 Andréia Vasconcelos-dos-Santos1
Andréia Vasconcelos-dos-Santos1 Isadora A. Oliveira1
Isadora A. Oliveira1 Miguel Clodomiro Lucena1
Miguel Clodomiro Lucena1 Natalia Rodrigues Mantuano1
Natalia Rodrigues Mantuano1 Stephen A. Whelan2
Stephen A. Whelan2 Wagner Barbosa Dias1
Wagner Barbosa Dias1 Adriane Regina Todeschini1*
Adriane Regina Todeschini1*- 1Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brasil
- 2Department of Biochemistry, Cardiovascular Proteomics Center, Boston University School of Medicine, Boston, MA, USA
Cancer cells depend on altered metabolism and nutrient uptake to generate and keep the malignant phenotype. The hexosamine biosynthetic pathway is a branch of glucose metabolism that produces UDP-GlcNAc and its derivatives, UDP-GalNAc and CMP-Neu5Ac and donor substrates used in the production of glycoproteins and glycolipids. Growing evidence demonstrates that alteration of the pool of activated substrates might lead to different glycosylation and cell signaling. It is already well established that aberrant glycosylation can modulate tumor growth and malignant transformation in different cancer types. Therefore, biosynthetic machinery involved in the assembly of aberrant glycans are becoming prominent targets for anti-tumor drugs. This review describes three classes of glycosylation, O-GlcNAcylation, N-linked, and mucin type O-linked glycosylation, involved in tumor progression, their biosynthesis and highlights the available inhibitors as potential anti-tumor drugs.
Introduction
Glycans constitute the most complex and abundant group of molecules in living organisms. Besides playing important roles in energy storage and supply, they often serve as essential biosynthetic precursors or structural elements needed to sustain all forms of life. The complex glycans are frequently attached to proteins, forming glycoproteins and proteoglycans, or to lipids, forming glycosphingolipids and glycosylphosphatidylinositol anchors (Figure 1a). The majority of glycoconjugates are expressed on the cell surface, where they form a thick layer known as glycocalyx. Glycans can also be secreted to the extracellular medium in order to be incorporated into the extracellular matrix (ECM). Such location places glycoconjugates as major players in cell-to-cell interactions and motility. In addition, glycosylation is analogous to phosphorylation in that it can be found on many, cytoplasmic, nuclear and mitochondrial proteins (Figure 1b). It is a dynamic post-translational modification (PTM) and it regulates many cellular functions as well (1).
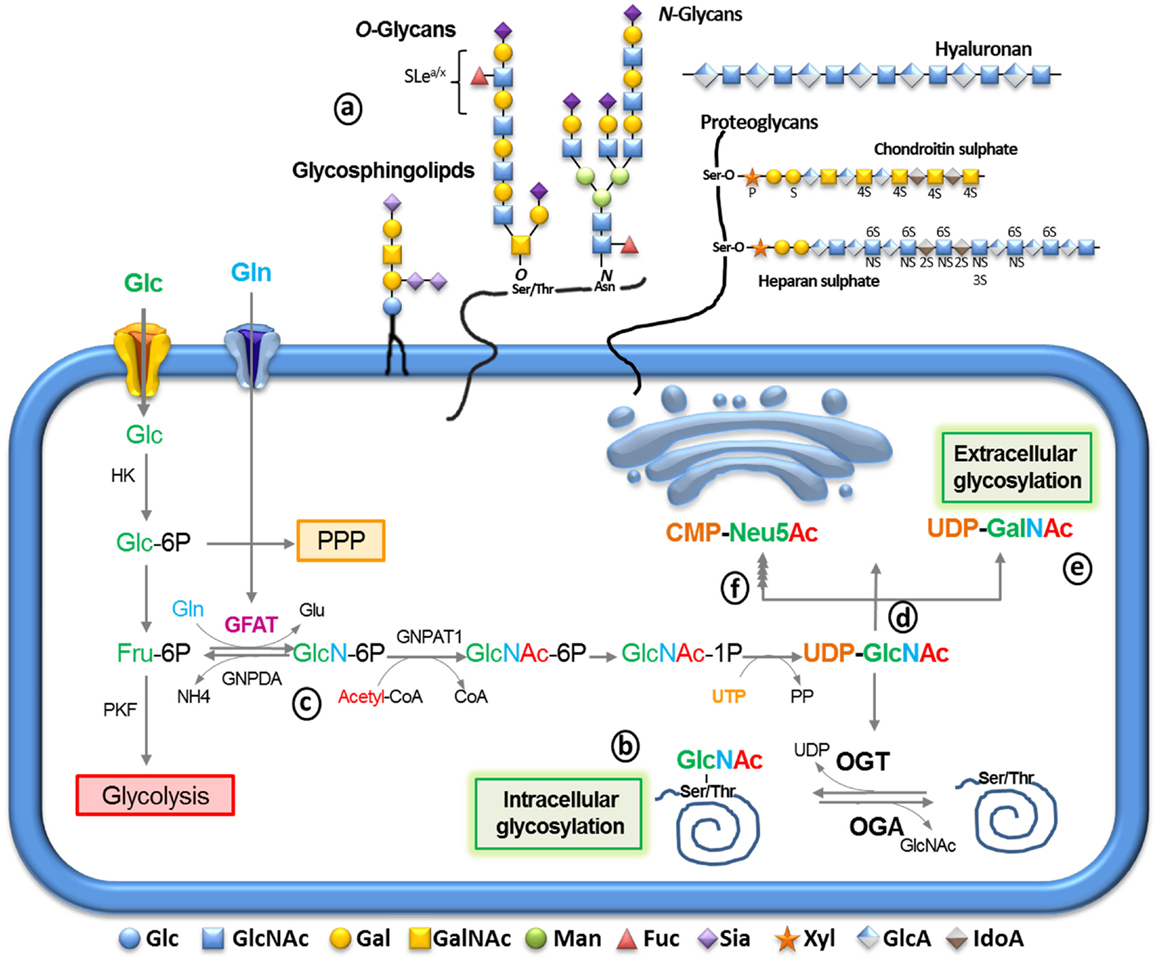
Figure 1. The synthesis of glycoconjugates from glucose through the hexosamine biosynthetic pathway (HBP). After glucose entry into the cell via the glucose transport, it is phosphorylated into glucose-6-phosphate (Glc-6P) by hexokinase (HK), mainly proceeding into glycolysis through conversion into fructose-6-phosphate (Fru-6P) by Glc-6P isomerase. Alternatively, Glc-6P may be utilized by the pentose phosphate pathway (PPP). Glc-6P can also be diverted to glucosamine-6-phosphate by the rate-limiting enzyme glutamine:fructose-6-phosphate amidotransferase (GFAT) (c). The end product of this pathway, uridinediphosphoglucose-N-acetylglucosamine (UDP-GlcNAc) (d) serves to build extracellular glycoconjugates (a), as well as, it is used for the biosynthesis of intracellular O-linked glycoproteins (b) by the enzyme O-GlcNAc transferase. Alternatively, UDP-GlcNAc can undergo epimerization to generate UDP-GalNAc (e) and CMP-Neu5Ac (f) which can be used for the extracellular biosynthesis of glycoproteins and glycolipids (a) UDP-GlcNAc and its derivatives are extremely responsive to variations in cell nutrients as its synthesis depends on products of the metabolism of glucose (green), amino acids (blue), fatty acids (red), and nucleotides (orange). Thus, glycosylation can serve as a reporter for the functional status of multiple pathways and considered a metabolic sensor.
Glycoconjugates participate in many key biological processes including cellular adhesion, migration, growth, differentiation, signal transduction, receptor activation, immune response modulation, quality control of protein folding, and host–pathogen interactions (2–4).
Glycans play several roles in different steps of tumor progression regulating tumor proliferation, invasion, metastasis, and angiogenesis (5). Therefore, aberrant glycosylation exhibits prominent candidates for cancer biomarkers, and their biosynthetic machinery have become targets for designing and synthesizing anti-tumors drugs.
Glycan structures do not depend only on genes, but also on the activities of glycosyltransferases and glycosidases and the availability of the donor substrates at the needed location. The donor substrates are derived from extracellular glucose (Glc) and from intracellular degradation of glycoconjugates in lysosomes, through the action of glycosidases and others enzymes as epimerases. Tumor cells have altered Glc metabolism, producing ATP primarily through glycolysis even under normoxic condition, thereby upregulating the Glc uptake approximately 10 times more than adjacent normal tissue (6) in order to sustain a highly demanding metabolism. This metabolic shift was termed “Warburg effect” and is critical for supporting the malignant phenotype (6). The high rate of glycolytic flux is a central metabolic hallmark of tumors and cancer cells support this rate by increasing the expression of Glc transporters (Glut) (7). This phenomenon of elevated Glc uptake has been clinically exploited to detect tumor cells by positron emission tomography (PET) scan (8). In addition, a stable Glc analog 2-deoxi-d-glucose (2-DG) has been suggested as a tumor therapeutic drug (9, 10). Various small molecule inhibitors of the glycolytic pathway have been used effectively in the past to halt the progression of cancer (11–13). The 2DG is a well-known glycolytic inhibitor, which inhibits the key glycolytic enzyme hexokinase. Recently, Muley et al. (10) evaluated the additional cellular effects of 2DG, apart from inhibiting glycolysis. Their findings indicate that 2DG increases the expression of p21 and p53 in colorectal cancer cell lines leading to cell cycle arrest at the G0/G1 phase.
Hexosamine Biosynthetic Pathway
Upon entering cells, Glc is rapidly converted to glucose-6-phosphate (Glc-6P) by hexokinase (Figure 1) and then to fructose-6-phosphate (Fru-6P) by glucose-6-phosphate isomerase. Despite the majority of Fru-6P being metabolized by phosphofructokinase (PFK) entering in to glycolysis, approximately 2–5% of Glc influx is directed by the hexosamine biosynthetic pathway (HBP) (14). The first and rate limiting step of the HBP is catalyzed by glutamine, fructose-6-phosphate amidotransferase (GFAT), which converts Fru-6P to glucosamine-6-phosphate (GlcN-6P) using glutamine (Gln) as an amine donor (Figure 1c).
GlcN-6P is further metabolized to uridine-5'-diphosphate-N-acetylglucosamine (UDP-GlcNAc) that serves as a major substrate for several kinds of glycosylation including O-linked N-acetylglucosamine (O-GlcNAc), O-glycans, N-glycans, glycosaminoglycans, and glycolipids (Figure 1d). UDP-GlcNAc can be epimerized to uridine-5′-diphospho-N-acetylgalactosamine (UDP-GalNAc; Figure 1e) or further metabolized to generate cytidine-5´-monophosphate-5-N-acetylneuraminic acid (CMP-Neu5Ac; Figure 1f). UDP-GlcNAc and its derivatives are considered sensors of the metabolic status of the cell, as it requires components of all four major classes of macromolecules: Glc, Gln, acetyl-coenzyme-A, and the nucleotide UDP. Gln is a key nutrient for tumor cells, being a major source of nitrogen and energy in rapidly dividing cells (15). Although the cause of increased flux through the HBP is not clear in tumor cells, it is likely to occur as a result of increased Glc and Gln uptake. To support this hypothesis, Itkonen et al. recently showed that several HBP genes were overexpressed in human prostate cancers (16). Thus, the link between altered metabolism and the up-regulation of glycosylation through the HBP provides a mechanism for cancer cells to sense and respond to a variety of environmental conditions.
How the HBP induces the malignancy process is not completely understood yet. One hypothesis is that the HBP exerts its effects by transforming growth factor-β (TGF-β) secretion. Many manuscripts have described that elevated Glc levels induce TGF-β production by different cell lines (17, 18). TGF-β is a known potent inductor of epithelial mesenchymal transition (EMT). The EMT involves a striking decline in epithelial markers, such as E-cadherin, ocludins, claudins, cytokeratin, and consequently cell polarity, accompanied by enhanced expression of mesenchymal markers, such as N-cadherin, vimentin, and fibronectin (FN), culminating in cell morphology alteration and increased cell motility (19). Besides, recent studies bring to light the involvement of a key O-glycosylation in the IIICS, a variant splicing domain of human FN, forming the oncofetal fibronectin (onfFN) during the EMT process (20). The importance of glycosylation in this process was supported by data showing that ppGalNAc-T6 knockdown inhibits onfFN biosynthesis and EMT in human prostate epithelial cells (20). In addition, a recent manuscript indicated that high Glc or GFAT2 overexpression induces EMT, onfFN production and increased ppGalNAc-T6 mRNA levels in human alveolar epithelial adenocarcinoma cells. Those factors imply that metabolite availability to the HBP exerts control over gene expression and modulates cell surface glycosylation, suggesting that changes in Glc uptake alters epithelial cell communication with neighboring cells and the ECM, which results in loss of tissue organization and contributes to tumor formation and progression (21). Thus, it is reasonable to think that glycan structures are changed by the metabolic status of the cells, and the aberrant glycosylation observed in tumors is a consequence of altered expression of glycosyltransferases combined with substrates availability. Therefore, the metabolic pathways, especially the HBP, can be directly implicated in alterations observed in O-GlcNAcylation (1), N-glycans (22), and O-glycans (21) in cancer cells.
O-Linked N-Acetylglucosamine
The O-GlcNAc PTM is characterized by the linkage of a β-N-acetylglucosamine moiety to the hydroxyl group of threonine (Thr) or serine (Ser) residues found in nuclear, cytoplasmic, and mitochondrial proteins (1). The addition of O-GlcNAc to proteins is catalyzed by O-GlcNAc transferase (OGT), and its removal is catalyzed by O-GlcNAcase (OGA). Deletion of OGT is lethal in mice at embryonic and single-cell level, highlighting the importance of O-GlcNAcylation in regulating basic cellular events (23). Aberrant O-GlcNAcylation has been linked to major diseases, including cancer, diabetes, and Alzheimer’s disease (24, 25). This dynamic glycosylation is analogous to phosphorylation and more than 1000 proteins have been described to be O-GlcNAcylated to date (26). The relationship between phosphorylation and O-GlcNAcylation has proved more complex than initially thought, since their function is not limited to site occupancy alone, but both PTMs can modulate each other at the same site or adjacent sites (27). Our group has also shown that O-GlcNAc can modulate tyrosine (Tyr) phosphorylation, indicating that the interplay between these PTMs at the substrate level is not limited to Ser and Thr residues (28). Growing evidence suggests that O-GlcNAcylation and phosphorylation not only compete for substrates (at the same or proximal sites), but also that O-GlcNAcylation regulates kinases and/or phosphatases. In one example, we showed that O-GlcNAcylation directly regulates the kinase activity of calcium/calmodulin-dependent protein kinase type IV (CaMKIV) toward cAMP response element-binding protein (CREB) (29). In addition, we recently showed that 39% of the kinases of the functional protein array are O-GlcNAcylated in vitro by recombinant OGT. Interestingly, the majority of identified kinases play a role in cancer (30).
Like phosphorylation, O-GlcNAcylation can modulate protein function, turnover, interactions, subcellular localization, enzyme activity, DNA affinity, and transcription activity (27). Several transcription factors involved in cancer biology, such as p53, c-Myc, NF-kB, and Sp1 are modified by O-GlcNAc (31). Over 60 papers were published in the past 3 years describing the relationship between O-GlcNAc and cancer, with a substantial portion of them related to the increase of O-GlcNAc and OGT in several types of tumors (32, 33). Increased protein O-GlcNAcylation and changes in OGT expression have been described in breast cancer, lung cancer, prostate cancer, pancreatic cancer, and colorectal cancer (16, 34–38). In addition, OGT silencing inhibits tumor growth in different models including breast cancer, prostate cancer, and pancreatic cancer (34, 37, 38), indicating that O-GlcNAcylation is important for tumorigenesis and suggesting that OGT represents a novel therapeutic target for these types of cancers (39). Interestingly, a low expression of OGA is suggested as a prognostic marker for hepatocellular carcinoma tumor recurrence (40). Another study suggests that the urinary content of OGT and OGA may be useful for bladder cancer diagnostics (41).
Recently, Hsieh-Wilson’s group showed that PFK is O-GlcNAcylated at Ser529 during hypoxia (42). This glycosylation inhibited the PFK activity and redirected the Glc flux through the pentose phosphate pathway (PPP), increasing the reducing power of the cell by the production of nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH). Such a shift in metabolic flux confers a selective growth advantage for cancer cells, since blocking the glycosylation of PFK at Ser529 reduced cancer cell proliferation in vitro and impaired tumor formation in vivo (42). Thus, blocking PFK1 glycosylation would provide a new strategy to combat cancer.
Despite evidence linking aberrant O-GlcNAcylation to cancer (1, 33, 43), only a few studies show how O-GlcNAc participates in the molecular mechanism involved in EMT. O-GlcNAcylation at serine 112 of Snail, the repressor of E-cadherin, blocks its phosphorylation by GSK3β and protects Snail from ubiquitylation and degradation. Hyperglycemic condition enhances O-GlcNAc modification and initiates EMT by transcriptional suppression of E-cadherin through Snail (44). Moreover, treatment of low metastatic human ovarian cancer cells (OVCAR-3) with the OGA inhibitors Thiamet-G and PUGNAc enhances their migration potential and decreases the expression of E-cadherin, consequently blocking the formation of the E-cadherin/catenin complex reducing intracellular adhesion. Whereas, when a high metastatic ovarian cancer cell lineage (HO-8910PM) are subjected to OGT silencing, the expression of E-cadherin is recovered and their potential migration ability is diminished (45). Taken together, the data demonstrate that O-GlcNAc plays an important role in EMT events, cell migration, and gain of malignancy and metastasis, which suggests it could be a potential target for cancer treatment.
Although OGT is a promising target against different types of tumors, OGT is an essential glycosyltransferase that targets specific sites on hundreds of protein substrates, making its inhibition a difficult task. Studies indicate that the specificity of OGT toward different substrates is modulated by transient associations with binding partners (46, 47). Recently, the structure of human OGT and the mechanism of action have been reported (48–50). Despite a series of new advances in structural and mechanistic features of OGT, the existing inhibitors for this enzyme are not as specific and efficient as the OGA inhibitors (51). In addition, whereas almost all OGT inhibitors target the UDP-GlcNAc binding site, they are not as potent as the reaction product UDP, which displays a Kd of 0.5 μM. Although studies have only been performed in cell culture, we highlight three OGT inhibitors: (i) ST045849 (1; Scheme 1 in Supplementary Material ) that has been used successfully in the inhibition of prostate cancer cell lines (16); (ii) ST060266 or BZX (2; Scheme 1), an irreversible inhibitor of OGT used in different cell types (49); and (iii) 4Ac-5S-GlcNAc, a cell-permeable compound that enters the HBP to be synthesized into UDP-5S-GlcNAc (3; Scheme 1), a potent OGT inhibitor (52). Interestingly, a recent OGT bisubstrate inhibitor, presenting an acceptor peptide linked to UDP has emerged as a new scaffold for the development of more specific inhibitors (53).
N-Linked Glycans
Many cell surface, lysosomal, and secreted proteins are post-translationaly modified by the addition of a β-GlcNAc to the asparagine (Asn) residues (N-linked) of the evolutionary conserved “sequon” Asn-X-Ser/Thr, where X is any amino acid except proline. N-linked glycans consist of a conserved pentasaccharide core Manα1-6(Manα1-3)Manβ1-4GlcNacβ1-4GlcNacβ1-Asn trimmed with different sugars, organized in up to five antennae branches. Such variable structures create an array of glycoforms with different physical and biochemical properties that confer functional diversity to the glycoprotein. N-glycosylation affects protein folding, playing a central role in protein quality control within the endoplasmic reticulum (ER), in metastatic potential and the spread of tumors (54).
Changes in the oligosaccharide structure of N-glycans have been described in breast, colon, prostate, lung, renal cell, hepatocellular carcinoma, pancreatic, and gastric cancer (55–67). Most growth factor receptors on the cell surface are N-glycosylated, including epithelial growth factor receptor (EGFR) (68), integrins (69), and TGF β receptor (TGFβR) (70). N-glycans are ligands for galectin 1 and 3 (71) and siglecs (72) at the cell surface, forming lattices that enhance the residence time of receptors (73–75). Oncogenesis has been shown to upregulate the expression of N-glycans with higher affinity for galectins and to increase the residence time of the receptors (76–79). These studies open the possibility to target surface distribution of growth factor receptors by modulating N-glycan branching. Such strategies might be useful in cancer therapy. Interestingly, inhibition of the N-glycan biosynthetic pathway is emerging as an exciting target to inhibit cancer progression.
The first steps of N-glycan biosynthesis are conserved among eukaryotic cells (80–82). The pathway begins at the cytoplasmic face of the ER membrane with the assembly of a 7-residue oligosaccharide precursor linked to the lipid dolichol-phosphate. Two GlcNAc molecules from the highly energetic donor GlcNAc-PP-dolichol are the first residues incorporated into dolichol-pyrophosphate. The activated monosaccharide donor is synthesized by the GlcNAc phosphotransferase, which transfers the GlcNAc-1-phosphate from UDP-GlcNAc (83). The oligosaccharide is further assembled, step by step, by specific glycosyltransferases that add other five mannoses (Man) to the disaccharide. The dolichol oligosaccharide chain anchored to the cytosolic side is moved into the lumen of the ER. Within this organelle, four Man and three Glc residues are added to the growing glycolipid.
The mature lipid-linked precursor is further transferred as a single block from the dolichol to Asn residues of nascent peptide by the transmembrane oligosaccharyl transferase protein complex (82). In the next step, glucosidases trim two terminal Glc from the glycoprotein Asn-GlcNAc2Man9Glc3, a crucial process for proper protein folding. The final Glc undergoes several cycles of removal and reintroduction while the process of protein folding is assisted by two lectin chaperones, known as calnexin and calreticulin, which bind to terminal Glc. After correct folding, the Glc and Man unities are removed by α-glucosidases and α-mannosidases, respectively.
Inhibition of protein folding in the lumen of the ER results in ER stress (84). ER stress activates various intracellular signaling pathways, known as the unfolded protein response (UPR), which is comprised by the inhibition of general protein translation, the induction of genes such as ER chaperones that increase the protein-folding capacity of the ER, and upregulation of aberrant protein degradation. The severe stress induces apoptosis and elimination of the damaged cell. In recent years, many groups have been seeking to pharmacologically aggravate chronic ER stress in cancer cells in order to enhance apoptosis and achieve tumor cell death.
Inhibition of N-glycan biosynthesis can impact protein folding and is emerging as an interesting strategy to reduce receptor tyrosine kinase (EGFR, ERβB2, insulin-like growth factor-I receptor, and Met) maturation and its cell surface expression in multiple cancers, including squamous cell carcinomas, adenocarcinomas of the breast, prostate and pancreas, and malignant gliomas (85–87). Along this line, it has been reported that ER stress induced by tunicamycin (4; Scheme 1) influences chemosensitivity of tumor cells to anticancer drugs (88) and to radiotherapy (85, 86, 89, 90) and influences tumor induced angiogenesis (91). Tunicamycin is a mixture of homologous nucleoside antibiotics that competitively inhibits (Ki 5 × 10–8 M) the enzyme GlcNAc phosphotransferase, which prevents the formation of GlcNAc-PP-dolichol (83, 92). Tunicamycin inhibits cell invasion and tumorigenicity both in vitro and in vivo (93–99). Other antibiotics that inhibit the lipid-linked saccharide pathway are amphomycin (5; Scheme 1), tridecaptin (6; Scheme 1), flavomycin (7; Scheme 1), diumycin (8; Scheme 1), and tsushimycin (9; Scheme 1) (83, 100).
Inhibition of protein folding can also be achieved by inhibiting glycoprotein-processing enzymes ensuing anti-tumoral activity (101–104). Castanopermine (10; Scheme 1) inhibits glucosidase I and leads to altered glycoproteins with Glc3Man7GlcNAc2 structures (105). The recent report of the first structural model of eukaryotic α-glucosidase (106) will improve the design and synthesis of novel enzyme inhibitors, which will hopefully be more effective against cancer.
The processed glycoprotein, therefore, moves to the Golgi where it is demannosylated by the Golgi α-mannosidase I forming the Man5GlcNAc2 structure, which is a substrate for the acetylglucosaminyltrasferase-I (GnT-I), a key enzyme in the development of multicellular organisms (107, 108). The addition of the first GlcNAc residue by GnT-I generates the GlcNAcMan5GlcNAc2, substrate for GnT-III or α-mannosidase II (Figure 2). α-Mannosidase II trims two Man residues from the intermediate to form the core GlcNAcMan3GlcNAc2 precursor of complex N-glycans (109). Conversely, the action of GnT-III on the GnT-I product before α-mannosidase II directs the pathway to hybrid structures (110). Inhibition of α-mannosidase II by 1-deoxymannojirimycin (11; Scheme 1) generates “high mannose” type of N-glycans. As the pathway progresses through the Golgi, the core GlcNAcMan3GlcNAc2 can be further modified by a series of GnTs (GnT-II, IV, V, and VI) that substitute a distinct position of the trimannosyl core generating different branches of complex N-glycans (Figure 2) (109).
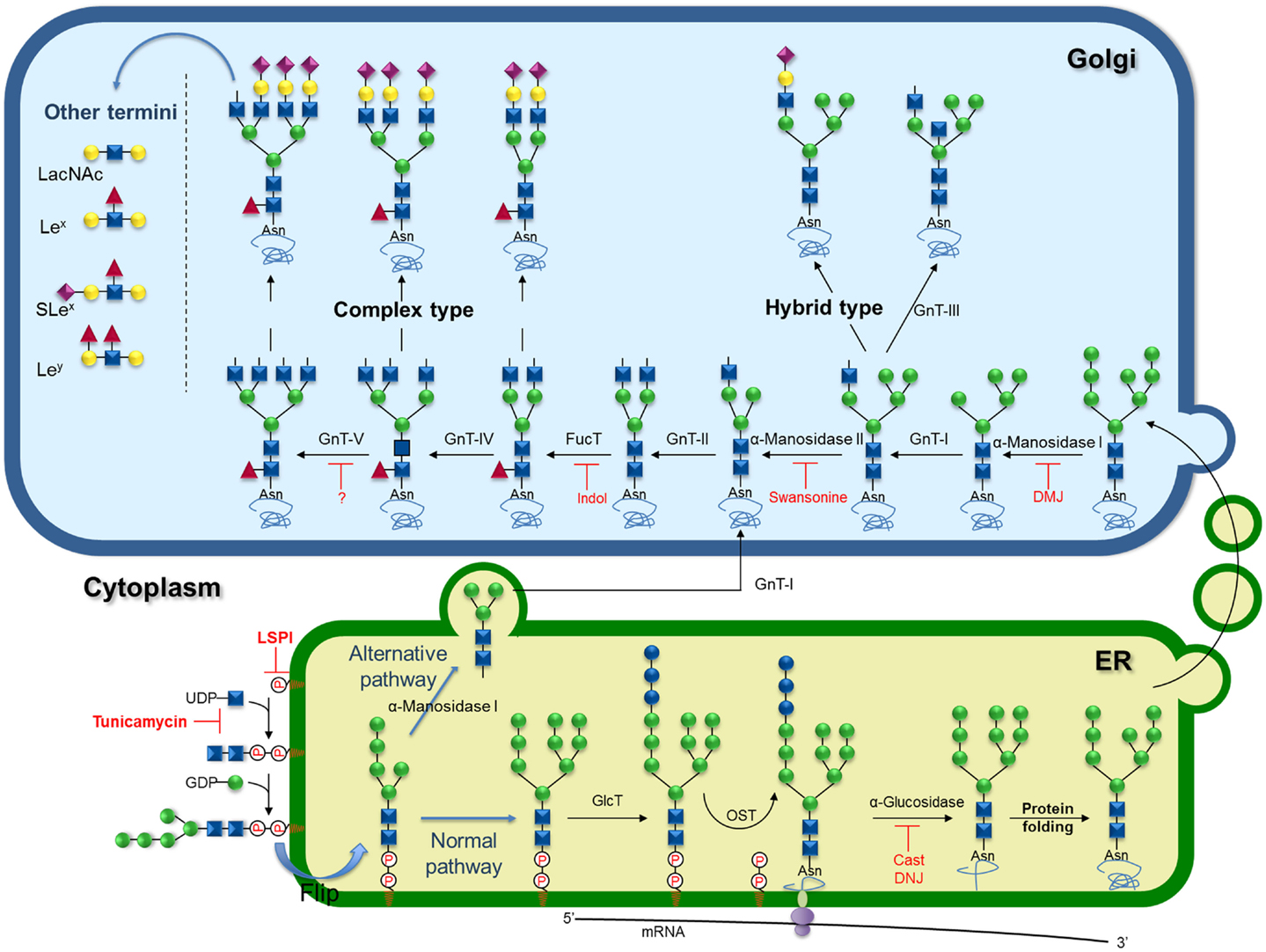
Figure 2. Schematic representation of biosynthesis and processing of N-linked oligosaccharides showing known inhibitors and key targets for inhibition. Inhibitors of the lipid-linked saccharide pathway (LSPi), tunicamycin, castanopermine (Cast), 1-deoxynojirimycin (DNJ), 1-deoxymannojirimycin (DMJ), swansonine, and indolizidine (Indol).
GnT-V expression is increased during oncogenic transformation (111–113), consequently, cancer cells commonly show increased β1-6G1cNAc-branching at the trimannosyl core of N-linked carbohydrates (55, 56, 58–61, 65–67, 114). GnT-III has been found to play an important role in the suppression of metastasis (115–121) as the introduction of bisecting GlcNAc suppresses β1-6 GlcNAc branching formation catalyzed by GnT-V. In the same line, GnT-I knockdown suppresses N-glycan branching and the invasive phenotype in HeLa and PC-3-Yellow cells (122).
GnT-V expression correlates with increased cell migration (77, 123, 124) and sensitivity to acute stimulation of growth factors (73). The latter effect is most probably due to the β1-6G1cNAc-branching increasing galectin binding and retention of growth receptors on the cell surface (75, 124, 125). Furthermore, during the development of the malignant phenotype, E-cadherin expression is accompanied by an increase in its β1-6 branched N-glycan structures (116, 126). Pinho et al. (116) demonstrated the importance of the N-glycan structures of E-cadherin in the development of gastric tumors. The authors demonstrated that modifications in N-glycan chains of E-cadherin by GnT-III lends stability to the protein in the cell membrane, contributing to the formation of stable adherent junctions. On the other hand, modifications catalyzed by GnT-V promotes the destabilization of E-cadherin, formation of unstable adherent junctions and inhibition of cell–cell interactions, which promotes gastric tumor invasiveness (116).
Despite the key role GnT-V plays in cancer progression, no inhibitors have been described thus far. However, a very well-known inhibitor of the production of complex β1,6-branched N-linked glycans is the alkaloid swainsonine (12; Scheme 1), a transition-state inhibitor of α-mannosidase II that removes α-1,3 and α-1,6 Man residues from the GlcNAc-Man5GlcNAc2-peptide, causing the formation of hybrid structures (127). Inhibition of N-linked oligosaccharide processing by swainsonine in tumor cells has generated attention in its use as an anticancer agent, since reports indicate that this drug inhibits tumor growth and metastasis, augments natural killer and macrophage-mediated tumor cell killing, and stimulates bone marrow cell proliferation (93, 128–130). This drug effect was also associated with the enhanced expression of the metalloproteinases gene (131). Recently, it was demonstrated that swainsonine inhibited cancer cell growth through the activation of the mitochondria-mediated caspase-dependent pathway (132).
Elongation of hybrid and complex N-glycans occurs by the addition of a β-Gal to the β1-2 linked GlcNAc yielding the Galβ1-4GlcNAc (LacNAc) moiety, which can be further elongated by the sequential addition of β1-3-linked GlcNAc and β1-4 linked Gal resulting in a poly-LacNAc extention. In addition, Gal can be substituted by a GalNAc forming the GalNAcβ1-4GlcNAc (LacdiNAc) sequence.
N-Glycans can be further decorated by the action of a number of transferases that add Gal, Fuc, sialic acid, and sulfate to the antennae (see below) resulting in the mature structure on the nascent protein (82). Those enzymes involved in N-glycans elongation and decoration steps act on O-glycans and glycolipids as well, so they are described below in the text.
The GlcNAc adjacent to Asn in the core can be modified by the action of α1-6-fucosyltransferase (FucT-VIII) (133). The FucT-VIII is overexpressed in several types of tumors as colorectal, hepatoma, ovarian, lung, and thyroid (134–137) cancer. Muinelo-Romay et al. showed higher activity and increase of FucT-VIII expression in human colorectal tissues correlating with the degree of infiltration through the intestinal and malignant transformation (134). FucT-VIII is also upregulated in non-small cell lung, correlating with tumor metastasis, disease recurrence, and poor survival of patients (137). These correlations have prompted many groups to pursue inhibitors of FucT-VIII as potential antitumorals. Inhibitors of a recombinant α1-6FucT from Rhizobium sp. have been described. Several racemic polyhydroxylated indolizidines have been tested as potential inhibitors of this enzyme. One of the castanopermine stereoisomers was the best inhibitor with an IC50 of 0.045 mM. Interestingly, this compound turned out to be the best mimic for the structural features of the fucose moiety in the presumed transition state (138, 139).
O-Glycosylation in Cancer
O-Glycosylation is a common type of PTM that consists of the attachment of a αGalNAc on Thr or Ser residues from specific sequences of target proteins. The newborn O-GalNAc glycan can be further modified by several glycosyltransferases acting in a sequential manner in order to extend the glycan chain either branched or linearly according to substrate specificity. O-Glycans are abundantly found attached on the cell surface and on the ECM proteins, especially in the form of mucins, heavily O-glycosylated proteins in which carbohydrate amount can account for 50% of the molecule by weight (140). The biological function of mucins and mucin-like glycoproteins is deeply dependent on its carbohydrate chains (141). Since the synthesis of O-glycans is controlled by the availability of substrates and enzymes in subcellular compartments, without any correcting mechanisms, such as N-glycosylation and protein folding, it is easy to understand why aberrant O-glycans are usually observed in tumorigenesis and metastasis. Altered O-glycosylation is a universal feature of cancer cells, but only specific glycan changes are frequently associated with tumors. The specific O-glycan changes commonly found in cancer cells, as well as its biosynthesis and potential as a drug target are depicted in the following items.
In cancer, truncated glycan mucin related tumor-associated carbohydrate antigens (TACA), are abnormally expressed in several epithelial cancers (i.e., gastric, pancreatic, colorectal, ovarian, and breast cancers) (142–144). In many types of cancers, enzymes from poly-peptidyl-αGalNAc transferase (ppGalNAcT) family are reported to be located throughout the Golgi, instead of being restricted to cis-Golgi as in normal conditions. The ppGalNAcT catalyzes the transfer of a α-GalNAc from UDP-GalNAc to a Ser or Thr residue of a glycoprotein, producing the Tn antigen. When the Tn antigen is generated, it can have three different fates: (i) it can be sialylated on C6 by a ST6GalNAcT; (ii) it can be substituted on C3 or C6 by a β-GlcNAc which gives rise to core 3 or core 6, respectively; or (iii) it can be galactosylated on C3 by the C1GalT1 in order to form core 1, also known as Thomsen-Friedenreich (TF) or T antigen. The emergence of Tn antigen can be a consequence of some abnormality with ppGalNAc-T, C1GalT1 (also known as T-synthase), or with a chaperone responsible for its proper folding or an imbalance in the ratio of the competing downstream core 1 processing enzymes (Figure 3) (145, 146).
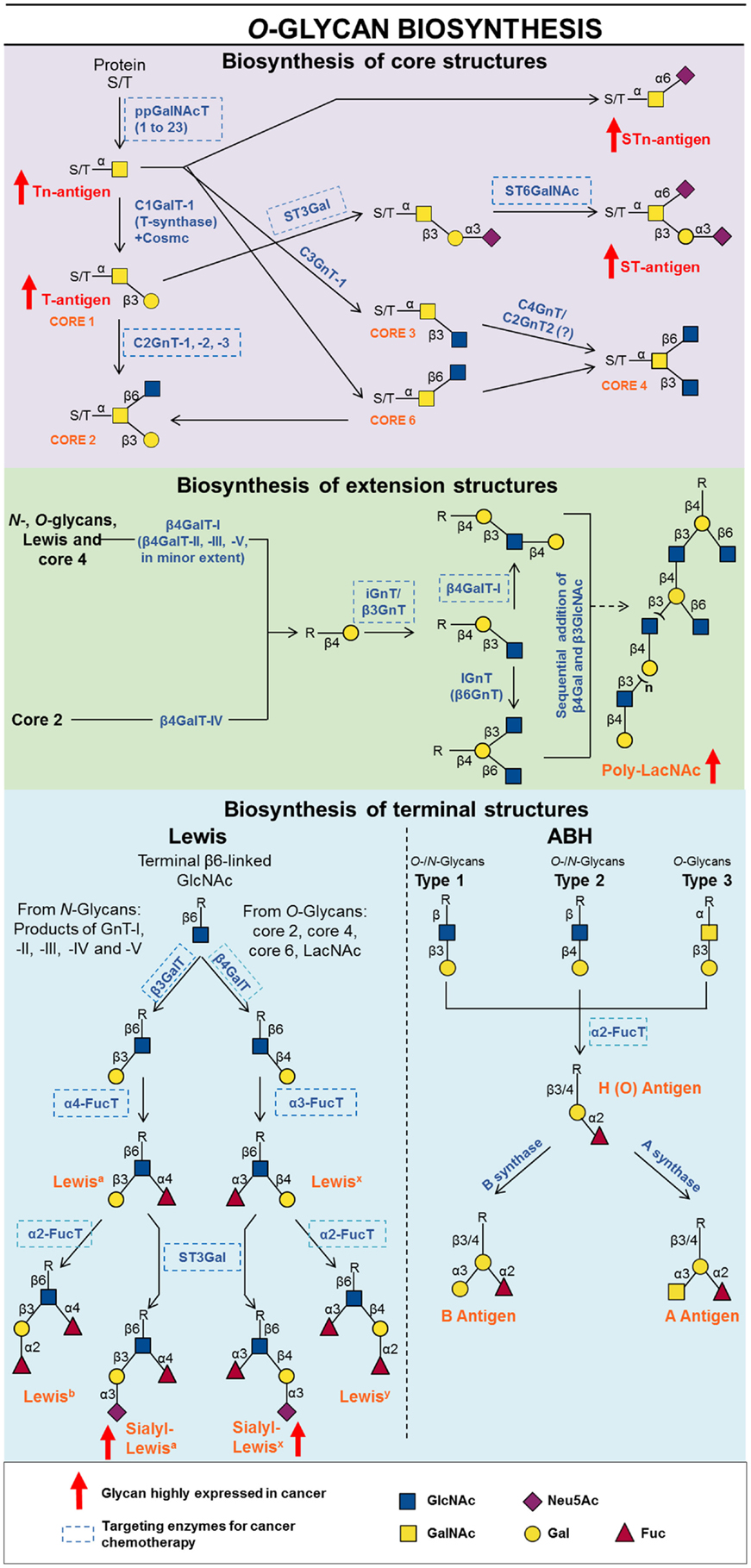
Figure 3. Schematic representation of biosynthesis and processing of O-linked oligosaccharides showing known inhibitors and key targets for inhibition.
Several enzymes from the T-synthase family are reported to be involved in many cancers in both positive and negative fashion. For example, ppGalNAc-T2 is overexpressed in gastric cancer and glioma cells, which is reported to decrease invasion and metastasis (147, 148), while ppGalNAc-T12 is considered to be a negative marker for gastric and colorectal cancer metastasis (149). On the other hand, other ppGalNAc-Ts are shown to be bad prognosis for tumor progression. Bladder cancer tissues contain high levels of ppGalNAc-T1 mRNA and silencing this gene significantly inhibits tumor growth in vivo (150). High expression of ppGalNAc-T3 is found in high grade tumors and correlates to poor prognosis in renal carcinomas, gastric carcinomas (151, 152) and in pancreas adenocarcinoma (153). The ppGalNAc-T6 is upregulated in the vast majority of breast cancers and it is suggested to disrupt mammalian cell carcinogenesis (154, 155). The ppGalNAc-T7 has been shown to play important roles in cervical cancer pathogenesis (156) and enhances metastatic process of melanoma cells (157). The ppGalNAc-T13 gene was upregulated in highly metastatic lung cancers (158). The ppGalNAc-T14 has been reported to contribute to ovarian cancer carcinogenesis through aberrant glycosylation of MUC13 (159) and may be a potential biomarker for breast cancer (160). Therefore, the ppGalNAcTs have been proposed as tumor diagnosis and prognosis markers, as well as foranti-cancer immunotherapy. In particular, ppGalNAc-T3 was shown to be recognized as an antigen by cytotoxic T lymphocytes from patients with brain tumor (161), while ppGalNAc-T6 is a marker for the prognosis of pancreatic cancer (162) and breast cancer metastasis (163). Besides the use of ppGalNAcTs as tools for diagnosis and prognosis, this class of enzymes is a potential target for anti-cancer chemotherapy.
Primary attempts to perform kinetic analysis of ppGalNAcT yielded a peptide inhibitor. The EPO-G (13; Scheme 1) consists of a 12-residue peptide, which differs from EPO-T, a commercial peptide usually utilized as an acceptor substrate of the enzyme by a single residue. Although EPO-G was observed to inhibit ppGalNAc and helped to define its mechanism of enzyme action, it presented an inexpressive Ki of 0.77 mM (164).
Another strategy to inhibit ppGalNAc, and consequently whole O-glycosylation, is to use derivatives from UDP-GalNAc. Hatanaka and coworkers had proposed UDP-GalNAc-based inhibitors using the model of tunicamycin, a well-known inhibitor of N-glycosylation. The authors had synthesized compounds substituting UMP with different length fatty acid chains (14; Scheme 1), which resulted in the 16 carbons-substituted UMP derivatives having a slight inhibition (IC50 of 0.63), of ppGalNAcT (165). Aryl-glycosides of GalNAc (166) were found to competitively inhibit the elongation of O-GalNAc chains and have been extensively used for investigating the role of mucin chemical composition on its biological functions and its biosynthesis (167). The derivative benzyl-αGalNAc (15; Scheme 1) was shown to decrease the level of sialylation and sulfation of mucins secreted by the human colon carcinoma cell line HT-29 MTX (168), and also decreased the binding of treated HM7 colon cancer cells to E-selectin [ELAM-1, recognizes sialyl Lea and sialyl Lex (169)], which would be a desirable effect in modifying the immunological and biological properties of colon cancer cells. However, the use of benzyl-αGalNAc as a drug is unlikely, because the desired effect is only reached in cell culture with a millimolar range. Such a compound also behaves as a competitive inhibitor of C1GalT1 (166), causing treated cells to express mucins with a high level of the cancer-associated Tn (168, 169).
Synthesis of C-glycosidic UDP-GalNAc mimics (16; Scheme 1) were reported, but to our knowledge, their biological activity were not tested (170). In vitro evaluation of the 3-, 4-, and 6-methylated UDP-GalNAc compounds (17; Scheme 1) effect had shown an inhibition pattern similar to UDP-GlcNAc (171).
Promising compounds targeted to ppGalNAcTs came from the work of Hang and coworkers (172), by screening an uridine-based library designed to target enzymes that utilize UDP-sugar substrates (173). Through this approach, compound 18 (Scheme 1) inhibited a series of ppGalNAcTs (ppGalNAcT-1 to T5, T7, T10, and T11) in micromolar range, suggesting selectivity against this enzyme family, since inverting and retaining GalTs or other UDP-sugar utilizing enzymes were not significantly inhibited. In addition, these compounds inhibited O-glycosylation but not N-glycosylation and induced apoptosis in two different cell types (Jurkat, a lymphoma cell line; and HEK293T). Therefore, compound 18 (Scheme 1) is a promising scaffold for O-glycan inhibition in cancer cells.
Recently, Pouilly and coworkers have employed metabolic engineering in order to label highly active metabolic cancer cells with UDP-αGalNAc analogs (19; Scheme 1). They observed that all compounds could be recognized by ppGalNAcT1, and that peracetylated GalNAc analogs with hydrophilic substitutions on the N-acetyl group, such as azidoacetyl and glycolyl, could be incorporated into cell surface glycoproteins at only slightly lower levels compared to the natural GalNAc. In addition, when mice were immunized with glycopeptides carrying the GalNAc analogs linked to Ser or Thr, some of them had produced an antiserum, which was specifically directed against GalNAzaSer/Thr, without cross-reactivity toward GalNAcSer/Thr. Such a result brings to light that this approach could be used in O-glycan biosynthesis research and also for passive immunotherapy of cancer directed at cell surface tumor-associated O-glycans (174).
The epitope O-GalNAc can be further sialylated on C6 by a ST6GalNAcT (175–178). The disaccharide STn is overexpressed in pancreas, gastric, bladder, breast, and ovarian cancer (142, 179–182). The appearance of STn is explained by the abnormal expression of ST6GalNAc together with its aberrant localization throughout the Golgi apparatus, contributing to the overpass of complex O-glycan processing enzymes resulting with expression of short STn-modified O-glycans (176). Overexpression of this sialylated antigen reduces cell–cell interactions and increases cell migration (183) most likely by altering cell adhesion to ECM (180, 184). Positive correlations of STn antigen expression with cancer aggressiveness and poor prognosis of the patients has provoked great interest in the functional analyses of STn. In this respect, Ozaki et al. (185) demonstrated that MUC1 carries a large proportion of STn in human advanced gastric cancer tissues.
ST6GalNAcT can also modify core 1 structures and its sialylated derivative comprises the sialyl T (ST) antigen (see below). Such a feature is found in breast cancer due to the increased expression of ST3GalT-1 combined with the low expression or absence of C2GnT-1 (186).
Core 1, T-Synthase, and Comsc
The peptide carrying O-linked αGalNAc can be converted to core 1 (Galβ1-3GalNAcα-S/T) structure by core 1 β1-3 galactosyltransferase, termed T-synthase or C1GalT. Cancer cells exhibit a high level of mucins with T and ST antigen (187). T disaccharide represents a cancer-associated antigen in colorectal carcinoma and it is suggested to act as a prognostic marker (188). T antigen is associated with adhesion and metastasis of human breast cancer cells through the binding to galectin-3 (189). Moreover, the overexpression of MUC1 gene in human and murine mammary carcinoma cells correlates with the increase of T antigen assumedly by the concomitant down regulation of both C2GnT1 and ST3Gal (190), which also causes the loss of adhesive properties to E-selectin of the cells studied, but favors the binding of MUC1-overexpressing cells to galectin-3.
The use of T antigen as a target for anti-cancer immunotherapy is in progress. The T antigen or its difluoro analog were coupled to a glycosynthetic peptide and to a tetanus toxoid (20; Scheme 1), affording synthetic vaccines, which induced very strong immune responses in mice overriding the natural tolerance of the immune system. The induced antibodies were selectively directed against the tumor-associated MUC1 structures and strongly bind to breast cancer cells of the MCF-7 cell line (191). This approach can be used for anti-cancer immunotherapy and the difluoro analog of T antigen may serve as an inhibitor for O-glycan synthesis. Recently, Sclerotium rolfsii lectin was shown to bind to T antigen and inhibit growth of human colon cancer HT29 and DLD-1 cells by binding to cell surface glycans and inducting apoptosis through both the caspase-8 and -9 mediated signaling (192), being an interesting possibility for therapy.
High levels of T antigen have been commonly associated with overexpression of T-synthase (146), or imbalance in the ratio of the competing downstream core 1 processing enzymes, C2GnT and ST6GalNAc-II (145). Remarkably, there is only a single gene encoding T-synthase in humans and other mammals (146), and enzymatic activity of this protein is dependent on its correct folding promoted by a unique chaperone. Cosmc is a molecular chaperone located in ER that promotes correct T-synthase folding and guiding to Golgi (193, 194). Somatic loss-of-function mutations of the chaperone gene Cosmc was shown to abolish the T-synthase activity, leading to the appearance of Tn antigen on the surface of many types of tumor cells, like the highly aggressive fibrosarcoma cell line (195), colon cancer cell line and melanoma-derived cell lines (196). Since Cosmc is encoded by a single gene in the X chromosome, its susceptibility to epigenetic silencing through hypermethylation of its promoter (197) also leads to Tn antigen increase in mucins.
The role of T-synthase in tumors varies according to the type of cancer tissue. Whereas in breast cancer there are multiple reports indicating that T-synthase does not have its expression or activity altered, there are O-glycan modifications other than T antigen present, and hepatocellular carcinomas show an overexpression of this enzyme which is associated with poor survival of the affected patients. In addition, the T-synthase plays a role on HGF/MET signaling pathway in hepatocarcinoma cells, leading to a proliferation pattern in this cell type (198). In human colon cancer cells, the suppression of T-synthase by iRNA was shown to be associated with an increased presence of Tn, STn and core 3 glycans in this cells surface, since T-synthase competes with ST6Gal and C3GnT for substrate (199). Based on all the evidence, it would be ideal to either completely or partially inhibit O-glycan biosynthesis without altering the core 1 formation.
Core 2 and C2GnT
Core 2 β1-6 N-acetylglucosaminyltransferase (C2GnT) competes with ST3GalNAc for the common core 1 substrate, and the control of mucins decoration with whether ST or core 2-derivated antigens depends on the relative expression of both glycosyltransferases and their subcellular localization. The classification of human C2GnTs were primarily based on the differential enzyme activities observed for C2GnT extracted from leukocytes and mucin-producing cells, known as C2GnT-L and C2GnT-M, respectively. Lately, the human C2GnT variants were classified as -1, -2, and -3, of which C2GnT-1 corresponds to L and C2GnT-2 to M.
Increased levels of C2GnT determine the increased branching of O-glycans and it is associated with the acquisition of invasive and metastatic potential rather than simply transformation of cancer cells (200). The expression of C2GnT-1 positively correlates with the progression of prostate cancer in human patients (201), testicular germ cell tumor (202), endometrial carcinoma (203) and with bladder tumor progression (204). In addition, high levels of C2GnT does not necessarily indicate that O-glycan chains of mucins will be terminated on core 2, but that there is an increase in the combination of possible glyco-structures, since core 2 is a substrate to further modifications, such as poly-LacNAc, sialic acid, fucose, and sulfate unities, increasing the variety of O-glycan chains on the cell surface and expanding the roles for these different glycans. Thus, high levels of C2GnT contributes to increases in core 2 O-glycan structures that also serve as substrates to ST3Gal, corroborated by the hypersialylation observed in leukemic leukocytes from chronic myelogenous leukemia and acute myeloblastic leukemia (205). Likewise, core 2 O-glycans of MUC1 from bladder tumors are modified with poly-LacNAc, which allows them to bind to galectin-3 and hamper the access of NK cells to the TNF-related apoptosis-inducing ligand present in the tumor cells surface (204). Therefore, core 2 O-glycans modified with poly-LacNAc can protect bladder tumor cells from NK cell mediated death, which highly favors metastasis.
On the other hand, C2GnT expression and surface decoration of T cells with O-glycans rich in core 2 structures renders them susceptible to galectin-1-induced cell death. It is suggested that this fact may be due to oligosaccharide-mediated clustering of CD45 molecules on the cell membrane, facilitating their binding to galectin-1 (206). Therefore, unlike in other cancer cells aforementioned, C2GnT expression favors T-lymphoma cell death by a particular mechanism and the haploinsufficiency of this enzyme is sufficient for loss of core 2 O-glycan expression and galectin-1-induced cell death resistance (207). Likewise, the downregulation of C2GnT-2 was observed in primary tumors from colorectal cancer patients as well as in colon cancer cell lines contrasting with normal colon tissues. The protective role of C2GnT-2 was demonstrated by the suppression of cell invasiveness and tumor growth of C2GnT-2-transfected colon cancer cells in vitro and in vivo (208).
Inhibiting C2GnT activity is desirable in cancer types in which core 2 is highly expressed and affecting in cancer progression, as well as in tumor cells containing the poly-LacNAc or hypersialylation phenotype, since C2GnT has been shown to act as a key regulator for such modifications (209, 210). However, a C2GnT inhibitor must be carefully designed, because such an enzyme shares homology with many other UDP-GlcNAc:N-acetyl-glutaminyl transferases. In addition, the use of UDP-GlcNAc analogs could potentially interfere with biosynthetic pathways involved in the interconversion, metabolism, and transport of these sugar nucleotides (211). For this reason, the first attempt to inhibit C2GnT was made by targeting its acceptor substrate with deoxy analogs (21; Scheme 1). However, they displayed a poor inhibitory effect, with a Ki of 0.56 mM, much higher than the enzyme Km (211). Kuhns and coworkers had also used several deoxy-analogs of core 1 to inhibit C2GnTs extracted from a series of tissues. Although they also obtained disappointing results, they at least observed differential binding requirements for the hydroxyl groups in the substrate from the enzymes explored (212). All types of C2GnT seem to have an absolute requirement for the 4- and 6-hydroxyls of GalNAc, and the 6-hydroxyl of Gal in the substrate, but the recognition of the 3- and 4-hydroxyls of galactose and the acetamido group of GalNAc is variable between C2GnT-1 and C2GnT-2. Molecules coupled to the chemically inert, but UV-detectable p-nitrophenol (pNP) group would serve as photoaffinity labeling reagents. Thus, a PnP substituted core 1, Galβ1-3GalNAcα-pNP (22; Scheme 1), an acceptor substrate analog, became a potent inhibitor upon 350 nm irradiation (213) with little inhibition of other O-glycan processing enzymes, even in higher doses.
Despite its potential application in research and clinical practice, there is no recent progress in the design of C2GnT inhibitors, but interesting scaffolds may arise from the studies of UDP-GlcNAc epimerase inhibitors and other UDP-GlcNAc transferases. Meanwhile, inhibitors of core 2 or poly-LacNAc-modified core 2 interactions with galectins are under review and may be another avenue to combat tumor progression promoted by these O-glycans.
Poly-N-acetyllactosamines
N-Glycans, core 1, and core 2 O-glycans can also be extended by N-acetylglucosaminyltransferases and galactosyltransferases to form sequences that represent the little i antigen. Linear poly-LacNAc units can be branched by members of the I-branching β-1,6-N-acetylglucosaminyl transferase (IGnT), resulting in the large I antigen. Moreover, poly-LacNAc chains can be synthesized from core 4, which arises from the addition of a β1-6GlcNAc on O-GalNAc from core 3 (Figure 3).
The control of chain length depends primarily on β4galactosyltransferase (β4GalT). There are seven β4GalTs characterized to date (214–219), among which β4GalT-I and β4GalT-III variants are the most widely expressed (217). The others are expressed tissue-specifically to a minor extent (217). β4GalT-I was the first isolated galactosyltransferase and it is known to act over a variety of substrates, being responsible for poly-LacNAc synthesis in N- and O-glycans, Lewis structures and core 4 O-linked carbohydrates. β4GalT-II, -III, and -V can also use these types of substrates but to a lower extent. In turn, β4GalT-IV is the one responsible for galactosylation of core 2 terminus from O-glycans (220). Furthermore, β4GalT-IV, but not β4GalT-I, drastically reduces its efficiency as the acceptors become longer (220), consistent with the fact that poly-LacNAcs on core 2 branched oligosaccharides are shorter (only two LacNAc repeats) than those in N-glycans (220). β4GalT-VI and β4GalT-VII do not act on N- or O-glycans, but were shown to synthesize Galβ(1-4)Glcβ-ceramide from Glcβ-ceramide (219) and to participate on proteoglycan biosynthesis (221), respectively.
The β3-N-acetylglucosaminyltransferase, which acts over β4Gal, was first characterized and called extension IGnT because it creates a new terminus for β4GalT to act upon (222). Besides IGnT, other β3GnTs were shown to be capable of both initiating and elongating poly-LacNAc chains. β3GnT-1, -2, -3, and -4 enzymes were found to act the same as IGnT, even though they do not share structural similarity with IGnT, but they do share conserved motifs with the β3GalT family (223, 224). These β3GnT enzymes were shown to act preferentially over poly-LacNAc and are suggested to be involved in the biosynthesis of poly-LacNAc sugar chains (224). In addition, their expression profiles were different among human tissues (223, 224).
The IGnT, creates multiple branches on the poly-LacNAc chain, which may serve as a mechanism for amplifying the functional potency of cell surface glycoproteins (225). There are three human isoforms of IGnT characterized thus far (226, 227), from which IGnT-1 and IGnT-2 showed similar tissue expression profiles, with the transcript expression of IGnT2 greater than that of IGnT1 and IGnT-3 (227). It is noteworthy that C2GnT-2 is also considered as an I-branching enzyme, although it exhibits only weak peridistal I-branching activity (228).
Increasing evidence demonstrates the association of poly-LacNAc chains found both in O- and N-glycans with cancer. Thyroid papillary carcinomas were observed to present high heterogeneity in their poly-LacNAc chain length and branching status, different from those produced in other thyroid neoplasms (229). Poly-LacNAc substituted oligosaccharides were shown to be expressed in a metastasis-dependent manner on melanoma cells (230) and also to be important for the interaction of carcinoma cells with hepatic cells in the process of liver metastasis (231). In addition, poly-LacNAc substitutions on O-glycans render prostate cancer cells resistant to NK cell toxicity, cooperating for prostate cancer cells to survive longer in host blood circulation and favoring the metastasis process (232).
Most of the associations between increased expression of poly-LacNAc and cancer occur through the binding of these oligosaccharides to galectins. Such interactions have been shown to mediate lung metastasis of melanoma cells through the adhesion of poly-LacNAc from N-glycans on galectin-3 (233) and to be important for tumor immune evasion, as demonstrated by the increase in tumor lymphocytic infiltration and tumor-specific cytotoxic T cells and decrease on melanoma growth in vivo after the treatment with a metabolic inhibitor of LacNAc biosynthesis (234).
Given the role of poly-LacNAc in cancer, it would be interesting to inhibit its binding to galectins, which can be achieved in two different manners: by inhibiting the binding itself, through blocking galectins, or by inhibiting enzymes involved in the LacNAc biosynthesis. In fact, poly-LacNAc processing enzymes have been shown to have a direct correlation with cancer.
For example, the overexpression of β4GalT-I, -II, and -V was suggested to cause the increase of galactosylation in human astrocytomas, which is associated with its malignancy (235). Particularly, β4GalT-IV was shown to behave as a strong predictor for tumor metastasis and is associated with poor overall survival of colorectal cancer patients (236). The upregulation of the expression of β4GalT-III in neuroblastoma (NB), correlates with advanced stage, unfavorable histology, and predicts a poor prognosis in NB patients. In addition, β4GalT-III expression increased cell migration, invasion, and tumor growth of NB cells possibly due to modified glycosylation of β1-integrin through increasing terminal Gal (237). Interestingly, there are an increasing number of reports showing that metastatic cells contain elevated levels of β4GalT-I on the cell surface, where it serves as an adhesion molecule (238, 239), whereas the Golgi levels of that protein remains the same between non-metastatic and metastatic cells. Elevated expression of surface β4GalT-I, characteristic of highly metastatic murine melanoma cells, contributes to their invasive phenotype in vitro and to their metastatic phenotype in vivo (240). The expression of β4GalT-I on the cell surface plays a crucial role in the proliferation of MCF-7 cells through its activity as a membrane receptor (241). In addition, cell surface β4GalT-I is suggested to induce multidrug resistance through the hedgehog pathway in the human leukemic cell line (242). These results reinforce the β4GalT-I as an unexpected target for inhibition due to its role in the carcinogenic process at intra and extracellular levels.
The design of the first β4GalT-I activity inhibitors were guided by the substrate specificity and mutagenesis studies of commercially available bovine β4GalT-I. Phosphonate donor substrate analogs were proposed to be interesting scaffolds since the phosphonic group would mimic the biphosphate of UDP and complex with the Mn+2 divalent ion present in the binding pocket of the enzyme. Nevertheless, of the synthesized adenosine 5´-phosphoric α-d-glucopyranosylphosphonic anhydride, guanosine 5´-phosphoric α-d-mannopyranosylphosphonic anhydride, and uridine-5´-phosphoric-α-galactopyranosylphosphonic anhydride (23; Scheme 1), only the last one inhibited β4GalT-I, with an apparent Ki of 165 μM. The compounds carrying hexose isomers other than Gal bound to purines did not display β4GalT-I inhibitory activity but showed slight cytotoxicity against B- and T-lymphoblastic leukemia cells (243), probably by a mechanism independent of β4GalT. The substitution of the diphosphate anhydride group by a hydroxylphosphinylmethylphosphate (24; Scheme 1) lowered the Ki value from 165 to 97 μM, but still did not show any in vitro antitumor activity (244). In addition, other molecular groups were substituted to mimic the interaction between the pyrophosphate of UDP and the metal ion of β4GalT active site. The substitution of pyrophosphate by a malonyl group yielded a compound (25; Scheme 1) with poor inhibitory activity, whereas the replacement by a Glc was more effective, resulting in a compound (26; Scheme 1) with satisfactory inhibitory activity (Ki = 119.6 μM) (245).
A novel compound was designed based on the model of an SN2-like transition state of two substrates for β4GalT activity. This model has two strategic characteristics: the use of a natural UDP as the leaving group instead of phosphonate and the linking of the acceptor (GlcNAcβ-OMe) and the donor (Gal moiety) via a methylene ether. The resulting tricomponent bisubstrate analog 27 (Scheme 1) showed a remarkably potent inhibitory activity toward bovine β4GalT-I, displaying Ki values of 1.35 μM for acceptor and 3.3 μM for donor substrate (246).
In an effort to avoid nonspecific binding of UDP-Gal-based inhibitors to other galactosyltransferases, Chung and coworkers developed selective β4GalT inhibitors based on the acceptor substrate. Such compounds had GlcNAc attached to aromatic aglycone moieties (28; Scheme 1) and exhibited Ki of 3.5–9.5 μM. The replacement of the aromatic group to other aglycones resulted in poor inhibition, suggesting that the aromatic ring is responsible for the drastic increase in the binding affinity of inhibitors (247).
Takayama and coworkers have used rapid electrospray mass spectrometry to identify selective inhibitors from a library based on the donor-sugar nucleotide UDP-Gal. From the iminocyclitols and phosphonates screened the compounds (29; Scheme 1) and UDP-2-fluoro-Gal (UDP-2-F-Gal, 30; Scheme 1) were the most effective, displaying 95% and 90% of enzyme inhibition at 1 mM. The UDP-2-fluoro-Gal also exhibits an IC50 of 202 μM (248).
The publication of bovine β4GalT-I crystal structures presenting the unbound (249) and the UDP-Gal bound forms (250) was of outstanding importance in demonstrating that a loop containing the Trp314 (equivalent to Trp310 in human β4GalT-I) moves toward the active site upon donor substrate binding. Based on this information, Takaya and coworkers have substituted C2 or C6 of Gal from UDP-Gal with a flexible trioxadecanyl group linked to a terminal naphthyl group in order to enhance the stacking interaction with Trp314 and to block the acceptor substrate entrance. Indeed, modification at the C6 position (31; Scheme 1) was more effective than the C2 position (32; Scheme 1) of Gal residue, yielding the strongest competitive inhibitor (Ki of 1.86 μM) against UDP-Gal (Km of 4.91 μM) thus far known (251).
Aiming to improve the design of biologically applicable inhibitors of galactosylation, a large series of modifications was made on GlcNAc to optimize the acceptor inhibitor geometry (252). From these studies, Brockhausen and coworkers made several observations that must be taken into account on the design of β4GalT targeted inhibitors. However, the best Ki values obtained, 0.06 mM for 1-thioGlcNAcβ-(2-naphthyl) (33; Scheme 1) and 0.01 mM for 1-thio-N-butyrylGlcNAcβ-(2-naphthyl) (34; Scheme 1), were almost 10-fold higher than inhibitors reported previously (251). The compound 1-thio-N-butyrylGlcNAcβ-(2-naphthyl) (34; Scheme 1) was further observed to inhibit 68–95% of human β4GalT activity from a series of tumor cell line lysates, without compromising the activity of other enzymes (253). In addition, the specificity of (34; Scheme 1) β4GalT was confirmed by testing the enzyme activity of recombinant glycosyltransferases with and without the compound.
In contrast with galactosyltransferase, the relation of IGnTs or β3GnTs expression in cancer has not been investigated well. Only recently, a new member of IGnT family, β3GnT-VIII, was cloned, characterized, and shown to have its transcript levels significantly increased on colorectal cancer tissues compared to normal tissues (254). The same scenario can be observed for I-branching IGnTs. The pioneering work from Zhang et al. (255) has demonstrated that IGnT-2 is overexpressed in highly metastatic breast cancer cell lines of human and mouse origin and basal-like breast tumor samples. Moreover, IGnT-2 expression was significantly correlated to the metastatic phenotype in breast tumor samples and its ectopic expression enhanced cell detachment, adhesion to endothelial cells, cell migration, and invasion in vitro and lung metastasis of breast cancer cells in vivo. The knockdown of IGnT-2 resulted in the elimination of metastatic aspects making this enzyme a promising target for metastatic breast cancer (255).
Information on IGnTs and IGnTs inhibitors are more limited than reports about their biological roles. In fact, there are no inhibitors that target extension or branching of N-acetylglucosaminyltransferases by acting on O-glycans. Features for developing inhibitors were based on studying the characterrization of the isolated and purified enzymes activity, from which β3GnT was observed to be inhibited by the product UDP (~70% inhibition at 1 mM) and by 4-thiouridine diphosphate (256). I-branching β6GnT was also inhibited by UDP and UTP, indicating that the uracil moiety and the number of phosphodiesters appear to be important for the enzyme binding and activity (257). Nonetheless, potent inhibitors can come from tests with UDP-GlcNAc analogs (258), used as inhibitors of others N-acetylglucosaminyltransferases, or from acceptor substrate analogs, which provide potent scaffolds for N-glycan branching GnT-V inhibition.
Lewis and ABH Blood Group Antigens
Lewis and ABH antigens are terminal oligosaccharide structures that can be attached to βGal, found in O- and N-linked glycans and glycolipids (Figure 3). They are termed “blood group antigens” because they were initially discovered on red cells, although they were afterward observed on the surface of many other cell types, such as epithelial and endothelial cells (259).
There are three main types of disaccharide precursors of ABH and Lewis antigens found in glycoproteins: (i) type 1, Galβl-3GlcNAcβ; (ii) type 2, Galβl-4GlcNAcβ; and (iii) type 3, Galβl-3GalNAcα. Type 1 and type 2 are found in lacto series of glycolipids and in N- and O-glycans. In O-glycans, type 1 originated by the activity of β3-galactosyltransferase 5 (β3GalT-5), the unique isoform known in humans, and type 2 consists of terminal LacNAc moieties, synthesized by a set of β4-GalTs as already mentioned in the previous section. The disaccharide of type 3 represents the core 1 or T antigen of O-glycans and is found exclusively in these structures (Figure 3) (260).
The H antigen is generated by α1-2 fucosyltransferases (α1-2FucT), encoded in humans by FUT1 and FUT2 genes, which adds an αFuc on terminal βGal residues of the 3 types of disaccharide precursors. The A and B antigens are originate from the same H precursor by the action of A and B enzymes that branch from the Galβ at C3 by adding an GalNAcα or Galα residue, respectively (261).
The increase of H-antigen correlates with the loss of A- and B-transferases expression and activity in red blood cells at the preleukemic stage (262), bladder malignant urothelium cells (263), and oral epithelia (264). The loss of blood group antigen A expression has a negative prognostic impact in stage I non-small cell lung cancer, especially in patients with adenocarcinoma (265). The increase in H epitope in cancer cells is related to α1-2FucT overexpression. Upregulated expression of FUT1 and β-N-acetylgalactosaminyltransferase and prostate-specific antigen (PSA) levels are biomarkers for prostatic cancer (266, 267). Moreover, 1,2-fucosylated glycans, at the surface of rat colon carcinoma cells, were associated with increased tumorigenicity, resistance to natural killer cytotoxicity and apoptosis (268, 269). Besides, a glucose analog of H-antigen, the 2-fucosyl lactose (H-2g), stimulates angiogenesis in endothelial cells (270).
Lewis antigens can be derivated only from type 1 and 2 precursors, by the addition of a αFuc to the position 4 or 3 of GlcNAc, giving the Lea or Lex antigens, respectively. Addition of other Fuc in the same position of the H types 1 and 2 antigens give the Leb and Ley antigens, respectively. The difucosylated Leb and Ley can also be synthesized from A and B antigens by the action of the same N-acetylglucosaminide: 3/4-αfucosyltransferases (α1-3/α1-4FucT) that participate in Lea and Lex biosynthesis (Figure 3) (271).
Cancers that are known to express LeY include ovarian, pancreatic, esophageal, stomach, colon, rectal, and lung cancers (272–276). Because the expression of LeY in normal tissues is low and it is highly expressed in cancer cells it is a good potential therapeutic target (276, 277). In fact, the use of LeY in cancer vaccines (278, 279) and immunoconjugated chemotherapy (280) are in progress. In addition, LeY, together with FUT1 gene upregulation, is suggested to be involved in cell migration required for the early steps of tumor angiogenesis (281). Yan et al. (272) found that overexpression of α1-2FucT in ovarian cancer cell line correlates with overexpression of LeY, increased invasiveness and poor prognosis for such types of cancer. FUT1 also modulates cell proliferation in the HER2-positive cancer cell line NCI-N87. Authors suggest that knockdown of FUT1 down regulates HER2 signaling via EGFR down regulation (282). Thus, FUT1 may serve as a new molecular target for HER2-overexpressing human cancers with activated EGFR signaling. Consequently, to inhibiting α1-2FucT-mediated cancer processes is an important matter, which has been the aim of several researchers. Palcic et al. (283) designed the first bisubstrate analog inhibitor (35; Scheme 1), containing structural elements of both, donor and acceptor of α1-2FucTs. The analog of the postulated transition-state was a potent inhibitor of porcine submaxillary α1-2FucTs in cell free systems, Ki = 2.3 μM. The same group demonstrated the inhibition of porcine submaxillary α1-2FucTs by deoxygenated oligosaccharide acceptor analogs (36; Scheme 1) with a Ki = 0.80 mM, but was not tested in cell systems (211).
The sialylated variants of Lea and Lex, sialyl-Lea (SLea) and sialyl-Lex (SLex), can be synthetized by the addition of a sialic acid in α2-3 linkage either onto Lea or Lex antigens or onto type 1 and type 2 precursors followed by its fucosylation on C3 or C4 of the GlcNAc moiety (Figure 3). Cancer cells have the ability to mimic immune cells, where they migrate to inflamed sites by expressing SLea and sialyl SLex on its surface which allows them to attach onto E- and P-selectins expressed by the endothelium (284, 285). Such sialylated oligosaccharides may also mediate metastasis by forming cellular thrombus through the binding to platelet-borne P-selectins, which can block leukocyte infiltration into tumors (285).
The expression of SLea was shown to be useful as a marker for colorectal carcinoma aggressiveness and prognosis (286) and is associated with metastasis of pancreas carcinoma cells (287), lung cancer cells, and liver cancer cells by the property of the saccharide to adhere to ELAM-1 of the endothelium (288). Overexpression of SLex correlates to tumor aggressiveness and prognosis in another set of cancer cells, consisting of prostate (289), colorectal (290), liver, and lung cancers (288, 291). In some types of tumors, as in breast cancer and melanoma, the appearance of both SLea and SLex is associated with the tumor emergence and higher expression of these epitopes correlates to the degree of malignancy (292, 293).
The increased expression of SLex and SLea antigens on tumor cells could be due to the upregulation of the genes encoding the enzymes responsible for the saccharide biosynthesis, or due to deficiency in the enzymes responsible for sulfation, which normally lead to the generation of Sialyl 6-Sulfo Lewisx antigen and α2-6-sialylation resulting in the production of disialyl Lea, present on the normal epithelium (294). In fact, mRNA levels of α1-3/1-4 FucT and ST3Gal are shown to be higher in colon adenocarcinoma cells (295). Therefore, the enzymes that participate on the biosynthesis of SLe antigens are targets for drug design.
There are six sialyltransferases reported to act upon C3 from terminal βGal residues, called ST3Gal-I to VI. Like α1-3/1-4 FucT, sialyltransferases also exhibits preferences toward substrates: ST3Gal-I and -II have a clear preference for the type 3. ST3Gal-III works on types 1 and 2 with a preference for type 1, whereas the opposite is observed for ST3Gal-IV. ST3Gal-V uses lactosylceramide as a substrate to generate the glycolipid GM3, while ST3Gal-VI works on type 2 precursors exclusively (296–298).
ST3Gal III is directly implicated in the enhancement of surface SLex levels in pancreatic ductal adenocarcinoma and plays a key role in several steps of tumor progression such as E-selectin adhesion, migration and metastasis formation (299). Increased expression of ST3Gal III in pancreatic ductal adenocarcinoma is concurrent with the increase of ST3Gal IV, suggesting their involvement in this pathology is probably due to the promotion of SLex or SLea biosynthesis (144).
Sialyltransferases are an evident target for drug design strategies and their expression can be a useful prognostic marker of malignant disease. In 1975, Bernacki (300) indicated that CMP inhibits the transfer of Neu5Ac from CMP-Neu5Ac to appropriate acceptor substrates by using rat liver microsommes as the source of sialyltransferase. From this finding, several donor, transition-states, bisubstrate, and acceptor analogs based on CMP-Neu5Ac have been synthesized and evaluated for inhibitory activity against sialyltransferase (301, 302). Schmidt and colleagues have extensively studied inhibitors of sialyltransferase based on CMP-Neu5Ac and its oxocarbenium transition state in ST-catalyzed reactions. They revealed that R-hydroxyphosphonate esters of CMP (37; Scheme 1), with a flattened ring system, are very strong sialyltransferase inhibitors, having Ki values in the nanomolar range (303). Concerning the disaccharide nucleoside, two compounds, inhibited sialyltransferase on the lymphocyte surface, which resulted in the decrease incorporation of sialic acid into endogenous cellular acceptors or into exogenous desialylated glycoconjugates (304). Sialyltransferase inhibition by the sialic acid-nucleoside analog (38; Scheme 1) (301) in a colon adenocarcinoma mouse model resulted in a significant prevention of lung metastasis and prolonged the survival.
Non-substrate-like inhibitors have also been published as inhibitors of sialyltransferase activity. A lithocholic acid analog, Lith-O-Asp (39; Scheme 1), inhibited the sialylation of integrin-β1. In addition, Lith-O-Asp altered protein expression levels and the phosphorylation state of various proteins involved in crucial metastasis and angiogenesis pathways, such as vimentin and RNH1, which inhibited angiogenesis and tumor growth in vivo, through angiogenin inhibition (305). Soyasaponin I (40; Scheme 1), a natural compound purified from soybean, has been shown to be a ST3Gal I inhibitor (306). It effectively inhibited breast cancer cells and murine melanoma cells from metastasis in vivo (307, 308). Since, soyasaponin I is difficult to obtain amounts large enough for cancer treatment, a series of lithocholic acid analogs derived from soyasaponin I were synthesized (41; Scheme 1) (309). The compound AL10 (42; Scheme 1), an ST3Gal and ST6Gal inhibitor, inhibited cell migration and invasion in vitro and suppressed lung metastasis in animal studies (310).
Whereas ST3Gal is the most obvious target among the enzymes that participate in Lewis biosynthesis, there is evidence that the inhibition of glycosyltransferases downstream can be an alternative for chemotherapy focused on Lewis glycans. The suppression of β3GalT5 gene was shown to reduce SLea expression in pancreas adenocarcinoma cell line (311). Other studies have correlated the expression of β3GalT5 with cancer epitopes, such as extended type 1 chains on lactosylceramides of human colon carcinoma (312) and CA19.9 in human pancreatic cancer tissue (313). Recently, Gao and coworkers have reached almost 100% inhibition of β3GalT5 activity with bivalent imizadolium salts (43; Scheme 1), nevertheless, these compounds presented little specificity against the enzyme (314). Conversely, the variety and number of inhibitors targeted to α1-3/1-4FucT is far greater than those targeted to β3GalT considering the extensive correlation between those enzymes and cancer progression.
Currently, the products of six FUT genes able to catalyze the addition of a Fuc residue in α1-3 or α1-4 position are known in humans. The FUT3 and FUT5 enzymes can use both type 1 and 2 precursors as substrates; FUT3 has a marked preference for type 1 and FUT5 for type 2. The FUT4, FUT6, FUT7, and FUT9 enzymes catalyze the addition of a Fuc exclusively onto type 2 precursor. In addition, FUT3, FUT4, FUT5, and FUT6 are able to fucosylate internal LacNAc motifs from poly-LacNAc structures (315), giving rise to more complex structures like dimeric Lex or trimeric Ley that can be further modified.
For instance, α1-3/1-4FucT protein expression was associated with poor prognosis in various types of cancer (316) and increased E-selectin adhesion and metastatic potential of human lung adenocarcinoma cells (317, 318). Gene knockdown approaches in leukemia T cell that decreased cell adhesion to E-selectin, reinforced this correlation (319). Besides, downregulation of FucT-3 and -5 through shRNA decreased levels of Lewis antigens, adhesion and binding capacities of gastric cells MKN45 (320). Carvalho et al. (321) revealed an increased expression of SLe, FUT5 and FUT6 during cell confluence of MKN45, which associated these enzymes with tumor progression. Guo et al. found that FUT6 was capable in promoting hepatocellular cell growth in vivo and in vitro, by modulating P13K, and also suggesting FUT6 as a promising biomarker and a potential therapeutic target for hepatocellular carcinoma (322). Moreover, another study showed that FUT6 was also increased in prostate cancer. A significant reduction of bone metastasis in a FUT6-induced bone metastasis mouse model of prostate cancer was achieved by using a Fuc mimetic inhibitor 2-F-peracetyl-Fuc to inhibit fucosyltransferase enzyme activity (323). In a recent study, Okeley and coworkers demonstrated that oral treatment with 2-F-Fuc provided complete protection from tumor engraftment in a syngeneic tumor vaccine model. The compound inhibited neutrophil extravasation, and delayed the outgrowth of tumor xenografts in immune-deficient mice (324). Zandberg and colleagues revealed that the mimetic inhibitor, 5T-Fuc (a peracetyletaed 5-thio-Fuc, 44; Scheme 1), blocked the α1-3/1-4FucT activity and decreased SLex on HepG2 cells. Furthermore, the group demonstrated that treatment with 5T-Fuc impaired selectin-mediated cell adhesion (325). The non-substrate related Panosialins A and B (45; Scheme 1) were used as an inhibitor of FUT7 and suppressed the expression of selectin ligands, on human leukemic lymphoma cell line (U937), which inhibits selectin-mediated cell adhesion (326). On the same cell line, a series of peracetylated N-acetyllactosamine (LacNAc) analogs (46; Scheme 1) exhibited an enhanced affinity by FucT- VI caused by a 90% inhibition of SLex expression (327).
Donor substrate mimetic compounds were widely explored as inhibitors of human FucTs. Unnatural sugar nucleotides, UDP-Fuc, ADP-Fuc and CDP-Fuc were tested against FucT-III. Unexpectedly, the enzyme does not only tolerate the exchange of guanosine for adenine but may also accept a pyrimidine base. UDP-Fuc and CDP-Fuc were utilized with lower efficiency than UDP-Fuc, nevertheless, they could act as Fuc donors. A series of GDP-Fuc derivatives was synthesized, purified and characterized in detail for their inhibition kinetics. Compound 47 (Scheme 1) had a Ki of 29 nM for human FucT-VI (328). Addition of hydrophobic moieties to the Fuc C6 seems to yield potent inhibitors. This strategy was explored to identify selective inhibitors for human recombinant FucT-V. It has been shown that both GDP-2F-Fuc and GDP-6F-Fuc (48; Scheme 1) act as competitive inhibitors of FUTs 3, 5, 6, and 7 with Ki values in the low micromolar range (329). Peracetylation of GDP-2F-Fuc improved its cell permeability and dramatically reduced cell surface fucosylation. Treatment of human HL-60 cells with the permethylated GDP-2F-Fuc nearly abolished synthesis of LeX and SLeX and led to significant decreases in E- and P-selectin binding (330).
Several C-glycosides were synthesized as potential inhibitors of FucT (331). Among the compounds tested, the αManp1-3CH2GalNAc (49; Scheme 1) displayed a mixed inhibition of FucT-VI, with respect to both the donor sugar GDP-Fuc and the acceptor LacNAc (332). However, a C-glycosyl ethylphosphonophosphate analog of GDP-Fuc (50; Scheme 1) presented only a weak inhibition against FUT3 (IC50 value of 2 mM). The modest activity was attributed to the α-anomeric configuration of this C-glycosyl analog, which is opposite to the β-configuration of the natural donor substrate GDP-Fuc (333).
Many other molecules synthesized have inhibitory activity: gallic acid (51; Scheme 1) and its derivates were identified as FucT-VII inhibitor (IC50 of 60 nM) (334); stachybotrydial (52; Scheme 1), isolated from the culture broth of the fungus Stachybotrys cylindrospora, were identified to be a potent FucT-V inhibitor (Ki of 10.7 μM) (335). Together these results point to the potential therapeutic applications for molecules that selectively block the endogenous generation of fucosylated glycan structures.
Conclusion
The potential of glycans as tools in cancer diagnosis and prognosis is unquestionable. This overview presents the importance of glycan inhibitors as possible anti-cancer drug targets. Several hundred targets exist for the development of inhibitors. The repertoire of available compounds, although extensive, contains few agents that have the affinity and specificity required for converting a laboratory reagent into a drug. However, the few drugs that have been developed have already proven their value as therapeutic agents. These success stories only represent the beginning of the importance of the glycobiology field to anti-cancer chemotherapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fonc.2015.00138
Scheme 1. Inhibitors of enzymes involved in the biosynthesis of glycans derived from the HBP.
References
1. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem (2011) 80:825–58. doi: 10.1146/annurev-biochem-060608-102511
2. Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature (2014) 511(7509):319–25. doi:10.1038/nature13535
3. Alisson-Silva F, de Carvalho Rodrigues D, Vairo L, Asensi KD, Vasconcelos-dos-Santos A, Mantuano NR, et al. Evidences for the involvement of cell surface glycans in stem cell pluripotency and differentiation. Glycobiology (2014) 24(5):458–68. doi:10.1093/glycob/cwu012
4. Freire-de-Lima L, Alisson-Silva F, Carvalho ST, Takiya CM, Rodrigues MM, DosReis GA, et al. Trypanosoma cruzi subverts host cell sialylation and may compromise antigen-specific CD8+ T cell responses. J Biol Chem (2010) 285(18):13388–96. doi:10.1074/jbc.M109.096305
5. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer (2005) 5(7):526–42. doi:10.1038/nrc1649
6. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi:10.1126/science.123.3191.309
7. Carvalho KC, Cunha IW, Rocha RM, Ayala FR, Cajaíba MM, Begnami MD, et al. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics (2011) 66(6):965–72. doi:10.1590/S1807-59322011000600008
8. Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer (2002) 2(9):683–93. doi:10.1038/nrc882
9. Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene (2006) 25(34):4633–46. doi:10.1038/sj.onc.1209597
10. Muley P, Olinger A, Tummala H. 2-Deoxyglucose induces cell cycle arrest and apoptosisin colorectal cancer cells independent of its glycolysis inhibition. Nutr Cancer (2015) 67(3):514–22. doi:10.1080/01635581.2015.1002626
11. Jung DW, Kim WH, Park SH, Lee J, Kim J, Su D, et al. A unique small molecule inhibitor of enolase clarifies its role in fundamental biological processes. ACS Chem Biol (2013) 8(6):1271–82. doi:10.1021/cb300687k
12. Li S, Li Y, Hu R, Li W, Qiu H, Cai H, et al. The mTOR inhibitor AZD8055 inhibits proliferation and glycolysis in cervical cancer cells. Oncol Lett (2013) 5(2):717–21. doi:10.3892/ol.2012.1058
13. Datta S, Li J, Mahdi F, Jekabsons MB, Nagle DG, Zhou YD. Glycolysis inhibitor screening identifies the bis-geranylacylphloroglucinol protonophore moronone from Moronobea coccinea. J Nat Prod (2012) 75(12):2216–22. doi:10.1021/np300711e
14. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem (1991) 266(8):4706–12.
15. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7(1):11–20. doi:10.1016/j.cmet.2007.10.002
16. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res (2013) 73(16):5277–87. doi:10.1158/0008-5472.CAN-13-0549
17. Kolm-Litty V, Sauer U, Nerlich A, Lehmann R, Schleicher ED. High glucose-induced transforming growth factor beta 1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J Clin Invest (1998) 101(1):160–9. doi:10.1172/JCI119875
18. Zhou L, Xue H, Yuan P, Ni J, Yu C, Huang Y, et al. Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin Exp Pharmacol Physiol (2010) 37(9):e152–7. doi:10.1111/j.1440-1681.2010.05421.x
19. Hakomori SI. Glycosynaptic microdomains controlling tumor cell phenotype through alteration of cell growth, adhesion, and motility. FEBS Lett (2010) 584(9):1901–6. doi:10.1016/j.febslet.2009.10.065
20. Freire-de-Lima L, Gelfenbeyn K, Ding Y, Mandel U, Clausen H, Handa K, et al. Involvement of O-glycosylation defining oncofetal fibronectin in epithelial-mesenchymal transition process. Proc Natl Acad Sci U S A (2011) 108(43):17690–5. doi:10.1073/pnas.1115191108
21. Alisson-Silva F, Freire-de-Lima L, Donadio JL, Lucena MC, Penha L, Sá-Diniz JN, et al. Increase of O-glycosylated oncofetal fibronectin in high glucose-induced epithelial-mesenchymal transition of cultured human epithelial cells. PLoS One (2013) 8(4):e60471. doi:10.1371/journal.pone.0060471
22. Dennis JW, Nabi IR, Demetriou M. Metabolism, cell surface organization, and disease. Cell (2009) 139(7):1229–41. doi:10.1016/j.cell.2009.12.008
23. O’Donnell N, Zachara NE, Hart GW, Marth JD. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol (2004) 24(4):1680–90. doi:10.1128/MCB.24.4.1680-1690.2004
24. Dias WB, Hart GW. O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol Biosyst (2007) 3(11):766–72. doi:10.1039/b704905f
25. Lowe DA, Emre M, Frey P, Kelly PH, Malanowski J, McAllister KH, et al. Progress in pharmacology of cerebral ischemia. Pharmazie (1994) 49(12):939–41.
26. Ostrowski A, van Aalten DM. Chemical tools to probe cellular O-GlcNAc signalling. Biochem J (2013) 456(1):1–12. doi:10.1042/BJ20131081
27. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature (2007) 446(7139):1017–22. doi:10.1038/nature05815
28. Whelan SA, Dias WB, Thiruneelakantapillai L, Lane MD, Hart GW. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked beta-N-acetylglucosamine in 3T3-L1 adipocytes. J Biol Chem (2010) 285(8):5204–11. doi:10.1074/jbc.M109.077818
29. Dias WB, Cheung WD, Wang Z, Hart GW. Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J Biol Chem (2009) 284(32):21327–37. doi:10.1074/jbc.M109.007310
30. Dias WB, Cheung WD, Hart GW. O-GlcNAcylation of kinases. Biochem Biophys Res Commun (2012) 422(2):224–8. doi:10.1016/j.bbrc.2012.04.124
31. Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer (2011) 11(9):678–84. doi:10.1038/nrc3114
32. Ma Z, Vosseller K. O-GlcNAc in cancer biology. Amino Acids (2013) 45(4):719–33. doi:10.1007/s00726-013-1543-8
33. de Queiroz RM, Carvalho E, Dias WB. O-GlcNAcylation: the sweet side of the cancer. Front Oncol (2014) 4:132. doi:10.3389/fonc.2014.00132
34. Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene (2010) 29(19):2831–42. doi:10.1038/onc.2010.41
35. Krześlak A, Forma E, Bernaciak M, Romanowicz H, Bryś M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin Exp Med (2012) 12(1):61–5. doi:10.1007/s10238-011-0138-5
36. Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X, et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta (2011) 1812(4):514–9. doi:10.1016/j.bbadis.2011.01.009
37. Ma ZY, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappa B activity in pancreatic cancer cells. J Biol Chem (2013) 288(21):15121–30. doi:10.1074/jbc.M113.470047
38. Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem (2012) 287(14):11070–81. doi:10.1074/jbc.M111.302547
39. Lynch TP, Reginato MJ. O-GlcNAc transferase a sweet new cancer target. Cell Cycle (2011) 10(11):1712–3. doi:10.4161/cc.10.11.15561
40. Zhu Q, Zhou L, Yang Z, Lai M, Xie H, Wu L, et al. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol (2012) 29(2):985–93. doi:10.1007/s12032-011-9912-1
41. Rozanski W, Krzeslak A, Forma E, Brys M, Blewniewski M, Wozniak P, et al. Prediction of bladder cancer based on urinary content of MGEA5 and OGT mRNA level. Clin Lab (2012) 58(5–6):579–83.
42. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (2012) 337(6097):975–80. doi:10.1126/science.1222278
43. Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci (2010) 35(10):547–55. doi:10.1016/j.tibs.2010.04.005
44. Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J (2010) 29(22):3787–96. doi:10.1038/emboj.2010.254
45. Jin FZ, Yu C, Zhao DZ, Wu MJ, Yang Z. A correlation between altered O-GlcNAcylation, migration and with changes in E-cadherin levels in ovarian cancer cells. Exp Cell Res (2013) 319(10):1482–90. doi:10.1016/j.yexcr.2013.03.013
46. Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J Biol Chem (2008) 283(49):33935–41. doi:10.1074/jbc.M806199200
47. Clarke AJ, Hurtado-Guerrero R, Pathak S, Schüttelkopf AW, Borodkin V, Shepherd SM, et al. Structural insights into mechanism and specificity of O-GlcNAc transferase. EMBO J (2008) 27(20):2780–8. doi:10.1038/emboj.2008.186
48. Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature (2011) 469(7331):564–7. doi:10.1038/nature09638
49. Jiang J, Lazarus MB, Pasquina L, Sliz P, Walker S. A neutral diphosphate mimic crosslinks the active site of human O-GlcNAc transferase. Nat Chem Biol (2012) 8(1):72–7. doi:10.1038/nchembio.711
50. Schimpl M, Zheng X, Borodkin VS, Blair DE, Ferenbach AT, Schüttelkopf AW, et al. O-GlcNAc transferase invokes nucleotide sugar pyrophosphate participation in catalysis. Nat Chem Biol (2012) 8(12):969–74. doi:10.1038/nchembio.1108
51. Macauley MS, Vocadlo DJ. Increasing O-GlcNAc levels: an overview of small-molecule inhibitors of O-GlcNAcase. Biochim Biophys Acta (2010) 1800(2):107–21. doi:10.1016/j.bbagen.2009.07.028
52. Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol (2011) 7(3):174–81. doi:10.1038/nchembio.520
53. Borodkin VS, Schimpl M, Gundogdu M, Rafie K, Dorfmueller HC, Robinson DA, et al. Bisubstrate UDP-peptide conjugates as human O-GlcNAc transferase inhibitors. Biochem J (2014) 457(3):497–502. doi:10.1042/BJ20131272
54. Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A (2002) 99(16):10231–3. doi:10.1073/pnas.172380699
55. Laferte S, Dennis JW. Purification of two glycoproteins expressing beta 1-6 branched Asn-linked oligosaccharides from metastatic tumour cells. Biochem J (1989) 259(2):569–76.
56. Li WP, Zuber C, Heitz PU, Roth J. Cytochemical staining for beta 1,6 branching of asparagine-linked oligosaccharides in variants of metastatic human colon carcinoma cells. Am J Pathol (1994) 145(2):470–80.
57. Arnold JN, Saldova R, Galligan MC, Murphy TB, Mimura-Kimura Y, Telford JE, et al. Novel glycan biomarkers for the detection of lung cancer. J Proteome Res (2011) 10(4):1755–64. doi:10.1021/pr101034t
58. Saldova R, Royle L, Radcliffe CM, Abd Hamid UM, Evans R, Arnold JN, et al. Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology (2007) 17(12):1344–56. doi:10.1093/glycob/cwm100
59. Saldova R, Reuben JM, Abd Hamid UM, Rudd PM, Cristofanilli M. Levels of specific serum N-glycans identify breast cancer patients with higher circulating tumor cell counts. Ann Oncol (2011) 22(5):1113–9. doi:10.1093/annonc/mdq570
60. Saldova R, Fan Y, Fitzpatrick JM, Watson RW, Rudd PM. Core fucosylation and alpha2-3 sialylation in serum N-glycome is significantly increased in prostate cancer comparing to benign prostate hyperplasia. Glycobiology (2011) 21(2):195–205. doi:10.1093/glycob/cwq147
61. Saldova R, Struwe WB, Wynne K, Elia G, Duffy MJ, Rudd PM. Exploring the glycosylation of serum CA125. Int J Mol Sci (2013) 14(8):15636–54. doi:10.3390/ijms140815636
62. Zhao YP, Ruan CP, Wang H, Hu ZQ, Fang M, Gu X, et al. Identification and assessment of new biomarkers for colorectal cancer with serum N-glycan profiling. Cancer (2012) 118(3):639–50. doi:10.1002/cncr.26342
63. Liu L, Yan B, Huang J, Gu Q, Wang L, Fang M, et al. The identification and characterization of novel N-glycan-based biomarkers in gastric cancer. PLoS One (2013) 8(10):e77821. doi:10.1371/journal.pone.0077821
64. Liu X, Nie H, Zhang Y, Yao Y, Maitikabili A, Qu Y, et al. Cell surface-specific N-glycan profiling in breast cancer. PLoS One (2013) 8(8):e72704. doi:10.1371/journal.pone.0072704
65. Kamiyama T, Yokoo H, Furukawa J, Kurogochi M, Togashi T, Miura N, et al. Identification of novel serum biomarkers of hepatocellular carcinoma using glycomic analysis. Hepatology (2013) 57(6):2314–25. doi:10.1002/hep.26262
66. Nouso K, Amano M, Ito YM, Miyahara K, Morimoto Y, Kato H, et al. Clinical utility of high-throughput glycome analysis in patients with pancreatic cancer. J Gastroenterol (2013) 48(10):1171–9. doi:10.1007/s00535-012-0732-7
67. Wang X, He H, Zhang H, Chen W, Ji Y, Tang Z, et al. Clinical and prognostic implications of beta1, 6-N-acetylglucosaminyltransferase V in patients with gastric cancer. Cancer Sci (2013) 104(2):185–93. doi:10.1111/cas.12049
68. Sato C, Kim JH, Abe Y, Saito K, Yokoyama S, Kohda D. Characterization of the N-oligosaccharides attached to the atypical Asn-X-Cys sequence of recombinant human epidermal growth factor receptor. J Biochem (2000) 127(1):65–72. doi:10.1093/oxfordjournals.jbchem.a022585
69. Zheng M, Fang H, Hakomori S. Functional role of N-glycosylation in alpha 5 beta 1 integrin receptor. De-N-glycosylation induces dissociation or altered association of alpha 5 and beta 1 subunits and concomitant loss of fibronectin binding activity. J Biol Chem (1994) 269(16):12325–31.
70. Purchio AF, Cooper JA, Brunner AM, Lioubin MN, Gentry LE, Kovacina KS, et al. Identification of mannose 6-phosphate in two asparagine-linked sugar chains of recombinant transforming growth factor-beta 1 precursor. J Biol Chem (1988) 263(28):14211–5.
71. Patnaik SK, Potvin B, Carlsson S, Sturm D, Leffler H, Stanley P. Complex N-glycans are the major ligands for galectin-1, -3, and -8 on Chinese hamster ovary cells. Glycobiology (2006) 16(4):305–17. doi:10.1093/glycob/cwj063
72. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol (2007) 7(4):255–66. doi:10.1038/nri2056
73. Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, et al. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science (2004) 306(5693):120–4. doi:10.1126/science.1102109
74. Lajoie P, Partridge EA, Guay G, Goetz JG, Pawling J, Lagana A, et al. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol (2007) 179(2):341–56. doi:10.1083/jcb.200611106
75. Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature (2001) 409(6821):733–9. doi:10.1038/35055582
76. Fernandes B, Sagman U, Auger M, Demetrio M, Dennis JW. Beta-1-6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res (1991) 51(2):718–23.
77. Boscher C, Zheng YZ, Lakshminarayan R, Johannes L, Dennis JW, Foster LJ, et al. Galectin-3 protein regulates mobility of N-cadherin and GM1 ganglioside at cell-cell junctions of mammary carcinoma cells. J Biol Chem (2012) 287(39):32940–52. doi:10.1074/jbc.M112.353334
78. Boscher C, Nabi IR. Galectin-3-and phospho-caveolin-1-dependent outside-in integrin signaling mediates the EGF motogenic response in mammary cancer cells. Mol Biol Cell (2013) 24(13):2134–45. doi:10.1091/mbc.E13-02-0095
79. Saldova R, Asadi Shehni A, Haakensen VD, Steinfeld I, Hilliard M, Kifer I, et al. Association of N-glycosylation with breast carcinoma and systemic features using high-resolution quantitative UPLC. J Proteome Res (2014) 13(5):2314–27. doi:10.1021/pr401092y
80. Banerjee DK. N-glycans in cell survival and death: cross-talk between glycosyltransferases. Biochim Biophys Acta (2012) 1820(9):1338–46. doi:10.1016/j.bbagen.2012.01.013
81. Schachter H. Complex N-glycans: the story of the “yellow brick road”. Glycoconj J (2014) 31(1):1–5. doi:10.1007/s10719-013-9507-5
82. Yan Q, Lennarz WJ. Oligosaccharyltransferase: a complex multisubunit enzyme of the endoplasmic reticulum. Biochem Biophys Res Commun (1999) 266(3):684–9. doi:10.1006/bbrc.1999.1886
83. Elbein AD. Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains. Annu Rev Biochem (1987) 56:497–534. doi:10.1146/annurev.bi.56.070187.002433
84. Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci (2010) 35(2):74–82. doi:10.1016/j.tibs.2009.10.001
85. Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res (2008) 68(10):3803–9. doi:10.1158/0008-5472.CAN-07-6389
86. Contessa JN, Bhojani MS, Freeze HH, Ross BD, Rehemtulla A, Lawrence TS. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin Cancer Res (2010) 16(12):3205–14. doi:10.1158/1078-0432.CCR-09-3331
87. Siddals KW, Allen J, Sinha S, Canfield AE, Kalra PA, Gibson JM. Apposite insulin-like growth factor (IGF) receptor glycosylation is critical to the maintenance of vascular smooth muscle phenotype in the presence of factors promoting osteogenic differentiation and mineralization. J Biol Chem (2011) 286(19):16623–30. doi:10.1074/jbc.M110.202929
88. Hou H, Sun H, Lu P, Ge C, Zhang L, Li H, et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse xenograft models of human hepatocellular carcinoma. Mol Cancer Ther (2013) 12(12):2874–84. doi:10.1158/1535-7163.MCT-13-0201
89. de-Freitas-Junior JC, Bastos LG, Freire-Neto CA, Rocher BD, Abdelhay ES, Morgado-Díaz JA. N-glycan biosynthesis inhibitors induce in vitro anticancer activity in colorectal cancer cells. J Cell Biochem (2012) 113(9):2957–66. doi:10.1002/jcb.24173
90. Yamamori T, Meike S, Nagane M, Yasui H, Inanami O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett (2013) 587(20):3348–53. doi:10.1016/j.febslet.2013.08.030
91. Banerjee A, Lang JY, Hung MC, Sengupta K, Banerjee SK, Baksi K, et al. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J Biol Chem (2011) 286(33):29127–38. doi:10.1074/jbc.M110.169771
92. Elbein AD. Glycosylation inhibitors for N-linked glycoproteins. Methods Enzymol (1987) 138:661–709.
93. Irimura T, Gonzalez R, Nicolson GL. Effects of tunicamycin on B16-metastatic melanoma cell-surface glycoproteins and blood-borne arrest and survival properties. Cancer Res (1981) 41(9):3411–8.
94. Mareel MM, Dragonetti CH, Hooghe RJ, Bruyneel EA. Effect of inhibitors of glycosylation and carbohydrate processing on invasion of malignant mouse Mo4 cells in organ-culture. Clin Exp Metastasis (1985) 3(3):197–207. doi:10.1007/BF01786763
95. Pulverer G, Beuth J, Ko HL, Yassin A, Ohshima Y, Roszkowski K, et al. Glycoprotein modifications of sarcoma L-1 tumor-cells by tunicamycin, swainsonine, bromoconduritol or 1-desoxynojirimycin treatment inhibits their metastatic lung colonization in Balb/C-mice. J Cancer Res Clin Oncol (1988) 114(2):217–20. doi:10.1007/BF00417842
96. Guo R, Cheng L, Zhao Y, Zhang J, Liu C, Zhou H, et al. Glycogenes mediate the invasive properties and chemosensitivity of human hepatocarcinoma cells. Int J Biochem Cell Biol (2013) 45(2):347–58. doi:10.1016/j.biocel.2012.10.006
97. Matsumura K, Sakai C, Kawakami S, Yamashita F, Hashida M. Inhibition of cancer cell growth by GRP78 siRNA lipoplex via activation of unfolded protein response. Biol Pharm Bull (2014) 37(4):648–53. doi:10.1248/bpb.b13-00930
98. Humphries MJ, Matsumoto K, White SL, Olden K. Inhibition of experimental metastasis by castanospermine in mice – blockage of 2 distinct stages of tumor colonization by oligosaccharide processing inhibitors. Cancer Res (1986) 46(10):5215–22.
99. Leppa S, Heino J, Jalkanen M. Increased glycosylation of beta(1) integrins affects the interaction of transformed s115 mammary epithelial-cells with laminin-1. Cell Growth Differ (1995) 6(7):853–61.
100. Kang MS, Spencer JP, Elbein AD. Amphomycin inhibition of mannose and glcnac incorporation into lipid-linked saccharides. J Biol Chem (1978) 253(24):8860–6.
101. Nishimura Y. gem-diamine 1-N-iminosugars and related iminosugars, candidate of therapeutic agents for tumor metastasis. Curr Top Med Chem (2003) 3(5):575–91. doi:10.2174/1568026033452492
102. Schraen-Maschke S, Zanetta JP. Role of oligomannosidic N-glycans in the proliferation, adhesion and signalling of C6 glioblastoma cells. Biochimie (2003) 85(1–2):219–29. doi:10.1016/S0300-9084(03)00018-X
103. Sánchez-Fernández EM, Rísquez-Cuadro R, Chasseraud M, Ahidouch A, Ortiz Mellet C, Ouadid-Ahidouch H, et al. Synthesis of N-, S-, and C-glycoside castanospermine analogues with selective neutral alpha-glucosidase inhibitory activity as antitumour agents. Chem Commun (Camb) (2010) 46(29):5328–30. doi:10.1039/c0cc00446d
104. Allan G, Ouadid-Ahidouch H, Sanchez-Fernandez EM, Risquez-Cuadro R, Fernandez JM, Ortiz-Mellet C, et al. New castanospermine glycoside analogues inhibit breast cancer cell proliferation and induce apoptosis without affecting normal cells. PLoS One (2013) 8(10):e76411. doi:10.1371/journal.pone.0076411
105. Saul R, Chambers JP, Molyneux RJ, Elbein AD. Castanospermine, a tetrahydroxylated alkaloid that inhibits beta-glucosidase and beta-glucocerebrosidase. Arch Biochem Biophys (1983) 221(2):593–7. doi:10.1016/0003-9861(83)90181-9
106. Barker MK, Rose DR. Specificity of processing alpha-glucosidase I is guided by the substrate conformation crystallographic and in silico studies. J Biol Chem (2013) 288(19):13563–74. doi:10.1074/jbc.M113.460436
107. Sarkar M, Iliadi KG, Leventis PA, Schachter H, Boulianne GL. Neuronal expression of Mgat1 rescues the shortened life span of Drosophila Mgat11 null mutants and increases life span. Proc Natl Acad Sci U S A (2010) 107(21):9677–82. doi:10.1073/pnas.1004431107
108. Schachter H. Mgat1-dependent N-glycans are essential for the normal development of both vertebrate and invertebrate metazoans. Semin Cell Dev Biol (2010) 21(6):609–15. doi:10.1016/j.semcdb.2010.02.010
109. Schachter H. Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochem Cell Biol (1986) 64(3):163–81. doi:10.1139/o86-026
110. Harpaz N, Schachter H. Control of glycoprotein synthesis. bovine colostrum UDP-N-acetylglucosamine:alpha-D-mannoside beta 2-N-acetylglucosaminyltransferase I. Separation from UDP-N-acetylglucosamine:alpha-D-mannoside beta 2-N-acetylglucosaminyltransferase II, partial purification, and substrate specificity. J Biol Chem (1980) 255(10):4885–93.
111. Yamashita K, Tachibana Y, Ohkura T, Kobata A. Enzymatic basis for the structural-changes of asparagine-linked sugar chains of membrane-glycoproteins of baby hamster-kidney cells induced by polyoma transformation. J Biol Chem (1985) 260(7):3963–9.
112. Buckhaults P, Chen L, Fregien N, Pierce M. Transcriptional regulation of N-acetylglucosaminyltransferase V by the src oncogene. J Biol Chem (1997) 272(31):19575–81. doi:10.1074/jbc.272.31.19575
113. Kang R, Saito H, Ihara Y, Miyoshi E, Koyama N, Sheng Y, et al. Transcriptional regulation of the N-acetylglucosaminyltransferase V gene in human bile duct carcinoma cells (HuCC-T1) is mediated by Ets-1. J Biol Chem (1996) 271(43):26706–12. doi:10.1074/jbc.271.43.26706
114. Hatakeyama S, Amano M, Tobisawa Y, Yoneyama T, Tsuchiya N, Habuchi T, et al. Serum N-glycan alteration associated with renal cell carcinoma detected by high throughput glycan analysis. J Urol (2014) 191(3):805–13. doi:10.1016/j.juro.2013.10.052
115. Pinho SS, Seruca R, Gärtner F, Yamaguchi Y, Gu J, Taniguchi N, et al. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol Life Sci (2011) 68(6):1011–20. doi:10.1007/s00018-010-0595-0
116. Pinho SS, Figueiredo J, Cabral J, Carvalho S, Dourado J, Magalhães A, et al. E-cadherin and adherens-junctions stability in gastric carcinoma: functional implications of glycosyltransferases involving N-glycan branching biosynthesis, N-acetylglucosaminyltransferases III and V. Biochim Biophys Acta (2013) 1830(3):2690–700. doi:10.1016/j.bbagen.2012.10.021
117. Isaji T, Kariya Y, Xu Q, Fukuda T, Taniguchi N, Gu J. Functional roles of the bisecting GlcNAc in integrin-mediated cell adhesion. Methods Enzymol (2010) 480:445–59. doi:10.1016/S0076-6879(10)80019-9
118. Okada T, Ihara H, Ito R, Taniguchi N, Ikeda Y. Bidirectional N-acetylglucosamine transfer mediated by beta-1,4-N-acetylglucosaminyltransferase III. Glycobiology (2009) 19(4):368–74. doi:10.1093/glycob/cwn145
119. Zhao Y, Nakagawa T, Itoh S, Inamori K, Isaji T, Kariya Y, et al. N-acetylglucosaminyltransferase III antagonizes the effect of N-acetyl-glucosaminyltransferase V on alpha3beta1 integrin-mediated cell migration. J Biol Chem (2006) 281(43):32122–30. doi:10.1074/jbc.M607274200
120. Iijima J, Zhao Y, Isaji T, Kameyama A, Nakaya S, Wang X, et al. Cell-cell interaction-dependent regulation of N-acetylglucosaminyltransferase III and the bisected N-glycans in GE11 epithelial cells. Involvement of E-cadherin-mediated cell adhesion. J Biol Chem (2006) 281(19):13038–46. doi:10.1074/jbc.M601961200
121. Shigeta M, Shibukawa Y, Ihara H, Miyoshi E, Taniguchi N, Gu J. beta1,4-N-acetylglucosaminyltransferase III potentiates beta1 integrin-mediated neuritogenesis induced by serum deprivation in Neuro2a cells. Glycobiology (2006) 16(6):564–71. doi:10.1093/glycob/cwj100
122. Beheshti Zavareh R, Sukhai MA, Hurren R, Gronda M, Wang X, Simpson CD, et al. Suppression of cancer progression by MGAT1 shRNA knockdown. PLoS One (2012) 7(9):e43721. doi:10.1371/journal.pone.0043721
123. Isaji T, Gu J, Nishiuchi R, Zhao Y, Takahashi M, Miyoshi E, et al. Introduction of bisecting GlcNAc into integrin alpha(5)beta(1) reduces ligand binding and down-regulates cell adhesion and cell migration. J Biol Chem (2004) 279(19):19747–54. doi:10.1074/jbc.M311627200
124. Lau KS, Dennis JW. N-Glycans in cancer progression. Glycobiology (2008) 18(10):750–60. doi:10.1093/glycob/cwn071
125. Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell (2007) 129(1):123–34. doi:10.1016/j.cell.2007.01.049
126. Pinho SS, Osório H, Nita-Lazar M, Gomes J, Lopes C, Gärtner F, et al. Role of E-cadherin N-glycosylation profile in a mammary tumor model. Biochem Biophys Res Commun (2009) 379(4):1091–6. doi:10.1016/j.bbrc.2009.01.024
127. Elbein AD. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit Rev Biochem (1984) 16(1):21–49. doi:10.3109/10409238409102805
128. Olden K, Breton P, Grzegorzewski K, Yasuda Y, Gause BL, Oredipe OA, et al. The potential importance of swainsonine in therapy for cancers and immunology. Pharmacol Ther (1991) 50(3):285–90. doi:10.1016/0163-7258(91)90046-O
129. Dennis JW. Effects of swainsonine and polyinosinic:polycytidylic acid on murine tumor cell growth and metastasis. Cancer Res (1986) 46(10):5131–6.
130. Humphries MJ, Matsumoto K, White SL, Olden K. Oligosaccharide modification by swainsonine treatment inhibits pulmonary colonization by B16-F10 murine melanoma cells. Proc Natl Acad Sci U S A (1986) 83(6):1752–6. doi:10.1073/pnas.83.6.1752
131. Korczak B, Dennis JW. Inhibition of N-linked oligosaccharide processing in tumor-cells is associated with enhanced tissue inhibitor of metalloproteinases (Timp) gene-expression. Int J Cancer (1993) 53(4):634–9. doi:10.1002/ijc.2910530418
132. Li Z, Huang Y, Dong F, Li W, Ding L, Yu G, et al. Swainsonine promotes apoptosis in human oesophageal squamous cell carcinoma cells in vitro and in vivo through activation of mitochondrial pathway. J Biosci (2012) 37(6):1005–16. doi:10.1007/s12038-012-9265-8
133. de Vries T, Knegtel RM, Holmes EH, Macher BA. Fucosyltransferases: structure/function studies. Glycobiology (2001) 11(10):119r–28r. doi:10.1093/glycob/11.10.119R
134. Muinelo-Romay L, Vázquez-Martín C, Villar-Portela S, Cuevas E, Gil-Martín E, Fernández-Briera A. Expression and enzyme activity of alpha(1,6)fucosyltransferase in human colorectal cancer. Int J Cancer (2008) 123(3):641–6. doi:10.1002/ijc.23521
135. Miyoshi E, Noda K, Ko JH, Ekuni A, Kitada T, Uozumi N, et al. Overexpression of alpha 1-6 fucosyltransferase in hepatoma cells suppresses intrahepatic metastasis after splenic injection in athymic mice. Cancer Res (1999) 59(9):2237–43.
136. Takahashi T, Ikeda Y, Miyoshi E, Yaginuma Y, Ishikawa M, Taniguchi N. alpha 1,6Fucosyltransferase is highly and specifically expressed in human ovarian serous adenocarcinomas. Int J Cancer (2000) 88(6):914–9. doi:10.1002/1097-0215(20001215)88:6<914::AID-IJC12>3.0.CO;2-1
137. Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, et al. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci U S A (2013) 110(2):630–5. doi:10.1073/pnas.1220425110
138. Calderon H, Carpintero M, Garcia-Junceda E, Fernandez-Mayoralas A, Bastida A. Structure/activity relationship of carba- and C-fucopyranosides as inhibitors of an a 1,6-fucosyltransferase by molecular modeling and kinetic studies. Lett Org Chem (2005) 2(3):247–51. doi:10.2174/1570178053765393
139. Bastida A, Fernández-Mayoralas A, Gómez Arrayás R, Iradier F, Carretero JC, García-Junceda E. Heterologous over-expression of alpha-1,6-fucosyltransferase from Rhizobium sp.: application to the synthesis of the trisaccharide beta-D-GlcNAc(1 -> 4)-[alpha-L-Fuc-(1 -> 6)]-D-GlcNAc, study of the acceptor specificity and evaluation of polyhydroxylated indolizidines as inhibitors. Chemistry (2001) 7(11):2390–7. doi:10.1002/1521-3765(20010601)7:11<2390::AID-CHEM23900>3.0.CO;2-0
140. Brockhausen I. Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep (2006) 7(6):599–604. doi:10.1038/sj.embor.7400705
141. Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer (2004) 4(1):45–60. doi:10.1038/nrc1251
142. Akita K, Yoshida S, Ikehara Y, Shirakawa S, Toda M, Inoue M, et al. Different levels of sialyl-Tn antigen expressed on MUC16 in patients with endometriosis and ovarian cancer. Int J Gynecol Cancer (2012) 22(4):531–8. doi:10.1097/IGC.0b013e3182473292
143. Akamine S, Nakagoe T, Sawai T, Tsuji T, Tanaka K, Hidaka S, et al. Differences in prognosis of colorectal cancer patients based on the expression of sialyl Lewisa, sialyl Lewisx and sialyl Tn antigens in serum and tumor tissue. Anticancer Res (2004) 24(4):2541–6.
144. Pérez-Garay M, Arteta B, Llop E, Cobler L, Pagès L, Ortiz R, et al. alpha2,3-sialyltransferase ST3Gal IV promotes migration and metastasis in pancreatic adenocarcinoma cells and tends to be highly expressed in pancreatic adenocarcinoma tissues. Int J Biochem Cell Biol (2013) 45(8):1748–57. doi:10.1016/j.biocel.2013.05.015
145. Schneider F, Kemmner W, Haensch W, Franke G, Gretschel S, Karsten U, et al. Overexpression of sialyltransferase CMP-sialic acid:Galbeta1,3GalNAc-R alpha6-sialyltransferase is related to poor patient survival in human colorectal carcinomas. Cancer Res (2001) 61(11):4605–11.
146. Ju T, Otto VI, Cummings RD. The Tn antigen-structural simplicity and biological complexity. Angew Chem Int Ed Engl (2011) 50(8):1770–91. doi:10.1002/anie.201002313
147. Hua D, Shen L, Xu L, Jiang Z, Zhou Y, Yue A, et al. Polypeptide N-acetylgalactosaminyltransferase 2 regulates cellular metastasis-associated behavior in gastric cancer. Int J Mol Med (2012) 30(6):1267–74. doi:10.3892/ijmm.2012.1130
148. Liu J, Yang L, Jin M, Xu L, Wu S. Regulation of the invasion and metastasis of human glioma cells by polypeptide N-acetylgalactosaminyltransferase 2. Mol Med Rep (2011) 4(6):1299–305. doi:10.3892/mmr.2011.569
149. Guo JM, Chen HL, Wang GM, Zhang YK, Narimatsu H. Expression of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase-12 in gastric and colonic cancer cell lines and in human colorectal cancer. Oncology (2004) 67(3–4):271–6. doi:10.1159/000081328
150. Ding MX, Wang HF, Wang JS, Zhan H, Zuo YG, Yang DL, et al. ppGalNAc T1 as a potential novel marker for human bladder cancer. Asian Pac J Cancer Prev (2012) 13(11):5653–7. doi:10.7314/APJCP.2012.13.11.5653
151. Onitsuka K, Shibao K, Nakayama Y, Minagawa N, Hirata K, Izumi H, et al. Prognostic significance of UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase-3 (GalNAc-T3) expression in patients with gastric carcinoma. Cancer Sci (2003) 94(1):32–6. doi:10.1111/j.1349-7006.2003.tb01348.x
152. Kitada S, Yamada S, Kuma A, Ouchi S, Tasaki T, Nabeshima A, et al. Polypeptide N-acetylgalactosaminyl transferase 3 independently predicts high-grade tumours and poor prognosis in patients with renal cell carcinomas. Br J Cancer (2013) 109(2):472–81. doi:10.1038/bjc.2013.331
153. Yamamoto S, Nakamori S, Tsujie M, Takahashi Y, Nagano H, Dono K, et al. Expression of uridine diphosphate N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyl transferase 3 in adenocarcinoma of the pancreas. Pathobiology (2004) 71(1):12–8. doi:10.1159/000072957
154. Park JH, Nishidate T, Kijima K, Ohashi T, Takegawa K, Fujikane T, et al. Critical roles of mucin 1 glycosylation by transactivated polypeptide N-acetylgalactosaminyltransferase 6 in mammary carcinogenesis. Cancer Res (2010) 70(7):2759–69. doi:10.1158/0008-5472.CAN-09-3911
155. Park JH, Katagiri T, Chung S, Kijima K, Nakamura Y. Polypeptide N-acetylgalactosaminyltransferase 6 disrupts mammary acinar morphogenesis through O-glycosylation of fibronectin. Neoplasia (2011) 13(4):320–6. doi:10.1593/neo.101440
156. Peng RQ, Wan HY, Li HF, Liu M, Li X, Tang H. MicroRNA-214 suppresses growth and invasiveness of cervical cancer cells by targeting UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 7. J Biol Chem (2012) 287(17):14301–9. doi:10.1074/jbc.M111.337642
157. Gaziel-Sovran A, Segura MF, Di Micco R, Collins MK, Hanniford D, Vega-Saenz de Miera E, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell (2011) 20(1):104–18. doi:10.1016/j.ccr.2011.05.027
158. Matsumoto Y, Zhang Q, Akita K, Nakada H, Hamamura K, Tokuda N, et al. pp-GalNAc-T13 induces high metastatic potential of murine Lewis lung cancer by generating trimeric Tn antigen. Biochem Biophys Res Commun (2012) 419(1):7–13. doi:10.1016/j.bbrc.2012.01.086
159. Wang R, Yu C, Zhao D, Wu M, Yang Z. The mucin-type glycosylating enzyme polypeptide N-acetylgalactosaminyltransferase 14 promotes the migration of ovarian cancer by modifying mucin 13. Oncol Rep (2013) 30(2):667–76. doi:10.3892/or.2013.2493
160. Wu C, Guo X, Wang W, Wang Y, Shan Y, Zhang B, et al. N-Acetylgalactosaminyltransferase-14 as a potential biomarker for breast cancer by immunohistochemistry. BMC Cancer (2010) 10:123. doi:10.1186/1471-2407-10-123
161. Tsuda N, Nonaka Y, Shichijo S, Yamada A, Ito M, Maeda Y, et al. UDP-Gal: betaGlcNAc beta1, 3-galactosyltransferase, polypeptide 3 (GALT3) is a tumour antigen recognised by HLA-A2-restricted cytotoxic T lymphocytes from patients with brain tumour. Br J Cancer (2002) 87(9):1006–12. doi:10.1038/sj.bjc.6600593
162. Li Z, Yamada S, Inenaga S, Imamura T, Wu Y, Wang KY, et al. Polypeptide N-acetylgalactosaminyltransferase 6 expression in pancreatic cancer is an independent prognostic factor indicating better overall survival. Br J Cancer (2011) 104(12):1882–9. doi:10.1038/bjc.2011.166
163. Freire T, Berois N, Sóñora C, Varangot M, Barrios E, Osinaga E. UDP-N-acetyl-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 6 (ppGalNAc-T6) mRNA as a potential new marker for detection of bone marrow-disseminated breast cancer cells. Int J Cancer (2006) 119(6):1383–8. doi:10.1002/ijc.21959
164. Wragg S, Hagen FK, Tabak LA. Kinetic analysis of a recombinant UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase. J Biol Chem (1995) 270(28):16947–54. doi:10.1074/jbc.270.28.16947
165. Hatanaka K, Slama JT, Elbein AD. Synthesis of a new inhibitor of the UDP-GalNAc:polypeptide galactosaminyl transferase. Biochem Biophys Res Commun (1991) 175(2):668–72. doi:10.1016/0006-291X(91)91617-L
166. Kuan SF, Byrd JC, Basbaum C, Kim YS. Inhibition of mucin glycosylation by aryl-N-acetyl-alpha-galactosaminides in human colon cancer cells. J Biol Chem (1989) 264(32):19271–7.
167. Vavasseur F, Dole K, Yang J, Matta KL, Myerscough N, Corfield A, et al. O-glycan biosynthesis in human colorectal adenoma cells during progression to cancer. Eur J Biochem (1994) 222(2):415–24. doi:10.1111/j.1432-1033.1994.tb18880.x
168. Delannoy P, Kim I, Emery N, De Bolos C, Verbert A, Degand P, et al. Benzyl-N-acetyl-alpha-D-galactosaminide inhibits the sialylation and the secretion of mucins by a mucin secreting HT-29 cell subpopulation. Glycoconj J (1996) 13(5):717–26. doi:10.1007/BF00702335
169. Huang J, Byrd JC, Yoon WH, Kim YS. Effect of benzyl-alpha-GalNAc, an inhibitor of mucin glycosylation, on cancer-associated antigens in human colon cancer cells. Oncol Res (1992) 4(11–12):507–15.
170. Schafer A, Thiem J. Synthesis of novel donor mimetics of UDP-Gal, UDP-GlcNAc, and UDP-GalNAc as potential transferase inhibitors. J Org Chem (2000) 65(1):24–9. doi:10.1021/jo990766l
171. Busca P, Piller V, Piller F, Martin OR. Synthesis and biological evaluation of new UDP-GalNAc analogues for the study of polypeptide-alpha-GalNAc-transferases. Bioorg Med Chem Lett (2003) 13(11):1853–6. doi:10.1016/S0960-894X(03)00287-7
172. Hang HC, Yu C, Ten Hagen KG, Tian E, Winans KA, Tabak LA, et al. Small molecule inhibitors of mucin-type O-linked glycosylation from a uridine-based library. Chem Biol (2004) 11(3):337–45. doi:10.1016/j.chembiol.2004.02.023
173. Winans KA, Bertozzi CR. An inhibitor of the human UDP-GlcNAc 4-epimerase identified from a uridine-based library: a strategy to inhibit O-linked glycosylation. Chem Biol (2002) 9(1):113–29. doi:10.1016/S1074-5521(02)00093-5
174. Pouilly S, Piller V, Piller F. Metabolic glycoengineering through the mammalian GalNAc salvage pathway. FEBS J (2012) 279(4):586–98. doi:10.1111/j.1742-4658.2011.08448.x
175. Marcos NT, Pinho S, Grandela C, Cruz A, Samyn-Petit B, Harduin-Lepers A, et al. Role of the human ST6GalNAc-I and ST6GalNAc-II in the synthesis of the cancer-associated sialyl-Tn antigen. Cancer Res (2004) 64(19):7050–7. doi:10.1158/0008-5472.CAN-04-1921
176. Sewell R, Bäckström M, Dalziel M, Gschmeissner S, Karlsson H, Noll T, et al. The ST6GalNAc-I sialyltransferase localizes throughout the golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J Biol Chem (2006) 281(6):3586–94. doi:10.1074/jbc.M511826200
177. Ikehara Y, Kojima N, Kurosawa N, Kudo T, Kono M, Nishihara S, et al. Cloning and expression of a human gene encoding an N-acetylgalactosamine-alpha2,6-sialyltransferase (ST6GalNAc I): a candidate for synthesis of cancer-associated sialyl-Tn antigens. Glycobiology (1999) 9(11):1213–24. doi:10.1093/glycob/9.11.1213
178. Marcos NT, Bennett EP, Gomes J, Magalhaes A, Gomes C, David L, et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front Biosci (Elite Ed) (2011) 3:1443–55. doi:10.2741/345
179. Schuessler MH, Pintado S, Welt S, Real FX, Xu M, Melamed MR, et al. Blood group and blood-group-related antigens in normal pancreas and pancreas cancer: enhanced expression of precursor type 1, Tn and sialyl-Tn in pancreas cancer. Int J Cancer (1991) 47(2):180–7. doi:10.1002/ijc.2910470204
180. Pinho S, Marcos NT, Ferreira B, Carvalho AS, Oliveira MJ, Santos-Silva F, et al. Biological significance of cancer-associated sialyl-Tn antigen: modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett (2007) 249(2):157–70. doi:10.1016/j.canlet.2006.08.010
181. Ferreira JA, Videira PA, Lima L, Pereira S, Silva M, Carrascal M, et al. Overexpression of tumour-associated carbohydrate antigen sialyl-Tn in advanced bladder tumours. Mol Oncol (2013) 7(3):719–31. doi:10.1016/j.molonc.2013.03.001
182. Leivonen M, Nordling S, Lundin J, von Boguslawski K, Haglund C. STn and prognosis in breast cancer. Oncology (2001) 61(4):299–305. doi:10.1159/000055337
183. Julien S, Krzewinski-Recchi MA, Harduin-Lepers A, Gouyer V, Huet G, Le Bourhis X, et al. Expression of sialyl-Tn antigen in breast cancer cells transfected with the human CMP-Neu5Ac: GalNAc alpha2,6-sialyltransferase (ST6GalNac I) cDNA. Glycoconj J (2001) 18(11–12):883–93. doi:10.1023/A:1022200525695
184. Julien S, Lagadec C, Krzewinski-Recchi MA, Courtand G, Le Bourhis X, Delannoy P. Stable expression of sialyl-Tn antigen in T47-D cells induces a decrease of cell adhesion and an increase of cell migration. Breast Cancer Res Treat (2005) 90(1):77–84. doi:10.1007/s10549-004-3137-3
185. Ozaki H, Matsuzaki H, Ando H, Kaji H, Nakanishi H, Ikehara Y, et al. Enhancement of metastatic ability by ectopic expression of ST6GalNAcI on a gastric cancer cell line in a mouse model. Clin Exp Metastasis (2012) 29(3):229–38. doi:10.1007/s10585-011-9445-1
186. Dalziel M, Whitehouse C, McFarlane I, Brockhausen I, Gschmeissner S, Schwientek T, et al. The relative activities of the C2GnT1 and ST3Gal-I glycosyltransferases determine O-glycan structure and expression of a tumor-associated epitope on MUC1. J Biol Chem (2001) 276(14):11007–15. doi:10.1074/jbc.M006523200
187. Hakomori S. Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol (2001) 491:369–402. doi:10.1007/978-1-4615-1267-7_24
188. Baldus SE, Zirbes TK, Hanisch FG, Kunze D, Shafizadeh ST, Nolden S, et al. Thomsen-Friedenreich antigen presents as a prognostic factor in colorectal carcinoma: a clinicopathologic study of 264 patients. Cancer (2000) 88(7):1536–43. doi:10.1002/(SICI)1097-0142(20000401)88:7<1536::AID-CNCR6>3.3.CO;2-N
189. Khaldoyanidi SK, Glinsky VV, Sikora L, Glinskii AB, Mossine VV, Quinn TP, et al. MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J Biol Chem (2003) 278(6):4127–34. doi:10.1074/jbc.M209590200
190. Solatycka A, Owczarek T, Piller F, Piller V, Pula B, Wojciech L, et al. MUC1 in human and murine mammary carcinoma cells decreases the expression of core 2 beta1,6-N-acetylglucosaminyltransferase and beta-galactoside alpha2,3-sialyltransferase. Glycobiology (2012) 22(8):1042–54. doi:10.1093/glycob/cws075
191. Hoffmann-Roder A, Kaiser A, Wagner S, Gaidzik N, Kowalczyk D, Westerlind U, et al. Synthetic antitumor vaccines from tetanus toxoid conjugates of MUC1 glycopeptides with the Thomsen-Friedenreich antigen and a fluorine-substituted analogue. Angew Chem Int Ed Engl (2010) 49(45):8498–503. doi:10.1002/anie.201003810
192. Inamdar SR, Savanur MA, Eligar SM, Chachadi VB, Nagre NN, Chen C, et al. The TF-antigen binding lectin from Sclerotium rolfsii inhibits growth of human colon cancer cells by inducing apoptosis in vitro and suppresses tumor growth in vivo. Glycobiology (2012) 22(9):1227–35. doi:10.1093/glycob/cws090
193. Aryal RP, Ju T, Cummings RD. The endoplasmic reticulum chaperone Cosmc directly promotes in vitro folding of T-synthase. J Biol Chem (2010) 285(4):2456–62. doi:10.1074/jbc.M109.065169
194. Aryal RP, Ju T, Cummings RD. Tight complex formation between Cosmc chaperone and its specific client non-native T-synthase leads to enzyme activity and client-driven dissociation. J Biol Chem (2012) 287(19):15317–29. doi:10.1074/jbc.M111.312587
195. Schietinger A, Philip M, Yoshida BA, Azadi P, Liu H, Meredith SC, et al. A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science (2006) 314(5797):304–8. doi:10.1126/science.1129200
196. Ju T, Lanneau GS, Gautam T, Wang Y, Xia B, Stowell SR, et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res (2008) 68(6):1636–46. doi:10.1158/0008-5472.CAN-07-2345
197. Mi R, Song L, Wang Y, Ding X, Zeng J, Lehoux S, et al. Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing tn antigen. J Biol Chem (2012) 287(49):41523–33. doi:10.1074/jbc.M112.371989
198. Wu YM, Liu CH, Huang MJ, Lai HS, Lee PH, Hu RH, et al. C1GALT1 enhances proliferation of hepatocellular carcinoma cells via modulating MET glycosylation and dimerization. Cancer Res (2013) 73(17):5580–90. doi:10.1158/0008-5472.CAN-13-0869
199. Barrow H, Tam B, Duckworth CA, Rhodes JM, Yu LG. Suppression of core 1 Gal-transferase is associated with reduction of TF and reciprocal increase of Tn, sialyl-Tn and core 3 glycans in human colon cancer cells. PLoS One (2013) 8(3):e59792. doi:10.1371/journal.pone.0059792
200. Yousefi S, Higgins E, Daoling Z, Pollex-Krüger A, Hindsgaul O, Dennis JW. Increased UDP-GlcNAc:Gal beta 1-3GaLNAc-R (GlcNAc to GaLNAc) beta-1, 6-N-acetylglucosaminyltransferase activity in metastatic murine tumor cell lines. Control of polylactosamine synthesis. J Biol Chem (1991) 266(3):1772–82.
201. Hagisawa S, Ohyama C, Takahashi T, Endoh M, Moriya T, Nakayama J, et al. Expression of core 2 beta1,6-N-acetylglucosaminyltransferase facilitates prostate cancer progression. Glycobiology (2005) 15(10):1016–24. doi:10.1093/glycob/cwi086
202. Hatakeyama S, Kyan A, Yamamoto H, Okamoto A, Sugiyama N, Suzuki Y, et al. Core 2 N-acetylglucosaminyltransferase-1 expression induces aggressive potential of testicular germ cell tumor. Int J Cancer (2010) 127(5):1052–9. doi:10.1002/ijc.25117
203. Miyamoto T, Suzuki A, Asaka R, Ishikawa K, Yamada Y, Kobara H, et al. Immunohistochemical expression of core 2 beta1,6-N-acetylglucosaminyl transferase 1 (C2GnT1) in endometrioid-type endometrial carcinoma: a novel potential prognostic factor. Histopathology (2013) 62(7):986–93. doi:10.1111/his.12107
204. Suzuki Y, Sutoh M, Hatakeyama S, Mori K, Yamamoto H, Koie T, et al. MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int J Oncol (2012) 40(6):1831–8. doi:10.3892/ijo.2012.1411
205. Brockhausen I, Kuhns W, Schachter H, Matta KL, Sutherland DR, Baker MA. Biosynthesis of O-glycans in leukocytes from normal donors and from patients with leukemia: increase in O-glycan core 2 UDP-GlcNAc:Gal beta 3 GalNAc alpha-R (GlcNAc to GalNAc) beta(1-6)-N-acetylglucosaminyltransferase in leukemic cells. Cancer Res (1991) 51(4):1257–63.
206. Nguyen JT, Evans DP, Galvan M, Pace KE, Leitenberg D, Bui TN, et al. CD45 modulates galectin-1-induced T cell death: regulation by expression of core 2 O-glycans. J Immunol (2001) 167(10):5697–707. doi:10.4049/jimmunol.167.10.5697
207. Cabrera PV, Amano M, Mitoma J, Chan J, Said J, Fukuda M, et al. Haploinsufficiency of C2GnT-I glycosyltransferase renders T lymphoma cells resistant to cell death. Blood (2006) 108(7):2399–406. doi:10.1182/blood-2006-04-018556
208. Huang MC, Chen HY, Huang HC, Huang J, Liang JT, Shen TL, et al. C2GnT-M is downregulated in colorectal cancer and its re-expression causes growth inhibition of colon cancer cells. Oncogene (2006) 25(23):3267–76. doi:10.1038/sj.onc.1209350
209. Maemura K, Fukuda M. Poly-N-acetyllactosaminyl O-glycans attached to leukosialin. The presence of sialyl Le(x) structures in O-glycans. J Biol Chem (1992) 267(34):24379–86.
210. Bierhuizen MF, Maemura K, Fukuda M. Expression of a differentiation antigen and poly-N-acetyllactosaminyl O-glycans directed by a cloned core 2 beta-1,6-N-acetylglucosaminyltransferase. J Biol Chem (1994) 269(6):4473–9.
211. Hindsgaul O, Kaur KJ, Srivastava G, Blaszczyk-Thurin M, Crawley SC, Heerze LD, et al. Evaluation of deoxygenated oligosaccharide acceptor analogs as specific inhibitors of glycosyltransferases. J Biol Chem (1991) 266(27):17858–62.
212. Kuhns W, Rutz V, Paulsen H, Matta KL, Baker MA, Barner M, et al. Processing O-glycan core 1, Gal beta 1-3GalNAc alpha-R. Specificities of core 2, UDP-GlcNAc: Gal beta 1-3 GalNAc-R(GlcNAc to GalNAc) beta 6-N-acetylglucosaminyltransferase and CMP-sialic acid: Gal beta 1-3GalNAc-R alpha 3-sialyltransferase. Glycoconj J (1993) 10(5):381–94. doi:10.1007/BF00731043
213. Toki D, Granovsky MA, Reck F, Kuhns W, Baker MA, Matta KL, et al. Inhibition of UDP-GlcNAc:Gal beta 1-3GalNAc-R (GlcNAc to GalNAc) beta 6-N-acetylglucosaminyltransferase from acute myeloid leukaemia cells by photoreactive nitrophenyl substrate derivatives. Biochem Biophys Res Commun (1994) 198(2):417–23. doi:10.1006/bbrc.1994.1061
214. Almeida R, Amado M, David L, Levery SB, Holmes EH, Merkx G, et al. A family of human beta4-galactosyltransferases. Cloning and expression of two novel UDP-galactose:beta-n-acetylglucosamine beta1, 4-galactosyltransferases, beta4Gal-T2 and beta4Gal-T3. J Biol Chem (1997) 272(51):31979–91. doi:10.1074/jbc.272.51.31979
215. Shaper NL, Meurer JA, Joziasse DH, Chou TD, Smith EJ, Schnaar RL, et al. The chicken genome contains two functional nonallelic beta1,4-galactosyltransferase genes. Chromosomal assignment to syntenic regions tracks fate of the two gene lineages in the human genome. J Biol Chem (1997) 272(50):31389–99. doi:10.1074/jbc.272.50.31389
216. Sato T, Furukawa K, Bakker H, Van den Eijnden DH, Van Die I. Molecular cloning of a human cDNA encoding beta-1,4-galactosyltransferase with 37% identity to mammalian UDP-Gal:GlcNAc beta-1,4-galactosyltransferase. Proc Natl Acad Sci U S A (1998) 95(2):472–7. doi:10.1073/pnas.95.2.472
217. Lo NW, Shaper JH, Pevsner J, Shaper NL. The expanding beta 4-galactosyltransferase gene family: messages from the databanks. Glycobiology (1998) 8(5):517–26. doi:10.1093/glycob/8.5.517
218. Schwientek T, Almeida R, Levery SB, Holmes EH, Bennett E, Clausen H. Cloning of a novel member of the UDP-galactose:beta-N-acetylglucosamine beta1,4-galactosyltransferase family, beta4Gal-T4, involved in glycosphingolipid biosynthesis. J Biol Chem (1998) 273(45):29331–40. doi:10.1074/jbc.273.45.29331
219. Nomura T, Takizawa M, Aoki J, Arai H, Inoue K, Wakisaka E, et al. Purification, cDNA cloning, and expression of UDP-Gal: glucosylceramide beta-1,4-galactosyltransferase from rat brain. J Biol Chem (1998) 273(22):13570–7. doi:10.1074/jbc.273.22.13570
220. Ujita M, McAuliffe J, Schwientek T, Almeida R, Hindsgaul O, Clausen H, et al. Synthesis of poly-N-acetyllactosamine in core 2 branched O-glycans. The requirement of novel beta-1,4-galactosyltransferase IV and beta-1,3-n-acetylglucosaminyltransferase. J Biol Chem (1998) 273(52):34843–9. doi:10.1074/jbc.273.52.34843
221. Almeida R, Levery SB, Mandel U, Kresse H, Schwientek T, Bennett EP, et al. Cloning and expression of a proteoglycan UDP-galactose:beta-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J Biol Chem (1999) 274(37):26165–71. doi:10.1074/jbc.274.37.26165
222. Sasaki K, Kurata-Miura K, Ujita M, Angata K, Nakagawa S, Sekine S, et al. Expression cloning of cDNA encoding a human beta-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc Natl Acad Sci U S A (1997) 94(26):14294–9. doi:10.1073/pnas.94.26.14294
223. Zhou D, Dinter A, Gutiérrez Gallego R, Kamerling JP, Vliegenthart JF, Berger EG, et al. A beta-1,3-N-acetylglucosaminyltransferase with poly-N-acetyllactosamine synthase activity is structurally related to beta-1,3-galactosyltransferases. Proc Natl Acad Sci U S A (1999) 96(2):406–11. doi:10.1073/pnas.96.2.406
224. Shiraishi N, Natsume A, Togayachi A, Endo T, Akashima T, Yamada Y, et al. Identification and characterization of three novel beta 1,3-N-acetylglucosaminyltransferases structurally related to the beta 1,3-galactosyltransferase family. J Biol Chem (2001) 276(5):3498–507. doi:10.1074/jbc.M004800200
225. Beum PV, Cheng PW. Biosynthesis and function of beta 1,6 branched mucin-type glycans. Adv Exp Med Biol (2001) 491:279–312.
226. Bierhuizen MF, Mattei MG, Fukuda M. Expression of the developmental I antigen by a cloned human cDNA encoding a member of a beta-1,6-N-acetylglucosaminyltransferase gene family. Genes Dev (1993) 7(3):468–78. doi:10.1101/gad.7.3.468
227. Inaba N, Hiruma T, Togayachi A, Iwasaki H, Wang XH, Furukawa Y, et al. A novel I-branching beta-1,6-N-acetylglucosaminyltransferase involved in human blood group I antigen expression. Blood (2003) 101(7):2870–6. doi:10.1182/blood-2002-09-2838
228. Yeh JC, Ong E, Fukuda M. Molecular cloning and expression of a novel beta-1, 6-N-acetylglucosaminyltransferase that forms core 2, core 4, and I branches. J Biol Chem (1999) 274(5):3215–21. doi:10.1074/jbc.274.5.3215
229. Ito N, Yokota M, Nagaike C, Morimura Y, Hatake K, Matsunaga T. Histochemical demonstration and analysis of poly-N-acetyllactosamine structures in normal and malignant human tissues. Histol Histopathol (1996) 11(1):203–14.
230. Krishnan V, Bane SM, Kawle PD, Naresh KN, Kalraiya RD. Altered melanoma cell surface glycosylation mediates organ specific adhesion and metastasis via lectin receptors on the lung vascular endothelium. Clin Exp Metastasis (2005) 22(1):11–24. doi:10.1007/s10585-005-2036-2
231. Kawakami H, Ito M, Miura Y, Hirano H. Involvement of N-acetyl-lactosamine-containing sugar structures in the liver metastasis of mouse colon carcinoma (colon 26) cells. J Gastroenterol Hepatol (1994) 9(6):567–71. doi:10.1111/j.1440-1746.1994.tb01562.x
232. Okamoto T, Yoneyama MS, Hatakeyama S, Mori K, Yamamoto H, Koie T, et al. Core2 O-glycan-expressing prostate cancer cells are resistant to NK cell immunity. Mol Med Rep (2013) 7(2):359–64. doi:10.3892/mmr.2012.1189
233. Srinivasan N, Bane SM, Ahire SD, Ingle AD, Kalraiya RD. Poly N-acetyll-actosamine substitutions on N- and not O-oligosaccharides or Thomsen-Friedenreich antigen facilitate lung specific metastasis of melanoma cells via galectin-3. Glycoconj J (2009) 26(4):445–56. doi:10.1007/s10719-008-9194-9
234. Cedeno-Laurent F, Opperman MJ, Barthel SR, Hays D, Schatton T, Zhan Q, et al. Metabolic inhibition of galectin-1-binding carbohydrates accentuates antitumor immunity. J Invest Dermatol (2012) 132(2):410–20. doi:10.1038/jid.2011.335
235. Xu S, Zhu X, Zhang S, Yin S, Zhou L, Chen C, et al. Over-expression of beta-1,4-galactosyltransferase I, II, and V in human astrocytoma. J Cancer Res Clin Oncol (2001) 127(8):502–6. doi:10.1007/s004320100246
236. Chen WS, Chang HY, Li CP, Liu JM, Huang TS. Tumor beta-1,4-galactosyltransferase IV overexpression is closely associated with colorectal cancer metastasis and poor prognosis. Clin Cancer Res (2005) 11(24 Pt 1):8615–22. doi:10.1158/1078-0432.CCR-05-1006
237. Chang HH, Chen CH, Chou CH, Liao YF, Huang MJ, Chen YH, et al. beta-1,4-galactosyltransferase III enhances invasive phenotypes via beta1-integrin and predicts poor prognosis in neuroblastoma. Clin Cancer Res (2013) 19(7):1705–16. doi:10.1158/1078-0432.CCR-12-2367
238. Lopez LC, Maillet CM, Oleszkowicz K, Shur BD. Cell surface and golgi pools of beta-1,4-galactosyltransferase are differentially regulated during embryonal carcinoma cell differentiation. Mol Cell Biol (1989) 9(6):2370–7.
239. Shur BD. Glycosyltransferases as cell adhesion molecules. Curr Opin Cell Biol (1993) 5(5):854–63. doi:10.1016/0955-0674(93)90035-O
240. Johnson FM, Shur BD. The level of cell surface beta1,4-galactosyltransferase I influences the invasive potential of murine melanoma cells. J Cell Sci (1999) 112(Pt 16):2785–95.
241. Choi HJ, Chung TW, Kim CH, Jeong HS, Joo M, Youn B, et al. Estrogen induced beta-1,4-galactosyltransferase 1 expression regulates proliferation of human breast cancer MCF-7 cells. Biochem Biophys Res Commun (2012) 426(4):620–5. doi:10.1016/j.bbrc.2012.08.140
242. Zhou H, Zhang Z, Liu C, Jin C, Zhang J, Miao X, et al. B4GALT1 gene knockdown inhibits the hedgehog pathway and reverses multidrug resistance in the human leukemia K562/adriamycin-resistant cell line. IUBMB Life (2012) 64(11):889–900. doi:10.1002/iub.1080
243. Vaghefi MM, Bernacki RJ, Dalley NK, Wilson BE, Robins RK. Synthesis of glycopyranosylphosphonate analogues of certain natural nucleoside diphosphate sugars as potential inhibitors of glycosyltransferases. J Med Chem (1987) 30(8):1383–91. doi:10.1021/jm00391a021
244. Vaghefi MM, Bernacki RJ, Hennen WJ, Robins RK. Synthesis of certain nucleoside methylenediphosphonate sugars as potential inhibitors of glycosyltransferases. J Med Chem (1987) 30(8):1391–9. doi:10.1021/jm00391a021
245. Wang R, Steensma DH, Takaoka Y, Yun JW, Kajimoto T, Wong CH. A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases. Bioorg Med Chem (1997) 5(4):661–72. doi:10.1016/S0968-0896(97)00005-9
246. Hashimoto H, Endo T, Kajihara Y. Synthesis of the first tricomponent bisubstrate analogue that exhibits potent inhibition against GlcNAc:beta-1,4-galactosyltransferase. J Org Chem (1997) 62(7):1914–5. doi:10.1021/jo962235s
247. Chung SJ, Takayama S, Wong CH. Acceptor substrate-based selective inhibition of galactosyltransferases. Bioorg Med Chem Lett (1998) 8(23):3359–64. doi:10.1016/S0960-894X(98)00618-0
248. Takayama S, Chung SJ, Igarashi Y, Ichikawa Y, Sepp A, Lechler RI, et al. Selective inhibition of beta-1,4- and alpha-1,3-galactosyltransferases: donor sugar-nucleotide based approach. Bioorg Med Chem (1999) 7(2):401–9. doi:10.1016/S0968-0896(98)00249-1
249. Gastinel LN, Cambillau C, Bourne Y. Crystal structures of the bovine beta4galactosyltransferase catalytic domain and its complex with uridine diphosphogalactose. EMBO J (1999) 18(13):3546–57. doi:10.1093/emboj/18.13.3546
250. Ramakrishnan B, Balaji PV, Qasba PK. Crystal structure of beta1,4-galactosyltransferase complex with UDP-Gal reveals an oligosaccharide acceptor binding site. J Mol Biol (2002) 318(2):491–502. doi:10.1016/S0022-2836(02)00020-7
251. Takaya K, Nagahori N, Kurogochi M, Furuike T, Miura N, Monde K, et al. Rational design, synthesis, and characterization of novel inhibitors for human beta1,4-galactosyltransferase. J Med Chem (2005) 48(19):6054–65. doi:10.1021/jm0504297
252. Brockhausen I, Benn M, Bhat S, Marone S, Riley JG, Montoya-Peleaz P, et al. UDP-Gal: GlcNAc-R beta1,4-galactosyltransferase – a target enzyme for drug design. Acceptor specificity and inhibition of the enzyme. Glycoconj J (2006) 23(7–8):525–41. doi:10.1007/s10719-006-7153-x
253. Gao Y, Lazar C, Szarek WA, Brockhausen I. Specificity of beta1,4-galactosyltransferase inhibition by 2-naphthyl 2-butanamido-2-deoxy-1-thio-beta-D-glucopyranoside. Glycoconj J (2010) 27(7–9):673–84. doi:10.1007/s10719-010-9312-3
254. Ishida H, Togayachi A, Sakai T, Iwai T, Hiruma T, Sato T, et al. A novel beta1,3-N-acetylglucosaminyltransferase (beta3Gn-T8), which synthesizes poly-N-acetyllactosamine, is dramatically upregulated in colon cancer. FEBS Lett (2005) 579(1):71–8. doi:10.1016/j.febslet.2004.11.037
255. Zhang H, Meng F, Wu S, Kreike B, Sethi S, Chen W, et al. Engagement of I-branching beta-1, 6-N-acetylglucosaminyltransferase 2 in breast cancer metastasis and TGF-beta signaling. Cancer Res (2011) 71(14):4846–56. doi:10.1158/0008-5472.CAN-11-0414
256. Kawashima H, Yamamoto K, Osawa T, Irimura T. Purification and characterization of UDP-GlcNAc:Gal beta 1-4Glc(NAc) beta-1,3-N-acetylglucosaminyltransferase (poly-N-acetyllactosamine extension enzyme) from calf serum. J Biol Chem (1993) 268(36):27118–26.
257. Sakamoto Y, Taguchi T, Tano Y, Ogawa T, Leppänen A, Kinnunen M, et al. Purification and characterization of UDP-GlcNAc:Galbeta1-4GlcNAcbeta1-3*Galbeta1-4Glc(NAc)-R(GlcNAc to *Gal) beta1,6N-acetylglucosaminyltransferase from hog small intestine. J Biol Chem (1998) 273(42):27625–32. doi:10.1074/jbc.273.42.27625
258. Tedaldi LM, Pierce M, Wagner GK. Optimised chemical synthesis of 5-substituted UDP-sugars and their evaluation as glycosyltransferase inhibitors. Carbohydr Res (2012) 364:22–7. doi:10.1016/j.carres.2012.10.009
259. Ravn V, Dabelsteen E. Tissue distribution of histo-blood group antigens. APMIS (2000) 108(1):1–28. doi:10.1034/j.1600-0463.2000.d01-1.x
260. Le Pendu J, Marionneau S, Cailleau-Thomas A, Rocher J, Le Moullac-Vaidye B, Clément M. ABH and Lewis histo-blood group antigens in cancer. APMIS (2001) 109(1):9–31. doi:10.1111/j.1600-0463.2001.tb00011.x
261. Hakomori S. Antigen structure and genetic basis of histo-blood groups A, B and O: their changes associated with human cancer. Biochim Biophys Acta (1999) 1473(1):247–66. doi:10.1016/S0304-4165(99)00183-X
262. Yoshida A, Kumazaki T, Davé V, Blank J, Dzik WH. Suppressed expression of blood group B antigen and blood group galactosyltransferase in a preleukemic subject. Blood (1985) 66(4):990–2.
263. Orntoft TF, Wolf H, Watkins WM. Activity of the human blood group ABO, Se, H, Le, and X gene-encoded glycosyltransferases in normal and malignant bladder urothelium. Cancer Res (1988) 48(15):4427–33.
264. Mandel U, Langkilde NC, Orntoft TF, Therkildsen MH, Karkov J, Reibel J, et al. Expression of histo-blood-group-A/B-gene-defined glycosyltransferases in normal and malignant epithelia: correlation with A/B-carbohydrate expression. Int J Cancer (1992) 52(1):7–12. doi:10.1002/ijc.2910520103
265. León-Atance P, Moreno-Mata N, González-Aragoneses F, Cañizares-Carretero MÁ, Poblet-Martínez E, Genovés-Crespo M, et al. Prognostic influence of loss of blood group A antigen expression in pathologic stage I non-small-cell lung cancer. Arch Bronconeumol (2012) 48(2):49–54. doi:10.1016/j.arbres.2011.10.003
266. Fukushima K, Satoh T, Baba S, Yamashita K. alpha 1,2-fucosylated and beta-N-acetylgalactosaminylated prostate-specific antigen as an efficient marker of prostatic cancer. Glycobiology (2010) 20(4):452–60. doi:10.1093/glycob/cwp197
267. Moehler TM, Sauer S, Witzel M, Andrulis M, Garcia-Vallejo JJ, Grobholz R, et al. Involvement of alpha 1-2-fucosyltransferase I (FUT1) and surface-expressed Lewis(y) (CD174) in first endothelial cell-cell contacts during angiogenesis. J Cell Physiol (2008) 215(1):27–36. doi:10.1002/jcp.21285
268. Goupille C, Hallouin F, Meflah K, Le Pendu J. Increase of rat colon carcinoma cells tumorigenicity by alpha(1-2) fucosyltransferase gene transfection. Glycobiology (1997) 7(2):221–9. doi:10.1093/glycob/7.2.221
269. Marionneau S, Bureau V, Goupille C, Hallouin F, Rocher J, Vaydie B, et al. Susceptibility of rat colon carcinoma cells to lymphokine activated killer-mediated cytotoxicity is decreased by alpha 1,2-fucosylation. Int J Cancer (2000) 86(5):713–7. doi:10.1002/(SICI)1097-0215(20000601)86:5<713::AID-IJC17>3.0.CO;2-V
270. Zhu K, Gerbino E, Beaupre DM, Mackley PA, Muro-Cacho C, Beam C, et al. Farnesyltransferase inhibitor R115777 (Zarnestra, Tipifarnib) synergizes with paclitaxel to induce apoptosis and mitotic arrest and to inhibit tumor growth of multiple myeloma cells. Blood (2005) 105(12):4759–66. doi:10.1182/blood-2004-11-4307
271. Mollicone R, Candelier JJ, Mennesson B, Couillin P, Venot AP, Oriol R. Five specificity patterns of (1 – – 3)-alpha-L-fucosyltransferase activity defined by use of synthetic oligosaccharide acceptors. Differential expression of the enzymes during human embryonic development and in adult tissues. Carbohydr Res (1992) 228(1):265–76. doi:10.1016/S0008-6215(00)90564-0
272. Yan L, Lin B, Gao L, Gao S, Liu C, Wang C, et al. Lewis (y) antigen overexpression increases the expression of MMP-2 and MMP-9 and invasion of human ovarian cancer cells. Int J Mol Sci (2010) 11(11):4441–52. doi:10.3390/ijms11114441
273. Isla Larrain M, Demichelis S, Crespo M, Lacunza E, Barbera A, Cretón A, et al. Breast cancer humoral immune response: involvement of Lewis y through the detection of circulating immune complexes and association with mucin 1 (MUC1). J Exp Clin Cancer Res (2009) 28:121. doi:10.1186/1756-9966-28-121
274. Myers RB, Srivastava S, Grizzle WE. Lewis Y antigen as detected by the monoclonal antibody BR96 is expressed strongly in prostatic adenocarcinoma. J Urol (1995) 153(5):1572–4. doi:10.1097/00005392-199505000-00061
275. Waldock A, Ellis IO, Armitage N, Turner DR, Hardcastle JD, Embleton J. Differential expression of the Lewis Y antigen defined by monoclonal antibody C14/1/46/10 in colonic polyps. Cancer (1989) 64(2):414–21. doi:10.1002/1097-0142(19890715)64:2<414::AID-CNCR2820640213>3.0.CO;2-0
276. Westwood JA, Murray WK, Trivett M, Haynes NM, Solomon B, Mileshkin L, et al. The Lewis-Y carbohydrate antigen is expressed by many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. J Immunother (2009) 32(3):292–301. doi:10.1097/CJI.0b013e31819b7c8e
277. Yuriev E, Farrugia W, Scott AM, Ramsland PA. Three-dimensional structures of carbohydrate determinants of Lewis system antigens: implications for effective antibody targeting of cancer. Immunol Cell Biol (2005) 83(6):709–17. doi:10.1111/j.1440-1711.2005.01374.x
278. Sabbatini PJ, Kudryashov V, Ragupathi G, Danishefsky SJ, Livingston PO, Bornmann W, et al. Immunization of ovarian cancer patients with a synthetic Lewis(y)-protein conjugate vaccine: a phase 1 trial. Int J Cancer (2000) 87(1):79–85. doi:10.1002/1097-0215(20000701)87:1<79::AID-IJC12>3.0.CO;2-L
279. Sabbatini PJ, Ragupathi G, Hood C, Aghajanian CA, Juretzka M, Iasonos A, et al. Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clin Cancer Res (2007) 13(14):4170–7. doi:10.1158/1078-0432.CCR-06-2949
280. Saleh MN, Sugarman S, Murray J, Ostroff JB, Healey D, Jones D, et al. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-doxorubicin in patients with Lewis Y-expressing epithelial tumors. J Clin Oncol (2000) 18(11):2282–92.
281. Motzer RJ, Reuter VE, Cordon-Cardo C, Bosl GJ. Blood group-related antigens in human germ cell tumors. Cancer Res (1988) 48(18):5342–7.
282. Kawai S, Kato S, Imai H, Okada Y, Ishioka C. Suppression of FUT1 attenuates cell proliferation in the HER2-overexpressing cancer cell line NCI-N87. Oncol Rep (2013) 29(1):13–20. doi:10.3892/or.2012.2120
283. Palcic MM, Heerze LD, Srivastava OP, Hindsgaul O. A bisubstrate analog inhibitor for alpha-(1-]2)-fucosyltransferase. J Biol Chem (1989) 264(29):17174–81.
284. Kannagi R. Carbohydrate-mediated cell adhesion involved in hematogenous metastasis of cancer. Glycoconj J (1997) 14(5):577–84. doi:10.1023/A:1018532409041
285. Kim YJ, Varki A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J (1997) 14(5):569–76. doi:10.1023/A:1018580324971
286. Nakayama T, Watanabe M, Katsumata T, Teramoto T, Kitajima M. Expression of sialyl Lewis(a) as a new prognostic factor for patients with advanced colorectal carcinoma. Cancer (1995) 75(8):2051–6. doi:10.1002/1097-0142(19950415)75:8<2051::AID-CNCR2820750804>3.0.CO;2-4
287. Iwai K, Ishikura H, Kaji M, Sugiura H, Ishizu A, Takahashi C, et al. Importance of E-selectin (ELAM-1) and sialyl Lewis(a) in the adhesion of pancreatic carcinoma cells to activated endothelium. Int J Cancer (1993) 54(6):972–7. doi:10.1002/ijc.2910540618
288. Takada A, Ohmori K, Yoneda T, Tsuyuoka K, Hasegawa A, Kiso M, et al. Contribution of carbohydrate antigens sialyl Lewis A and sialyl Lewis X to adhesion of human cancer cells to vascular endothelium. Cancer Res (1993) 53(2):354–61.
289. Jørgensen T, Berner A, Kaalhus O, Tveter KJ, Danielsen HE, Bryne M. Up-regulation of the oligosaccharide sialyl LewisX: a new prognostic parameter in metastatic prostate cancer. Cancer Res (1995) 55(9):1817–9.
290. Nakamori S, Kameyama M, Imaoka S, Furukawa H, Ishikawa O, Sasaki Y, et al. Increased expression of sialyl Lewisx antigen correlates with poor survival in patients with colorectal carcinoma: clinicopathological and immunohistochemical study. Cancer Res (1993) 53(15):3632–7.
291. Ogawa J, Tsurumi T, Yamada S, Koide S, Shohtsu A. Blood vessel invasion and expression of sialyl Lewisx and proliferating cell nuclear antigen in stage I non-small cell lung cancer. Relation to postoperative recurrence. Cancer (1994) 73(4):1177–83. doi:10.1002/1097-0142(19940215)73:4<1177::AID-CNCR2820730409>3.0.CO;2-0
292. Renkonen J, Paavonen T, Renkonen R. Endothelial and epithelial expression of sialyl Lewis(x) and sialyl Lewis(a) in lesions of breast carcinoma. Int J Cancer (1997) 74(3):296–300. doi:10.1002/(SICI)1097-0215(19970620)74:3<296::AID-IJC11>3.0.CO;2-A
293. Ravindranath MH, Amiri AA, Bauer PM, Kelley MC, Essner R, Morton DL. Endothelial-selectin ligands sialyl Lewis(x) and sialyl Lewis(a) are differentiation antigens immunogenic in human melanoma. Cancer (1997) 79(9):1686–97. doi:10.1002/(SICI)1097-0142(19970501)79:9<1686::AID-CNCR8>3.0.CO;2-A
294. Kannagi R. Carbohydrate antigen sialyl Lewis a – its pathophysiological significance and induction mechanism in cancer progression. Chang Gung Med J (2007) 30(3):189–209.
295. Majuri ML, Niemelä R, Tiisala S, Renkonen O, Renkonen R. Expression and function of alpha 2,3-sialyl- and alpha 1,3/1,4-fucosyltransferases in colon adenocarcinoma cell lines: role in synthesis of E-selectin counter-receptors. Int J Cancer (1995) 63(4):551–9. doi:10.1002/ijc.2910630416
296. Tsuji S, Datta AK, Paulson JC. Systematic nomenclature for sialyltransferases. Glycobiology (1996) 6(7):v–vii.
297. Kono M, Takashima S, Liu H, Inoue M, Kojima N, Lee YC, et al. Molecular cloning and functional expression of a fifth-type alpha 2,3-sialyltransferase (mST3Gal V: GM3 synthase). Biochem Biophys Res Commun (1998) 253(1):170–5. doi:10.1006/bbrc.1998.9768
298. Okajima T, Fukumoto S, Ito H, Kiso M, Hirabayashi Y, Urano T, et al. Molecular cloning of brain-specific GD1alpha synthase (ST6GalNAc V) containing CAG/Glutamine repeats. J Biol Chem (1999) 274(43):30557–62. doi:10.1074/jbc.274.43.30557
299. Pérez-Garay M, Arteta B, Pagès L, de Llorens R, de Bolòs C, Vidal-Vanaclocha F, et al. alpha2,3-sialyltransferase ST3Gal III modulates pancreatic cancer cell motility and adhesion in vitro and enhances its metastatic potential in vivo. PLoS One (2010) 5(9):e12524. doi:10.1371/journal.pone.0012524
300. Bernacki RJ. Regulation of rat-liver glycoprotein: N-acetylneuraminic acid transferase activity by pyrimidine nucleotides. Eur J Biochem (1975) 58(2):477–81. doi:10.1111/j.1432-1033.1975.tb02395.x
301. Kijima-Suda I, Miyamoto Y, Toyoshima S, Itoh M, Osawa T. Inhibition of experimental pulmonary metastasis of mouse colon adenocarcinoma 26 sublines by a sialic acid:nucleoside conjugate having sialyltransferase inhibiting activity. Cancer Res (1986) 46(2):858–62.
302. Kijima I, Ezawa K, Toyoshima S, Furuhata K, Ogura H, Osawa T. Induction of suppressor T cells by neuraminic acid derivatives. Chem Pharm Bull (Tokyo) (1982) 30(9):3278–83. doi:10.1248/cpb.30.3278
303. Skropeta D, Schwörer R, Haag T, Schmidt RR. Asymmetric synthesis and affinity of potent sialyltransferase inhibitors based on transition-state analogues. Glycoconj J (2004) 21(5):205–19. doi:10.1023/B:GLYC.0000045094.71207.22
304. Kijima-Suda I, Toyoshima S, Itoh M, Furuhata K, Ogura H, Osawa T. Inhibition of sialyltransferases of murine lymphocytes by disaccharide nucleosides. Chem Pharm Bull (Tokyo) (1985) 33(2):730–9. doi:10.1248/cpb.33.730
305. Chen JY, Tang YA, Huang SM, Juan HF, Wu LW, Sun YC, et al. A novel sialyltransferase inhibitor suppresses FAK/paxillin signaling and cancer angiogenesis and metastasis pathways. Cancer Res (2011) 71(2):473–83. doi:10.1158/0008-5472.CAN-10-1303
306. Wu CY, Hsu CC, Chen ST, Tsai YC. Soyasaponin I, a potent and specific sialyltransferase inhibitor. Biochem Biophys Res Commun (2001) 284(2):466–9. doi:10.1006/bbrc.2001.5002
307. Hsu CC, Lin TW, Chang WW, Wu CY, Lo WH, Wang PH, et al. Soyasaponin-I-modified invasive behavior of cancer by changing cell surface sialic acids. Gynecol Oncol (2005) 96(2):415–22. doi:10.1016/j.ygyno.2004.10.010
308. Chang WW, Yu CY, Lin TW, Wang PH, Tsai YC. Soyasaponin I decreases the expression of alpha2,3-linked sialic acid on the cell surface and suppresses the metastatic potential of B16F10 melanoma cells. Biochem Biophys Res Commun (2006) 341(2):614–9. doi:10.1016/j.bbrc.2005.12.216
309. Chang KH, Lee L, Chen J, Li WS. Lithocholic acid analogues, new and potent alpha-2,3-sialyltransferase inhibitors. Chem Commun (Camb) (2006) 6:629–31. doi:10.1039/b514915k
310. Chiang CH, Wang CH, Chang HC, More SV, Li WS, Hung WC. A novel sialyltransferase inhibitor AL10 suppresses invasion and metastasis of lung cancer cells by inhibiting integrin-mediated signaling. J Cell Physiol (2010) 223(2):492–9. doi:10.1002/jcp.22068
311. Mare L, Trinchera M. Suppression of beta 1,3galactosyltransferase beta 3Gal-T5 in cancer cells reduces sialyl-Lewis a and enhances poly N-acetyllactosamines and sialyl-Lewis x on O-glycans. Eur J Biochem (2004) 271(1):186–94. doi:10.1046/j.1432-1033.2003.03919.x
312. Lin CH, Fan YY, Chen YY, Wang SH, Chen CI, Yu LC, et al. Enhanced expression of beta 3-galactosyltransferase 5 activity is sufficient to induce in vivo synthesis of extended type 1 chains on lactosylceramides of selected human colonic carcinoma cell lines. Glycobiology (2009) 19(4):418–27. doi:10.1093/glycob/cwn156
313. Hayashi N, Nakamori S, Okami J, Nagano H, Dono K, Umeshita K, et al. Association between expression levels of CA 19-9 and N-acetylglucosamine-beta;1,3-galactosyltransferase 5 gene in human pancreatic cancer tissue. Pathobiology (2004) 71(1):26–34. doi:10.1159/000072959
314. Gao Y, Vlahakis JZ, Szarek WA, Brockhausen I. Selective inhibition of glycosyltransferases by bivalent imidazolium salts. Bioorg Med Chem (2013) 21(5):1305–11. doi:10.1016/j.bmc.2012.12.034
315. Nishihara S, Iwasaki H, Kaneko M, Tawada A, Ito M, Narimatsu H. Alpha1,3-fucosyltransferase 9 (FUT9; Fuc-TIX) preferentially fucosylates the distal GlcNAc residue of polylactosamine chain while the other four alpha1,3FUT members preferentially fucosylate the inner GlcNAc residue. FEBS Lett (1999) 462(3):289–94. doi:10.1016/S0014-5793(99)01549-5
316. Ogawa J, Inoue H, Koide S. Expression of alpha-1,3-fucosyltransferase type IV and VII genes is related to poor prognosis in lung cancer. Cancer Res (1996) 56(2):325–9.
317. Martín-Satué M, Marrugat R, Cancelas JA, Blanco J. Enhanced expression of alpha(1,3)-fucosyltransferase genes correlates with E-selectin-mediated adhesion and metastatic potential of human lung adenocarcinoma cells. Cancer Res (1998) 58(7):1544–50.
318. Martin-Satue M, de Castellarnau C, Blanco J. Overexpression of alpha(1,3)-fucosyltransferase VII is sufficient for the acquisition of lung colonization phenotype in human lung adenocarcinoma HAL-24Luc cells. Br J Cancer (1999) 80(8):1169–74. doi:10.1038/sj.bjc.6690482
319. Hiraiwa N, Dohi T, Kawakami-Kimura N, Yumen M, Ohmori K, Maeda M, et al. Suppression of sialyl Lewis X expression and E-selectin-mediated cell adhesion in cultured human lymphoid cells by transfection of antisense cDNA of an alpha1 – >3 fucosyltransferase (Fuc-T VII). J Biol Chem (1996) 271(49):31556–61. doi:10.1074/jbc.271.49.31556
320. Padró M, Cobler L, Garrido M, de Bolós C. Down-regulation of FUT3 and FUT5 by shRNA alters Lewis antigens expression and reduces the adhesion capacities of gastric cancer cells. Biochim Biophys Acta (2011) 1810(12):1141–9. doi:10.1016/j.bbagen.2011.09.011
321. Carvalho AS, Harduin-Lepers A, Magalhães A, Machado E, Mendes N, Costa LT, et al. Differential expression of alpha-2,3-sialyltransferases and alpha-1,3/4-fucosyltransferases regulates the levels of sialyl Lewis a and sialyl Lewis x in gastrointestinal carcinoma cells. Int J Biochem Cell Biol (2010) 42(1):80–9. doi:10.1016/j.biocel.2009.09.010
322. Guo Q, Guo B, Wang Y, Wu J, Jiang W, Zhao S, et al. Functional analysis of alpha 1,3/4-fucosyltransferase VI in human hepatocellular carcinoma cells. Biochem Biophys Res Commun (2012) 417(1):311–7. doi:10.1016/j.bbrc.2011.11.106
323. Li J, Guillebon AD, Hsu JW, Barthel SR, Dimitroff CJ, Lee YF, et al. Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br J Cancer (2013) 109(12):3014–22. doi:10.1038/bjc.2013.690
324. Okeley NM, Alley SC, Anderson ME, Boursalian TE, Burke PJ, Emmerton KM, et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc Natl Acad Sci U S A (2013) 110(14):5404–9. doi:10.1073/pnas.1222263110
325. Zandberg WF, Kumarasamy J, Pinto BM, Vocadlo DJ. Metabolic inhibition of sialyl-Lewis X biosynthesis by 5-thiofucose remodels the cell surface and impairs selectin-mediated cell adhesion. J Biol Chem (2012) 287(47):40021–30. doi:10.1074/jbc.M112.403568
326. Shinoda K, Shitara K, Yoshihara Y, Kusano A, Uosaki Y, Ohta S, et al. Panosialins, inhibitors of an alpha1,3-fucosyltransferase Fuc-TVII, suppress the expression of selectin ligands on U937 cells. Glycoconj J (1998) 15(11):1079–83. doi:10.1023/A:1006953626578
327. Mong TK, Lee LV, Brown JR, Esko JD, Wong CH. Synthesis of N-acetyllactosamine derivatives with variation in the aglycon moiety for the study of inhibition of sialyl Lewis x expression. Chembiochem (2003) 4(9):835–40. doi:10.1002/cbic.200300650
328. Yu-Nong L, Daniel S, Sheng-Wei L, Sue-Ming C, Ting-Chien L, Yu-Ruei C. Chemoenzymatic synthesis of GDP-L-fucose derivatives as potent and selective alpha-1,3-fucosyltransferase inhibitors. Adv Synth Catal (2012) 354(9):1750–8. doi:10.1002/adsc.201100940
329. Burkart MD, Vincent SP, Düffels A, Murray BW, Ley SV, Wong CH. Chemo-enzymatic synthesis of fluorinated sugar nucleotide: useful mechanistic probes for glycosyltransferases. Bioorg Med Chem (2000) 8(8):1937–46. doi:10.1016/S0968-0896(00)00139-5
330. Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, et al. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat Chem Biol (2012) 8(7):661–8. doi:10.1038/nchembio.999
331. Luengo JI, Gleason JG. Synthesis of C-fucopyranosyl analogs of Gdp-L-fucose as inhibitors of fucosyltransferases. Tetrahedron Lett (1992) 33(46):6911–4. doi:10.1016/S0040-4039(00)60893-6
332. Pasquarello C, Picasso S, Demange R, Malissard M, Berger EG, Vogel P. The C-disaccharide alpha-C(1 -> 3)-mannopyranoside of N-acetylgalactosamine is an inhibitor of glycohydrolases and of human alpha-1,3-fucosyltransferase VI. its epimer alpha-(1 -> 3)-mannopyranoside of N-acetyltalosamine is not. J Org Chem (2000) 65(14):4251–60. doi:10.1021/jo991952u
333. Vidal S, Bruyère I, Malleron A, Augé C, Praly JP. Non-isosteric C-glycosyl analogues of natural nucleotide diphosphate sugars as glycosyltransferase inhibitors. Bioorg Med Chem (2006) 14(21):7293–301. doi:10.1016/j.bmc.2006.06.057
334. Niu X, Fan X, Sun J, Ting P, Narula S, Lundell D. Inhibition of fucosyltransferase VII by gallic acid and its derivatives. Arch Biochem Biophys (2004) 425(1):51–7. doi:10.1016/j.abb.2004.02.039
Keywords: glycans, glycosyltransferases, inhibitors, cancer, hexosamine biosynthetic pathway, O-linked glycan, N-linked glycan, glycoconjugate
Citation: Vasconcelos-dos-Santos A, Oliveira IA, Lucena MC, Mantuano NR, Whelan SA, Dias WB and Todeschini AR (2015) Biosynthetic machinery involved in aberrant glycosylation: promising targets for developing of drugs against cancer. Front. Oncol. 5:138. doi: 10.3389/fonc.2015.00138
Received: 04 May 2015; Accepted: 02 June 2015;
Published: 25 June 2015
Edited by:
Ala-Eddin Al Moustafa, McGill and Concordia Universities, Canada; Syrian Research Cancer Center of the Syrian Society Against Cancer, SyriaReviewed by:
Daniel Christian Hoessli, International Center for Chemical and Biological Sciences, SwitzerlandOlivier Micheau, Institut national de la santé et de la recherche médicale, France
Copyright: © 2015 Vasconcelos-dos-Santos, Oliveira, Lucena, Mantuano, Whelan, Dias and Todeschini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriane Regina Todeschini, Instituto de Biofísica Carlos Chagas Filho,Universidade Federal do Rio de Janeiro, Avenida Carlos Chagas Filho, 373 - Cidade Universitária, Rio de Janeiro, 21941902, Rio de Janeiro, Brazil,YWRyaWFuZXRAYmlvZi51ZnJqLmJy