however, are currently lacking. Cox and co-workers reported in a series of publications that the ADRB2 agonists epinephrine and isoproterenol caused a rise in the intracellular cAMP levels, followed by increased activity of cAMP-dependent PKA and a higher number of neuroendocrine-like cancer cells (70, 84). cAMP has been shown to induce neuroendocrine differentiation to various extent in multiple prostate cancer cell lines; namely LNCaP, PC-3, and PC-3-M (70, 85, 87–89). Furthermore, neuroendocrine differentiation of LNCaP cells was observed when the cells were transfected with a plasmid expressing a constitutive active PKA catalytic subunit (84). The induction of a neuroendocrine-like morphology was inhibited after transfection of the LNCaP cells with a PKA regulatory subunit containing mutations that rendered the PKA holoenzyme complex in an inactive state despite increased cAMP levels. Moreover, cAMP-signaling has been reported to up-regulate neuropeptides like PTHrP and neurotensin in LNCaP cells (70).
Interestingly, the first evidence of different substrate specificity between the various isoforms of the catalytic subunit of PKA was observed in prostate cancer cells (90). Prostate cancer cells express both the ubiquitously expressed Cα subunit and the cell-type specific Cβ isoforms (Cβ1, Cβ2, Cβ3, and Cβ4) of PKA (91). The PKA Cβ2 subunit has previously been shown to be up-regulated in the more proliferating prostate epithelial cells present in malignant compared to benign prostate tissue (90). Up-regulation of MYC is an early event during prostate tumorigenesis and PKA Cβ2 has been shown to be a MYC target gene and to participate in a positive feedback loop whereby MYC is stabilized (90). Prolonged activation of PKA Cα, however, represses MYC transcription and may thereby promote growth arrest and neuroendocrine differentiation. In contrast, the PKA Cβ2 splice variant has only a minor effect on MYC transcription and is supposed to be linked to the growth stimulatory effect of MYC. Although it is unknown whether any specific PKA isoforms act downstream of ADRB2, the overall effect of adrenergic stimulation of LNCaP cells is inhibition of proliferation; indicating that Cα is mediating the effect (70, 92). Similarly, growth arrest was observed after cAMP treatment in PC-3-M cells, suggesting that Cα plays a dominating role (85). Generally, the anti-mitogenic effect of ADRB stimulation involving cAMP is in agreement with the non-mitotic characteristic of most neuroendocrine cells.
Neurite outgrowth, a dynamic process in which actin rearrangements cause the cells to obtain a more neuronal phenotype, can be observed in LNCaP cells after 3–5 days of incubation in charcoal-stripped serum (mimicking androgen deprivation) (93). These morphological changes occur simultaneously with a rise in cAMP (6), linking neurite outgrowth to the before-mentioned cAMP-induced neuroendocrine differentiation. Upon adrenergic stimulation, neurite outgrowth is observed as early as after 1 hour (70). Activation of ADRB has been shown to induce an immediate increase in cAMP, which could explain the more rapid appearance of a neuronal phenotype (70) as compared to the delayed increase following androgen-depletion.
Cytoskeletal rearrangements are essential in the process of neurite outgrowth, and are regulated by small Rho GTPases like CDC42, Rac1, and RhoA, each controlling distinct morphogenic pathways. Inactivation of RhoA promotes neurite outgrowth in neuronal cells (94, 95). One possible mechanism by which ADRB/cAMP/PKA regulate these cytoskeletal rearrangements involved in neurite outgrowth is through direct inactivation of RhoA (80, 94), as illustrated in Figure 1. In LNCaP cells, the RhoA inhibitor C3 transferase was reported to induce trans-differentiation to neuroendocrine-like cells (77, 96). Furthermore, inhibition of the RhoA downstream effector ROCK has been shown to induce neurite outgrowth in PC-3 cells and to a lesser extent also in LNCaP cells (97). A similar effect is seen through PAK4-induced activation of RhoA as shown in Figure 1 (80). PAK4 may also mediate the effect of ADRB2/cAMP/PKA on neuroendocrine differentiation in prostate cancer cells by regulating the activity of the transcription factor CREB (78). PKA has been shown to activate PAK4 through phosphorylation, which induced the transcriptional activity of CREB and thereby the expression of NSE/ENO2.
In general, assembly of stress fibers plays an important role in adhesion and motility of eukaryotic cells and loss of stress fibers is associated with neurite outgrowth and reduced migratory capacity (98). Upon destabilization of stress fibers, the cell experiences cytoskeletal alterations and loss of focal adhesions, both required for the cell to migrate. Maintenance of stress fiber integrity is ensured through inhibition of actin filament depolymerization and is regulated by the RhoA/ROCK pathway (99). In addition, PKA has been shown to phosphorylate actin monomers directly, thereby destabilizing the stress fibers (100). These mechanisms have not been explored in prostate cancer models.
ADRB2 Expression and Effects on Metastasis
Most prostate cancer metastases are detected in bone, lymph nodes, lung, and liver (101). Metastasis is a complex multi-step process involving the ability of cancer cells to detach from the primary tumor site, degrade extracellular matrix, migrate to other parts of the body, and to invade and settle at the metastatic site (102). The requirement for different properties is constantly changing during the metastatic process, favoring cells with high plasticity. Whereas, de-differentiation like epithelial–mesenchymal transition (EMT) promotes detachment and migration, re-differentiation, or mesenchymal–epithelial transition favors homing to metastatic sites.
In the work by Yu and colleagues, it was shown that the expression level of ADRB2 changes during the metastatic process in prostate cancer (58). Although up-regulation of ADRB2 is observed in malignant compared to benign prostate tissue (57), a decrease in ADRB2 expression is observed in aggressive relative to indolent prostate cancer (58). Interestingly, knockdown of ADRB2 was shown to induce EMT of transformed prostatic epithelial cells (RWPE-1). Expressional analyses revealed that the ADRB2 knockdown cells acquired an increased expression of vimentin (VIM) and N-cadherin (CDH2), as well as lowered expression of β-catenin (CTNNB1) and integrin β4 (ITGB4) suggesting that the cells harbor a mesenchymal-like phenotype. The ADRB2 knockdown cells, as well as cells treated with an ADRB2 antagonist (ICI 118,551), showed increased ability to migrate and invade. Conversely, treatment with an ADRB agonist, isoproterenol, reduced invasion in these cells as well as in DU145 cells (58).
In a PC-3 xenograft mouse model, however, norepinephrine promoted metastasis (59). This might be due to increased migration, as suggested in a study by Lang et al. where increased migratory activity in PC-3 cells was observed upon norepinephrine stimulation (103). The effect was partially inhibited by treating the cells with the β1-specific β-blocker atenolol, and fully inhibited with the β2-specific blocker ICI 118,551. Furthermore, in a xenograft model using ADRB2 and ADRB3 double knockout mice (ADRB2−/−, ADRB3−/−), lowered human tumor cell dissemination to lymph nodes and distant organs was observed (42). Whether stromal ADRB2 and ADRB3 affect the metastatic process could not be addressed in this model system as tumor development was severely compromised in ADRB2−/−, ADRB3−/− mice.
The ADRB2 expression level affects the phenotype of the prostate cells and thereby their ability to migrate and invade (58), and probably also their ability to settle at the metastatic site, which would indicate a role of ADRB2 in the whole metastatic process. Low expression of ADRB2 in prostatic epithelial cells is associated with a mesenchymal-like phenotype (58). These cells may have the potential to re-differentiate into epithelial cells adapted to the microenvironment at the metastatic site. To what extent this involves up-regulation of ADRB2 and development of neuroendocrine-like tumors at the metastatic site is currently not known. Interestingly, adrenergic stimulation has been linked to pro-angiogenic processes in different cancer models (81, 104), and may thus aid in providing the cancer cells with another mean to escape the primary tumor site.
Stress-Induced Regulation of Angiogenesis
Neuroendocrine cells are the primary site of VEGF production within the prostate (105). It is therefore compelling that the number of neuroendocrine cells present in high-grade prostatic carcinoma correlates with the degree of neovascularization (106, 107). Studies have shown that neuroendocrine cells promote growth of neighboring cancer cells through secretion of neuropeptides (108–111), and consequently an increased energy supply through the blood stream is required by the tumor. The fact that several factors are involved in both angiogenesis and neuroendocrine differentiation is intriguing, and points at a possible linkage between the two processes.
Chronic stress has been reported to increase tissue norepinephrine levels in an ovarian carcinoma mouse model, resulting in the formation of new blood vessels, and increased expression of the pro-angiogenic VEGF (104). Similar observations have been reported in the androgen-sensitive LNCaP cell line, where VEGF-expression increased in a dose-dependent manner upon ADRB2-mediated epinephrine stimulation (112). Furthermore, in androgen-insensitive PC-3 cells, norepinephrine, and isoproterenol stimulation induced VEGF expression through increased activity of the cAMP/PKA pathway (81). Interleukin 6 (IL6), a well-known inducer of neuroendocrine differentiation, and a putative downstream target of adrenergic signaling, also functions as a pro-angiogenic factor (92, 113, 114). Moreover, conditioned media from norepinephrine stimulated PC-3 cells induced HUVEC capillary tube formation in an in vitro angiogenesis assay (81). In concordance with the adrenergic stimulatory effects on VEGF and capillary tube formation, treatment of rats with propranolol resulted in a reduction in ventral prostate blood vessel volume (115). Hassan and colleagues were, however, not able to detect a significant up-regulation of plasma-VEGF levels in stressed compared to calm mice (116). They also measured the micro-vessel density in Hi-Myc mice after stress-induced adrenergic stimulation, and also here they did not observe any significant difference between calm and stressed Hi-Myc mice.
Although there are very few studies that have addressed the effects of ADRB stimulation on angiogenesis in prostate cancer, the strong evidence from other model systems (117–119) warrants further investigation into this field in different prostate cancer models.
Adrenergic Regulation of Apoptosis
Stress has been reported to reduce apoptotic activity (116, 120), whereas sympathectomy increases apoptosis in mouse prostate cancer models (42). Thus, prolonged elevation of catecholamines may promote prostate cancer progression by inducing resistance to apoptosis. This is in agreement with the observation that epinephrine protects LNCaP and C4-2 cells from apoptosis induced by the PI3K inhibitor LY294002, and thapsigargin (79).
Adrenergic signaling regulates apoptotic activity by multiple mechanisms as indicated in Figure 1. The best characterized mechanism in prostate cancer cells is the PKA-mediated phosphorylation of BAD on Ser112 and Ser155 (79, 121, 122). Increased phosphorylation of BAD at Ser112 was also observed in mice, and may explain the stress-induced resistance to apoptosis observed in mouse models (116). PAK4 has also been shown to phosphorylate BAD on Ser112 in HeLa cells over-expressing PAK4 (123). Furthermore, PKA induces apoptosis-resistance by directly or indirectly activating CREB and thereby up-regulating the level of BCL2 (78). The anti-apoptotic BCL2 protein acts downstream of ADRB2 in pancreatic cancer cells (124).
Repeated immobilization stress, which elevates the plasma level of epinephrine and inhibits apoptosis, has been shown to accelerate cancer development in mice through stimulation of β-adrenergic receptors (116). Prolonged elevation of catecholamines is also observed in obese (125, 126) and chronically stressed (127, 128) individuals, and may represent one mechanism by which obesity, and perhaps stress, promote development of aggressive prostate cancer (129–131). As a negative feedback mechanism, enhanced levels of catecholamines may lead to down-regulation of ADRB2. Interestingly, expression of ADRB2 in prostatectomy specimens is inversely correlated with biochemical recurrence (BCR) (58), suggesting that chronic stress and low levels of ADRB2 are associated with disease progression. Studies are needed to enlighten this hypothesis.
Controversies, Clinical Implications, and Conclusions
In normal prostate physiology, the sympathetic nervous system regulates prostate differentiation and secretory activity of luminal cells, predominantly through ADRB2 (34, 35, 40, 41). We know from in vitro and in vivo prostate cancer models that chronic elevation of ADRB activity by exposing mice to repeated stress or by adding ADRB agonists promotes neuroendocrine differentiation (70, 84–86), metastasis (58, 103), angiogenesis (78, 81, 112, 115), and apoptosis-resistance (116, 120); together indicating that adrenergic signaling promotes prostate cancer progression (Figure 2).
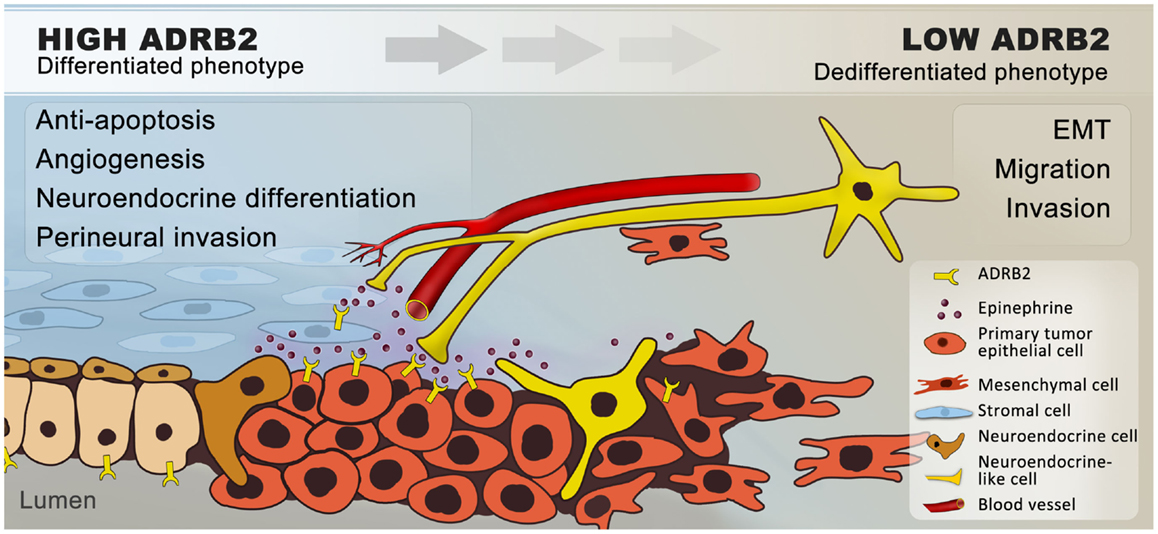
Figure 2. Hypothetical model of how β2-adrenergic signaling may promote progression of prostate cancer. In summary, we hypothesize that ADRB2s expressed on luminal cells are activated by catecholamines, which are secreted by nerves and transported through blood vessels in response to stress. Catecholamines are possibly also secreted by proximal chromaffin-like cells and macrophages (not shown) that also can produce epinephrine and norepinephrine, respectively. In addition, ADRB2s expressed on stromal cells are activated by sympathetic stimuli. Upon ligand-binding, the expression of anti-apoptotic and pro-angiogenic factors is increased and a number of cancer cells undergo trans-differentiation to neuroendocrine-like cells. Together this will favor tumor growth. Angiogenesis and neurogenesis are closely linked (132) and sympathetic activation may stimulate perineural invasion through chemotaxis. In general, chronic ADRB2 activation down-regulates the ADRB2-level, leading to de-differentiation and epithelial–mesenchymal transition, with a subsequent increase in the migratory and invasive potential of the cells. Cancer cells expressing low levels of ADRB2 will thereby follow the nerves and blood vessels to metastatic sites.
There are, however, several controversies in the field that challenge this hypothetical model. To begin with, the collective evidence fails to point in any obvious direction in terms of whether adrenergic signaling is beneficial or disadvantageous for prostate cancer patients. Both stimulatory and inhibitory effects of adrenergic stimulation on proliferation have been observed in cell line studies (70, 73, 85, 92). The majority of publications involving prostate cancer cell lines, however, claim that elevated β-adrenergic receptor activity induces growth arrest in vitro and has no effect in mouse models, alongside undergoing neuroendocrine differentiation in cell lines (116). Furthermore, adrenergic signaling up-regulates VEGF expression (112) and promotes HUVEC capillary tube formation in cell line experiments (81). Induction of anti-apoptotic mechanisms through ADRB2 stimulation has been seen in both cell lines and in prostate cancer xenograft models (116). To what extent these mechanisms are involved in development of human prostate cancer is unknown, but reduced apoptotic activity and stimulation of angiogenesis may be consequences of up-regulated ADRB2 levels, which are seen in malignant compared to benign prostatic epithelial cells (Figure 3).
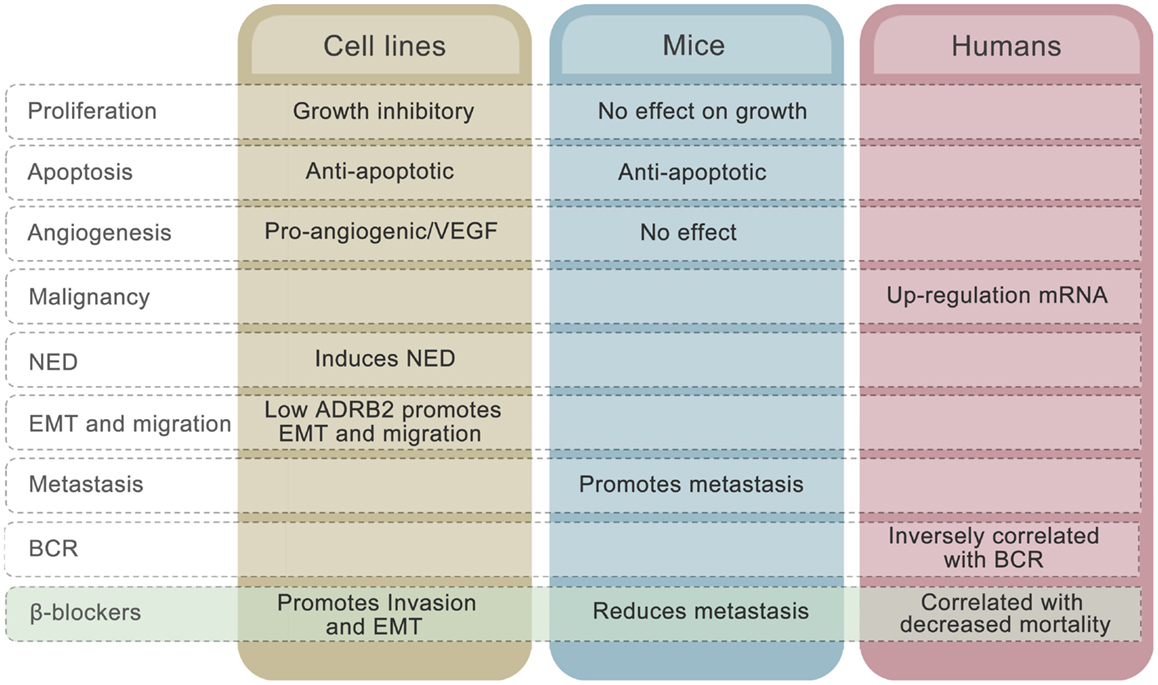
Figure 3. Effects of ADRB2 on tumor characteristics in cell lines, mouse models, and human prostate cancer. The effects of β-blockers on the different characteristics in each model system is also shown.
The mechanisms described above cannot explain why low rather than high level of ADRB2 is associated with disease progression measured as BCR. The inverse correlation between ADRB2 and BCR may relate to the observation that, whereas high ADRB2 activity induces neuroendocrine differentiation, low ADRB2 activity/level promotes EMT in prostate cell lines (Figure 3). Cancer cells expressing low levels of ADRB2 have a mesenchymal-like phenotype and have a higher probability of being in the circulation at time of prostate removal (radical prostatectomy). These cells may also have a higher degree of plasticity and will therefore more easily adapt to environmental changes and thereby produce recurrent tumors in both humans and mice. The fact that reduction in adrenergic activity induces EMT in prostatic epithelial cells gives rise to another conflicting observation, since adrenergic stimulation promoted metastasis in a PC-3 xenograft model (59). A plausible explanation to this is that circulating tumor cells are already present in the xenograft model due to the mesenchymal-like phenotype of PC-3 cells and that binding of ligand to ADRB promotes mesenchymal to epithelial transition (MET) and homing to metastatic sites. Along the same line, we may explain the β-blocker paradox. In cell line experiments, β-blockers have been shown to promote EMT (58) whereas use of β-blocker is associated with reduced mortality in prostate cancer patients (21, 22). Again, inhibition of MET by β-blockers is one hypothesis that needs to be unraveled.
The reports on effects of β-blockers on mortality in other cancer types brings forth an important question: are the in vivo effects of β-blockers mediated by common tissue specific/non-specific attributes, or are the effects indirect (i.e., systemic or neural effects facilitated by other local or distant tissue expressing ADRBs)? β-blockers probably have an effect on immune responses, hormone levels, angiogenesis, neurogenesis, and at the metastatic niche. In the prostate, stromal cells proximal to tumor tissue express ADRBs, and may exert the effect, which may also explain the discrepancy between cell line results and in vivo data. It is also worth noting that the majority of β-blockers are targeting β1-adrenergic receptors or both β1- and β2-adrenergic receptors, whereas ADRB2 has been the receptor mediating the effects on cancer cells. Another plausible explanation lies in the antagonistic mechanism of action. Propranolol, for example, a commonly used antagonist in vitro, has been shown to function as an inverse agonist (133), and can thus lower the β-adrenergic receptor’s activity below its’ basal level. In clinical practice, however, numerous β-blockers are used, and their mechanisms of action vary. Furthermore, the differences observed could be dose-dependent, as it is difficult to measure the dose in patient tissue, whereas this parameter can be controlled in cell lines and animal models. We anticipate that ADRB antagonists will reduce the development of neuroendocrine prostate cancers, but this has not yet been addressed in any publications. More studies are needed to unravel whether β-blockers can play a role in future tailored prostate cancer therapy.
ADRB2 may play a role both as a prognostic and as a predictive biomarker in prostate cancer. We do not know, however, whether the expression level of ADRB2 is a driver of progression. Still, it is plausible to hypothesize that the receptor may be involved in maintenance of a differentiated phenotype, an attribute that is lost when the cells gain plasticity and metastasize, and the disease reaches an incurable stage. We know that ADRB2 is inversely correlated with time to BCR and that it acts independently of Gleason score, surgical margin status and preoperative PSA as a prognostic marker (58). Actually, ADRB2 was the strongest predictor of clinical failure in the study by Yu et al. Validation studies also addressing a potential association with metastasis, development of castration resistance, and survival is warranted to determine whether ADRB2 is a clinically relevant prognostic marker in prostate cancer. The fact that β-adrenergic signaling induces neuroendocrine differentiation and apoptosis-resistance of prostate cancer cells suggest that ADRB2 could play a role in predicting responsiveness to pro-apoptotic drugs.
Author Contributions
All authors (Peder Rustøen Braadland, Håkon Ramberg, Helene Hartvedt Grytli, and Kristin Austlid Taskén) have contributed to the design, drafting, and approval of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to many colleagues for extensive discussions and give particular thanks to Turid Eide, Finn Olav Levy, Kurt Allen Krobert, Aud Svindland, Lars Magne Eri, Viktor Berge, Nishtman Dizeyi, Martina Tinzl, and Anders Bjartell. We apologize to the authors and colleagues whose work could not be included or cited directly due to the broad scope of this review.
References
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer (2013). Available from: http://globocan.iarc.fr
2. Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res (2004) 64(24):9209–16. doi:10.1158/0008-5472.CAN-04-2442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Tanaka M, Suzuki Y, Takaoka K, Suzuki N, Murakami S, Matsuzaki O, et al. Progression of prostate cancer to neuroendocrine cell tumor. Int J Urol (2001) 8(8):431–6. doi:10.1046/j.1442-2042.2001.00347.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Turbat-Herrera EA, Herrera GA, Gore I, Lott RL, Grizzle WE, Bonnin JM. Neuroendocrine differentiation in prostatic carcinomas. A retrospective autopsy study. Arch Pathol Lab Med (1988) 112(11):1100–5.
5. Frigo DE, McDonnell DP. Differential effects of prostate cancer therapeutics on neuroendocrine transdifferentiation. Mol Cancer Ther (2008) 7(3):659–69. doi:10.1158/1535-7163.MCT-07-0480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Burchardt T, Burchardt M, Chen MW, Cao Y, de la Taille A, Shabsigh A, et al. Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J Urol (1999) 162(5):1800–5. doi:10.1016/S0022-5347(05)68241-9
7. Sargos P, Ferretti L, Gross-Goupil M, Orre M, Cornelis F, Henriques de Figueiredo B, et al. Characterization of prostate neuroendocrine cancers and therapeutic management: a literature review. Prostate Cancer Prostatic Dis (2014) 17(3):220–6. doi:10.1038/pcan.2014.17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Alberti C. Neuroendocrine differentiation in prostate carcinoma: focusing on its pathophysiologic mechanisms and pathological features. G Chir (2010) 31(11–12):568–74.
9. McKeithen D, Graham T, Chung LW, Odero-Marah V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate (2010) 70(9):982–92. doi:10.1002/pros.21132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Wang T, Carraway R, Chen H, Briggs J, FitzGerald TJ. ERK1 mediated epithelial-mesenchymal transition and neuroendocrine development in prostate cancer cells that survive high-dose ionizing radiation. Int J Radiat Oncol Biol Phys (2012) 84(3):S667–8. doi:10.1016/j.ijrobp.2012.07.1782
11. Conteduca V, Aieta M, Amadori D, De Giorgi U. Neuroendocrine differentiation in prostate cancer: current and emerging therapy strategies. Crit Rev Oncol Hematol (2014) 92(1):11–24. doi:10.1016/j.critrevonc.2014.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Lee GT, Kwon SJ, Lee JH, Jeon SS, Jang KT, Choi HY, et al. Macrophages induce neuroendocrine differentiation of prostate cancer cells via BMP6-IL6 Loop. Prostate (2011) 71(14):1525–37. doi:10.1002/pros.21369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol (2004) 45(5):586–92. doi:10.1016/j.eururo.2003.11.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res (2008) 68(23):9663–70. doi:10.1158/0008-5472.CAN-08-2229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol (2011) 29(19):2645–52. doi:10.1200/JCO.2010.33.4441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol (2011) 29(19):2635–44. doi:10.1200/JCO.2010.33.5422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, et al. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget (2010) 1(7):628–38.
18. De Giorgi V, Grazzini M, Gandini S, Benemei S, Lotti T, Marchionni N, et al. Treatment with beta-blockers and reduced disease progression in patients with thick melanoma. Arch Intern Med (2011) 171(8):779–81. doi:10.1001/archinternmed.2011.131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lemeshow S, Sorensen HT, Phillips G, Yang EV, Antonsen S, Riis AH, et al. Beta-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev (2011) 20(10):2273–9. doi:10.1158/1055-9965.EPI-11-0249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Diaz ES, Karlan BY, Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol (2012) 127(2):375–8. doi:10.1016/j.ygyno.2012.07.102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Grytli HH, Fagerland MW, Fossa SD, Tasken KA, Haheim LL. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate (2013) 73(3):250–60. doi:10.1002/pros.22564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Grytli HH, Fagerland MW, Fossa SD, Tasken KA. Association between use of beta-blockers and prostate cancer-specific survival: a cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol (2014) 65(3):635–41. doi:10.1016/j.eururo.2013.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Choi CH, Song T, Kim TH, Choi JK, Park JY, Yoon A, et al. Meta-analysis of the effects of beta blocker on survival time in cancer patients. J Cancer Res Clin Oncol (2014) 140(7):1179–88. doi:10.1007/s00432-014-1658-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Wu JW, Boudreau DM, Park Y, Simonds NI, Freedman AN. Commonly used diabetes and cardiovascular medications and cancer recurrence and cancer-specific mortality: a review of the literature. Expert Opin Drug Saf (2014) 13(8):1071–99. doi:10.1517/14740338.2014.926887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Cardwell CR, Coleman HG, Murray LJ, O’Sullivan JM, Powe DG. Beta-blocker usage and prostate cancer survival: a nested case-control study in the UK Clinical Practice Research Datalink cohort. Cancer Epidemiol (2014) 38(3):279–85. doi:10.1016/j.canep.2014.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Spector S, Gordon R, Sjoerdsma A, Udenfriend S. End-product inhibition of tyrosine hydroxylase as a possible mechanism for regulation of norepinephrine synthesis. Mol Pharmacol (1967) 3(6):549–55.
27. Ostrowski ML, Wheeler TM. Paraganglia of the prostate. Location, frequency, and differentiation from prostatic adenocarcinoma. Am J Surg Pathol (1994) 18(4):412–20. doi:10.1097/00000478-199404000-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Nordenstam H, Adams-Ray J. Chromaffin granules and their cellular location in human skin. Z Zellforsch Mikrosk Anat (1957) 45(4):435–43. doi:10.1007/BF00338886
29. Grailer J, Haggadone M, Ward P, editors. Catecholamines promote an M2 macrophage activation phenotype. Proceedings of the 100th Annual Meeting of the American-Association-of-Immunologists (2013).
30. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480(7375):104–8. doi:10.1038/nature10653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene (2014) 33(19):2423–31. doi:10.1038/onc.2013.191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Lanciotti M, Masieri L, Raspollini MR, Minervini A, Mari A, Comito G, et al. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. Biomed Res Int (2014) 2014:486798. doi:10.1155/2014/486798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Rodrigues AO, Machado MT, Wroclawski ER. Prostate innervation and local anesthesia in prostate procedures. Rev Hosp Clin Fac Med Sao Paulo (2002) 57(6):287–92. doi:10.1590/S0041-87812002000600008
34. White CW, Xie JH, Ventura S. Age-related changes in the innervation of the prostate gland: implications for prostate cancer initiation and progression. Organogenesis (2013) 9(3):206–15. doi:10.4161/org.24843
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Guthrie PD, Freeman MR, Liao ST, Chung LW. Regulation of gene expression in rat prostate by androgen and beta-adrenergic receptor pathways. Mol Endocrinol (1990) 4(9):1343–53. doi:10.1210/mend-4-9-1343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Ganzer R, Stolzenburg JU, Wieland WF, Brundl J. Anatomic study of periprostatic nerve distribution: immunohistochemical differentiation of parasympathetic and sympathetic nerve fibres. Eur Urol (2012) 62(6):1150–6. doi:10.1016/j.eururo.2012.03.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Baumgarten HG, Falck B, Holstein AF, Owman C, Owman T. Adrenergic innervation of the human testis, epididymis, ductus deferens and prostate: a fluorescence microscopic and fluorimetric study. Z Zellforsch Mikrosk Anat (1968) 90(1):81–95. doi:10.1007/BF00496704
38. Owman C, Sjoestrand NO. Short adrenergic neurons and catecholamine-containing cells in vas deferens and accessory male genital glands of different mammals. Z Zellforsch Mikrosk Anat (1965) 66(2):300–20. doi:10.1007/BF00344342
39. McVary KT, Razzaq A, Lee C, Venegas MF, Rademaker A, McKenna KE. Growth of the rat prostate gland is facilitated by the autonomic nervous system. Biol Reprod (1994) 51(1):99–107. doi:10.1095/biolreprod51.1.99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Lepor H, Gup DI, Baumann M, Shapiro E. Laboratory assessment of terazosin and alpha-1 blockade in prostatic hyperplasia. Urology (1988) 32(6 Suppl):21–6.
41. Goepel M, Wittmann A, Rubben H, Michel MC. Comparison of adrenoceptor subtype expression in porcine and human bladder and prostate. Urol Res (1997) 25(3):199–206. doi:10.1007/BF00941983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science (2013) 341(6142):1236361. doi:10.1126/science.1236361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ayala GE, Wheeler TM, Shine HD, Schmelz M, Frolov A, Chakraborty S, et al. In vitro dorsal root ganglia and human prostate cell line interaction: redefining perineural invasion in prostate cancer. Prostate (2001) 49(3):213–23. doi:10.1002/pros.1137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Andersen S, Richardsen E, Nordby Y, Ness N, Storkersen O, Al-Shibli K, et al. Disease-specific outcomes of radical prostatectomies in Northern Norway; a case for the impact of perineural infiltration and postoperative PSA-doubling time. BMC Urol (2014) 14(1):49. doi:10.1186/1471-2490-14-49
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Katz B, Srougi M, Dall’Oglio M, Nesrallah AJ, Sant’anna AC, Pontes J Jr, et al. Perineural invasion detection in prostate biopsy is related to recurrence-free survival in patients submitted to radical prostatectomy. Urol Oncol (2013) 31(2):175–9. doi:10.1016/j.urolonc.2010.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res (2008) 14(23):7593–603. doi:10.1158/1078-0432.CCR-08-1164
47. Ayala GE, Dai H, Ittmann M, Li R, Powell M, Frolov A, et al. Growth and survival mechanisms associated with perineural invasion in prostate cancer. Cancer Res (2004) 64(17):6082–90. doi:10.1158/0008-5472.CAN-04-0838
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Kakies C, Hakenberg OW, Gunia S, Erbersdobler A. Prostate cancer with Paneth cell-like neuroendocrine differentiation and extensive perineural invasion: coincidence or causal relationship? Pathol Res Pract (2011) 207(11):715–7. doi:10.1016/j.prp.2011.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Guo K, Ma Q, Li J, Wang Z, Shan T, Li W, et al. Interaction of the sympathetic nerve with pancreatic cancer cells promotes perineural invasion through the activation of STAT3 signaling. Mol Cancer Ther (2013) 12(3):264–73. doi:10.1158/1535-7163.MCT-12-0809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Slater M, Barden JA, Murphy CR. Tyrosine kinase A, autonomic and transmitter receptors, but not innervation, are upregulated in the aging rat prostate. Acta Histochem (2000) 102(4):427–38. doi:10.1078/0065-1281-00565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Penn RB, Frielle T, McCullough JR, Aberg G, Benovic JL. Comparison of R-, S-, and RS-albuterol interaction with human beta 1- and beta 2-adrenergic receptors. Clin Rev Allergy Immunol (1996) 14(1):37–45. doi:10.1007/BF02772201
52. Nagmani R, Pasco DS, Salas RD, Feller DR. Evaluation of beta-adrenergic receptor subtypes in the human prostate cancer cell line-LNCaP. Biochem Pharmacol (2003) 65(9):1489–94. doi:10.1016/S0006-2952(03)00105-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Ramos-Jimenez J, Soria-Jasso LE, Lopez-Colombo A, Reyes-Esparza JA, Camacho J, Arias-Montano JA. Histamine augments beta2-adrenoceptor-induced cyclic AMP accumulation in human prostate cancer cells DU-145 independently of known histamine receptors. Biochem Pharmacol (2007) 73(6):814–23. doi:10.1016/j.bcp.2006.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Berkowitz DE, Nardone NA, Smiley RM, Price DT, Kreutter DK, Fremeau RT, et al. Distribution of beta 3-adrenoceptor mRNA in human tissues. Eur J Pharmacol (1995) 289(2):223–8. doi:10.1016/0922-4106(95)90098-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Haynes JM. Beta(2) and beta(3)-adrenoceptor inhibition of alpha(1)-adrenoceptor-stimulated Ca(2+) elevation in human cultured prostatic stromal cells. Eur J Pharmacol (2007) 570(1–3):18–26. doi:10.1016/j.ejphar.2007.05.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Chamberlain PD, Jennings KH, Paul F, Cordell J, Berry A, Holmes SD, et al. The tissue distribution of the human beta3-adrenoceptor studied using a monoclonal antibody: direct evidence of the beta3-adrenoceptor in human adipose tissue, atrium and skeletal muscle. Int J Obes Relat Metab Disord (1999) 23(10):1057–65. doi:10.1038/sj.ijo.0801039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ramberg H, Eide T, Krobert KA, Levy FO, Dizeyi N, Bjartell AS, et al. Hormonal regulation of beta2-adrenergic receptor level in prostate cancer. Prostate (2008) 68(10):1133–42. doi:10.1002/pros.20778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Yu J, Cao Q, Mehra R, Laxman B, Yu J, Tomlins SA, et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell (2007) 12(5):419–31. doi:10.1016/j.ccr.2007.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Palm D, Lang K, Niggemann B, Drell TL IV, Masur K, Zaenker KS, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer (2006) 118(11):2744–9. doi:10.1002/ijc.21723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2(5):401–4. doi:10.1158/2159-8290.CD-12-0095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Zepp EA, Thomas JA. Effect of androgens on isoproterenol-induced increases in mouse accessory sex organ cyclic AMP in vitro. Biochem Pharmacol (1978) 27(4):465–8. doi:10.1016/0006-2952(78)90377-5
62. Sun T, Wang X, He HH, Sweeney CJ, Liu SX, Brown M, et al. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene (2014) 33(21):2790–800. doi:10.1038/onc.2013.230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Collins S, Quarmby VE, French FS, Lefkowitz RJ, Caron MG. Regulation of the beta 2-adrenergic receptor and its mRNA in the rat ventral prostate by testosterone. FEBS Lett (1988) 233(1):173–6. doi:10.1016/0014-5793(88)81378-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE, et al. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep (2007) 8(9):871–8. doi:10.1038/sj.embor.7401046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J (2011) 30(13):2719–33. doi:10.1038/emboj.2011.158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell (2010) 17(5):443–54. doi:10.1016/j.ccr.2010.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Mounir Z, Lin F, Lin VG, Korn JM, Yu Y, Valdez R, et al. TMPRSS2:ERG blocks neuroendocrine and luminal cell differentiation to maintain prostate cancer proliferation. Oncogene (2014) 1–11. doi:10.1038/onc.2014.308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Hagglof C, Hammarsten P, Stromvall K, Egevad L, Josefsson A, Stattin P, et al. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS One (2014) 9(2):e86824. doi:10.1371/journal.pone.0086824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Qi M, Yang X, Zhang F, Lin T, Sun X, Li Y, et al. ERG rearrangement is associated with prostate cancer-related death in Chinese prostate cancer patients. PLoS One (2014) 9(2):e84959. doi:10.1371/journal.pone.0084959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Cox ME, Deeble PD, Lakhani S, Parsons SJ. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res (1999) 59(15):3821–30.
71. Mitra SP, Carraway RE. Synergistic effects of neurotensin and beta-adrenergic agonist on 3,5-cyclic adenosine monophosphate accumulation and DNA synthesis in prostate cancer PC3 cells. Biochem Pharmacol (1999) 57(12):1391–7. doi:10.1016/S0006-2952(99)00064-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Kasbohm EA, Guo R, Yowell CW, Bagchi G, Kelly P, Arora P, et al. Androgen receptor activation by G(s) signaling in prostate cancer cells. J Biol Chem (2005) 280(12):11583–9. doi:10.1074/jbc.M414423200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Zhang P, He X, Tan J, Zhou X, Zou L. Beta-arrestin2 mediates beta-2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncol Rep (2011) 26(6):1471–7. doi:10.3892/or.2011.1417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Grandoch M, Rose A, ter Braak M, Jendrossek V, Rubben H, Fischer JW, et al. EPAC inhibits migration and proliferation of human prostate carcinoma cells. Br J Cancer (2009) 101(12):2038–42. doi:10.1038/sj.bjc.6605439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Misra UK, Pizzo SV. Upregulation of mTORC2 activation by the selective agonist of EPAC, 8-CPT-2Me-cAMP, in prostate cancer cells: assembly of a multiprotein signaling complex. J Cell Biochem (2012) 113(5):1488–500. doi:10.1002/jcb.24018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Misra UK, Pizzo SV. Evidence for a pro-proliferative feedback loop in prostate cancer: the role of Epac1 and COX-2-dependent pathways. PLoS One (2013) 8(4):e63150. doi:10.1371/journal.pone.0063150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Jones SE, Palmer TM. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell Signal (2012) 24(8):1504–14. doi:10.1016/j.cellsig.2012.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Park MH, Lee HS, Lee CS, You ST, Kim DJ, Park BH, et al. p21-activated kinase 4 promotes prostate cancer progression through CREB. Oncogene (2013) 32(19):2475–82. doi:10.1038/onc.2012.255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Sastry KS, Karpova Y, Prokopovich S, Smith AJ, Essau B, Gersappe A, et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J Biol Chem (2007) 282(19):14094–100. doi:10.1074/jbc.M611370200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Wells CM, Whale AD, Parsons M, Masters JR, Jones GE. PAK4: a pluripotent kinase that regulates prostate cancer cell adhesion. J Cell Sci (2010) 123(Pt 10):1663–73. doi:10.1242/jcs.055707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Park SY, Kang JH, Jeong KJ, Lee J, Han JW, Choi WS, et al. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer (2011) 128(10):2306–16. doi:10.1002/ijc.25589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Merkle D, Hoffmann R. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: cross-talk with the androgen receptor. Cell Signal (2011) 23(3):507–15. doi:10.1016/j.cellsig.2010.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci (2001) 2(10):727–33. doi:10.1038/35094577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Cox ME, Deeble PD, Bissonette EA, Parsons SJ. Activated 3’,5’-cyclic AMP-dependent protein kinase is sufficient to induce neuroendocrine-like differentiation of the LNCaP prostate tumor cell line. J Biol Chem (2000) 275(18):13812–8. doi:10.1074/jbc.275.18.13812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Bang YJ, Pirnia F, Fang WG, Kang WK, Sartor O, Whitesell L, et al. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proc Natl Acad Sci U S A (1994) 91(12):5330–4. doi:10.1073/pnas.91.12.5330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer (2007) 14(3):531–47. doi:10.1677/ERC-07-0061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Farini D, Puglianiello A, Mammi C, Siracusa G, Moretti C. Dual effect of pituitary adenylate cyclase activating polypeptide on prostate tumor LNCaP cells: short- and long-term exposure affect proliferation and neuroendocrine differentiation. Endocrinology (2003) 144(4):1631–43. doi:10.1210/en.2002-221009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Zelivianski S, Verni M, Moore C, Kondrikov D, Taylor R, Lin MF. Multipathways for transdifferentiation of human prostate cancer cells into neuroendocrine-like phenotype. Biochim Biophys Acta (2001) 1539(1–2):28–43. doi:10.1016/S0167-4889(01)00087-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Goodin JL, Rutherford CL. Identification of differentially expressed genes during cyclic adenosine monophosphate-induced neuroendocrine differentiation in the human prostatic adenocarcinoma cell line LNCaP. Mol Carcinog (2002) 33(2):88–98. doi:10.1002/mc.10025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Padmanabhan A, Li X, Bieberich CJ. Protein kinase A regulates MYC protein through transcriptional and post-translational mechanisms in a catalytic subunit isoform-specific manner. J Biol Chem (2013) 288(20):14158–69. doi:10.1074/jbc.M112.432377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kvissel AK, Ramberg H, Eide T, Svindland A, Skalhegg BS, Tasken KA. Androgen dependent regulation of protein kinase A subunits in prostate cancer cells. Cell Signal (2007) 19(2):401–9. doi:10.1016/j.cellsig.2006.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Deeble PD, Murphy DJ, Parsons SJ, Cox ME. Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol (2001) 21(24):8471–82. doi:10.1128/MCB.21.24.8471-8482.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Shen R, Dorai T, Szaboles M, Katz AE, Olsson CA, Buttyan R. Transdifferentiation of cultured human prostate cancer cells to a neuroendocrine cell phenotype in a hormone-depleted medium. Urol Oncol (1997) 3(2):67–75. doi:10.1016/S1078-1439(97)00039-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Caron E. Rac signalling: a radical view. Nat Cell Biol (2003) 5(3):185–7. doi:10.1038/ncb0303-185
95. Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex (2000) 10(10):927–38. doi:10.1093/cercor/10.10.927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Regnauld K, Nguyen QD, Vakaet L, Bruyneel E, Launay JM, Endo T, et al. G-protein alpha(olf) subunit promotes cellular invasion, survival, and neuroendocrine differentiation in digestive and urogenital epithelial cells. Oncogene (2002) 21(25):4020–31. doi:10.1038/sj.onc.1205498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Somlyo AV, Bradshaw D, Ramos S, Murphy C, Myers CE, Somlyo AP. Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem Biophys Res Commun (2000) 269(3):652–9. doi:10.1006/bbrc.2000.2343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Troller U, Larsson C. Cdc42 is involved in PKCepsilon- and delta-induced neurite outgrowth and stress fibre dismantling. Biochem Biophys Res Commun (2006) 349(1):91–8. doi:10.1016/j.bbrc.2006.07.200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol (2006) 173(3):383–94. doi:10.1083/jcb.200511093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta (2004) 1692(2–3):159–74. doi:10.1016/j.bbamcr.2004.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol (2000) 31(5):578–83. doi:10.1053/hp.2000.6698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
103. Lang K, Drell TL IV, Lindecke A, Niggemann B, Kaltschmidt C, Zaenker KS, et al. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int J Cancer (2004) 112(2):231–8. doi:10.1002/ijc.20410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med (2006) 12(8):939–44. doi:10.1038/nm1447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Borre M, Nerstrom B, Overgaard J. Association between immunohistochemical expression of vascular endothelial growth factor (VEGF), VEGF-expressing neuroendocrine-differentiated tumor cells, and outcome in prostate cancer patients subjected to watchful waiting. Clin Cancer Res (2000) 6(5):1882–90.
106. Grobholz R, Bohrer MH, Siegsmund M, Junemann KP, Bleyl U, Woenckhaus M. Correlation between neovascularisation and neuroendocrine differentiation in prostatic carcinoma. Pathol Res Pract (2000) 196(5):277–84. doi:10.1016/S0344-0338(00)80056-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Heinrich E, Trojan L, Friedrich D, Voss M, Weiss C, Michel MS, et al. Neuroendocrine tumor cells in prostate cancer: evaluation of the neurosecretory products serotonin, bombesin, and gastrin – impact on angiogenesis and clinical follow-up. Prostate (2011) 71(16):1752–8. doi:10.1002/pros.21392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Bologna M, Festuccia C, Muzi P, Biordi L, Ciomei M. Bombesin stimulates growth of human prostatic cancer cells in vitro. Cancer (1989) 63(9):1714–20. doi:10.1002/1097-0142(19900501)63:9<1714::AID-CNCR2820630912>3.0.CO;2-H
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Iwamura M, Abrahamsson PA, Foss KA, Wu G, Cockett AT, Deftos LJ. Parathyroid hormone-related protein: a potential autocrine growth regulator in human prostate cancer cell lines. Urology (1994) 43(5):675–9. doi:10.1016/0090-4295(94)90183-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Abrahamsson PA. Neuroendocrine cells in tumour growth of the prostate. Endocr Relat Cancer (1999) 6(4):503–19. doi:10.1677/erc.0.0060503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Amorino GP, Deeble PD, Parsons SJ. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene (2007) 26(5):745–56. doi:10.1038/sj.onc.1209814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Bavadekar S, Budajaja F, Patel K, Vansal S. Epinephrine stimulates secretion of VEGF by human prostate cancer cells, LNCaP, through a beta2-adrenergic receptor-mediated pathway. FASEB J (2013) 27:1105.11.
113. Delk NA, Farach-Carson MC. Interleukin-6: a bone marrow stromal cell paracrine signal that induces neuroendocrine differentiation and modulates autophagy in bone metastatic PCa cells. Autophagy (2012) 8(4):650–63. doi:10.4161/auto.19226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Nilsson MB, Armaiz-Pena G, Takahashi R, Lin YG, Trevino J, Li Y, et al. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J Biol Chem (2007) 282(41):29919–26. doi:10.1074/jbc.M611539200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Plecas B, Glavaski A, Solarovic T. Propranolol treatment affects ventral prostate blood vessels and serum testosterone concentrations in adult rats. Andrologia (1997) 29(2):109–14. doi:10.1111/j.1439-0272.1997.tb00472.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, Flores A, et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Invest (2013) 123(2):874–86. doi:10.1172/JCI63324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang S, et al. Adrenergic signaling promotes angiogenesis through endothelial cell-tumor cell crosstalk. Endocr Relat Cancer (2014) 21(5):783–95. doi:10.1530/ERC-14-0236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Yang EV, Kim SJ, Donovan EL, Chen M, Gross AC, Webster Marketon JI, et al. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun (2009) 23(2):267–75. doi:10.1016/j.bbi.2008.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Jiang Q, Ding S, Wu J, Liu X, Wu Z. Norepinephrine stimulates mobilization of endothelial progenitor cells after limb ischemia. PLoS One (2014) 9(7):e101774. doi:10.1371/journal.pone.0101774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Hassan S, Karpova Y, Flores A, D’Agostino R Jr, Kulik G. Surgical stress delays prostate involution in mice. PLoS One (2013) 8(11):e78175. doi:10.1371/journal.pone.0078175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer (2014) 14(1):13–25. doi:10.1038/nrc3645
122. Sun XQ, Bao JG, Nelson KC, Li KC, Kulik G, Zhou XB. Systems modeling of anti-apoptotic pathways in prostate cancer: psychological stress triggers a synergism pattern switch in drug combination therapy. PLoS Comput Biol (2013) 9(12):e1003358. doi:10.1371/journal.pcbi.1003358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Gnesutta N, Qu J, Minden A. The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J Biol Chem (2001) 276(17):14414–9. doi:10.1074/jbc.M011046200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Zhang D, Ma Q, Wang Z, Zhang M, Guo K, Wang F, et al. beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Mol Cancer (2011) 10:146. doi:10.1186/1476-4598-10-146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Currie G, Freel EM, Perry CG, Dominiczak AF. Disorders of blood pressure regulation-role of catecholamine biosynthesis, release, and metabolism. Curr Hypertens Rep (2012) 14(1):38–45. doi:10.1007/s11906-011-0239-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Kargi AY, Iacobellis G. Adipose tissue and adrenal glands: novel pathophysiological mechanisms and clinical applications. Int J Endocrinol (2014) 2014:614074. doi:10.1155/2014/614074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Miller GE, Chen E. Life stress and diminished expression of genes encoding glucocorticoid receptor and beta2-adrenergic receptor in children with asthma. Proc Natl Acad Sci U S A (2006) 103(14):5496–501. doi:10.1073/pnas.0506312103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Pervanidou P, Chrousos GP. Metabolic consequences of stress during childhood and adolescence. Metabolism (2012) 61(5):611–9. doi:10.1016/j.metabol.2011.10.005
129. Allott EH, Masko EM, Freedland SJ. Obesity and prostate cancer: weighing the evidence. Eur Urol (2013) 63(5):800–9. doi:10.1016/j.eururo.2012.11.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Chalfin HJ, Lee SB, Jeong BC, Freedland SJ, Alai H, Feng Z, et al. Obesity and long-term survival after radical Prostatectomy. J Urol (2014) 192(4):1100–4. doi:10.1016/j.juro.2014.04.086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Sigurdardottir LG, Markt SC, Rider JR, Haneuse S, Fall K, Schernhammer ES, et al. Urinary melatonin levels, sleep disruption, and risk of prostate cancer in elderly men. Eur Urol (2014). doi:10.1016/j.eururo.2014.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Carmeliet P. Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet (2003) 4(9):710–20. doi:10.1038/nrg1158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol (2003) 64(6):1357–69. doi:10.1124/mol.64.6.1357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ADRB2, β-adrenergic receptor, prostate cancer, neuroendocrine differentiation, angiogenesis, apoptosis, metastasis, β-blocker
Citation: Braadland PR, Ramberg H, Grytli HH and Taskén KA (2015) β-adrenergic receptor signaling in prostate cancer. Front. Oncol. 4:375. doi: 10.3389/fonc.2014.00375
Received: 20 October 2014; Accepted: 16 December 2014;
Published online: 12 January 2015.
Edited by:
Mercedes Salido, University of Cadiz, SpainReviewed by:
Anthony Joshua, University Health Network, CanadaBrock O’Neil, University of Utah, USA
George Kulik, Wake Forest University, USA
Copyright: © 2015 Braadland, Ramberg, Grytli and Taskén. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristin Austlid Taskén, Department of Tumor Biology, Institute of Cancer Research, Division of Cancer Medicine, Transplantation and Surgery, Oslo University Hospital, P.O. Box 4953 Nydalen, Oslo NO-0424, Norway e-mail:ay5hLnRhc2tlbkBtZWRpc2luLnVpby5ubw==