 Takaaki Tsunematsu
Takaaki Tsunematsu Rieko Arakaki
Rieko Arakaki Akiko Yamada
Akiko Yamada Yasusei Kudo
Yasusei Kudo- Department of Oral Molecular Pathology, Institute of Biomedical Sciences, Tokushima University Graduate School, Tokushima, Japan
Aurora-A is a well-known mitotic kinase that regulates mitotic entry, spindle formation, and chromosome maturation as a canonical role. During mitosis, Aurora-A protein is stabilized by its phosphorylation at Ser51 via blocking anaphase-promoting complex/cyclosome-mediated proteolysis. Importantly, overexpression and/or hyperactivation of Aurora-A is involved in tumorigenesis via aneuploidy and genomic instability. Recently, the novel function of Aurora-A for DNA replication has been revealed. In mammalian cells, DNA replication is strictly regulated for preventing over-replication. Pre-replication complex (pre-RC) formation is required for DNA replication as an initiation step occurring at the origin of replication. The timing of pre-RC formation depends on the protein level of geminin, which is controlled by the ubiquitin–proteasome pathway. Aurora-A phosphorylates geminin to prevent its ubiquitin-mediated proteolysis at the mitotic phase to ensure proper pre-RC formation and ensuing DNA replication. In this review, we introduce the novel non-canonical role of Aurora-A in DNA replication.
Introduction
Cyclin-dependent kinases (CDKs) acquire catalytic activity by forming complexes with the cyclins and promote cell cycle progression via phosphorylation of crucial target proteins (1). In mitosis, other kinases such as Aurora-A, Aurora-B, and Aurora-C tightly regulate drastic and rapid morphological changes (2). Aurora-A, the serine/threonine kinase, is essential for several events during mitosis including entry of mitosis, duplication of centrosome, spindle formation, segregation of chromosome, and cytokinesis (3). Aurora-A protein expression peaks during mitosis and decreases at G1 phase in mammalian cells (4). Expression of Aurora-A protein is reduced in late mitosis as a consequence of ubiquitin-mediated proteolysis by anaphase-promoting complex/cyclosome (APC/C) and its co-activator Cdh1 (5–7). It is well known that protein level of various cell cycle regulators is regulated by the ubiquitin–proteasome system (UPS) for proper regulation of cell cycle (1, 8). Aurora-A protein is ubiquitylated via recognition of destruction box (D-box) in the C-terminal by Cdh1 (5) and an additional A-box/DAD motif (9, 10). Furthermore, Ser53 (equivalent to Ser51 in human Aurora-A) of the A-box is phosphorylated during mitosis and this phosphorylation is important for the protein stabilization of Xenopus and human Aurora-A (4, 11, 12).
DNA replication is strictly restricted to occur only once per cell cycle in eukaryotes. To prevent over-replication, replication origins are restricted to activate only once per cell cycle by a mechanism called “licensing.” The assembly of the pre-replication complex (pre-RC) mediates licensing at the origins of replication (13, 14). The assembly of the pre-RC at replication origins can only occur from late mitosis to early G1 with low CDK activity and high activity of APC/C (13, 14). Once pre-RC complexes are assembled, origins are licensed for replication in the ensuing S phase. Geminin is known as a repressor of re-replication and directly binds to chromatin licensing and DNA replication factor 1 (Cdt1) to prevent pre-RC formation (15). Recently, we found that Aurora-A phosphorylates geminin to prevent its ubiquitin-mediated proteolysis at the mitotic phase to ensure proper pre-RC formation and ensuing DNA replication. In this review, we introduce the novel non-canonical role of Aurora-A in DNA replication, notably its initiation process called “licensing.”
Ubiquitin–Proteasome Pathway
The UPS marks proteins for destruction by attaching a polyubiquitin chain and subsequently degrading these proteins via the activity of a multicatalytic enzyme, 26S proteasome (8). Ubiquitin in its monomeric form is a small protein that contains only 76 amino acids. Attachment of a polyubiquitin chain to a substrate requires the concerted action of three enzymes, E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) (8). E1 forms a high-energy thioester bond with ubiquitin in an ATP-dependent reaction, and then the ubiquitin molecule is transferred from E1 to E2. E3 is classified into two distinct classes based on the homology domain: HECT and RING domains. The HECT-type E3s form covalent linkages with ubiquitin from E2 by using a conserved cysteine and subsequently transfer ubiquitin to substrates. On the contrary, the RING-type E3s function as adaptors to facilitate the positioning and transfer of ubiquitin from E2 directly onto the substrate (16). A number of E3s have been found to physically bind to the substrate. Both E2 and E3 proteins exist as large families and the substrate specificity is thought to be defined by different combinations of E2s with different E3 proteins. The human genome encodes only two E1s and less than 40 E2s. Moreover, more than 600 different E3 ligases have been identified in the human genome, allowing for tremendous diversity in substrates (17).
Cell Cycle Control by APC/C Ubiquitin Ligase
The specific, rapid, and timely proteolysis of cell cycle regulators by the UPS represents an important mechanism that ensures proper progression via the cell division cycle in a unidirectional and irreversible manner. The proteolysis of many core components of the cell cycle machinery is controlled by two major classes of ubiquitin ligases, the SCF complex and the APC/C complex, which are RING-type E3s. SCF complexes represent an evolutionarily conserved class of E3 enzymes containing four subunits: Skp1, Cul1, one of many F-box proteins, and Roc1/Rbx1 (18). APC/C is composed of at least a dozen different subunits, namely APC1, APC2, Cdc27/APC3, APC4 APC5, Cdc16/APC6, APC7, Cdc23/APC8, Doc1/APC10, APC11, CDC26, and APC13, but it can only ubiquitylate substrates with the help of a co-activator protein (19). In mammalian cells, APC/C activity is regulated by its binding with the co-activator proteins Cdc20 and Cdh1 during specific periods of the cell cycle (19) (Figure 1). All of these proteins are characterized by the presence of sequence elements, known as the C-box and the IR-tail, which mediate their binding to APC/C (20–22). Cdc20 and Cdh1 contain a C-terminal WD40 domain that is predicted to fold into a propeller-like structure and that is believed to recognize APC/C substrates by interacting with specific recognition elements in these substrates called D-box (RxxL) and KEN-box (KEN) (23–25). In addition to D-box and KEN-box, other motifs, including A-box (RxLxPSN), CRY-box (CRYxPS), GxEN-box (GxEN), Spo13 D-box (LxExxN), and O-box (unknown sequence), are also recognized by APC/C (10, 11, 26–29). The APC/CCdc20 complex is necessary for progression through mitosis and it facilitates the exit from mitosis by inactivating CDK1, and the APC/CCdh1 complex helps to maintain low CDK activity and the G0/G1 state (19, 30) (Figure 1). The APC/CCdh1 and APC/CCdc20 complexes target distinctive specific substrates. Although several recent studies have indicated that both co-activators and APC/C have important roles in substrate recognition, the mechanism by which APC/C recognizes its substrates is unclear. As inappropriate activation of APC/C could cause fatal errors in cell cycle progression, protein degradation via APC/C activation is tightly controlled. APC/C activation is also regulated by APC/C inhibiting proteins, such as mitotic arrest-deficient 2 (Mad2), budding uninhibited by benzimidazole-related 1 (BubR1), budding uninhibited by benzimidazole 1 (Bub1), and early mitotic inhibitor 1 and 2 (Emi1 and Emi2) (19). However, it is also unclear how the timing of degradation of numerous APC/C substrates is regulated. Indeed, substrates are not degraded at the same time by APC/C in spite of activation of APC/C during mitosis. It is unclear why the timing of the ubiquitylation of substrates is different. It was recently demonstrated that (i) phosphorylation and acetylation interfere with ubiquitylation of substrates by APC/C (4, 31–33), (ii) intrinsic regulation of APC/C by substrate ordering is attributable to kinetic differences in the ubiquitylation process (34), and (iii) ubiquitylation of the substrate is inhibited by the binding protein of APC/C (35). Thus, the timing of the ubiquitylation by APC/C may be regulated by protein modification, the processing of ubiquitylation, and binding by an inhibitor.
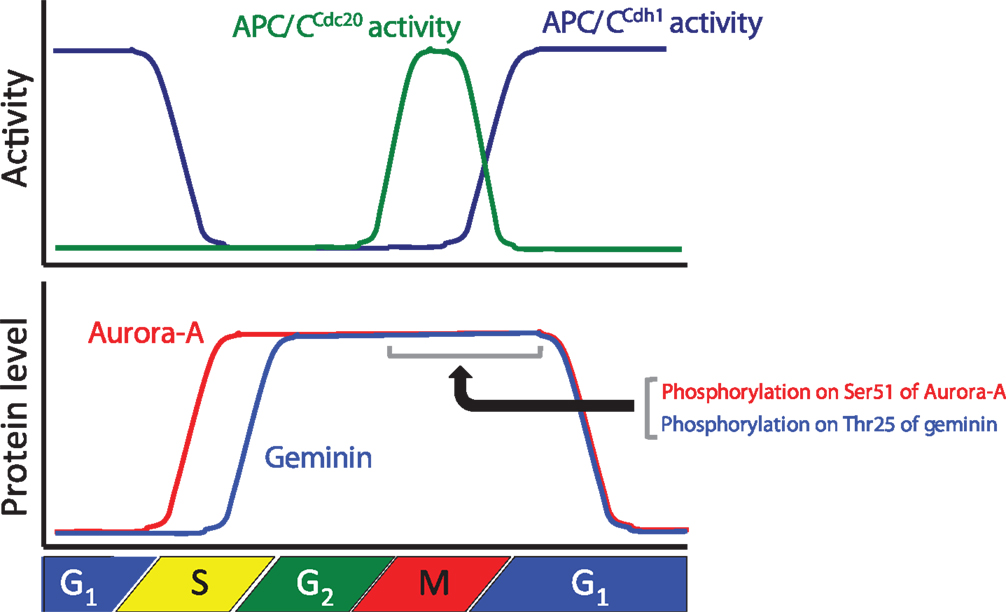
Figure 1. Relationships between the protein levels of Aurora-A and geminin and anaphase-promoting complex/cyclosome (APC/C) activity during cell cycle progression. The graph shows APC/CCdc20 and APC/CCdh1 activities and the protein levels of Aurora-A and geminin during cell cycle progression.
Aurora-A Kinase
Aurora-A is one of the Aurora kinases (Aurora-A, Aurora-B, and Aurora-C), which are highly conserved serine/threonine kinases (36). Aurora-A plays an important role in chromosomal alignment and segregation during mitosis and meiosis (36). Indeed, Aurora-A phosphorylates a large number of substrates, including p53, polo-like kinase-1 (PLK1), CDC25B, BRCA1, centrin, LATS2, GEF-H1, TACC3, NDEL1, HDAC6, Ski, HURP, PP1, TPX2, Eg5, histone H3, CENP-A, CENP-E, CEP192, CEP192, CPEB, LIMK1, LIMK2, SRC, RalA, AKT, and PC2 (37). Aurora-A-mediated phosphorylation of substrates contributes to the activation of kinase activity, protein degradation, protein stabilization, targeting of the centrosome, maturation and separation of centrosome, translocation, and negative regulation of protein function (37). For example, phosphorylation of p53 is involved in its protein degradation (38). Aurora-A activates Plk-1 in G2 phase via the direct phosphorylation of Thr210 (39). Phosphorylation of LATS2, NDEL1, and TACC3 promotes centrosome maturation (40–42). Aurora-A shares high homology between species and it is evolutionarily ancient, with Aurora-A sharing 82% sequence identity between the human and rodent genes. Aurora-A contains a key threonine, the T-loop residue Thr288, within its kinase domain, and Thr 288 is phosphorylated to allow for kinase activity via autophosphorylation (9, 43, 44). The expression level of Aurora-A mRNA and protein is controlled in a cell cycle-dependent manner. Expression of Aurora-A mRNA peaks at G2/M, with protein expression peaking slightly later (45, 46). The promoter of Aurora-A contains specific sequences required for transcription in G2 phase (46–48). Expression of Aurora-A protein peaks during mitosis and decreases in G1 phase as a consequence of ubiquitylation by APC/CCdh1 (4–7) (Figure 1).
The APC/CCdh1 ubiquitin ligase complex recognizes its substrates with either D-Box and/or KEN-box motifs (19, 24, 25). Although Aurora-A has four D-Box motifs and one KEN-box motif, the one of four D-box (D-box at C-terminal) and N-terminal A-box (47RxLxPSN52) are required for the ubiquitylation of human Aurora-A protein (4, 5, 9, 10). Moreover, Xenopus Ser53 (or Ser51 in humans) within the A-box is phosphorylated during mitosis, and this phosphorylation is essential for mitotic-specific stabilization (4, 11, 12) (Figure 2). Similarly as Aurora-A regulation via phosphorylation, CDC6 protein is protected from APC/CCdh1-mediated degradation by virtue of its phosphorylation (31). The phosphorylation sites of CDC6 by cyclin E/CDK2 are located directly adjacent to the D-box, therefore preventing the recognition of CDC6 by APC/CCdh1. In the case of human Aurora-A protein, Ser51 is located far from the D-box, but Ser51 is located in the A-box. However, phosphorylated Aurora-A at Ser51 can bind to Cdh1 (4). The mechanism by which Aurora-A degradation is prevented by phosphorylation on Ser51 is unclear. Other regulators, such as Cdc4/Fbxw7, checkpoint with forkhead and ring finger domain (Chfr), and Aurora-A–interacting protein 1 (AIP), are involved in degradation of Aurora-A protein (49–51).
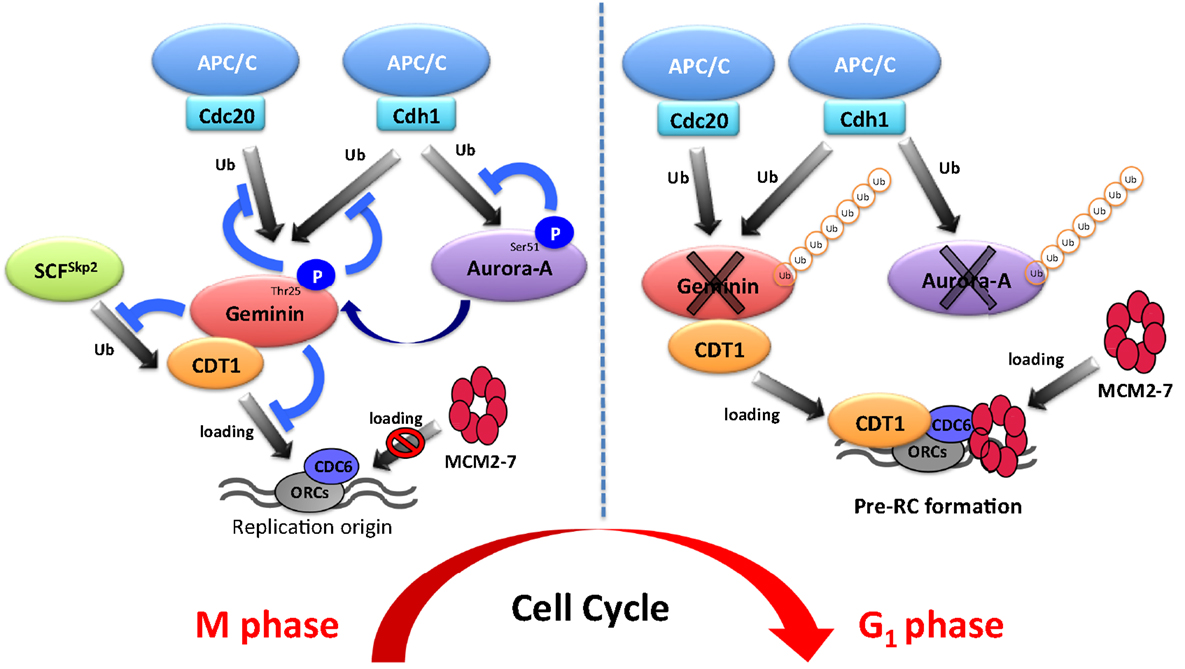
Figure 2. Schematic model of DNA replication via the aurora-A–geminin–CDT1 axis.
It is well known that overexpression of Aurora-A protein is frequently observed in various human cancers, and that aneuploidy, centrosome amplification, and tumorigenic transformation are induced by its overexpression in cultured human and rodent cells (3, 45, 52). Indeed, Aurora-A is mapped to chromosome 20q13.2, a region commonly amplified in human cancers (45, 52, 53). Therefore, Aurora-A overexpression is believed to be caused by gene amplification or transcriptional activation. However, a previous report illustrated that Aurora-A amplification was detected in only 3% of cases, but overexpression of Aurora-A mRNA and protein was observed in more than 60% of cases in hepatocellular carcinomas (54). Similar discrepancies between gene amplification and protein overexpression rates of Aurora-A are reported in other types of cancers, including head and neck, breast, gastric, and ovarian (4, 52, 55, 56). Interestingly, constitutive phosphorylation of Ser51 is observed in head and neck cancer cells with overexpression of Aurora-A protein. As Ser51 phosphorylation inhibits APC/CCdh1-mediated degradation, it is possible that constitutive phosphorylation on Ser51 may induce protein stabilization and its consequent accumulation in cancer cells that exhibit overexpression of Aurora-A protein (4). Importantly, Aurora-A overexpression is considered to promote tumorigenesis via disruption of maintenance of the normal centrosome or chromosome number (3, 57, 58).
DNA Replication and Pre-RC Formation
The ability of a eukaryotic cell to precisely and accurately replicate its DNA is crucial to maintain genome stability. Eukaryotic chromosomes need to be replicated by numerous replication forks that are initiated from replication origins spaced throughout the genome because of the sizes of the chromosomes. Therefore, eukaryotic cells are continually exposed to a risk of over-replication. As described previously, licensing is restricted to occur only once per cell cycle to prevent over-replication. Licensing is the assembly of the pre-RC on replication origins (13, 14). Pre-RC is composed of the origin recognition complex (ORC), cell division cycle 6 (Cdc6), CDT1, and the mini-chromosome maintenance (MCM) proteins (59). Cdc6 and CDT1 are loaded onto replication origins in an ORC-dependent manner during late M and early G1 phase, after which they subsequently recruit MCM proteins to the origins. Pre-RC formation occurs from late mitosis to early G1. The pre-RC is a protein complex composed of ORC, CDC6, CDT1, and MCM2–7, known as putative DNA helicase (13, 14). During late M and early G1, CDC6 and CDT1 bind to replication origins and subsequently induce the recruitment of MCMs to the origins (13, 14). Pre-RC formation is needed for replication in the subsequent S phase. Therefore, it is necessary to prevent re-assembly of the pre-RC during S, G2, and M phase. Two major inhibitory pathways exist to prevent pre-RC re-assembly, namely CDK1- and CDK2-based pathways. CDK1 inactivation during G2 phase induces re-replication through re-assembly of MCMs (60). Consistently, silencing of cyclin A, but not cyclin B, causes re-replication in Drosophila cells (61). Taken together, CDKs suppress re-replication by preventing pre-RC re-assembly. To explain this phenomenon, multiple mechanisms are considered in S and G2 phases. For example, CDT1 and ORC1 are phosphorylated by CDKs, resulting in their degradation in an SCFSkp2-dependent manner (62–65). Additionally, CDKs phosphorylate CDC6 and induce its nuclear export in mammalian cells (66–68). Another pathway involves geminin, known as an inhibitor of DNA replication. Geminin functionally inhibits pre-RC re-assembly through direct binding to CDT1 during S, G2, and M phases, which ensures genome stability and prevents aneuploidy (15). Indeed, ectopic overexpression of geminin suppresses pre-RC formation and subsequently blocks DNA replication (69). In addition, geminin knockdown in mammalian cells induces re-replication (70, 71), indicating that geminin has critical roles in the regulation of replication. Although it seemingly sounds contradictory, geminin stabilizes CDT1 protein during mitosis via preventing its ubiquitin-mediated proteolysis (69). Furthermore, the mitotic depletion of geminin induces CDT1 downregulation and prevents MCM loading in the ensuing G1 phase (69, 72). Thereby, the negative and positive roles of geminin are essential for pre-RC formation, indicating that the protein level of geminin must be strictly controlled for proper DNA replication.
Involvement of Aurora-A in Pre-RC Formation
To ensure pre-RC assembly during late mitosis and early G1 phase, cell cycle-dependent degradation of geminin is caused by the UPS (73). The geminin protein level oscillates during the cell cycle via APC/C-mediated ubiquitylation (69, 73) (Figure 1). Recent analyses at the single-cell level by time-lapse fluorescence microscopy analysis revealed that geminin degradation takes place following cyclin B degradation in late anaphase (74). Although Geminin is a substrate of APC/C, geminin is stable even in mitosis in spite of active APC/C. Indeed, geminin is phosphorylated by Aurora-A on Thr25 to prevent its APC/C-dependent proteolysis in mitosis (69) (Figure 1). Geminin contains the consensus sequences (R-X-S/T-L/V) recognized by Aurora-A as observed in amino acids 23–26 (RRTL) within the D-box of geminin (69). Interestingly, immunoprecipitation analysis revealed that both HA-tagged Cdh1 and HA-tagged Cdc20 interacted with wild-type geminin and Thr25 phospho-defective mutant (gemininT25A) but not with Thr25 phospho-mimicking mutant (gemininT25D), indicating that the inability of gemininT25D to interact with APC/CCdh1 and APC/CCdc20 may explain its resistance to APC/C-dependent proteolysis (69). In general, distinct substrates are specifically recognized by APC/C complex and are tightly degraded to adjust the critical timing (19). In fact, all of substrates of APC/C are not degraded at same time even though APC/C is active. Phosphorylation in CDC6, Aurora-A, and Skp2 as well as geminin protects from APC/C-mediated ubiquitylation (4, 31, 32). In particular, phosphorylation in CDC6, Skp2, or geminin interferes with the binding of APC/CCdh1 (31, 32, 69). We previously have shown that the phosphorylation of human Aurora-A on Ser51 interferes with its ubiquitylation by APCCdh1. Interestingly, constitutive phosphorylation on Ser51 is well correlated with protein overexpression and stabilization in cancer cells (4). As geminin is frequently overexpressed in certain types of human cancer (75, 76), it is interesting to examine if constitutive phosphorylation at Thr25 induces its protein overexpression in cancer. Importantly, stabilized geminin during mitosis ensures pre-RC formation via protecting CDT1 ubiquitylation by SCFSkp2 (69). Aurora-A–geminin–CDT1 axis regulates proper DNA replication (Figure 2).
Conclusion
Aurora-A is a well-known mitotic kinase that regulates mitotic entry, spindle formation, and chromosome maturation as a canonical role. In this review, we shed light on a novel function of Aurora-A for regulating DNA replication via proper formation of the pre-RC. Indeed, Aurora-A phosphorylates geminin to prevent APC/C-mediated proteolysis in mitosis. To ensure pre-RC formation, stabilized mitotic geminin protects CDT1 from SCFSkp2-dependent proteolysis. This novel mechanism controlled by the Aurora-A–geminin–CDT1 axis is essential for proper regulation of DNA replication (Figure 2). Emi1 was identified as a factor inhibiting the function of APC/CCdh1 and it is degraded by SCFβTrcp at early M phase (77–80). It was recently revealed that Emi1 silencing prevents the transition from S to G2 phase by downregulating geminin via APC/C activation (81, 82). Therefore, the protein level of geminin is also regulated by the Emi1-mediated inhibition of APC/CCdh1 activity. During cell cycle progression, strict regulation of the amount of geminin protein is essential for proper DNA replication. The protein level of geminin is strictly determined by Emi1- and Aurora-A-mediated protection from ubiquitylation by APC/C.
A series of periodic kinase reactions by CDKs promote cell cycle progression and the fidelity of cell division is dependent on the accumulation and ordered destruction of critical protein regulators (1). Thus, the UPS contributes to the precise regulation of the cell cycle. The UPS also contributes to the precise regulation of DNA replication via the Aurora-A–geminin–CDT1 axis (Figure 2). Interestingly, Aurora-A protein is also ubiquitylated by APC/CCdh1. It is well known that overexpression and/or hyperactivation of Aurora-A is involved in tumorigenesis via aneuploidy and genomic instability (3). Moreover, Aurora-A is frequently overexpressed in various cancers (3, 43, 52–54). As DNA replication is strictly regulated to prevent over-replication in mammalian cells, disruption of this mechanism may be involved in Aurora-A-mediated tumorigenesis. We suggest that deregulation of DNA replication via Aurora-A–geminin–CDT1 axis can be used as a potential diagnostic and therapeutic target in cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by Grants-in-Aid from the Ministry of Education, Science and Culture of Japan (to YK, NI, and TT) and grants from the Takeda Science Foundation, Tokushima University (pilot study program), and Uehara Memorial Foundation to YK.
References
1. Murray AW. Recycling the cell cycle: cyclins revisited. Cell (2004) 116:221–34. doi:10.1016/S0092-8674(03)01080-8
2. Carmena M, Earnshaw WC. The cellular geography of Aurora kinases. Nat Rev Mol Cell Biol (2003) 4:842–54. doi:10.1038/nrm1245
3. Marumoto T, Zhang D, Saya H. Aurora-A-a guardian of poles. Nat Rev Cancer (2005) 5:42–50. doi:10.1038/nrc1526
4. Kitajima S, Kudo Y, Ogawa I, Tatsuka M, Kawai H, Pagano M, et al. Constitutive phosphorylation of Aurora-A on Ser51 induces its protein overexpression through the inhibition of APC/C-Cdh1 mediated degradation in cancer. PLoS One (2007) 2:e944. doi:10.1371/journal.pone.0000944
5. Arlot-Bonnemains Y, Klotzbucher A, Giet R, Uzbekov R, Bihan R, Prigent C. Identification of a functional destruction box in the Xenopus laevis Aurora-A kinase pEg2. FEBS Lett (2001) 508:149–52. doi:10.1016/S0014-5793(01)03048-4
6. Castro A, Arlot-Bonnemains Y, Vigneron S, Labbe JC, Prigent C, Lorca T. APC/fizzy-related targets Aurora-A kinase for proteolysis. EMBO Rep (2002) 3:457–62. doi:10.1093/embo-reports/kvf095
7. Taguchi S, Honda K, Sugiura K, Yamaguchi A, Furukawa K, Urano T. Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS Lett (2002) 519:59–65. doi:10.1016/S0014-5793(02)02711-4
8. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem (1998) 67:425–79. doi:10.1146/annurev.biochem.67.1.425
9. Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev (2002) 16:2274–85. doi:10.1101/gad.1007302
10. Castro A, Vigneron S, Bernis C, Labbe JC, Prigent C, Lorca T. The D-Box-activating domain (DAD) is a new proteolysis signal that stimulates the silent D-Box sequence of Aurora-A. EMBO Rep (2002) 3:1209–14. doi:10.1093/embo-reports/kvf241
11. Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci U S A (2002) 99:15440–5. doi:10.1073/pnas.202606599
12. Crane R, Kloepfer A, Ruderman JV. Requirements for the destruction of human Aurora-A. J Cell Sci (2004) 117:5975–83. doi:10.1242/jcs.01418
13. Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev (2007) 21:497–518. doi:10.1101/gad.1508907
14. Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol (2005) 6:476–86. doi:10.1038/nrm1663
15. Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science (2000) 290:2309–12. doi:10.1126/science.290.5500.2309
16. Skaar JR, Pagano M. Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol (2009) 21:816–24. doi:10.1016/j.ceb.2009.08.004
17. Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One (2008) 3:e1487. doi:10.1371/journal.pone.0001487
18. Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol (2004) 5:739–51. doi:10.1038/nrm1471
19. Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol (2006) 7:644–56. doi:10.1038/nrm1988
20. Schwab M, Neutzner M, Mocker D, Seufert W. Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J (2001) 20:5165–75. doi:10.1093/emboj/20.18.5165
21. Passmore LA, McCormack EA, Au SW, Paul A, Willison KR, Harper JW, et al. Doc1 mediates the activity of the anaphase-promoting complex by contributing to substrate recognition. EMBO J (2003) 22:786–96. doi:10.1093/emboj/cdg084
22. Vodermaier HC, Gieffers C, Maurer-Stroh S, Eisenhaber F, Peters JM. TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr Biol (2003) 13:1459–68. doi:10.1016/S0960-9822(03)00581-5
23. Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters JM. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell (2005) 18:543–53. doi:10.1016/j.molcel.2005.04.023
24. Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature (1991) 349:132–8. doi:10.1038/349132a0
25. Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev (2000) 14:655–65. doi:10.1101/gad.14.6.655
26. Reis A, Levasseur M, Chang HY, Elliott DJ, Jones KT. The CRY box: a second APCcdh1-dependent degron in mammalian cdc20. EMBO Rep (2006) 7:1040–5. doi:10.1038/sj.embor.7400772
27. Castro A, Vigneron S, Bernis C, Labbe JC, Lorca T. Xkid is degraded in a D-box, KEN-box, and A-box-independent pathway. Mol Cell Biol (2003) 23:4126–38. doi:10.1128/MCB.23.12.4126-4138.2003
28. Sullivan M, Morgan DO. A novel destruction sequence targets the meiotic regulator Spo13 dor anaphase-promoting complex-dependent degradation in anaphase I. J Biol Chem (2007) 282:19710–5. doi:10.1074/jbc.M701507200
29. Araki M, Wharton RP, Tang Z, Yu H, Asano M. Degradation of origin recognition complex large subunit by the anaphase-promoting complex in Drosophila. EMBO J (2003) 22:6115–26. doi:10.1093/emboj/cdg573
30. Guardavaccaro D, Pagano M. Stabilizers and destabilizers controlling cell cycle oscillators. Mol Cell (2006) 22:1–4. doi:10.1016/j.molcel.2006.03.017
31. Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell (2005) 122:915–26. doi:10.1016/j.cell.2005.08.013
32. Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol (2009) 11:420–32. doi:10.1038/ncb1849
33. Choi E, Choe H, Min J, Choi JY, Kim J, Lee H. BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J (2009) 28:2077–89. doi:10.1038/emboj.2009.123
34. Rape M, Reddy SK, Kirschner MW. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell (2006) 124:89–103. doi:10.1016/j.cell.2005.10.032
35. Wolthuis R, Clay-Farrace L, van Zon W, Yekezare M, Koop L, Ogink J, et al. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell (2008) 30:290–302. doi:10.1016/j.molcel.2008.02.027
36. Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell (1995) 81:95–105. doi:10.1016/0092-8674(95)90374-7
37. Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol. Life Sci. (2013) 70:661–87. doi:10.1007/s00018-012-1073-7
38. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by Aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet (2004) 36:55–62. doi:10.1038/ng1279
39. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by Aurora A to promote checkpoint recovery. Nature (2008) 455:119–23. doi:10.1038/nature07185
40. Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, et al. The centrosomeal protein Lats2 is a phosphorylateion target of Aurora-A kinase. Genes Cells (2004) 9:383–97. doi:10.1111/j.1356-9597.2004.00732.x
41. Mori D, Yano Y, Toyo-oka K, Yoshida N, Yamada M, Muramatsu M, et al. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Mol Cell Biol (2007) 27:352–67. doi:10.1128/MCB.00878-06
42. Glet R, McLean D, Descamps S, Lee MJ, Raff JW, Prigent C, et al. Drosophila Aurora A kinase is required to localized D-TACC to centrosomes and to regulate astral microtubules. J Cell Biol (2002) 156:437–51. doi:10.1083/jcb.200108135
43. Ferrari S, Marin O, Pagano MA, Meggio F, Hess D, El-Shemerly M, et al. Aurora-A site specificity: a study with synthetic peptide substrates. Biochem J (2005) 390:293–302. doi:10.1042/BJ20050343
44. Haydon CE, Eyers PA, Aveline-Wolf LD, Resing KA, Maller JL, Ahn NG. Identification of novel phosphorylation sites on Xenopus laevis Aurora A and analysis of phosphopeptide enrichment by immobilized metal-affinity chromatography. Mol Cell Proteomics (2003) 2:1055–67. doi:10.1074/mcp.M300054-MCP200
45. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of Drosophila Aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J (1998) 17:3052–65. doi:10.1093/emboj/17.11.3052
46. Kimura M, Kotani S, Hattori T, Sumi N, Yoshioka T, Todokoro K, et al. Cell cycle-dependent expression and spindle pole localization of a novel human protein kinase, Aik, related to Aurora of Drosophila and yeast Ipl1. J Biol Chem (1997) 272:13766–71. doi:10.1074/jbc.272.21.13766
47. Kimura M, Uchida C, Takano Y, Kitagawa M, Okano Y. Cell cycle-dependent regulation of the human aurora B promoter. Biochem Biophys Res Commun (2004) 316:930–6. doi:10.1016/j.bbrc.2004.01.178
48. Tanaka M, Ueda A, Kanamori H, Ideguchi H, Yang J, Kitajima S, et al. Cell-cycle-dependent regulation of human Aurora A transcription is mediated by periodic repression of E4TF1. J Biol Chem (2002) 277:10719–26. doi:10.1074/jbc.M108252200
49. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature (2004) 432:775–9. doi:10.1038/nature03155
50. Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, et al. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet (2005) 37:401–6. doi:10.1038/ng1538
51. Lim SK, Gopalan G. Aurora-A kinase interacting protein 1 (AURKAIP1) promotes Aurora-A degradation through an alternative ubiquitin-independent pathway. Biochem J (2007) 403:119–27. doi:10.1042/BJ20061272
52. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet (1998) 20:189–93. doi:10.1038/2496
53. Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, et al. Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res (2000) 6:1833–9.
54. Jeng YM, Peng SY, Lin CY, Hsu HC. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res (2004) 10:2065–71. doi:10.1158/1078-0432.CCR-1057-03
55. Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer (2001) 84:824–31. doi:10.1054/bjoc.2000.1684
56. Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res (2003) 9:1420–6.
57. Goepfert TM, Brinkley BR. The centrosome-associated Aurora/Ipl-like kinase family. Curr Top Dev Biol (2000) 49:331–42. doi:10.1016/S0070-2153(99)49016-7
58. Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J (2002) 21:483–92. doi:10.1093/emboj/21.4.483
59. Dutta A, Bell SP. Initiation of DNA replication in eukaryotic cells. Annu Rev Cell Dev Biol (2002) 13:293–332. doi:10.1146/annurev.cellbio.13.1.293
60. Hochegger H, Dejsuphong D, Sonoda E, Saberi A, Rajendra E, Krik J, et al. An essentiall role for Cdk1 in S phase control is revealed via chemicall genetics in vertebrate cells. J Cell Biol (2007) 178:257–68. doi:10.1083/jcb.200702034
61. Mihaylov IS, Kondo T, Jones L, Ryzhikov S, Tanaka J, Zheng J, et al. Control of DNA replication and chromosome ploidy by geminin and cyclin A. Mol Cell Biol (2002) 22:1868–80. doi:10.1128/MCB.22.6.1868-1880.2002
62. Fujita M, Ishimi Y, Nakamura H, Kiyono T, Tsurumi T. Nuclear organization of DNA replication initiation proteins in mammalian cells. J Biol Chem (2002) 277:10354–61. doi:10.1074/jbc.M111398200
63. Méndez J, Zou-Yang XH, Kim SY, Hidaka M, Tansey WP, Stillman B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol Cell (2002) 9:481–91. doi:10.1016/S1097-2765(02)00467-7
64. Liu E, Li X, Yan F, Zhao Q, Wu X. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J Biol Chem (2004) 279:17283–8. doi:10.1074/jbc.C300549200
65. Sugimoto N, Tatsumi Y, Tsurumi T, Matsukage A, Kiyono T, Nishitani H, et al. Cdt1 phosphorylation by cyclin A-dependent kinases negatively regulates its function without affecting geminin binding. J Biol Chem (2004) 279:19691–7. doi:10.1074/jbc.M313175200
66. Pelizon C, Madine MA, Romanowski P, Laskey RA. Unphosphorylatable mutants of Cdc6 disrupt its nuclear export but still support DNA replication once per cell cycle. Genes Dev (2000) 14:2526–33. doi:10.1101/gad.176300
67. Petersen BO, Lukas J, Sorensen CS, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J (1999) 18:396–410. doi:10.1093/emboj/18.2.396
68. Jiang W, Wells NJ, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci U S A (1999) 96:6193–8. doi:10.1073/pnas.96.11.6193
69. Tsunematsu T, Takihara Y, Ishimaru N, Pagano M, Takata T, Kudo Y. Aurora-A controls pre-replicative complex formation and DNA replication by promoting the stabilization of geminin and Cdt1 in mitosis. Nat Commun (2013) 4:1885. doi:10.1038/ncomms2859
70. Ballabeni A, Melixetian M, Zamponi R, Masiero L, Marinoni F, Helin K. Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO J (2004) 23:3122–32. doi:10.1038/sj.emboj.7600314
71. McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell (1998) 93:1043–53. doi:10.1016/S0092-8674(00)81209-X
72. Zhu W, Chen Y, Dutta A. Rereplication by depletion of geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol Cell Biol (2004) 24:7140–50. doi:10.1128/MCB.24.16.7140-7150.2004
73. Melixetian M, Ballabeni A, Masiero L, Gasparini P, Zamponi R, Bartek J, et al. Loss of geminin induces rereplication in the presence of functional p53. J Cell Biol (2004) 165:473–82. doi:10.1083/jcb.200403106
74. Clijsters L, Ogink J, Wolthuis R. The spindle checkpoint, APC/CCdc20, and APC/CCdh1 play distinct roles in connecting mitosis to S phase. J Cell Biol (2013) 201:1013–26. doi:10.1083/jcb.201211019
75. Montanari M, Boninsegna A, Faraglia B, Coco C, Giordano A, Cittadini A, et al. Increased expression of geminin stimulates the growth of mammary epithelial cells and is a frequent event in human tumors. J Cell Physiol (2005) 202:215–22. doi:10.1002/jcp.20120
76. Bravou V, Nishitani H, Song SY, Taraviras S, Varakis J. Expression of the licensing factors, Cdt1 and geminin, in human colon cancer. Int J Oncol (2005) 27:1511–8.
77. Reimann JD, Gardner BE, Margottin-Goguet F, Jackson PK. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev (2001) 15:3278–85. doi:10.1101/gad.945701
78. Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell (2001) 105:645–55. doi:10.1016/S0092-8674(01)00361-0
79. Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JD, Jackson PK. Prophase destruction of Emi1 by the SCFβTrCP/Slimb ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev Cell (2003) 4:813–26. doi:10.1016/S1534-5807(03)00153-9
80. Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, et al. Control of meiotic and mitotic progression by the F box protein β-Trcp1 in vivo. Dev Cell (2003) 4:799–812. doi:10.1016/S1534-5807(03)00154-0
81. Machida YJ, Dutta A. The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev (2007) 21:184–94. doi:10.1101/gad.1495007
Keywords: Aurora-A, geminin, DNA replication, pre-RC, ubiquitin, proteasome, degradation
Citation: Tsunematsu T, Arakaki R, Yamada A, Ishimaru N and Kudo Y (2015) The non-canonical role of Aurora-A in DNA replication. Front. Oncol. 5:187. doi: 10.3389/fonc.2015.00187
Received: 26 May 2015; Accepted: 05 August 2015;
Published: 25 August 2015
Edited by:
Ignacio Perez De Castro, Spanish National Cancer Research Centre, SpainReviewed by:
Daniele Guardavaccaro, Hubrecht Institute, NetherlandsJuan Mendez, Centro Nacional de Investigaciones Oncológicas, Spain
Copyright: © 2015 Tsunematsu, Arakaki, Yamada, Ishimaru and Kudo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasusei Kudo, Department of Oral Molecular Pathology, Institute of Biomedical Sciences, Tokushima University Graduate School, 3-18-15 Kuramoto, Tokushima 770-8504, Japan,eWFzdXNlaUB0b2t1c2hpbWEtdS5hYy5qcA==