 Loredana Mauro
Loredana Mauro Giuseppina Daniela Naimo
Giuseppina Daniela Naimo Emilia Ricchio
Emilia Ricchio Maria Luisa Panno
Maria Luisa Panno Sebastiano Andò
Sebastiano Andò- Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Cosenza, Italy
Obesity is a chronic and multifactorial disorder that is reaching epidemic proportions. It is characterized by an enlarged mass of adipose tissue caused by a combination of size increase of preexisting adipocytes (hypertrophy) and de novo adipocyte differentiation (hyperplasia). Obesity is related to many metabolic disorders like hypertension, type 2 diabetes, metabolic syndrome, and cardiovascular disease, and it is associated with an increased risk of cancer development in different tissues including breast. Adipose tissue is now regarded as not just a storage reservoir for excess energy, but rather as an endocrine organ, secreting a large number of bioactive molecules called adipokines. Among these, adiponectin represents the most abundant adipose tissue-excreted protein, which exhibits insulin sensitizing, anti-inflammatory, and antiatherogenic properties. The serum concentrations of adiponectin are inversely correlated with body mass index. Recently, low levels of plasma adiponectin have been associated with an increased risk for obesity-related cancers and development of more aggressive phenotype, concomitantly with alterations in the bioavailability of insulin-like growth factor-I (IGF-I) and IGF-I receptor (IGF-IR) signaling pathways. In this review, we discuss the cross-talk between adiponectin/AdipoR1 and IGF-I/IGF-IR in breast cancer.
Introduction
Breast cancer is one of the most common forms of female malignancy in the world. Many epidemiological studies suggest an important, but still controversial, role for obesity and adipose tissue mass in breast cancer risk and an association with tumor phenotypes (1). The latter event has been suggested to relay on the increased estrogen production in peripheral fat deposits, through the aromatization of androgens, secreted by the adrenal gland (2). However, on the basis of many recent studies, the contribution of obesity to the development of breast carcinoma cannot be ascribed to the increased estrogen levels only. In more recent years, it has been demonstrated that autocrine, endocrine, and paracrine-acting adipocytes-derived factors, known as adipocytokines, may contribute to the regulation of breast cancer development and progression (3). As a member of the adipocytokine family, adiponectin is synthesized and secreted almost exclusively by the adipose tissue (4), and its plasma concentration is inversely correlated with adiposity (5). Adiponectin exhibits anti-inflammatory activity, a protective effect against metabolic disorders, such as insulin resistance, fatty-acid oxidation, and insulin sensitivity.
Furthermore, recent clinical studies have implicated that the reduction of circulating adiponectin levels is a risk factor not only for type 2 diabetes and cardiovascular diseases but also for several types of cancers, including breast cancer (6, 7). However, conflicting observations have been reported on the effects elicited by adiponectin on breast cancer cell growth. It has been extensively demonstrated that adiponectin induces an anti-proliferative response in human estrogen receptor alpha (ERα)-negative breast cancer cells (8–10), while controversial data are reported in ERα-positive cells (11–16). The possible mechanisms through which adiponectin exerts anticancer effects may include activation of AMP-activated kinase and decrease of mTOR signaling (12). This addresses how a network of different signaling pathways is involved in the adiponectin-mediated effects in breast cancer, and the response to adiponectin seems to be dependent on breast cancer phenotypes.
Obesity and Breast Carcinogenesis
Breast carcinoma represents the highest incidence of cancers affecting women, and it is the second most fatal cancer type (17, 18). Aside from the genetic predisposition, a myriad of other factors can contribute to the pathogenesis of breast cancer (19). Among the modifiable factors, obesity not only represents a serious risk for breast carcinogenesis but also positively correlated with a poor outcome (20).
It is now widely accepted that obesity may promote breast tumor through several mechanisms (Figure 1). Traditionally, the adverse effect of obesity on breast cancer prognosis has been linked to the higher estrogen levels produced, consequent to a greater aromatase activity due to the excess of adipose tissue (21). Despite the well documented relationship between obesity and estrogenic activity, it is evident that this cannot fully explain the association between body weight and breast cancer risk and prognosis.
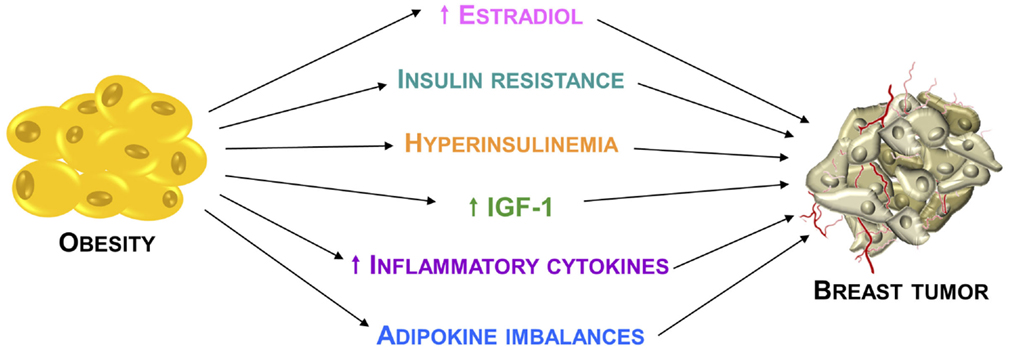
Figure 1. Relationship between obesity and breast cancer. Principal mechanisms through which the obesity condition may promote breast cancer development and progression.
Estrogen-independent mechanisms for which there are both experimental and epidemiological supports involve insulin resistance, hyperinsulinemia, greater bioavailability of insulin-like growth factor-I (IGF-I), which represents the more relevant obesity-related growth factor, and dysregulation of insulin-like growth factor-I receptor (IGF-IR) downstream signaling pathways (22).
Apart from these mechanisms, another important element in obesity-mediated breast carcinogenesis is represented by the interaction between tumor cells and the surrounding microenvironment, which comprises stromal cells, soluble factors, signaling molecules, and extracellular matrix that can promote tumorigenesis, and make the tumor resistant from host immunity and therapeutic response. Importantly, obesity is also characterized by multiple changes in the adipose tissue biology.
Particularly, white adipose tissue in obese individuals exhibits chronic mild inflammatory status, mostly defined by infiltration of leukocytes, including macrophages (23). For instance, stromal adipocytes directly influence breast cancer cells growth and progression through the secretion of several biologically active polypeptides known as adipokines (24, 25), which have a chemo-attractant action, causing the recruitment of macrophages. The activated macrophages release proinflammatory molecules, including TNFα, IL-1β, and IL-6. These cytokines play both local and systemic actions, contributing to insulin resistance and breast cancer tumorigenesis (26, 27).
Adiponectin
Adiponectin, one of the most important adipokines, is produced exclusively in white adipocytes. The human gene encoding adiponectin maps to chromosome 3q27, a region associated with susceptibility for developing metabolic syndrome and type 2 diabetes in Caucasians (28). Adiponectin gene spans 16 kb and contains three exons and two introns (29). Several single nucleotide polymorphisms (SNPs) in the coding region were identified, which are associated with alterations of adiponectin function and important clinical conditions. In particular, SNPs are associated with the strengthening of adiponectin effects on insulin resistance, type 2 diabetes, obesity, dyslipidemia, and many obesity-related malignancies (30).
Structurally, the molecule of human adiponectin consists of 244 amino acid residues and contains four distinct domains; at the N-terminus, there is an 18 amino acid long-signal peptide followed by a short-hypervariable region without homology to any known sequences and a collagen domain with 22 repeated motifs; C-terminal contains globular domain homologous to C1q molecule of complement cascade (Figure 2A). The C-terminal globular domain also shows homology with TNF-α trimeric cytokines family (31, 32).
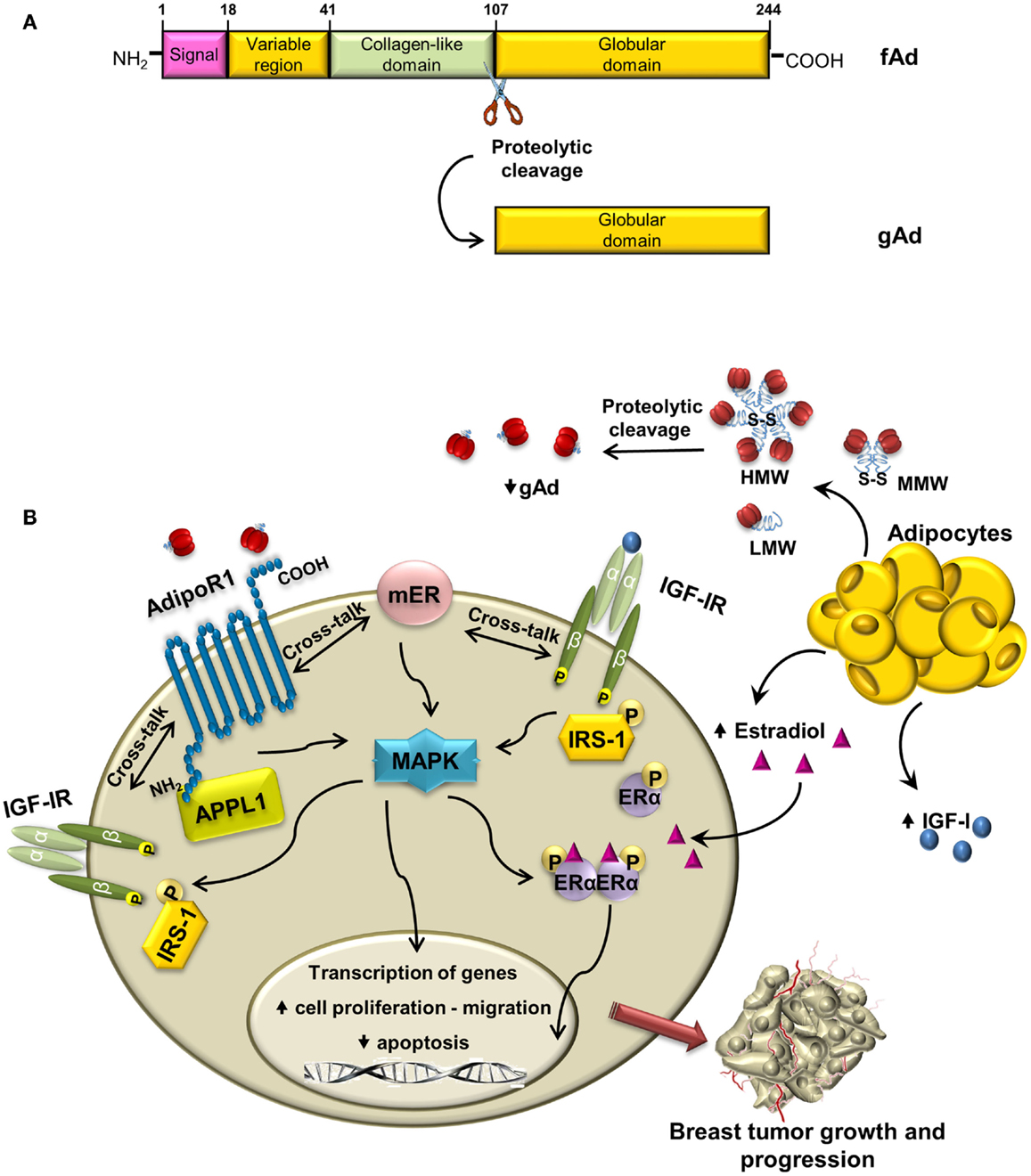
Figure 2. Cross-talk between adiponectin and IGF-IR in breast cancer: overview of mechanisms. (A) Schematic structure of adiponectin monomer with different domains and proteolytic cleavage site indicated. fAd and gAd indicate full length and globular adiponectin, respectively. (B) In ERα-positive breast cancer cells, adiponectin, produced by adipocytes, binds to AdipoR1, and establishes cross-talk with membrane ERα and with IGF-IR. The enhanced phosphorylation of MAPK may allow the activation of IGF-IR, IRS-1, and ERα, which contribute to breast tumor growth and progression.
Once synthesized, adiponectin undergoes posttranslational hydroxylation and glycosylation modifications (33) and before secretion it forms trimers (low-molecular weight, LMW) that oligomerize to produce hexame-rich middle molecular weight (MMW) and high-molecular weight (HMW) forms. Adiponectin circulating in plasma exists as the full-length protein (fAd) or a proteolytic cleavage fragment known as globular adiponectin (gAd) (34–36), which is probably generated by elastase digestion (37, 38) (Figure 2A).
The levels of this adipokine are abundant in human plasma, with concentrations ranging from 3 to 30 μg/ml (39), and they are about two to three times lower in male compared to female, due to the lower amounts of HMW form (40, 41). Its concentration is inversely correlated with body mass index (42). Indeed, unlike most of the other adipokines, plasma adiponectin levels are found to be lower in obese than in lean individuals (3). Moreover, low-circulating levels are found in type 2 diabetes (43), and mice lacking adiponectin develop metabolic syndrome, with insulin resistance, glucose intolerance, hyperglycemia, and hypertension (44, 45). The mechanisms responsible for the adiponectin downregulation are still unclear. It has been speculated that its reduced levels in obesity may be caused by the enhanced production of proinflammatory cytokines, in particular, by the tumor necrosis factor α (TNFα) (46). However, another potential explanation indicates a negative feedback of adiponectin on its own production and probably on the expression of its receptors during the development of obesity (47).
Adiponectin Receptors
Adiponectin exerts many of its cellular effects through binding to two receptor isoforms, the adiponectin receptor 1 (AdipoR1) and 2 (AdipoR2) (48). AdipoR1 and AdipoR2 contain seven transmembrane domains with internal N-terminus and external C-terminus regions; thus, they are both structurally and functionally distinct from G-protein-coupled receptors (GPCR) (49). AdipoR1 presents high affinity for gAd and low affinity for the full-size ligand (50), and it is expressed ubiquitously but abundantly in skeletal muscle and endothelial cells. AdipoR2 has intermediate affinity for both forms of adiponectin and is predominantly expressed in the liver (51). Since AdipoR1 is the predominant receptor in skeletal muscle, while AdipoR2 is predominantly expressed in liver, this correlated with the fact that gAd exerts its insulin mimetic and insulin-sensitizing effect more effectively compared to fAd in skeletal muscle and vice versa (52).
A non-classical third potential adiponectin receptor is T-cadherin, which was also found to competitively bind only the hexameric and HMW forms of adiponectin (53–55). Various studies have suggested the involvement of T-cadherin, which plays an important role in cell adhesion and in calcium-mediated cell to cell interactions and signaling (53), in mediating functional effects of adiponectin. T-cadherin lacks an intracellular domain needed for signal transduction; thus, it has been suggested that it may function as a coreceptor by competing with AdipoR1 and AdipoR2 receptors for adiponectin binding or interfering with adiponectin signal transduction (56).
Adiponectin Signaling Pathways
Adiponectin activates various signaling molecules when bound to its receptors. Several adiponectin receptor binding proteins have now been identified (57). Among these, the first and best characterized is the adaptor protein APPL1, containing a pleckstrin homology domain, a phosphotyrosine binding (PTB) domain, and a leucine zipper motif (57, 58). AdipoR1 and AdipoR2 interact with the PTB domain of APPL-1 through their N-terminal intracellular region, thereby inducing adiponectin actions through the sequential activation of downstream signaling. It has emerged that APPL1 plays an important role in mediating many of adiponectin’s effects, including metabolic, antiinflammation, antiatherogenic, and cytoprotection responses (59, 60).
Adiponectin exerts its effects through the activation of AMPK, mTOR, PI3K/Akt, MAPK, PPAR-α, STAT3, and NF-kB (47, 59).
Most of its effects are mediated through the cellular energy sensor AMPK (61, 62), which promotes glucose utilization that results in an increased fatty-acid oxidation, increased glucose uptake at the skeletal muscle level, and reduced gluconeogenesis in the liver (47). In addition, adiponectin exerts its insulin-sensitizing effects through the activation of PPAR-α, thereby enhancing fatty-acid combustion and energy consumption, leading to a tissue decrease content of triglycerides in the liver and skeletal muscle, and improving insulin sensitivity in vivo (63).
Adiponectin and Breast Cancer
Adiponectin is emerging as an important factor in carcinogenesis. Many clinical investigations suggested that low-adiponectin concentrations are associated with an increased risk for obesity-related cancer, such as prostate, colon, endometrial, and breast cancer (47). Epidemiological studies address that, in women with low-circulating adiponectin levels, breast tumors may present a more aggressive phenotype, exemplified by large tumor size, high-histological grade, estrogen receptor negativity, and increased angiogenesis and metastasis (6). Recently, through a transcriptomic profiling, Merdad and co-workers evidenced a downregulation of adiponectin and other molecules involved in lipid metabolism in surgically resected breast tumors patients. The results address how some ethnic groups are more susceptible to breast cancer occurrence (64).
It has been well documented that the pathogenesis of mammary cancer is not only dependent on genetic alterations but also largely on the interactions between malignant cells and components of the breast microenvironment, which exerts an important influence on the phenotype of the neoplastic cells and on tumor progression. Microenvironmental molecules, such as chemokines, cytokines, and growth factors, could influence tumor progression by three major, non-mutually exclusive, mechanisms. The first is by further increasing the genetic instability of tumor cells. The second is by inducing signaling cascades in tumor cells via tumor-associated receptors thereby controlling gene expression in these cells. The third mechanism is by exerting selective pressures on the cells (24, 65).
Thus, the malignant cell phenotype is regulated not only by autonomous signals originating from cancer cells but also by the effects of the surrounding stromal cells, which influence mammary epithelial cell growth and differentiation (24). The close association between mammary epithelial cells and adipocytes may favor a more direct action of adipokines on such tissue (11).
However, the role of this adipokine on breast tumorigenesis is still unclear, and it seems to be dependent on cell types. Low-adiponectin doses mediate an anti-proliferative response in ERα-negative breast cancer cells through the regulation of genes involved in cell cycle, such as p53, Bax, Bcl-2, c-myc, and Cyclin D1 (66). In addition, in these cells, the adipokine is able to activate the AMPK, which in turn inhibits mTOR, through tuberous sclerosis complex 2 (TSC2), thus counteracting carcinogenesis (67, 68). Moreover, activated AMPK plays a crucial role in the regulation of growth arrest and apoptosis by stimulating p21 and p53 (69). On the other hand, it is emerging that adiponectin, at low concentrations, increases proliferation in ERα-positive breast cancer cells (15, 16). This occurs through the activation of ERα at both genomic and non-genomic levels (70), together with the positive regulation of cyclin D1 expression (71).
On the basis of these observations, adiponectin may represent a promising diagnostic and prognostic biomarker to identify high-susceptibility individuals for developing obesity-related tumors. Recently, it has developed an adiponectin-based short peptide, ADP 355, acting as AdipoR1 agonist, and able to modulate several signaling pathways (AMPK, Akt, STAT3, and ERK1/2) in a manner similar to gAd. In breast cancer cells, ADP 355 reproduces the adiponectin-induced anti-proliferative activity both in vitro and in vivo (3, 72, 73).
IGF-IR in Breast Cancer
Insulin-like growth factor-I receptor is an evolutionary conserved, ubiquitous transmembrane tyrosine kinase structurally similar to the insulin receptor. IGF-IR regulates different biological processes, such as proliferation, survival, differentiation, transformation, cell-substrate, and cell–cell interactions (74–79). More recently, it has been highlighted that the role of the IGF-IR in the development and maintenance of the cancer stem cells, in epithelial–mesenchymal transition, and in the regulation of the tumor microenvironment (80). The first evidence that IGF-IR is critical in tumorigenesis was provided by the observation that constitutive overexpression of IGF-IR induced the transformed phenotype in cultured cells (81, 82).
It is now quite evident that IGF-IR plays a multifaceted and complex role in the development and progression of a malignant disease. Numerous clinical and experimental data indicated that IGF-IR is overexpressed in several subtypes of breast cancer (83), and many conditions lead to the activation of IGF-IR tyrosine kinase activity (84), allowing interaction with its main substrates, such as insulin receptor substrates (IRS) and the Src-homology-2-containing protein SH2 (SHC) (74). Once phosphorylated, these proteins act as docking molecules that bind to and activate cellular kinases, initiating different downstream signaling pathways. In this regard, IGF-IR induces activation of Ras/raf/MAPK and PI3K/Akt signaling, which alter the expression of genes involved in cell proliferation and survival (85), thus contributing to breast carcinogenesis.
In hormone-dependent breast cancer cells, the IGF-IR and ERα are co-expressed and IGF-I acts in synergy with estradiol to stimulate proliferation (74). Indeed, estradiol up-regulates IGF-IR mRNA and protein levels as well as its tyrosine phosphorylation (73, 74, 86). Furthermore, estradiol significantly stimulates the expression of IRS-1 in ERα-positive cells (87).
Elevated expression of IGF-IR or IRS-1 appears to increase drug and radio resistance of breast cancer cells and favor cancer recurrence in patients (88, 89). In addition to promote cell growth, IGF-IR counteracts apoptosis in ERα-positive cells, activating the IRS-1/PI3K pathway (74, 89). For these reasons, IGF axis has been validated for developing targeted therapy for cancer prevention and treatment (80, 90, 91).
Adiponectin and IGF-IR Interaction in Breast Cancer
Insulin-like growth factors and IGF-IR are involved in the metabolic cellular response to adiponectin in many cell types (63). It has been widely demonstrated that IGF-IR increases growth and survival of neoplastic cells, altering the expression of specific genes regulated through the activation of Akt and MAPK pathways (92).
Emerging evidences address the existence of a cross-talk between adiponectin/AdipoR1 and IGF-IR in breast cancer. It has been previously demonstrated that adiponectin increases IGF-IR-β subunit tyrosine phosphorylation and the downstream MAPK activation in granulosa cells (93). More recently, it has been shown that low concentrations of adiponectin rapidly increase IGF-IR phosphorylation in ERα-positive breast cancer cells, concomitantly with ligand-independent activation of ERα (70). It is worth to note that in MCF-7 cells, upon adiponectin treatment, knockdown of ERα reduces IGF-IR phosphorylation, whereas a specific siRNA for IGF-IR prevents adiponectin-induced ERα transactivation (70).
Moreover, IGF-IR takes part in the formation of a protein complex involved in the induction of ERα-positive breast cancer cell growth. Indeed, adiponectin enhances the coimmunoprecipitation of AdipoR1, APPL1, ERα, IGF-IR, and c-Src in MCF-7 cells. The formation of this multiprotein complex leads, via c-Src, to MAPK activation, which is abrogated in the presence of specific RNA silencers targeting ERα or IGF-IR (70). On the other hand, in ERα-negative MDA-MB-231 cells adiponectin is no longer able to induce MAPK phosphorylation, but it increases activation of AMPK, which mediates anti-proliferative effect in these cells (70, 71). In this concern, it has been well documented how in breast cancer cells the adiponectin-induced activation of AMPK is concomitant with inactivation of MAPK (12).
Interestingly, our unpublished data demonstrate that adiponectin enhances phosphorylation and protein expression of IRS-1 in ERα-positive breast cancer cells, thus amplifying the IGF-I/IGF-IR growth signaling. All these data address the existence of a possible cross-talk between Adiponectin/AdipoR1, ERα, and IGF-IR involved in the positive regulation of ERα-positive breast cancer cell growth. These data are supported by other papers showing that adiponectin, via its adapter protein APPL1, is able to induce phosphorylation of IRS-1, which mediates the effect of activated IR/IGF-IR in normal and tumoral cells (63, 94).
Our preliminary results surprisingly show that adiponectin at low concentrations is able to potentiate IGF-I-induced anchorage-independent growth in ERα-positive breast cancer cells. In contrast, adiponectin is able to elicit opposite action in antagonizing the stimulatory effects induced by IGF-I in ERα-negative breast cancer cells. In the same vein, wound healing assays evidence that in ERα-positive breast cancer cells, adiponectin, and IGF-I enhance cell motility and the co-treatment results in an additive effect, addressing how adiponectin may synergize with IGF-IR signaling. On the contrary, in ERα-negative breast cancer cells adiponectin counteracts the IGF-I-induced cell migration (manuscript in preparation). These results well fit with recent findings demonstrating that adiponectin is able to regulate migration and invasion in different breast cancer cells (95–97).
Conclusion
Adiponectin is emerging as a crucial adipokine involved in breast carcinogenesis in women with obesity. In ERα-positive breast cancer cells, the interaction of adiponectin with its specific receptor induces the activation of multiple pathways, through the interplay between ERα and IGF-IR. This leads to (i) increased activation of MAPK and upregulation of genes involved in proliferation and inhibition of apoptosis, (ii) induction of cell migration (Figure 2B). On the basis of these findings, we may conclude that adiponectin differently modulates IGF-I stimulatory effect in breast cancer cells in relationship to ERα status. Indeed, the antagonistic effects exerted by adiponectin on IGF-IR signaling are evident only in ERα-negative breast cancer cells. Thus, only in the latter circumstance, adiponectin sounds to be exploited in novel therapeutic strategies for breast cancer treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Associazione Italiana Ricerca sul Cancro [AIRC; IG grant 11595 and My First AIRC Grant (MFAG) 6180]. We are grateful to Dr. Sturino (University of Calabria, Italy) for his assistance with English revision.
References
1. James FR, Wootton S, Jackson A, Wiseman M, Copson ER, Cutress RI. Obesity in breast cancer – what is the risk factor? Eur J Cancer (2015) 51(6):705–20. doi: 10.1016/j.ejca.2015.01.057
2. Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem (2004) 279:19908–15. doi:10.1074/jbc.M313191200
3. Surmacz E. Leptin and adiponectin: emerging therapeutic targets in breast cancer. J Mammary Gland Biol Neoplasia (2013) 18:321–32. doi:10.1007/s10911-013-9302-8
4. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev (2005) 26:439–51. doi:10.1210/er.2005-0005
5. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab (2001) 86:1930–5. doi:10.1210/jcem.86.5.7463
6. Mantzoros C, Petridou E, Dessypris N, Chavelas C, Dalamaga M, Alexe DM, et al. Adiponectin and breast cancer risk. J Clin Endocrinol Metab (2004) 89:1102–7. doi:10.1210/jc.2003-031804
7. Miyoshi Y, Funahashi T, Kihara S, Taguchi T, Tamaki Y, Matsuzawa Y, et al. Association of serum adiponectin levels with breast cancer risk. Clin Cancer Res (2003) 9:5699–704.
8. Kang JH, Lee YY, Yu BY, Yang BS, Cho KH, Yoon DK, et al. Adiponectin induces growth arrest and apoptosis of MDA-MB-231 breast cancer cell. Arch Pharm Res (2005) 28:1263–9. doi:10.1007/BF02978210
9. Dos Santos E, Benaitreau D, Dieudonne MN, Leneveu MC, Serazin V, Giudicelli Y, et al. Adiponectin mediates an antiproliferative response in human MDA-MB 231 breast cancer cells. Oncol Rep (2008) 20:971–7. doi:10.3892/or_00000098
10. Grossmann ME, Nkhata KJ, Mizuno NK, Ray A, Cleary MP. Effects of adiponectin on breast cancer cell growth and signaling. Br J Cancer (2008) 98:370–9. doi:10.1038/sj.bjc.6604166
11. Körner A, Pazaitou-Panayiotou K, Kelesidis T, Kelesidis I, Williams CJ, Kaprara A, et al. Total and high-molecular-weight adiponectin in breast cancer: in vitro and in vivo studies. J Clin Endocrinol Metab (2007) 92:1041–8. doi:10.1210/jc.2006-1858
12. Dieudonne MN, Bussiere M, Dos Santos E, Leneveu MC, Giudicelli Y, Pecquery R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem Biophys Res Commun (2006) 345:271–9. doi:10.1016/j.bbrc.2006.04.076
13. Jardé T, Caldefie-Chézet F, Goncalves-Mendes N, Mishellany F, Buechler C, Penault-Llorca F, et al. Involvement of adiponectin and leptin in breast cancer: clinical and in vitro studies. Endocr Relat Cancer (2009) 16:1197–210. doi:10.1677/ERC-09-0043
14. Treeck O, Lattrich C, Juhasz-Boess I, Buchholz S, Pfeiler G, Ortmann O. Adiponectin differentially affects gene expression in human mammary epithelial and breast cancer cells. Br J Cancer (2008) 99:1246–50. doi:10.1038/sj.bjc.6604692
15. Pfeiler GH, Buechler C, Neumeier M, Schäffler A, Schmitz G, Ortmann O, et al. Adiponectin effects on human breast cancer cells are dependent on 17-beta estradiol. Oncol Rep (2008) 19:787–93. doi:10.3892/or.19.3.787
16. Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, et al. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res (2009) 15:3265–76. doi:10.1158/1078-0432.CCR-08-2649
17. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin (2012) 62:10–29. doi:10.3322/caac.20138
18. DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin (2011) 61:409–18. doi:10.3322/caac.20134
19. Singletary SE. Rating the risk factors for breast cancer. Ann Surg (2003) 237:474–82. doi:10.1097/00000658-200304000-00007
20. Loi S, Milne RL, Friedlander ML, McCredie MR, Giles GG, Hopper JL, et al. Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol Biomarkers Prev (2005) 14:1686–91. doi:10.1158/1055-9965.EPI-05-0042
21. McTiernan A, Rajan KB, Tworoger SS, Irwin M, Bernstein L, Baumgartner R, et al. Adiposity and sex hormones in postmenopausal breast cancer survivors. J Clin Oncol (2003) 21:1961–6. doi:10.1200/JCO.2003.07.057
22. Sundaram S, Johnson AR, Makowski L. Obesity, metabolism and the microenvironment: links to cancer. J Carcinog (2013) 12:19. doi:10.4103/1477-3163.119606
23. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med (2012) 18:363–74. doi:10.1038/nm.2627
24. Andò S, Catalano S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol (2011) 8:263–75. doi:10.1038/nrendo.2011.184
25. Vona-Davis L, Rose DP. Adipokines as endocrine, paracrine and autocrine factors in breast cancer risk and progression. Endocr Relat Cancer (2007) 14:189–206. doi:10.1677/ERC-06-0068
26. Wolin KY, Carson K, Colditz GA. Obesity and cancer. Oncologist (2010) 15:556–65. doi:10.1634/theoncologist.2009-0285
27. Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res (2013) 19:6074–83. doi:10.1158/1078-0432.CCR-12-2603
28. Comuzzie AG, Funahashi T, Sonnenberg G, Martin LJ, Jacob HJ, Black AE, et al. The genetic basis of plasma variation in adiponectin, a global endophenotype for obesity and the metabolic syndrome. J Clin Endocrinol Metab (2001) 86:4321–5. doi:10.1210/jcem.86.9.7878
29. Saito K, Tobe T, Minoshima S, Aakawa S, Sumiya J, Yoda M, et al. Organization of the gene for gelatin-binding protein (GBP28). Gene (1999) 229:67–73. doi:10.1016/S0378-1119(99)00041-4
30. Takahashi M, Arita Y, Yamagata K, Matsukawa Y, Okutomi K, Horie M, et al. Genomic structure and mutations in adipose-specific gene, adiponectin. Int J Obes Relat Metab Disord (2000) 24:861–8. doi:10.1038/sj.ijo.0801244
31. Shapiro L, Scherer PE. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr Biol (1998) 8:335–8. doi:10.1016/S0960-9822(98)70133-2
32. Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood (2000) 96:1723–32.
33. Wang Y, Xu A, Knight C, Xu LY, Cooper GJ. Hydroxylation and glycosylation of the four conserved lysine residues in the collagenous domain of adiponectin: potential role in the modulation of its insulin-sensitizing activity. J Biol Chem (2002) 277:19521–9. doi:10.1074/jbc.M200601200
34. Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, et al. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem (2003) 278:9073–85. doi:10.1074/jbc.M207198200
35. Tsao TS, Murrey HE, Hug C, Lee DH, Lodish HF. Oligomerization state-dependent activation of NF-kappa B signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30). J Biol Chem (2002) 277:29359–62. doi:10.1074/jbc.C200312200
36. Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem (2003) 278:40352–63. doi:10.1074/jbc.M300365200
37. Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A (2001) 98:2005–10. doi:10.1073/pnas.98.4.2005
38. Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, et al. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology (2005) 146:790–6. doi:10.1210/en.2004-1096
39. Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care (2003) 26:2442–50. doi:10.2337/diacare.26.8.2442
40. Combs TP, Berg AH, Rajala MW, Klebanov S, Iyengar P, Jimenez-Chillaron JC, et al. Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes (2003) 52:268–76. doi:10.2337/diabetes.52.2.268
41. Xu A, Chan KW, Hoo RL, Wang Y, Tan KC, Zhang J, et al. Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J Biol Chem (2005) 280:18073–80. doi:10.1074/jbc.M414231200
42. Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol (2010) 316:129–39. doi:10.1016/j.mce.2009.08.018
43. Okamoto Y, Kihara S, Funahashi T, Matsuzawa Y, Libby P. Adiponectin: a key adipocytokine in metabolic syndrome. Clin Sci (2006) 110:267–78. doi:10.1042/CS20050182
44. Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem (2002) 277:25863–6. doi:10.1074/jbc.C200251200
45. Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med (2002) 8:731–7. doi:10.1038/nm724
46. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol (2006) 6:772–83. doi:10.1038/nri1937
47. Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev (2012) 33:547–94. doi:10.1210/er.2011-1015
48. Brochu-Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, Palin MF. Adiponectin action from head to toe. Endocrine (2010) 37:11–32. doi:10.1007/s12020-009-9278-8
49. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature (2003) 423:762–9. doi:10.1038/nature01705
50. Wang H, Zhang H, Jia Y, Zhang Z, Craig R, Wang X, et al. Adiponectin receptor 1 gene (ADIPOR1) as a candidate for type 2 diabetes and insulin resistance. Diabetes (2004) 53:2132–6. doi:10.2337/diabetes.53.8.2132
51. Chen X, Wang Y. Adiponectin and breast cancer. Med Oncol (2011) 28:1288–95. doi:10.1007/s12032-010-9617-x
52. Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem (2003) 278:2461–8. doi:10.1074/jbc.M209033200
53. Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A (2004) 101:10308–13. doi:10.1073/pnas.0403382101
54. Asada K, Yoshiji H, Noguchi R, Ikenaka Y, Kitade M, Kaji K, et al. Crosstalk between high-molecular-weight adiponectin and T-cadherin during liver fibrosis development in rats. Int J Mol Med (2007) 20:725–9. doi:10.3892/ijmm.20.5.725
55. Chan DW, Lee JM, Chan PC, Ng IO. Genetic and epigenetic inactivation of T-cadherin in human hepatocellular carcinoma cells. Int J Cancer (2008) 123:1043–52. doi:10.1002/ijc.23634
56. Lee MH, Klein RL, El-Shewy HM, Luttrell DK, Luttrell LM. The adiponectin receptors AdipoR1 and AdipoR2 activate ERK1/2 through a Src/Ras-dependent pathway and stimulate cell growth. Biochemistry (2008) 47:11682–92. doi:10.1021/bi801451f
57. Buechler C, Wanninger J, Neumeier M. Adiponectin receptor binding proteins – recent advances in elucidating adiponectin signalling pathways. FEBS Lett (2010) 584:4280–6. doi:10.1016/j.febslet.2010.09.035
58. Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol (2006) 8:516–23. doi:10.1038/ncb1404
59. Obeid S, Hebbard L. Role of adiponectin and its receptors in cancer. Cancer Biol Med (2012) 9:213–20. doi:10.7497/j.issn.2095-3941.2012.04.001
60. Deepa SS, Dong LQ. APPL1: role in adiponectin signaling and beyond. Am J Physiol Endocrinol Metab (2009) 296:E22–36. doi:10.1152/ajpendo.90731.2008
61. Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes (2003) 52:1355–63. doi:10.2337/diabetes.52.6.1355
62. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med (2007) 13:332–9. doi:10.1038/nm1557
63. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med (2001) 7:941–6. doi:10.1038/90984
64. Merdad A, Karim S, Schulten HJ, Jayapal M, Dallol A, Buhmeida A, et al. Transcriptomics profiling study of breast cancer from Kingdom of Saudi Arabia revealed altered expression of adiponectin and fatty acid binding protein4: is lipid metabolism associated with breast cancer? BMC Genomics (2015) 16:S11. doi:10.1186/1471-2164-16-S1-S11
65. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest (2011) 121:3804–9. doi:10.1172/JCI57099
66. Wang Y. Adiponectin and breast cancer. Med Oncol (2011) 28:1288–95. doi:10.1007/s12032-010-9617-x
67. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115:577–90. doi:10.1016/S0092-8674(03)00929-2
68. Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci (2005) 26:69–76. doi:10.1016/j.tips.2004.12.011
69. Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, et al. Adenosine monophosphateactivated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res (2005) 97:837–44. doi:10.1161/01.RES.0000185823.73556.06
70. Mauro L, Pellegrino M, De Amicis F, Ricchio E, Giordano F, Rizza P, et al. Evidences that estrogen receptor α interferes with adiponectin effects on breast cancer cell growth. Cell Cycle (2014) 13:553–64. doi:10.4161/cc.27455
71. Mauro L, Pellegrino M, Giordano F, Ricchio E, Rizza P, De Amicis F, et al. Estrogen receptor-α drives adiponectin effects on cyclin D1 expression in breast cancer cells. FASEB J (2015) 29:2150–60. doi:10.1096/fj.14-262808
72. Khan S, Shukla S, Sinha S, Meeran SM. Role of adipokines and cytokines in obesity-associated breast cancer: therapeutic targets. Cytokine Growth Factor Rev (2013) 24:503–13. doi:10.1016/j.cytogfr.2013.10.001
73. Surmacz E, Otvos L. Molecular targeting of obesity pathways in cancer. Horm Mol Biol Clin Investig (2015) 22(2):53–62. doi:10.1515/hmbci-2015-0007
74. Surmacz E. Function of the IGF-I receptor in breast cancer. J Mammary Gland Biol Neoplasia (2000) 5:95–105. doi:10.1023/A:1009523501499
75. Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene (2000) 19:5574–81. doi:10.1038/sj.onc.1203854
76. Mauro L, Bartucci M, Morelli C, Andò S, Surmacz E. IGF-I receptor-induced cell-cell adhesion of MCF-7 breast cancer cells requires the expression of junction protein ZO-1. J Biol Chem (2001) 276:39892–7. doi:10.1074/jbc.M106673200
77. Le Roith D. Regulation of proliferation and apoptosis by the insulin-like growth factor I receptor. Growth Horm IGF Res (2000) 10:S12–3. doi:10.1016/S1096-6374(00)90005-4
78. Mauro L, Salerno M, Morelli C, Boterberg T, Bracke ME, Surmacz E. Role of the IGF-I receptor in the regulation of cell-cell adhesion: implications in cancer development and progression. J Cell Physiol (2003) 194:108–16. doi:10.1002/jcp.10207
79. Mauro L, Surmacz E. IGF-I receptor, cell-cell adhesion, tumour development and progression. J Mol Histol (2004) 35:247–53. doi:10.1023/B:HIJO.0000032356.98363.a2
80. Seccareccia E, Brodt P. The role of the insulin-like growth factor-I receptor in malignancy: an update. Growth Horm IGF Res (2012) 22:193–9. doi:10.1016/j.ghir.2012.09.003
81. Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol (1990) 10:464–73.
82. Pietrzkowski Z, Lammers R, Carpenter G, Soderquist AM, Limardo M, Phillips PD, et al. Constitutive expression of insulin-like growth factor 1 and insulin-like growth factor 1 receptor abrogates all requirements for exogenous growth factors. Cell Growth Differ (1992) 3:199–205.
83. Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, et al. Phosphorylated insulin-like growth factor-1/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res (2008) 68:10238–46. doi:10.1158/0008-5472.CAN-08-2755
84. Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer (2004) 4:505–18. doi:10.1038/nrc1387
85. Stewart AJ, Johnson MD, May FE, Westley BR. Role of insulin-like growth factors and the type I insulin-like growth factor receptor in the estrogen-stimulated proliferation of human breast cancer cells. J Biol Chem (1990) 265:21172–8.
86. Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, et al. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol (1999) 13:787–96. doi:10.1210/mend.13.5.0274
87. Mauro L, Salerno M, Panno ML, Bellizzi D, Sisci D, Miglietta A, et al. Estradiol increases IRS-1 gene expression and insulin signaling. Biochem Biophys Res Commun (2001) 288:685–9. doi:10.1006/bbrc.2001.5815
88. Salerno M, Sisci D, Mauro L, Guvakova MA, Andò S, Surmacz E. Insulin receptor substrate 1 is a target for the pure antiestrogen ICI 182,780 in breast cancer cells. Int J Cancer (1999) 81(299–304):1999. doi:10.1002/(SICI)1097-0215(19990412)81:2<299::AID-IJC21>3.3.CO;2-#
89. Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Andò S, et al. Interaction between estrogen receptor alpha and insulin/IGF signaling in breast cancer. Curr Cancer Drug Targets (2008) 8:597–610. doi:10.2174/156800908786241104
90. Gao J, Chang YS, Jallal B, Viner J. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res (2012) 72(1):3–12. doi:10.1158/0008-5472.CAN-11-0550
91. Janssen JA, Varewijck AJ. IGF-IR targeted therapy: past, present and future. Front Endocrinol (2014) 5:224. doi:10.3389/fendo.2014.00224
92. LeRoith D, Roberts CT. The insulin-like growth factor system and cancer. Cancer Lett (2003) 195:127–37. doi:10.1016/S0304-3835(03)00159-9
93. Chabrolle C, Tosca L, Dupont J. Regulation of adiponectin and its receptors in rat ovary by human chorionic gonadotrophin treatment and potential involvement of adiponectin in granulosa cell steroidogenesis. Reproduction (2007) 133:719–31. doi:10.1530/REP-06-0244
94. Ryu J, Galan AK, Xin X, Dong F, Abdul-Ghani MA, Zhou L, et al. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep (2014) 7:1227–38. doi:10.1016/j.celrep.2014.04.006
95. Jia Z, Liu Y, Cui S. Adiponectin induces breast cancer cell migration and growth factor expression. Cell Biochem Biophys (2014) 70:1239–45. doi:10.1007/s12013-014-0047-9
96. Saxena NK, Sharma D. Metastasis suppression by adiponectin: LKB1 rises up to the challenge. Cell Adh Migr (2010) 4:358–62. doi:10.4161/cam.4.3.11541
Keywords: breast cancer, obesity, adiponectin, insulin-like growth factor-I receptor, estrogen receptor
Citation: Mauro L, Naimo GD, Ricchio E, Panno ML and Andò S (2015) Cross-talk between adiponectin and IGF-IR in breast cancer. Front. Oncol. 5:157. doi: 10.3389/fonc.2015.00157
Received: 05 May 2015; Accepted: 29 June 2015;
Published: 15 July 2015
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Giovanni Vitale, Istituto Auxologico Italiano and Università degli Studi di Milano, ItalyWen Zhou, Columbia University, USA
Copyright: © 2015 Mauro, Naimo, Ricchio, Panno and Andò. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastiano Andò, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Via P. Bucci, Arcavacata di Rende, Cosenza 87036, Italy,c2ViYXN0aWFuby5hbmRvQHVuaWNhbC5pdA==