 Michele Milella
Michele Milella Italia FalconeFabiana ConciatoriUrsula Cesta Incani
Italia FalconeFabiana ConciatoriUrsula Cesta Incani Anais Del Curatolo
Anais Del Curatolo Nicola InzerilliCarmen M. A. NuzzoVanja Vaccaro
Nicola InzerilliCarmen M. A. NuzzoVanja Vaccaro Sabrina VariFrancesco Cognetti
Sabrina VariFrancesco Cognetti Ludovica Ciuffreda*
Ludovica Ciuffreda*- Division of Medical Oncology A, Regina Elena National Cancer Institute, Rome, Italy
PTEN is the most important negative regulator of the PI3K signaling pathway. In addition to its canonical, PI3K inhibition-dependent functions, PTEN can also function as a tumor suppressor in a PI3K-independent manner. Indeed, the PTEN network regulates a broad spectrum of biological functions, modulating the flow of information from membrane-bound growth factor receptors to nuclear transcription factors, occurring in concert with other tumor suppressors and oncogenic signaling pathways. PTEN acts through its lipid and protein phosphatase activity and other non-enzymatic mechanisms. Studies conducted over the past 10 years have expanded our understanding of the biological role of PTEN, showing that in addition to its ability to regulate proliferation and cell survival, it also plays an intriguing role in regulating genomic stability, cell migration, stem cell self-renewal, and tumor microenvironment. Changes in PTEN protein levels, location, and enzymatic activity through various molecular mechanisms can generate a continuum of functional PTEN levels in inherited syndromes, sporadic cancers, and other diseases. PTEN activity can indeed, be modulated by mutations, epigenetic silencing, transcriptional repression, aberrant protein localization, and post-translational modifications. This review will discuss our current understanding of the biological role of PTEN, how PTEN expression and activity are regulated, and the consequences of PTEN dysregulation in human malignant tumors.
What is PTEN?
PTEN stands for Phosphatase and TENsin homolog deleted on chromosome 10 and is a classical tumor suppressor gene located in the 10q23 region of chromosome 10 encoding for a 403-aminoacid multifunctional protein (predicted MW 47 kDa), which possesses lipid and protein phosphatase activities. PTEN was identified in 1977 by three independent groups: two groups used a positional-cloning approach, whereas the third group identified PTEN by a biochemical approach, which aimed to identify a gene encoding for a phosphatase with tensin and auxilin homology (1–3). The crystal structure of PTEN was resolved by Lee et al. in 1999, although two flexible regions of unknown function had to be deleted for technical reasons; crystal structure revealed the presence of a phosphatase domain, a C2 lipid membrane-binding domain, and a class I PDZ binding motif at the C-terminus, which recognizes target proteins (4). The PTEN gene is almost ubiquitously expressed in mammals throughout early embryogenesis (5); although the main protein subcellular localization is cytoplasmic and/or membrane bound, nuclear localization has been described and bears important functional consequences (6). In addition, it has been reported that the recently identified PTEN protein variants, such as PTEN-Long, can exit, exist, and function outside the cell in a paracrine type fashion (7).
PTEN acts as a classical tumor suppressor, mainly involved in the homeostatic maintenance of the phosphatidylinositol 3 kinase (PI3K)/AKT cascade (Figure 1A). PI3K, a lipid kinase activated by receptor tyrosine kinases, G protein-coupled receptors, and RAS activation, converts the lipid second messenger phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3); PIP3 recruits phosphatidylinositol-dependent kinase 1 (PDK1) and AKT to the plasma membrane, where AKT is phosphorylated on Thr308 by PDK1 and on Ser473 by the mammalian target of rapamycin (mTOR) complex 2 (mTORC2) (8, 9). By dephosphorylating PIP3 to PIP2, PTEN reverses the action of PI3K, thereby hampering all downstream functions controlled by the AKT/mTOR axis, such as cycle progression, induction of cell death, transcription, translation, stimulation of angiogenesis, and stem cell self-renewal (10–17). Even though the biological effects of PTEN are dominated by its ability to dephosphorylate lipid substrates, PTEN has also been reported to exhibit protein phosphatase activity, responsible for some of its biological effects, including inhibition of cell migration and cell-cycle arrest (18, 19). More recently, a growing list of functionally relevant, phosphatase-independent activities have been described (Table 1) (20, 21).
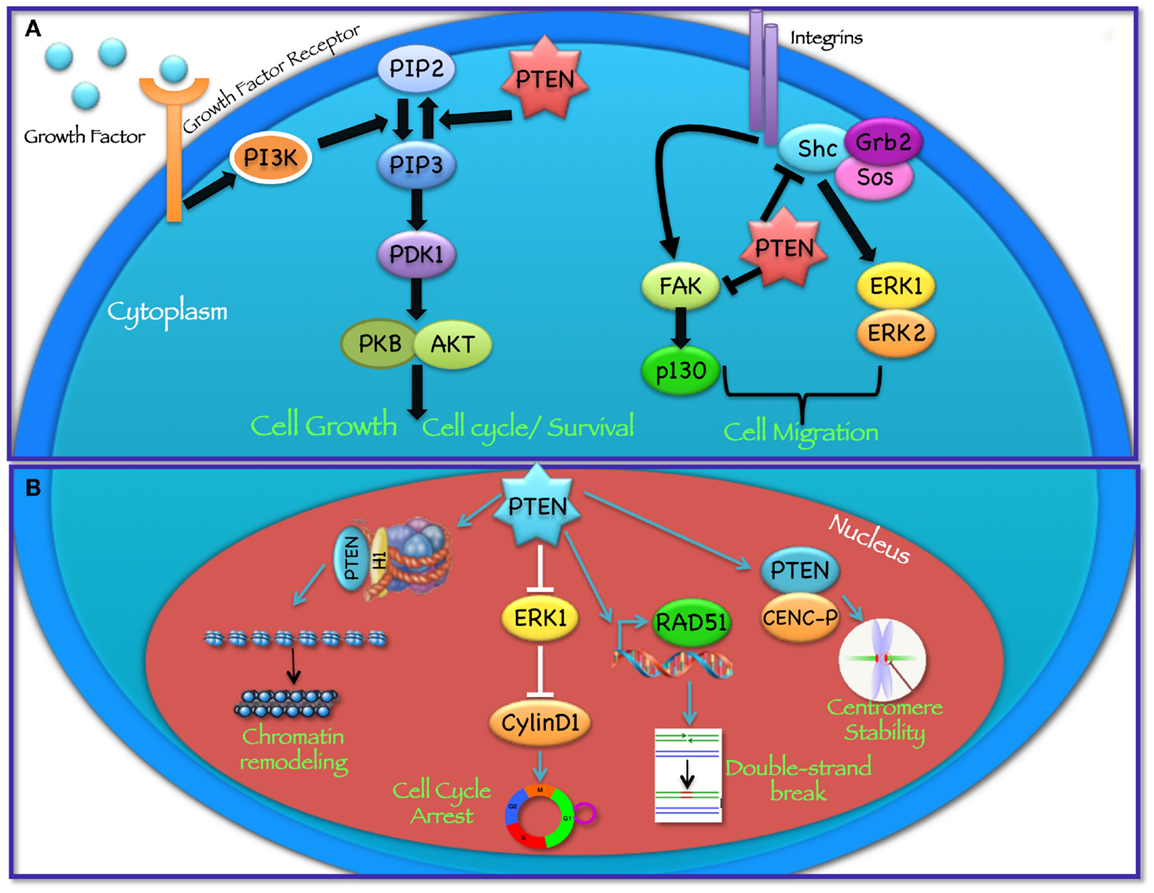
Figure 1. Cytoplasmic and nuclear PTEN functions. PTEN acts in regulating a wide spectrum of biological functions, at least in part determined by its subcellular localization. (A) In the cytoplasm PTEN dephosphorylates PIP3 to PIP2, thereby reversing the action of PI3K and hampering all downstream functions controlled by the AKT/mTOR axis, such as cycle progression, induction of cell death, transcription, translation, stimulation of angiogenesis, and stem cell self-renewal. In addition, through its protein phosphatase activity directed against FAK and SHC, PTEN modulates complex pathways affecting cell migration. (B) In the nucleus, PTEN cooperates in maintaining genomic integrity, repairing DNA double-strand breaks, controlling homologous recombination, and promoting ubiquitin-dependent degradation of oncoproteins such as PLK1 and AURK. In addition, PTEN controls cell-cycle progression by modulating ERK phosphorylation and cyclin D1 levels and regulates chromatin remodeling by binding to histone H1.
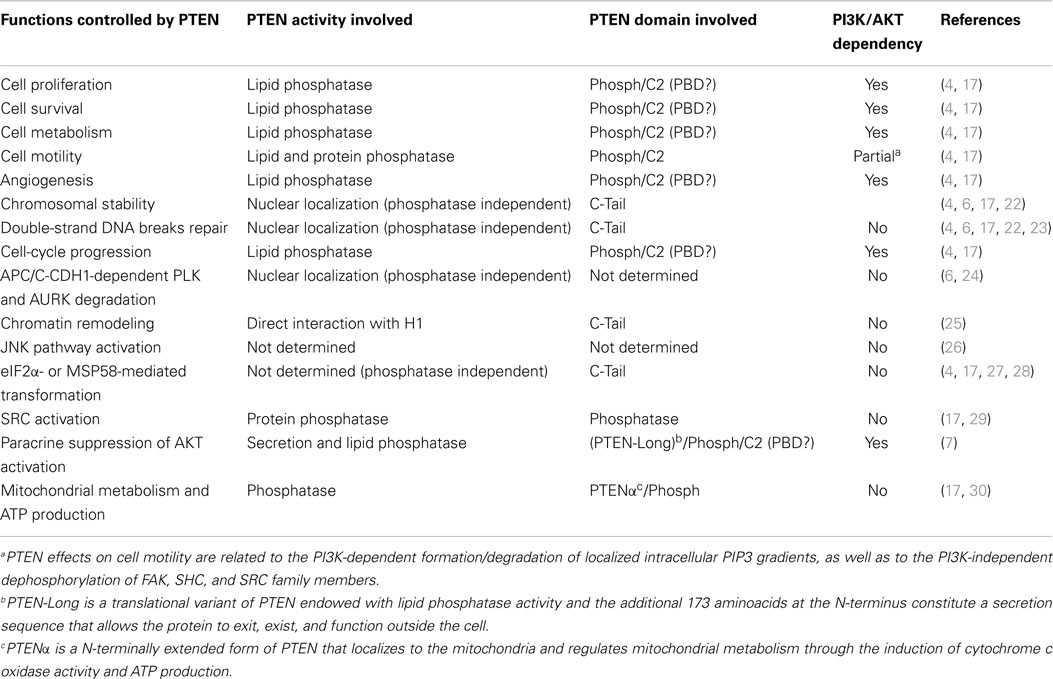
Table 1. Selected cell functions controlled by specific PTEN activities/domains and their dependency on regulation of the PI3K pathway.
PTEN function is commonly lost in a large proportion of human cancers through somatic mutations, gene silencing, or epigenetic mechanisms (Table 2). Tumor-associated mutations may occur in all PTEN domains, thus implying that each distinct protein region (and each related PTEN activity) may be pathologically relevant to cancer initiation and progression. In addition, emerging evidence shows that PTEN gene/protein dosage is quantitatively relevant during tumor development, as partial loss of PTEN function (haploinsufficiency) is sufficient to promote growth in some human malignancies. The mechanisms involved in regulation of PTEN dosage include methylation, micro-RNA (miR) or pseudogene expression, and protein phosphorylation (31–33). In this review, we will discuss the cellular functions controlled by PTEN, the molecular mechanisms of the subtle regulation of PTEN expression and function, and the role of its mutational/expression status in cancer.
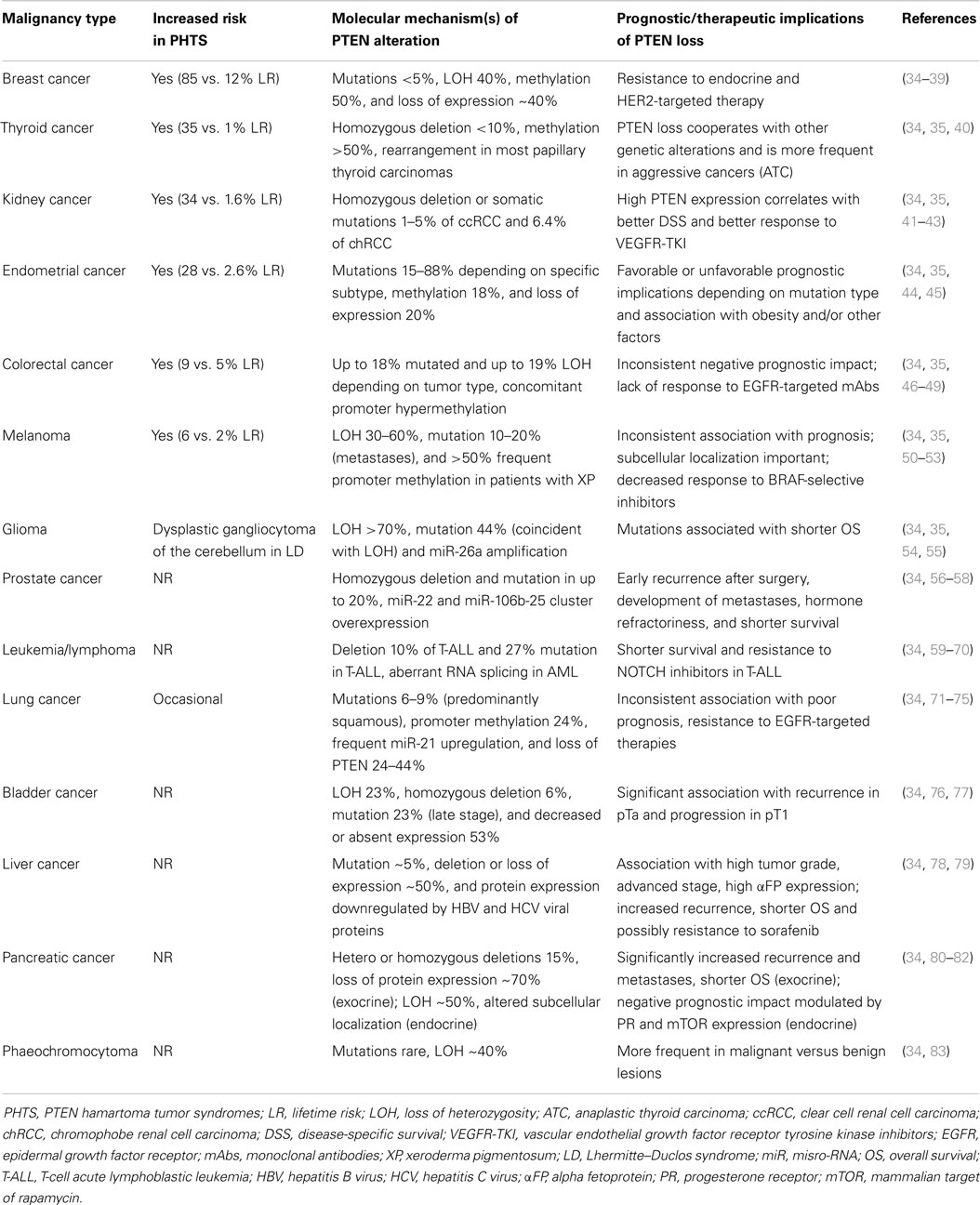
Table 2. Incidence and prognostic significance of PTEN alterations in PHTS and sporadic human cancers.
Cellular Functions Controlled by PTEN
PTEN-Mediated Regulation of Metabolic Pathways
The PTEN/PI3K pathway may influence key steps in metabolic pathways during cell proliferation and tumorigenesis. Recent data from two independently generated transgenic mouse models, based on two similar PTEN-containing bacterial artificial chromosomes (BAC), have shown that PTEN is involved in the control of metabolic pathways through PI3K-dependent and -independent functions (30, 84). Garcia-Cao et al. demonstrated that transgenic mice overexpressing PTEN show reduced body size, due to decreased cell number, increased energy expenditure, and reduced body fat accumulation. Cells derived from these mice show reduced glucose and glutamine uptake, increased mitochondrial oxidative phosphorylation, and resistance to oncogenic transformation. Ortega Molina et al. showed that mice carrying additional genomic copies of PTEN have increased energy expenditure and are protected from metabolic pathologies and cancer. Moreover, a recent human study demonstrates that PTEN haploinsufficiency is a monogenic cause of profound constitutive insulin sensitization that is apparently obesogenic. In particular, the authors demonstrated that patients who are heterozygous carriers of PTEN mutations, which cause Cowden syndrome, are at increased risk of obesity and cancer, but at decreased risk of diabetes due to enhanced insulin sensitivity (85). The role of PTEN in insulin-stimulated glucose uptake is controversial in the literature. Substantial evidence suggests a role for PTEN in the regulation of glucose uptake, due to its ability to modulate insulin signaling (86, 87). For example, Nakashima et al. have shown that PTEN overexpression in adipocytes inhibits insulin-stimulated, PI3K activation-dependent 2-deoxyglucose uptake, and glucose transporter type 4 (GLUT4) translocation, a key event in insulin signaling (88), which ultimately leads to decreased glucose cellular uptake. Conversely, Mosser et al. suggest that PTEN does not modulate GLUT4 translocation and metabolic functions of insulin under normal physiological conditions (89). However, Morani et al. using genetic manipulations of PTEN expression, have shown the involvement of PTEN in the regulation of membrane expression of glucose transporter type 1 (GLUT1), suggesting that PTEN regulates glucose uptake, at least in transformed cells, such as thyroid cancer cells (90). On the other hand, PTEN is involved in the modulation of gluconeogenesis through inhibition of forkhead box O (FOXO) 1, peroxisome proliferator activated receptor (PPAR)γ, and PPARγ-coactivator 1a (84).
PTEN Role in Cell Motility
Among its many functions, PTEN plays an important role in the regulation of cell motility, particularly, in controlling the directionality of chemotaxis. Studies conducted in Dictyostelium over the past years suggest that both PI3K and PTEN activities, and the resulting formation of localized intracellular PIP3 gradients, are required during chemotaxis (91–93). In these studies, the authors show that during chemotaxis PIP3 levels are enriched at the leading edge of migrating cells, while PTEN relocalizes at the opposite side of the cell; as a result, when PTEN is lost, Dictyostelium cells have defects in polarization and chemotaxis (94, 95). Several in vitro studies have shown that PTEN phosphatase activity is required to regulate cellular migration with some reports indicating the involvement of the C2 domain and another implicating PTEN’s protein phosphatase activity (96–99). In particular, it has been shown that PTEN overexpression is able to inhibit the spreading of glioblastoma cells and the migration and spreading of fibroblasts. On the other hand, knockdown of endogenous PTEN expression enhanced cell migration in fibroblasts possibly through the involvement of focal adhesion kinase (FAK) (100): PTEN dephosphorylates FAK, a cytoplasmic phosphoprotein activated by integrins, thereby inhibiting cell migration. PTEN may also dephosphorylate SHC, thus inhibiting downstream pathways, including the mitogen activated protein kinase (MAPK) pathway, which leads to the modulation of cell motility (Figure 1A). Recent studies, aimed at understanding the role of PTEN in cell motility, suggest that both lipid and protein phosphatase activities contribute to PTEN-mediated regulation of migration, through interactions with Rac1 and the SRC family kinase, FYN (101).
PTEN and Angiogenesis
Recent studies performed in glioblastoma cell lines suggest that PTEN is potentially important in the control of angiogenesis within brain tumors. In fact, reconstitution of PTEN expression in the PTEN−/− U87 MG glioma model caused a dramatic decrease in tumor growth in vivo and increase in mice survival, without significantly effecting the proliferation of these cells in vitro. The observed effect was due to the induction of thrombospondin-1, a negative regulator of angiogenesis, consequently reducing recruitment of blood vessels to the tumor (102). In addition, PTEN loss can also facilitate glioma growth by promoting HIF1 expression and activity (103). In both cases, PTEN’s ability to counteract neo-angiogenesis appears to be related to its lipid phosphatase activity and to suppression of signaling through the PI3K/AKT/mTOR axis. Our group and others have indeed demonstrated that the mTOR/eIF4E axis downstream of PI3K, plays a crucial role in the regulation of vascular endothelial growth factor (VEGF) production in breast cancer cells through increased hypoxia inducible factor 1α (HIF1α) expression and transcriptional activation. This suggests that the anti-tumor activity observed after PI3K (and especially mTOR) cascade inhibition may be due, at least in part, to the anti-angiogenic effects observed with mTORC1 inhibitors, such as temsirolimus (104). Along these lines, recent studies from our group have shown that the anti-angiogenic activity observed with MEK inhibitors in melanoma models in vitro and in vivo (105) is due, at least in part, to MEK inhibition-induced upregulation of PTEN expression. Indeed, in cell lines in which PTEN function is offset by the expression of a dominant negative PTEN mutant (DN-PTEN), the effects observed on VEGF production after MEK inhibitor treatment are partially blocked (106).
Role of PTEN in the Nucleus
PTEN plays an important tumor suppressor role in the nucleus (Figure 1B) and the absence of nuclear PTEN is associated with more aggressive cancers (107–109). Recent findings have shown that the PTEN protein is present in the nucleus of cell lines and tissue cells, even if the mechanisms of PTEN nuclear localization have yet to be fully elucidated (109–112). Given the absence of a classical nuclear localization signal sequence (NLS) or nuclear export sequences (NES), numerous molecular mechanisms have been proposed, including simple diffusion, active shuttling by the RAN GTPase, or phosphorylation-dependent shuttling and monoubiquitylation-dependent import (6, 113). Trotman et al. have demonstrated that PTEN nuclear import is mediated by its ubiquitination by the E3 ligase, neural precursor cell expressed, developmentally downregulated-4-1 (NEDD4-1) (114). Recently, sumoylation has also been reported to mediate PTEN nuclear retention (115). Within the nucleus, PTEN has been shown to interact with the centromere specific binding protein C (CENP-C) to modulate centrosome stability (22). Moreover, PTEN is involved in DNA-damage responses through upregulation of the transcription of Rad51, a key component of the homologous recombination system, which repairs DNA double-strand breaks (22, 23). In addition, nuclear PTEN controls cell-cycle progression by decreasing cyclin D1 levels in the nucleus and regulates cellular senescence through anaphase promoting complex (APC)-CDH1-mediated protein degradation (6, 24) (Figure 1B). Several reports suggest that nuclear PTEN functions may be independent of its catalytic activity and may be regulated by PTEN physical interaction with nuclear target proteins, such as p53, microspherule protein 1 (MSP58; also known as MCRS1) or HIF1α (27, 116, 117). Recent evidence also demonstrates that PTEN is involved in transcriptional regulation through the control of chromatin remodeling. Chen et al. have shown that PTEN physically associates with histone H1, through its C-terminal domain, to maintain a condensed chromatin structure. Loss of PTEN or its C-terminal portion promotes chromatin decondensation and gene activation (25).
Alternative PTEN Variants
Recent studies have led to the identification of new PTEN variants: PTEN-Long and PTENα. Hopkins et al. identified an alternative initiation codon, as compared to the canonical start site on the PTEN promoter, which predicts the expression of a longer PTEN variant. PTEN-Long contains a 173-amino acid domain at its N-terminus followed by the classical 403 amino acids of PTEN. PTEN-Long is a translational variant of PTEN endowed with lipid phosphatase activity and the additional amino acids constitute a secretion sequence that allows the protein to exit, exist, and function outside the cell (7, 118). Recent studies have reported that PTEN-Long can be detected in human serum and plasma and can act as a therapeutic factor involved in tumor regression (7, 119). Hopkins et al. have demonstrated that the purified PTEN-Long protein injected in mice is taken up by several tissues and antagonizes AKT signaling inducing an increase in blood glucose levels. In addition, they found PTEN-Long has tumor-suppressive function inducing tumor cell death in vitro and in vivo (7). The molecular mechanisms involved in PTEN-Long regulation have yet to be defined, but the long variant can be coexpressed with the classical PTEN isoform. PTEN-Long is expressed in normal tissues such as the breast and brain, whereas its expression levels are lower in human breast tumors and mouse models of glioblastoma (7, 20, 118).
Recently, Liang et al. have identified another N-terminally extended form of PTEN (PTENα) that localizes to the cytoplasm and the mitochondria and is involved in the regulation of mitochondrial metabolism through the induction of cytochrome c oxidase activity and ATP production. Moreover, PTENα promotes energy production and interacts with canonical PTEN to increase PTEN-induced putative kinase 1 (PINK1) protein levels (120).
Fine Tuning of PTEN Activity
PTEN dysfunction plays a crucial role in the pathogenesis of hereditary and sporadic tumors (34, 121). During tumor development, mutations, and deletions of PTEN lead to the inactivation of its enzymatic activity, with consequently increased cell proliferation and reduced cell death. In addition to genomic inactivation, many other pathogenic mechanisms involved in the repression of PTEN gene expression or in the aberrant subcellular compartmentalization of the protein are associated with tumorigenesis. Several different mechanisms are known to fine tune PTEN expression and function, including transcriptional regulation, post-transcriptional regulation by non-coding RNAs, post-translational modifications, and protein–protein interactions.
Transcriptional Regulation
PTEN transcription is negatively and positively regulated by different transcription factors. p53 can upregulate PTEN, resulting in a complex interplay between these two tumor suppressors (122). EGR1 binds to the PTEN promoter and upregulates PTEN expression in response to insulin-like growth factor-2 (IGF-2) stimulation or radiation (123, 124). PPARγ can also upregulate PTEN gene expression (125).
On the contrary, SNAIL, c-Jun, and nuclear factor kappa-B (NF-kB) have been reported to negatively regulate PTEN transcription (106, 126, 127). Active NOTCH1 has been reported to act as both a negative and a positive regulator of PTEN transcription through interacting with MYC and CBF-1, respectively (128, 129). Our group and others have shown that c-Jun is involved in the regulation of PTEN expression (106, 130, 131). In particular, our group has demonstrated that PTEN is a target of a re-wired MEK–ERK–c-Jun survival pathway, occurring in BRAF-mutant melanoma, but also in other malignant tumors, as well as in normal fibroblasts. Indeed, upon genetic and/or pharmacologic modulation of ERK activity, c-Jun and PTEN are counter-regulated with strict time/dose dependency. Moreover, ERK-independent genetic modulation of c-Jun expression exerts the same effects on PTEN regulation and expression of a constitutively active MEK/ERK fusion protein in non-transformed fibroblasts concomitantly upregulating c-Jun and downregulating PTEN expression. These data reveal that ERK-dependent regulation of PTEN expression, occurring at least in part through c-Jun-mediated transcriptional repression, is a physiological regulatory mechanism, which takes place in both normal and cancer cells of different histological origin (106).
Post-Transcriptional Regulation
micro-RNA contribute to the regulation of PTEN expression in many tumors. In fact, it has been demonstrated that the oncogenic miR-21, one of the most frequently upregulated miRs in cancer, directly targets and downregulates PTEN in specific cancers, including hepatocellular, ovarian, and lung cancer (132, 133). Recently, it has been shown that miR-25 controls PTEN levels in human tumors and contributes to experimental tumorigenesis (106, 134). As it has been discussed above for the involvement of c-Jun, miR-25 provides another interesting link between the MEK/ERK and PI3K/PTEN/AKT/mTOR pathways. Our group has recently shown that miR-25 expression levels are controlled by ERK activation status, which in turn regulates PTEN protein levels in melanoma cells. The MYC oncogene also may downregulate PTEN through increased expression of miR-19 (135).
In recent years, new findings have lead to hypothesize that both non-coding and protein-coding genes possess a novel mRNA-dependent non-coding function that enables them to act as a decoy to block the effect of specific miR on other RNA in a model termed the competing endogenous RNA (ceRNA) hypothesis (17, 134). This appears to be the case for the PTEN pseudogene 1 (PTENP1) that shares significant sequence identity with PTEN mRNA in regions that harbor miR target sites. PTENP1 was found to regulate PTEN expression through sequestration of PTEN-targeting miR, thereby increasing the half-life of PTEN mRNA and increasing PTEN protein levels (17, 134).
Epigenetic Silencing
Epigenetic silencing of PTEN expression through aberrant methylation of the gene promoter or of histone modifications has been observed in many types of cancers (17, 34). Several studies have shown that silencing of PTEN transcription is often due to the presence of hypermethylated CpG islands in the PTEN promoter. Hypermethylation has been observed in colorectal, endometrial, breast, gastric, prostate, melanoma, and lung cancer (19, 50, 71, 136, 137). Lu et al. have demonstrated that the zinc-finger transcription factor sal-like protein 4 (SALL4) represses PTEN transcription by recruiting an epigenetic repressor complex (Mi-2/NuRD) (138). Histone deacetylase inhibitors, on the other hand, are able to increase PTEN transcription in fibroblasts (139).
Post-Translational Modifications
PTEN function is also regulated by post-translational modifications, including phosphorylation, acetylation, oxidation, and ubiquitination. PTEN has six phosphorylation sites, which have been implicated in the modulation of its tumor suppressor functions, stability, and subcellular compartmentalization (140). Caseine kinase 2 (CK2) is a protein kinase that phosphorylates PTEN on Thr366, Ser370, Ser380, Thr382, Thr383, and Ser385. CK2-mediated phosphorylation stabilizes PTEN, but creates a closer conformation that decreases interactions with binding partners and reduces its plasma membrane localization (111, 141). PTEN can be also phosphorylated by LKB1 on Ser385, resulting in its inactivation (142). Glycogen Synthase Kinase 3β (GSK3β) phosphorylates PTEN at Ser362 and Thr366, decreasing its phosphatase activity (143). PTEN phosphorylation can also be mediated by the kinases RAK, ROCK, JNK, JNKK, and SRC (141, 143–145).
Another mechanism involved in PTEN regulation is through the ubiquitin/proteasome pathway. NEDD4-1 is an E3 ubiquitin–protein ligase involved in promoting PTEN ubiquitin-mediated degradation (146). Recent evidence has identified WWP2 as an additional E3 ubiquitin ligase, which mediates PTEN ubiquitination-dependent degradation. Amodio et al. have shown that in some tumors, such as non-small-cell lung cancer (NSCLC), PTEN downregulation via ubiquitin-mediated degradation is an important mechanism leading to loss of PTEN activity (147). Previous studies have suggested that PTEN functions can be regulated by acetylation. The acetyltransferase P300/CBP-associated factor (PCAF) has been demonstrated to acetylate PTEN at Lys125 and Lys128 sites, which, in turn, negatively regulate PTEN catalytic activity (148). Catalytic PTEN activity is also controlled by reactive oxygen species (ROS). In particular, recent studies have shown that ROS regulate PTEN activity by oxidative-stress-induced formation of a disulfide bond between the active site Cys124 and Cys7 (149).
Protein-Protein Interactions
Some cellular proteins, including phosphatases, can affect PTEN functions either directly or indirectly via protein–protein interactions. Such protein–protein interactions may influence PTEN activity by modifying its conformation, stability, subcellular compartmentalization, and lipid membrane binding. For example, membrane-associated guanylate kinase inverted 2 (MAGI2) binding to PTEN increases its activity (150). Conversely, DJ1 (also known as PARK7: Parkinson protein 7) binds PTEN under oxidative conditions, thereby inhibiting its activity. DJ1 expression is associated with increased AKT activity and poor prognosis in different tumor types (151, 152). Some proteins that interact with PTEN are involved in its translocation across the cytoplasm and subcellular localization (21). Microtubule-associated Ser/Thr kinase 2 (MAST2) is another PTEN regulator. Terrien et al. have recently demonstrated that when MAST2 and PTEN form a complex, the phosphorylation of PTEN by MAST2 drastically increases and destabilization of this interaction promotes neuronal cell survival, through the alteration of PTEN intracellular trafficking (153). Zmajkovicova et al., have demonstrated that interaction between PTEN, MEK1, and MAGI1 is necessary for PTEN membrane recruitment and PIP3 turnover and AKT signaling (154). MEK1 and MAGI1 are both essential for complex formation. In fact, MEK1 binding to MAGI1 promotes both complex formation and PTEN translocation onto the membrane.
PTEN Loss in Human Cancer
Germline mutations of PTEN are the underlying genetic causes of related disorders clinically referred to as PTEN hamartoma syndromes (PHTS) including: Cowden syndrome, Bannayan–Zonana syndrome, Lhermitte–Duclos syndrome, Proteus syndrome, and Proteus-like syndrome. Mutations responsible for these syndromes result in a non-functional or absent protein, which causes uncontrolled cell growth, leading to tumor (either benign or malignant) growth (34, 155–157). Cowden syndrome is the best-described syndrome within PHTS, with approximately 80% of patients having germline PTEN mutations. Patients with Cowden syndrome have a high risk for benign and malignant tumors of the breast (lifetime risk – LR-85%), thyroid (LR 35%), kidney (renal cell carcinoma – RCC-, LR 33%), and endometrium (LR 28%), which correspond to sporadic tumor types that commonly exhibit somatic PTEN inactivation (155, 157, 158) (Table 2). In addition, increased risk for colorectal cancer (9%) and melanoma (6%), previously not believed to be part of PHTS, have also been described (35). In addition to the well-characterized role of PTEN mutations in PHTS syndromes, new evidence shows that PTEN mutation is one of the most validated causes of autism spectrum disorders, intellectual disability, and extreme macrocephaly (159, 160).
The PTEN tumor suppressor is frequently lost, either partially or fully, from many sporadic tumor types. Somatic inactivation of PTEN occurs in a wide range of neoplastic diseases, including melanoma, glioblastoma, colon, and endometrial cancers (161, 162) (Table 2). More than a decade of research has expanded our knowledge on PTEN’s role in cancer. Experiments performed in transgenic mice have demonstrated that loss of both copies of the PTEN gene results in embryonic lethality, whereas PTEN heterozygous mutants develop a diverse range of dysplasias in a wide spectrum of tissues with high incidence of prostate and colon cancer (163, 164). Interestingly, recent studies highlighted a crucial, dose-dependent role of PTEN in cancer, showing that subtle reductions in active PTEN levels dictate cancer susceptibility in a dose-dependent manner (31, 165). However, PTEN loss alone is sufficient to cause tumorigenesis in some tissues but not in others, making the role of PTEN more ambiguous (34). Accordingly, these data strongly support the hypothesis that PTEN is haploinsufficient tumor suppressor and, although PTEN deletion alone has minimal effects, it frequently contributes to tumorigenesis in the context of other genetic alterations (31, 166, 167).
PTEN Role in the Regulation of Stem Cell Biology
Recent data have begun to shed light on the critical role of PTEN in stem cell maintenance and cancer-initiating cell biology (11, 168). The effects of PTEN loss in stem cells may be tissue-dependent. PTEN deletion in neuronal stem cells increases proliferation and maintains their self-renewing capacity. Similarly, PTEN loss causes increased proliferation, de-differentiation, and progression toward prostatic intraepithelial neoplasia in prostate stem cells (30, 59, 168, 169). On the contrary, in both melanocytes and hematopoietic cells, deletion of PTEN leads to normal stem cell exhaustion (170, 171). PTEN loss has been shown to enhance the number of tumor initiating cells in a mouse model of leukemia and in solid tumors, such as breast cancer (172, 173). The role of PTEN expression and function in the maintenance of lung and colorectal cancer stem cells (CSC) has not been extensively studied. However, it is worthy to note that PTEN expression is usually very low in both lung and colon CSC. Moreover, in colon CSC, PTEN expression is also strikingly upregulated during differentiation and by stimuli that inhibit CSC growth and tumorigenic activity, such as such as BMP4 treatment or thymosin β4 targeting (11, 174, 175).
PTEN in Melanoma
Loss of PTEN plays an important role in the development of 30–60% of melanomas, however, the mechanisms by which loss of this gene leads to tumor formation remain uncertain. Recent evidence has shown that decreased PTEN transcript levels were associated with PTEN promoter methylation in melanoma (34, 50). Wang et al. showed that more than 50% of the melanomas from patients with xeroderma pigmentosum display PTEN mutations, typically related to ultraviolet radiation exposure, highlighting the link between DNA-damage and PTEN mutations in this disease (51). Dankort et al. have demonstrated the ability of PTEN silencing to cooperate with BRAFV600E mutations in the genesis of metastatic melanoma (52). Several research groups have recently demonstrated that MEK blockade may induce compensatory signaling through PI3K pathway in melanoma, inducing its own resistance factors and explaining the highly variable clinical responses to MEK inhibition in different cellular contexts (106, 131, 176–178). Without a doubt, PTEN status is critical in determining the functional outcome of pharmacologic MEK inhibition in melanoma and in cellular contexts in which PTEN is genetically unaltered, MEK blockade induces a cross-talk mechanism that leads to PTEN protein induction, playing an important, albeit not exclusive, role in the anti-tumor and anti-angiogenic activities of MEK inhibitors (106). Consistently, PTEN loss impairs the anti-tumor activity of MEK inhibitors in preclinical models (106, 179, 180) and correlates with decreased efficacy of BRAF-targeted treatments in metastatic melanoma patients (53).
PTEN in Pancreatic Cancer
Mouse genetic studies, supported a potential role for PTEN as a haploinsufficient tumor suppressor. Indeed, homozygous deletion of PTEN in the pancreas leads to metaplasia that may progress toward frank carcinoma in approximately 20% of cases in transgenic mice. Decreased PTEN expression has been demonstrated in pancreatic tumor cell lines, although deletion or mutations that cause PTEN loss of activity have not been detected with significant frequency in human pancreatic ductal adenocarcinoma (PDAC) (80). In particular, Perren and collaborators have shown that, although PTEN is not mutated in pancreatic cancers, its subcellular localization may decrease its function (81). Ying et al. have recently documented a strong cooperative interaction between KRASG12D and PTEN loss in promoting metastatic PDAC (82).
PTEN in Colorectal Cancer
PTEN mutations are relatively prevalent in colorectal cancer and constitute potential markers of response to EGFR and MAPK inhibitor-based therapies. In fact, PTEN loss or inactivating mutations are found in a variable proportion (5–30%) of sporadic colorectal cancers (46, 47, 181). Interestingly, studies performed in in vivo model systems show that PTEN reactivation in a colorectal cancer (CRC) cell line exhibiting PTEN loss reduces its metastatic capability without affecting primary tumor formation. Moreover, PTEN reactivation also changed the organotropic homing from liver and lung metastasis to liver-only metastasis (182). Importantly, Razis et al. have shown that PTEN levels are predictive of cetuximab efficacy in CRC models with activated EGFR signaling and wild type KRAS/BRAF status and in the presence of an intact PI3K/AKT pathway (48).
PTEN in Lung Cancer
PTEN mutations occur at a low frequency in NSCLC and in small-cell lung cancer (SCLC), with the notable exception of squamous cell carcinoma of the lung, in which PTEN is mutated in 6–9% of the cases and significantly altered in up to 15% of cases, taking into account loss of expression as well (72). However, when detected, PTEN mutations appear to exert an effect on the therapeutic response to EGFR/PI3K pathway inhibitors (73). Since loss of PTEN protein expression is found in 24–44% of NSCLC (74), other mechanisms to decrease PTEN expression and function could be relevant in lung cancer. For example, epigenetic silencing may partially explain PTEN loss in cases when PTEN mutations or homozygous deletions are absent (71, 75). Soria et al. have suggested that 24% of early NSCLC samples lack PTEN expression, which correlated with PTEN promoter methylation (71). In addition, Zhang et al. have shown that levels of miR-21 were upregulated in lung cancer compared with normal lung tissue and correlate with a reduction in PTEN mRNA levels in advanced tumor stage (133).
PTEN in Breast Cancer
Germline PTEN mutation in Cowden syndrome has a predisposition to breast cancer, where female CS patients have up to 85% LR of developing breast cancer (155, 157, 158). In sporadic breast carcinomas, the frequency of PTEN loss is 30–40% (36). Epigenetic aberrations also may cause a decrease in PTEN levels, strongly correlating with tumor stage and grade with complete loss occurring more frequently in metastatic than in primary tumors (76, 163, 167, 183). In addition, PTEN mutations may overlap with other mutations including human epidermal growth factor receptor 2 (HER2) and loss of a single PTEN allele has been shown to accelerate tumorigenesis in HER2-overexpressing breast tumors (184). Early studies by Nagata et al. show that, in addition to antagonizing HER2-driven tumorigenesis, PTEN also sensitizes breast cancer to trastuzumab treatment (37). In fact, PTEN loss and PTEN-independent activation of the PI3K pathway were identified as a major determinant of trastuzumab resistance in preclinical models and clinical samples as well (37–39, 185).
PTEN in Leukemia
Although mutations of the PTEN gene appear to be generally rare in hematological tumors (59), its functional inactivation is frequently observed in several hematopoietic neoplasms (60–62, 186). Experiments conducted in mice with inducible PTEN deletions have demonstrated that loss of PTEN drives proliferation of leukemia-initiating cells and promotes leukemogenesis. PI3K pathway blockade prevents leukemogenesis and restores the normal self-renewing capacity of hematopoietic stem cells (63, 64). Cheong et al. have shown that reduction of PTEN phosphorylation, associated with its inactivity, is observed in approximately 75% of acute myeloid leukemia (AML) patients (65). Another study has shown that aberrant PTEN transcripts are present in 24% of AML patients, 80% of cell lines, and 13% of normal controls analyzed (66). In chronic myeloid leukemia (CML), the BCR–ABL fusion protein mediates the exclusion of PTEN from the nucleus. Such effect is reversed upon inactivation of BCR–ABL by imatinib treatment, which restores PTEN physiological localization localization (67). Recently, it has been shown that PTEN loss accelerates T-cell acute lymphoblastic leukemia (T-ALL) onset, producing multiclonal tumors (68, 69). NOTCH1 receptor may inhibit PTEN expression through the HES-1 transcription factor and this may in turn lead to AKT activation and resistance to glucocorticoids (70). Indeed, T-ALL with PTEN loss are resistant to NOTCH1 inhibitors while they are sensitive to AKT inhibitors (68).
Discussion and Conclusion
PTEN is an extremely powerful and multifaceted tumor suppressor functionally involved in many different “hallmarks” of cancer. The main mechanism by which PTEN activity restrains cancer development and progression remains its ability to downmodulate signaling through the PI3K pathway, thereby indirectly inhibiting AKT downstream targets, such as GSK3, FOXO, B cell lymphoma 2 (BCL-2) antagonist of cell death (BAD), the E3 ubiquitin–protein ligase MDM2 and p27, which control survival, cell proliferation, angiogenesis, and cellular metabolism (187). On the other hand, the mTORC1 arm of the PI3K/AKT/mTOR pathway is also activated in response to the loss of PTEN inhibitory activity, resulting in the phosphorylation of p70 ribosomal protein S6 kinase (S6K; also known as RPS6K) and inhibition of 4E-binding protein 1 (4EBP1; also known as eIF4EBP1) to activate protein translation (188, 189) leading to the enhanced translation of specific mRNAs that are crucial for cell growth and proliferation. Recently, 4EBP1 has indeed emerged as a key negative regulator of cell proliferation downstream of mTORC1, and its inactivation may directly promote the growth of sporadic cancers (190, 191).
As discussed above, novel tumor-suppressive functions of PTEN, independent of its lipid phosphatase activity and ability to keep the PI3K/AKT/mTOR pathway at bay, have recently emerged. Among these, functions related to PTEN nuclear localization appear to be particularly interesting (Figure 1B). Indeed, PTEN cooperates in maintaining genomic integrity, repairing DNA double-strand breaks, controlling homologous recombination, and promoting ubiquitin-dependent degradation of oncoproteins such as polo-like kinase 1 (PLK1) and Aurora kinases (AURK). This is particularly important, as loss of these PTEN activities may actually crucially sensitize tumor cells to the cytotoxic action of inhibitors of the DNA repair enzyme poly (ADP-ribose) polymerase (PARP) and have important implications in tumor cell sensitivity to PLK and AURK inhibitors (24, 192–194). Similarly, the phosphatase-independent role played by PTEN in controlling non-canonical signaling pathways, such as the JNK pathway (26), the eukaryotic translation initiation factor 2α kinase 2 (eIF2αK2; also known as PKR) – eIF2α phosphorylation cascade (28) or MSP58-mediated cellular transformation (27), and its ability to dephosphorylate protein substrates, such as FAK (195), cAMP responsive-element-binding protein (CREB) (196), and the non-receptor Tyr kinase SRC (29), may have important implications for the development of rational pharmacological combinations simultaneously targeting the PI3K/AKT and other relevant pathways.
Along these lines, another interesting indication toward the possible therapeutic exploitation of PTEN loss in human cancer is the observation that PTEN critically lies at the intersection of two major survival/proliferation pathways, the canonical PI3K/AKT/mTOR pathway (of which PTEN is an integral part), and the RAS/MEK/ERK pathway (11). Our group has recently shown that MEK inhibition restores PTEN expression in tumor cells with an intact PTEN gene by inhibiting a re-wired MEK–ERK–c-Jun/miR-25 survival pathway. Under these conditions, combined MEK/mTOR blockade may exert frank antagonistic effects in terms of inhibition of VEGF production and tumor cell growth (106). Conversely, loss of PTEN activity marks a functional state of relative resistance to the growth-inhibitory and anti-angiogenic activity of MEK inhibitors (53, 106, 179, 180), raising the interesting hypothesis that combined pharmacological inhibition of the MEK/ERK and PI3K/AKT/mTOR may result in highly synergistic anti-tumor activity selectively in PTEN-null tumors (11, 106, 197).
Synthetic lethality-based and rational combinatorial strategies highlighted above are two of the possible approaches to address the problem that usually happens with loss-of-function “oncogenic drivers,” restoring the oncosuppressive activity of a missing PTEN gene/protein by pharmacological means may be difficult. However, given the importance of non-genomic mechanisms regulating PTEN expression in those cases where the PTEN gene is conserved, epigenetic therapy through pharmacological modulation of histone acetylation status or promoter methylation, as well as inhibition of signaling pathways that are known to regulate PTEN expression, are particularly attractive, alone or in combination with inhibitors of other signaling pathways, whose activation is known to cooperate with PTEN loss in driving the different aspects of the neoplastic phenotype (198).
Finally, identification of clinical situations in which loss of PTEN activity is a major driving force, and hence PTEN-based therapeutics may be exquisitely effective, is cumbersome. The exceptionally complex regulation of PTEN activity calls for a more comprehensive assessment of PTEN status in human tumors. This should encompass sequencing the entire PTEN gene, transcriptional, and protein expression analysis, as well as assessment of post-translational modifications and subcellular localization. An alternative possibility could be to develop functional (genomic, transcriptional, or proteomic) “signatures” of loss of PTEN function. Although, such approach would require extensive validation to identify reliable surrogate(s) for PTEN loss, preliminary evidence suggests that “signatures” of PTEN loss may be developed and may identify patients/tissues with loss of PTEN function more efficiently than immunohistochemistry for the PTEN protein (26, 76).
Loss of the many PTEN activities remains a crucial event in the development and progression of a vast and ever increasing proportion of human cancers. Even though little progress has been made so far in developing agents to therapeutically enhance the tumor-suppressive functions of PTEN, there is little doubt that with the current rapid expansion of knowledge on PTEN functions and regulation, its exploitation for therapeutic purposes will soon become a reality for many cancer patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work presented has been supported in part by grants from the Italian Association for Cancer Research (AIRC): IG 14362 and Special Program Molecular Clinical Oncology 5 per mille 9979. Ursula Cesta Incani is Ph.D. student at the Doctoral School in Immunological, Hematological and Reumatological Sciences (UCI) and Anais Del Curatolo and Fabiana Conciatori are Ph.D. students at the Doctoral School of Oncology and Digestive Oncology (FB and ADC) of the University of Rome “La Sapienza.” The authors wish to thank Mrs. Tania Merlino for revising the English form of the manuscript.
References
1. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275(5308):1943–7. doi: 10.1126/science.275.5308.1943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res (1997) 57(11):2124–9.
3. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet (1997) 15(4):356–62. doi:10.1038/ng0497-356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell (1999) 99(3):323–34. doi:10.1016/S0092-8674(00)81663-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A (2012) 109(10):3921–6. doi:10.1073/pnas.1117135109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci (2008) 121(Pt 3):249–53. doi:10.1242/jcs.022459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science (2013) 341(6144):399–402. doi:10.1126/science.1234907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307(5712):1098–101. doi:10.1126/science.1106148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol (1997) 7(10):776–89. doi:10.1016/S0960-9822(06)00336-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol (2004) 22(14):2954–63. doi:10.1200/JCO.2004.02.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Ciuffreda L, Falcone I, Incani UC, Del Curatolo A, Conciatori F, Matteoni S, et al. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv Biol Regul (2014) 56:66–80. doi:10.1016/j.jbior.2014.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet (2006) 7(8):606–19. doi:10.1038/nrg1879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Blind RD. Disentangling biological signaling networks by dynamic coupling of signaling lipids to modifying enzymes. Adv Biol Regul (2014) 54:25–38. doi:10.1016/j.jbior.2013.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Laurent PA, Severin S, Gratacap MP, Payrastre B. Class I PI 3-kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv Biol Regul (2014) 54:162–74. doi:10.1016/j.jbior.2013.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273(22):13375–8. doi:10.1074/jbc.273.22.13375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A (1998) 95(23):13513–8. doi:10.1073/pnas.95.23.13513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi:10.1038/nrm3330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hlobilkova A, Guldberg P, Thullberg M, Zeuthen J, Lukas J, Bartek J. Cell cycle arrest by the PTEN tumor suppressor is target cell specific and may require protein phosphatase activity. Exp Cell Res (2000) 256(2):571–7. doi:10.1006/excr.2000.4867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Leslie NR, Foti M. Non-genomic loss of PTEN function in cancer: not in my genes. Trends Pharmacol Sci (2011) 32(3):131–40. doi:10.1016/j.tips.2010.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: the long and the short of it. Trends Biochem Sci (2014) 39(4):183–90. doi:10.1016/j.tibs.2014.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. De Melo J, He L, Tang D. The protein-protein interaction-mediated inactivation of PTEN. Curr Mol Med (2014) 14(1):22–33. doi:10.2174/1566524013666131118100542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell (2007) 128(1):157–70. doi:10.1016/j.cell.2006.11.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Sun Z, Huang C, He J, Lamb KL, Kang X, Gu T, et al. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep (2014) 6(5):844–54. doi:10.1016/j.celrep.2014.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell (2011) 144(2):187–99. doi:10.1016/j.cell.2010.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Chen ZH, Zhu M, Yang J, Liang H, He J, He S, et al. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep (2014) 8(6):2003–14. doi:10.1016/j.celrep.2014.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell (2007) 11(6):555–69. doi:10.1016/j.ccr.2007.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Okumura K, Zhao M, Depinho RA, Furnari FB, Cavenee WK. Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proc Natl Acad Sci U S A (2005) 102(8):2703–6. doi:10.1073/pnas.0409370102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Mounir Z, Krishnamoorthy JL, Robertson GP, Scheuner D, Kaufman RJ, Georgescu MM, et al. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal (2009) 2(102):ra85. doi:10.1126/scisignal.2000389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med (2011) 17(4):461–9. doi:10.1038/nm.2309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell (2012) 149(1):49–62. doi:10.1016/j.cell.2012.02.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in PTEN dose determine cancer susceptibility. Nat Genet (2010) 42(5):454–8. doi:10.1038/ng.556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Alimonti A. PTEN breast cancer susceptibility: a matter of dose. Ecancermedicalscience (2010) 4:192. doi:10.3332/ecancer.2010.192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Tamguney T, Stokoe D. New insights into PTEN. J Cell Sci (2007) 120(Pt 23):4071–9. doi:10.1242/jcs.015230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer (2011) 11(4):289–301. doi:10.1038/nrc3037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res (2012) 18(2):400–7. doi:10.1158/1078-0432.CCR-11-2283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Zhang HY, Liang F, Jia ZL, Song ST, Jiang ZF. PTEN mutation, methylation and expression in breast cancer patients. Oncol Lett (2013) 6(1):161–8. doi:10.3892/ol.2013.1331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell (2004) 6(2):117–27. doi:10.1016/j.ccr.2004.06.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Pandolfi PP. Breast cancer – loss of PTEN predicts resistance to treatment. N Engl J Med (2004) 351(22):2337–8. doi:10.1056/NEJMcibr043143
39. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell (2007) 12(4):395–402. doi:10.1016/j.ccr.2007.08.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kouniavsky G, Zeiger MA. Thyroid tumorigenesis and molecular markers in thyroid cancer. Curr Opin Oncol (2010) 22(1):23–9. doi:10.1097/CCO.0b013e328333846f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) 499(7456):43–9. doi:10.1038/nature12222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet (2015) 47(1):13–21. doi:10.1038/ng.3146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Muriel Lopez C, Esteban E, Astudillo A, Pardo P, Berros JP, Izquierdo M, et al. Predictive factors for response to treatment in patients with advanced renal cell carcinoma. Invest New Drugs (2012) 30(6):2443–9. doi:10.1007/s10637-012-9836-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Dellas A, Jundt G, Sartorius G, Schneider M, Moch H. Combined PTEN and p27kip1 protein expression patterns are associated with obesity and prognosis in endometrial carcinomas. Clin Cancer Res (2009) 15(7):2456–62. doi:10.1158/1078-0432.CCR-08-1732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Matias-Guiu X, Prat J. Molecular pathology of endometrial carcinoma. Histopathology (2013) 62(1):111–23. doi:10.1111/his.12053
46. Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, et al. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol (2002) 161(2):439–47. doi:10.1016/S0002-9440(10)64200-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Frattini M, Signoroni S, Pilotti S, Bertario L, Benvenuti S, Zanon C, et al. Phosphatase protein homologue to tensin expression and phosphatidylinositol-3 phosphate kinase mutations in colorectal cancer. Cancer Res (2005) 65(23):11227. doi:10.1158/0008-5472.CAN-05-2780
48. Razis E, Pentheroudakis G, Rigakos G, Bobos M, Kouvatseas G, Tzaida O, et al. EGFR gene gain and PTEN protein expression are favorable prognostic factors in patients with KRAS wild-type metastatic colorectal cancer treated with cetuximab. J Cancer Res Clin Oncol (2014) 140(5):737–48. doi:10.1007/s00432-014-1626-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Molinari F, Frattini M. Functions and regulation of the PTEN gene in colorectal cancer. Front Oncol (2013) 3:326. doi:10.3389/fonc.2013.00326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res (2006) 66(13):6546–52. doi:10.1158/0008-5472.CAN-06-0384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Wang Y, Digiovanna JJ, Stern JB, Hornyak TJ, Raffeld M, Khan SG, et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A (2009) 106(15):6279–84. doi:10.1073/pnas.0812401106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf(V600E) cooperates with PTEN loss to induce metastatic melanoma. Nat Genet (2009) 41(5):544–52. doi:10.1038/ng.356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Nathanson KL, Martin AM, Wubbenhorst B, Greshock J, Letrero R, D’Andrea K, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res (2013) 19(17):4868–78. doi:10.1158/1078-0432.CCR-13-0827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Koul D. PTEN signaling pathways in glioblastoma. Cancer Biol Ther (2008) 7(9):1321–5. doi:10.4161/cbt.7.9.6954
55. Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene (2008) 27(41):5416–30. doi:10.1038/onc.2008.239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi:10.1038/nature11125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature (2011) 470(7333):214–20. doi:10.1038/nature09744
58. Roychowdhury S, Chinnaiyan AM. Advancing precision medicine for prostate cancer through genomics. J Clin Oncol (2013) 31(15):1866–73. doi:10.1200/JCO.2012.45.3662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol (2009) 4:127–50. doi:10.1146/annurev.pathol.4.110807.092311
60. Gronbaek K, Zeuthen J, Guldberg P, Ralfkiaer E, Hou-Jensen K. Alterations of the MMAC1/PTEN gene in lymphoid malignancies. Blood (1998) 91(11):4388–90.
61. Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M. PTEN gene alterations in lymphoid neoplasms. Blood (1998) 92(9):3410–5.
62. Scarisbrick JJ, Woolford AJ, Russell-Jones R, Whittaker SJ. Loss of heterozygosity on 10q and microsatellite instability in advanced stages of primary cutaneous T-cell lymphoma and possible association with homozygous deletion of PTEN. Blood (2000) 95(9):2937–42.
63. Cheung AM, Mak TW. PTEN in the haematopoietic system and its therapeutic indications. Trends Mol Med (2006) 12(11):503–5. doi:10.1016/j.molmed.2006.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Chang H, Qi XY, Claudio J, Zhuang L, Patterson B, Stewart AK. Analysis of PTEN deletions and mutations in multiple myeloma. Leuk Res (2006) 30(3):262–5. doi:10.1016/j.leukres.2005.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Cheong JW, Eom JI, Maeng HY, Lee ST, Hahn JS, Ko YW, et al. Phosphatase and tensin homologue phosphorylation in the C-terminal regulatory domain is frequently observed in acute myeloid leukaemia and associated with poor clinical outcome. Br J Haematol (2003) 122(3):454–6. doi:10.1046/j.1365-2141.2003.04452.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Liu TC, Lin PM, Chang JG, Lee JP, Chen TP, Lin SF. Mutation analysis of PTEN/MMAC1 in acute myeloid leukemia. Am J Hematol (2000) 63(4):170–5. doi:10.1002/(SICI)1096-8652(200004)63:4<170::AID-AJH2>3.0.CO;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Panuzzo C, Crivellaro S, Carra G, Guerrasio A, Saglio G, Morotti A. BCR-ABL promotes PTEN downregulation in chronic myeloid leukemia. PLoS One (2014) 9(10):e110682. doi:10.1371/journal.pone.0110682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med (2007) 13(10):1203–10. doi:10.1038/nm1636
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Martelli AM, Lonetti A, Buontempo F, Ricci F, Tazzari PL, Evangelisti C, et al. Targeting signaling pathways in T-cell acute lymphoblastic leukemia initiating cells. Adv Biol Regul (2014) 56:6–21. doi:10.1016/j.jbior.2014.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell (2013) 24(6):766–76. doi:10.1016/j.ccr.2013.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res (2002) 8(5):1178–84.
72. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol (2013) 24(9):2371–6. doi:10.1093/annonc/mdt205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Yokomizo A, Tindall DJ, Drabkin H, Gemmill R, Franklin W, Yang P, et al. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene (1998) 17(4):475–9. doi:10.1038/sj.onc.1201956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol (2005) 36(7):768–76. doi:10.1016/j.humpath.2005.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Jin G, Kim MJ, Jeon HS, Choi JE, Kim DS, Lee EB, et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer (2010) 69(3):279–83. doi:10.1016/j.lungcan.2009.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A (2007) 104(18):7564–9. doi:10.1073/pnas.0702507104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Cordes I, Kluth M, Zygis D, Rink M, Chun F, Eichelberg C, et al. PTEN deletions are related to disease progression and unfavourable prognosis in early bladder cancer. Histopathology (2013) 63(5):670–7. doi:10.1111/his.12209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Hu TH, Wang CC, Huang CC, Chen CL, Hung CH, Chen CH, et al. Down-regulation of tumor suppressor gene PTEN, overexpression of p53, plus high proliferating cell nuclear antigen index predict poor patient outcome of hepatocellular carcinoma after resection. Oncol Rep (2007) 18(6):1417–26. doi:10.3892/or.18.6.1417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Zhong Y, Yan J, Deng M, Hu K, Yao Z, Zou Y, et al. Impaired phosphate and tension homologue deleted on chromosome 10 expression and its prognostic role in radical surgery for hepatocellular carcinoma with family aggregation resulting from hepatitis B and liver cirrhosis. Exp Biol Med (2013) 238(8):866–73. doi:10.1177/1535370213494654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Aguirre AJ, Brennan C, Bailey G, Sinha R, Feng B, Leo C, et al. High-resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci U S A (2004) 101(24):9067–72. doi:10.1073/pnas.0402932101
81. Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol (2000) 157(4):1097–103. doi:10.1016/S0002-9440(10)64624-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov (2011) 1(2):158–69. doi:10.1158/2159-8290.CD-11-0031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. van Nederveen FH, Perren A, Dannenberg H, Petri BJ, Dinjens WN, Komminoth P, et al. PTEN gene loss, but not mutation, in benign and malignant phaeochromocytomas. J Pathol (2006) 209(2):274–80. doi:10.1002/path.1968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Munoz-Martin M, Gomez-Lopez G, Canamero M, et al. PTEN positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab (2012) 15(3):382–94. doi:10.1016/j.cmet.2012.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Pal A, Barber TM, Van de Bunt M, Rudge SA, Zhang Q, Lachlan KL, et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med (2012) 367(11):1002–11. doi:10.1056/NEJMoa1113966
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Vellai T, McCulloch D, Gems D, Kovacs AL. Effects of sex and insulin/insulin-like growth factor-1 signaling on performance in an associative learning paradigm in Caenorhabditis elegans. Genetics (2006) 174(1):309–16. doi:10.1534/genetics.106.061499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Wong JT, Kim PT, Peacock JW, Yau TY, Mui AL, Chung SW, et al. PTEN (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia (2007) 50(2):395–403. doi:10.1007/s00125-006-0531-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Nakashima N, Sharma PM, Imamura T, Bookstein R, Olefsky JM. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3-L1 adipocytes. J Biol Chem (2000) 275(17):12889–95. doi:10.1074/jbc.275.17.12889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Mosser VA, Li Y, Quon MJ. PTEN does not modulate GLUT4 translocation in rat adipose cells under physiological conditions. Biochem Biophys Res Commun (2001) 288(4):1011–7. doi:10.1006/bbrc.2001.5876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Morani F, Phadngam S, Follo C, Titone R, Aimaretti G, Galetto A, et al. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J Mol Endocrinol (2014) 53(2):247–58. doi:10.1530/JME-14-0118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Funamoto S, Milan K, Meili R, Firtel RA. Role of phosphatidylinositol 3’ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in dictyostelium. J Cell Biol (2001) 153(4):795–810. doi:10.1083/jcb.153.4.795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Huang X, Czerwinski E, Mellgren RL. Purification and properties of the Dictyostelium calpain-like protein, Cpl. Biochemistry (2003) 42(6):1789–95. doi:10.1021/bi026461+
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell (2002) 109(5):599–610. doi:10.1016/S0092-8674(02)00745-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene (2008) 27(41):5464–76. doi:10.1038/onc.2008.243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science (2000) 287(5455):1037–40. doi:10.1126/science.287.5455.1037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Leslie NR, Downes CP. PTEN: the down side of PI 3-kinase signalling. Cell Signal (2002) 14(4):285–95. doi:10.1016/S0898-6568(01)00234-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Leslie NR. PTEN: an intercellular peacekeeper? Sci Signal (2012) 5(250):e50. doi:10.1126/scisignal.2003685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol (2007) 17(2):115–25. doi:10.1016/j.cub.2006.12.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science (2004) 303(5661):1179–81. doi:10.1126/science.1092089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Park MJ, Kim MS, Park IC, Kang HS, Yoo H, Park SH, et al. PTEN suppresses hyaluronic acid-induced matrix metalloproteinase-9 expression in U87MG glioblastoma cells through focal adhesion kinase dephosphorylation. Cancer Res (2002) 62(21):6318–22.
101. Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, et al. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res (2008) 68(6):1862–71. doi:10.1158/0008-5472.CAN-07-1182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, et al. PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci U S A (2001) 98(8):4622–7. doi:10.1073/pnas.081063798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev (2000) 14(4):391–6. doi:10.1101/gad.14.4.391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Del Bufalo D, Ciuffreda L, Trisciuoglio D, Desideri M, Cognetti F, Zupi G, et al. Antiangiogenic potential of the mammalian target of rapamycin inhibitor temsirolimus. Cancer Res (2006) 66(11):5549–54. doi:10.1158/0008-5472.CAN-05-2825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Ciuffreda L, Del Bufalo D, Desideri M, Di Sanza C, Stoppacciaro A, Ricciardi MR, et al. Growth-inhibitory and antiangiogenic activity of the MEK inhibitor PD0325901 in malignant melanoma with or without BRAF mutations. Neoplasia (2009) 11(8):720–31.
106. Ciuffreda L, Di Sanza C, Cesta Incani U, Eramo A, Desideri M, Biagioni F, et al. The mitogen-activated protein kinase (MAPK) cascade controls phosphatase and tensin homolog (PTEN) expression through multiple mechanisms. J Mol Med (Berl) (2012) 90(6):667–79. doi:10.1007/s00109-011-0844-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, et al. Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. J Cell Sci (2006) 119(Pt 24):5160–8. doi:10.1242/jcs.000133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol (2000) 156(5):1693–700. doi:10.1016/S0002-9440(10)65040-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Lachyankar MB, Sultana N, Schonhoff CM, Mitra P, Poluha W, Lambert S, et al. A role for nuclear PTEN in neuronal differentiation. J Neurosci (2000) 20(4):1404–13.
111. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem (2001) 276(2):993–8. doi:10.1074/jbc.M009134200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res (2003) 63(2):282–6.
113. Chung JH, Ginn-Pease ME, Eng C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res (2005) 65(10):4108–16. doi:10.1158/0008-5472.CAN-05-0124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell (2007) 128(1):141–56. doi:10.1016/j.cell.2006.11.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science (2013) 341(6144):395–9. doi:10.1126/science.1236188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell (2003) 3(2):117–30. doi:10.1016/S1535-6108(03)00021-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc Natl Acad Sci U S A (2008) 105(7):2622–7. doi:10.1073/pnas.0706790105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J, et al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal (2012) 5(243):ra70. doi:10.1126/scisignal.2003084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Malaney P, Uversky VN, Dave V. The PTEN Long N-tail is intrinsically disordered: increased viability for PTEN therapy. Mol Biosyst (2013) 9(11):2877–88. doi:10.1039/c3mb70267g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Liang H, He S, Yang J, Jia X, Wang P, Chen X, et al. PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab (2014) 19(5):836–48. doi:10.1016/j.cmet.2014.03.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell (2008) 133(3):403–14. doi:10.1016/j.cell.2008.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Stambolic V, Woodgett JR. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol (2006) 16(9):461–6. doi:10.1016/j.tcb.2006.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Moorehead RA, Hojilla CV, De Belle I, Wood GA, Fata JE, Adamson ED, et al. Insulin-like growth factor-II regulates PTEN expression in the mammary gland. J Biol Chem (2003) 278(50):50422–7. doi:10.1074/jbc.M306894200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol (2001) 3(12):1124–8. doi:10.1038/ncb1201-1124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol (2001) 11(10):764–8. doi:10.1016/S0960-9822(01)00225-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Escriva M, Peiro S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentis B, et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol (2008) 28(5):1528–40. doi:10.1128/MCB.02061-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell (2007) 11(5):447–60. doi:10.1016/j.ccr.2007.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Whelan JT, Forbes SL, Bertrand FE. CBF-1 (RBP-J kappa) binds to the PTEN promoter and regulates PTEN gene expression. Cell Cycle (2007) 6(1):80–4. doi:10.4161/cc.6.1.3648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature (1998) 395(6698):125–6. doi:10.1038/25870
130. Hettinger K, Vikhanskaya F, Poh MK, Lee MK, de Belle I, Zhang JT, et al. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ (2007) 14(2):218–29. doi:10.1038/sj.cdd.4401946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Vasudevan KM, Burikhanov R, Goswami A, Rangnekar VM. Suppression of PTEN expression is essential for antiapoptosis and cellular transformation by oncogenic Ras. Cancer Res (2007) 67(21):10343–50. doi:10.1158/0008-5472.CAN-07-1827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology (2007) 133(2):647–58. doi:10.1053/j.gastro.2007.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin Chim Acta (2010) 411(11–12):846–52. doi:10.1016/j.cca.2010.02.074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b ∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal (2010) 3(117):ra29. doi:10.1126/scisignal.2000594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17 92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev (2009) 23(24):2806–11. doi:10.1101/gad.1872909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Khan S, Kumagai T, Vora J, Bose N, Sehgal I, Koeffler PH, et al. PTEN promoter is methylated in a proportion of invasive breast cancers. Int J Cancer (2004) 112(3):407–10. doi:10.1002/ijc.20447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Salvesen HB, MacDonald N, Ryan A, Jacobs IJ, Lynch ED, Akslen LA, et al. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int J Cancer (2001) 91(1):22–6. doi:10.1002/1097-0215(20010101)91:1<22::AID-IJC1002>3.0.CO;2-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Lu J, Jeong HW, Kong N, Yang Y, Carroll J, Luo HR, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One (2009) 4(5):e5577. doi:10.1371/journal.pone.0005577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Pan L, Lu J, Wang X, Han L, Zhang Y, Han S, et al. Histone deacetylase inhibitor trichostatin a potentiates doxorubicin-induced apoptosis by up-regulating PTEN expression. Cancer (2007) 109(8):1676–88. doi:10.1002/cncr.22585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Vazquez S, Aquilina JA, Jamie JF, Sheil MM, Truscott RJ. Novel protein modification by kynurenine in human lenses. J Biol Chem (2002) 277(7):4867–73. doi:10.1074/jbc.M107529200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG. Direct identification of PTEN phosphorylation sites. FEBS Lett (2002) 528(1–3):145–53. doi:10.1016/S0014-5793(02)03274-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Mehenni H, Lin-Marq N, Buchet-Poyau K, Reymond A, Collart MA, Picard D, et al. LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum Mol Genet (2005) 14(15):2209–19. doi:10.1093/hmg/ddi225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem (2005) 280(42):35195–202. doi:10.1074/jbc.M503045200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Correia NC, Girio A, Antunes I, Martins LR, Barata JT. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur J Cancer (2014) 50(1):216–25. doi:10.1016/j.ejca.2013.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Fragoso R, Barata JT. Kinases, tails and more: regulation of PTEN function by phosphorylation. Methods (2014). doi:10.1016/j.ymeth.2014.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell (2007) 128(1):129–39. doi:10.1016/j.cell.2006.11.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Amodio N, Scrima M, Palaia L, Salman AN, Quintiero A, Franco R, et al. Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative regulator, in non-small-cell lung carcinomas. Am J Pathol (2010) 177(5):2622–34. doi:10.2353/ajpath.2010.091075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem (2006) 281(36):26562–8. doi:10.1074/jbc.M605391200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem (2002) 277(23):20336–42. doi:10.1074/jbc.M111899200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci U S A (2000) 97(8):4233–8. doi:10.1073/pnas.97.8.4233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell (2005) 7(3):263–73. doi:10.1016/j.ccr.2005.02.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Kim YC, Kitaura H, Taira T, Iguchi-Ariga SM, Ariga H. Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. Int J Oncol (2009) 35(6):1331–41. doi:10.3892/ijo_00000451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Terrien E, Chaffotte A, Lafage M, Khan Z, Prehaud C, Cordier F, et al. Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci Signal (2012) 5(237):ra58. doi:10.1126/scisignal.2002941
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, Baccarini M. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell (2013) 50(1):43–55. doi:10.1016/j.molcel.2013.01.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Eng C. PTEN: one gene, many syndromes. Hum Mutat (2003) 22(3):183–98. doi:10.1002/humu.10257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet (1997) 16(1):64–7. doi:10.1038/ng0597-64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Mester JL, Moore RA, Eng C. PTEN germline mutations in patients initially tested for other hereditary cancer syndromes: would use of risk assessment tools reduce genetic testing? Oncologist (2013) 18(10):1083–90. doi:10.1634/theoncologist.2013-0174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet (1998) 7(3):507–15. doi:10.1093/hmg/7.3.507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A. Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am J Med Genet A (2007) 143A(6):589–93. doi:10.1002/ajmg.a.31619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Garcia-Junco-Clemente P, Golshani P. PTEN: a master regulator of neuronal structure, function, and plasticity. Commun Integr Biol (2014) 7(1):e28358. doi:10.4161/cib.28358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat (2000) 16(2):109–22. doi:10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0
162. Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A (1999) 96(8):4240–5. doi:10.1073/pnas.96.8.4240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. PTEN is essential for embryonic development and tumour suppression. Nat Genet (1998) 19(4):348–55. doi:10.1038/1235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, et al. Mutation of PTEN/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A (1999) 96(4):1563–8. doi:10.1073/pnas.96.4.1563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: how much is too little? Cancer Res (2011) 71(3):629–33. doi:10.1158/0008-5472.CAN-10-2488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, et al. Haploinsufficiency of the PTEN tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A (2001) 98(20):11563–8. doi:10.1073/pnas.201167798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. PTEN dose dictates cancer progression in the prostate. PLoS Biol (2003) 1(3):E59. doi:10.1371/journal.pbio.0000059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Hill R, Wu H. PTEN, stem cells, and cancer stem cells. J Biol Chem (2009) 284(18):11755–9. doi:10.1074/jbc.R800071200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Groszer M, Erickson R, Scripture-Adams DD, Dougherty JD, Le Belle J, Zack JA, et al. PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc Natl Acad Sci U S A (2006) 103(1):111–6. doi:10.1073/pnas.0509939103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Inoue-Narita T, Hamada K, Sasaki T, Hatakeyama S, Fujita S, Kawahara K, et al. PTEN deficiency in melanocytes results in resistance to hair graying and susceptibility to carcinogen-induced melanomagenesis. Cancer Res (2008) 68(14):5760–8. doi:10.1158/0008-5472.CAN-08-0889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature (2006) 441(7092):518–22. doi:10.1038/nature04747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. PTEN dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature (2006) 441(7092):475–82. doi:10.1038/nature04703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A (2007) 104(41):16158–63. doi:10.1073/pnas.0702596104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Lombardo Y, Scopelliti A, Cammareri P, Todaro M, Iovino F, Ricci-Vitiani L, et al. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology (2011) 140(1):297–309. doi:10.1053/j.gastro.2010.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Ricci-Vitiani L, Mollinari C, di Martino S, Biffoni M, Pilozzi E, Pagliuca A, et al. Thymosin beta4 targeting impairs tumorigenic activity of colon cancer stem cells. FASEB J (2010) 24(11):4291–301. doi:10.1096/fj.10-159970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Friday BB, Yu C, Dy GK, Smith PD, Wang L, Thibodeau SN, et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res (2008) 68(15):6145–53. doi:10.1158/0008-5472.CAN-08-1430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest (2008) 118(9):3065–74. doi:10.1172/JCI34739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene (2008) 27(41):5527–41. doi:10.1038/onc.2008.247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Byron SA, Loch DC, Wellens CL, Wortmann A, Wu JY, Wang J, et al. Sensitivity to the MEK inhibitor E6201 in melanoma cells is associated with mutant BRAF and wildtype PTEN status. Mol Cancer (2012) 11:75. doi:10.1186/1476-4598-11-75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene (2012) 31(4):446–57. doi:10.1038/onc.2011.250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Naguib A, Cooke JC, Happerfield L, Kerr L, Gay LJ, Luben RN, et al. Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: associations with clinicopathological and dietary factors. BMC Cancer (2011) 11:123. doi:10.1186/1471-2407-11-123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Chowdhury S, Ongchin M, Wan G, Sharratt E, Brattain MG, Rajput A. Restoration of PTEN activity decreases metastases in an orthotopic model of colon cancer. J Surg Res (2013) 184(2):755–60. doi:10.1016/j.jss.2013.03.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell (2000) 100(4):387–90. doi:10.1016/S0092-8674(00)80674-1
184. Schade B, Rao T, Dourdin N, Lesurf R, Hallett M, Cardiff RD, et al. PTEN deficiency in a luminal ErbB-2 mouse model results in dramatic acceleration of mammary tumorigenesis and metastasis. J Biol Chem (2009) 284(28):19018–26. doi:10.1074/jbc.M109.018937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Fabi A, Metro G, Di Benedetto A, Nistico C, Vici P, Melucci E, et al. Clinical significance of PTEN and p-Akt co-expression in HER2-positive metastatic breast cancer patients treated with trastuzumab-based therapies. Oncology (2010) 78(2):141–9. doi:10.1159/000312656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Basecke J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia (2008) 22(4):686–707. doi:10.1038/leu.2008.26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129(7):1261–74. doi:10.1016/j.cell.2007.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Ciuffreda L, Di Sanza C, Incani UC, Milella M. The mTOR pathway: a new target in cancer therapy. Curr Cancer Drug Targets (2010) 10(5):484–95. doi:10.2174/156800910791517172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol (2009) 10(5):307–18. doi:10.1038/nrm2672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science (2010) 328(5982):1172–6. doi:10.1126/science.1187532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell (2010) 18(1):39–51. doi:10.1016/j.ccr.2010.05.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med (2009) 1(6–7):315–22. doi:10.1002/emmm.200900041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res (2010) 70(13):5457–64. doi:10.1158/0008-5472.CAN-09-4295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, et al. Polo-like kinase 1 facilitates loss of PTEN tumor suppressor-induced prostate cancer formation. J Biol Chem (2011) 286(41):35795–800. doi:10.1074/jbc.C111.269050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science (1998) 280(5369):1614–7. doi:10.1126/science.280.5369.1614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res (2011) 71(8):2821–5. doi:10.1158/0008-5472.CAN-10-3399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Ciuffreda L, Incani UC, Steelman LS, Abrams SL, Falcone I, Curatolo AD, et al. Signaling intermediates (MAPK and PI3K) as therapeutic targets in NSCLC. Curr Pharm Des (2014) 20(24):3944–57. doi:10.2174/13816128113196660763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Boosani CS, Agrawal DK. PTEN modulators: a patent review. Expert Opin Ther Pat (2013) 23(5):569–80. doi:10.1517/13543776.2013.768985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: PTEN, cancer, subcellular localization, PHTS, PI3K
Citation: Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CMA, Vaccaro V, Vari S, Cognetti F and Ciuffreda L (2015) PTEN: multiple functions in human malignant tumors. Front. Oncol. 5:24. doi: 10.3389/fonc.2015.00024
Received: 06 November 2014; Accepted: 22 January 2015;
Published online: 16 February 2015.
Edited by:
Alexandre Arcaro, University of Bern, SwitzerlandReviewed by:
Daniel Christian Hoessli, University of Karachi, PakistanBernd E. R. Nuernberg, University Hospital Tübingen, Germany
Copyright: © 2015 Milella, Falcone, Conciatori, Cesta Incani, Del Curatolo, Inzerilli, Nuzzo, Vaccaro, Vari, Cognetti and Ciuffreda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludovica Ciuffreda, Division of Medical Oncology A, Regina Elena National Cancer Institute, Via Elio Chianesi 53, Rome 00144, Italy e-mail:bHVkb3ZpY2FjaXVmZnJlZGFAaG90bWFpbC5jb20=