 Santiago Rello-Varona1
Santiago Rello-Varona1 David Herrero-Martín1Laura Lagares-Tena1
David Herrero-Martín1Laura Lagares-Tena1 Roser López-Alemany1Núria Mulet-Margalef1Juan Huertas-Martínez1Silvia Garcia-Monclús1Xavier García del Muro1
Roser López-Alemany1Núria Mulet-Margalef1Juan Huertas-Martínez1Silvia Garcia-Monclús1Xavier García del Muro1 Cristina Muñoz-Pinedo2
Cristina Muñoz-Pinedo2 Oscar Martínez Tirado1*
Oscar Martínez Tirado1*- 1Sarcoma Research Group, Molecular Oncology Laboratory, Bellvitge Biomedical Research Institute (IDIBELL), L’Hospitalet de Llobregat, Barcelona, Spain
- 2Cell Death Regulation Group, Molecular Oncology Laboratory, Bellvitge Biomedical Research Institute (IDIBELL), L’Hospitalet de Llobregat, Barcelona, Spain
Cell death can occur through different mechanisms, defined by their nature and physiological implications. Correct assessment of cell death is crucial for cancer therapy success. Sarcomas are a large and diverse group of neoplasias from mesenchymal origin. Among cell death types, apoptosis is by far the most studied in sarcomas. Albeit very promising in other fields, regulated necrosis and other cell death circumstances (as so-called “autophagic cell death” or “mitotic catastrophe”) have not been yet properly addressed in sarcomas. Cell death is usually quantified in sarcomas by unspecific assays and in most cases the precise sequence of events remains poorly characterized. In this review, our main objective is to put into context the most recent sarcoma cell death findings in the more general landscape of different cell death modalities.
Introduction
Facts
• Sarcomas are a highly heterogeneous group of mesenchymal tumors.
• Among cell death mechanisms, only apoptosis has been extensively studied in sarcomas.
• Fusion proteins, actors of translocation-derived sarcomagenesis, play an anti-apoptotic role in sarcomas.
• Proper and deeper assessment of cell death in sarcomas is mandatory.
Challenges
• Can we improve the current therapeutic protocols in sarcomas through a better knowledge of cell death mechanisms?
• Can we assess more accurately the sequence of events of every type of cell death?
• Which are the key molecules that determine tumor cell death after therapy?
• Do translocation-bearing sarcomas have specific weaknesses in their cell death signaling networks?
Cancer therapies are aimed to induce the specific destruction of tumor cells without compromising patient health. This makes cell death mechanisms a central point of any therapeutic approach (1, 2). However, no every death is equally desirable in terms of therapy (3). The need of theoretical arrangement in the field has become evident during the past years. Our knowledge on cell death mechanisms has increased enormously and the available methodology has become more and more sophisticated. Therefore, a clear nomenclature based on reliable markers has been proposed (1, 4). Additionally, the growing number of cell death participants have been organized in clear hierarchic frameworks (5).
Sarcomas are a rare and heterogeneous group (more than 50 different clinical and molecular entities) of malignant tumors with mesenchymal origin. Molecular biology of sarcomas has remained elusive until recently, and a better knowledge remains as an unmet need (6). New drugs against potential targets in tumor cells with a crucial role in their metabolism or pro-survival fitness could improve the prognosis of these patients. Indeed, the relatively high rate of therapeutic failure and tumor relapse demands a better assessment of cell death induction. But scientific efforts in this discipline are historically undermined by the relative low investments and isolated work (7).
The scientific landscape involving cell death mechanisms in sarcomas can be improved. The majority of articles included in the present review focused on apoptosis (mostly) and necrosis, whose morphological characters (Figure 1) and signaling players (Figure 2) are better described. Many studies about cell death in sarcomas just describe the occurrence of cell death without a proper characterization of the sequence-of-events leading to a particular form of death. The aim of the present review is to help sarcoma researchers to face new knowledge on cell death mechanisms in order to routinely include it in their assessments.
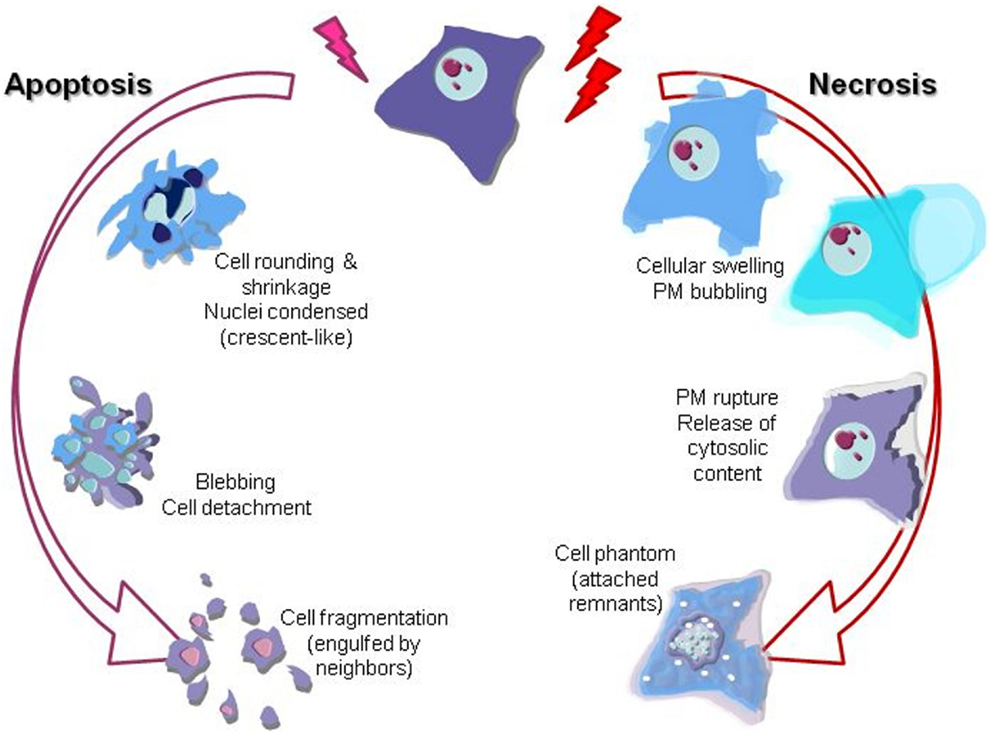
Figure 1. Graphical illustration of the most prominent morphological features of apoptotic and necrotic cell death mechanisms. Nuclei changes (karyorrhexis), cytoplasm shrinkage, and blebbing are the most evident descriptors of apoptosis. On the other hand, necrosis is clearly recognizable by cell swelling (loss of osmotic barrier) leading to the plasma membrane (PM) breakage and final release of the inner soluble content and nuclei karyolysis.
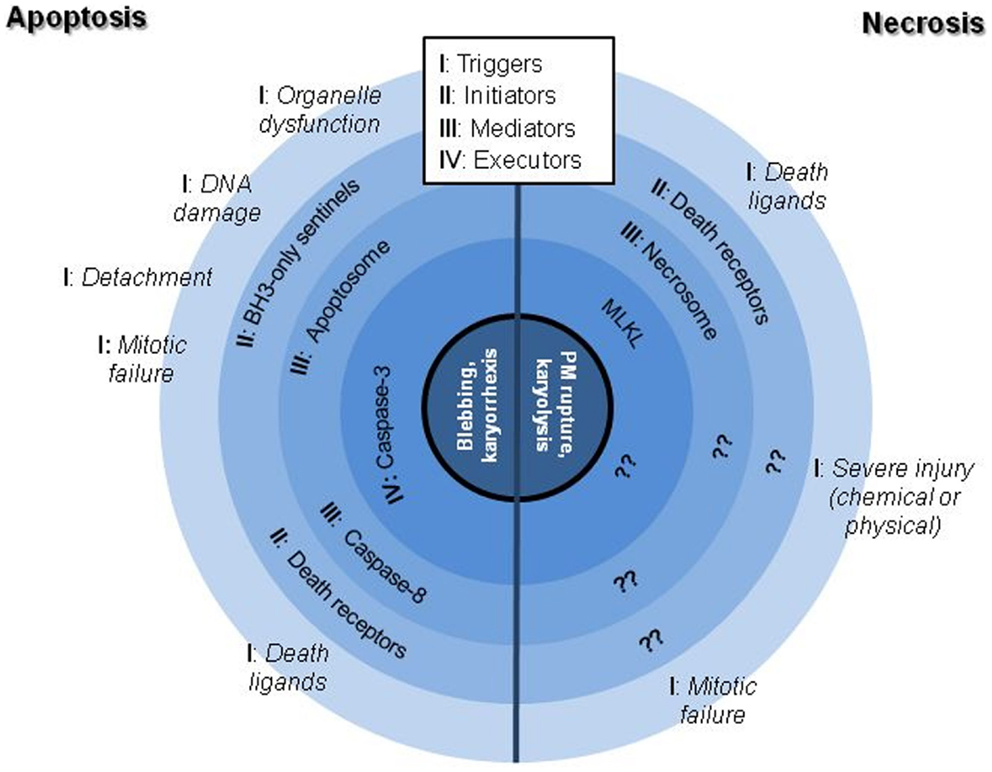
Figure 2. Schematic representation of the better characterized signaling hubs of apoptotic and necrotic cell death mechanisms. Note that necrotic processes are substantially worse described than apoptotic ones, being still controversial if the execution phase is protein-driven or result of a massive metabolic failure.
Cell Death Mechanisms
Apoptosis
Apoptosis involves a cellular controlled demolition process. Signaling cascades are finely orchestrated and secured, to ensure its perfect onset only when it is required (8). Caspases are the major actors in cellular demolition; once triggered, caspases can cross-activate each other and thus amplify the apoptotic signal (8). Apoptosis is by far the most studied form of cell death in sarcoma research. Nevertheless, researchers either employ uninformative methods about the form of death (i.e., Trypan Blue assay), or the mechanisms leading to such death are not always fully analyzed. Apoptosis recognition is easy by simple morphological features visible under the microscope: nuclear condensation and fragmentation, blebbing etc. (Figure 1). Other techniques (immunofluorescence or western blotting of cleaved caspases and/or caspase substrates, etc.) can be used to monitor specific mediators and executors of the process (9–11). Based on their biochemical features, we can describe two major pathways in apoptotic signaling: the intrinsic or mitochondrial pathway and the death receptor pathway (Figure 2).
Mitochondrial apoptosis
The “intrinsic pathway” is defined by the role of the mitochondria as encounter point of most of its initiators and mediators. The Bcl-2 family of proteins controls this pathway by regulating the formation of a pore in the mitochondrial outer membrane (12). Several signaling pathways converge in the regulation of Bcl-2 proteins, from DNA-damage sensor system to organelle stress and malfunction or growth factor signaling (Figure 2) (13, 14). In order to demonstrate that a drug or physiological input induces apoptosis through the mitochondrial pathway, exogenous overexpression of anti-apoptotic Bcl-2 family members can be performed; this should either prevent cell death or switch the mechanism to necrosis.
Some sarcomas rely on the presence of specific aberrant fusion proteins, generated after chromosomal rearrangements. Deregulation of gene expression in sarcomas driven by these chimeric oncoproteins can occur at different levels (epigenetic silencing, transcription activity, messenger processing, etc.) affecting every cellular process, including apoptosis (Figure 3). In the case of Ewing Sarcoma (ES), the fusion proteins EWS-FLI1 or EWS-ERG have an inhibitory effect on part of the apoptotic machinery (15, 16). This effect is mediated by direct or indirect interactions with several signaling pathways modulating apoptosis repression and inducing sustained growth (17–20). Alveolar rhabdomyosarcoma (aRMS) is also dependent on fusion proteins involving different PAX proteins with FOXO1, which also targets different signaling networks in order to ensure evasion of apoptosis (21, 22). SYT-SSX chimera proteins are present in the majority of synovial sarcoma tumors. They are involved in resistance to pro-apoptotic stimuli by modulating the levels and the activity of key apoptotic players of the Bcl-2 family of proteins (23). Furthermore, certain translocation-bearing sarcomas are also characterized by failure to complete tissue differentiation (i.e., RMS to skeletal muscle, liposarcoma to adipocytes) in a process mediated by their specific fusion protein and linked to the inhibition of apoptosis induction (24–26). Several recent studies have linked miRNAs status with apoptosis regulation in chromosome translocation-bearing sarcomas. Hence, mitochondrial apoptotic resistance in ES correlates with miR-125b upregulation through p53 and Bak (27) but overexpression of miR-206 in RMS promotes proliferation arrest and some sort of cell death (28). Overexpression of miR-145 and miR-451 in liposarcoma cell lines decreases cellular proliferation, impairs cell cycle progression, and boosts cell death (29), whereas overexpression of miR-26a-2 has the opposite effects (30).
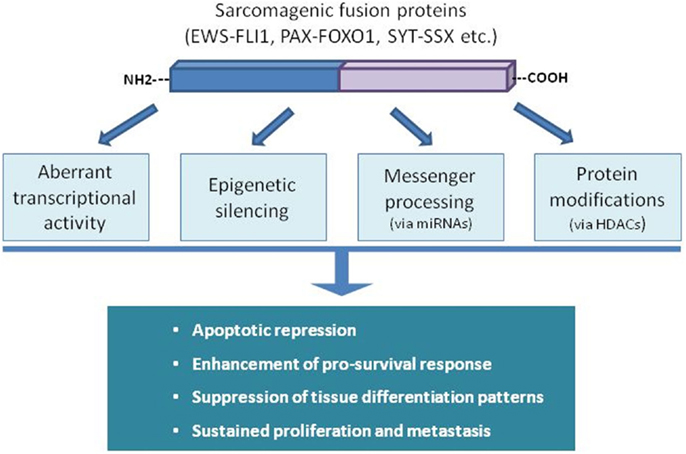
Figure 3. Fusion proteins in sarcomas disturb the natural physiological balance between pro-survival and death signaling inputs through different ways. The panoply of mechanisms and cellular targets disturbed demonstrates the powerful tumorigenic effect of a single event of genomic rearrangement.
The levels and status of key pro- and anti-apoptotic proteins are also crucial for understanding the differential sensitivity of cells toward apoptosis. Most ES cells have both the p53 downstream pathway and the DNA-damage signaling pathway functionally intact. The resistance of some ES cell lines to p53-induced apoptosis has been linked to a high Bcl-2/Bax ratio and low levels of Apaf-1 (31). However, the influence of fusion proteins inactivates p53 by deacetylation at Lys-382 driven by both EWS-FLI1 and HDAC1 (32), meaning that re-expression or re-activation of p53 could be a good strategy against these tumors. Similar phenomena occur in other fusion-positive sarcomas and accordingly, histone deacetylase inhibitors have been successfully tested as apoptotic inducers in different sarcoma types (33, 34). p53 re-activator agents as Nutlin-3 and/or PRIMA-1 are able to induce apoptosis successfully through Noxa, Puma, or p21 upregulation in both mutant and wild-type p53 sarcoma cell lines (35–37). Among downstream p53 targets p21, c-Myc, Bax, MDM2, DRAL, Bcl-2, and Bcl-xL have been suggested as key apoptotic regulators in different sarcoma models (38–43). Plasma membrane-anchored growth receptors such as NGFR or IGF-1R have an anti-apoptotic role (44, 45). In contrast, distinct behaviors have been suggested for the closely related receptors PDGFR α and β (46). Thus, PDGFR α favors cellular stemness and PDGFR β promotes angiogenesis in the tumor stroma. Hepatocyte-growth-factor activator inhibitors (HAI-1 and HAI-2) act as tumor suppressors leading to apoptosis and necrosis in leiomyosarcoma (47). Also, inhibition of endogenous tyrosine kinase B (TrkB) signaling suppresses cell proliferation and increases apoptosis in cultured leiomyosarcoma cells (48). In this context, tyrosine kinase inhibitors like Sorafenib induce apoptosis on many leiomyosarcoma or synovial sarcoma cell lines by inhibiting the RAF/MEK/ERK signaling pathway, among others (49, 50). Apoptotic cascades induced by other kinase inhibitors like JAK1 and 2 have been analyzed in detail in RMS and ES cells. These inhibitors lead to the alteration of the balance between the pro-apoptotic Bax and the anti-apoptotic proteins Bcl-2 and Bcl-xL, the release of cytochrome c, and the activation of caspase-9, -8, and -3 (51, 52).
Many other different strategies have been used in sarcomas to induce mitochondrial apoptosis. Betulinic acid is able to target the mitochondria in ES, promoting the permeabilization of the outer membrane resulting in the release, from the mitochondria to the cytosol, of soluble factors such as AIF and cytochrome c, who ultimately leads to caspase activation (53). Direct targeting of mitochondrial physiology was also explored in RMS with photodynamic therapy (54) and ROS-generation agents (55). Proteasome inhibitors as Bortezomib generate a major stress in the cell machinery, triggering a number of different reactions, many of them aimed to induce apoptosis. Bortezomib has been successfully employed in different pre-clinical models (56, 57). Heat shock proteins are among the most important actors against protein stress in cells. Accordingly, Hsp-90 antagonists had been shown to induce transient growth arrest and apoptosis in RMS cells (58). Likewise, some metabolic disruptors like 2-deoxyglucose, Lovastatin, and Catechins have been successfully tested as promoters of mitochondrial apoptosis by unbalancing the equilibrium of Bcl-2 family of proteins (59–61). Furthermore, down-regulation of inhibitor of apoptosis proteins (IAPs) also leads to apoptosis, identified by PARP cleavage, in pediatric sarcomas (62).
To keep their correct physiology, cells rely in their interaction with neighbors and microenvironment, meaning that detachment is a major apoptotic trigger. The process of detachment-induced apoptosis is termed anoikis (4). The lack of attachment activates signals from the plasma membrane, mostly by integrins and the focal adhesion kinase (FAK) that regulate the BH3-only proteins through the mitochondrial commitment to cell suicide (Figure 2) (63). Cell culture in non-adherent conditions, like soft-agar, is the better way to study this process. Suppression of anoikis cell death is considered an important hallmark of transformed cells and thus, a pre-metastatic key process (64).
Anoikis resistance in sarcomas has been described to be associated with integrins, Bcl-2 and caspase-8, CD99 isoforms, RANK, and ERK (65–68). ES cells survival in non-adherent conditions is mediated by E-cadherin dependent spheroid formation, avoiding apoptotic triggering by means of the PI3K/Akt pathway (69). Scotlandi et al. demonstrated the relevance of IGF-1R in the anoikis-resistant ES cell line TC71. Impairment of IGF-1R signaling (by neutralizing antibodies or siRNAs expression) led to a lower survival in anchorage-independent growth conditions and a decrease on metastatic ability (70). In synovial sarcoma, the increased IGF-2 synthesis protects cells from anoikis and is required for tumor formation in vivo (71). Another trans-membrane growth factor receptor, the ErbB4 Tyrosine kinase, gets phosphorylated in ES spheroids and its expression is linked to anoikis avoidance, metastatic disease, and bad outcome (72). In RMS, spheroids obtained after cell culture enrichment express stem cell gene markers such as oct4, pax3, sox2, c-myc, and nanog. It was also found that CD133 was upregulated in these spheres, conferring cells higher resistance to Cisplatin and Chlorambucil in vivo (73). In osteosarcoma (OS) cells, anoikis can be induced by zoledronic acid, DNA methylation inhibitors as decitabine or cyclooxygenase-2 inhibitors via PI3K/Akt pathway inhibiting β-catenin, TrkB, and E-cadherin (74–76).
Several of the aforementioned reports present indeed interesting data for a number of plausible targets concerning mitochondrial apoptosis. However, it is worth noting that in most of these cases, apoptotic analyses rely only in AnnexinV (AnnV) tests or caspase-3 activation kits, being uninformative about the precise processes involved. Although extended in the community, when the end-points of AnnV-PI tests are not carefully selected, this could lead to the misidentification of late apoptotic and necrotic cells; similarly, caspase-3 is a common final step in apoptotic cell death that does not imply a single precise activation pathway (Figure 2) (11).
The death receptor pathway
Caspase-8 is the most characteristic mediator of the “death receptor pathway” (Figure 2). In this case, the triggers of the apoptotic process are extracellular signals (mostly from the TNF family) and the initiators and mediators encounter not in the mitochondrial outer membrane but rather close to the plasma membrane (77). Besides direct stimulation of cell death, death receptors can also induce specific protein synthesis by means of the NF-κB pathway that balances and even counteracts the apoptotic signaling (78).
TRAIL is a death ligand that has been studied in several sarcomas for therapeutic purposes (79–81). TRAIL-induced apoptosis is regulated by other receptors and downstream effectors including cFLIP and the Bcl-2 family (82–84). The TRAIL receptor, death receptor 5, has been identified as a mediator of chemically induced apoptosis in RMS, synovial sarcoma and leiomyosarcoma, activating several apoptosis triggers (85–87). TNFα and FasL receptors play also a significant role in the survival/apoptotic balance with p21 as critical mediator of the anti-apoptotic effect of TNFα-induced NF-κB (88, 89). Bad, a pro-apoptotic member of the Bcl-2 gene family, has been linked to FasL induced apoptosis in ES (90). Activation of death receptors could be combined with other challenges like doxorubicin, interleukin-12, or immunotoxins (91–93). Some other TNF receptor-related proteins, like NGFR, have been proposed to be crucial in specific sarcomas (94). Thus, there is still a need for a better understanding of the role of the other cell death receptors in sarcomas.
Besides the death receptors themselves, the best strategy to enhance extrinsic apoptosis is repressing NF-κB activation. This rationale has been employed with success against ES and synovial sarcoma (95, 96). Sensitization to apoptosis has also been achieved by re-expressing caspase-8 through demethylation or gene transfer (97).
Necrosis
Necrosis, in contrast to apoptosis, has been viewed classically as a form of accidental death brought about by injury to the cell by pathogens or toxins. Despite the extended pre-judice, necrosis is more than a mere accidental death (5). Loss of plasma membrane integrity, the “cellular explosion”, is the major morphological feature and characteristic element of necrosis (Figure 1) (9, 98). Non-accidental or “regulated” necrosis has attracted a growing interest in the scientific community in the last years. Necroptosis is the best known phenotype in this group. It is induced by either the activation of death receptors or specific injuries that are followed by the recruitment of the so-called necrosome of which the principal participants are the receptor-interacting protein kinases (RIPK1 and RIPK3), which finally activate the executor MLKL (Figure 2) (99).
Necroptosis is just starting to be studied in sarcomas. It can be distinguished from apoptosis by its distinct morphology and the inability of caspase inhibitors to prevent it (10, 11). In an OS model, RIPK1-mediated necroptosis was confirmed as the main cell death mechanism involved in Shikonin therapy, as only Necrostatin-1 (an inhibitor of RIPK1) was able to induce treatment reversion (100). Basit et al. found that Obatoclax (a Bcl-2 inhibitor) treatment in RMS cell lines promoted necroptosis rather than autophagic cell death, being autophagy only a necessary event required for the necrosome assembly (101). So, it becomes clear that there is still a big room for improvement in the accurate characterization of regulated necrosis responses in anti-sarcoma therapy.
Other Scenarios for the Cell Death Drama
The long-standing dichotomy apoptosis-necrosis is in part nothing but a classification artifact. Many times the exact nature of the mechanism triggered relies simply on the intensity of the injury or on the available energy (102). Furthermore, in the cell death landscape, there are other “circumstances” worth of some additional explanation.
A classical example of “double-edged sword” is autophagy, sometimes included as a cell death mechanism, although it usually proceeds as a pro-survival process. Autophagy targets apoptotic-signaling mitochondria for isolation and degradation, thus interrupting the apoptotic outcome. Several proteins cross-link autophagy and apoptosis signaling pathways, being mTOR one of the most studied (103). As a process impacting the energy availability, autophagy also dialogs with necrotic signaling and some reports point to a close relationship with necroptotic triggering (101, 102). Again, it seems to be a question of threshold. In many cases, an excessive autophagy can lead to cell death but this death follows a mixed pattern with parallel apoptotic or necrotic phenotypes. Only when inhibition of autophagy can impede cell death and the final phenotype is considered non-apoptotic cell death, we can classify it as “autophagic cell death” (4, 102). Among the different techniques available, autophagy can be better followed by microscopy assessment of autophagosome formation (11, 104).
To our knowledge, except for some interesting report showing autophagic triggering of necroptosis in RMS (101), no instances of true autophagic cell death have been described in sarcomas yet. Indeed, its role in cancer therapy is still controversial (102). In ES and OS, the protective role of autophagy was insufficient to block apoptotic cell death when triggered by either the intrinsic or the death receptor pathways (105, 106). Autophagy has also been described to be actively removing micronuclei in OS cells, generating an interesting connection with the stabilization of cells recovering from failures during mitosis (107).
Mitotic catastrophe (MC), previously classified as a form of cell death, constitutes a crossroad that could drive cells to die with either apoptotic or necrotic features, go into senescence, or even survive (108). Again, the precise features of the final death phenotype depend on cell context and energy availability (108, 109). The clearest triggers of MC are the dysfunctions of the mitotic spindle. Those dangers are monitored by specific checkpoint proteins determining the final outcome. Thus, cells evading the mitotic arrest have an increase in chromosome instability (110). MC can be easily followed by means of microscopy observation, usually aided with fluorescent markers, video-microscopy, and cell fate imaging analysis.
Proper metaphase arrangement is required for mitosis and is a key process monitored by several checkpoint regulators (Figure 2). BubR1, involved in the mitotic spindle checkpoint, has been shown to be necessary for survival in some RMS cell lines and its knockdown promoted growth suppression and “mitotic catastrophe” but the final outcome was not elucidated (111). Plk1 is another major component of MC signaling: siRNA inhibition of Plk1 killed RMS cells and the chemical inhibitor BI 2536 induced G2/M arrest and cell death in OS cell lines (112, 113). Inhibitors of Aurora kinases block the formation of the cleavage furrow, disrupting cytokinesis, and killing leiomyosarcoma and synovial sarcoma cells (114, 115). Chk1 blockade with CEP-3891 caused an abrogation of the S and G2 checkpoints after ionizing radiation, giving rise to nuclear fragmentation as a consequence of defective chromosome segregation and promoting cell death (116). Many active drugs tested in sarcoma cells have been described to disrupt normal cell cycle. Those compounds range from small molecules or plant derivatives, to cell cycle kinase inhibitors, viral proteins etc. Several studies showed cell cycle arrest and changes in the levels of MC mediators as Survivin. For example, Keyomarsi’s group showed that combined therapy with doxorubicin and roscovitine in synovial sarcoma and leiomyosarcoma induced a synergistic increase in autophagy in addition to a marked arrest in G2/M (117). Links between MC and autophagy have also been commented previously for OS (107). In any case, it would be desirable to perform an exhaustive mitotic study or cell fate analysis together with the proper assessment of the nature of cell cycle blockade (metaphase arrest, G2 stop, or even senescence).
Cell Death Mechanisms in Anti-Sarcoma Clinical Trials
New targeted therapies linked to key cell death mechanisms are continuously being developed (118). Preferred to cytostatic alternatives, cell death induction is the goal of the vast majority of cancer treatments. And among the known mechanisms, apoptosis is the center of therapeutic developments (118). As a non-inflammatory mechanism, apoptosis is traditionally considered cleaner than necrosis, but its exact relevance in overall therapeutic success is uncertain. Necrosis, due to its pro-inflammatory nature, has been regarded as a back door for metastatic cells to escape from the primary tumor (3, 119). But, depending on the circumstances, necrosis could be effective enough to induce tumor clearance (120). Conversely, a particular apoptotic phenotype with the ability to trigger immune response against cancer cells has been described (119). Moreover, classic chemotherapeutic agents are shown to induce apoptosis by interfering with the normal cell division processes and this could lead to the triggering of MC (108, 109, 121). Induction of MC vs. direct apoptosis triggering depends of the effective drug concentration within the cells and thus, could be different among the tumor mass (122). MC drives most of the cells to major death mechanisms but opens the gates for the appearance of new stable karyotypes translating into perhaps new resistant cancer clones (108, 123, 124).
The treatment of advanced sarcomas is based on classic chemotherapeutic agents: anthracyclines and ifosfamide as first option and, after progression, other agents like gemcitabine in combination with docetaxel (or Dacarbazine) and trabectedin. The benefit of chemotherapy is well-known, but limited, because a high percentage of patients die due to the disease in approximately 1 year from diagnosis (125, 126).
In the past years, several sarcoma-focused clinical trials have evaluated the activity in monotherapy of novel drugs with known connections to a particular cell death mechanism (Table 1). So far, only two phase III trials have been reported, reflecting that targeted therapies have been mostly developed in recent years and remain in a pre-clinical stage (127–137). The first trial was focused on the mTOR signaling pathway, which links apoptosis with autophagy (102, 103). The study evaluated the role of ridaforolimus as maintenance therapy after clinical benefit to chemotherapy (133). The other trial analyzed the activity of Pazopanib (a multitargeted kinase inhibitor) in pre-treated soft-tissue sarcoma patients (136).
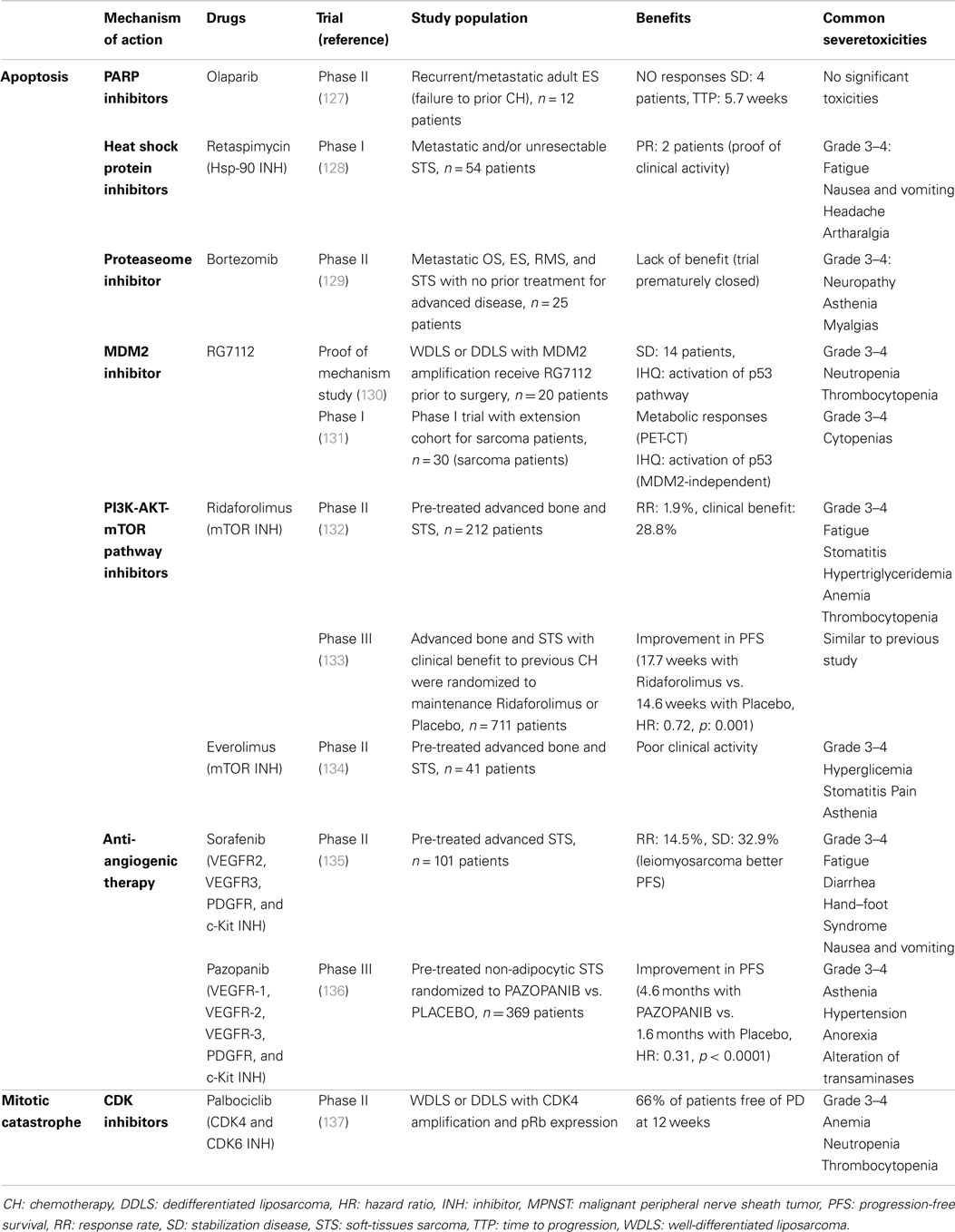
Table 1. Summary of already published clinical trials that evaluate target therapies in sarcomas, classified regarding the mechanism of action.
It is easily noticeable that many of the targets mentioned above have still not reached the clinical trial stage in sarcomas. Further research should be aimed to fill that gap by a better description of the pre-clinical effects in terms of quantity and quality (type, characterization, assessment of resistant phenotypes, etc.) of the induced cell death. A summary of the ongoing clinical trials in sarcomas are included in Table 2.
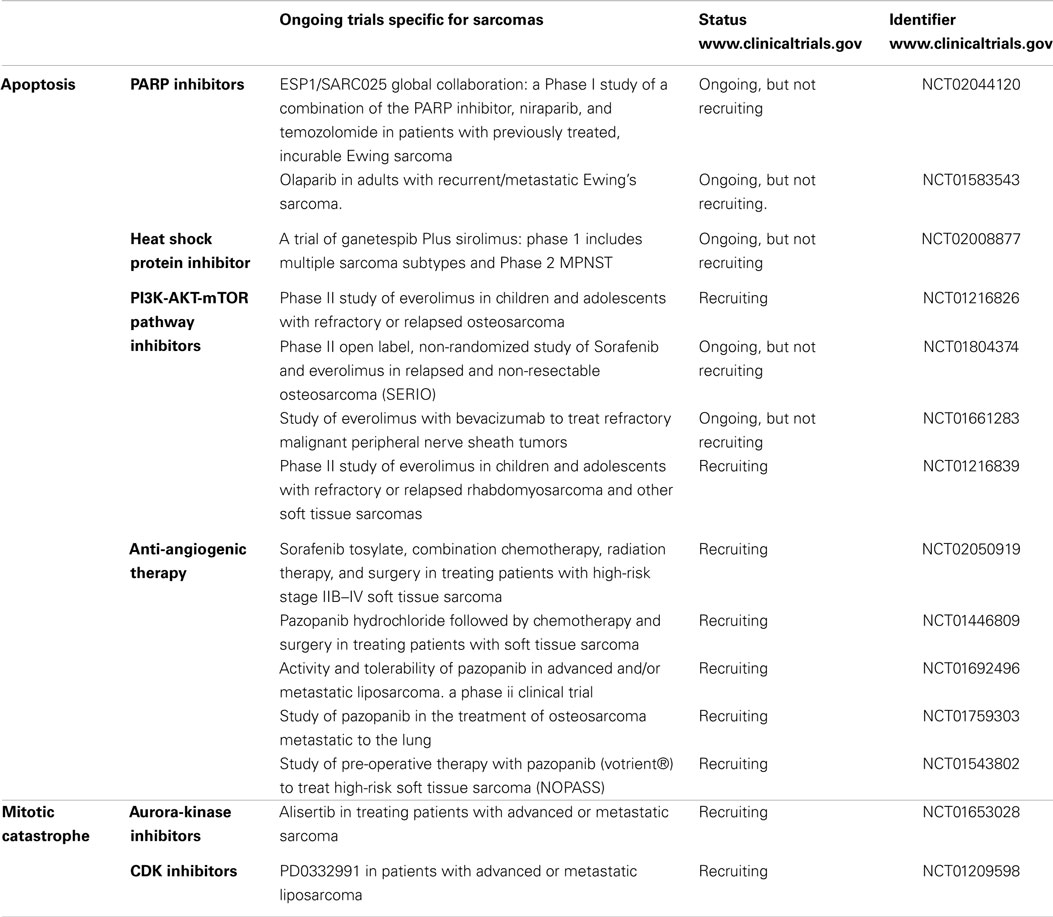
Table 2. Summary of clinical trials that are ongoing and evaluate target therapies in sarcomas, classified regarding the mechanism of action.
Concluding Remarks
As often happens with research on rare diseases, sarcoma research suffers from funding shortage and delayed implementation of technical advances. But there is also an urgent need to improve current therapeutic modalities in sarcomas and reduce their burden. Additionally, due to their heterogeneity, sarcoma models are very difficult to compare among them. Those constrains define sarcoma research today. Cell death induction is the basis of cancer therapy, but we are still far from understanding the mechanisms of cell death signaling in sarcomas. The relatively low attention paid to particular phenomena like autophagy or MC, with crucial roles in therapy success, is symptomatic that we need to get back to the laboratory benches and improve our methods (3, 118, 124). We abuse too often of indirect tests, easy to read-out in flow cytometers, or high-content analyzers. And perhaps, we rely too much in bibliographic data, not looking for the actual connections between our treatments and the specific cell death trigger.
Sarcoma research needs the implementation of a better determination of cell death mechanisms. The definition of the nature of cell death is not a vain effort as the differences in mechanisms could have tremendous consequences in terms of chemo-resistance or in immunogenic potential (108, 119, 123, 124). We need to dedicate more time to define cell death circumstances, but sometimes it seems that this attention only happens when researchers are faced with unusual/specific cell death signals (death receptors, MC, necroptosis etc.) while relying in the bulk caspase-3 or AnnV-PI kits for the rest of the occasions.
The extra work we are proposing is neither difficult nor exhausting, as it requires only to spend a little time looking “what” actually happens to our cells (and “when”). Cell death is evident to the trained eye by merely observing the cells in the cell culture room’s inverted microscope (Figure 1). Then, there are enough valuable tests, clear and easy to perform, for the major cell death pathways (138). Performed in the correct set of end-points a simple DAPI staining would serve to determine whether we are facing apoptosis, necrosis, or MC (10, 11). Therefore, we encourage researchers to perform those tests and include their results in their publications prior to embark themselves into more complex analysis about the intimacy of cell physiology. Let’s concentrate on describing better “what” is happening before moving on solving “how” it is happening.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SR-V is a MarieCurieCOFUND-BeatriuDePinòs Researcher (The European Union 7th Framework Program for RD and the Generalitat de Catalunya’s Department for Economy and Knowledge: Secretary for Universities and Research). DH-M is funded by Asociación Española contra el Cáncer-AECC. LL-T is funded by the Comissionat per a Universitats i Recerca (CUR) from Departament d’Innovació, Universitats I Empresa (DIUE) de la Generalitat de Catalunya i del Fons Social Europeu. SG-M is funded by Asociación Alba Pérez lucha contra el cáncer infantil. This work was funded by Fondo de Investigaciones Sanitarias-ISCIII (CP06/00151; CES12/021; PI11/00038; PI12/01908). Studies in CMP’s lab related to the topic of this review are supported by Fondo de Investigaciones Sanitarias-ISCIII grant PI13/00139. We apologize to those whose valuable work could not be covered in this issue for space reasons.
Abbreviations
AIF, apoptosis inducing factor; Akt, protein kinase B; Apaf-1, apoptotic protease activating factor 1; Bak, Bcl-2 homologous antagonist/killer; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; BubR1, mitotic checkpoint serine/threonine-protein kinase BUB1 beta protein; CD133, prominin-1; CD99, cluster of differentiation 99 protein; cFLIP, cellular FLICE inhibitory protein; Chk1, checkpoint kinase 1; c-Myc, cellular avian myelocytomatosis viral oncogene homolog; DAPI, 4′,6-diamidino-2-phenylindole; DRAL, downregulated in rhabdomyosarcoma LIM protein; ERG, protein encoded by erythroblast transformation-specific related gene; ERK, extracellular signal-regulated protein kinase; EWS, Ewing sarcoma RNA-binding protein; FasL, fas ligand protein; FLI1, Friend leukemia integration 1 transcription factor; FOXO1, forkhead box protein O1; HDAC1, histone deacetylase 1; IGF-1R, insulin-like growth factor 1 receptor; IHQ, immunohistochemistry; JAK, Janus kinase; MC, mitotic catastrophe; MDM2, mouse double minute 2 homolog protein; MEK, mitogen-activated protein kinase kinase; miRNA, micro RNA; MLKL, mixed lineage kinase domain-like protein; mTOR, mammalian target of rapamycin protein; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells protein; NGFR, low-affinity nerve growth factor receptor; Noxa, Phorbol-12-myristate-13-acetate-induced protein 1; oct4, octamer-binding transcription factor 4; p21, cyclin-dependent kinase inhibitor 1; PARP, poly (ADP-ribose) polymerase; PAX, protein encoded by paired box gene; PD, progression disease; PDGFR, platelet-derived growth factor receptor; PET-CT, positron emission tomography; PI, propidium iodide; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; Plk1, polo-like kinase 1; PM, plasma membrane; PR, partial response; pRb, retinoblastoma protein; puma, p53 upregulated modulator of apoptosis; RAF, raf proto-oncogene serine/threonine-protein kinase; RANK, receptor activator of nuclear factor κB; ROS, reactive oxygen species; sox2, sex determining region Y-box 2; SSX, protein encoded by synovial sarcoma X breakpoint gene; STS, soft tissue sarcoma; SYT, protein encoded by synovial sarcoma translocation on chromosome 18 gene; TNFR, tumor necrosis factor receptor; TRAIL, TNF-related apoptosis-inducing ligand; VEGFR-2, vascular endothelial growth factor receptor 2.
References
1. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ (2009) 16:3–11. doi: 10.1038/cdd.2008.150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Fulda S. Exploiting apoptosis pathways for the treatment of pediatric cancers. Pediatr Blood Cancer (2009) 53:533–6. doi:10.1002/pbc.21922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Ricci MS, Zong W. Chemotherapeutic approaches for targeting cell death pathways. Oncologist (2006) 11:342–57. doi:10.1634/theoncologist.11-4-342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ (2012) 19:107–20. doi:10.1038/cdd.2011.96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15:135–47. doi:10.1038/nrm3737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Ordóñez JL, Osuna D, García-Domínguez DJ, Amaral AT, Otero-Motta AP, Mackintosh C, et al. The clinical relevance of molecular genetics in soft tissue sarcomas. Adv Anat Pathol (2010) 17:162–81. doi:10.1097/PAP.0b013e3181d98cbf
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Martín Liberal J, Lagares-Tena L, Sáinz-Jaspeado M, Mateo-Lozano S, García Del Muro X, Tirado OM. Targeted therapies in sarcomas: challenging the challenge. Sarcoma (2012) 2012:626094. doi:10.1155/2012/626094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol (2013) 23:620–33. doi:10.1016/j.tcb.2013.07.006
9. Rello S, Stockert JC, Moreno V, Gámez A, Pacheco M, Juarranz A, et al. Morphological criteria to distinguish cell death induced by apoptotic and necrotic treatments. Apoptosis (2005) 10:201–8. doi:10.1007/s10495-005-6075-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Krysko DV, Vanden Berghe T, D’Herde K, Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods (2008) 44:205–21. doi:10.1016/j.ymeth.2007.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov (2011) 10:221–37. doi:10.1038/nrd3373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Braun F, de Carné Trécesson S, Bertin-Ciftci J, Juin P. Protect and serve: Bcl-2 proteins as guardians and rulers of cancer cell survival. Cell Cycle (2013) 12:2937–47. doi:10.4161/cc.25972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Galluzzi L, Bravo-San Pedro JM, Kroemer G. Organelle-specific initiation of cell death. Nat Cell Biol (2014) 16:728–36. doi:10.1038/ncb3005
14. Vousden KH, Lane DP. P53 in health and disease. Nat Rev Mol Cell Biol (2007) 8:275–83. doi:10.1038/nrm2147
15. Yi H, Fujimura Y, Ouchida M, Prasad DD, Rao VN, Reddy ES. Inhibition of apoptosis by normal and aberrant Fli-1 and erg proteins involved in human solid tumors and leukemias. Oncogene (1997) 14:1259–68. doi:10.1038/sj.onc.1201099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Soldatenkov VA, Trofimova IN, Rouzaut A, McDermott F, Dritschilo A, Notario V. Differential regulation of the response to DNA damage in Ewing’s sarcoma cells by ETS1 and EWS/FLI-1. Oncogene (2002) 21:2890–5. doi:10.1038/sj.onc.1205393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med (2009) 15:750–6. doi:10.1038/nm.1983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Tirado OM, MacCarthy CM, Fatima N, Villar J, Mateo-Lozano S, Notario V. Caveolin-1 promotes resistance to chemotherapy-induced apoptosis in Ewing’s sarcoma cells by modulating PKCalpha phosphorylation. Int J Cancer (2010) 126:426–36. doi:10.1002/ijc.24754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Surdez D, Benetkiewicz M, Perrin V, Han Z-Y, Pierron G, Ballet S, et al. Targeting the EWSR1-FLI1 oncogene-induced protein kinase PKC-β abolishes ewing sarcoma growth. Cancer Res (2012) 72:4494–503. doi:10.1158/0008-5472.CAN-12-0371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Stoll G, Surdez D, Tirode F, Laud K, Barillot E, Zinovyev A, et al. Systems biology of Ewing sarcoma: a network model of EWS-FLI1 effect on proliferation and apoptosis. Nucleic Acids Res (2013) 41:8853–71. doi:10.1093/nar/gkt678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Ahn EH, Mercado GE, Laé M, Ladanyi M. Identification of target genes of PAX3-FOXO1 in alveolar rhabdomyosarcoma. Oncol Rep (2013) 30:968–78. doi:10.3892/or.2013.2513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Jothi M, Mal M, Keller C, Mal AK. Small molecule inhibition of PAX3-FOXO1 through AKT activation suppresses malignant phenotypes of alveolar rhabdomyosarcoma. Mol Cancer Ther (2013) 12:2663–74. doi:10.1158/1535-7163.MCT-13-0277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Jones KB, Su L, Jin H, Lenz C, Randall RL, Underhill TM, et al. SS18-SSX2 and the mitochondrial apoptosis pathway in mouse and human synovial sarcomas. Oncogene (2013) 32:.e1–5. doi:10.1038/onc.2012.247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Yang Z, Zhang J, Cong H, Huang Z, Sun L, Liu C, et al. A retrovirus-based system to stably silence GDF-8 expression and enhance myogenic differentiation in human rhabdomyosarcoma cells. J Gene Med (2008) 10:825–33. doi:10.1002/jgm.1216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Taylor BS, DeCarolis PL, Angeles CV, Brenet F, Schultz N, Antonescu CR, et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov (2011) 1:587–97. doi:10.1158/2159-8290.CD-11-0181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Wu YV, Okada T, DeCarolis P, Socci N, O’Connor R, Geha RC, et al. Restoration of C/EBPα in dedifferentiated liposarcoma induces G2/M cell cycle arrest and apoptosis. Genes Chromosomes Cancer (2012) 51:313–27. doi:10.1002/gcc.21917
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Iida K, Fukushi J-I, Matsumoto Y, Oda Y, Takahashi Y, Fujiwara T, et al. miR-125b develops chemoresistance in Ewing sarcoma/primitive neuroectodermal tumor. Cancer Cell Int (2013) 13:21. doi:10.1186/1475-2867-13-21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Missiaglia E, Shepherd CJ, Patel S, Thway K, Pierron G, Pritchard-Jones K, et al. MicroRNA-206 expression levels correlate with clinical behaviour of rhabdomyosarcomas. Br J Cancer (2010) 102:1769–77. doi:10.1038/sj.bjc.6605684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Gits CMM, van Kuijk PF, Jonkers MBE, Boersma AWM, Smid M, van Ijcken WF, et al. MicroRNA expression profiles distinguish liposarcoma subtypes and implicate miR-145 and miR-451 as tumor suppressors. Int J Cancer (2014) 135:348–61. doi:10.1002/ijc.28694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Lee DH, Amanat S, Goff C, Weiss LM, Said JW, Doan NB, et al. Overexpression of miR-26a-2 in human liposarcoma is correlated with poor patient survival. Oncogenesis (2013) 2:e47. doi:10.1038/oncsis.2013.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Kovar H, Pospisilova S, Jug G, Printz D, Gadner H. Response of Ewing tumor cells to forced and activated p53 expression. Oncogene (2003) 22:3193–204. doi:10.1038/sj.onc.1206391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Li Y, Li X, Fan G, Fukushi J-I, Matsumoto Y, Iwamoto Y, et al. Impairment of p53 acetylation by EWS-Fli1 chimeric protein in Ewing family tumors. Cancer Lett (2012) 320:14–22. doi:10.1016/j.canlet.2012.01.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Hecker RM, Amstutz RA, Wachtel M, Walter D, Niggli FK, Schäfer BW. p21 downregulation is an important component of PAX3/FKHR oncogenicity and its reactivation by HDAC inhibitors enhances combination treatment. Oncogene (2010) 29:3942–52. doi:10.1038/onc.2010.145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Su L, Sampaio AV, Jones KB, Pacheco M, Goytain A, Lin S, et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell (2012) 21:333–47. doi:10.1016/j.ccr.2012.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Pishas KI, Al-Ejeh F, Zinonos I, Kumar R, Evdokiou A, Brown MP, et al. Nutlin-3a is a potential therapeutic for ewing sarcoma. Clin Cancer Res (2011) 17:494–504. doi:10.1158/1078-0432.CCR-10-1587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Miyachi M, Kakazu N, Yagyu S, Katsumi Y, Tsubai-Shimizu S, Kikuchi K, et al. Restoration of p53 pathway by nutlin-3 induces cell cycle arrest and apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res (2009) 15:4077–84. doi:10.1158/1078-0432.CCR-08-2955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Aryee DNT, Niedan S, Ban J, Schwentner R, Muehlbacher K, Kauer M, et al. Variability in functional p53 reactivation by PRIMA-1(Met)/APR-246 in Ewing sarcoma. Br J Cancer (2013) 109:2696–704. doi:10.1038/bjc.2013.635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Leiser AL, Anderson SE, Nonaka D, Chuai S, Olshen AB, Chi DS, et al. Apoptotic and cell cycle regulatory markers in uterine leiomyosarcoma. Gynecol Oncol (2006) 101:86–91. doi:10.1016/j.ygyno.2005.09.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Taylor AC, Schuster K, McKenzie PP, Harris LC. Differential cooperation of oncogenes with p53 and Bax to induce apoptosis in rhabdomyosarcoma. Mol Cancer (2006) 5:53. doi:10.1186/1476-4598-5-53
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Cheney MD, McKenzie PP, Volk EL, Fan L, Harris LC. MDM2 displays differential activities dependent upon the activation status of NFkappaB. Cancer Biol Ther (2008) 7:38–44. doi:10.4161/cbt.7.1.5125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Scholl FA, McLoughlin P, Ehler E, de Giovanni C, Schäfer BW. DRAL is a p53-responsive gene whose four and a half LIM domain protein product induces apoptosis. J Cell Biol (2000) 151:495–506. doi:10.1083/jcb.151.3.495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Armistead PM, Salganick J, Roh JS, Steinert DM, Patel S, Munsell M, et al. Expression of receptor tyrosine kinases and apoptotic molecules in rhabdomyosarcoma: correlation with overall survival in 105 patients. Cancer (2007) 110:2293–303. doi:10.1002/cncr.23038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Margue CM, Bernasconi M, Barr FG, Schäfer BW. Transcriptional modulation of the anti-apoptotic protein BCL-XL by the paired box transcription factors PAX3 and PAX3/FKHR. Oncogene (2000) 19:2921–9. doi:10.1038/sj.onc.1203607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Astolfi A, Nanni P, Landuzzi L, Ricci C, Nicoletti G, Rossi I, et al. An anti-apoptotic role for NGF receptors in human rhabdomyosarcoma. Eur J Cancer (2001) 37:1719–25. doi:10.1016/S0959-8049(01)00190-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Mayeenuddin LH, Yu Y, Kang Z, Helman LJ, Cao L. Insulin-like growth factor 1 receptor antibody induces rhabdomyosarcoma cell death via a process involving AKT and Bcl-x(L). Oncogene (2010) 29:6367–77. doi:10.1038/onc.2010.364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ehnman M, Missiaglia E, Folestad E, Selfe J, Strell C, Thway K, et al. Distinct effects of ligand-induced PDGFRα and PDGFRβ signaling in the human rhabdomyosarcoma tumor cell and stroma cell compartments. Cancer Res (2013) 73:2139–49. doi:10.1158/0008-5472.CAN-12-1646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Nakamura K, Abarzua F, Hongo A, Kodama J, Nasu Y, Kumon H, et al. Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2) are potential targets in uterine leiomyosarcoma. Int J Oncol (2010) 37:605–14. doi:10.3892/ijo_00000709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Makino K, Kawamura K, Sato W, Kawamura N, Fujimoto T, Terada Y. Inhibition of uterine sarcoma cell growth through suppression of endogenous tyrosine kinase B signaling. PLoS One (2012) 7:e41049. doi:10.1371/journal.pone.0041049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Pula G, Pistilli A, Montagnoli C, Stabile AM, Rambotti MG, Rende M. The tricyclic antidepressant amitriptyline is cytotoxic to HTB114 human leiomyosarcoma and induces p75(NTR)-dependent apoptosis. Anticancer Drugs (2013) 24:899–910. doi:10.1097/CAD.0b013e328364312f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Michels S, Trautmann M, Sievers E, Kindler D, Huss S, Renner M, et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer Res (2013) 73:2518–28. doi:10.1158/0008-5472.CAN-12-3023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Cen L, Hsieh F-C, Lin H-J, Chen C-S, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer (2007) 97:785–91. doi:10.1038/sj.bjc.6603952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Yan S, Li Z, Thiele CJ. Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in Neuroblastoma and Pediatric Sarcomas In vitro and In vivo. Oncotarget (2013) 4:433–45.
53. Fulda S, Jeremias I, Pietsch T, Debatin KM. Betulinic acid: a new chemotherapeutic agent in the treatment of neuroectodermal tumors. Klin Pädiatrie (1999) 211:319–22. doi:10.1055/s-2008-1043808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Seitz G, Warmann SW, Armeanu S, Heitmann H, Ruck P, Hoffman RM, et al. In vitro photodynamic therapy of childhood rhabdomyosarcoma. Int J Oncol (2007) 30:615–20. doi:10.3892/ijo.30.3.615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Herrero Martín D, Boro A, Schäfer BW. Cell-based small-molecule compound screen identifies fenretinide as potential therapeutic for translocation-positive rhabdomyosarcoma. PLoS One (2013) 8:e55072. doi:10.1371/journal.pone.0055072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Nakamura T, Tanaka K, Matsunobu T, Okada T, Nakatani F, Sakimura R, et al. The mechanism of cross-resistance to proteasome inhibitor bortezomib and overcoming resistance in Ewing’s family tumor cells. Int J Oncol (2007) 31:803–11. doi:10.3892/ijo.31.4.803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Bersani F, Taulli R, Accornero P, Morotti A, Miretti S, Crepaldi T, et al. Bortezomib-mediated proteasome inhibition as a potential strategy for the treatment of rhabdomyosarcoma. Eur J Cancer (2008) 44:876–84. doi:10.1016/j.ejca.2008.02.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Lesko E, Gozdzik J, Kijowski J, Jenner B, Wiecha O, Majka M. HSP90 antagonist, geldanamycin, inhibits proliferation, induces apoptosis and blocks migration of rhabdomyosarcoma cells in vitro and seeding into bone marrow in vivo. Anticancer Drugs (2007) 18:1173–81. doi:10.1097/CAD.0b013e3282ef532d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Dimitroulakos J, Ye LY, Benzaquen M, Moore MJ, Kamel-Reid S, Freedman MH, et al. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res (2001) 7:158–67.
60. Sun Y, Wang H, Lin F, Hua J, Zhou G. Inhibition of proliferation and gene expression regulation by (-)-epigallocatechin-3-gallate in human synovial sarcoma cells. Med Oncol (2011) 28:1463–8. doi:10.1007/s12032-010-9560-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Ramírez-Peinado S, Alcázar-Limones F, Lagares-Tena L, El Mjiyad N, Caro-Maldonado A, Tirado OM, et al. 2-deoxyglucose induces Noxa-dependent apoptosis in alveolar rhabdomyosarcoma. Cancer Res (2011) 71:6796–806. doi:10.1158/0008-5472.CAN-11-0759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Holt SV, Brookes KE, Dive C, Makin GWJ. Down-regulation of XIAP by AEG35156 in paediatric tumour cells induces apoptosis and sensitises cells to cytotoxic agents. Oncol Rep (2011) 25:1177–81. doi:10.3892/or.2011.1167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta (2013) 1833:3481–98. doi:10.1016/j.bbamcr.2013.06.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Crompton BD, Carlton AL, Thorner AR, Christie AL, Du J, Calicchio ML, et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res (2013) 73:2873–83. doi:10.1158/0008-5472.CAN-12-1944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Díaz-Montero CM, McIntyre BW. Acquisition of anoikis resistance in human osteosarcoma cells. Eur J Cancer (2003) 39:2395–402. doi:10.1016/S0959-8049(03)00575-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Lin D, Feng J, Chen W. Bcl-2 and caspase-8 related anoikis resistance in human osteosarcoma MG-63 cells. Cell Biol Int (2008) 32:1199–206. doi:10.1016/j.cellbi.2008.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Scotlandi K, Zuntini M, Manara MC, Sciandra M, Rocchi A, Benini S, et al. CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene (2007) 26:6604–18. doi:10.1038/sj.onc.1210481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Akiyama T, Choong PFM, Dass CRRANK-. Fc inhibits malignancy via inhibiting ERK activation and evoking caspase-3-mediated anoikis in human osteosarcoma cells. Clin Exp Metastasis (2010) 27:207–15. doi:10.1007/s10585-010-9319-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Kang H-G, Jenabi JM, Zhang J, Keshelava N, Shimada H, May WA, et al. E-cadherin cell-cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res (2007) 67:3094–105. doi:10.1158/0008-5472.CAN-06-3259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Scotlandi K, Maini C, Manara MC, Benini S, Serra M, Cerisano V, et al. Effectiveness of insulin-like growth factor I receptor antisense strategy against Ewing’s sarcoma cells. Cancer Gene Ther (2002) 9:296–307. doi:10.1038/sj.cgt.7700442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Sun Y, Gao D, Liu Y, Huang J, Lessnick S, Tanaka S. IGF2 is critical for tumorigenesis by synovial sarcoma oncoprotein SYT-SSX1. Oncogene (2006) 25:1042–52. doi:10.1038/sj.onc.1209143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Mendoza-Naranjo A, El-Naggar A, Wai DH, Mistry P, Lazic N, Ayala FRR, et al. Tavares Guerreiro Fregnani JH, et al. EMBO Mol Med (2013) 5:1019–34. doi:10.1002/emmm.201202343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Walter D, Satheesha S, Albrecht P, Bornhauser BC, D’Alessandro V, Oesch SM, et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS One (2011) 6:e19506. doi:10.1371/journal.pone.0019506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Evdokiou A, Labrinidis A, Bouralexis S, Hay S, Findlay DM. Induction of cell death of human osteogenic sarcoma cells by zoledronic acid resembles anoikis. Bone (2003) 33:216–28. doi:10.1016/S8756-3282(03)00223-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Al-Romaih K, Somers GR, Bayani J, Hughes S, Prasad M, Cutz J-C, et al. Modulation by decitabine of gene expression and growth of osteosarcoma U2OS cells in vitro and in xenografts: identification of apoptotic genes as targets for demethylation. Cancer Cell Int (2007) 7:14. doi:10.1186/1475-2867-7-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Liu B, Qu L, Yang Z, Tao H. Cyclooxygenase-2 inhibitors induce anoikis in osteosarcoma via PI3K/Akt pathway. Med Hypotheses (2012) 79:98–100. doi:10.1016/j.mehy.2012.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Oikonomou E, Pintzas A. The TRAIL of oncogenes to apoptosis. Biofactors (2013) 39:343–54. doi:10.1002/biof.1112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi:10.1186/1476-4598-12-86
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Wang S, Ren W, Liu J, Lahat G, Torres K, Lopez G, et al. TRAIL and doxorubicin combination induces proapoptotic and antiangiogenic effects in soft tissue sarcoma in vivo. Clin Cancer Res (2010) 16:2591–604. doi:10.1158/1078-0432.CCR-09-2443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Gasparini C, Vecchi Brumatti L, Monasta L, Zauli G. TRAIL-based therapeutic approaches for the treatment of pediatric malignancies. Curr Med Chem (2013) 20:2254–71. doi:10.2174/0929867311320170009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Karlisch C, Harati K, Chromik AM, Bulut D, Klein-Hitpass L, Goertz O, et al. Effects of TRAIL and taurolidine on apoptosis and proliferation in human rhabdomyosarcoma, leiomyosarcoma and epithelioid cell sarcoma. Int J Oncol (2013) 42:945–56. doi:10.3892/ijo.2013.1772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Petak I, Douglas L, Tillman DM, Vernes R, Houghton JA. Pediatric rhabdomyosarcoma cell lines are resistant to Fas-induced apoptosis and highly sensitive to TRAIL-induced apoptosis. Clin Cancer Res (2000) 6:4119–27.
83. Cheong H-J, Lee KS, Woo IS, Won J-H, Byun JH. Up-regulation of the DR5 expression by proteasome inhibitor MG132 augments TRAIL-induced apoptosis in soft tissue sarcoma cell lines. Cancer Res Treat (2011) 43:124–30. doi:10.4143/crt.2011.43.2.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Nair P, Lu M, Petersen S, Ashkenazi A. Apoptosis initiation through the cell-extrinsic pathway. Methods Enzymol (2014) 544:99–128. doi:10.1016/B978-0-12-417158-9.00005-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Sakai T, Eskander RN, Guo Y, Kim KJ, Mefford J, Hopkins J, et al. Flavokawain B, a kava chalcone, induces apoptosis in synovial sarcoma cell lines. J Orthop Res (2012) 30:1045–50. doi:10.1002/jor.22050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Eskander RN, Randall LM, Sakai T, Guo Y, Hoang B, Zi X. Flavokawain B, a novel, naturally occurring chalcone, exhibits robust apoptotic effects and induces G2/M arrest of a uterine leiomyosarcoma cell line. J Obstet Gynaecol Res (2012) 38:1086–94. doi:10.1111/j.1447-0756.2011.01841.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Kang Z, Chen J-J, Yu Y, Li B, Sun S-Y, Zhang B, et al. Drozitumab, a human antibody to death receptor 5, has potent antitumor activity against rhabdomyosarcoma with the expression of caspase-8 predictive of response. Clin Cancer Res (2011) 17:3181–92. doi:10.1158/1078-0432.CCR-10-2874
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Javelaud D, Wietzerbin J, Delattre O, Besançon F. Induction of p21Waf1/Cip1 by TNFalpha requires NF-kappaB activity and antagonizes apoptosis in Ewing tumor cells. Oncogene (2000) 19:61–8. doi:10.1038/sj.onc.1203246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Mitsiades N, Poulaki V, Leone A, Tsokos M. Fas-mediated apoptosis in Ewing’s sarcoma cell lines by metalloproteinase inhibitors. J Natl Cancer Inst (1999) 91:1678–84. doi:10.1093/jnci/91.19.1678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Lee B, Galli S, Tsokos M. Sensitive Ewing sarcoma and neuroblastoma cell lines have increased levels of BAD expression and decreased levels of BAR expression compared to resistant cell lines. Cancer Lett (2007) 247:110–4. doi:10.1016/j.canlet.2006.03.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Ricci C, Polito L, Nanni P, Landuzzi L, Astolfi A, Nicoletti G, et al. HER/erbB receptors as therapeutic targets of immunotoxins in human rhabdomyosarcoma cells. J Immunother (2002) 25:314–23. doi:10.1097/00002371-200207000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Zhou Z, Lafleur EA, Koshkina NV, Worth LL, Lester MS, Kleinerman ES. Interleukin-12 up-regulates Fas expression in human osteosarcoma and Ewing’s sarcoma cells by enhancing its promoter activity. Mol Cancer Res (2005) 3:685–91. doi:10.1158/1541-7786.MCR-05-0092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Kuçi S, Rettinger E, Voss B, Weber G, Stais M, Kreyenberg H, et al. Efficient lysis of rhabdomyosarcoma cells by cytokine-induced killer cells: implications for adoptive immunotherapy after allogeneic stem cell transplantation. Haematologica (2010) 95:1579–86. doi:10.3324/haematol.2009.019885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Fanburg-Smith JC, Miettinen M. Low-affinity nerve growth factor receptor (p75) in dermatofibrosarcoma protuberans and other nonneural tumors: a study of 1,150 tumors and fetal and adult normal tissues. Hum Pathol (2001) 32:976–83. doi:10.1053/hupa.2001.27602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Horiuchi K, Morioka H, Nishimoto K, Suzuki Y, Susa M, Nakayama R, et al. Growth suppression and apoptosis induction in synovial sarcoma cell lines by a novel NF-kappaB inhibitor, dehydroxymethylepoxyquinomicin (DHMEQ). Cancer Lett (2008) 272:336–44. doi:10.1016/j.canlet.2008.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Mathieu J, Besançon F. Clinically tolerable concentrations of arsenic trioxide induce p53-independent cell death and repress NF-kappa B activation in Ewing sarcoma cells. Int J Cancer (2006) 119:1723–7. doi:10.1002/ijc.21970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Fulda S, Küfer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene (2001) 20:5865–77. doi:10.1038/sj.onc.1204750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci (2007) 32:37–43. doi:10.1016/j.tibs.2006.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer (2013) 13:580. doi:10.1186/1471-2407-13-580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ (2013) 20:1161–73. doi:10.1038/cdd.2013.45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta (2013) 1833:3448–59. doi:10.1016/j.bbamcr.2013.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol (2014) 15:81–94. doi:10.1038/nrm3735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol (2010) 221:117–24. doi:10.1002/path.2694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Lorin S, Borges A, Ribeiro Dos Santos L, Souquère S, Pierron G, Ryan KM, et al. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res (2009) 69:6924–31. doi:10.1158/0008-5472.CAN-09-1270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Souquère S, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem (2006) 281:30373–82. doi:10.1074/jbc.M602097200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Mariño G, et al. Autophagic removal of micronuclei. Cell Cycle (2012) 11:170–6. doi:10.4161/cc.11.1.18564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Vitale I, Galluzzi L, Senovilla L, Criollo A, Jemaà M, Castedo M, et al. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ (2011) 18:1403–13. doi:10.1038/cdd.2010.145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Blank M, Shiloh Y. Programs for cell death: apoptosis is only one way to go. Cell Cycle (2007) 6:686–95. doi:10.4161/cc.6.6.3990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Morales AG, Brassesco MS, Pezuk JA, Oliveira JC, Montaldi AP, Sakamoto-Hojo ET, et al. BI 2536-mediated PLK1 inhibition suppresses HOS and MG-63 osteosarcoma cell line growth and clonogenicity. Anticancer Drugs (2011) 22:995–1001. doi:10.1097/CAD.0b013e32834a16d4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Wan X, Yeung C, Kim SY, Dolan JG, Ngo VN, Burkett S, et al. Identification of FoxM1/Bub1b signaling pathway as a required component for growth and survival of rhabdomyosarcoma. Cancer Res (2012) 72:5889–99. doi:10.1158/0008-5472.CAN-12-1991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Hu K, Lee C, Qiu D, Fotovati A, Davies A, Abu-Ali S, et al. Small interfering RNA library screen of human kinases and phosphatases identifies polo-like kinase 1 as a promising new target for the treatment of pediatric rhabdomyosarcomas. Mol Cancer Ther (2009) 8:3024–35. doi:10.1158/1535-7163.MCT-09-0365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Lund-Andersen C, Patzke S, Nähse-Kumpf V, Syljuåsen RG. PLK1-inhibition can cause radiosensitization or radioresistance dependent on the treatment schedule. Radiother Oncol (2014) 110:355–61. doi:10.1016/j.radonc.2013.12.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Shan W, Akinfenwa PY, Savannah KB, Kolomeyevskaya N, Laucirica R, Thomas DG, et al. A small-molecule inhibitor targeting the mitotic spindle checkpoint impairs the growth of uterine leiomyosarcoma. Clin Cancer Res (2012) 18:3352–65. doi:10.1158/1078-0432.CCR-11-3058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Arai R, Tsuda M, Watanabe T, Ose T, Obuse C, Maenaka K, et al. Simultaneous inhibition of Src and aurora kinases by SU6656 induces therapeutic synergy in human synovial sarcoma growth, invasion and angiogenesis in vivo. Eur J Cancer (2012) 48:2417–30. doi:10.1016/j.ejca.2011.12.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Syljuåsen RG, Sørensen CS, Nylandsted J, Lukas C, Lukas J, Bartek J. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation. Cancer Res (2004) 64:9035–40. doi:10.1158/0008-5472.CAN-04-2434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Lambert LA, Qiao N, Hunt KK, Lambert DH, Mills GB, Meijer L, et al. Autophagy: a novel mechanism of synergistic cytotoxicity between doxorubicin and roscovitine in a sarcoma model. Cancer Res (2008) 68:7966–74. doi:10.1158/0008-5472.CAN-08-1333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol (2006) 3:388–98. doi:10.1038/ncponc0538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol (2009) 9:353–63. doi:10.1038/nri2545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Guerriero JL, Ditsworth D, Fan Y, Zhao F, Crawford HC, Zong W-X. Chemotherapy induces tumor clearance independent of apoptosis. Cancer Res (2008) 68:9595–600. doi:10.1158/0008-5472.CAN-08-2452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ (2012) 19:369–77. doi:10.1038/cdd.2011.197
122. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6:583–92. doi:10.1038/nrc1893
123. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol (2009) 10:478–87. doi:10.1038/nrm2718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet (2012) 13:795–806. doi:10.1038/nrg3317
125. Italiano A, Mathoulin-Pelissier S, Cesne AL, Terrier P, Bonvalot S, Collin F, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer (2011) 117:1049–54. doi:10.1002/cncr.25538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Billingsley KG, Lewis JJ, Leung DH, Casper ES, Woodruff JM, Brennan MF. Multifactorial analysis of the survival of patients with distant metastasis arising from primary extremity sarcoma. Cancer (1999) 85:389–95. doi:10.1002/(SICI)1097-0142(19990115)85:2<389::AID-CNCR17>3.3.CO;2-A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Choy E, Butrynski J, Harmon D, Morgan J, George S, Wagner A, et al. Translation of preclinical predictive sensitivity of Ewing sarcoma to PARP inhibition: phase II study of olaparib in adult patients with recurrent/metastatic Ewing sarcoma following failure of prior chemotherapy. Proceedings of the 104th Annual Meeting of the American Association for Cancer Research. Washington DC (2013). LB–174.
128. Wagner AJ, Chugh R, Rosen LS, Morgan JA, George S, Gordon M, et al. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin Cancer Res (2013) 19:6020–9. doi:10.1158/1078-0432.CCR-13-0953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Maki RG, Kraft AS, Scheu K, Yamada J, Wadler S, Antonescu CR, et al. A multicenter phase II study of bortezomib in recurrent or metastatic sarcomas. Cancer (2005) 103:1431–8. doi:10.1002/cncr.20968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Ray-Coquard I, Blay J-Y, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol (2012) 13:1133–40. doi:10.1016/S1470-2045(12)70474-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Kurzock R, Blay J-Y, Bui-Nguyen B, Wagner A, Maki RG, Schwartz GK, et al. A phase I study of MDM2 antagonist RG7112 in patients (pts) with relapsed/refractory solid tumors. Proceedings of the 2012 ASCO Meeting. Chicago, IL (2012). e13600.
132. Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D’Amato GZ, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol (2012) 30:78–84. doi:10.1200/JCO.2011.35.6329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Demetri GD, Chawla SP, Ray-Coquard I, Le Cesne A, Staddon AP, Milhem MM, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol (2013) 31:2485–92. doi:10.1200/JCO.2012.45.5766
134. Yoo C, Lee J, Rha SY, Park KH, Kim TM, Kim YJ, et al. Multicenter phase II study of everolimus in patients with metastatic or recurrent bone and soft-tissue sarcomas after failure of anthracycline and ifosfamide. Invest New Drugs (2013) 31:1602–8. doi:10.1007/s10637-013-0028-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Santoro A, Comandone A, Basso U, Soto Parra H, De Sanctis R, Stroppa E, et al. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann Oncol (2013) 24:1093–8. doi:10.1093/annonc/mds607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Van der Graaf WTA, Blay J-Y, Chawla SP, Kim D-W, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2012) 379:1879–86. doi:10.1016/S0140-6736(12)60651-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol (2013) 31:2024–8. doi:10.1200/JCO.2012.46.5476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Rello-Varona S, Herrero-Martin D, Lopez-Alemany R, Munoz-Pinedo C, Tirado OM. “(Not) All (Dead) Things Share the Same Breath”: identification of cell death mechanisms in anticancer therapy. Cancer Res (2015) 75(6):913–7. doi:10.1158/0008-5472.CAN-14-3494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cell death mechanisms, sarcoma, translocation-bearing sarcomas, apoptosis, necrosis, autophagic cell death, mitotic catastrophe
Citation: Rello-Varona S, Herrero-Martín D, Lagares-Tena L, López-Alemany R, Mulet-Margalef N, Huertas-Martínez J, Garcia-Monclús S, García del Muro X, Muñoz-Pinedo C and Tirado OM (2015) The importance of being dead: cell death mechanisms assessment in anti-sarcoma therapy. Front. Oncol. 5:82. doi: 10.3389/fonc.2015.00082
Received: 16 January 2015; Accepted: 21 March 2015;
Published online: 07 April 2015.
Edited by:
Thomas Grunewald, Ludwig-Maximilians-Universität München, GermanyCopyright: © 2015 Rello-Varona, Herrero-Martín, Lagares-Tena, López-Alemany, Mulet-Margalef, Huertas-Martínez, Garcia-Monclús, García del Muro, Muñoz-Pinedo and Tirado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oscar Martínez Tirado, Grup de Recerca en Sarcomas, Institut de Investigaciò Biomédica de Bellvitge, Hospital Duran i Reynals 3a planta, Gran Via de l’Hospitalet 199, L’Hospitalet de Llobregat, Barcelona 08908, Spain e-mail:b21hcnRpbmV6QGlkaWJlbGwuY2F0
Dedication: To the memory of Alba Pérez.